User login
Asymptomatic Nodules on Hand, Forearm, and Index Finger
Emergency Department Evaluation of Patients With Intrathecal Pumps
When to Suspect Ischemic Colitis
PE and AMI in Undiagnosed PFO
Acute pulmonary embolism (PE) and acute myocardial infarction (AMI) are common inpatient diagnoses, and are frequently in the differential diagnosis of patients evaluated for chest pain and dyspnea. We present a case with 1 unifying explanation for these entities to coexist. Acute PE with subsequent embolism to the coronary arteries via a patent foramen ovale (PFO) is rare, but the underlying disorder and anatomical variant are common. Of practical significance, hospitalized patients with acute PE and PFO may have up to a 5‐fold increase in morbidity compared to patients with isolated PE.
CASE REPORT
A 79‐year‐old male smoker underwent resection of a recurrent high‐grade liposarcoma of the right upper extremity. He had no antecedent history of coronary artery disease (CAD) or atrial fibrillation, and had no additional vascular risk factors. On postoperative day 2, he developed acute chest pain, dyspnea, and hypoxia. He appeared alert but was diaphoretic and in moderate distress. Pulse was 84 beats per minute, blood pressure 230/120 mm Hg, and oxygen saturation 59% on room air (93% on supplemental oxygen). Heart and lung exam were unremarkable. Neck veins were not distended. Extremity exam was negative for edema, asymmetry, or calf tenderness, and pedal pulses were palpable bilaterally.
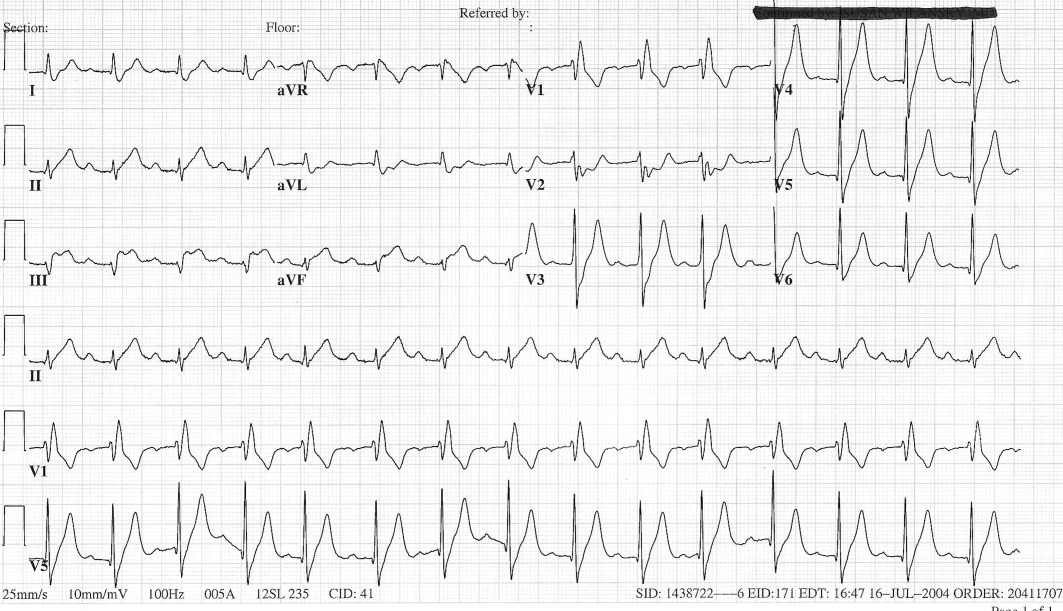
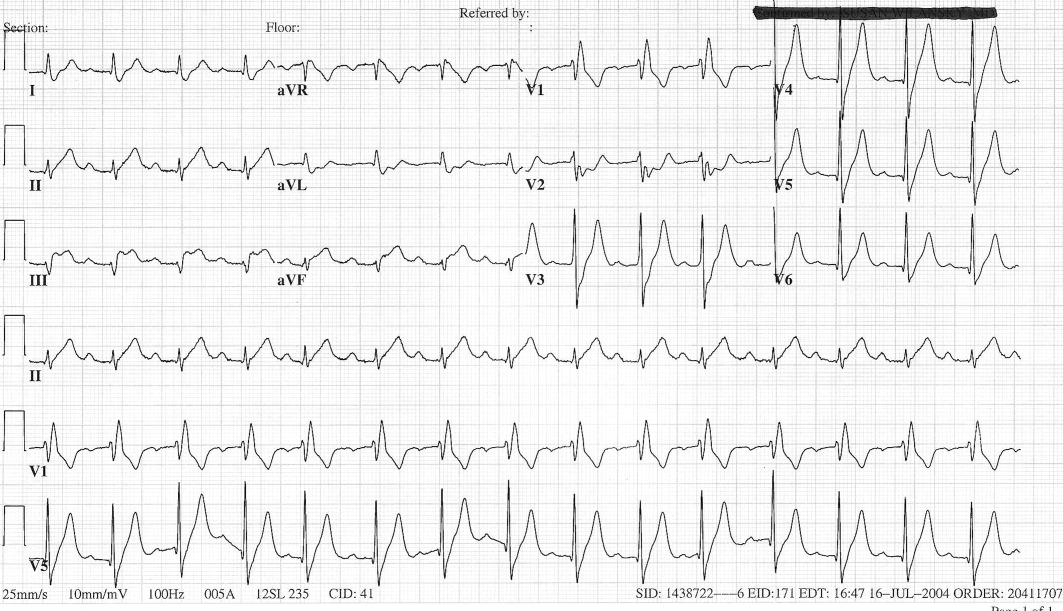
The patient's initial complete blood count, metabolic panel, and cardiac enzymes were within normal limits. Arterial blood gas (on 4‐L nasal cannula) revealed pH of 7.35, partial pressure of arterial oxygen (PaO2) of 65.5 mm Hg, partial pressure of arterial CO2 (PaCO2) of 45.4 mm Hg, and an alveolar‐arterial gradient of 131.3 mm Hg. Electrocardiogram (ECG) (Figure 1) showed an unchanged right bundle branch block, but new 2.5‐mm ST segment elevation in leads V4‐6, III, and aVF, and ST depression in aVL. At this point, the available data suggested either PE with secondary ECG changes or acute ST‐elevation MI with hypoxia. Given the ST elevation in 2 coronary distributions and concern for multivessel CAD, the patient was referred for emergent coronary angiography.
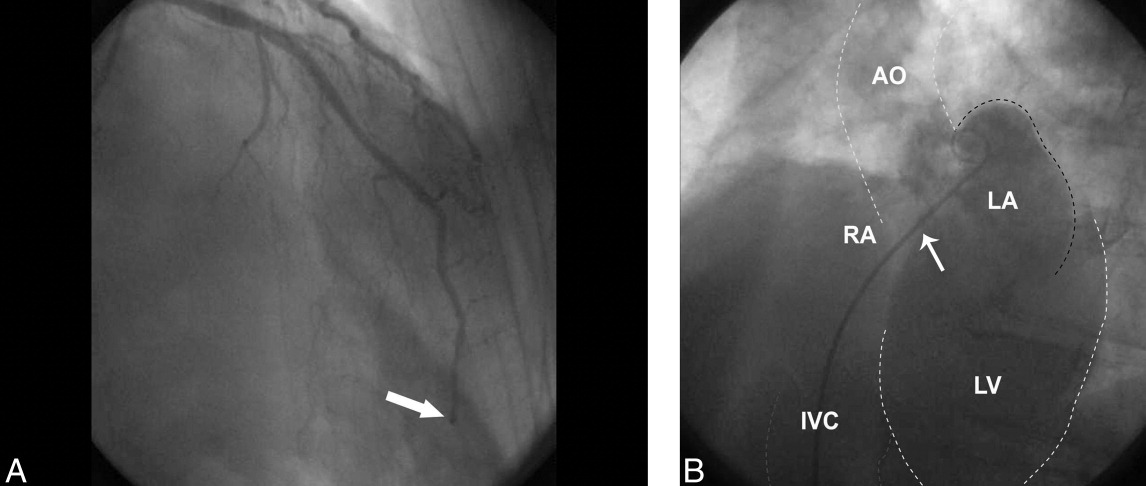
The patient was given aspirin, intravenous unfractionated heparin, and morphine. Left heart catheterization showed an abrupt cutoff in the distal left anterior descending artery (LAD), suggestive of thrombosis secondary to coronary embolism (Figure 2A); angioplasty was not attempted due to the distal location of the occlusion. The posterior descending artery from the right coronary artery was relatively short and the inferior apex was supplied by the distal LAD. The left ventriculogram demonstrated preserved ejection fraction but severe apical hypokinesis, correlating with the occluded vascular territory. The remaining coronary arteries were without significant stenosis. Based on the angiogram findings, paradoxical embolism was suspected. Right heart catheterization identified a previously undiagnosed PFO (Figure 2B); no thrombus was visualized. Right to left shunt was not identified; right ventricular systolic pressure was 46 mm Hg (normal, 15‐25 mm Hg). Subsequent spiral computed tomography (CT) revealed bilateral PEs. It was concluded that the patient had suffered an acute PE, from which the thrombus was able to traverse the PFO and left heart, ultimately entering the LAD, causing the acute embolic MI (Figure 3). ST elevation was present in the inferior leads secondary to the wraparound LAD that supplied the inferior apex, as demonstrated by the wall motion abnormality present on ventriculography.

The patient was felt to be in a hypercoagulable state due to his malignancy and recent surgery; given the diagnosis of an acute PE, no ultrasound was performed to search for a deep vein thrombosis. The patient was transferred to the intensive care unit, continued on intravenous heparin and oxygen and started on oral warfarin. Subsequent hospital course was complicated by aspiration pneumonia and worsening hypoxia, but after a 17‐day hospital stay he was weaned off supplemental oxygen and transferred to an extended care facility with a therapeutic international normalized ratio (INR).
DISCUSSION
PFOs are congenital cardiac lesions that may persist through adulthood1 and are found incidentally in 19% to 36% of the normal population.2 They may range from 1 to 19 mm in size.2, 3 Contrast echocardiography has enabled a simple, accurate, and safe procedure for diagnosis (>5 microbubbles in the left heart cavities within three cardiac cycles after their appearance in the right atrium is considered diagnostic), though transesophageal echocardiography (TEE) is the gold standard.4
First described by Cohnheim in 1877,5 paradoxical embolism refers to the passage of embolic material from the venous to arterial circulation through a cardiac defect such as a PFO. However, definite confirmation of paradoxical embolism essentially requires catching the thrombus in the act of crossing the foramen ovale. Direct observation of this during life is rarely possible, and remains confined to isolated echocardiographic reports.6‐13
In clinical practice, the diagnosis of paradoxical embolism is almost always presumptive and relies on: (1) the occurrence of an arterial thromboembolic event in the absence of atrial fibrillation, left‐sided heart disease, or severe atherosclerosis; (2) the detection of right‐to‐left shunt, usually through a PFO or an atrial septal defect (ASD); and (3) the presence of venous thrombosis or PE.14
Although most patients are asymptomatic, during the past 20 years an association of PFO with stroke, migraine headache, peripheral arterial occlusion, and decompression induced neurologic dysfunction has been suggested.1 The neurological symptoms are proposed to be secondary to passage of small thrombi from the venous system through the PFO into arterial circulation during a transient right‐to‐left shunt. The source of clots cannot be established in most patients; fewer than 10% will have detectable deep vein thrombosis.15 Even less common are paradoxical emboli to the coronary arteries,1 estimated at 5% to 10% of all paradoxical emboli.16
In order for a thrombus to paradoxically embolize across a PFO, an atrial right‐to‐left pressure gradient must be present. Such a gradient occurs in normal individuals during early ventricular systole and with a Valsalva maneuver.17‐19 In a community‐based cohort study conducted to evaluate potential stroke risk, 148 (out of 581) subjects were found to have a PFO by TEE; 84 (57%) had right‐to‐left shunting at rest, and 136 (92%) had right‐to‐left shunting with Valsalva.2 Pathologic instances of pulmonary hypertension such as PE further elevate right‐heart pressures, further promoting intracardiac shunt.
Acute PE in the setting of PFO carries important prognostic implications. Konstantinides et al.20 prospectively evaluated 139 patients with large acute PE, all of whom had pulmonary hypertension and 96% of whom had right ventricular dilatation. They found a high prevalence of PFOs (35%) in this population. Furthermore, the subgroup with both PE and PFO had a high mortality (33% death rate compared with 14% in those without PFO; P = 0.015). When logistic regression analysis was performed, only arterial hypotension (odds ratio [OR] 26.3; P 0.001) and the presence of PFO (OR 11.4; P 0.001) remained significantly correlated with mortality. The authors reported that the presence of a PFO was associated with more than a 5‐fold increase in the adjusted risk of major in‐hospital complications (P 0.001); no specific etiologic factors were proposed for this association.
In general, MI in the absence of CAD is uncommon, comprising 1% to 6% of all cases.21 No cause is found for the majority, but reported etiologies include coronary spasm in 15%, hypercoagulable states in 13%, collagen vascular diseases in 2%, and paradoxical embolism in 2%.22 AMI due to coronary embolism is uncommon, and when it does occur, left‐to‐left emboli in the setting of atrial fibrillation or prosthetic valves are far more common than paradoxical emboli. In an autopsy series of 1,050 patients with MIs, Prizel et al.23 identified only 55 patients with coronary embolism, none of which was right‐sided in origin.
A handful of published case reports documented paradoxical embolism as a cause for AMI.24‐26 Reported cases more often involved an ASD rather than PFO.27, 28 In most cases, diagnosis was made postmortem, though in a comprehensive review of the literature, Meier‐Ewert et al.13 identified 8 cases of AMI due to paradoxical embolism being diagnosed antemortem. Paradoxical emboli have been identified in all major divisions of the epicardial circulation, though involvement of the left coronary circulation is more common than the right.16
It is well‐established that the prevalence of PFO in patients with cryptogenic stroke is significantly higher than in the general population,1 and Crump et al.21 examined a case‐matched series of 18 patients with AMI who had little to no CAD (30% stenosis) to see if the frequency of PFO was similarly higher in this group. Each group had identical frequency of PFO (28%; P = NS). The authors concluded that PFO is unlikely to contribute significantly to AMI. However, this study was limited by the small number of patients and the fact that transthoracic echocardiography (TTE) was used instead of TEE for the diagnosis of PFO.
The definitive diagnosis of AMI due to paradoxical embolism requires angiographic findings consistent with embolic occlusion (such as the cutoff sign in a distal coronary artery we observed in Figure 2A), cardiac defects predisposing to paradoxical emboli (such as PFO), and evidence of a venous source for thromboembolism. Alternatively, diagnosis can be made via direct visualization of emboli in the coronary arteries by TEE, or by autopsy.
Although electrocardiography is essential in the diagnosis and treatment of MI, it has the potential to be deceptive. Acute pulmonary hypertension caused by PE may be accompanied by ST elevation in inferior leads II, III, and aVF in a pseudoinfarction pattern mimicking AMI.29 This ECG abnormality probably reflects reciprocal changes of inferoposterior ischemia from right ventricular pressure overloading. However, clearly distinguishing between pseudoinfarction and true inferior infarction in the setting of PE requires coronary angiography.
Regarding therapy, acute treatment of PE is well‐established and consists of at least 5 days of therapeutically‐dosed heparin product that overlaps with therapeutic warfarin anticoagulation. Management of the PFO and coronary embolism is less clear; there are no guidelines for treatment of coronary embolism. Management strategies should focus on treatment of acute ischemia as well as prevention of future emboli, principally anticoagulation. Because the pathogenesis of AMI in this setting is drastically different from MI secondary to atherosclerosis, there is neither a biological basis nor clinical data to suggest benefit from initiation of beta‐blockers, aspirin, angiotensin converting enzyme inhibitors, or statins.
While studies have been done, and are underway to address optimal management of PFO in the setting of both stroke and migraine headache, to our knowledge, no such trials have addressed PFO and MI. Mehan et al.30 reported 2 cases of AMI caused by suspected paradoxical embolism, and in both cases, instant percutaneous closure of PFO was undertaken. However, there are no data to support or refute such an intervention in this particular setting.
- ,,, et al.Patent foramen ovale: current pathology, pathophysiology, and clinical status.J Am Coll Cardiol.2005;46:1768–1776.
- ,,, et al.Prevalence of potential risk factors for stroke assessed by transesophageal echocardiography and carotid ultrasonography: the SPARC Study.Mayo Clin Proc.1999;74:862–869.
- ,,.Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts.Mayo Clin Proc.1984;59:17–20.
- ,,.Diagnosis of an anatomically and physiologically significant patent foramen ovale.Echocardiography.2006;23:810–815.
- .Thrombose und Embolie: Vorlesung über allgemeine Pathologie. Vol.1.Berlin:Hirschwald;1877;134.
- ,,,,,.Impending paradoxical embolism from atrial thrombus: correct diagnosis by transesophageal echocardiography and prevention by surgery.J Am Coll Cardiol.1985;5:1002–1004.
- ,,.Diagnosis of paradoxic embolism by transesophageal echocardiography.Am Heart J.1991;121:1552–1554.
- ,,,,,.Impending paradoxical embolism: echocardiographic diagnosis of an intracardiac thrombus crossing a patent foramen ovale.Am Heart J.1991;122(3 Pt 1):859–862.
- ,,,,,.Cryptogenic ischemic stroke and paradoxical embolism: should a patent foramen ovale be closed? Case report and literature review.Angiology.2001;52:793–799.
- ,,,,.Thrombus‐in‐transit and paradoxical embolism.J Am Soc Echocardiogr.2002;15:1021–1022.
- ,,,.Caught in the act: impending paradoxical embolism.Asian Cardiovasc Thorac Ann.2002;10:342–343.
- .Paradoxical embolism to the left main coronary artery: visualization by transesophageal echocardiography.J Am Soc Echocardiogr.2002;15:1417–1418.
- ,,,,,.Paradoxical embolism in the left main coronary artery: diagnosis by transesophageal echocardiography.Mayo Clin Proc.2003;78:103–106.
- .Specifics of patent foramen ovale.Adv Neurol.2003;92:197–202.
- ,,, et al.Frequency of deep vein thrombosis in patients with patent foramen ovale and ischemic stroke or transient ischemic attack.Am J Cardiol.1997;80:1066–1069.
- ,.Paradoxical coronary embolism: a rare cause of acute myocardial infarction.Rev Cardiovasc Med.2003;4:107–111.
- ,.Positive contrast echocardiography in patients with patent foramen ovale and normal right heart hemodynamics.Am J Cardiol.1982;49:1806–1809.
- ,,, et al.Contrast echocardiographic visualization of cough‐induced right to left shunt through a patent foramen ovale.J Am Coll Cardiol.1984;4:587–594.
- ,,,,.Transesophageal echocardiographic demonstration of distinct mechanisms for right to left shunting across a patent foramen ovale in the absence of pulmonary hypertension.J Am Coll Cardiol.1991;18:1112–1117.
- ,,,,,.Patent foramen ovale is an important predictor of adverse outcome in patients with major acute pulmonary embolism.Circulation.1998;97:1946–1951.
- ,,,.Prevalence of patent foramen ovale in patients with acute myocardial infarction and angiographically normal coronary arteries.Am J Cardiol.2000;85:1368–1370.
- ,,,,,.Clinical characteristics, aetiological factors and long‐term prognosis of myocardial infarction with an absolutely normal coronary angiogram; a 3‐year follow‐up study of 91 patients.Eur Heart J.2001;22:1459–1465.
- ,,.Coronary artery embolism and myocardial infarction.Ann Intern Med.1978;88:155–161.
- ,..Myocardial infarction due to a paradoxical embolism.Am J Med.1969;47:995–998.
- ,,,,,.[Myocardial infarction caused by probable paradoxical embolism and aneurysm of the interatrial septum].Presse Med.1996;25:907 [French].
- ,,.Acute myocardial infarction probably caused by paradoxical embolus in a pregnant woman.Heart.2004;90:e12.
- ,,,,.A case of acute pulmonary embolism and acute myocardial infarction with suspected paradoxical embolism after laparoscopic surgery.Heart Vessels.1999;4:197–200.
- ,,,,,.Acute myocardial infarction caused by paradoxical coronary embolization in a patient with a patent foramen ovale.J Am Soc Echocardiogr.2001;14:1227–1229.
- .ECG manifestations of selected extracardiac diseases.Emerg Med Clin N Am.2006;24:133–143.
- ,,,.Instant percutaneous closure of patent foramen ovale in patients with acute myocardial infarction and normal coronary arteries.Catheter Cardiovasc Interv.2006;67:279–282.
Acute pulmonary embolism (PE) and acute myocardial infarction (AMI) are common inpatient diagnoses, and are frequently in the differential diagnosis of patients evaluated for chest pain and dyspnea. We present a case with 1 unifying explanation for these entities to coexist. Acute PE with subsequent embolism to the coronary arteries via a patent foramen ovale (PFO) is rare, but the underlying disorder and anatomical variant are common. Of practical significance, hospitalized patients with acute PE and PFO may have up to a 5‐fold increase in morbidity compared to patients with isolated PE.
CASE REPORT
A 79‐year‐old male smoker underwent resection of a recurrent high‐grade liposarcoma of the right upper extremity. He had no antecedent history of coronary artery disease (CAD) or atrial fibrillation, and had no additional vascular risk factors. On postoperative day 2, he developed acute chest pain, dyspnea, and hypoxia. He appeared alert but was diaphoretic and in moderate distress. Pulse was 84 beats per minute, blood pressure 230/120 mm Hg, and oxygen saturation 59% on room air (93% on supplemental oxygen). Heart and lung exam were unremarkable. Neck veins were not distended. Extremity exam was negative for edema, asymmetry, or calf tenderness, and pedal pulses were palpable bilaterally.
The patient's initial complete blood count, metabolic panel, and cardiac enzymes were within normal limits. Arterial blood gas (on 4‐L nasal cannula) revealed pH of 7.35, partial pressure of arterial oxygen (PaO2) of 65.5 mm Hg, partial pressure of arterial CO2 (PaCO2) of 45.4 mm Hg, and an alveolar‐arterial gradient of 131.3 mm Hg. Electrocardiogram (ECG) (Figure 1) showed an unchanged right bundle branch block, but new 2.5‐mm ST segment elevation in leads V4‐6, III, and aVF, and ST depression in aVL. At this point, the available data suggested either PE with secondary ECG changes or acute ST‐elevation MI with hypoxia. Given the ST elevation in 2 coronary distributions and concern for multivessel CAD, the patient was referred for emergent coronary angiography.
The patient was given aspirin, intravenous unfractionated heparin, and morphine. Left heart catheterization showed an abrupt cutoff in the distal left anterior descending artery (LAD), suggestive of thrombosis secondary to coronary embolism (Figure 2A); angioplasty was not attempted due to the distal location of the occlusion. The posterior descending artery from the right coronary artery was relatively short and the inferior apex was supplied by the distal LAD. The left ventriculogram demonstrated preserved ejection fraction but severe apical hypokinesis, correlating with the occluded vascular territory. The remaining coronary arteries were without significant stenosis. Based on the angiogram findings, paradoxical embolism was suspected. Right heart catheterization identified a previously undiagnosed PFO (Figure 2B); no thrombus was visualized. Right to left shunt was not identified; right ventricular systolic pressure was 46 mm Hg (normal, 15‐25 mm Hg). Subsequent spiral computed tomography (CT) revealed bilateral PEs. It was concluded that the patient had suffered an acute PE, from which the thrombus was able to traverse the PFO and left heart, ultimately entering the LAD, causing the acute embolic MI (Figure 3). ST elevation was present in the inferior leads secondary to the wraparound LAD that supplied the inferior apex, as demonstrated by the wall motion abnormality present on ventriculography.
The patient was felt to be in a hypercoagulable state due to his malignancy and recent surgery; given the diagnosis of an acute PE, no ultrasound was performed to search for a deep vein thrombosis. The patient was transferred to the intensive care unit, continued on intravenous heparin and oxygen and started on oral warfarin. Subsequent hospital course was complicated by aspiration pneumonia and worsening hypoxia, but after a 17‐day hospital stay he was weaned off supplemental oxygen and transferred to an extended care facility with a therapeutic international normalized ratio (INR).
DISCUSSION
PFOs are congenital cardiac lesions that may persist through adulthood1 and are found incidentally in 19% to 36% of the normal population.2 They may range from 1 to 19 mm in size.2, 3 Contrast echocardiography has enabled a simple, accurate, and safe procedure for diagnosis (>5 microbubbles in the left heart cavities within three cardiac cycles after their appearance in the right atrium is considered diagnostic), though transesophageal echocardiography (TEE) is the gold standard.4
First described by Cohnheim in 1877,5 paradoxical embolism refers to the passage of embolic material from the venous to arterial circulation through a cardiac defect such as a PFO. However, definite confirmation of paradoxical embolism essentially requires catching the thrombus in the act of crossing the foramen ovale. Direct observation of this during life is rarely possible, and remains confined to isolated echocardiographic reports.6‐13
In clinical practice, the diagnosis of paradoxical embolism is almost always presumptive and relies on: (1) the occurrence of an arterial thromboembolic event in the absence of atrial fibrillation, left‐sided heart disease, or severe atherosclerosis; (2) the detection of right‐to‐left shunt, usually through a PFO or an atrial septal defect (ASD); and (3) the presence of venous thrombosis or PE.14
Although most patients are asymptomatic, during the past 20 years an association of PFO with stroke, migraine headache, peripheral arterial occlusion, and decompression induced neurologic dysfunction has been suggested.1 The neurological symptoms are proposed to be secondary to passage of small thrombi from the venous system through the PFO into arterial circulation during a transient right‐to‐left shunt. The source of clots cannot be established in most patients; fewer than 10% will have detectable deep vein thrombosis.15 Even less common are paradoxical emboli to the coronary arteries,1 estimated at 5% to 10% of all paradoxical emboli.16
In order for a thrombus to paradoxically embolize across a PFO, an atrial right‐to‐left pressure gradient must be present. Such a gradient occurs in normal individuals during early ventricular systole and with a Valsalva maneuver.17‐19 In a community‐based cohort study conducted to evaluate potential stroke risk, 148 (out of 581) subjects were found to have a PFO by TEE; 84 (57%) had right‐to‐left shunting at rest, and 136 (92%) had right‐to‐left shunting with Valsalva.2 Pathologic instances of pulmonary hypertension such as PE further elevate right‐heart pressures, further promoting intracardiac shunt.
Acute PE in the setting of PFO carries important prognostic implications. Konstantinides et al.20 prospectively evaluated 139 patients with large acute PE, all of whom had pulmonary hypertension and 96% of whom had right ventricular dilatation. They found a high prevalence of PFOs (35%) in this population. Furthermore, the subgroup with both PE and PFO had a high mortality (33% death rate compared with 14% in those without PFO; P = 0.015). When logistic regression analysis was performed, only arterial hypotension (odds ratio [OR] 26.3; P 0.001) and the presence of PFO (OR 11.4; P 0.001) remained significantly correlated with mortality. The authors reported that the presence of a PFO was associated with more than a 5‐fold increase in the adjusted risk of major in‐hospital complications (P 0.001); no specific etiologic factors were proposed for this association.
In general, MI in the absence of CAD is uncommon, comprising 1% to 6% of all cases.21 No cause is found for the majority, but reported etiologies include coronary spasm in 15%, hypercoagulable states in 13%, collagen vascular diseases in 2%, and paradoxical embolism in 2%.22 AMI due to coronary embolism is uncommon, and when it does occur, left‐to‐left emboli in the setting of atrial fibrillation or prosthetic valves are far more common than paradoxical emboli. In an autopsy series of 1,050 patients with MIs, Prizel et al.23 identified only 55 patients with coronary embolism, none of which was right‐sided in origin.
A handful of published case reports documented paradoxical embolism as a cause for AMI.24‐26 Reported cases more often involved an ASD rather than PFO.27, 28 In most cases, diagnosis was made postmortem, though in a comprehensive review of the literature, Meier‐Ewert et al.13 identified 8 cases of AMI due to paradoxical embolism being diagnosed antemortem. Paradoxical emboli have been identified in all major divisions of the epicardial circulation, though involvement of the left coronary circulation is more common than the right.16
It is well‐established that the prevalence of PFO in patients with cryptogenic stroke is significantly higher than in the general population,1 and Crump et al.21 examined a case‐matched series of 18 patients with AMI who had little to no CAD (30% stenosis) to see if the frequency of PFO was similarly higher in this group. Each group had identical frequency of PFO (28%; P = NS). The authors concluded that PFO is unlikely to contribute significantly to AMI. However, this study was limited by the small number of patients and the fact that transthoracic echocardiography (TTE) was used instead of TEE for the diagnosis of PFO.
The definitive diagnosis of AMI due to paradoxical embolism requires angiographic findings consistent with embolic occlusion (such as the cutoff sign in a distal coronary artery we observed in Figure 2A), cardiac defects predisposing to paradoxical emboli (such as PFO), and evidence of a venous source for thromboembolism. Alternatively, diagnosis can be made via direct visualization of emboli in the coronary arteries by TEE, or by autopsy.
Although electrocardiography is essential in the diagnosis and treatment of MI, it has the potential to be deceptive. Acute pulmonary hypertension caused by PE may be accompanied by ST elevation in inferior leads II, III, and aVF in a pseudoinfarction pattern mimicking AMI.29 This ECG abnormality probably reflects reciprocal changes of inferoposterior ischemia from right ventricular pressure overloading. However, clearly distinguishing between pseudoinfarction and true inferior infarction in the setting of PE requires coronary angiography.
Regarding therapy, acute treatment of PE is well‐established and consists of at least 5 days of therapeutically‐dosed heparin product that overlaps with therapeutic warfarin anticoagulation. Management of the PFO and coronary embolism is less clear; there are no guidelines for treatment of coronary embolism. Management strategies should focus on treatment of acute ischemia as well as prevention of future emboli, principally anticoagulation. Because the pathogenesis of AMI in this setting is drastically different from MI secondary to atherosclerosis, there is neither a biological basis nor clinical data to suggest benefit from initiation of beta‐blockers, aspirin, angiotensin converting enzyme inhibitors, or statins.
While studies have been done, and are underway to address optimal management of PFO in the setting of both stroke and migraine headache, to our knowledge, no such trials have addressed PFO and MI. Mehan et al.30 reported 2 cases of AMI caused by suspected paradoxical embolism, and in both cases, instant percutaneous closure of PFO was undertaken. However, there are no data to support or refute such an intervention in this particular setting.
Acute pulmonary embolism (PE) and acute myocardial infarction (AMI) are common inpatient diagnoses, and are frequently in the differential diagnosis of patients evaluated for chest pain and dyspnea. We present a case with 1 unifying explanation for these entities to coexist. Acute PE with subsequent embolism to the coronary arteries via a patent foramen ovale (PFO) is rare, but the underlying disorder and anatomical variant are common. Of practical significance, hospitalized patients with acute PE and PFO may have up to a 5‐fold increase in morbidity compared to patients with isolated PE.
CASE REPORT
A 79‐year‐old male smoker underwent resection of a recurrent high‐grade liposarcoma of the right upper extremity. He had no antecedent history of coronary artery disease (CAD) or atrial fibrillation, and had no additional vascular risk factors. On postoperative day 2, he developed acute chest pain, dyspnea, and hypoxia. He appeared alert but was diaphoretic and in moderate distress. Pulse was 84 beats per minute, blood pressure 230/120 mm Hg, and oxygen saturation 59% on room air (93% on supplemental oxygen). Heart and lung exam were unremarkable. Neck veins were not distended. Extremity exam was negative for edema, asymmetry, or calf tenderness, and pedal pulses were palpable bilaterally.
The patient's initial complete blood count, metabolic panel, and cardiac enzymes were within normal limits. Arterial blood gas (on 4‐L nasal cannula) revealed pH of 7.35, partial pressure of arterial oxygen (PaO2) of 65.5 mm Hg, partial pressure of arterial CO2 (PaCO2) of 45.4 mm Hg, and an alveolar‐arterial gradient of 131.3 mm Hg. Electrocardiogram (ECG) (Figure 1) showed an unchanged right bundle branch block, but new 2.5‐mm ST segment elevation in leads V4‐6, III, and aVF, and ST depression in aVL. At this point, the available data suggested either PE with secondary ECG changes or acute ST‐elevation MI with hypoxia. Given the ST elevation in 2 coronary distributions and concern for multivessel CAD, the patient was referred for emergent coronary angiography.
The patient was given aspirin, intravenous unfractionated heparin, and morphine. Left heart catheterization showed an abrupt cutoff in the distal left anterior descending artery (LAD), suggestive of thrombosis secondary to coronary embolism (Figure 2A); angioplasty was not attempted due to the distal location of the occlusion. The posterior descending artery from the right coronary artery was relatively short and the inferior apex was supplied by the distal LAD. The left ventriculogram demonstrated preserved ejection fraction but severe apical hypokinesis, correlating with the occluded vascular territory. The remaining coronary arteries were without significant stenosis. Based on the angiogram findings, paradoxical embolism was suspected. Right heart catheterization identified a previously undiagnosed PFO (Figure 2B); no thrombus was visualized. Right to left shunt was not identified; right ventricular systolic pressure was 46 mm Hg (normal, 15‐25 mm Hg). Subsequent spiral computed tomography (CT) revealed bilateral PEs. It was concluded that the patient had suffered an acute PE, from which the thrombus was able to traverse the PFO and left heart, ultimately entering the LAD, causing the acute embolic MI (Figure 3). ST elevation was present in the inferior leads secondary to the wraparound LAD that supplied the inferior apex, as demonstrated by the wall motion abnormality present on ventriculography.
The patient was felt to be in a hypercoagulable state due to his malignancy and recent surgery; given the diagnosis of an acute PE, no ultrasound was performed to search for a deep vein thrombosis. The patient was transferred to the intensive care unit, continued on intravenous heparin and oxygen and started on oral warfarin. Subsequent hospital course was complicated by aspiration pneumonia and worsening hypoxia, but after a 17‐day hospital stay he was weaned off supplemental oxygen and transferred to an extended care facility with a therapeutic international normalized ratio (INR).
DISCUSSION
PFOs are congenital cardiac lesions that may persist through adulthood1 and are found incidentally in 19% to 36% of the normal population.2 They may range from 1 to 19 mm in size.2, 3 Contrast echocardiography has enabled a simple, accurate, and safe procedure for diagnosis (>5 microbubbles in the left heart cavities within three cardiac cycles after their appearance in the right atrium is considered diagnostic), though transesophageal echocardiography (TEE) is the gold standard.4
First described by Cohnheim in 1877,5 paradoxical embolism refers to the passage of embolic material from the venous to arterial circulation through a cardiac defect such as a PFO. However, definite confirmation of paradoxical embolism essentially requires catching the thrombus in the act of crossing the foramen ovale. Direct observation of this during life is rarely possible, and remains confined to isolated echocardiographic reports.6‐13
In clinical practice, the diagnosis of paradoxical embolism is almost always presumptive and relies on: (1) the occurrence of an arterial thromboembolic event in the absence of atrial fibrillation, left‐sided heart disease, or severe atherosclerosis; (2) the detection of right‐to‐left shunt, usually through a PFO or an atrial septal defect (ASD); and (3) the presence of venous thrombosis or PE.14
Although most patients are asymptomatic, during the past 20 years an association of PFO with stroke, migraine headache, peripheral arterial occlusion, and decompression induced neurologic dysfunction has been suggested.1 The neurological symptoms are proposed to be secondary to passage of small thrombi from the venous system through the PFO into arterial circulation during a transient right‐to‐left shunt. The source of clots cannot be established in most patients; fewer than 10% will have detectable deep vein thrombosis.15 Even less common are paradoxical emboli to the coronary arteries,1 estimated at 5% to 10% of all paradoxical emboli.16
In order for a thrombus to paradoxically embolize across a PFO, an atrial right‐to‐left pressure gradient must be present. Such a gradient occurs in normal individuals during early ventricular systole and with a Valsalva maneuver.17‐19 In a community‐based cohort study conducted to evaluate potential stroke risk, 148 (out of 581) subjects were found to have a PFO by TEE; 84 (57%) had right‐to‐left shunting at rest, and 136 (92%) had right‐to‐left shunting with Valsalva.2 Pathologic instances of pulmonary hypertension such as PE further elevate right‐heart pressures, further promoting intracardiac shunt.
Acute PE in the setting of PFO carries important prognostic implications. Konstantinides et al.20 prospectively evaluated 139 patients with large acute PE, all of whom had pulmonary hypertension and 96% of whom had right ventricular dilatation. They found a high prevalence of PFOs (35%) in this population. Furthermore, the subgroup with both PE and PFO had a high mortality (33% death rate compared with 14% in those without PFO; P = 0.015). When logistic regression analysis was performed, only arterial hypotension (odds ratio [OR] 26.3; P 0.001) and the presence of PFO (OR 11.4; P 0.001) remained significantly correlated with mortality. The authors reported that the presence of a PFO was associated with more than a 5‐fold increase in the adjusted risk of major in‐hospital complications (P 0.001); no specific etiologic factors were proposed for this association.
In general, MI in the absence of CAD is uncommon, comprising 1% to 6% of all cases.21 No cause is found for the majority, but reported etiologies include coronary spasm in 15%, hypercoagulable states in 13%, collagen vascular diseases in 2%, and paradoxical embolism in 2%.22 AMI due to coronary embolism is uncommon, and when it does occur, left‐to‐left emboli in the setting of atrial fibrillation or prosthetic valves are far more common than paradoxical emboli. In an autopsy series of 1,050 patients with MIs, Prizel et al.23 identified only 55 patients with coronary embolism, none of which was right‐sided in origin.
A handful of published case reports documented paradoxical embolism as a cause for AMI.24‐26 Reported cases more often involved an ASD rather than PFO.27, 28 In most cases, diagnosis was made postmortem, though in a comprehensive review of the literature, Meier‐Ewert et al.13 identified 8 cases of AMI due to paradoxical embolism being diagnosed antemortem. Paradoxical emboli have been identified in all major divisions of the epicardial circulation, though involvement of the left coronary circulation is more common than the right.16
It is well‐established that the prevalence of PFO in patients with cryptogenic stroke is significantly higher than in the general population,1 and Crump et al.21 examined a case‐matched series of 18 patients with AMI who had little to no CAD (30% stenosis) to see if the frequency of PFO was similarly higher in this group. Each group had identical frequency of PFO (28%; P = NS). The authors concluded that PFO is unlikely to contribute significantly to AMI. However, this study was limited by the small number of patients and the fact that transthoracic echocardiography (TTE) was used instead of TEE for the diagnosis of PFO.
The definitive diagnosis of AMI due to paradoxical embolism requires angiographic findings consistent with embolic occlusion (such as the cutoff sign in a distal coronary artery we observed in Figure 2A), cardiac defects predisposing to paradoxical emboli (such as PFO), and evidence of a venous source for thromboembolism. Alternatively, diagnosis can be made via direct visualization of emboli in the coronary arteries by TEE, or by autopsy.
Although electrocardiography is essential in the diagnosis and treatment of MI, it has the potential to be deceptive. Acute pulmonary hypertension caused by PE may be accompanied by ST elevation in inferior leads II, III, and aVF in a pseudoinfarction pattern mimicking AMI.29 This ECG abnormality probably reflects reciprocal changes of inferoposterior ischemia from right ventricular pressure overloading. However, clearly distinguishing between pseudoinfarction and true inferior infarction in the setting of PE requires coronary angiography.
Regarding therapy, acute treatment of PE is well‐established and consists of at least 5 days of therapeutically‐dosed heparin product that overlaps with therapeutic warfarin anticoagulation. Management of the PFO and coronary embolism is less clear; there are no guidelines for treatment of coronary embolism. Management strategies should focus on treatment of acute ischemia as well as prevention of future emboli, principally anticoagulation. Because the pathogenesis of AMI in this setting is drastically different from MI secondary to atherosclerosis, there is neither a biological basis nor clinical data to suggest benefit from initiation of beta‐blockers, aspirin, angiotensin converting enzyme inhibitors, or statins.
While studies have been done, and are underway to address optimal management of PFO in the setting of both stroke and migraine headache, to our knowledge, no such trials have addressed PFO and MI. Mehan et al.30 reported 2 cases of AMI caused by suspected paradoxical embolism, and in both cases, instant percutaneous closure of PFO was undertaken. However, there are no data to support or refute such an intervention in this particular setting.
- ,,, et al.Patent foramen ovale: current pathology, pathophysiology, and clinical status.J Am Coll Cardiol.2005;46:1768–1776.
- ,,, et al.Prevalence of potential risk factors for stroke assessed by transesophageal echocardiography and carotid ultrasonography: the SPARC Study.Mayo Clin Proc.1999;74:862–869.
- ,,.Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts.Mayo Clin Proc.1984;59:17–20.
- ,,.Diagnosis of an anatomically and physiologically significant patent foramen ovale.Echocardiography.2006;23:810–815.
- .Thrombose und Embolie: Vorlesung über allgemeine Pathologie. Vol.1.Berlin:Hirschwald;1877;134.
- ,,,,,.Impending paradoxical embolism from atrial thrombus: correct diagnosis by transesophageal echocardiography and prevention by surgery.J Am Coll Cardiol.1985;5:1002–1004.
- ,,.Diagnosis of paradoxic embolism by transesophageal echocardiography.Am Heart J.1991;121:1552–1554.
- ,,,,,.Impending paradoxical embolism: echocardiographic diagnosis of an intracardiac thrombus crossing a patent foramen ovale.Am Heart J.1991;122(3 Pt 1):859–862.
- ,,,,,.Cryptogenic ischemic stroke and paradoxical embolism: should a patent foramen ovale be closed? Case report and literature review.Angiology.2001;52:793–799.
- ,,,,.Thrombus‐in‐transit and paradoxical embolism.J Am Soc Echocardiogr.2002;15:1021–1022.
- ,,,.Caught in the act: impending paradoxical embolism.Asian Cardiovasc Thorac Ann.2002;10:342–343.
- .Paradoxical embolism to the left main coronary artery: visualization by transesophageal echocardiography.J Am Soc Echocardiogr.2002;15:1417–1418.
- ,,,,,.Paradoxical embolism in the left main coronary artery: diagnosis by transesophageal echocardiography.Mayo Clin Proc.2003;78:103–106.
- .Specifics of patent foramen ovale.Adv Neurol.2003;92:197–202.
- ,,, et al.Frequency of deep vein thrombosis in patients with patent foramen ovale and ischemic stroke or transient ischemic attack.Am J Cardiol.1997;80:1066–1069.
- ,.Paradoxical coronary embolism: a rare cause of acute myocardial infarction.Rev Cardiovasc Med.2003;4:107–111.
- ,.Positive contrast echocardiography in patients with patent foramen ovale and normal right heart hemodynamics.Am J Cardiol.1982;49:1806–1809.
- ,,, et al.Contrast echocardiographic visualization of cough‐induced right to left shunt through a patent foramen ovale.J Am Coll Cardiol.1984;4:587–594.
- ,,,,.Transesophageal echocardiographic demonstration of distinct mechanisms for right to left shunting across a patent foramen ovale in the absence of pulmonary hypertension.J Am Coll Cardiol.1991;18:1112–1117.
- ,,,,,.Patent foramen ovale is an important predictor of adverse outcome in patients with major acute pulmonary embolism.Circulation.1998;97:1946–1951.
- ,,,.Prevalence of patent foramen ovale in patients with acute myocardial infarction and angiographically normal coronary arteries.Am J Cardiol.2000;85:1368–1370.
- ,,,,,.Clinical characteristics, aetiological factors and long‐term prognosis of myocardial infarction with an absolutely normal coronary angiogram; a 3‐year follow‐up study of 91 patients.Eur Heart J.2001;22:1459–1465.
- ,,.Coronary artery embolism and myocardial infarction.Ann Intern Med.1978;88:155–161.
- ,..Myocardial infarction due to a paradoxical embolism.Am J Med.1969;47:995–998.
- ,,,,,.[Myocardial infarction caused by probable paradoxical embolism and aneurysm of the interatrial septum].Presse Med.1996;25:907 [French].
- ,,.Acute myocardial infarction probably caused by paradoxical embolus in a pregnant woman.Heart.2004;90:e12.
- ,,,,.A case of acute pulmonary embolism and acute myocardial infarction with suspected paradoxical embolism after laparoscopic surgery.Heart Vessels.1999;4:197–200.
- ,,,,,.Acute myocardial infarction caused by paradoxical coronary embolization in a patient with a patent foramen ovale.J Am Soc Echocardiogr.2001;14:1227–1229.
- .ECG manifestations of selected extracardiac diseases.Emerg Med Clin N Am.2006;24:133–143.
- ,,,.Instant percutaneous closure of patent foramen ovale in patients with acute myocardial infarction and normal coronary arteries.Catheter Cardiovasc Interv.2006;67:279–282.
- ,,, et al.Patent foramen ovale: current pathology, pathophysiology, and clinical status.J Am Coll Cardiol.2005;46:1768–1776.
- ,,, et al.Prevalence of potential risk factors for stroke assessed by transesophageal echocardiography and carotid ultrasonography: the SPARC Study.Mayo Clin Proc.1999;74:862–869.
- ,,.Incidence and size of patent foramen ovale during the first 10 decades of life: an autopsy study of 965 normal hearts.Mayo Clin Proc.1984;59:17–20.
- ,,.Diagnosis of an anatomically and physiologically significant patent foramen ovale.Echocardiography.2006;23:810–815.
- .Thrombose und Embolie: Vorlesung über allgemeine Pathologie. Vol.1.Berlin:Hirschwald;1877;134.
- ,,,,,.Impending paradoxical embolism from atrial thrombus: correct diagnosis by transesophageal echocardiography and prevention by surgery.J Am Coll Cardiol.1985;5:1002–1004.
- ,,.Diagnosis of paradoxic embolism by transesophageal echocardiography.Am Heart J.1991;121:1552–1554.
- ,,,,,.Impending paradoxical embolism: echocardiographic diagnosis of an intracardiac thrombus crossing a patent foramen ovale.Am Heart J.1991;122(3 Pt 1):859–862.
- ,,,,,.Cryptogenic ischemic stroke and paradoxical embolism: should a patent foramen ovale be closed? Case report and literature review.Angiology.2001;52:793–799.
- ,,,,.Thrombus‐in‐transit and paradoxical embolism.J Am Soc Echocardiogr.2002;15:1021–1022.
- ,,,.Caught in the act: impending paradoxical embolism.Asian Cardiovasc Thorac Ann.2002;10:342–343.
- .Paradoxical embolism to the left main coronary artery: visualization by transesophageal echocardiography.J Am Soc Echocardiogr.2002;15:1417–1418.
- ,,,,,.Paradoxical embolism in the left main coronary artery: diagnosis by transesophageal echocardiography.Mayo Clin Proc.2003;78:103–106.
- .Specifics of patent foramen ovale.Adv Neurol.2003;92:197–202.
- ,,, et al.Frequency of deep vein thrombosis in patients with patent foramen ovale and ischemic stroke or transient ischemic attack.Am J Cardiol.1997;80:1066–1069.
- ,.Paradoxical coronary embolism: a rare cause of acute myocardial infarction.Rev Cardiovasc Med.2003;4:107–111.
- ,.Positive contrast echocardiography in patients with patent foramen ovale and normal right heart hemodynamics.Am J Cardiol.1982;49:1806–1809.
- ,,, et al.Contrast echocardiographic visualization of cough‐induced right to left shunt through a patent foramen ovale.J Am Coll Cardiol.1984;4:587–594.
- ,,,,.Transesophageal echocardiographic demonstration of distinct mechanisms for right to left shunting across a patent foramen ovale in the absence of pulmonary hypertension.J Am Coll Cardiol.1991;18:1112–1117.
- ,,,,,.Patent foramen ovale is an important predictor of adverse outcome in patients with major acute pulmonary embolism.Circulation.1998;97:1946–1951.
- ,,,.Prevalence of patent foramen ovale in patients with acute myocardial infarction and angiographically normal coronary arteries.Am J Cardiol.2000;85:1368–1370.
- ,,,,,.Clinical characteristics, aetiological factors and long‐term prognosis of myocardial infarction with an absolutely normal coronary angiogram; a 3‐year follow‐up study of 91 patients.Eur Heart J.2001;22:1459–1465.
- ,,.Coronary artery embolism and myocardial infarction.Ann Intern Med.1978;88:155–161.
- ,..Myocardial infarction due to a paradoxical embolism.Am J Med.1969;47:995–998.
- ,,,,,.[Myocardial infarction caused by probable paradoxical embolism and aneurysm of the interatrial septum].Presse Med.1996;25:907 [French].
- ,,.Acute myocardial infarction probably caused by paradoxical embolus in a pregnant woman.Heart.2004;90:e12.
- ,,,,.A case of acute pulmonary embolism and acute myocardial infarction with suspected paradoxical embolism after laparoscopic surgery.Heart Vessels.1999;4:197–200.
- ,,,,,.Acute myocardial infarction caused by paradoxical coronary embolization in a patient with a patent foramen ovale.J Am Soc Echocardiogr.2001;14:1227–1229.
- .ECG manifestations of selected extracardiac diseases.Emerg Med Clin N Am.2006;24:133–143.
- ,,,.Instant percutaneous closure of patent foramen ovale in patients with acute myocardial infarction and normal coronary arteries.Catheter Cardiovasc Interv.2006;67:279–282.
PRESsed for time
A 36‐year‐old woman was admitted after new‐onset Hseizures. She had been diagnosed with breast cancer 5 years prior to admission. At that time, she underwent left radical mastectomy and lymph node dissection. Lymph nodes were positive for metastatic disease with negative HER‐2‐Neu and positive estrogen and progesterone receptors. She was treated with docetaxel and tamoxifen but subsequently developed metastatic left hip lesions and was treated with letrozole and anastrozole. Three years later, scans revealed further metastatic disease to the liver, lung, and vertebral column. She was subsequently treated with capecitabine, until further disease progression led to the use of carboplatin and paclitaxel. Seven months prior to admission, her cancer was progressing and she was switched to doxorubicin, gemcitabine, and bevacizumab. Six weeks prior to admission, both positron emission tomography (PET) and computed tomography (CT) scan of her whole body and magnetic resonance imaging (MRI) of the brain illustrated significant improvement. Her last dose of bevacizumab was given 3 weeks prior to her admission.
Two weeks prior to admission, patient reported new‐onset daily headache. These were often localized in the occipital region. She reported some associated nausea and occasional emesis. Subsequently, she developed photophobia and phonophobia. On seeking outpatient treatment for her headache, it was noted that her systolic blood pressure had increased from a baseline of 100 mm Hg to 170 mm Hg. On the day prior to admission, she reported severe headache and several episodes of emesis and later that evening had a witnessed tonic‐clonic seizure.
The patient presented to an outside hospital and had an unremarkable noncontrast CT scan of her brain. An examination of her cerebrospinal fluid revealed negative gram stain, and a normal white blood cell count and protein level. She was treated with lorazepam, phenytoin, and decadron. On becoming more alert, she insisted on going home, where she later developed recurrent headache and presented to our emergency room.
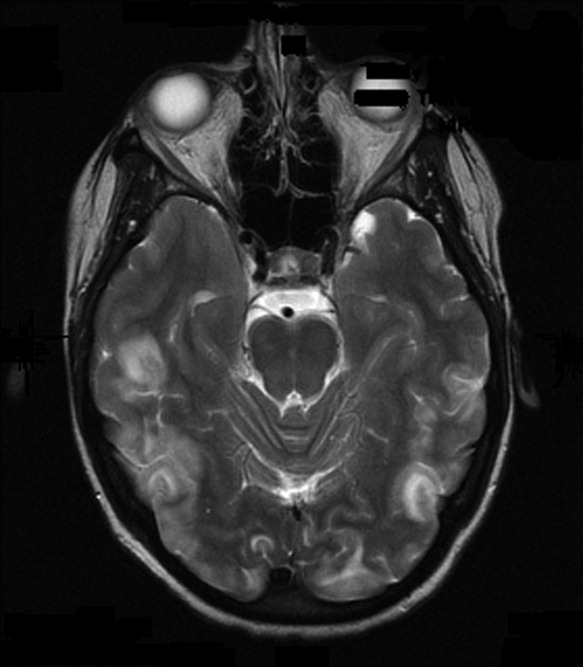
On admission to our service, she was noted to be confused and irritable, and unable to provide any history. Her exam revealed a blood pressure of 143/102 mmHg. No localizing neurologic signs were noted and her laboratory values were normal. After sedation, MRI of the brain was obtained (Figure 1). This revealed diffuse and patchy gyriform hyperintensity of the white matter, most consistent with posterior reversible encephalopathy syndrome (PRES).
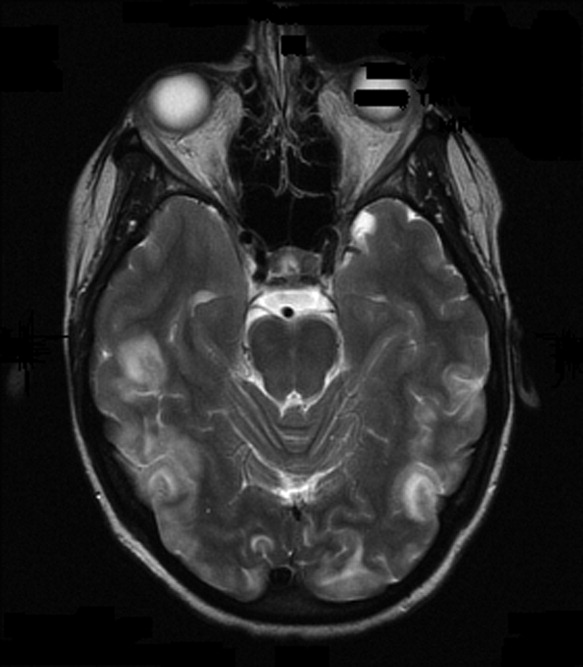
Upon reflection, the patient had new onset hypertension that coincided with the initiation and dosing of bevacizumab. Bevacizumab, an antineoplastic agent, is a recombinant humanized monoclonal antibody that binds to and neutralizes vascular endothelial growth factor, thereby preventing angiogenesis.1 It is known to cause grade 3 hypertension in a minority of patients. Therefore, it was postulated that the patient's persistent blood pressure elevation resulted in vasogenic brain edema, precipitating her seizure. Subsequent to the diagnosis, her blood pressure was aggressively controlled with oral enalapril, metoprolol, triamterene/hydrochlorothiazide, and hydralazine. By hospital day 7, her headache had subsided and her altered mental status had resolved. She had no further episodes of seizures and bevacizumab was discontinued.
PRES has a distinct constellation of clinical symptoms and radiologic findings. The name PRES is a misnomer, as this syndrome is not always reversible, nor is it restricted to the white matter or to the posterior areas of the brain.2 It is hypothesized that a sudden rise in blood pressure leads to elevations in intracranial pressure, which exceeds the brain's autoregulatory mechanisms. This subsequently leads to transudation of fluid into the brain parenchyma. Interestingly, it appears that it is not the absolute level of systolic blood pressure that is critical in the development of PRES, but the rate of change in blood pressure. Hence, patients with chronic hypertension have developed adaptive vascular changes that protect them from this type of parenchymal damage.
PRES has gained increasing recognition due to the use of immunosuppressive and chemotherapeutic medications in organ transplant and oncology patients. Drugs such as cyclosporine, tacrolimus, fludarabine, vincristine, cisplatin, cytarabine, interferon‐alpha, interleukin, antiretroviral therapy, erythropoietin, granulocyte stimulating factor, and intravenous immunoglobulin have all been implicated.3 In addition to increasing blood pressure, these agents likely cause direct toxic injury to the brain, disrupting the blood‐brain barrier and resulting in subsequent edema. Other conditions associated with PRES include renal disease, vasculitis, endocrine disorders, porphyria, cocaine or amphetamine abuse, and stimulant abuse.
Clinically, PRES can present as headache, altered mental status, confusion, drowsiness progressing to stupor, emesis, abnormal visual perceptions, visual neglect, cortical blindness, difficulty with memory and concentration, brisk deep tendon reflexes, weakness, ataxia, and seizure activity. PRES has a characteristic appearance on neuroimaging that differentiates it from other forms of hypertensive encephalopathy. Edema of the white or gray matter in the posterior cerebral hemispheres, particularly the bilateral parietooccipital regions, is seen. PRES can also diffusely involve the brain stem, cerebellum, basal ganglia, and the frontal lobes. Abnormalities on neuroimaging are often symmetric but clinical manifestations can be asymmetric. MRI and CT scans can both be utilized for characterization of PRES.4
There are currently no published guidelines for the management of PRES. Expert opinion suggests removing the underlying cause and aggressively treating the hypertension.5 Furthermore, initiation and duration of antiepileptics remains controversial. After aggressive blood pressure control, resolution of findings on neuroimaging studies are expected anywhere from 8 days to 17 months.
Timely recognition of PRES is critical for prevention of further neurologic compromise. Immediate discontinuation of offending agents, as well as aggressive treatment of blood pressure, is the cornerstone treatment for PRES. In the future, a better understanding of the pathophysiology of PRES can lead to improved diagnostic and management options.
- ,,.Reversible posterior leukoencephalopathy syndrome and bevacizumab.N Engl J Med.2006;354(9):980–982.
- ,,, et al.A reversible posterior leukoencephalopathy syndrome.N Engl J Med.1996;334(8):494–500.
- ,,,,,.Reversible posterior leukoencephalopathy syndrome complicating cytotoxic chemotherapy for hematologic malignancies.Am J Hematol.2004;77(1):72–76.
- ,,, et al.Posterior leukoencephalopathy without severe hypertension: utility of diffusion‐weighted MRI.Neurology.1998;51(5):1369–1376.
- .Posterior leukoencephalopathy syndrome.Postgrad Med J.2001;77(903):24–28.
A 36‐year‐old woman was admitted after new‐onset Hseizures. She had been diagnosed with breast cancer 5 years prior to admission. At that time, she underwent left radical mastectomy and lymph node dissection. Lymph nodes were positive for metastatic disease with negative HER‐2‐Neu and positive estrogen and progesterone receptors. She was treated with docetaxel and tamoxifen but subsequently developed metastatic left hip lesions and was treated with letrozole and anastrozole. Three years later, scans revealed further metastatic disease to the liver, lung, and vertebral column. She was subsequently treated with capecitabine, until further disease progression led to the use of carboplatin and paclitaxel. Seven months prior to admission, her cancer was progressing and she was switched to doxorubicin, gemcitabine, and bevacizumab. Six weeks prior to admission, both positron emission tomography (PET) and computed tomography (CT) scan of her whole body and magnetic resonance imaging (MRI) of the brain illustrated significant improvement. Her last dose of bevacizumab was given 3 weeks prior to her admission.
Two weeks prior to admission, patient reported new‐onset daily headache. These were often localized in the occipital region. She reported some associated nausea and occasional emesis. Subsequently, she developed photophobia and phonophobia. On seeking outpatient treatment for her headache, it was noted that her systolic blood pressure had increased from a baseline of 100 mm Hg to 170 mm Hg. On the day prior to admission, she reported severe headache and several episodes of emesis and later that evening had a witnessed tonic‐clonic seizure.
The patient presented to an outside hospital and had an unremarkable noncontrast CT scan of her brain. An examination of her cerebrospinal fluid revealed negative gram stain, and a normal white blood cell count and protein level. She was treated with lorazepam, phenytoin, and decadron. On becoming more alert, she insisted on going home, where she later developed recurrent headache and presented to our emergency room.
On admission to our service, she was noted to be confused and irritable, and unable to provide any history. Her exam revealed a blood pressure of 143/102 mmHg. No localizing neurologic signs were noted and her laboratory values were normal. After sedation, MRI of the brain was obtained (Figure 1). This revealed diffuse and patchy gyriform hyperintensity of the white matter, most consistent with posterior reversible encephalopathy syndrome (PRES).
Upon reflection, the patient had new onset hypertension that coincided with the initiation and dosing of bevacizumab. Bevacizumab, an antineoplastic agent, is a recombinant humanized monoclonal antibody that binds to and neutralizes vascular endothelial growth factor, thereby preventing angiogenesis.1 It is known to cause grade 3 hypertension in a minority of patients. Therefore, it was postulated that the patient's persistent blood pressure elevation resulted in vasogenic brain edema, precipitating her seizure. Subsequent to the diagnosis, her blood pressure was aggressively controlled with oral enalapril, metoprolol, triamterene/hydrochlorothiazide, and hydralazine. By hospital day 7, her headache had subsided and her altered mental status had resolved. She had no further episodes of seizures and bevacizumab was discontinued.
PRES has a distinct constellation of clinical symptoms and radiologic findings. The name PRES is a misnomer, as this syndrome is not always reversible, nor is it restricted to the white matter or to the posterior areas of the brain.2 It is hypothesized that a sudden rise in blood pressure leads to elevations in intracranial pressure, which exceeds the brain's autoregulatory mechanisms. This subsequently leads to transudation of fluid into the brain parenchyma. Interestingly, it appears that it is not the absolute level of systolic blood pressure that is critical in the development of PRES, but the rate of change in blood pressure. Hence, patients with chronic hypertension have developed adaptive vascular changes that protect them from this type of parenchymal damage.
PRES has gained increasing recognition due to the use of immunosuppressive and chemotherapeutic medications in organ transplant and oncology patients. Drugs such as cyclosporine, tacrolimus, fludarabine, vincristine, cisplatin, cytarabine, interferon‐alpha, interleukin, antiretroviral therapy, erythropoietin, granulocyte stimulating factor, and intravenous immunoglobulin have all been implicated.3 In addition to increasing blood pressure, these agents likely cause direct toxic injury to the brain, disrupting the blood‐brain barrier and resulting in subsequent edema. Other conditions associated with PRES include renal disease, vasculitis, endocrine disorders, porphyria, cocaine or amphetamine abuse, and stimulant abuse.
Clinically, PRES can present as headache, altered mental status, confusion, drowsiness progressing to stupor, emesis, abnormal visual perceptions, visual neglect, cortical blindness, difficulty with memory and concentration, brisk deep tendon reflexes, weakness, ataxia, and seizure activity. PRES has a characteristic appearance on neuroimaging that differentiates it from other forms of hypertensive encephalopathy. Edema of the white or gray matter in the posterior cerebral hemispheres, particularly the bilateral parietooccipital regions, is seen. PRES can also diffusely involve the brain stem, cerebellum, basal ganglia, and the frontal lobes. Abnormalities on neuroimaging are often symmetric but clinical manifestations can be asymmetric. MRI and CT scans can both be utilized for characterization of PRES.4
There are currently no published guidelines for the management of PRES. Expert opinion suggests removing the underlying cause and aggressively treating the hypertension.5 Furthermore, initiation and duration of antiepileptics remains controversial. After aggressive blood pressure control, resolution of findings on neuroimaging studies are expected anywhere from 8 days to 17 months.
Timely recognition of PRES is critical for prevention of further neurologic compromise. Immediate discontinuation of offending agents, as well as aggressive treatment of blood pressure, is the cornerstone treatment for PRES. In the future, a better understanding of the pathophysiology of PRES can lead to improved diagnostic and management options.
A 36‐year‐old woman was admitted after new‐onset Hseizures. She had been diagnosed with breast cancer 5 years prior to admission. At that time, she underwent left radical mastectomy and lymph node dissection. Lymph nodes were positive for metastatic disease with negative HER‐2‐Neu and positive estrogen and progesterone receptors. She was treated with docetaxel and tamoxifen but subsequently developed metastatic left hip lesions and was treated with letrozole and anastrozole. Three years later, scans revealed further metastatic disease to the liver, lung, and vertebral column. She was subsequently treated with capecitabine, until further disease progression led to the use of carboplatin and paclitaxel. Seven months prior to admission, her cancer was progressing and she was switched to doxorubicin, gemcitabine, and bevacizumab. Six weeks prior to admission, both positron emission tomography (PET) and computed tomography (CT) scan of her whole body and magnetic resonance imaging (MRI) of the brain illustrated significant improvement. Her last dose of bevacizumab was given 3 weeks prior to her admission.
Two weeks prior to admission, patient reported new‐onset daily headache. These were often localized in the occipital region. She reported some associated nausea and occasional emesis. Subsequently, she developed photophobia and phonophobia. On seeking outpatient treatment for her headache, it was noted that her systolic blood pressure had increased from a baseline of 100 mm Hg to 170 mm Hg. On the day prior to admission, she reported severe headache and several episodes of emesis and later that evening had a witnessed tonic‐clonic seizure.
The patient presented to an outside hospital and had an unremarkable noncontrast CT scan of her brain. An examination of her cerebrospinal fluid revealed negative gram stain, and a normal white blood cell count and protein level. She was treated with lorazepam, phenytoin, and decadron. On becoming more alert, she insisted on going home, where she later developed recurrent headache and presented to our emergency room.
On admission to our service, she was noted to be confused and irritable, and unable to provide any history. Her exam revealed a blood pressure of 143/102 mmHg. No localizing neurologic signs were noted and her laboratory values were normal. After sedation, MRI of the brain was obtained (Figure 1). This revealed diffuse and patchy gyriform hyperintensity of the white matter, most consistent with posterior reversible encephalopathy syndrome (PRES).
Upon reflection, the patient had new onset hypertension that coincided with the initiation and dosing of bevacizumab. Bevacizumab, an antineoplastic agent, is a recombinant humanized monoclonal antibody that binds to and neutralizes vascular endothelial growth factor, thereby preventing angiogenesis.1 It is known to cause grade 3 hypertension in a minority of patients. Therefore, it was postulated that the patient's persistent blood pressure elevation resulted in vasogenic brain edema, precipitating her seizure. Subsequent to the diagnosis, her blood pressure was aggressively controlled with oral enalapril, metoprolol, triamterene/hydrochlorothiazide, and hydralazine. By hospital day 7, her headache had subsided and her altered mental status had resolved. She had no further episodes of seizures and bevacizumab was discontinued.
PRES has a distinct constellation of clinical symptoms and radiologic findings. The name PRES is a misnomer, as this syndrome is not always reversible, nor is it restricted to the white matter or to the posterior areas of the brain.2 It is hypothesized that a sudden rise in blood pressure leads to elevations in intracranial pressure, which exceeds the brain's autoregulatory mechanisms. This subsequently leads to transudation of fluid into the brain parenchyma. Interestingly, it appears that it is not the absolute level of systolic blood pressure that is critical in the development of PRES, but the rate of change in blood pressure. Hence, patients with chronic hypertension have developed adaptive vascular changes that protect them from this type of parenchymal damage.
PRES has gained increasing recognition due to the use of immunosuppressive and chemotherapeutic medications in organ transplant and oncology patients. Drugs such as cyclosporine, tacrolimus, fludarabine, vincristine, cisplatin, cytarabine, interferon‐alpha, interleukin, antiretroviral therapy, erythropoietin, granulocyte stimulating factor, and intravenous immunoglobulin have all been implicated.3 In addition to increasing blood pressure, these agents likely cause direct toxic injury to the brain, disrupting the blood‐brain barrier and resulting in subsequent edema. Other conditions associated with PRES include renal disease, vasculitis, endocrine disorders, porphyria, cocaine or amphetamine abuse, and stimulant abuse.
Clinically, PRES can present as headache, altered mental status, confusion, drowsiness progressing to stupor, emesis, abnormal visual perceptions, visual neglect, cortical blindness, difficulty with memory and concentration, brisk deep tendon reflexes, weakness, ataxia, and seizure activity. PRES has a characteristic appearance on neuroimaging that differentiates it from other forms of hypertensive encephalopathy. Edema of the white or gray matter in the posterior cerebral hemispheres, particularly the bilateral parietooccipital regions, is seen. PRES can also diffusely involve the brain stem, cerebellum, basal ganglia, and the frontal lobes. Abnormalities on neuroimaging are often symmetric but clinical manifestations can be asymmetric. MRI and CT scans can both be utilized for characterization of PRES.4
There are currently no published guidelines for the management of PRES. Expert opinion suggests removing the underlying cause and aggressively treating the hypertension.5 Furthermore, initiation and duration of antiepileptics remains controversial. After aggressive blood pressure control, resolution of findings on neuroimaging studies are expected anywhere from 8 days to 17 months.
Timely recognition of PRES is critical for prevention of further neurologic compromise. Immediate discontinuation of offending agents, as well as aggressive treatment of blood pressure, is the cornerstone treatment for PRES. In the future, a better understanding of the pathophysiology of PRES can lead to improved diagnostic and management options.
- ,,.Reversible posterior leukoencephalopathy syndrome and bevacizumab.N Engl J Med.2006;354(9):980–982.
- ,,, et al.A reversible posterior leukoencephalopathy syndrome.N Engl J Med.1996;334(8):494–500.
- ,,,,,.Reversible posterior leukoencephalopathy syndrome complicating cytotoxic chemotherapy for hematologic malignancies.Am J Hematol.2004;77(1):72–76.
- ,,, et al.Posterior leukoencephalopathy without severe hypertension: utility of diffusion‐weighted MRI.Neurology.1998;51(5):1369–1376.
- .Posterior leukoencephalopathy syndrome.Postgrad Med J.2001;77(903):24–28.
- ,,.Reversible posterior leukoencephalopathy syndrome and bevacizumab.N Engl J Med.2006;354(9):980–982.
- ,,, et al.A reversible posterior leukoencephalopathy syndrome.N Engl J Med.1996;334(8):494–500.
- ,,,,,.Reversible posterior leukoencephalopathy syndrome complicating cytotoxic chemotherapy for hematologic malignancies.Am J Hematol.2004;77(1):72–76.
- ,,, et al.Posterior leukoencephalopathy without severe hypertension: utility of diffusion‐weighted MRI.Neurology.1998;51(5):1369–1376.
- .Posterior leukoencephalopathy syndrome.Postgrad Med J.2001;77(903):24–28.