User login
Pulmonary Embolism After Knee Arthroscopy
Fibula Stress Fracture Mimicking a Malignancy
Septic Trochanteric Bursitis in an Adolescent
Painless, Atraumatic, Isolated Lateral Compartment Syndrome of the Leg: An Unusual Triad of Atypical Findings
Chondromyxoid Fibroma of the Radial Shaft Treated With Nonvascularized Fibular Autograft
Bilateral Adrenal Hemorrhage Complication
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
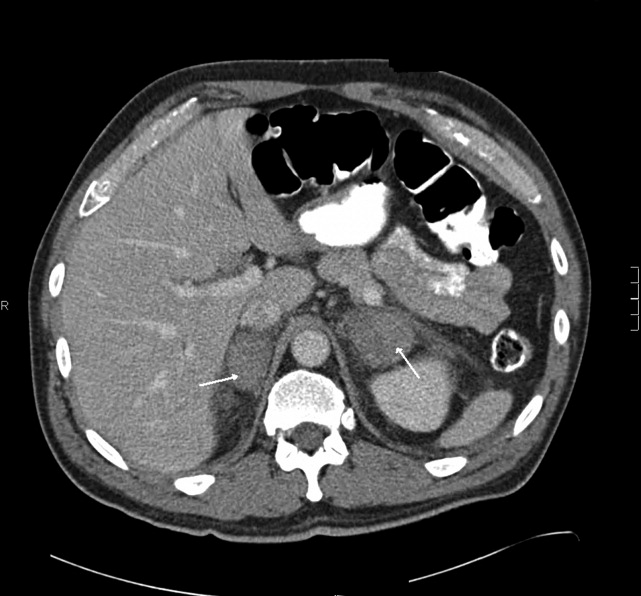
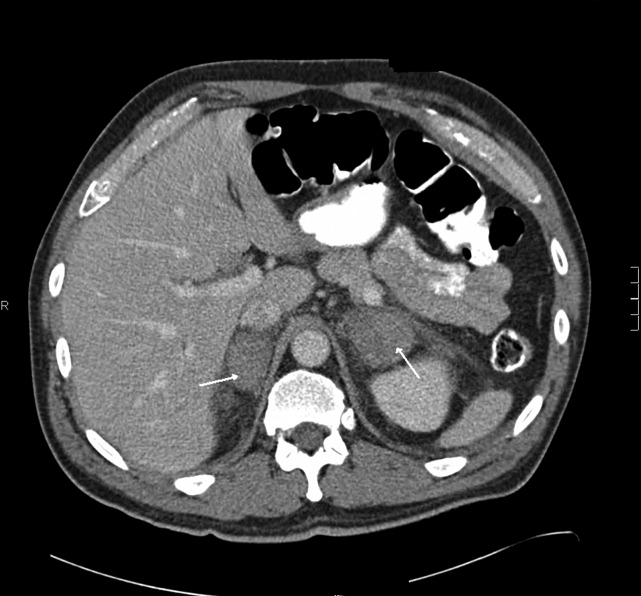
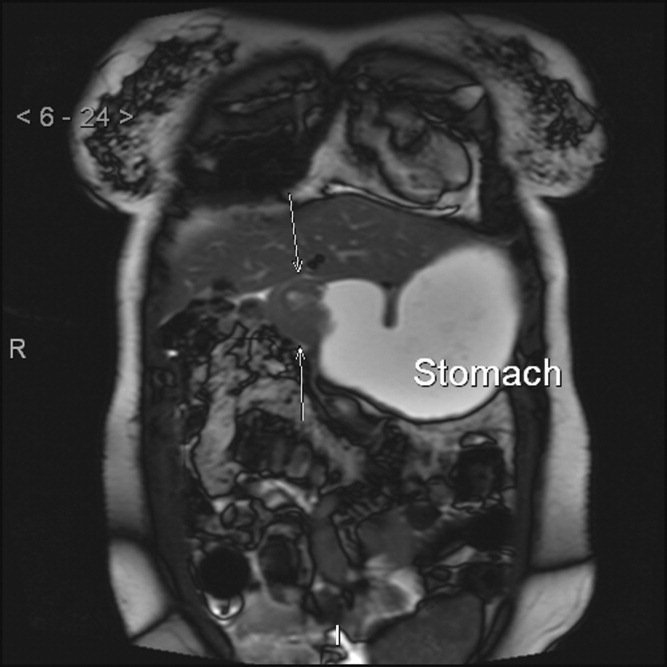
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
A 52‐year‐old man presented to the emergency department (ED) from a skilled nursing facility with a complaint of bilateral upper‐quadrant abdominal pain of 48 hours' duration. The pain was sharp, nonradiating, constant, and was associated with nausea, vomiting, and constipation. The patient denied any fever, back pain, dysuria, melena, or hematochezia. In the rehabilitation facility the patient had been initially evaluated for this pain. He was given laxatives and stool softeners for presumed constipation but these measures had not been effective. A computed tomography (CT) scan of the abdomen had only showed stool in the colon and he was sent to the ED for further evaluation.
Apart from severe degenerative joint disease in both his knees he was in good health. He was in the skilled nursing facility (SNF) for rehabilitation for bilateral knee replacement surgery done 9 days prior to this presentation. His postoperative course was unremarkable. He had been maintained on prophylaxis for venous thromboembolism with enoxaparin since postoperative day 1 at a daily dose of 40 mg subcutaneously, and was transferred to the SNF on postoperative day 6 on the same dose. His was receiving oxycodone and Tylenol for pain. He was on no other medications.
Vital signs on presentation revealed a temperature of 97.5F, a heart rate of 100 beats per minute, a respiratory rate of 16 breaths per minute, and a blood pressure of 136/69 mmHg. He was alert and oriented and in mild distress from the abdominal pain. Examination was normal except for tenderness in the upper quadrants of the abdomen though no rigidity or rebound tenderness were noted. Routine chemistries were normal except for sodium of 134 mg/dL. His white count, hemoglobin, hematocrit, and platelet levels were noted to be at 17.5K/L, 10 g/dL, 30%, and 345K/L, respectively, and were stable with regard to his discharge laboratory values. His serum eosinophil level was normal. A complete workup for hypercoagulable state and bleeding disorders including assays for antibodies associated with heparin‐induced thrombocytopenia were negative. He was admitted for further evaluation and treatment.
The patient had another CT scan of the abdomen (Figure 1), which when compared to the one done at the SNF 2 days prior showed markedly enlarged bilateral adrenal glands suggestive of bilateral acute adrenal hemorrhage. The enoxaparin was discontinued and empiric steroid replacement therapy was begun. A random cortisol level was normal but a cosyntropin stimulation test showed an absolute increase in cortisol level of only 0.8 g/dL at both 30 and 60 minutes after administration of 250 g of cosyntropin. An investigation was undertaken to determine if the patient had any prior risk factors for bleeding. There was no evidence of infection and a comprehensive evaluation for bleeding, and coagulation disorders was normal. The bilateral adrenal hemorrhage was attributed to the use of enoxaparin in the postoperative setting. Unfortunately, the patient subsequently developed a deep venous thrombosis in his lower extremity and an inferior vena cava (IVC) filter was placed before discharge. He was doing well 6 months later, and is still continued on glucocorticoid and mineralocorticoid replacement therapy and follows up with endocrinology as an outpatient.
Discussion
Bilateral adrenal hemorrhage is usually associated with massive sepsis from Gram‐negative organisms such as Neisseria meningitides, Pseudomonas aeroginosa, Escherichia coli, and Bacteroides fragilis. Rupert Waterhouse, in 1911, was the first person to describe a patient with severe meningococcal sepsis resulting in acute adrenal hemorrhage and collapse. This was also later described independently by Carl Friderichsen in 1918, and is now referred to as the Waterhouse‐Friderichsen syndrome. Other causes include antiphospholipid antibody syndrome, heparin‐associated thrombocytopenia (HIT), and severe physical stress. Bilateral adrenal hemorrhage can also spontaneously occur in the postoperative period, especially after cardiothoracic or orthopedic surgery. This phenomenon may be related to the frequent use of prophylactic anticoagulants after these types of procedures.
The first case report of bilateral adrenal hemorrhage secondary to use of anticoagulants was described in 1947, and the first case report of successful resuscitation after corticosteroid administration in a patient with bilateral adrenal hemorrhage secondary to anticoagulant use was described by Thorn in 1956.1 A review of the literature demonstrates multiple case reports of adrenal hemorrhage reported in the postoperative period, particularly after joint arthroplasty, and especially after knee replacement surgeries. Most of the recent cases have been associated with use of prophylactic low‐dose heparin or low‐molecular‐weight heparin at the time of adrenal hemorrhage. In a study of 157 case reports of individuals with bilateral hemorrhage (including 22 autopsies), 48 cases were associated with administration of anticoagulants, although the dose and effect were not specified.2 Amador et al.1 showed that out of 4325 autopsies performed from 1949 to 1962 in their institution, 30 cases were found of bilateral hemorrhage, of which 10 were receiving heparin at presumably prophylactic doses; 5 of these patients were also receiving dicumarol.
Mayo Clinic investigators performed a retrospective review of all cases of adrenal hemorrhage over a period of 25 years at their hospital, and found 141 cases of adrenal hemorrhage, of which 78 were bilateral and 63 were unilateral,3 and in 67 patients the condition was diagnosed at autopsy. In this study 14 patients had adrenal hemorrhage in the postoperative period in the absence of lupus anticoagulant or HIT; there was no specific mention in this study of the use of postoperative anticoagulants. Finally, a multicenter case control study was undertaken by Kovacs et al.4 to assess putative risk factors for development of bilateral massive adrenal hemorrhage. In the multivariate analysis, thrombocytopenia, exposure to heparin, and sepsis were found to be strongly associated with risk of hemorrhage. Of 23 patients with bilateral, massive adrenal hemorrhage, 16 had been exposed to heparin, and at least 6 were on exclusively subcutaneous heparin. The authors concluded that heparin exposure was a much bigger risk factor than other coagulopathies, and those exposed to heparin of any route or type for 4 to 6 days and those exposed for more than 6 days were about 17 and 34 times, respectively, more likely to develop bilateral hemorrhage than those who had less than 4 days or no exposure.
The clinical presentation of adrenal insufficiency due to bilateral adrenal hemorrhage is often nonspecific. Symptoms may include abdominal pain, back pain, fever, nausea, vomiting, weakness, obtundation, confusion, and hypotensionall of which are also common postoperative symptoms and can be missed or ignored.5 Rao et al.6 profiled the clinical presentation of 64 cases of bilateral hemorrhage and found the following: abdominal, flank, back, or chest pain (86%); anorexia, nausea, or vomiting (47%); psychiatric symptoms (42%); fever (66%); hypotension recognized before shock episode (19%); and abdominal rigidity or rebound (22%). Adrenal insufficiency becomes clinically evident once 90% of the gland is destroyed. About 50% of patients do not manifest typical laboratory abnormalities, so a high degree of suspicion is necessary to diagnose the condition.3 Also, the laboratory diagnosis of adrenal insufficiency using random cortisol levels is unreliable, as reference ranges in patients experiencing stress (as in the postoperative period) have not been well studied or established. In patients with bilateral hemorrhage postoperatively on prophylactic anticoagulants, the coagulation profile is usually within normal limits and there is typically no evidence of spontaneous bleeding elsewhere. In later stages, the typical laboratory findings of abnormal adrenal function such as hypokalemia, hyponatremia, declining cortisol levels, and an inappropriate response to adrenocorticotropic hormone stimulation test may be seen. A significant drop in hemoglobin secondary to hemorrhage may also be encountered in some patients secondary to the bleed.
CT is the most reliable and extensively used imaging modality for making the diagnosis, although magnetic resonance imaging (MRI) or ultrasound may also be utilized. Early in the course of adrenal hemorrhage, CT findings may be negative, and repeated imaging is appropriate when clinical suspicion is high. The presence of bilateral adrenal enlargement with increased signal attenuation suggests bilateral adrenal hemorrhage. MRI can both characterize adrenal hematomas, and estimate their age.7, 8
Postoperative adrenal hemorrhage and insufficiency is easily treatable and has excellent outcomes; survivors will need lifelong corticosteroid replacement (and usually mineralocorticoid replacement as well). In the Mayo Clinic study, survival was 100% with treatment vs. 17% without treatment. In comparison, sepsis‐induced or stress‐induced adrenal insufficiency has poor outcomes despite adequate treatment (9% survival with treatment vs. 6% survival without treatment).3 Death can occur within hours to days of symptoms if untreated. Treatment includes timely initiation of adrenal hormone replacement and reversal of coagulopathies.
Postoperative venous thromboembolism (VTE) prophylaxis with anticoagulants is the appropriate care in many cases, but, along with the postoperative state itself, also appears to be a risk factor for this unusual condition. Postoperative bilateral adrenal hemorrhage is rare and potentially fatal. Early identification and prompt initiation of steroid replacement therapy and reversal of coagulopathies can prove to be lifesaving. Making this diagnosis can be very challenging, as the clinical presentation and laboratory findings of adrenal hemorrhage are vague and nonspecific and mimic many nonlife threatening postoperative complications. Radiological diagnosis by CT may initially be normal and thus further confound the diagnosis. Hence, providers should remain vigilant for associated complications even with low‐dose prophylactic heparin or low‐molecular‐weight heparin in postoperative patients, and prompt, presumptive treatment with corticosteroids should be started while awaiting confirmation by imaging and laboratory testing.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
- .Adrenal hemorrhage during anticoagulant therapy. A clinical and pathological study of ten cases.Ann Intern Med.1965;63(4):559–571.
- ,,,,,.Adrenal hemorrhage in the adult.Medicine.1978;57(3):211–221.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76(2):161–168.
- ,,.Bilateral massive adrenal hemorrhage. Assessment of putative risk factors by the case‐control method.Medicine.2001;80(1):45–53.
- .Bilateral massive adrenal hemorrhage.Med Clin North Am.1995;79(1):107–129.
- ,,.Bilateral massive adrenal hemorrhage: early recognition and treatment.Ann Intern Med.1989;110(3):227–235.
- ,,, et al.Imaging of nontraumatic hemorrhage of the adrenal gland.Radiographics.1999;19(4):949–963.
- ,,,,.Spontaneous unilateral adrenal hemorrhage: computerized tomography and magnetic resonance imaging findings in 8 cases.J Urol.1995;154(5):1647–1651.
Metastatic Lobular Breast Carcinoma
Gastric outlet obstruction (GOO) is frequently a diagnostic dilemma as malignancies have surpassed benign diseases as etiologies of GOO.1, 2 A case is presented of a previously healthy patient with persistent vomiting who was sequentially diagnosed with peptic ulcer disease (PUD), pancreatitis, and cholecystitis. Unfortunately, the diagnostic workup was less straightforward than initially suspected, as she was eventually diagnosed with a malignant GOO.
To the best of our knowledge, this is the second case in which GOO was the presenting manifestation of a previously undiagnosed metastatic lobular breast carcinoma.1 Although gastrointestinal involvement may be a late manifestation of metastatic breast carcinoma that almost always follows after spread to other sites, this case illustrates GOO as a presenting manifestation.3 Perhaps the most salient teaching point, consistent with recent literature, is that endoscopic biopsies in the workup of malignant GOO are often misleading and delay diagnosis.4
Case Report
A 44‐year‐old female with a history of fibrocystic breast disease presented with 1 month of right upper quadrant abdominal pain and nonbilious emesis of undigested food occurring several hours postprandially. Gallstones were demonstrated on an abdominal sonogram, and esophagogastroduodenoscopy revealed a normal proximal esophagus, distal esophagitis, and a Schatzki ring. An 8‐mm duodenal bulb ulcer was biopsied with benign results, but no duodenal obstruction or narrowing was visualized. Therapy for presumptive Helicobacter pylori infection and PUD was initiated, but she was hospitalized shortly later with persistent vomiting and acute renal failure. An abdominal computed tomography (CT) scan showed evidence of pancreatitis but a normal biliary system and normal small and large bowels. There was no clinical jaundice, and hepatic function tests were normal. After medical therapy, an open cholecystectomy was performed because of dense adhesions of the large and small bowels to the liver and gallbladder. No gross abnormalities of the common bile duct or intestines were described.
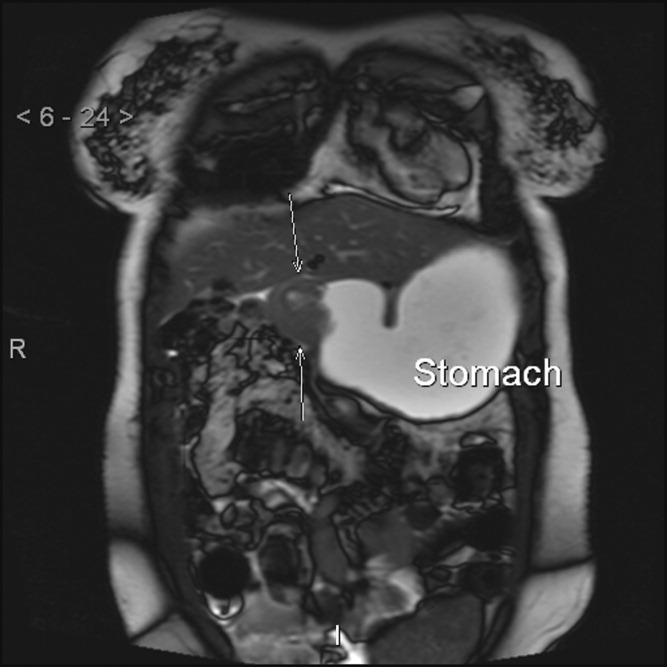
With the persistence of intractable vomiting, abdominal and pelvic CT scans were repeated and revealed duodenal thickening and luminal narrowing. Follow‐up abdominal magnetic resonance imaging (Figure 1) demonstrated an infiltrating duodenal mass with a resultant high‐grade GOO. Histological examination from repeat endoscopic biopsies of the duodenal mass revealed signet ring cells infiltrating the lamina propria with positive estrogen and progesterone receptors. Upon presentation to the surgical service, a physical examination revealed a right breast mass, and a subsequent breast biopsy diagnosed lobular breast carcinoma with 10% estrogen receptor and 5% progesterone receptor and histology identical to that of the duodenal mass. Immunohistochemical assays confirmed primary breast carcinoma with duodenal metastasis. A lumpectomy was performed, and docetaxel was initiated for stage IV invasive lobular breast carcinoma. A gastrostomy and duodenal stents were placed for the GOO with resolution of the vomiting, and she received hospice and palliative care.
Discussion
Prior to the advent of histamine blockers, PUD was the most common cause of GOO.1 Currently, malignancy accounts for 60% of cases of GOO.13 To the best of our knowledge, this patient's presentation represents the second case in which the initial manifestation of an undiagnosed metastatic breast disease was GOO.1 The diagnosis was both challenging and unusual because of the patient's other organic diseases (gallbladder disease, pancreatic disease, and PUD), which distracted from the underlying diagnosis of malignant GOO.
GOO is a clinical syndrome of diverse etiologies and pathogenesis that is characterized by mechanical impediment of gastric emptying. GOO is divided into 2 well‐defined groups: benign and malignant causes.14 Benign causes include: PUD, gastric polyps, pyloric stenosis, congenital duodenal webs, gallstone obstruction, pancreatic pseudocysts, and bezoars. Gastric cancer is the most common malignant cause and is followed by duodenal carcinoma, pancreatic carcinoma, cholangiocarcinoma, and metastatic disease of the gastric outlet.
Because of the stomach's significant capacity to distend, malignant GOO is often undetected clinically until a high‐grade obstruction develops.13 In patients with a previous diagnosis of carcinoma, nausea and vomiting can be mistakenly attributed to radiation or chemotherapy. Characteristic symptoms include nonbilious emesis of undigested food, early satiety, epigastric fullness, and abdominal pain.13
This case presentation is consistent with recent literature, which reports a surprising lack of reliability in diagnosing malignant GOO by endoscopy.4 In 1 study, endoscopic biopsy for detection of malignant GOO was associated with poor sensitivity (37%) in comparison with surgical biopsy.4 The authors recommended that patients who have a high clinical suspicion of malignant GOO (older patients and those without PUD) and who have initially benign biopsies be considered for surgical exploration prior to medical therapy or undergo repeat endoscopy with jumbo biopsies.4
Alternatively, endoscopic ultrasound has been advocated for detecting early mucosal gastric or duodenal cancer in patients without ulcerous changes on endoscopy. Radiographic features associated with submucosal tumor infiltration include irregular narrowing and budding signs.5 As conventional CT is considered suboptimal for the detection of gastric or intestinal carcinomas, multidetector‐row CT with multiplanar reconstruction has enhanced the overall ability to detect early gastric cancer and advanced gastric cancers with a sensitivity of 96.2%.6
Conclusion
The preceding case demonstrates valuable teaching points encountered in diagnosing malignant GOO. In patients with intractable vomiting severe enough to produce renal failure, other organic causes should be considered before one proceeds directly to cholecystectomy. This case further confirms that endoscopic biopsy is often alarmingly inadequate for diagnosing malignant GOO.46 Thus, if worrisome symptoms persist, the provider should not be comforted by normal endoscopy and should escalate investigations accordingly. More clinical trials should evaluate the roles of endoscopic ultrasound and multidetector‐row CT in the early detection of gastric and duodenal carcinoma in patients with normal endoscopic biopsies, which may have led to a more timely diagnosis in this patient.5, 6
- ,,, et al.Previously undiagnosed infiltrating lobular carcinoma of the breast presenting as a gastric outlet obstruction.Am J Gastroenterol.2001;12:3475–3477.
- ,,, et al.Breast cancer masquerading as primary gastric carcinoma.Aust N Z J Surg.1986;56:398–399.
- ,,, et al.The spectrum of gastrointestinal metastases of breast carcinoma: stomach.Gastrointest Endosc.1992;38:130–135.
- ,,.Gastric outlet obstruction with benign endoscopic biopsy should be further explored for malignancy.Gastrointest Endosc.1998;48:497–502.
- ,.The accuracy of endoscopic ultrasonography in differentiating mucosal from deeper gastric cancer.Am J Gastroenterol.2008;103:1801–1809.
- ,,, et al.Diagnosis of gastric cancer with MDCT using the water filling method and multiplanar reconstruction: CT histologic correlation.Am J Roentgenol.2005;185:1152–1158.
Gastric outlet obstruction (GOO) is frequently a diagnostic dilemma as malignancies have surpassed benign diseases as etiologies of GOO.1, 2 A case is presented of a previously healthy patient with persistent vomiting who was sequentially diagnosed with peptic ulcer disease (PUD), pancreatitis, and cholecystitis. Unfortunately, the diagnostic workup was less straightforward than initially suspected, as she was eventually diagnosed with a malignant GOO.
To the best of our knowledge, this is the second case in which GOO was the presenting manifestation of a previously undiagnosed metastatic lobular breast carcinoma.1 Although gastrointestinal involvement may be a late manifestation of metastatic breast carcinoma that almost always follows after spread to other sites, this case illustrates GOO as a presenting manifestation.3 Perhaps the most salient teaching point, consistent with recent literature, is that endoscopic biopsies in the workup of malignant GOO are often misleading and delay diagnosis.4
Case Report
A 44‐year‐old female with a history of fibrocystic breast disease presented with 1 month of right upper quadrant abdominal pain and nonbilious emesis of undigested food occurring several hours postprandially. Gallstones were demonstrated on an abdominal sonogram, and esophagogastroduodenoscopy revealed a normal proximal esophagus, distal esophagitis, and a Schatzki ring. An 8‐mm duodenal bulb ulcer was biopsied with benign results, but no duodenal obstruction or narrowing was visualized. Therapy for presumptive Helicobacter pylori infection and PUD was initiated, but she was hospitalized shortly later with persistent vomiting and acute renal failure. An abdominal computed tomography (CT) scan showed evidence of pancreatitis but a normal biliary system and normal small and large bowels. There was no clinical jaundice, and hepatic function tests were normal. After medical therapy, an open cholecystectomy was performed because of dense adhesions of the large and small bowels to the liver and gallbladder. No gross abnormalities of the common bile duct or intestines were described.
With the persistence of intractable vomiting, abdominal and pelvic CT scans were repeated and revealed duodenal thickening and luminal narrowing. Follow‐up abdominal magnetic resonance imaging (Figure 1) demonstrated an infiltrating duodenal mass with a resultant high‐grade GOO. Histological examination from repeat endoscopic biopsies of the duodenal mass revealed signet ring cells infiltrating the lamina propria with positive estrogen and progesterone receptors. Upon presentation to the surgical service, a physical examination revealed a right breast mass, and a subsequent breast biopsy diagnosed lobular breast carcinoma with 10% estrogen receptor and 5% progesterone receptor and histology identical to that of the duodenal mass. Immunohistochemical assays confirmed primary breast carcinoma with duodenal metastasis. A lumpectomy was performed, and docetaxel was initiated for stage IV invasive lobular breast carcinoma. A gastrostomy and duodenal stents were placed for the GOO with resolution of the vomiting, and she received hospice and palliative care.
Discussion
Prior to the advent of histamine blockers, PUD was the most common cause of GOO.1 Currently, malignancy accounts for 60% of cases of GOO.13 To the best of our knowledge, this patient's presentation represents the second case in which the initial manifestation of an undiagnosed metastatic breast disease was GOO.1 The diagnosis was both challenging and unusual because of the patient's other organic diseases (gallbladder disease, pancreatic disease, and PUD), which distracted from the underlying diagnosis of malignant GOO.
GOO is a clinical syndrome of diverse etiologies and pathogenesis that is characterized by mechanical impediment of gastric emptying. GOO is divided into 2 well‐defined groups: benign and malignant causes.14 Benign causes include: PUD, gastric polyps, pyloric stenosis, congenital duodenal webs, gallstone obstruction, pancreatic pseudocysts, and bezoars. Gastric cancer is the most common malignant cause and is followed by duodenal carcinoma, pancreatic carcinoma, cholangiocarcinoma, and metastatic disease of the gastric outlet.
Because of the stomach's significant capacity to distend, malignant GOO is often undetected clinically until a high‐grade obstruction develops.13 In patients with a previous diagnosis of carcinoma, nausea and vomiting can be mistakenly attributed to radiation or chemotherapy. Characteristic symptoms include nonbilious emesis of undigested food, early satiety, epigastric fullness, and abdominal pain.13
This case presentation is consistent with recent literature, which reports a surprising lack of reliability in diagnosing malignant GOO by endoscopy.4 In 1 study, endoscopic biopsy for detection of malignant GOO was associated with poor sensitivity (37%) in comparison with surgical biopsy.4 The authors recommended that patients who have a high clinical suspicion of malignant GOO (older patients and those without PUD) and who have initially benign biopsies be considered for surgical exploration prior to medical therapy or undergo repeat endoscopy with jumbo biopsies.4
Alternatively, endoscopic ultrasound has been advocated for detecting early mucosal gastric or duodenal cancer in patients without ulcerous changes on endoscopy. Radiographic features associated with submucosal tumor infiltration include irregular narrowing and budding signs.5 As conventional CT is considered suboptimal for the detection of gastric or intestinal carcinomas, multidetector‐row CT with multiplanar reconstruction has enhanced the overall ability to detect early gastric cancer and advanced gastric cancers with a sensitivity of 96.2%.6
Conclusion
The preceding case demonstrates valuable teaching points encountered in diagnosing malignant GOO. In patients with intractable vomiting severe enough to produce renal failure, other organic causes should be considered before one proceeds directly to cholecystectomy. This case further confirms that endoscopic biopsy is often alarmingly inadequate for diagnosing malignant GOO.46 Thus, if worrisome symptoms persist, the provider should not be comforted by normal endoscopy and should escalate investigations accordingly. More clinical trials should evaluate the roles of endoscopic ultrasound and multidetector‐row CT in the early detection of gastric and duodenal carcinoma in patients with normal endoscopic biopsies, which may have led to a more timely diagnosis in this patient.5, 6
Gastric outlet obstruction (GOO) is frequently a diagnostic dilemma as malignancies have surpassed benign diseases as etiologies of GOO.1, 2 A case is presented of a previously healthy patient with persistent vomiting who was sequentially diagnosed with peptic ulcer disease (PUD), pancreatitis, and cholecystitis. Unfortunately, the diagnostic workup was less straightforward than initially suspected, as she was eventually diagnosed with a malignant GOO.
To the best of our knowledge, this is the second case in which GOO was the presenting manifestation of a previously undiagnosed metastatic lobular breast carcinoma.1 Although gastrointestinal involvement may be a late manifestation of metastatic breast carcinoma that almost always follows after spread to other sites, this case illustrates GOO as a presenting manifestation.3 Perhaps the most salient teaching point, consistent with recent literature, is that endoscopic biopsies in the workup of malignant GOO are often misleading and delay diagnosis.4
Case Report
A 44‐year‐old female with a history of fibrocystic breast disease presented with 1 month of right upper quadrant abdominal pain and nonbilious emesis of undigested food occurring several hours postprandially. Gallstones were demonstrated on an abdominal sonogram, and esophagogastroduodenoscopy revealed a normal proximal esophagus, distal esophagitis, and a Schatzki ring. An 8‐mm duodenal bulb ulcer was biopsied with benign results, but no duodenal obstruction or narrowing was visualized. Therapy for presumptive Helicobacter pylori infection and PUD was initiated, but she was hospitalized shortly later with persistent vomiting and acute renal failure. An abdominal computed tomography (CT) scan showed evidence of pancreatitis but a normal biliary system and normal small and large bowels. There was no clinical jaundice, and hepatic function tests were normal. After medical therapy, an open cholecystectomy was performed because of dense adhesions of the large and small bowels to the liver and gallbladder. No gross abnormalities of the common bile duct or intestines were described.
With the persistence of intractable vomiting, abdominal and pelvic CT scans were repeated and revealed duodenal thickening and luminal narrowing. Follow‐up abdominal magnetic resonance imaging (Figure 1) demonstrated an infiltrating duodenal mass with a resultant high‐grade GOO. Histological examination from repeat endoscopic biopsies of the duodenal mass revealed signet ring cells infiltrating the lamina propria with positive estrogen and progesterone receptors. Upon presentation to the surgical service, a physical examination revealed a right breast mass, and a subsequent breast biopsy diagnosed lobular breast carcinoma with 10% estrogen receptor and 5% progesterone receptor and histology identical to that of the duodenal mass. Immunohistochemical assays confirmed primary breast carcinoma with duodenal metastasis. A lumpectomy was performed, and docetaxel was initiated for stage IV invasive lobular breast carcinoma. A gastrostomy and duodenal stents were placed for the GOO with resolution of the vomiting, and she received hospice and palliative care.
Discussion
Prior to the advent of histamine blockers, PUD was the most common cause of GOO.1 Currently, malignancy accounts for 60% of cases of GOO.13 To the best of our knowledge, this patient's presentation represents the second case in which the initial manifestation of an undiagnosed metastatic breast disease was GOO.1 The diagnosis was both challenging and unusual because of the patient's other organic diseases (gallbladder disease, pancreatic disease, and PUD), which distracted from the underlying diagnosis of malignant GOO.
GOO is a clinical syndrome of diverse etiologies and pathogenesis that is characterized by mechanical impediment of gastric emptying. GOO is divided into 2 well‐defined groups: benign and malignant causes.14 Benign causes include: PUD, gastric polyps, pyloric stenosis, congenital duodenal webs, gallstone obstruction, pancreatic pseudocysts, and bezoars. Gastric cancer is the most common malignant cause and is followed by duodenal carcinoma, pancreatic carcinoma, cholangiocarcinoma, and metastatic disease of the gastric outlet.
Because of the stomach's significant capacity to distend, malignant GOO is often undetected clinically until a high‐grade obstruction develops.13 In patients with a previous diagnosis of carcinoma, nausea and vomiting can be mistakenly attributed to radiation or chemotherapy. Characteristic symptoms include nonbilious emesis of undigested food, early satiety, epigastric fullness, and abdominal pain.13
This case presentation is consistent with recent literature, which reports a surprising lack of reliability in diagnosing malignant GOO by endoscopy.4 In 1 study, endoscopic biopsy for detection of malignant GOO was associated with poor sensitivity (37%) in comparison with surgical biopsy.4 The authors recommended that patients who have a high clinical suspicion of malignant GOO (older patients and those without PUD) and who have initially benign biopsies be considered for surgical exploration prior to medical therapy or undergo repeat endoscopy with jumbo biopsies.4
Alternatively, endoscopic ultrasound has been advocated for detecting early mucosal gastric or duodenal cancer in patients without ulcerous changes on endoscopy. Radiographic features associated with submucosal tumor infiltration include irregular narrowing and budding signs.5 As conventional CT is considered suboptimal for the detection of gastric or intestinal carcinomas, multidetector‐row CT with multiplanar reconstruction has enhanced the overall ability to detect early gastric cancer and advanced gastric cancers with a sensitivity of 96.2%.6
Conclusion
The preceding case demonstrates valuable teaching points encountered in diagnosing malignant GOO. In patients with intractable vomiting severe enough to produce renal failure, other organic causes should be considered before one proceeds directly to cholecystectomy. This case further confirms that endoscopic biopsy is often alarmingly inadequate for diagnosing malignant GOO.46 Thus, if worrisome symptoms persist, the provider should not be comforted by normal endoscopy and should escalate investigations accordingly. More clinical trials should evaluate the roles of endoscopic ultrasound and multidetector‐row CT in the early detection of gastric and duodenal carcinoma in patients with normal endoscopic biopsies, which may have led to a more timely diagnosis in this patient.5, 6
- ,,, et al.Previously undiagnosed infiltrating lobular carcinoma of the breast presenting as a gastric outlet obstruction.Am J Gastroenterol.2001;12:3475–3477.
- ,,, et al.Breast cancer masquerading as primary gastric carcinoma.Aust N Z J Surg.1986;56:398–399.
- ,,, et al.The spectrum of gastrointestinal metastases of breast carcinoma: stomach.Gastrointest Endosc.1992;38:130–135.
- ,,.Gastric outlet obstruction with benign endoscopic biopsy should be further explored for malignancy.Gastrointest Endosc.1998;48:497–502.
- ,.The accuracy of endoscopic ultrasonography in differentiating mucosal from deeper gastric cancer.Am J Gastroenterol.2008;103:1801–1809.
- ,,, et al.Diagnosis of gastric cancer with MDCT using the water filling method and multiplanar reconstruction: CT histologic correlation.Am J Roentgenol.2005;185:1152–1158.
- ,,, et al.Previously undiagnosed infiltrating lobular carcinoma of the breast presenting as a gastric outlet obstruction.Am J Gastroenterol.2001;12:3475–3477.
- ,,, et al.Breast cancer masquerading as primary gastric carcinoma.Aust N Z J Surg.1986;56:398–399.
- ,,, et al.The spectrum of gastrointestinal metastases of breast carcinoma: stomach.Gastrointest Endosc.1992;38:130–135.
- ,,.Gastric outlet obstruction with benign endoscopic biopsy should be further explored for malignancy.Gastrointest Endosc.1998;48:497–502.
- ,.The accuracy of endoscopic ultrasonography in differentiating mucosal from deeper gastric cancer.Am J Gastroenterol.2008;103:1801–1809.
- ,,, et al.Diagnosis of gastric cancer with MDCT using the water filling method and multiplanar reconstruction: CT histologic correlation.Am J Roentgenol.2005;185:1152–1158.
Brugada Syndrome Unmasked by a Mosquito
Two weeks after returning from missionary work in Haiti, a 53‐year‐old woman with no significant past medical history presented with 5 days of worsening fevers, chills, diaphoresis, myalgias, and severe nausea. Notably, she did not take malaria prophylaxis while in Haiti.
Her temperature was 40.1C, her blood pressure was 100/58 mm Hg, and her heart rate was 102 beats per minute. Physical examination was remarkable only for her ill appearance. Initial lab work revealed anemia (hemoglobin, 10.4 g/dL; hematocrit, 29.4%), thrombocytopenia (23,000/mm),3 and evidence of acute renal failure (blood urea nitrogen, 58 mg/dL; creatinine, 4.2 mg/dL). Other labs were within normal limits.
Malaria was considered high on the differential diagnosis. A parasite smear was therefore obtained, and the findings were consistent with Plasmodium falciparum infection (5.5% parasitemia).
She was admitted to the intensive care unit for hydration and initiation of antimalarial therapy. Her severe nausea prevented administration of oral medications; therefore, the infectious disease consultant recommended treatment with intravenous quinidine.
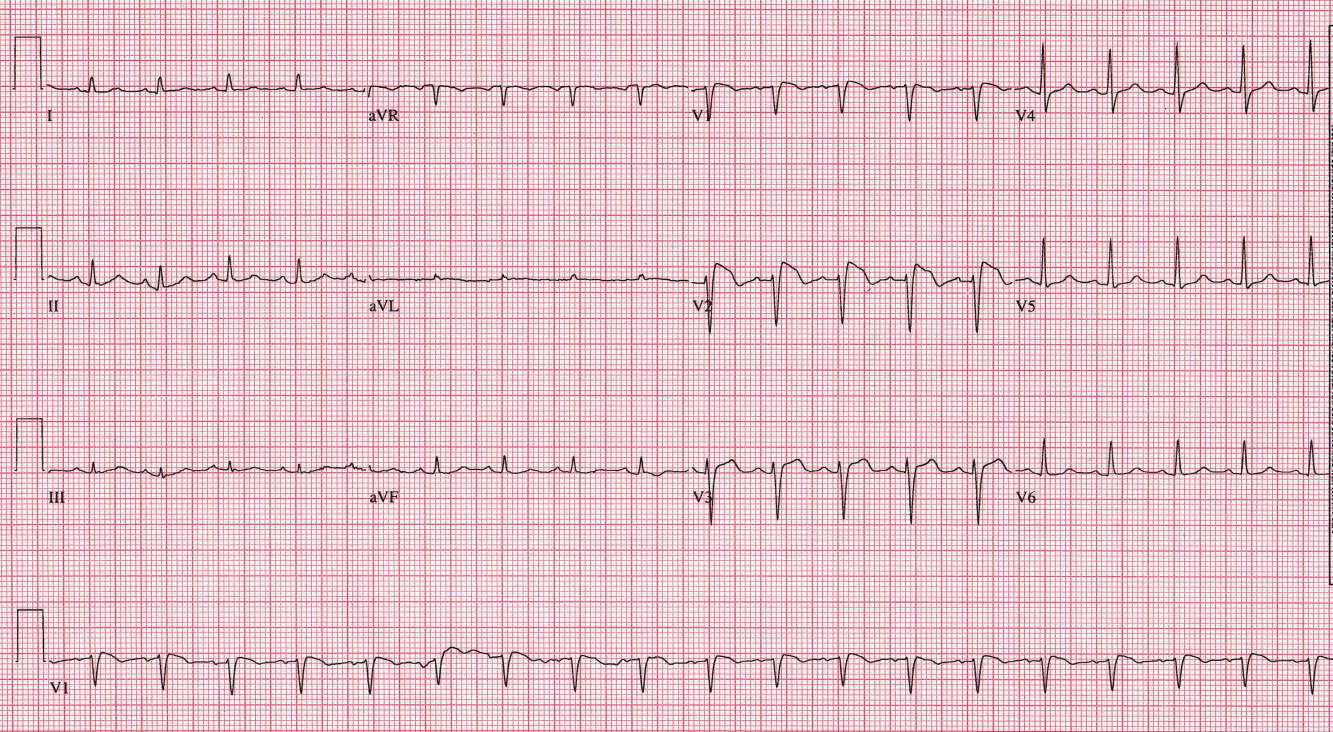
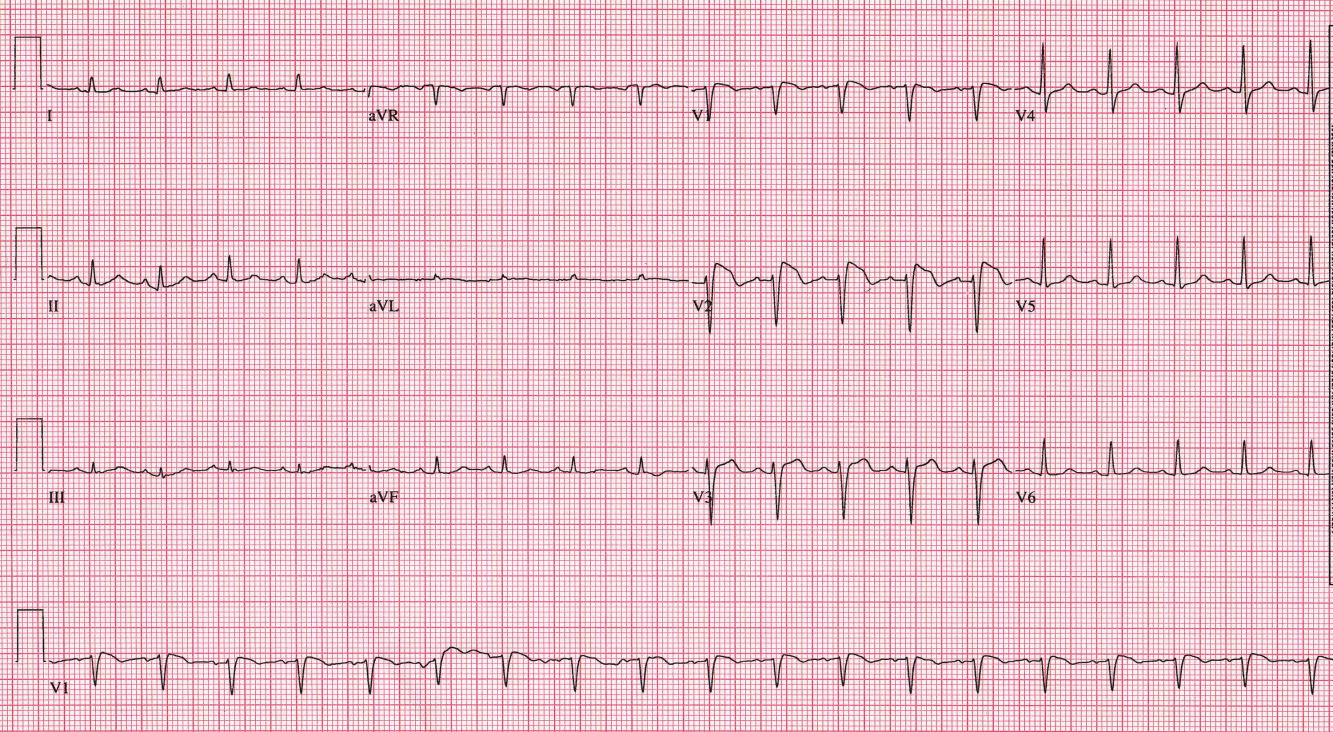
Prior to initiation of quinidine, an electrocardiogram (ECG) was obtained (Figure 1). No prior ECGs were available for comparison. Prominent ST segment elevation was noted, prompting reassessment of the patient. She denied chest pain. Cardiac enzymes were normal, and an urgent echocardiogram demonstrated normal ventricular function with mild mitral regurgitation. Given that suspicion for acute coronary syndrome was low, the ECG findings were managed conservatively.
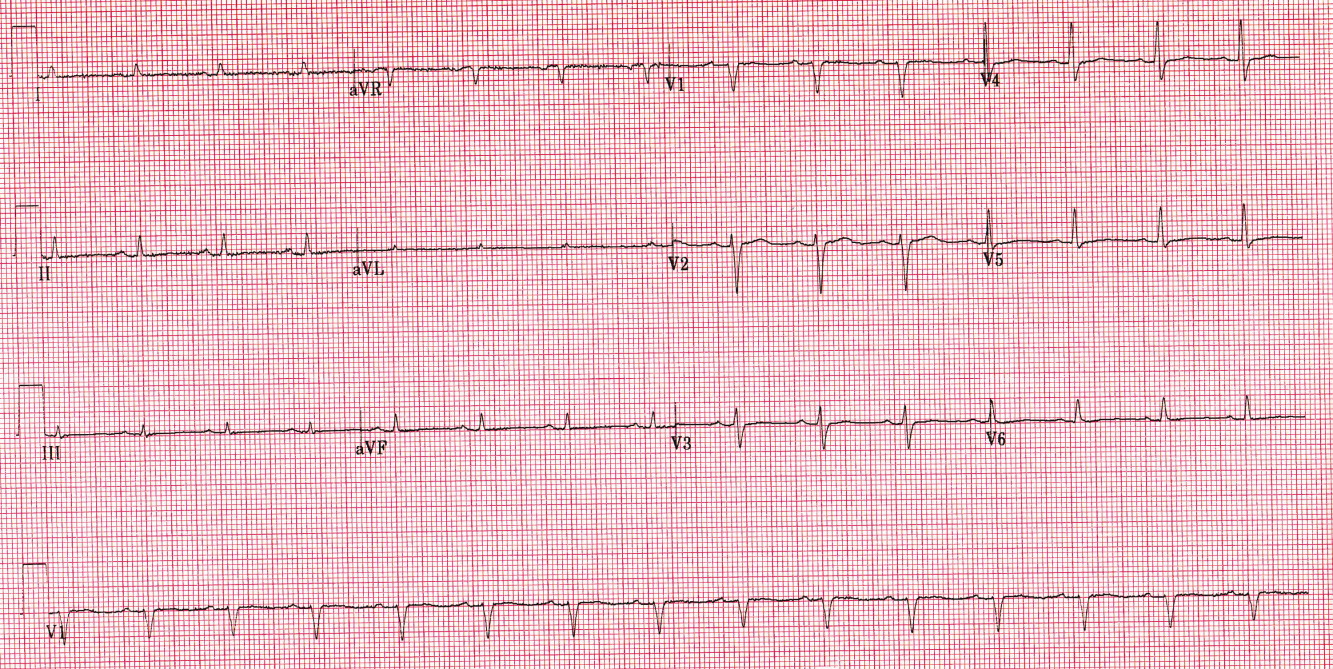
Overnight, she defervesced and appeared to improve clinically. Cardiac enzymes remained negative. A repeat ECG obtained several hours after admission revealed complete resolution of the ST elevation (Figure 2). Repeat ECGs remained normal through the time of discharge, and no ventricular arrhythmias were noted on telemetry.
On the basis of the characteristic ECG appearance, a presumptive diagnosis of Brugada syndrome was made. The patient did not have a history of presyncope, syncope, or agonal night‐time breathing or a family history of sudden death. Two weeks following discharge, she was seen in the outpatient electrophysiology clinic to discuss further risk stratification. A procainamide challenge, followed by programmed ventricular stimulation (electrophysiology study), was recommended. The procainamide challenge revealed ST segment changes consistent with Brugada syndrome. She was not inducible for ventricular arrhythmias during the electrophysiology study. On the basis of these findings as well as her lack of symptoms, there was no indication for an implantable cardioverter defibrillator.
Discussion
The finding of ST segment elevation in a critically ill patient raises concern for a variety of processes, including myocardial infarction, coronary vasospasm, myocarditis, pericarditis, and electrolyte abnormalities. Our patient's presentation was not consistent with any of these diagnoses, and the ST segment changes had the highly characteristic coved appearance seen in patients with Brugada syndrome.
Brugada syndrome, which was first described in 1992,1 is an inherited cardiac channelopathy. It is most commonly associated with loss‐of‐function mutations in SCN5A, the gene that encodes the subunit of the cardiac sodium channel. The syndrome displays autosomal dominant inheritance with variable penetrance, and affected individuals are at increased risk of sudden death due to ventricular fibrillation.
The classic ECG manifestations of Brugada syndrome consist of an RSR pattern (pseudo‐RBBB) with a 2‐mm convex (coved) ST segment elevation and T wave inversion in leads V1 to V3 (Figure 1). There are also 2 less common patterns that display a saddle‐back ST‐T configuration with lesser ST segment elevation and upright or biphasic T waves. All 3 patterns can be transient, and their expression can be modulated by a number of factors, including autonomic tone, electrolyte abnormalities, ischemia, drugs, and body temperature.
The ECG appearance of Brugada syndrome is the result of the decreased function of the cardiac sodium channel. The inward flow of sodium through this channel is what depolarizes the cell. When this flow is blunted, the repolarizing effect of the transient outward potassium current is left relatively unopposed, and the action potential duration (APD) is shortened. This effect is prominent in the right ventricular outflow tract epicardium (which is why the ECG changes are noted in the precordial leads overlying this territory). Because the APD determines the refractory period of a cell (ie, how soon the cell can be re‐excited), the shortening of the APD allows epicardial cells to return to an excitable state while neighboring cells in the other myocardial layers are still refractory. This phenomenon, which is known as transmural dispersion of refractoriness, creates a voltage gradient between cellular layers and provides an ideal substrate for the precipitation of sustained reentrant ventricular arrhythmias.2
Two issues related to our case bear further explanation. First, on the basis of quinidine's sodium channel blocking properties (it is a class I antiarrhythmic), one would predict that it would exacerbate Brugada syndrome. Although this is true of other class I drugs, quinidine also is a potent blocker of transient outward potassium current, and this effect can actually lead to normalization of the ECG.2 Second, febrile illness can cause premature inactivation of the sodium channel in patients with Brugada syndrome,3 and fever can unmask the ECG changes and even promote arrhythmias in susceptible patients.4 We postulate that our patient had her underlying Brugada syndrome unmasked by her febrile illness and that the initiation of quinidine (blockade of transient outward potassium current) and defervescence (improved sodium current) contributed to the normalization of her ECG.
Although the details of our patient's presentation are somewhat unusual, we hope that this case highlights the dilemma created by the incidental discovery of a Brugada‐pattern ECG. Clinicians need to be aware that the cornerstone of the evaluation centers on determining whether the patient has any risk factors for sudden death: ventricular arrhythmias, a family history of sudden death, or symptoms suggestive of aborted sudden death (syncope, seizures, or nocturnal agonal respiration). In the absence of any of these risk factors, asymptomatic individuals are likely at low risk and can be followed clinically. If the diagnosis is in question, the typical ECG pattern can be elicited by challenge with a sodium channel blocking agent (most commonly procainamide). Although many patients will often undergo further invasive risk stratification, the utility of this approach is the subject of controversy. Finally, screening of family members should be considered.
- ,.Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report.J Am Coll Cardiol.1992;20(6):1391–1396.
- .Brugada syndrome.Pacing Clin Electrophysiol.2006;29(10):1130–1159.
- ,,, et al.Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent.Circ Res.1999;85(9):803–809.
- ,.Fever and Brugada syndrome.Pacing Clin Electrophysiol.2002;25(11):1537–1539.
Two weeks after returning from missionary work in Haiti, a 53‐year‐old woman with no significant past medical history presented with 5 days of worsening fevers, chills, diaphoresis, myalgias, and severe nausea. Notably, she did not take malaria prophylaxis while in Haiti.
Her temperature was 40.1C, her blood pressure was 100/58 mm Hg, and her heart rate was 102 beats per minute. Physical examination was remarkable only for her ill appearance. Initial lab work revealed anemia (hemoglobin, 10.4 g/dL; hematocrit, 29.4%), thrombocytopenia (23,000/mm),3 and evidence of acute renal failure (blood urea nitrogen, 58 mg/dL; creatinine, 4.2 mg/dL). Other labs were within normal limits.
Malaria was considered high on the differential diagnosis. A parasite smear was therefore obtained, and the findings were consistent with Plasmodium falciparum infection (5.5% parasitemia).
She was admitted to the intensive care unit for hydration and initiation of antimalarial therapy. Her severe nausea prevented administration of oral medications; therefore, the infectious disease consultant recommended treatment with intravenous quinidine.
Prior to initiation of quinidine, an electrocardiogram (ECG) was obtained (Figure 1). No prior ECGs were available for comparison. Prominent ST segment elevation was noted, prompting reassessment of the patient. She denied chest pain. Cardiac enzymes were normal, and an urgent echocardiogram demonstrated normal ventricular function with mild mitral regurgitation. Given that suspicion for acute coronary syndrome was low, the ECG findings were managed conservatively.
Overnight, she defervesced and appeared to improve clinically. Cardiac enzymes remained negative. A repeat ECG obtained several hours after admission revealed complete resolution of the ST elevation (Figure 2). Repeat ECGs remained normal through the time of discharge, and no ventricular arrhythmias were noted on telemetry.
On the basis of the characteristic ECG appearance, a presumptive diagnosis of Brugada syndrome was made. The patient did not have a history of presyncope, syncope, or agonal night‐time breathing or a family history of sudden death. Two weeks following discharge, she was seen in the outpatient electrophysiology clinic to discuss further risk stratification. A procainamide challenge, followed by programmed ventricular stimulation (electrophysiology study), was recommended. The procainamide challenge revealed ST segment changes consistent with Brugada syndrome. She was not inducible for ventricular arrhythmias during the electrophysiology study. On the basis of these findings as well as her lack of symptoms, there was no indication for an implantable cardioverter defibrillator.
Discussion
The finding of ST segment elevation in a critically ill patient raises concern for a variety of processes, including myocardial infarction, coronary vasospasm, myocarditis, pericarditis, and electrolyte abnormalities. Our patient's presentation was not consistent with any of these diagnoses, and the ST segment changes had the highly characteristic coved appearance seen in patients with Brugada syndrome.
Brugada syndrome, which was first described in 1992,1 is an inherited cardiac channelopathy. It is most commonly associated with loss‐of‐function mutations in SCN5A, the gene that encodes the subunit of the cardiac sodium channel. The syndrome displays autosomal dominant inheritance with variable penetrance, and affected individuals are at increased risk of sudden death due to ventricular fibrillation.
The classic ECG manifestations of Brugada syndrome consist of an RSR pattern (pseudo‐RBBB) with a 2‐mm convex (coved) ST segment elevation and T wave inversion in leads V1 to V3 (Figure 1). There are also 2 less common patterns that display a saddle‐back ST‐T configuration with lesser ST segment elevation and upright or biphasic T waves. All 3 patterns can be transient, and their expression can be modulated by a number of factors, including autonomic tone, electrolyte abnormalities, ischemia, drugs, and body temperature.
The ECG appearance of Brugada syndrome is the result of the decreased function of the cardiac sodium channel. The inward flow of sodium through this channel is what depolarizes the cell. When this flow is blunted, the repolarizing effect of the transient outward potassium current is left relatively unopposed, and the action potential duration (APD) is shortened. This effect is prominent in the right ventricular outflow tract epicardium (which is why the ECG changes are noted in the precordial leads overlying this territory). Because the APD determines the refractory period of a cell (ie, how soon the cell can be re‐excited), the shortening of the APD allows epicardial cells to return to an excitable state while neighboring cells in the other myocardial layers are still refractory. This phenomenon, which is known as transmural dispersion of refractoriness, creates a voltage gradient between cellular layers and provides an ideal substrate for the precipitation of sustained reentrant ventricular arrhythmias.2
Two issues related to our case bear further explanation. First, on the basis of quinidine's sodium channel blocking properties (it is a class I antiarrhythmic), one would predict that it would exacerbate Brugada syndrome. Although this is true of other class I drugs, quinidine also is a potent blocker of transient outward potassium current, and this effect can actually lead to normalization of the ECG.2 Second, febrile illness can cause premature inactivation of the sodium channel in patients with Brugada syndrome,3 and fever can unmask the ECG changes and even promote arrhythmias in susceptible patients.4 We postulate that our patient had her underlying Brugada syndrome unmasked by her febrile illness and that the initiation of quinidine (blockade of transient outward potassium current) and defervescence (improved sodium current) contributed to the normalization of her ECG.
Although the details of our patient's presentation are somewhat unusual, we hope that this case highlights the dilemma created by the incidental discovery of a Brugada‐pattern ECG. Clinicians need to be aware that the cornerstone of the evaluation centers on determining whether the patient has any risk factors for sudden death: ventricular arrhythmias, a family history of sudden death, or symptoms suggestive of aborted sudden death (syncope, seizures, or nocturnal agonal respiration). In the absence of any of these risk factors, asymptomatic individuals are likely at low risk and can be followed clinically. If the diagnosis is in question, the typical ECG pattern can be elicited by challenge with a sodium channel blocking agent (most commonly procainamide). Although many patients will often undergo further invasive risk stratification, the utility of this approach is the subject of controversy. Finally, screening of family members should be considered.
Two weeks after returning from missionary work in Haiti, a 53‐year‐old woman with no significant past medical history presented with 5 days of worsening fevers, chills, diaphoresis, myalgias, and severe nausea. Notably, she did not take malaria prophylaxis while in Haiti.
Her temperature was 40.1C, her blood pressure was 100/58 mm Hg, and her heart rate was 102 beats per minute. Physical examination was remarkable only for her ill appearance. Initial lab work revealed anemia (hemoglobin, 10.4 g/dL; hematocrit, 29.4%), thrombocytopenia (23,000/mm),3 and evidence of acute renal failure (blood urea nitrogen, 58 mg/dL; creatinine, 4.2 mg/dL). Other labs were within normal limits.
Malaria was considered high on the differential diagnosis. A parasite smear was therefore obtained, and the findings were consistent with Plasmodium falciparum infection (5.5% parasitemia).
She was admitted to the intensive care unit for hydration and initiation of antimalarial therapy. Her severe nausea prevented administration of oral medications; therefore, the infectious disease consultant recommended treatment with intravenous quinidine.
Prior to initiation of quinidine, an electrocardiogram (ECG) was obtained (Figure 1). No prior ECGs were available for comparison. Prominent ST segment elevation was noted, prompting reassessment of the patient. She denied chest pain. Cardiac enzymes were normal, and an urgent echocardiogram demonstrated normal ventricular function with mild mitral regurgitation. Given that suspicion for acute coronary syndrome was low, the ECG findings were managed conservatively.
Overnight, she defervesced and appeared to improve clinically. Cardiac enzymes remained negative. A repeat ECG obtained several hours after admission revealed complete resolution of the ST elevation (Figure 2). Repeat ECGs remained normal through the time of discharge, and no ventricular arrhythmias were noted on telemetry.
On the basis of the characteristic ECG appearance, a presumptive diagnosis of Brugada syndrome was made. The patient did not have a history of presyncope, syncope, or agonal night‐time breathing or a family history of sudden death. Two weeks following discharge, she was seen in the outpatient electrophysiology clinic to discuss further risk stratification. A procainamide challenge, followed by programmed ventricular stimulation (electrophysiology study), was recommended. The procainamide challenge revealed ST segment changes consistent with Brugada syndrome. She was not inducible for ventricular arrhythmias during the electrophysiology study. On the basis of these findings as well as her lack of symptoms, there was no indication for an implantable cardioverter defibrillator.
Discussion
The finding of ST segment elevation in a critically ill patient raises concern for a variety of processes, including myocardial infarction, coronary vasospasm, myocarditis, pericarditis, and electrolyte abnormalities. Our patient's presentation was not consistent with any of these diagnoses, and the ST segment changes had the highly characteristic coved appearance seen in patients with Brugada syndrome.
Brugada syndrome, which was first described in 1992,1 is an inherited cardiac channelopathy. It is most commonly associated with loss‐of‐function mutations in SCN5A, the gene that encodes the subunit of the cardiac sodium channel. The syndrome displays autosomal dominant inheritance with variable penetrance, and affected individuals are at increased risk of sudden death due to ventricular fibrillation.
The classic ECG manifestations of Brugada syndrome consist of an RSR pattern (pseudo‐RBBB) with a 2‐mm convex (coved) ST segment elevation and T wave inversion in leads V1 to V3 (Figure 1). There are also 2 less common patterns that display a saddle‐back ST‐T configuration with lesser ST segment elevation and upright or biphasic T waves. All 3 patterns can be transient, and their expression can be modulated by a number of factors, including autonomic tone, electrolyte abnormalities, ischemia, drugs, and body temperature.
The ECG appearance of Brugada syndrome is the result of the decreased function of the cardiac sodium channel. The inward flow of sodium through this channel is what depolarizes the cell. When this flow is blunted, the repolarizing effect of the transient outward potassium current is left relatively unopposed, and the action potential duration (APD) is shortened. This effect is prominent in the right ventricular outflow tract epicardium (which is why the ECG changes are noted in the precordial leads overlying this territory). Because the APD determines the refractory period of a cell (ie, how soon the cell can be re‐excited), the shortening of the APD allows epicardial cells to return to an excitable state while neighboring cells in the other myocardial layers are still refractory. This phenomenon, which is known as transmural dispersion of refractoriness, creates a voltage gradient between cellular layers and provides an ideal substrate for the precipitation of sustained reentrant ventricular arrhythmias.2
Two issues related to our case bear further explanation. First, on the basis of quinidine's sodium channel blocking properties (it is a class I antiarrhythmic), one would predict that it would exacerbate Brugada syndrome. Although this is true of other class I drugs, quinidine also is a potent blocker of transient outward potassium current, and this effect can actually lead to normalization of the ECG.2 Second, febrile illness can cause premature inactivation of the sodium channel in patients with Brugada syndrome,3 and fever can unmask the ECG changes and even promote arrhythmias in susceptible patients.4 We postulate that our patient had her underlying Brugada syndrome unmasked by her febrile illness and that the initiation of quinidine (blockade of transient outward potassium current) and defervescence (improved sodium current) contributed to the normalization of her ECG.
Although the details of our patient's presentation are somewhat unusual, we hope that this case highlights the dilemma created by the incidental discovery of a Brugada‐pattern ECG. Clinicians need to be aware that the cornerstone of the evaluation centers on determining whether the patient has any risk factors for sudden death: ventricular arrhythmias, a family history of sudden death, or symptoms suggestive of aborted sudden death (syncope, seizures, or nocturnal agonal respiration). In the absence of any of these risk factors, asymptomatic individuals are likely at low risk and can be followed clinically. If the diagnosis is in question, the typical ECG pattern can be elicited by challenge with a sodium channel blocking agent (most commonly procainamide). Although many patients will often undergo further invasive risk stratification, the utility of this approach is the subject of controversy. Finally, screening of family members should be considered.
- ,.Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report.J Am Coll Cardiol.1992;20(6):1391–1396.
- .Brugada syndrome.Pacing Clin Electrophysiol.2006;29(10):1130–1159.
- ,,, et al.Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent.Circ Res.1999;85(9):803–809.
- ,.Fever and Brugada syndrome.Pacing Clin Electrophysiol.2002;25(11):1537–1539.
- ,.Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report.J Am Coll Cardiol.1992;20(6):1391–1396.
- .Brugada syndrome.Pacing Clin Electrophysiol.2006;29(10):1130–1159.
- ,,, et al.Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent.Circ Res.1999;85(9):803–809.
- ,.Fever and Brugada syndrome.Pacing Clin Electrophysiol.2002;25(11):1537–1539.