User login
Intraosseous Ganglion of the Glenoid
Advanced Shoulder Joint Tuberculosis Treated With Débridement and Closed Continuous Irrigation and Suction: A Report of Two Cases
Scapular Osteochondroma Treated With Arthroscopic Excision Using Prone Positioning
Anatomic Variations of the Palmaris Longus Muscle
Hypercalcemia and Milk‐Alkali Syndrome
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
Historically, the milk‐alkali syndrome developed as an adverse reaction to the Sippy regimen of frequent feedings of milk, cream, and alkaline powders as treatment for peptic ulcer disease.1 The classic description includes hypercalcemia, metabolic alkalosis, and renal failure. This syndrome seemingly disappeared when modern acid suppression therapies such as histamine‐2 blockers and proton pump inhibitors improved dyspepsia treatment. Over the past 20 years, milk‐alkali syndrome has had a resurgence, as consumption of supplements containing calcium has increased.2 Calcium carbonate supplements are a popular over‐the‐counter treatment for osteoporosis, dyspepsia, hypocalcemia, and hyperphosphatemia; these supplements provide both the calcium and alkali required for the development of milk‐alkali syndrome.
A 46‐year‐old man presented to the emergency department after his physician ordered outpatient laboratory tests to evaluate his fatigue. The patient was found to have acute renal failure and hypercalcemia. His serum creatinine was 3.6 mg/dL, increased from his baseline of 1.1 mg/dL several months prior, and his serum calcium was 14.9 mg/dL. Ten days prior to admission he developed increasing fatigue, decreased appetite, and decreased urine output, which he attributed to recent manual labor during summer. He reported taking an occasional calcium carbonate (Tums) for dyspepsia. He did not report pain or other complaints.
His medical history included hypertension and hyperlipidemia. He had a colonoscopy 1 year prior to presentation that was significant for a high‐grade dysplastic polyp and was currently due for repeat colonoscopy. His medications included clonidine, lisinopril, and aspirin. He had no recent medication changes. He had a 30 pack/year history of cigarette smoking and drank occasionally.
On physical exam, his temperature was 99.8F, blood pressure 97/48 mmHg, heart rate 89 beats per minute, respirations 20 breaths per minute, with a room air saturation of 97%. He had dry mucus membranes and the remainder of the physical exam was unremarkable.
In the emergency department laboratory testing revealed a creatinine of 4.6 mg/dL, serum total calcium of 15.9 mg/dL, serum bicarbonate level of 26 mmol/L, phosphate of 3.9 mg/dL, albumin of 4.4 gm/dL, and alkaline phosphatase of 92 IU/L. The urine specific gravity was 1.019 gm/mL (see Table 1 for the patient's complete admission laboratory values).
| Result (Normal Range) | |
|---|---|
| |
| At admission | |
| Sodium (mmol/L) | 135 (135‐145) |
| Potassium (mmol/L) | 4.4 (3.8‐5.0) |
| Chloride (mmol/L) | 97 (98‐107) |
| Bicarbonate (mmol/L) | 25 (22‐31) |
| BUN (mg/dL) | 64 (9‐20) |
| Creatinine (mg/dL) | 4.6 (0.5‐1.5) |
| Calcium (mg/dL) | 15.9 (8.4‐10.2) |
| Albumin (g/dL) | 4.4 (3.5‐5.0) |
| Alkaline phosphatase (U/L) | 92 (38‐126) |
| ALT (U/L) | 24 (11‐66) |
| AST (U/L) | 17 (15‐46) |
| Bilirubin, total (mg/dL) | 0.3 (0.3‐1.2) |
| Ionized calcium (mmol/L) | 1.58 (1.13‐1.32) |
| Creatinine kinase (U/L) | 83 (35‐232) |
| WBC (thousands/cm2) | 12.1 (4‐11) |
| Hemoglobin (g/dL) | 16.5 (14‐18) |
| Hematocrit (%) | 48.6 (40‐54) |
| Platelets (thousands/cm2) | 224 (150‐350) |
| Intact PTH (pg/mL) | 18.78 (15‐65) |
| PTHrP (pmol/L) | 2.5 (5) |
| 25‐OH‐Vitamin D (ng/mL) | 23 (16‐74) |
| H. pylori antibody | Negative |
| SPEP | No M‐spike |
| UPEP | No M‐spike |
| Hospital Day 4 (date of discharge) | |
| BUN (mg/dL) | 21 (9‐20) |
| Creatinine (mg/dL) | 1.6 (0.5‐1.5) |
| Calcium(mg/dL) | 8.4 (8.4‐10.2) |
| WBC (thousands/cm2) | 6.5 (4‐11) |
| Hemoglobin (g/dL) | 14.1 (14‐18) |
| Hematocrit (%) | 40.8 (40‐54) |
| Day 10 (at follow‐up) | |
| BUN (mg/dL) | 16 (9‐20) |
| Creatinine (mg/dL) | 0.9 (0.5‐1.5) |
| Calcium (mg/dL) | 8.1 (8.4‐10.2) |
| Intact PTH (pg/mL) | 240 (15‐65) |
His intact parathyroid (PTH) hormone was 18.8 pg/mL (normal, 15‐65). PTH hormone‐related peptide (PTHrP) was 2.5 pmol/L. After reviewing these laboratory test results, we proceeded with further questioning, during which he admitted to taking approximately 15 to 20 Tums (>7.5 gm of calcium carbonate) daily for dyspepsia rather than the occasional Tums he had originally reported. Over the next 3 days, his calcium decreased to 8.4 mg/dL and his creatinine decreased to 1.6 mg/dL with intravenous hydration. His fatigue improved. His 25‐hydroxy vitamin D (25‐OH‐Vitamin D) level was 23 ng/mL (normal, 16‐74 ng/mL).
At his 1 week follow‐up, his calcium was 8.1 mg/dL, and his creatinine had returned to normal at 0.9 mg/dL. His intact PTH level was elevated at 240 pg/mL.
Discussion
Milk‐alkali syndrome is now believed to be the third most common reason for hypercalcemia hospital admission.2, 3 Malignancy and primary hyperparathyroidism are the only 2 causes of hypercalcemia more common than milk‐alkali syndrome in hospitalized patients; these must be excluded before making a definitive diagnosis of milk‐alkali syndrome. The differential diagnosis for hypercalcemia also includes other less common etiologies such as medications (hydrochlorothiazide and lithium), as well as familial hypocalciuric hypercalcemia, hyperthyroidism, Addison's disease, acromegaly, tertiary hyperparathyroidism, and vitamin D intoxication. Physicians often discover hypercalcemia incidentally on routine laboratory tests, and diagnostic workup should include a thorough history and physical examination, as well as further laboratory evaluation.
The diagnosis of milk‐alkali syndrome requires a history of increased calcium and alkali intake, but is otherwise a diagnosis of exclusion. Given the increasing consumption of nonprescribed calcium supplements, one should have a high index of suspicion for the diagnosis of milk‐alkali syndrome, as patients may not consider calcium carbonate to be hazardous or even a medication and thus may not report calcium carbonate consumption. Patients often view calcium carbonate as a benign treatment for dyspepsia. Its over‐the‐counter availability and economical price make it a common self‐treatment for minor dyspepsia or as prevention of osteoporosis. Calcium supplementation is increasingly added to many products, making it easy for patients to consume large quantities of calcium unknowingly. Of note, without the absorbable alkali supplied by the carbonate in calcium carbonate (Tums), milk‐alkali syndrome does not occur. The amount of calcium carbonate necessary to cause milk‐alkali syndrome is not well known, though it is speculated to be as little as 5 to 10 g of calcium in the form of calcium carbonate, especially in those with other risk factors for hypercalcemia such as chronic renal insufficiency or vomiting.2 Workup of hypercalcemia should entail careful questioning about medications, as well as over‐the‐counter supplements, vitamins, and foods.
Manifestations of the milk‐alkali syndrome include renal failure, metabolic alkalosis, and volume contraction. Normally, the kidneys prevent hypercalcemia by excretion of excess calcium. Hypercalcemia can cause tubular damage and vasoconstriction of the renal afferent arteriole leading to acute renal failure.2, 4 Hypercalcemia can also cause nephrogenic diabetes insipidus, causing impaired renal concentrating ability, leading to increased sodium excretion and volume contraction.2 In addition, alkalosis further impairs calciuresis.2 Laboratory values usually reveal suppressed PTH and vitamin D levels due to hypercalcemia caused by exogenous intake of calcium.5 Hypercalcemia causes suppression of PTH, which can lead to hyperphosphatemia, as well as decreased conversion of vitamin D to the active 1,25‐dihyroxyvitamin‐D form.
The management of hypercalcemia due to milk‐alkali syndrome is supportive and includes saline hydration as well as withholding calcium carbonate. Management of hypercalcemia due to malignancy and hyperparathyroidism includes bisphosphonates with the addition of calcitonin if symptoms are severe.6, 7 There is no evidence that supports the use of bisphosphonates in the treatment of milk‐alkali syndrome. Loop diuretics are sometimes used to promote calciuresis, though evidence is lacking to support this, and it may worsen renal failure.6
In this case, a middle‐aged man took greater than the recommended dose of calcium carbonate for dyspepsia, which led to the development of acute renal failure and hypercalcemia. At first, the patient did not provide an accurate history of the extent of his calcium carbonate ingestion, leading us to focus on hyperparathyroidism or malignancy. With aggressive hydration and cessation of calcium carbonate, his renal function and serum calcium returned to baseline. Because we initially assumed occult malignancy as the most likely diagnosis, we gave the patient pamidronate. The patient did not have a significant alkalemia (serum bicarbonate level was normal). This was thought to be due to the patient's degree of renal failure causing a concomitant metabolic acidosis. The patient's follow‐up elevated PTH level may be explained by bisphosphonate administration or underlying primary hyperparathyroidism. Of note, decreasing calcium levels have also been speculated to be a cause of high PTH levels.8
In conclusion, physicians should have a high index of suspicion for milk‐alkali syndrome in patients with hypercalcemia. Calcium carbonate is responsible for most cases of milk‐alkali syndrome, and clinicians should inquire about the use of this supplement in all patients with hypercalcemia. Milk‐alkali syndrome is no longer a merely a historical curiosity; it is currently the third most common cause of hospital admissions for hypercalcemia.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
- .Landmark article May 15, 1915: Gastric and duodenal ulcer. Medical cure by an efficient removal of gastric juice corrosion. By Bertram W. Sippy.JAMA.1983;250(16):2192–2197.
- ,,,.Milk‐alkali syndrome: a historical review and description of the modern version of the syndrome.Am J Med Sci.2006;331(5):233–242.
- ,,.Milk‐alkali syndrome is a major cause of hypercalcaemia among non‐end‐stage renal disease (non‐ESRD) inpatients.Clin Endocrinol (Oxf).2005;63(5):566–576.
- ,.Milk‐alkali syndrome associated with calcium carbonate consumption. Report of 7 patients with parathyroid hormone levels and an estimate of prevalence among patients hospitalized with hypercalcemia.Medicine (Baltimore).1995;74(2):89–96.
- ,,,,,.The milk‐alkali syndrome. A reversible form of acute renal failure.Arch Intern Med.1993;153(8):1005–1010.
- ,,.Narrative review: furosemide for hypercalcemia: an unproven yet common practice.Ann Intern Med.2008;149(4):259–263.
- .Salmon calcitonin in the acute management of hypercalcemia.Calcif Tissue Int.1990;46(suppl):S26–S30.
- ,.Milk alkali syndrome. Does it exist and can it be differentiated from primary hyperparathyroidism?Ann Surg.1983;197(4):427–433.
Small Bowel Obstruction by Gallstone Ileus
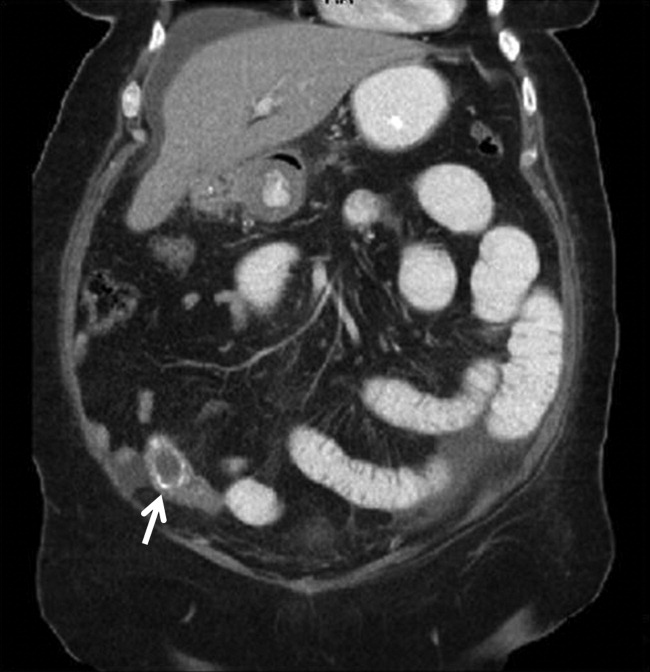
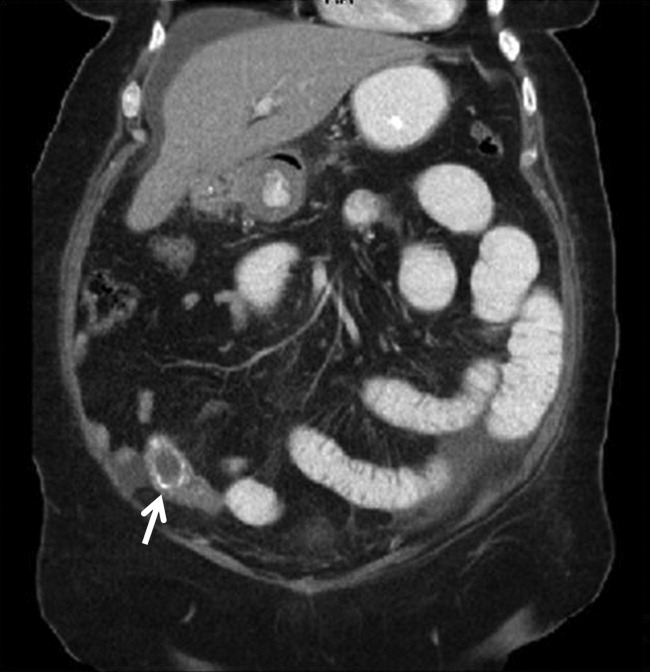
A 67‐year‐old female presented with nausea and nonspecific abdominal pain occurring intermittently for 2 weeks. Physical examination revealed no abdominal guarding or rigidity and was significant only for slightly hypoactive bowel sounds. Routine laboratory evaluation including liver panel was unremarkable. X‐ray of the abdomen showed air fluid levels, and a diagnosis of small bowel obstruction/emleus secondary to adhesions from a previous hysterectomy was established. Conservative management with nasogastric suction, intravenous (IV) fluids and nil‐by‐mouth was continued for 3 days but no clinical improvement was seen. Due to continued abdominal pain, an ultrasound of the abdomen was done, which revealed cholelithiasis and chronic cholecystitis. At this point the patient was transferred to our facility and a computerized axial tomography (CAT) scan of the abdomen was done, which revealed a gallstone in the ileum (Figure 1) and the diagnosis of gallstone ileus was made. Air in the biliary tract (pneumobilia) was noted, suggesting the presence of a fistula between the gallbladder and the gastrointestinal (GI) tract. The fistula itself could not be clearly visualized. The patient was immediately taken to surgery, where small bowel exploration was done. A large gallstone completely obstructing the terminal ileum was removed by enterotomy. The gallbladder was adherent to the stomach and a cholecystogastric fistula with a gallstone coming out on the gastric end was noted. This gallstone was removed and the cholecystogastric fistula was repaired. Cholecystectomy was done at the same time. The patient recovered without any complications and has been doing well.
Discussion
Gallstone ileus accounts for approximately 1% to 2% cases of small bowel obstruction, most of which are in the elderly population. It is much more common in females as compared to males.1 Although the morbidity and mortality associated with gallstone ileus has reduced in comparison to the previous decade, probably due to the more effective usage of imaging techniques like ultrasound, CAT scan, magnetic resonance imaging (MRI), and endoscopy in the diagnostic evaluation of abdominal pain, the numbers still range between 12% to 25%.1, 2 Early diagnosis plays a major role in reducing the mortality in these patients. Therefore, it is important to consider gallstone ileus in the differential diagnosis of an elderly patient presenting with bowel obstruction.
Gallstone ileus is usually associated with a biliaryenteric fistula that allows the passage of a gallstone from the gallbladder into the bowel. This gallstone gets impacted in the gastrointestinal lumen and causes mechanical bowel obstruction. The term gallstone ileus is a misnomer as the gallstone causes actual obstruction rather than just ileus. Considering the high prevalence of cholelithiasis, it must be realized that formation of cholecystoenteric fistula is relatively rare (about 2% in patients with cholecystitis). It is proposed that pericholecystic inflammation after an episode of cholecystitis results in the formation of adhesions between the biliary and gastrointestinal tracts. The gallstone causes pressure necrosis of the biliary wall and then erodes through it to form a fistulous communication with the adherent enteric system. Among these fistulous communications, cholecystoduodenal fistulas are the most common (60%) while cholecystocolonic and cholecystogastric fistulas are also seen. After the biliary stone has eroded through the enteric wall, air within the intestinal tract now freely enters the biliary system, leading to an appearance of pneumobilia on imaging studies. Mirizzi syndrome, described as common hepatic duct obstruction caused by an extrinsic compression from an impacted stone in the cystic duct, is often associated with gallstone ileus.3
Once the gallstone enters the enteric tract through the fistula and traverses down the gastrointestinal tract, it causes intermittent abdominal pain, nausea, and vomiting. The symptoms may be spread over multiple days as the gallstone causes transient obstruction with its impaction and disimpaction. These obstructing gallstones that cause luminal obstruction are usually larger than 2 cm. Majority of them will traverse the duodenum, jejunum, and small intestine, and finally get lodged in the terminal ileum (60%), the narrowest part of the small intestine. Other sites where obstruction may occur include jejunum (16%), duodenum, stomach, and colon.1 Bouveret's syndrome is a variant of gallstone ileus wherein the gallstone impacts in the pylorus of the stomach or the duodenum, leading to gastric outlet obstruction.3
The diagnosis of gallstone ileus is not always straightforward and requires a high index of suspicion. It is classically described by the Rigler's triadpneumobilia, partial or complete bowel obstruction, and ectopic gallstone,4 although often all 3 signs are not elicited. Most patients need open enterolithotomy for relief of bowel obstruction. Cholecystectomy may or may not be performed. The literature is controversial regarding the best approach of surgical management. Two surgical approaches are equally accepted: (1) a 1‐stage approach, which includes enterolithotomy, cholecystectomy, and fistula repair at the same time; and (2) the other option is a 2‐stage approach in which enterolithotomy is performed first and biliary surgery is performed later, if indicated. The patient's age, comorbidities, and the associated surgical risks are often used to decide between the 2 surgical approaches.
Gallstone ileus should be considered in the differential for the etiology of small bowel obstruction, especially in an elderly female known to have cholelithiasis.
- ,.Gallstone ileus (a review of 1001 reported cases).Am Surg.1994;60:441–446.
- ,,, et al.Improving the outcome in gallstone ileus.Am J Surg.1986;151:572–576.
- ,,.The relationship of Mirizzi syndrome and cholecystoenteric fistula: validation of a modified classification.World J Surg.2008;32(10):2237–2243.
- ,,.Gallstone obstruction: pathogenesis and roentgen manifestations.JAMA.1941;117:1753–1759.
A 67‐year‐old female presented with nausea and nonspecific abdominal pain occurring intermittently for 2 weeks. Physical examination revealed no abdominal guarding or rigidity and was significant only for slightly hypoactive bowel sounds. Routine laboratory evaluation including liver panel was unremarkable. X‐ray of the abdomen showed air fluid levels, and a diagnosis of small bowel obstruction/emleus secondary to adhesions from a previous hysterectomy was established. Conservative management with nasogastric suction, intravenous (IV) fluids and nil‐by‐mouth was continued for 3 days but no clinical improvement was seen. Due to continued abdominal pain, an ultrasound of the abdomen was done, which revealed cholelithiasis and chronic cholecystitis. At this point the patient was transferred to our facility and a computerized axial tomography (CAT) scan of the abdomen was done, which revealed a gallstone in the ileum (Figure 1) and the diagnosis of gallstone ileus was made. Air in the biliary tract (pneumobilia) was noted, suggesting the presence of a fistula between the gallbladder and the gastrointestinal (GI) tract. The fistula itself could not be clearly visualized. The patient was immediately taken to surgery, where small bowel exploration was done. A large gallstone completely obstructing the terminal ileum was removed by enterotomy. The gallbladder was adherent to the stomach and a cholecystogastric fistula with a gallstone coming out on the gastric end was noted. This gallstone was removed and the cholecystogastric fistula was repaired. Cholecystectomy was done at the same time. The patient recovered without any complications and has been doing well.
Discussion
Gallstone ileus accounts for approximately 1% to 2% cases of small bowel obstruction, most of which are in the elderly population. It is much more common in females as compared to males.1 Although the morbidity and mortality associated with gallstone ileus has reduced in comparison to the previous decade, probably due to the more effective usage of imaging techniques like ultrasound, CAT scan, magnetic resonance imaging (MRI), and endoscopy in the diagnostic evaluation of abdominal pain, the numbers still range between 12% to 25%.1, 2 Early diagnosis plays a major role in reducing the mortality in these patients. Therefore, it is important to consider gallstone ileus in the differential diagnosis of an elderly patient presenting with bowel obstruction.
Gallstone ileus is usually associated with a biliaryenteric fistula that allows the passage of a gallstone from the gallbladder into the bowel. This gallstone gets impacted in the gastrointestinal lumen and causes mechanical bowel obstruction. The term gallstone ileus is a misnomer as the gallstone causes actual obstruction rather than just ileus. Considering the high prevalence of cholelithiasis, it must be realized that formation of cholecystoenteric fistula is relatively rare (about 2% in patients with cholecystitis). It is proposed that pericholecystic inflammation after an episode of cholecystitis results in the formation of adhesions between the biliary and gastrointestinal tracts. The gallstone causes pressure necrosis of the biliary wall and then erodes through it to form a fistulous communication with the adherent enteric system. Among these fistulous communications, cholecystoduodenal fistulas are the most common (60%) while cholecystocolonic and cholecystogastric fistulas are also seen. After the biliary stone has eroded through the enteric wall, air within the intestinal tract now freely enters the biliary system, leading to an appearance of pneumobilia on imaging studies. Mirizzi syndrome, described as common hepatic duct obstruction caused by an extrinsic compression from an impacted stone in the cystic duct, is often associated with gallstone ileus.3
Once the gallstone enters the enteric tract through the fistula and traverses down the gastrointestinal tract, it causes intermittent abdominal pain, nausea, and vomiting. The symptoms may be spread over multiple days as the gallstone causes transient obstruction with its impaction and disimpaction. These obstructing gallstones that cause luminal obstruction are usually larger than 2 cm. Majority of them will traverse the duodenum, jejunum, and small intestine, and finally get lodged in the terminal ileum (60%), the narrowest part of the small intestine. Other sites where obstruction may occur include jejunum (16%), duodenum, stomach, and colon.1 Bouveret's syndrome is a variant of gallstone ileus wherein the gallstone impacts in the pylorus of the stomach or the duodenum, leading to gastric outlet obstruction.3
The diagnosis of gallstone ileus is not always straightforward and requires a high index of suspicion. It is classically described by the Rigler's triadpneumobilia, partial or complete bowel obstruction, and ectopic gallstone,4 although often all 3 signs are not elicited. Most patients need open enterolithotomy for relief of bowel obstruction. Cholecystectomy may or may not be performed. The literature is controversial regarding the best approach of surgical management. Two surgical approaches are equally accepted: (1) a 1‐stage approach, which includes enterolithotomy, cholecystectomy, and fistula repair at the same time; and (2) the other option is a 2‐stage approach in which enterolithotomy is performed first and biliary surgery is performed later, if indicated. The patient's age, comorbidities, and the associated surgical risks are often used to decide between the 2 surgical approaches.
Gallstone ileus should be considered in the differential for the etiology of small bowel obstruction, especially in an elderly female known to have cholelithiasis.
A 67‐year‐old female presented with nausea and nonspecific abdominal pain occurring intermittently for 2 weeks. Physical examination revealed no abdominal guarding or rigidity and was significant only for slightly hypoactive bowel sounds. Routine laboratory evaluation including liver panel was unremarkable. X‐ray of the abdomen showed air fluid levels, and a diagnosis of small bowel obstruction/emleus secondary to adhesions from a previous hysterectomy was established. Conservative management with nasogastric suction, intravenous (IV) fluids and nil‐by‐mouth was continued for 3 days but no clinical improvement was seen. Due to continued abdominal pain, an ultrasound of the abdomen was done, which revealed cholelithiasis and chronic cholecystitis. At this point the patient was transferred to our facility and a computerized axial tomography (CAT) scan of the abdomen was done, which revealed a gallstone in the ileum (Figure 1) and the diagnosis of gallstone ileus was made. Air in the biliary tract (pneumobilia) was noted, suggesting the presence of a fistula between the gallbladder and the gastrointestinal (GI) tract. The fistula itself could not be clearly visualized. The patient was immediately taken to surgery, where small bowel exploration was done. A large gallstone completely obstructing the terminal ileum was removed by enterotomy. The gallbladder was adherent to the stomach and a cholecystogastric fistula with a gallstone coming out on the gastric end was noted. This gallstone was removed and the cholecystogastric fistula was repaired. Cholecystectomy was done at the same time. The patient recovered without any complications and has been doing well.
Discussion
Gallstone ileus accounts for approximately 1% to 2% cases of small bowel obstruction, most of which are in the elderly population. It is much more common in females as compared to males.1 Although the morbidity and mortality associated with gallstone ileus has reduced in comparison to the previous decade, probably due to the more effective usage of imaging techniques like ultrasound, CAT scan, magnetic resonance imaging (MRI), and endoscopy in the diagnostic evaluation of abdominal pain, the numbers still range between 12% to 25%.1, 2 Early diagnosis plays a major role in reducing the mortality in these patients. Therefore, it is important to consider gallstone ileus in the differential diagnosis of an elderly patient presenting with bowel obstruction.
Gallstone ileus is usually associated with a biliaryenteric fistula that allows the passage of a gallstone from the gallbladder into the bowel. This gallstone gets impacted in the gastrointestinal lumen and causes mechanical bowel obstruction. The term gallstone ileus is a misnomer as the gallstone causes actual obstruction rather than just ileus. Considering the high prevalence of cholelithiasis, it must be realized that formation of cholecystoenteric fistula is relatively rare (about 2% in patients with cholecystitis). It is proposed that pericholecystic inflammation after an episode of cholecystitis results in the formation of adhesions between the biliary and gastrointestinal tracts. The gallstone causes pressure necrosis of the biliary wall and then erodes through it to form a fistulous communication with the adherent enteric system. Among these fistulous communications, cholecystoduodenal fistulas are the most common (60%) while cholecystocolonic and cholecystogastric fistulas are also seen. After the biliary stone has eroded through the enteric wall, air within the intestinal tract now freely enters the biliary system, leading to an appearance of pneumobilia on imaging studies. Mirizzi syndrome, described as common hepatic duct obstruction caused by an extrinsic compression from an impacted stone in the cystic duct, is often associated with gallstone ileus.3
Once the gallstone enters the enteric tract through the fistula and traverses down the gastrointestinal tract, it causes intermittent abdominal pain, nausea, and vomiting. The symptoms may be spread over multiple days as the gallstone causes transient obstruction with its impaction and disimpaction. These obstructing gallstones that cause luminal obstruction are usually larger than 2 cm. Majority of them will traverse the duodenum, jejunum, and small intestine, and finally get lodged in the terminal ileum (60%), the narrowest part of the small intestine. Other sites where obstruction may occur include jejunum (16%), duodenum, stomach, and colon.1 Bouveret's syndrome is a variant of gallstone ileus wherein the gallstone impacts in the pylorus of the stomach or the duodenum, leading to gastric outlet obstruction.3
The diagnosis of gallstone ileus is not always straightforward and requires a high index of suspicion. It is classically described by the Rigler's triadpneumobilia, partial or complete bowel obstruction, and ectopic gallstone,4 although often all 3 signs are not elicited. Most patients need open enterolithotomy for relief of bowel obstruction. Cholecystectomy may or may not be performed. The literature is controversial regarding the best approach of surgical management. Two surgical approaches are equally accepted: (1) a 1‐stage approach, which includes enterolithotomy, cholecystectomy, and fistula repair at the same time; and (2) the other option is a 2‐stage approach in which enterolithotomy is performed first and biliary surgery is performed later, if indicated. The patient's age, comorbidities, and the associated surgical risks are often used to decide between the 2 surgical approaches.
Gallstone ileus should be considered in the differential for the etiology of small bowel obstruction, especially in an elderly female known to have cholelithiasis.
- ,.Gallstone ileus (a review of 1001 reported cases).Am Surg.1994;60:441–446.
- ,,, et al.Improving the outcome in gallstone ileus.Am J Surg.1986;151:572–576.
- ,,.The relationship of Mirizzi syndrome and cholecystoenteric fistula: validation of a modified classification.World J Surg.2008;32(10):2237–2243.
- ,,.Gallstone obstruction: pathogenesis and roentgen manifestations.JAMA.1941;117:1753–1759.
- ,.Gallstone ileus (a review of 1001 reported cases).Am Surg.1994;60:441–446.
- ,,, et al.Improving the outcome in gallstone ileus.Am J Surg.1986;151:572–576.
- ,,.The relationship of Mirizzi syndrome and cholecystoenteric fistula: validation of a modified classification.World J Surg.2008;32(10):2237–2243.
- ,,.Gallstone obstruction: pathogenesis and roentgen manifestations.JAMA.1941;117:1753–1759.
Gastric Involvement in NSG
Necrotizing sarcoid granulomatosis (NSG) is an immune system disorder characterized by necrotizing granulomas, as opposed to noncaseating granulomas in classical sarcoidosis. Over the past 3 decades there have been over 120 reported cases of NSG with pulmonary and extrapulmonary involvement.1 We present a patient found to have histological evidence of necrotizing granuloma in her gastric antrum, and we believe this is the first Case Report of NSG involving the stomach.
Case Report
A 21‐year‐old African‐American female first presented to an outside hospital with fever, epigastric pain, shortness of breath, and headache. Two days later she complained of nonbloody, nonbilious vomiting, and was found to have leukocytosis (17,770 cells/m3), elevated lipase (224 U/L), elevated C‐reactive protein (14.7 mg/L), and an inflamed pancreas on computed tomography (CT). She was treated conservatively for pancreatitis and started on ampicillin/sulbactam. After 2 contrast CT scans on consecutive days, she developed acute renal failure (creatinine 2.0 mg/dL compared to baseline of 1.0 mg/dL), and was transferred to our hospital for further evaluation and management.
Upon transfer, the patient's temperature was 37.0C, pulse was 102 beats/minute, blood pressure was 141/84 mm Hg, and oxygen saturation was 94%. On examination, she was tender to palpation in her epigastrium and right upper quadrant, but the remainder of the physical exam was unremarkable. She was started on moxifloxacin and managed with intravenous (IV) fluid hydration and pain control. Within 3 days, CT showed resolving pancreatitis, magnetic resonance cholangiopancreatography (MRCP) was negative, and her creatinine began normalizing (1.3 mg/dL). Nonetheless, she continued to complain of abdominal pain, shortness of breath, and intermittent low‐grade fevers. She then also developed bilateral panuveitis requiring high‐dose steroid eye drops.
Chest x‐ray showed subtle bilateral nodular and bronchiolitic infiltrates with no evidence of enlarged hilar nodes, and subsequent bronchoscopy showed no abnormalities. Additional workup included negative blood and urine cultures, purified protein derivative (PPD), and Clostridium difficile assay; as well as negative human immunodeficiency virus (HIV), cryptococcus, Helicobacter pylori, Borellia burgdorferi, syphilis (fluorescent treponemal antibody), aspergillus, histoplasma, and rheumatological serologies. Her white blood count (20,900 cells/m3), C‐reactive protein (6.6 mg/L), and erythrocyte sedimentation rate (100 mm/hour) remained elevated.
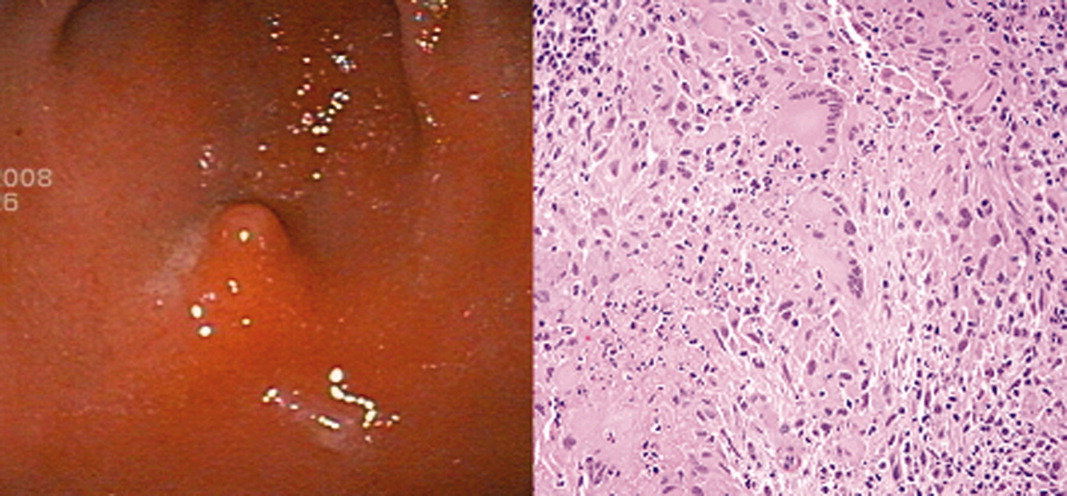
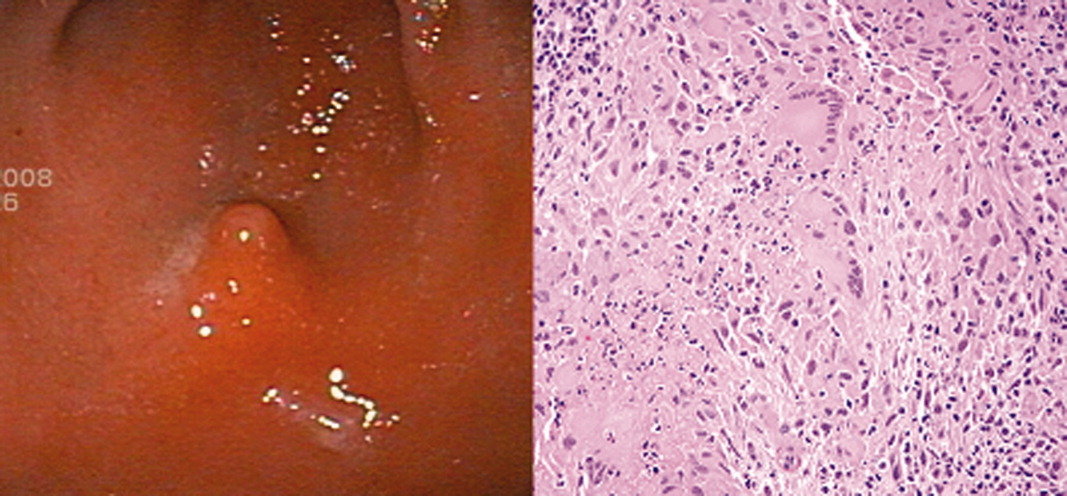
The patient continued to complain of epigastric pain. Repeat abdominal CT scan showed large retroperitoneal and mesenteric lymph nodes, and esophagogastroduodenoscopy (EGD) showed gastritis with an antral nodule (Figure 1). Biopsy of the nodule revealed a necrotizing granuloma with mixed cellular infiltrate. Biopsy stains were negative for bacteria, borellia, treponemes, acid‐fast bacilli, and fungi. The patient was diagnosed with necrotizing sarcoid granulomatosis and started on an oral prednisone taper. She responded to steroid treatment with prompt resolution of her uveitis, shortness of breath, abdominal pain, and fevers. She was discharged following treatment, has continued to do well, and is seen regularly at the sarcoid clinic for follow‐up.
Discussion
NSG was first characterized as a distinct variation from sarcoidosis by Liebow2 in 1973, and was noted to have 3 characteristic differences: (1) histological evidence of sarcoid‐appearing granuloma and necrosis, (2) pulmonary nodules without hilar lymphadenopathy on imaging, and (3) a clinically benign course. Over the past 3 decades, there have been more than 120 cases of reported NSG involving the lungs, gastrointestinal tract, kidney, skin, and central nervous system.1 Since Liebow's2 description, newly reported NSG cases have generally been found to be consistent with the aforementioned criteria, although hilar lymphadenopathy may be particularly more common than previously thought. One review noted a range of 8% to 79% prevalence of hilar lymphadenopathy in reported NSG series.3 Therefore, while hilar lymphadenopathy still currently appears to be less common in NSG than in typical sarcoidosis, its presence should not rule out the diagnosis.
Our patient's history is consistent with Liebow's2 criteria since she had histological evidence of necrotizing granuloma and pulmonary involvement without hilar lymphadenopathy, and responded promptly to steroid treatment. To our knowledge, this is the first case of NSG reported in the stomach.
While less than 1% of sarcoid patients are reported to have gastrointestinal involvement, all of these cases have either been noncaseating granulomas (classical sarcoidosis) or were found outside the stomach.1, 4, 5 Most of the data regarding sarcoid symptomatology and treatment are derived from reports on classical sarcoidosis. In classical sarcoidosis, there is gastric antral involvement in approximately 10% of patients with systemic disease.4 These patients may present with nausea, vomiting, and weight loss, and are often effectively treated with a single dose of prednisone 30 to 40 mg followed by a maintenance dose of 10 to 15 mg daily over 6 months.5 Less data are available regarding necrotizing sarcoid presentation and treatment, especially with regard to gastrointestinal involvement.
We hope to raise awareness regarding: (1) the variation in noncaseating versus necrotizing sarcoid‐type disorders, (2) the benefit of steroid treatment once infectious etiologies are ruled out, and (3) the potential for further extrapulmonary involvement in previously unreported organ systems.
- ,,.Necrotizing sarcoid granulomatosis mimicking an intracranial neoplasm: clinicopathologic features and review of the literature.Mod Pathol.2000;13(8)909–913.
- .Pulmonary angiitis and granulomatosis.Am J Respir Dis.1973;108:1–18.
- ,,,.Pulmonary angiitis and granulomatosis: radiologic‐pathologic correlation.Radiographics.1998;18(3):687–710.
- ,,, et al.Multiple antral ulcers in gastric sarcoid.J Clin Gastroenterol.1997;24(2):97–99.
- ,,.Gastric sarcoidosis: a case report and review of the literature.South Med J.2007;100(3):301–303.
Necrotizing sarcoid granulomatosis (NSG) is an immune system disorder characterized by necrotizing granulomas, as opposed to noncaseating granulomas in classical sarcoidosis. Over the past 3 decades there have been over 120 reported cases of NSG with pulmonary and extrapulmonary involvement.1 We present a patient found to have histological evidence of necrotizing granuloma in her gastric antrum, and we believe this is the first Case Report of NSG involving the stomach.
Case Report
A 21‐year‐old African‐American female first presented to an outside hospital with fever, epigastric pain, shortness of breath, and headache. Two days later she complained of nonbloody, nonbilious vomiting, and was found to have leukocytosis (17,770 cells/m3), elevated lipase (224 U/L), elevated C‐reactive protein (14.7 mg/L), and an inflamed pancreas on computed tomography (CT). She was treated conservatively for pancreatitis and started on ampicillin/sulbactam. After 2 contrast CT scans on consecutive days, she developed acute renal failure (creatinine 2.0 mg/dL compared to baseline of 1.0 mg/dL), and was transferred to our hospital for further evaluation and management.
Upon transfer, the patient's temperature was 37.0C, pulse was 102 beats/minute, blood pressure was 141/84 mm Hg, and oxygen saturation was 94%. On examination, she was tender to palpation in her epigastrium and right upper quadrant, but the remainder of the physical exam was unremarkable. She was started on moxifloxacin and managed with intravenous (IV) fluid hydration and pain control. Within 3 days, CT showed resolving pancreatitis, magnetic resonance cholangiopancreatography (MRCP) was negative, and her creatinine began normalizing (1.3 mg/dL). Nonetheless, she continued to complain of abdominal pain, shortness of breath, and intermittent low‐grade fevers. She then also developed bilateral panuveitis requiring high‐dose steroid eye drops.
Chest x‐ray showed subtle bilateral nodular and bronchiolitic infiltrates with no evidence of enlarged hilar nodes, and subsequent bronchoscopy showed no abnormalities. Additional workup included negative blood and urine cultures, purified protein derivative (PPD), and Clostridium difficile assay; as well as negative human immunodeficiency virus (HIV), cryptococcus, Helicobacter pylori, Borellia burgdorferi, syphilis (fluorescent treponemal antibody), aspergillus, histoplasma, and rheumatological serologies. Her white blood count (20,900 cells/m3), C‐reactive protein (6.6 mg/L), and erythrocyte sedimentation rate (100 mm/hour) remained elevated.
The patient continued to complain of epigastric pain. Repeat abdominal CT scan showed large retroperitoneal and mesenteric lymph nodes, and esophagogastroduodenoscopy (EGD) showed gastritis with an antral nodule (Figure 1). Biopsy of the nodule revealed a necrotizing granuloma with mixed cellular infiltrate. Biopsy stains were negative for bacteria, borellia, treponemes, acid‐fast bacilli, and fungi. The patient was diagnosed with necrotizing sarcoid granulomatosis and started on an oral prednisone taper. She responded to steroid treatment with prompt resolution of her uveitis, shortness of breath, abdominal pain, and fevers. She was discharged following treatment, has continued to do well, and is seen regularly at the sarcoid clinic for follow‐up.
Discussion
NSG was first characterized as a distinct variation from sarcoidosis by Liebow2 in 1973, and was noted to have 3 characteristic differences: (1) histological evidence of sarcoid‐appearing granuloma and necrosis, (2) pulmonary nodules without hilar lymphadenopathy on imaging, and (3) a clinically benign course. Over the past 3 decades, there have been more than 120 cases of reported NSG involving the lungs, gastrointestinal tract, kidney, skin, and central nervous system.1 Since Liebow's2 description, newly reported NSG cases have generally been found to be consistent with the aforementioned criteria, although hilar lymphadenopathy may be particularly more common than previously thought. One review noted a range of 8% to 79% prevalence of hilar lymphadenopathy in reported NSG series.3 Therefore, while hilar lymphadenopathy still currently appears to be less common in NSG than in typical sarcoidosis, its presence should not rule out the diagnosis.
Our patient's history is consistent with Liebow's2 criteria since she had histological evidence of necrotizing granuloma and pulmonary involvement without hilar lymphadenopathy, and responded promptly to steroid treatment. To our knowledge, this is the first case of NSG reported in the stomach.
While less than 1% of sarcoid patients are reported to have gastrointestinal involvement, all of these cases have either been noncaseating granulomas (classical sarcoidosis) or were found outside the stomach.1, 4, 5 Most of the data regarding sarcoid symptomatology and treatment are derived from reports on classical sarcoidosis. In classical sarcoidosis, there is gastric antral involvement in approximately 10% of patients with systemic disease.4 These patients may present with nausea, vomiting, and weight loss, and are often effectively treated with a single dose of prednisone 30 to 40 mg followed by a maintenance dose of 10 to 15 mg daily over 6 months.5 Less data are available regarding necrotizing sarcoid presentation and treatment, especially with regard to gastrointestinal involvement.
We hope to raise awareness regarding: (1) the variation in noncaseating versus necrotizing sarcoid‐type disorders, (2) the benefit of steroid treatment once infectious etiologies are ruled out, and (3) the potential for further extrapulmonary involvement in previously unreported organ systems.
Necrotizing sarcoid granulomatosis (NSG) is an immune system disorder characterized by necrotizing granulomas, as opposed to noncaseating granulomas in classical sarcoidosis. Over the past 3 decades there have been over 120 reported cases of NSG with pulmonary and extrapulmonary involvement.1 We present a patient found to have histological evidence of necrotizing granuloma in her gastric antrum, and we believe this is the first Case Report of NSG involving the stomach.
Case Report
A 21‐year‐old African‐American female first presented to an outside hospital with fever, epigastric pain, shortness of breath, and headache. Two days later she complained of nonbloody, nonbilious vomiting, and was found to have leukocytosis (17,770 cells/m3), elevated lipase (224 U/L), elevated C‐reactive protein (14.7 mg/L), and an inflamed pancreas on computed tomography (CT). She was treated conservatively for pancreatitis and started on ampicillin/sulbactam. After 2 contrast CT scans on consecutive days, she developed acute renal failure (creatinine 2.0 mg/dL compared to baseline of 1.0 mg/dL), and was transferred to our hospital for further evaluation and management.
Upon transfer, the patient's temperature was 37.0C, pulse was 102 beats/minute, blood pressure was 141/84 mm Hg, and oxygen saturation was 94%. On examination, she was tender to palpation in her epigastrium and right upper quadrant, but the remainder of the physical exam was unremarkable. She was started on moxifloxacin and managed with intravenous (IV) fluid hydration and pain control. Within 3 days, CT showed resolving pancreatitis, magnetic resonance cholangiopancreatography (MRCP) was negative, and her creatinine began normalizing (1.3 mg/dL). Nonetheless, she continued to complain of abdominal pain, shortness of breath, and intermittent low‐grade fevers. She then also developed bilateral panuveitis requiring high‐dose steroid eye drops.
Chest x‐ray showed subtle bilateral nodular and bronchiolitic infiltrates with no evidence of enlarged hilar nodes, and subsequent bronchoscopy showed no abnormalities. Additional workup included negative blood and urine cultures, purified protein derivative (PPD), and Clostridium difficile assay; as well as negative human immunodeficiency virus (HIV), cryptococcus, Helicobacter pylori, Borellia burgdorferi, syphilis (fluorescent treponemal antibody), aspergillus, histoplasma, and rheumatological serologies. Her white blood count (20,900 cells/m3), C‐reactive protein (6.6 mg/L), and erythrocyte sedimentation rate (100 mm/hour) remained elevated.
The patient continued to complain of epigastric pain. Repeat abdominal CT scan showed large retroperitoneal and mesenteric lymph nodes, and esophagogastroduodenoscopy (EGD) showed gastritis with an antral nodule (Figure 1). Biopsy of the nodule revealed a necrotizing granuloma with mixed cellular infiltrate. Biopsy stains were negative for bacteria, borellia, treponemes, acid‐fast bacilli, and fungi. The patient was diagnosed with necrotizing sarcoid granulomatosis and started on an oral prednisone taper. She responded to steroid treatment with prompt resolution of her uveitis, shortness of breath, abdominal pain, and fevers. She was discharged following treatment, has continued to do well, and is seen regularly at the sarcoid clinic for follow‐up.
Discussion
NSG was first characterized as a distinct variation from sarcoidosis by Liebow2 in 1973, and was noted to have 3 characteristic differences: (1) histological evidence of sarcoid‐appearing granuloma and necrosis, (2) pulmonary nodules without hilar lymphadenopathy on imaging, and (3) a clinically benign course. Over the past 3 decades, there have been more than 120 cases of reported NSG involving the lungs, gastrointestinal tract, kidney, skin, and central nervous system.1 Since Liebow's2 description, newly reported NSG cases have generally been found to be consistent with the aforementioned criteria, although hilar lymphadenopathy may be particularly more common than previously thought. One review noted a range of 8% to 79% prevalence of hilar lymphadenopathy in reported NSG series.3 Therefore, while hilar lymphadenopathy still currently appears to be less common in NSG than in typical sarcoidosis, its presence should not rule out the diagnosis.
Our patient's history is consistent with Liebow's2 criteria since she had histological evidence of necrotizing granuloma and pulmonary involvement without hilar lymphadenopathy, and responded promptly to steroid treatment. To our knowledge, this is the first case of NSG reported in the stomach.
While less than 1% of sarcoid patients are reported to have gastrointestinal involvement, all of these cases have either been noncaseating granulomas (classical sarcoidosis) or were found outside the stomach.1, 4, 5 Most of the data regarding sarcoid symptomatology and treatment are derived from reports on classical sarcoidosis. In classical sarcoidosis, there is gastric antral involvement in approximately 10% of patients with systemic disease.4 These patients may present with nausea, vomiting, and weight loss, and are often effectively treated with a single dose of prednisone 30 to 40 mg followed by a maintenance dose of 10 to 15 mg daily over 6 months.5 Less data are available regarding necrotizing sarcoid presentation and treatment, especially with regard to gastrointestinal involvement.
We hope to raise awareness regarding: (1) the variation in noncaseating versus necrotizing sarcoid‐type disorders, (2) the benefit of steroid treatment once infectious etiologies are ruled out, and (3) the potential for further extrapulmonary involvement in previously unreported organ systems.
- ,,.Necrotizing sarcoid granulomatosis mimicking an intracranial neoplasm: clinicopathologic features and review of the literature.Mod Pathol.2000;13(8)909–913.
- .Pulmonary angiitis and granulomatosis.Am J Respir Dis.1973;108:1–18.
- ,,,.Pulmonary angiitis and granulomatosis: radiologic‐pathologic correlation.Radiographics.1998;18(3):687–710.
- ,,, et al.Multiple antral ulcers in gastric sarcoid.J Clin Gastroenterol.1997;24(2):97–99.
- ,,.Gastric sarcoidosis: a case report and review of the literature.South Med J.2007;100(3):301–303.
- ,,.Necrotizing sarcoid granulomatosis mimicking an intracranial neoplasm: clinicopathologic features and review of the literature.Mod Pathol.2000;13(8)909–913.
- .Pulmonary angiitis and granulomatosis.Am J Respir Dis.1973;108:1–18.
- ,,,.Pulmonary angiitis and granulomatosis: radiologic‐pathologic correlation.Radiographics.1998;18(3):687–710.
- ,,, et al.Multiple antral ulcers in gastric sarcoid.J Clin Gastroenterol.1997;24(2):97–99.
- ,,.Gastric sarcoidosis: a case report and review of the literature.South Med J.2007;100(3):301–303.