User login
Case Studies in Toxicology: A Patchwork of Problems in Parkinson Patients
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?

What medications are used to treat PD? What are some associated complications?
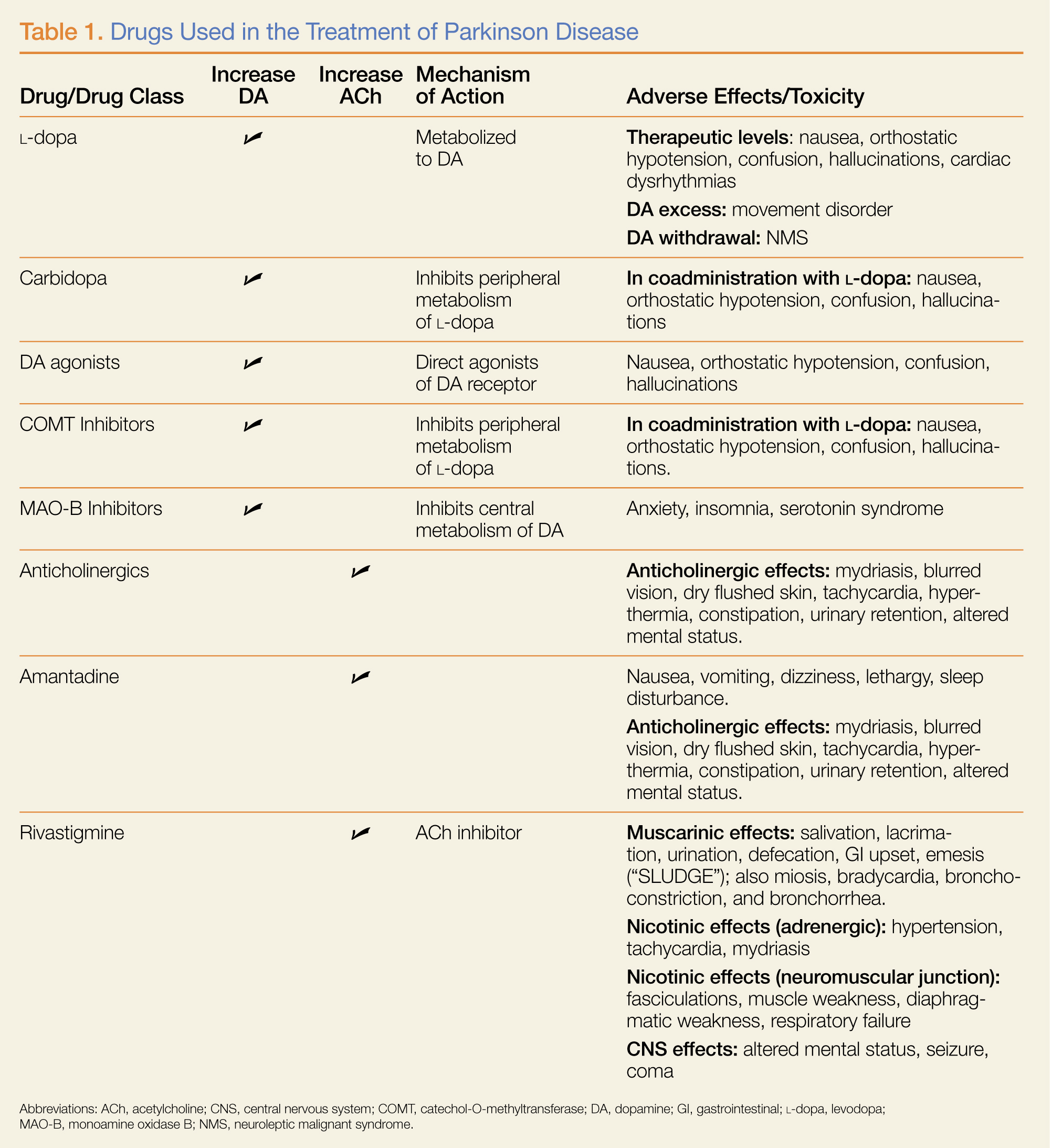
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 76-year-old man with Parkinson disease (PD) and hypertension presented to the ED with acute onset of severe tremulousness, blurred vision, salivation, lacrimation, diffuse muscle aches, and extremity weakness. His initial vital signs were: blood pressure, 175/74 mm Hg; heart rate, 62 beats/minute; respiratory rate, 16 breaths/minute; temperature, 37°C (98.6°F). Oxygen saturation was 100% on room air. On physical examination, the patient had excessive lacrimation and salivation, a coarse resting tremor, and 2/5 strength in both the upper and lower extremities. The remainder of the examination, including abdominal and pulmonary systems, was unremarkable compared with baseline findings.
How does the pathophysiology of PD explain how treatments are targeted?
What medications are used to treat PD? What are some associated complications?
Dopamine Precursors and Agonists
(L-dopa) can be combined with the L-amino acid decarboxylase inhibitor carbidopa to prevent peripheral metabolism by this enzyme and thereby increase brain concentrations of DA following metabolism by DA decarboxylase in the central nervous system (CNS).1 Dopamine agonists, including bromocriptine, ropinirole, and pramipexole, do not depend on endogenous conversion to DA and have substantially longer durations of action, limiting the dose-related fluctuations in motor function common in some PD patients taking L-dopa.1 For these reasons, DA agonists have often replaced L-dopa as initial treatment, especially in younger patients. Catechol-O-methyltransferase inhibitors (tolcapone, entacapone) prevent peripheral breakdown of DA, allowing a higher fraction to reach the CNS.
With respect to side effects, all of the dopaminergic medications can cause nausea, hallucinations, confusion, and orthostatic hypotension.
Anticholinergic Drugs
Although the precise mechanism by which anticholinergic drugs improve PD is not fully understood, agents such as trihexyphenidyl, benztropine mesylate, and diphenhydramine hydrochloride were prescribed even before the discovery of L-dopa and continue to be used today.1 Adverse effects are a function of the antimuscarinic (anticholinergic) properties of the drugs and may include mydriasis and blurred vision, dry flushed skin, tachycardia, hyperthermia, constipation, urinary retention, and altered mental status.
Amantadine
In addition to the anticholinergics, amantadine is also used to treat PD. This antiviral agent alters DA release in the brain, produces anticholinergic effects, and blocks N-methyl-D-aspartate glutamate receptors.1 Common adverse drug effects include anticholinergic signs as well as nausea, vomiting, dizziness, lethargy, and sleep disturbance, all of which are usually mild and reversible.
Case Continuation
A review of the patient’s medication history revealed he has been taking L-dopa/carbidopa. In addition to L-dopa/carbidopa, he was recently prescribed transdermal rivastigmine patches (13.3 mg/24 h). At bedtime the evening prior to presentation, the patient applied more than 20 rivastigmine patches. Approximately 5 hours later, he awoke with the previously described findings whereupon his wife removed the patches and brought him to the ED.
What is rivastigmine and what is its role in PD
Rivastigmine is a carbamate-type cholinesterase inhibitor (CEI) indicated for the treatment of mild-to-moderate dementia associated with PD and Alzheimer disease.2 Tacrine, a medicinal noncarbamate CEI, is also prescribed for this use.2 Both drugs increase ACh concentrations in relevant brain regions and foster the formation of new memory.
Cholinesterase inhibitors are mechanistically analogous to the insecticidal carbamates (eg, aldicarb) and the organophosphates (OPs) (eg, malathion). They inhibit the metabolism of ACh by acetylcholinesterase (AChE) in the various cholinergic synapses, increasing the intrasynaptic concentration of ACh.
Additional AChEs include physostigmine, a carbamate commonly used in the ED to treat anticholinergic toxicity. Physostigmine raises the local synaptic concentration of ACh to compete for the muscarinic ACh receptor with drugs such as diphenhydramine or atropine. Other CEIs (eg, neostigmine, pyridostigmine, edrophonium) are used to raise intrasynaptic ACh concentrations and overcome antibody blockade of nicotinic ACh receptors at the neuromuscular junction in patients with myasthenia gravis.
What is the toxidrome associated with carbamate overdose
Carbamate toxicity, as manifested by the cholinergic toxidrome, largely resembles OP toxicity but with an important difference: Both OPs and carbamates function by binding to and inhibiting AChE; however, the carbamate-AChE bond undergoes spontaneous hydrolysis, thereby reactivating the enzyme. Consequently, the clinical effects of carbamate toxicity, though potentially severe, are self-limited and usually only last 24 hours or less.4
How should this patient be managed?
The general approach to a patient with medical carbamate toxicity is similar to that of a patient with OP poisoning. Dermal exposure, as is the case with this patient, should prompt skin decontamination to minimize ongoing exposure. Patch removal is necessary but is not sufficient to prevent ongoing absorption, since a depot of medication typically forms in the dermal tissue. In the presence of significant or life-threatening muscarinic effects (eg, bronchorrhea, bronchospasm, seizure), an antimuscarinic agent such as atropine is indicated. Various dosing schemes of atropine exist; at our institution, we recommend an initial dose of 1 to 3 mg intravenously (IV), with escalating doses every 5 minutes until reversal of bronchorrhea and bronchospasm occur.4 This is followed by initiation of an atropine infusion at a rate of 10% to 20% of the total loading dose per hour (to a maximum of 2 mg/h).4
Pralidoxime (2-PAM) and other oximes, accelerate the reactivation of carbamate-inhibited AChE and have effects at both the nicotinic and muscarinic synapses. Reactivation results in the enhanced metabolism of intrasynaptic ACh and decreased clinical cholinergic effects. Since atropine is only effective at muscarinic receptors, oximes were administered in this case to reverse neuromuscular weakness.
Although early administration of 2-PAM is indicated in the setting of significant OP poisoning (due to irreversible inhibition of AChE), its use for medical carbamate toxicity is controversial. Early animal studies of carbamate toxicity suggested that treatment with oximes worsened outcomes; however, this has not been demonstrated in more recent studies.5,6 Therefore, although 2-PAM may be beneficial in treating cases of clinically significant carbamate poisoning (which can be prolonged and severe), these benefits should be weighed against the potential risks.
Case Conclusion
Upon arrival to the ED, the patient’s skin was cleansed thoroughly. As he did not exhibit muscarinic findings of bradycardia, bronchoconstriction, or bronchorrhea, atropine was not indicated. He was treated conservatively with IV fluid hydration and admitted to the medicine floor. Since he continued to exhibit profound extremity weakness with no improvement 12 hours from the onset of symptoms, pralidoxime 1 g IV was administered over a 30-minute period. Shortly thereafter, patient’s motor strength improved from 2/5 to 4/5 in both upper and lower extremities. No complications were noted, and the patient‘s weakness and tremulousness continued to resolve. He was transferred to a skilled nursing facility on hospital day 6.
Dr Laskowski is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.
- Standaert DG, Roberson ED. Treatment of central nervous system degenerative disorders. In: Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s The Pharmacologic Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill; 2011:609-628
- Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633-638.
- Exelon Patch [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013.
- Eddleston M, Clark RF. Insecticides: organic phosphorus compounds and carbamates. In: Nelson LS, Lewin NA, Howland MA, Hoffman RS, Goldfrank LR, Flomenbaum NE, eds. Goldfrank’s Toxicologic Emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:1450-1466.
- Natoff IL, Reiff B. Effect of oximes on the acute toxicity of anticholinesterase carbamates. Toxicol Appl Pharmacol. 1973;25(4):569-575.
- Mercurio-Zappala M, Hack JB, Salvador A, Hoffman RS. Pralidoxime in carbaryl poisoning: an animal model. Hum Exp Toxicol. 2007;26(2)125-129.

Case Report: Nasal Septal Abscess
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
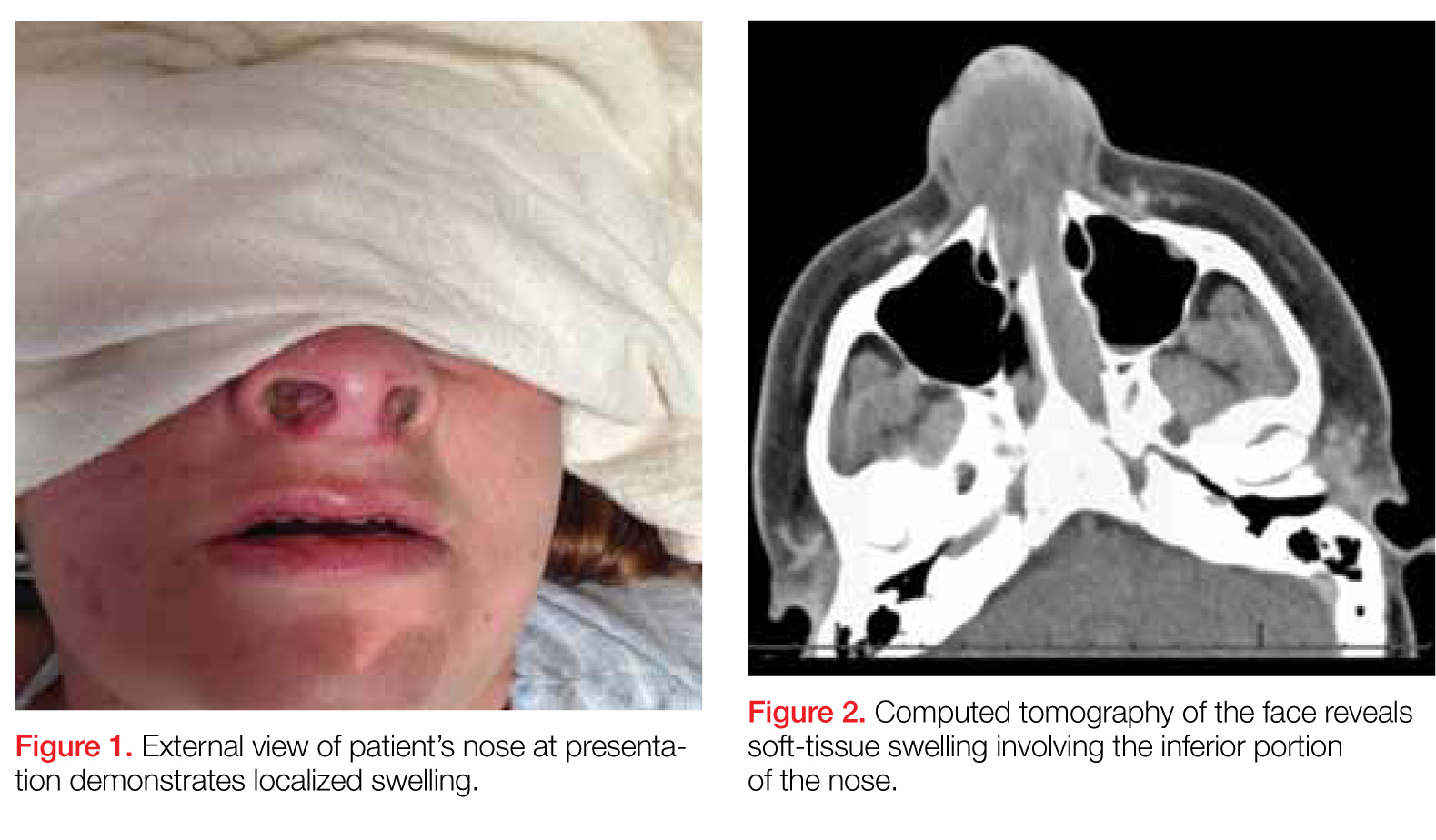
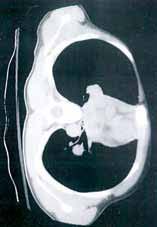
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
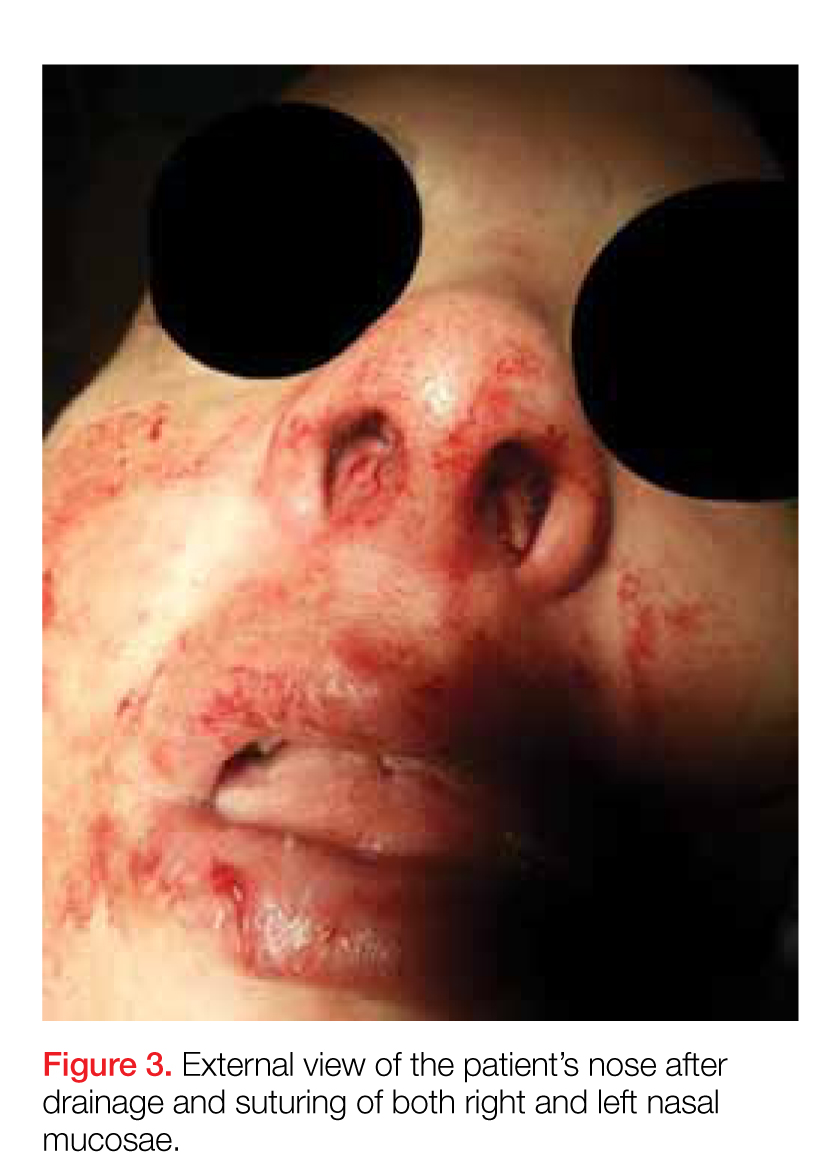
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
Case
A 28-year-old woman with history of bipolar disorder and methamphetamine abuse presented to the ED complaining of nasal swelling and pain. She was unable to provide any medical history regarding the onset of her symptoms or other details, which the ED team attributed to her underlying psychiatric disorder. She denied nasal trauma, insufflation, or insertion of foreign bodies into the nasal cavity. When the patient’s mother was contacted, she stated her daughter’s symptoms, which she believed were secondary to a domestic-violence-related injury, had been present and evolving over the past 2 weeks. She also related that the patient had been treated at another ED 4 days earlier and discharged with oral antibiotics.
On physical examination, the bilateral nares were entirely occluded by soft-tissue swelling, with fluctuance on palpation. The area was erythematous, and there were pustules scattered throughout the local region (Figure 1). There was no evidence of spreading cellulitis. During the examination, the patient had a labile level of alertness that fluctuated between somnolence and agitation; however, she was arousable and had satisfactory airway guarding. Patient’s vital signs remained stable throughout evaluation and treatment in the ED. On physical examination, her pupils were equal bilaterally, extraocular movements were intact, and no neurological deficits were detected. A complete blood cell count showed leukocytosis, with a white blood cell count of 18,240/uL and a predominance (88.2%) of neutrophils. All other laboratory values were within normal limits.
Computed tomography (CT) of the face revealed prominent soft-tissue swelling involving the inferior portion of the nose (Figure 2). In addition to swelling and obstruction of the bilateral nares, heterogeneity was also noted within the affected tissues and thought to represent a fluid component.
After the procedure, the patient was admitted to the hospital for observation on the medical psychiatric unit where she received additional IV antibiotic therapy as well as a psychiatric consultation. After a 24-hour observation period, she was discharged on a one-week regimen of oral clindamycin and instructions for outpatient follow-up with OMFS for septal repair. Cultures taken during exploration were positive for pan-sensitive Staphylococcus aureus. The working diagnosis at discharge was bilateral septal abscess from untreated bilateral septal hematoma due to an unreported facial trauma.
Discussion
Nasal septal abscess, a rare complication of a nasal septal hematoma, is defined as a collection of pus between the cartilaginous or bony nasal septum and its normally applied mucoperichondrium or mucoperiosteum. Patients most commonly present with fluctuant, tender, bilateral, or unilateral nasal obstruction as a result of anterior nasal septum swelling. Other symptoms include localized pain, swelling, fever, headache, or perinasal tenderness.1 The external portion of the nose is swollen, erythematous, and tender, and the anterior nasal cavities are occluded by a smooth, round, deep red or grey swelling.2 In a review of pediatric patients with nasal septal abscess, the most common complaint was nasal congestion (95%). Other significant complaints were nasal pain (50%), fever (50%), and headache (5%).3,4
Nasal septal abscess is most commonly caused by a hematoma. Although trauma is typically associated with this condition, it is not the sole cause. Other etiology includes nasal surgery, a furuncle of the nasal vestibule, sinusitis, or, in rare cases, infection from a dental extraction.3
Staphylococcus aureus is the most common pathogen. Streptococcus and other anaerobes are less common, and pediatric patients are more susceptible to Haemophilus influenza than adults. Although rare, Psuedomonas and Klebsiella have also been reported.3
When nasal septal abscess is suspected, prior to drainage, the diagnosis should be confirmed by CT of the face and include the paranasal sinuses. Computed tomography is an excellent imaging tool for abscess detection and is the community standard for evaluation. Magnetic resonance imaging is not usually utilized (especially in the acute or ED setting) as it is unlikely to affect or alter initial management. In radiographs, nasal septal abscess typically appears as fluid collection with thin rim enhancement in the cartilaginous nasal septum5 (Figure 2). These findings can be missed on brain CT alone.5
In patients presenting several days from a related trauma, distinguishing uncomplicated septal hematoma from nasal septal abscess can be very difficult—though nasal septal abscesses tend to be larger and more painful. In addition, there may be inflammation of the overlying mucosa, occasionally with exudates. In untreated cases, infection can extend into the cavernous sinus causing intracranial infections or cavernous sinus thrombosis. The most common complication of septal abscess is cartilage necrosis that can result in nasal structural collapse and “saddle-nose” deformity. Complications, including meningitis, can develop quickly (ie, within 3 to 4 days).6
The structural complications associated with septal abscess result from the avascular nature of the septal cartilage, which receives blood from the adherent mucoperichondrium. Hematoma and abscess can expand and obstruct the blood vessels that supply the nasal cartilage. Pressure of the hematoma on the septum causes progressive avascular necrosis.6
Patients with confirmed nasal septal abscess should obtain otolaryngology or OMFS consultation in the ED. Due to the high risk of complications and need for follow up, immediate drainage should also be directed by otolaryngology or OMFS. All patients should be discharged on oral broad-spectrum antibiotics, with a referral to an otolaryngologist or OMFS within 24 hours for evaluation and possible removal of nasal packs.7
Dr Yusuf is an academic chief resident, John Peter Smith Emergency Medicine Residency Program, Fort Worth, Texas. Dr Kirk is associate residency director and ultrasound director, department of emergency medicine, John Peter Smith Health System, Fort Worth, Texas.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.
- Huang PH, Chiang YC, Yang TH, Chao PZ, Lee FP. Nasal septal abscess. Otolaryngol Head Neck Surg. 2006;135(2):335,336.
- Shapiro RS. Nasal septal abscess. Can Med Assoc J. 1978;119(11):1321-1323.
- Lo SH, Wang PC. Nasal septal abscess as a complication of laser inferior turbinectomy. Chang Gung Med J. 2004;27(5):390-393.
- Canty PA, Berkowitz RG. Hematoma and abscess of the nasal septum in children. Arch Otolaryngol Head Neck Surg. 1996;122(12):1373-1376.
- Debnam JM, Gillenwater AM, Ginsberg LE. Nasal septal abscess in patients with immunosuppression. Am J Neuroradiol. 2007;28(10):1878,1879.
- Friedman M, Landsberg R, Chiampas G. Nasal septal hematoma evacuation. In: Reichman EF, Simon RR, eds. Emergency Medicine Procedures. New York, NY: McGraw-Hill; 2004. http://www.accessemergencymedicine.com/content.aspx?aID=45644. Accessed March 20, 2014.
- Summers SM, Bey T. Epistaxis, nasal fractures, and rhinosinusitis. In: Tintinalli JE, Stapczynski JS, Ma OJ, Cline DM, Cydulka RK, Meckler GD, eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide. 7th ed. New York, NY: McGraw-Hill; 2011. http://www.accessemergencymedicine.com/content.aspx?aID=6388080. Accessed March 20, 2014.

Poland Syndrome: A Congenital Abnormality Mimicking a Traumatic Injury
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.

Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
Case
A 12-year-old boy presented to the ED via emergency medical services after he was struck by motor vehicle while skateboarding without a helmet or other safety equipment. He was thrown approximately 10 feet, but experienced no loss of consciousness, pain, or active bleeding at the site of the accident. Unaccompanied by family, he arrived to the ED fully immobilized on a long back board. His field vital signs were stable: blood pressure (BP), 100/65 mm Hg; heart rate (HR) 105 beats/minute; respiratory rate (RR), 22 breaths/minute; temperature, afebrile. Oxygen saturation was 100% on room air. The patient had an estimated Glasgow Coma Scale (GCS) of 14, with one point removed due to confusion.
Primary examination showed an intact airway with equal breath sounds bilaterally, and pulses were equal in all extremities with audible heart sounds. The patient was able to move all extremities, and showed no obvious deformities or bleeding. He was neurologically intact, with equal strength and sensation. He did, however, elicit some confusion during the examination, continuously stating it was “all his fault” and asking the medical staff where he was. This confusion persisted even after repeated reorientation. His vital signs remained stable, with slight tachycardia (BP, 105/67 mm hg; HR 100 beats/minute; RR, 17 breaths/minute; temperature, afebrile; pulse oxygen saturation, 99%). An abbreviated history revealed no allergies, medications, or past medical history. When questioned, the patient had no recollection of the accident or the last time he had eaten.
A secondary survey was significant for a small contusion/abrasion on the patient’s forehead but an otherwise normal head, ear, eyes, nose, and throat examination and no cervical c-spine tenderness. The patient denied any chest wall tenderness, but there was a dramatic palpable defect in the right chest wall, with profound asymmetry when compared to the left chest wall. No sharp, bony edges could be palpated, nor could any crepitance be felt. Breath sounds were reexamined and remained equal and nonlabored, and the patient continued to have a stable oxygen saturation of 99% on room air. The rest of the secondary survey was negative, and c-spine, pelvic, and portable chest X-rays were all negative for acute findings.
Due to the physical examination findings on the chest wall, a computed tomography (CT) scan of the chest was performed with contrast (Figure). The chest CT was normal, except for a lack of musculature over the right anterior chest wall. The patient’s mother arrived shortly after imaging studies, at which time he was reexamined. When interviewing his mother for further history, she stated that her son had been diagnosed with mild Poland Syndrome as a child, and that he has always had a chest deformity. All other studies, including a noncontrast CT of the brain, were normal. The child quickly improved during his 6-hour observation in the ED, and he was subsequently discharged home with the diagnosis of a concussion.
Discussion
Poland syndrome, also known as hand and ipsilateral thorax syndrome, is a rare congenital disorder with unknown etiology.1,2 The condition was first officially described in 1841 by Alfred Poland at Guy’s Hospital in London, though reports exist as early as 1826. Poland, a medical student, made the discovery while examining the cadaver of a hanged convict.
The occurrence of Poland syndrome is estimated to be from 1 in 25,000 to 1 in 75,000 to 100,000 by some reports,1-4 with a higher incidence in males than females (3:1 ratio) and 75% right-sided dominance.2 The syndrome is primarily described as unilateral, but there is one case report of suspected bilateral involvement.1 The components of the syndrome consist of aplasia of the sternal head of the pectoralis major muscle, hypoplasia of the pectoralis minor muscle, decreased development of breast and subcutaneous tissue, and a variety of ipsilateral hand abnormalities, including shortened carpels and phalanges, and syndactyly. The syndrome is quite variable, with different individuals eliciting combinations of the above components.
Poland syndrome was initially believed to be a nonfamilial disorder due to its sporadic nature, as illustrated by a case report of an isolated affected identical twin.3 However, enough cases of familial involvement have been reported that there is a proposed theory of an inheritable trait. Although over 250 patients with this syndrome have been described, there is no clear cause.2 The current theory of etiology is felt to be due to a lack of blood flow in the subclavian artery, or one of its branches, early in the development of the fetus, around the end of the sixth week of development. Individuals can have mild to severe manifestations, ranging as mild (eg, only pectoralis involvement), to severe (eg, rib hypoplasia, complete absence of ipsilateral hand, dextrocardia, lung herniation). Case reports of high functioning athletes with the disorder show that there is not necessarily functional impairment.
In addition to Poland syndrome, there are a number of congenital abnormalities that can also mimic traumatic chest injuries. Historically, surgeons have classified congenital wall deformities into one of five categories: Poland syndrome, pectus excavatum, pectus carinatum, sternal clefts, and generic skeletal and cartilage dysplasias (eg, absent ribs, rib torsion, vertebral anomalies).5-7 Of these categories, Poland syndrome, pectus excavatum, and some skeletal dysplasias cause anterior chest wall depression.5,6 Although these are examples of congenital thoracic wall abnormalities, one must also remember postoperative changes, which may also appear to be traumatic in origin. Examples of specific procedures are lumpectomy, mastectomy, rib resection, lung resection, or even cardiac surgery—all of which can alter the physical findings of the chest wall.
Conclusion
This report is an interesting case of an impaired patient presenting to the ED after a traumatic incident and unable to describe a past medical history of a congenital disorder. Although the patient was high functioning, as exemplified by his ability to complete normal adolescent activities such as skateboarding, he had a significant physical finding which appeared to correspond to the mechanism of his injury. He was initially thought to have a significant injury involving his chest wall, since secondary examination revealed a palpable defect. Although the patient was oxygenating well, and in no apparent distress, his altered mental status raised concerns about the accuracy of his report, with confusion and perseveration.
When a rare congenital abnormality imitates a traumatic condition, merely having the name of the condition—as we did when the family arrived—does not necessarily rule out the absence of a related deficit or injury. To better differentiate acute from preexisting physical deformities or deficits, one must gather and process multiple diagnostic clues. This is best accomplished by combining the presence or absence of symptoms (in this case, pain, dyspnea, or hemoptysis), physical examination findings (eg, ecchymosis, crepitance, flail segment), and supportive diagnostic tests (radiographs, CT, and echocardiograms). This approach will systematically eliminate or suggest acute traumatic diagnoses. With specific traumatic causes such as rib fracture, pneumothorax, or pulmonary contusion eliminated, one can expand the (nontraumatic) differential, keeping in mind the possibility of a congenital disorder.
Dr Martin is an emergency physician at Emergency Medical Associates of NY and NJ; and emergency medicine education director, Monmouth Medical Center, Long Branch, NJ.
Dr Martin reports no conflict of interest or financial arrangements.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
- Fokin AA, Robicsek F. Poland syndrome revisited. Ann Thorac Surg. 2002;74(6):2218-2225
- Darian VB, Argenta LC, Pasyk KA. Familial Poland’s syndrome. Ann Plast Surg. 1989;23(6):531-537
- Stevens D, Fink B, Prevel C. Poland’s syndrome in one identical twin. J Pediatr Orthop. 2000;20(3):392-395.
- McGrath MH, Pomerantz J. Plastic surgery. In: Townsend CM Jr, Beauchamp RD, Evers BM, Mattox KL, eds. Sabiston Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 19th ed. Philadelphia, PA: Elsevier Saunders; 2012:1935
- Spear SL, Pelletiere CV, Lee ES, Grotting JC. Anterior thoracic hypoplasia: a separate entity from Poland syndrome. Plast Reconstr Surg. 2004;113(1):
- Hodgkinson, DJ. Chest wall implants: their use for pectus excavatum, pectoralis muscle tears, Poland’s syndrome, and muscular insufficiency. Aesthetic Plast Surg. 1997;21(1):7-15.
- Hodgkinson, DJ. The management of anterior chest wall deformity in patients presenting for breast augmentation. Plast Reconstr Surg. 2002;109(5): 1714-1723.
Failure of the Vari-Angle Hip Screw System: Two Cases
Delayed response in ipilimumab therapy
Metastatic melanoma is a deadly disease with a 5-year survival rate lower than 20%.1 In 2011, ipilimumab, a fully humanized antibody that binds to cytotoxic T-lymphocyte–associated antigen 4 (CTLA4) was approved by the US Food and Drug Administration based on improved survival in a pivotal trial.2 CTLA4 is a molecule on cytotoxic T-lymphocytes that plays a critical role in attenuating immune responses. Ipilimumab blocks the binding of B7, the ligand of CTLA4, thereby blocking the activation of CTLA4 and sustaining antitumor immune responses. The time course to response can be variable with immunotherapeutics. We report on a patient who experienced a considerable delay before responding to ipilimumab.
Click on the PDF icon at the top of this introduction to read the full article.
Metastatic melanoma is a deadly disease with a 5-year survival rate lower than 20%.1 In 2011, ipilimumab, a fully humanized antibody that binds to cytotoxic T-lymphocyte–associated antigen 4 (CTLA4) was approved by the US Food and Drug Administration based on improved survival in a pivotal trial.2 CTLA4 is a molecule on cytotoxic T-lymphocytes that plays a critical role in attenuating immune responses. Ipilimumab blocks the binding of B7, the ligand of CTLA4, thereby blocking the activation of CTLA4 and sustaining antitumor immune responses. The time course to response can be variable with immunotherapeutics. We report on a patient who experienced a considerable delay before responding to ipilimumab.
Click on the PDF icon at the top of this introduction to read the full article.
Metastatic melanoma is a deadly disease with a 5-year survival rate lower than 20%.1 In 2011, ipilimumab, a fully humanized antibody that binds to cytotoxic T-lymphocyte–associated antigen 4 (CTLA4) was approved by the US Food and Drug Administration based on improved survival in a pivotal trial.2 CTLA4 is a molecule on cytotoxic T-lymphocytes that plays a critical role in attenuating immune responses. Ipilimumab blocks the binding of B7, the ligand of CTLA4, thereby blocking the activation of CTLA4 and sustaining antitumor immune responses. The time course to response can be variable with immunotherapeutics. We report on a patient who experienced a considerable delay before responding to ipilimumab.
Click on the PDF icon at the top of this introduction to read the full article.
Delayed Spontaneous Reduction of Traumatic Pediatric Atlantoaxial Rotatory Subluxation
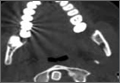
Fracture of a Dual-Modular Femoral Component at the Stem–Sleeve Junction in a Metal-on-Metal Total Hip Arthroplasty

Occlusion of Brachial Pseudoaneurysm After Periprosthetic Humerus Fracture

The Métaizeau Technique for Pediatric Radial Neck Fracture With Elbow Dislocation: Intraoperative Pitfalls and Associated Forearm Compartment Syndrome
There’s No Place Like Home… for Carbon Monoxide Poisoning
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?


Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?
Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
An 84-year-old woman with a history of hypertension and dyslipidemia and her husband, an 88-year-old man with a history of dementia and coronary artery disease, presented to the ED via EMS after neighbors discovered the woman lying on her living room floor, responding only to painful stimuli. Earlier in the evening, the same neighbors had helped the husband to bed after noticing that he had become lethargic. The EMS report indicated that a car had been left running in a closed garage of the patients’ home. The fire department identified an ambient carbon monoxide (CO) concentration of 88 ppm.
Upon arrival to the ED, the woman’s vital signs were: blood pressure (BP), 130/74 mm Hg; heart rate (HR), 63 beats/minute; respiratory rate (RR), 16 breaths/minute; temperature, 99°F. Oxygen saturation was 99% on room air. Her husband’s vital signs were: BP, 150/66 mm Hg; HR, 59 beats/minute; RR, 19 breaths/minute; temperature, 98°F; oxygen saturation was 98% on room air.
What is carbon monoxide poisoning?
Carbon monoxide is a colorless and odorless toxic gas produced by incomplete combustion of carbon-based fuel. Common sources in the United States include portable generators, gas-powered furnaces, cooking appliances, poorly ventilated home-heating systems, and motor vehicles (Box 1).1
Carbon monoxide is the leading cause of unintentional poisoning deaths in the United States,1 resulting in more than 20,000 ED visits and 2,000 hospital admissions. Nearly three-fourths of these deaths are due to exposures in the home, with more than half occurring during the months of November through February.2,3 The average cost of a hospital admission for confirmed CO poisoning is over $11,000, with a cumulative nationwide total cost of over $26 million per year. While the hospitalization rate for persons aged 18 to 44 years is only 6.7%, the admittance rates for persons aged 65 to 84 years and older than 85 years are 33% and 43%, respectively.3 Although there has been a slight decline in the incidence of CO poisoning over the past 10 years, it is still a public health concern (Figure 1).2
Who is most susceptible to motor vehicle-related carbon monoxide poisoning?
The US Centers for Disease Control and Prevention (CDC) reports that motor vehicles are the second most common source of CO exposure.4 A study of US news media reports covering a 2.5-year period revealed that 8% of such poisonings were the result of a motor vehicle left running in a garage—the overall mortality rate of which is suggested to be significantly higher than that of other sources of CO exposure.5
Approximately 430 deaths per year are caused by unintentional, nonfire-related CO poisoning,6 and the CDC reports the death rate is highest in persons older than age 65 years.1 The death rate from these exposures is more than three times higher in men than women (Figure 2).6 In addition, older patients are disproportionately affected: In US news media-reported cases of CO poisoning that included patient age, 29% occurred in persons older than age 80 years.5 Moreover, in approximately one-third of motor vehicle-related deaths due to CO poisoning, nearly all of patients older than age 80 years were found dead at the scene of exposure. These reports suggest that the elderly are at greater risk for CO exposure due to age-related cognitive changes, physical inability to escape a toxic environment once becoming symptomatic, and a greater susceptibility to poisoning due to comorbid conditions.5
Case Continued
The husband and wife’s initial carboxyhemoglobin concentrations in this case were 35% and 13%, respectively. Both were treated with hyperbaric oxygen (HBO) without complication. During their inpatient stay, the woman noted that their home did not have a CO detector.
What is the role of hyperbaric oxygen therapy as a treatment option for CO poisoning?
Hyperbaric oxygen therapy greatly accelerates the dissociation of hemoglobin from CO, reduces free radical-related cellular damage, and may have a role in preventing adverse neurological sequelae in the setting of CO poisoning. Although controversy exists, HBO therapy is generally indicated in select patients with elevated CO levels and abnormal neurological findings, cardiovascular findings, or persistent metabolic acidosis. While few ED patients with CO exposure receive HBO therapy, over 20% of patients requiring inpatient hospitalization receive treatment.3
What preventive measures can be taken to reduce motor vehicle-related CO poisoning?
The literature supports the enforcement of motor vehicle emissions standards and the proper use of home CO detectors as primary preventive strategies. Computerized data from the CDC, US Census Bureau, and US Environmental Protection Agency from 1968 to 1998 were used to evaluate the influence of national vehicle emissions policies on CO-related mortality. The Clean Air Act of 1970 set environmental limits on CO emissions from automobiles at 15.0 g/mile in 1975; the EPA further reduced this standard to 3.4 g/mile for automobiles manufactured after 1981. After the enforcement of standards set forth by the Clean Air Act and the introduction of the catalytic converter in 1975, CO emissions from automobiles decreased by an estimated 76.3%, and unintentional motor vehicle-related CO deaths declined by 81.3% (Figure 3).7 (Catalytic converters contain elements [eg, platinum] that catalyze the oxidation of CO to carbon dioxide.)
Since CO exposure occurs primarily in the home, the installation of battery-powered or battery-backed CO alarms—both in the home and garage—can prevent poisoning. These detectors are inexpensive and available at common retail stores. Unfortunately, despite the easy availability and access to CO detectors, only 39 states currently have legislation mandating their use, and approximately two-thirds of the states with existing legislation only require CO detectors in newly built structures.8
In 2010, the state of New York enacted legislation known as “Amanda’s Law,” (named after a teenaged girl whose death was caused by CO poisoning from a defective boiler) mandating CO detectors in all one- and two-family homes with heating sources that may emit CO or have attached garages. However, an industry survey in 2011 found that nearly half of New York families were not aware of this law.9 The two largest surveys on home CO detector use—those conducted by the US Census Bureau and CDC—estimate the national rate of having a working CO detector in a home is 32% to 40%, with a lower prevalence among those living in manufactured housing, renting a home, or living below the poverty level.10
What is the utilization of CO detectors by ED patients?
Case conclusion
After hospital admission and treatment, both patients were discharged on hospital day 2 with a return to a baseline mental status. Neither patient reported neurological sequelae or new cognitive changes when a follow up call was placed more than 6 months after HBO treatment. The couple furthermore reported that they installed a CO detector upon their return home.
Dr West is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr McGregor is a resident, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York.
Dr Touger is an associate professor of clinical emergency medicine, department of emergency medicine, Albert Einstein College of Medicine, Bronx, New York. He is also medical director of the Jacobi Medical Center hyperbaric chamber.
Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at New York University School of Medicine and New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.
- Centers for Disease Control and Prevention. Carbon monoxide-related deaths—United States, 1999-2004. MMWR Morb Mortal Wkly Rep. 2007;56(50):1309-1312.
- Centers for Disease Control and Prevention. Carbon mononoxide exposures—United States, 2000-2009. MMWR Morb Mortal Wkly Rep. 2011;60(30):1014-1017.
- Iqbal S, Law HZ, Clower JH, Yip FY, Elixhauser A. Hospital burden of unintentional carbon monoxide poisoning in the United States, 2007. Am J Emerg Med. 2012;30(5):657-664.
- Centers for Disease Control and Prevention. Nonfatal, unintentional, non-fire-related carbon monoxide exposures—United States, 2004-2006. MMWR Morb Mortal Wkly Rep. 2008;57(33):896-899.
- Hampson NB. Residential carbon monoxide poisoning from motor vehicles. Am J Emerg Med. 2011;29(1):75-77.
- Centers for Disease Control and Prevention. Average annual number of deaths and death rates from unintentional, non-fire-related carbon monoxide poisoning, by sex and age group—United States, 1999–2010. MMWR Morb Mortal Wkly Rep. 2014;63(3):65.
- Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. JAMA. 2002;288(8):988-995.
- Carbon monoxide detectors: state statutes. National Conference of State Legislatures Web site. http://www.ncsl.org/research/environment-and-natural-resources/carbon-monoxide-detectors-state-statutes.aspx. Accessed March 11, 2014.
- Survey results: New York homeowners and the risk of carbon monoxide poisoning. Kidde Web site. http://www.kidde.com/PressRoom/Pages/SurveyResultsNYHomeownersCORisks.aspx. Accessed March 11, 2014.
- Iqbal S, Clower JH, King M, Bell J, Yip YF. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012;127(5):486-496.
- Johnson-Arbor K, Liebman DL, Carter EM. A survey of residential carbon monoxide detector utilization among Connecticut Emergency Department patients. Clin Toxicol (Phila). 2012;50(5):384-389.
