User login
Atypical Presentation of Fat Embolism Syndrome After Gunshot Wound to the Foot
Fat embolism syndrome (FES) is a rare complication reported primarily after long bone fractures, with an incidence of 0.3% to 2.2%.1-3 It is most commonly caused by trauma and is thought to result from movement of bone fragments or to occur during intramedullary reaming.1 Both of these factors lead to a distortion of the bone marrow cavity, allowing marrow and fat to enter the circulatory system.1
Although the true pathophysiology remains poorly understood, it is possible that, once in systemic circulation, the fat particles become lodged in the vascular system, inciting an inflammatory response, leading to organ dysfunction via mechanical or biochemical processes.4 Typically, the diagnosis is made after clinical features are observed, including hypoxemia, petechial rash, and cerebral signs not related to a head injury or other conditions.5,6
Although FES is an uncommon complication after traumatic injuries, mortality after FES in a recent study was reported to be 10%.1 FES is most commonly seen after fractures of the femur and tibia, although cases have been described involving fractures of the radius, ulna, and humerus.1,3 We present an atypical case of cerebral FES after multiple fractures of the foot; to our knowledge, such a case has not been reported in the English-language literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 42-year-old man was hunting with his son when he was accidentally shot in the left foot with a .270-caliber rifle bullet at close range. The patient sought care at a local hospital and, in the ensuing 3 hours, his mentation appeared normal. He reported pain and numbness distal to the injury in the tibial nerve distribution, but he remained vascularly intact, alert, and oriented. He was given 7 mg of hydromorphone hydrochloride over 2 hours for pain control and was transferred to our hospital via ambulance approximately 6 hours after injury. Upon arrival, he was noted to be extremely sedated and obtunded, responding only to pain with spontaneous eye opening. He was unable to follow commands. He was given
1.2 mg of naloxone intravenously to reverse what was presumed to be acute opioid intoxication; however, his mental status did not improve.
On examination, the patient was noted to have a small entrance wound through the Achilles tendon (Figures 1A, 1B) and an exit wound on the plantar aspect of the foot near the heads of the first and second metatarsals (Figures 1C, 1D) with minimal bleeding and no gross contamination. There was significant edema on the medial and proximal aspects of the left foot, 3+ dorsalis pedis pulse, and a capillary refill of 4 seconds. Radiographs showed traumatic fracture deformities of the calcaneus, navicular, medial cuneiform, and first and second metatarsal bases, as well as an intra-articular fracture deformity of the left talus extending to the talar dome (Figures 2A-2C). Neurologic examination could not be reliably obtained because of the patient’s mental status. He was determined to be unstable for immediate surgery, and his left leg was splinted pending neurologic evaluation.


The patient’s oxygen saturation was 94%, and his temperature was 38.2°C (100.76°F). Although his heart rate was in the 90s upon arrival, he became tachycardic over the next 4 hours, with heart rate ranging from the 110s to 130s; he remained tachycardic for approximately 72 hours. Laboratory values upon arrival showed a hemoglobin value of 12.8 g/dL and platelets of 249,000/μL. He developed anemia and thrombocytopenia within 72 hours of the injury, with a low of 6.6 g/dL and 88,000/μL, respectively, by postinjury day 4. Computed tomography of the head, electroencephalography, urine drug screen, and lumbar puncture were unremarkable. The patient never became hypoxemic. Within 14 hours after injury, he was completely comatose with extensor posturing. In the intensive care unit (ICU), the patient was intubated for airway protection.
The next day, the patient underwent magnetic resonance imaging (MRI) of the brain, which showed innumerable tiny infarcts throughout cerebral hemispheres, cerebellum, and brainstem in a characteristic “starfield” pattern on T2-weighted images (Figure 3). This was radiographically consistent with fat emboli related to the left lower extremity gunshot wound. An echocardiogram showed small right-to-left shunt and a possible intrapulmonary shunt, although this was never confirmed. The echocardiogram was technically challenging secondary to his persistent tachycardia. He also developed a subtle petechial rash (Figure 4A).


The patient underwent percutaneous gastrostomy-tube placement for nutrition on postinjury day 4 and remained intubated, unable to protect his airway, and nonresponsive with extensor posturing (Figure 4B). He was also taken to the operating room for spanning external fixator placement on postinjury day 3 to restore calcaneal height and length as well as foot stability (Figures 5A, 5B).

The patient was treated with supportive care and was discharged from the hospital in a comatose state on hospital day 17 to a rehabilitation facility. He began to emerge from the coma 6 weeks after injury, and his external fixator was removed and a cast applied to his lower extremity. His entrance and exit wounds healed as expected. Initial agitation was treated with propranolol and quetiapine. Because he continued to have difficulty with spasticity and increased tone, he was given botulinum toxin type A injections in the pectoral muscles, biceps, and forearms. He made continued and rapid improvement in response to intensive multidisciplinary therapy and returned home 4½ months after injury. Eight months after the injury, he is now walking independently with a cane and independent with his activities of daily living. Unfortunately, he has substantial pain in his foot, which appears to be a combination of both neuropathic and posttraumatic arthrosis causes. He is undergoing consultation for a possible amputation. Radiographs show consolidation of the hind and midfoot fractures with retained bullet fragments (Figures 6A-6C). He continues to receive multidisciplinary care to address cognitive limitations and is making progress.

Discussion
FES is a life-threatening disease affecting multiple organ systems.7 Classically, the pulmonary, central nervous, and dermatologic systems are affected.5,6,8 While FES is most recognizable after long bone fractures and orthopedic procedures involving the intramedullary canal, to our knowledge, FES after gunshot wound and concomitant fractures of the foot has never been reported.
The syndrome is defined by major and minor criteria as outlined by Gurd.5 Major criteria include hypoxia, deteriorating mental status, and petechiae. This case represents a somewhat atypical presentation of FES, because dermatologic manifestations and pulmonary compromise were subtle. The minor criteria consisting of tachycardia, fever, anemia, and thrombocytopenia were present in our patient, although at different phases during the progression of the syndrome. This emphasizes the difficulty in diagnosing FES because the symptoms do not occur simultaneously.
In the classic syndrome, after an initial asymptomatic interval of 12 to 72 hours, pulmonary, neurologic, and/or dermatologic changes usually ensue.9 Altered mental status, including headache, confusion, stupor, coma, rigidity, or convulsions, has been documented in 86% of patients.10 In our case, the neurologic symptoms presented earlier, at around 6 hours after injury, and respiratory symptoms, including hypoxia, tachypnea, and dyspnea, reported in 75% of cases,2,11 did not occur at all. In fact, continued intubation was only required in this case for neuromuscular airway protection. Classic dermatologic manifestations, a reddish-brown nonpalpable petechial rash diffusely covering the upper torso and extremities, normally appears within 12 to 36 hours.12,13 Nevertheless, in our case, these findings were subtle compared with others previously reported.14,15 In fact, despite being seen by numerous physicians, including neurologists and ICU intensivists, only the orthopedists’ notes made reference to this modest finding (Figure 4A).
Further complicating the diagnosis is that, during the onset of symptoms, patients are typically victims of polytrauma and/or routinely given narcotics to help with significant pain. Therefore, it is appropriate to rule out opioid overdose and other metabolic sources of mental-status change. This can be done fairly expeditiously with laboratory testing and narcotic reversal. After these have been eliminated, FES should be considered in a patient with rapid neurologic deterioration, because a delay in treatment can affect outcomes.2,4,16
Because continuous showering of emboli to the brain and other organs occurs without fracture stabilization, rapid diagnosis with high clinical suspicion of FES is essential and can be aided immensely with MRI. In fact, MRI is the most sensitive test for this diagnosis and correlates with clinical severity of brain injury.17 T2-weighted images show regions of high-signal intensity and “starfield” pattern, which are sensitive markers for FES (Figure 3).18 These tests can be done concomitantly with a well-splinted extremity, and definitive stabilization should be carried out promptly because early splinting and fixation of orthopedic fractures improves outcomes.17
Perhaps the most important reason to make an expeditious diagnosis is to help counsel families, who are undoubtedly in shock and disbelief. Recovery times can vary widely, with the patient often continuing to regain cognitive and motor function over the course of months to years.2 Without knowledge of signs of improvement in neurologic outcome, families cannot be accurately counseled regarding potential for recovery. The practicing orthopedist should be aware of this disorder, because initial neurologic deterioration may seem hopeless. Furthermore, supportive care should be initiated early with multidisciplinary teams and extensive rehabilitation because these offer the best outcomes in patients with FES.4,18 Although our patient continues to have cognitive impairment, his recovery in the preceding 8 months has been aided by rapid diagnosis and multidisciplinary care and should offer hope to other patients faced with this situation.
1. Akhtar S. Fat embolism. Anesthesiol Clin. 2009;27(3):533-550.
2. Müller C, Rahn BA, Pfister U, Meinig RP. The incidence, pathogenesis, diagnosis, and treatment of fat embolism. Orthop Rev. 1994;23(2):107-117.
3. Stein PD, Yaekoub AY, Matta F, Kleerekoper M. Fat embolism syndrome. Am J Med Sci. 2008;336(6):472-477.
4. Parisi DM, Koval K, Egol K. Fat embolism syndrome. Am J Orthop. 2002;31(9):507-512.
5. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surb Br. 1970;52(4):732-737.
6. Lee SC, Yoon JY, Nam CH, Kim TK, Jung KA, Lee DW. Cerebral fat embolism syndrome after simultaneous bilateral total knee arthroplasty: a case series. J Arthroplasty. 2012;27(3):409-414.
7. Gurd AR, Wilson RI. Fat-embolism syndrome. Lancet. 1972;2(7770):
231-232.
8. Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury. 2006;37(Suppl 4):S68-S73.
9. Weiss W, Bardana D, Yen D. Delayed presentation of fat embolism syndrome after intramedullary nailing of a fractured femur: a case report. J Trauma. 2009;66(3):E42-E45.
10. Byrick RJ. Fat embolism and postoperative coagulopathy. Can J Anaesth. 2001;48(7):618-621.
11. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
12. Burgher LW. Fat embolism syndrome. Chest. 1981;79(2):131-132.
13. Burgher LW, Dines DE, Linscheid RL, Didier EP. Fat embolism and the adult respiratory distress syndrome. Mayo Clin Proc. 1974;49(2):107-109.
14. Liu DD, Hsieh NK, Chen HI. Histopathological and biochemical changes following fat embolism with administration of corn oil micelles: a new animal model for fat embolism syndrome. J Bone Joint Surg Br. 2008;90(11):
1517-1521.
15. Liu HK, Chen WC. Images in clinical medicine. Fat embolism syndrome. N Engl J Med. 2011;364(18):1761.
16. Pinney SJ, Keating JF, Meek RN. Fat embolism syndrome in isolated femoral fractures: does timing of nailing influence incidence? Injury. 1998;29(2):
131-133.
17. Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma. 1999;46(2):324-327.
18. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.
Fat embolism syndrome (FES) is a rare complication reported primarily after long bone fractures, with an incidence of 0.3% to 2.2%.1-3 It is most commonly caused by trauma and is thought to result from movement of bone fragments or to occur during intramedullary reaming.1 Both of these factors lead to a distortion of the bone marrow cavity, allowing marrow and fat to enter the circulatory system.1
Although the true pathophysiology remains poorly understood, it is possible that, once in systemic circulation, the fat particles become lodged in the vascular system, inciting an inflammatory response, leading to organ dysfunction via mechanical or biochemical processes.4 Typically, the diagnosis is made after clinical features are observed, including hypoxemia, petechial rash, and cerebral signs not related to a head injury or other conditions.5,6
Although FES is an uncommon complication after traumatic injuries, mortality after FES in a recent study was reported to be 10%.1 FES is most commonly seen after fractures of the femur and tibia, although cases have been described involving fractures of the radius, ulna, and humerus.1,3 We present an atypical case of cerebral FES after multiple fractures of the foot; to our knowledge, such a case has not been reported in the English-language literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 42-year-old man was hunting with his son when he was accidentally shot in the left foot with a .270-caliber rifle bullet at close range. The patient sought care at a local hospital and, in the ensuing 3 hours, his mentation appeared normal. He reported pain and numbness distal to the injury in the tibial nerve distribution, but he remained vascularly intact, alert, and oriented. He was given 7 mg of hydromorphone hydrochloride over 2 hours for pain control and was transferred to our hospital via ambulance approximately 6 hours after injury. Upon arrival, he was noted to be extremely sedated and obtunded, responding only to pain with spontaneous eye opening. He was unable to follow commands. He was given
1.2 mg of naloxone intravenously to reverse what was presumed to be acute opioid intoxication; however, his mental status did not improve.
On examination, the patient was noted to have a small entrance wound through the Achilles tendon (Figures 1A, 1B) and an exit wound on the plantar aspect of the foot near the heads of the first and second metatarsals (Figures 1C, 1D) with minimal bleeding and no gross contamination. There was significant edema on the medial and proximal aspects of the left foot, 3+ dorsalis pedis pulse, and a capillary refill of 4 seconds. Radiographs showed traumatic fracture deformities of the calcaneus, navicular, medial cuneiform, and first and second metatarsal bases, as well as an intra-articular fracture deformity of the left talus extending to the talar dome (Figures 2A-2C). Neurologic examination could not be reliably obtained because of the patient’s mental status. He was determined to be unstable for immediate surgery, and his left leg was splinted pending neurologic evaluation.
The patient’s oxygen saturation was 94%, and his temperature was 38.2°C (100.76°F). Although his heart rate was in the 90s upon arrival, he became tachycardic over the next 4 hours, with heart rate ranging from the 110s to 130s; he remained tachycardic for approximately 72 hours. Laboratory values upon arrival showed a hemoglobin value of 12.8 g/dL and platelets of 249,000/μL. He developed anemia and thrombocytopenia within 72 hours of the injury, with a low of 6.6 g/dL and 88,000/μL, respectively, by postinjury day 4. Computed tomography of the head, electroencephalography, urine drug screen, and lumbar puncture were unremarkable. The patient never became hypoxemic. Within 14 hours after injury, he was completely comatose with extensor posturing. In the intensive care unit (ICU), the patient was intubated for airway protection.
The next day, the patient underwent magnetic resonance imaging (MRI) of the brain, which showed innumerable tiny infarcts throughout cerebral hemispheres, cerebellum, and brainstem in a characteristic “starfield” pattern on T2-weighted images (Figure 3). This was radiographically consistent with fat emboli related to the left lower extremity gunshot wound. An echocardiogram showed small right-to-left shunt and a possible intrapulmonary shunt, although this was never confirmed. The echocardiogram was technically challenging secondary to his persistent tachycardia. He also developed a subtle petechial rash (Figure 4A).
The patient underwent percutaneous gastrostomy-tube placement for nutrition on postinjury day 4 and remained intubated, unable to protect his airway, and nonresponsive with extensor posturing (Figure 4B). He was also taken to the operating room for spanning external fixator placement on postinjury day 3 to restore calcaneal height and length as well as foot stability (Figures 5A, 5B).
The patient was treated with supportive care and was discharged from the hospital in a comatose state on hospital day 17 to a rehabilitation facility. He began to emerge from the coma 6 weeks after injury, and his external fixator was removed and a cast applied to his lower extremity. His entrance and exit wounds healed as expected. Initial agitation was treated with propranolol and quetiapine. Because he continued to have difficulty with spasticity and increased tone, he was given botulinum toxin type A injections in the pectoral muscles, biceps, and forearms. He made continued and rapid improvement in response to intensive multidisciplinary therapy and returned home 4½ months after injury. Eight months after the injury, he is now walking independently with a cane and independent with his activities of daily living. Unfortunately, he has substantial pain in his foot, which appears to be a combination of both neuropathic and posttraumatic arthrosis causes. He is undergoing consultation for a possible amputation. Radiographs show consolidation of the hind and midfoot fractures with retained bullet fragments (Figures 6A-6C). He continues to receive multidisciplinary care to address cognitive limitations and is making progress.
Discussion
FES is a life-threatening disease affecting multiple organ systems.7 Classically, the pulmonary, central nervous, and dermatologic systems are affected.5,6,8 While FES is most recognizable after long bone fractures and orthopedic procedures involving the intramedullary canal, to our knowledge, FES after gunshot wound and concomitant fractures of the foot has never been reported.
The syndrome is defined by major and minor criteria as outlined by Gurd.5 Major criteria include hypoxia, deteriorating mental status, and petechiae. This case represents a somewhat atypical presentation of FES, because dermatologic manifestations and pulmonary compromise were subtle. The minor criteria consisting of tachycardia, fever, anemia, and thrombocytopenia were present in our patient, although at different phases during the progression of the syndrome. This emphasizes the difficulty in diagnosing FES because the symptoms do not occur simultaneously.
In the classic syndrome, after an initial asymptomatic interval of 12 to 72 hours, pulmonary, neurologic, and/or dermatologic changes usually ensue.9 Altered mental status, including headache, confusion, stupor, coma, rigidity, or convulsions, has been documented in 86% of patients.10 In our case, the neurologic symptoms presented earlier, at around 6 hours after injury, and respiratory symptoms, including hypoxia, tachypnea, and dyspnea, reported in 75% of cases,2,11 did not occur at all. In fact, continued intubation was only required in this case for neuromuscular airway protection. Classic dermatologic manifestations, a reddish-brown nonpalpable petechial rash diffusely covering the upper torso and extremities, normally appears within 12 to 36 hours.12,13 Nevertheless, in our case, these findings were subtle compared with others previously reported.14,15 In fact, despite being seen by numerous physicians, including neurologists and ICU intensivists, only the orthopedists’ notes made reference to this modest finding (Figure 4A).
Further complicating the diagnosis is that, during the onset of symptoms, patients are typically victims of polytrauma and/or routinely given narcotics to help with significant pain. Therefore, it is appropriate to rule out opioid overdose and other metabolic sources of mental-status change. This can be done fairly expeditiously with laboratory testing and narcotic reversal. After these have been eliminated, FES should be considered in a patient with rapid neurologic deterioration, because a delay in treatment can affect outcomes.2,4,16
Because continuous showering of emboli to the brain and other organs occurs without fracture stabilization, rapid diagnosis with high clinical suspicion of FES is essential and can be aided immensely with MRI. In fact, MRI is the most sensitive test for this diagnosis and correlates with clinical severity of brain injury.17 T2-weighted images show regions of high-signal intensity and “starfield” pattern, which are sensitive markers for FES (Figure 3).18 These tests can be done concomitantly with a well-splinted extremity, and definitive stabilization should be carried out promptly because early splinting and fixation of orthopedic fractures improves outcomes.17
Perhaps the most important reason to make an expeditious diagnosis is to help counsel families, who are undoubtedly in shock and disbelief. Recovery times can vary widely, with the patient often continuing to regain cognitive and motor function over the course of months to years.2 Without knowledge of signs of improvement in neurologic outcome, families cannot be accurately counseled regarding potential for recovery. The practicing orthopedist should be aware of this disorder, because initial neurologic deterioration may seem hopeless. Furthermore, supportive care should be initiated early with multidisciplinary teams and extensive rehabilitation because these offer the best outcomes in patients with FES.4,18 Although our patient continues to have cognitive impairment, his recovery in the preceding 8 months has been aided by rapid diagnosis and multidisciplinary care and should offer hope to other patients faced with this situation.
Fat embolism syndrome (FES) is a rare complication reported primarily after long bone fractures, with an incidence of 0.3% to 2.2%.1-3 It is most commonly caused by trauma and is thought to result from movement of bone fragments or to occur during intramedullary reaming.1 Both of these factors lead to a distortion of the bone marrow cavity, allowing marrow and fat to enter the circulatory system.1
Although the true pathophysiology remains poorly understood, it is possible that, once in systemic circulation, the fat particles become lodged in the vascular system, inciting an inflammatory response, leading to organ dysfunction via mechanical or biochemical processes.4 Typically, the diagnosis is made after clinical features are observed, including hypoxemia, petechial rash, and cerebral signs not related to a head injury or other conditions.5,6
Although FES is an uncommon complication after traumatic injuries, mortality after FES in a recent study was reported to be 10%.1 FES is most commonly seen after fractures of the femur and tibia, although cases have been described involving fractures of the radius, ulna, and humerus.1,3 We present an atypical case of cerebral FES after multiple fractures of the foot; to our knowledge, such a case has not been reported in the English-language literature. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 42-year-old man was hunting with his son when he was accidentally shot in the left foot with a .270-caliber rifle bullet at close range. The patient sought care at a local hospital and, in the ensuing 3 hours, his mentation appeared normal. He reported pain and numbness distal to the injury in the tibial nerve distribution, but he remained vascularly intact, alert, and oriented. He was given 7 mg of hydromorphone hydrochloride over 2 hours for pain control and was transferred to our hospital via ambulance approximately 6 hours after injury. Upon arrival, he was noted to be extremely sedated and obtunded, responding only to pain with spontaneous eye opening. He was unable to follow commands. He was given
1.2 mg of naloxone intravenously to reverse what was presumed to be acute opioid intoxication; however, his mental status did not improve.
On examination, the patient was noted to have a small entrance wound through the Achilles tendon (Figures 1A, 1B) and an exit wound on the plantar aspect of the foot near the heads of the first and second metatarsals (Figures 1C, 1D) with minimal bleeding and no gross contamination. There was significant edema on the medial and proximal aspects of the left foot, 3+ dorsalis pedis pulse, and a capillary refill of 4 seconds. Radiographs showed traumatic fracture deformities of the calcaneus, navicular, medial cuneiform, and first and second metatarsal bases, as well as an intra-articular fracture deformity of the left talus extending to the talar dome (Figures 2A-2C). Neurologic examination could not be reliably obtained because of the patient’s mental status. He was determined to be unstable for immediate surgery, and his left leg was splinted pending neurologic evaluation.
The patient’s oxygen saturation was 94%, and his temperature was 38.2°C (100.76°F). Although his heart rate was in the 90s upon arrival, he became tachycardic over the next 4 hours, with heart rate ranging from the 110s to 130s; he remained tachycardic for approximately 72 hours. Laboratory values upon arrival showed a hemoglobin value of 12.8 g/dL and platelets of 249,000/μL. He developed anemia and thrombocytopenia within 72 hours of the injury, with a low of 6.6 g/dL and 88,000/μL, respectively, by postinjury day 4. Computed tomography of the head, electroencephalography, urine drug screen, and lumbar puncture were unremarkable. The patient never became hypoxemic. Within 14 hours after injury, he was completely comatose with extensor posturing. In the intensive care unit (ICU), the patient was intubated for airway protection.
The next day, the patient underwent magnetic resonance imaging (MRI) of the brain, which showed innumerable tiny infarcts throughout cerebral hemispheres, cerebellum, and brainstem in a characteristic “starfield” pattern on T2-weighted images (Figure 3). This was radiographically consistent with fat emboli related to the left lower extremity gunshot wound. An echocardiogram showed small right-to-left shunt and a possible intrapulmonary shunt, although this was never confirmed. The echocardiogram was technically challenging secondary to his persistent tachycardia. He also developed a subtle petechial rash (Figure 4A).
The patient underwent percutaneous gastrostomy-tube placement for nutrition on postinjury day 4 and remained intubated, unable to protect his airway, and nonresponsive with extensor posturing (Figure 4B). He was also taken to the operating room for spanning external fixator placement on postinjury day 3 to restore calcaneal height and length as well as foot stability (Figures 5A, 5B).
The patient was treated with supportive care and was discharged from the hospital in a comatose state on hospital day 17 to a rehabilitation facility. He began to emerge from the coma 6 weeks after injury, and his external fixator was removed and a cast applied to his lower extremity. His entrance and exit wounds healed as expected. Initial agitation was treated with propranolol and quetiapine. Because he continued to have difficulty with spasticity and increased tone, he was given botulinum toxin type A injections in the pectoral muscles, biceps, and forearms. He made continued and rapid improvement in response to intensive multidisciplinary therapy and returned home 4½ months after injury. Eight months after the injury, he is now walking independently with a cane and independent with his activities of daily living. Unfortunately, he has substantial pain in his foot, which appears to be a combination of both neuropathic and posttraumatic arthrosis causes. He is undergoing consultation for a possible amputation. Radiographs show consolidation of the hind and midfoot fractures with retained bullet fragments (Figures 6A-6C). He continues to receive multidisciplinary care to address cognitive limitations and is making progress.
Discussion
FES is a life-threatening disease affecting multiple organ systems.7 Classically, the pulmonary, central nervous, and dermatologic systems are affected.5,6,8 While FES is most recognizable after long bone fractures and orthopedic procedures involving the intramedullary canal, to our knowledge, FES after gunshot wound and concomitant fractures of the foot has never been reported.
The syndrome is defined by major and minor criteria as outlined by Gurd.5 Major criteria include hypoxia, deteriorating mental status, and petechiae. This case represents a somewhat atypical presentation of FES, because dermatologic manifestations and pulmonary compromise were subtle. The minor criteria consisting of tachycardia, fever, anemia, and thrombocytopenia were present in our patient, although at different phases during the progression of the syndrome. This emphasizes the difficulty in diagnosing FES because the symptoms do not occur simultaneously.
In the classic syndrome, after an initial asymptomatic interval of 12 to 72 hours, pulmonary, neurologic, and/or dermatologic changes usually ensue.9 Altered mental status, including headache, confusion, stupor, coma, rigidity, or convulsions, has been documented in 86% of patients.10 In our case, the neurologic symptoms presented earlier, at around 6 hours after injury, and respiratory symptoms, including hypoxia, tachypnea, and dyspnea, reported in 75% of cases,2,11 did not occur at all. In fact, continued intubation was only required in this case for neuromuscular airway protection. Classic dermatologic manifestations, a reddish-brown nonpalpable petechial rash diffusely covering the upper torso and extremities, normally appears within 12 to 36 hours.12,13 Nevertheless, in our case, these findings were subtle compared with others previously reported.14,15 In fact, despite being seen by numerous physicians, including neurologists and ICU intensivists, only the orthopedists’ notes made reference to this modest finding (Figure 4A).
Further complicating the diagnosis is that, during the onset of symptoms, patients are typically victims of polytrauma and/or routinely given narcotics to help with significant pain. Therefore, it is appropriate to rule out opioid overdose and other metabolic sources of mental-status change. This can be done fairly expeditiously with laboratory testing and narcotic reversal. After these have been eliminated, FES should be considered in a patient with rapid neurologic deterioration, because a delay in treatment can affect outcomes.2,4,16
Because continuous showering of emboli to the brain and other organs occurs without fracture stabilization, rapid diagnosis with high clinical suspicion of FES is essential and can be aided immensely with MRI. In fact, MRI is the most sensitive test for this diagnosis and correlates with clinical severity of brain injury.17 T2-weighted images show regions of high-signal intensity and “starfield” pattern, which are sensitive markers for FES (Figure 3).18 These tests can be done concomitantly with a well-splinted extremity, and definitive stabilization should be carried out promptly because early splinting and fixation of orthopedic fractures improves outcomes.17
Perhaps the most important reason to make an expeditious diagnosis is to help counsel families, who are undoubtedly in shock and disbelief. Recovery times can vary widely, with the patient often continuing to regain cognitive and motor function over the course of months to years.2 Without knowledge of signs of improvement in neurologic outcome, families cannot be accurately counseled regarding potential for recovery. The practicing orthopedist should be aware of this disorder, because initial neurologic deterioration may seem hopeless. Furthermore, supportive care should be initiated early with multidisciplinary teams and extensive rehabilitation because these offer the best outcomes in patients with FES.4,18 Although our patient continues to have cognitive impairment, his recovery in the preceding 8 months has been aided by rapid diagnosis and multidisciplinary care and should offer hope to other patients faced with this situation.
1. Akhtar S. Fat embolism. Anesthesiol Clin. 2009;27(3):533-550.
2. Müller C, Rahn BA, Pfister U, Meinig RP. The incidence, pathogenesis, diagnosis, and treatment of fat embolism. Orthop Rev. 1994;23(2):107-117.
3. Stein PD, Yaekoub AY, Matta F, Kleerekoper M. Fat embolism syndrome. Am J Med Sci. 2008;336(6):472-477.
4. Parisi DM, Koval K, Egol K. Fat embolism syndrome. Am J Orthop. 2002;31(9):507-512.
5. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surb Br. 1970;52(4):732-737.
6. Lee SC, Yoon JY, Nam CH, Kim TK, Jung KA, Lee DW. Cerebral fat embolism syndrome after simultaneous bilateral total knee arthroplasty: a case series. J Arthroplasty. 2012;27(3):409-414.
7. Gurd AR, Wilson RI. Fat-embolism syndrome. Lancet. 1972;2(7770):
231-232.
8. Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury. 2006;37(Suppl 4):S68-S73.
9. Weiss W, Bardana D, Yen D. Delayed presentation of fat embolism syndrome after intramedullary nailing of a fractured femur: a case report. J Trauma. 2009;66(3):E42-E45.
10. Byrick RJ. Fat embolism and postoperative coagulopathy. Can J Anaesth. 2001;48(7):618-621.
11. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
12. Burgher LW. Fat embolism syndrome. Chest. 1981;79(2):131-132.
13. Burgher LW, Dines DE, Linscheid RL, Didier EP. Fat embolism and the adult respiratory distress syndrome. Mayo Clin Proc. 1974;49(2):107-109.
14. Liu DD, Hsieh NK, Chen HI. Histopathological and biochemical changes following fat embolism with administration of corn oil micelles: a new animal model for fat embolism syndrome. J Bone Joint Surg Br. 2008;90(11):
1517-1521.
15. Liu HK, Chen WC. Images in clinical medicine. Fat embolism syndrome. N Engl J Med. 2011;364(18):1761.
16. Pinney SJ, Keating JF, Meek RN. Fat embolism syndrome in isolated femoral fractures: does timing of nailing influence incidence? Injury. 1998;29(2):
131-133.
17. Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma. 1999;46(2):324-327.
18. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.
1. Akhtar S. Fat embolism. Anesthesiol Clin. 2009;27(3):533-550.
2. Müller C, Rahn BA, Pfister U, Meinig RP. The incidence, pathogenesis, diagnosis, and treatment of fat embolism. Orthop Rev. 1994;23(2):107-117.
3. Stein PD, Yaekoub AY, Matta F, Kleerekoper M. Fat embolism syndrome. Am J Med Sci. 2008;336(6):472-477.
4. Parisi DM, Koval K, Egol K. Fat embolism syndrome. Am J Orthop. 2002;31(9):507-512.
5. Gurd AR. Fat embolism: an aid to diagnosis. J Bone Joint Surb Br. 1970;52(4):732-737.
6. Lee SC, Yoon JY, Nam CH, Kim TK, Jung KA, Lee DW. Cerebral fat embolism syndrome after simultaneous bilateral total knee arthroplasty: a case series. J Arthroplasty. 2012;27(3):409-414.
7. Gurd AR, Wilson RI. Fat-embolism syndrome. Lancet. 1972;2(7770):
231-232.
8. Habashi NM, Andrews PL, Scalea TM. Therapeutic aspects of fat embolism syndrome. Injury. 2006;37(Suppl 4):S68-S73.
9. Weiss W, Bardana D, Yen D. Delayed presentation of fat embolism syndrome after intramedullary nailing of a fractured femur: a case report. J Trauma. 2009;66(3):E42-E45.
10. Byrick RJ. Fat embolism and postoperative coagulopathy. Can J Anaesth. 2001;48(7):618-621.
11. Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56(3):408-416.
12. Burgher LW. Fat embolism syndrome. Chest. 1981;79(2):131-132.
13. Burgher LW, Dines DE, Linscheid RL, Didier EP. Fat embolism and the adult respiratory distress syndrome. Mayo Clin Proc. 1974;49(2):107-109.
14. Liu DD, Hsieh NK, Chen HI. Histopathological and biochemical changes following fat embolism with administration of corn oil micelles: a new animal model for fat embolism syndrome. J Bone Joint Surg Br. 2008;90(11):
1517-1521.
15. Liu HK, Chen WC. Images in clinical medicine. Fat embolism syndrome. N Engl J Med. 2011;364(18):1761.
16. Pinney SJ, Keating JF, Meek RN. Fat embolism syndrome in isolated femoral fractures: does timing of nailing influence incidence? Injury. 1998;29(2):
131-133.
17. Takahashi M, Suzuki R, Osakabe Y, et al. Magnetic resonance imaging findings in cerebral fat embolism: correlation with clinical manifestations. J Trauma. 1999;46(2):324-327.
18. Parizel PM, Demey HE, Veeckmans G, et al. Early diagnosis of cerebral fat embolism syndrome by diffusion-weighted MRI (starfield pattern). Stroke. 2001;32(12):2942-2944.

Aneurysmal Bone Cyst Involving the Metacarpal Bone in a Child
Less than 5% of aneurysmal bone cysts (ABCs) are located in the hand,1 and only a few cases have been reported in the literature.2-7 Unfortunately, it is impossible to predict when an ABC will exhibit aggressive behavior.4,8 Aneurysmal bone cysts and giant cell bone tumors have been considered benign9 lesions that can behave in a locally aggressive fashion.1 Optimal treatment has not been established because treatment is variable depending on the condition of the lesion. Several authors have recommended more radical treatment modalities, such as en bloc resection or excision diaphysectomy followed by strut bone grafting, which had a relatively low rate of recurrence. A relatively low rate of recurrence and other complications indicate that those techniques would serve as a good strategy for patients with expansile hand ABCs in terms of safety, simplicity, and reduced number of reoperations.3,7,10
This article reports a case of an ABC of the second metacarpal bone of the right hand in a 12-year-old boy treated with curettage and autologous morselized iliac bone grafting. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right hand–dominant 12-year-old-boy, who noticed the development of a lump in the dorsum of his right hand. On examination, we found a large, firm swelling of the dorsum of his right hand over the second metacarpal. Radiographic examination showed a symmetrical expansile lytic lesion (22×24×25 mm) involving the entire second metacarpal bone (Figure 1A). Magnetic resonance imaging (MRI) showed a well-defined expansile intramedullary lesion with preservation of the epiphyseal plate, shell-like periosteal reaction, and a multilocular appearance with a hemorrhagic compartment (fluid-fluid levels) (Figure 1B).
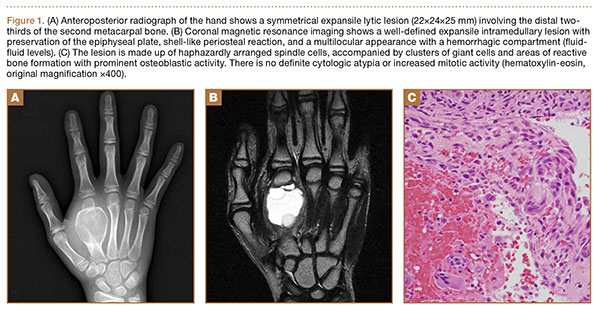
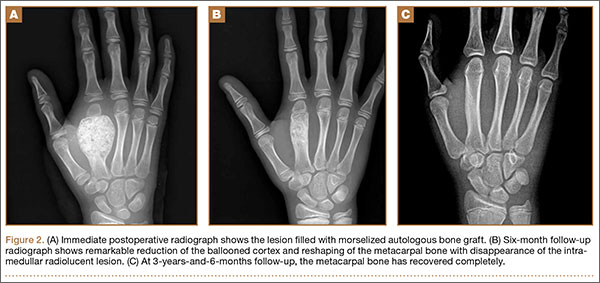
At surgery, we found a blood-filled cyst, and the cortex was very thin. The lesion extended to the distal two-thirds of the bone to the level of the physeal plate. We had considered using allograft or other bone substitutes. However, we did not have confidence in the bone-induction potential and power of osteogenesis of bone substitutes or allograft compared with autologous bone graft. Consequently, we performed autologous bone grafting, despite its being an invasive procedure, on the immature iliac crest. We performed thorough curettage of the intramedullary material without damaging the physeal plate, followed by impact morselized autologous bone grafting. Histologic examination confirmed that the final diagnosis was identical to the provisional diagnosis shown on MRI (Figure 1C). A thumb spica cast was applied for 4 weeks after surgery, and regular follow-up radiographs were taken for 3 years and 6 months until confirmation of complete normalization of the lesion without recurrence (Figures 2A-2C).
Discussion
Primary ABCs in the small tubular bones of the hands are rare. Less than 5% of aneurysmal cysts are located in the hand.1 Only a few small cases of this condition have been reported in the literature.2-7 Radiographic examination showed that, in all cases, the lesion was both expansile and completely lucent.7 Although radiographic finding of ABC in short tubular bone characteristically shows central symmetry with expansion into the diaphysis and subarticular bone, the appearance of an ABC on radiographs and angiograms is usually not diagnostic.8 Even though fluid-fluid levels are highly suggestive of ABC, only pathologic study confirms the diagnosis. MRI may be a good tool for postsurgery follow-up. On the basis of these ideas, we performed histological examination and confirmed the diagnosis of ABC of the metacarpus by radiograph and MRI.
The goals in the treatment of primary ABCs are preservation of function and avoidance of recurrence. Unfortunately, it is impossible to predict the possible aggressive behavior in ABCs. Active or aggressive character in certain localizations of ABC in children requires either curettage, which has a considerable recurrence rate, or radical segmental excision, which raises complex reconstructive challenges. Frassica and colleagues7 reported no recurrences in 3 patients treated by complete excision and bone grafting. Curettage and bone grafting in 7 cases were associated with 4 recurrences.7
Because optimal treatment has not been established,3 current recommendations vary, depending on the condition of the lesion. Several authors recommend more radical treatment modalities, such as en bloc resection, excision diaphysectomy, cryotherapy, and strut bone grafting, and a relatively low rate of recurrence and other complications indicates that those techniques would serve as a good strategy for patients with expansile ABCs in the hand.3,7,10 On the other hand, successful results with less aggressive procedures, such as curettage and autologous bone grafting, have been reported.4,5,8
In pediatric patients, surgery to preserve the growth plate is recommended.5 Ropars and colleagues4 suggested that aggressive treatment approaches, such as cryotherapy and resection with reconstruction, should be used only in cases when the articular surface is involved, when full-bone invasion of the phalanx or metacarpal has occurred, or in cases of more than 1 recurrence.
In conclusion, despite the high risk of recurrence of ABC treated with curettage with bone grafting, the findings of the present case show that ABC of the metacarpal bone in children can be treated successfully with curettage followed by morselized autologous bone grafting without recurrence.
1. Athanasian EA. Aneurysmal bone cyst and giant cell tumor of bone of the hand and distal radius. Hand Clin. 2004;20(3):269-281, vi.
2. Tarazona-Velutini P, Romo-Rodriguez R, Saleme-Cruz J. Aneurysmatic bone cyst in the proximal phalanx of a finger. Case report and literature review. Acta Ortop Mex. 2012;26(4):245-249.
3. Jafari D, Jamshidi K, Najdmazhar F, Shariatzade H, Liaghat O. Expansile aneurysmal bone cyst in the tubular bones of the hand treated with en bloc excision and autograft reconstruction: a report of 12 cases. J Hand Surg Eur Vol. 2011;36(8):648-655.
4. Ropars M, Kaila R, Briggs T, Cannon S. Aneurysmal bone cysts of the metacarpals and phalanges of the hand. A 6 case series and literature review. Chir Main. 2007;26(4-5):214-217.
5. Sproule JA, Salmo E, Mortimer G, O’Sullivan M. Aneursymal bone cyst of the proximal phalanx of the thumb in a child. Hand Surg. 2002;7(1):147-150.
6. Schwartz GB, Hammerman MZ. Aneurysmal bone cyst of the fifth metacarpal. Orthop Rev. 1989;18(12):1309-1314.
7. Frassica FJ, Amadio PC, Wold LE, Beabout JW. Aneurysmal bone cyst: clinicopathologic features and treatment of ten cases involving the hand. J Hand Surg Am. 1988;13(5):676-683.
8. Louahem D, Kouyoumdjian P, Ghanem I, et al. Active aneurysmal bone cysts in children: possible evolution after biopsy. J Child Orthop. 2012;6(4):333-338.
9. Lindfors NC. Treatment of a recurrent aneurysmal bone cyst with bioactive glass in a child allows for good bone remodelling and growth. Bone. 2009;45(2):398-400.
10. Salon A, Rémi J, Brunelle F, Drapé JL, Glorion Ch. Total replacement of a middle phalanx by free non-vascularized chondral graft, after failure of sclerotherapy for treatment of an aneurysmal bone cyst. Chir Main. 2005;24(3-4):187-192.
Less than 5% of aneurysmal bone cysts (ABCs) are located in the hand,1 and only a few cases have been reported in the literature.2-7 Unfortunately, it is impossible to predict when an ABC will exhibit aggressive behavior.4,8 Aneurysmal bone cysts and giant cell bone tumors have been considered benign9 lesions that can behave in a locally aggressive fashion.1 Optimal treatment has not been established because treatment is variable depending on the condition of the lesion. Several authors have recommended more radical treatment modalities, such as en bloc resection or excision diaphysectomy followed by strut bone grafting, which had a relatively low rate of recurrence. A relatively low rate of recurrence and other complications indicate that those techniques would serve as a good strategy for patients with expansile hand ABCs in terms of safety, simplicity, and reduced number of reoperations.3,7,10
This article reports a case of an ABC of the second metacarpal bone of the right hand in a 12-year-old boy treated with curettage and autologous morselized iliac bone grafting. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right hand–dominant 12-year-old-boy, who noticed the development of a lump in the dorsum of his right hand. On examination, we found a large, firm swelling of the dorsum of his right hand over the second metacarpal. Radiographic examination showed a symmetrical expansile lytic lesion (22×24×25 mm) involving the entire second metacarpal bone (Figure 1A). Magnetic resonance imaging (MRI) showed a well-defined expansile intramedullary lesion with preservation of the epiphyseal plate, shell-like periosteal reaction, and a multilocular appearance with a hemorrhagic compartment (fluid-fluid levels) (Figure 1B).
At surgery, we found a blood-filled cyst, and the cortex was very thin. The lesion extended to the distal two-thirds of the bone to the level of the physeal plate. We had considered using allograft or other bone substitutes. However, we did not have confidence in the bone-induction potential and power of osteogenesis of bone substitutes or allograft compared with autologous bone graft. Consequently, we performed autologous bone grafting, despite its being an invasive procedure, on the immature iliac crest. We performed thorough curettage of the intramedullary material without damaging the physeal plate, followed by impact morselized autologous bone grafting. Histologic examination confirmed that the final diagnosis was identical to the provisional diagnosis shown on MRI (Figure 1C). A thumb spica cast was applied for 4 weeks after surgery, and regular follow-up radiographs were taken for 3 years and 6 months until confirmation of complete normalization of the lesion without recurrence (Figures 2A-2C).
Discussion
Primary ABCs in the small tubular bones of the hands are rare. Less than 5% of aneurysmal cysts are located in the hand.1 Only a few small cases of this condition have been reported in the literature.2-7 Radiographic examination showed that, in all cases, the lesion was both expansile and completely lucent.7 Although radiographic finding of ABC in short tubular bone characteristically shows central symmetry with expansion into the diaphysis and subarticular bone, the appearance of an ABC on radiographs and angiograms is usually not diagnostic.8 Even though fluid-fluid levels are highly suggestive of ABC, only pathologic study confirms the diagnosis. MRI may be a good tool for postsurgery follow-up. On the basis of these ideas, we performed histological examination and confirmed the diagnosis of ABC of the metacarpus by radiograph and MRI.
The goals in the treatment of primary ABCs are preservation of function and avoidance of recurrence. Unfortunately, it is impossible to predict the possible aggressive behavior in ABCs. Active or aggressive character in certain localizations of ABC in children requires either curettage, which has a considerable recurrence rate, or radical segmental excision, which raises complex reconstructive challenges. Frassica and colleagues7 reported no recurrences in 3 patients treated by complete excision and bone grafting. Curettage and bone grafting in 7 cases were associated with 4 recurrences.7
Because optimal treatment has not been established,3 current recommendations vary, depending on the condition of the lesion. Several authors recommend more radical treatment modalities, such as en bloc resection, excision diaphysectomy, cryotherapy, and strut bone grafting, and a relatively low rate of recurrence and other complications indicates that those techniques would serve as a good strategy for patients with expansile ABCs in the hand.3,7,10 On the other hand, successful results with less aggressive procedures, such as curettage and autologous bone grafting, have been reported.4,5,8
In pediatric patients, surgery to preserve the growth plate is recommended.5 Ropars and colleagues4 suggested that aggressive treatment approaches, such as cryotherapy and resection with reconstruction, should be used only in cases when the articular surface is involved, when full-bone invasion of the phalanx or metacarpal has occurred, or in cases of more than 1 recurrence.
In conclusion, despite the high risk of recurrence of ABC treated with curettage with bone grafting, the findings of the present case show that ABC of the metacarpal bone in children can be treated successfully with curettage followed by morselized autologous bone grafting without recurrence.
Less than 5% of aneurysmal bone cysts (ABCs) are located in the hand,1 and only a few cases have been reported in the literature.2-7 Unfortunately, it is impossible to predict when an ABC will exhibit aggressive behavior.4,8 Aneurysmal bone cysts and giant cell bone tumors have been considered benign9 lesions that can behave in a locally aggressive fashion.1 Optimal treatment has not been established because treatment is variable depending on the condition of the lesion. Several authors have recommended more radical treatment modalities, such as en bloc resection or excision diaphysectomy followed by strut bone grafting, which had a relatively low rate of recurrence. A relatively low rate of recurrence and other complications indicate that those techniques would serve as a good strategy for patients with expansile hand ABCs in terms of safety, simplicity, and reduced number of reoperations.3,7,10
This article reports a case of an ABC of the second metacarpal bone of the right hand in a 12-year-old boy treated with curettage and autologous morselized iliac bone grafting. The patient’s guardian provided written informed consent for print and electronic publication of this case report.
Case Report
The patient was a right hand–dominant 12-year-old-boy, who noticed the development of a lump in the dorsum of his right hand. On examination, we found a large, firm swelling of the dorsum of his right hand over the second metacarpal. Radiographic examination showed a symmetrical expansile lytic lesion (22×24×25 mm) involving the entire second metacarpal bone (Figure 1A). Magnetic resonance imaging (MRI) showed a well-defined expansile intramedullary lesion with preservation of the epiphyseal plate, shell-like periosteal reaction, and a multilocular appearance with a hemorrhagic compartment (fluid-fluid levels) (Figure 1B).
At surgery, we found a blood-filled cyst, and the cortex was very thin. The lesion extended to the distal two-thirds of the bone to the level of the physeal plate. We had considered using allograft or other bone substitutes. However, we did not have confidence in the bone-induction potential and power of osteogenesis of bone substitutes or allograft compared with autologous bone graft. Consequently, we performed autologous bone grafting, despite its being an invasive procedure, on the immature iliac crest. We performed thorough curettage of the intramedullary material without damaging the physeal plate, followed by impact morselized autologous bone grafting. Histologic examination confirmed that the final diagnosis was identical to the provisional diagnosis shown on MRI (Figure 1C). A thumb spica cast was applied for 4 weeks after surgery, and regular follow-up radiographs were taken for 3 years and 6 months until confirmation of complete normalization of the lesion without recurrence (Figures 2A-2C).
Discussion
Primary ABCs in the small tubular bones of the hands are rare. Less than 5% of aneurysmal cysts are located in the hand.1 Only a few small cases of this condition have been reported in the literature.2-7 Radiographic examination showed that, in all cases, the lesion was both expansile and completely lucent.7 Although radiographic finding of ABC in short tubular bone characteristically shows central symmetry with expansion into the diaphysis and subarticular bone, the appearance of an ABC on radiographs and angiograms is usually not diagnostic.8 Even though fluid-fluid levels are highly suggestive of ABC, only pathologic study confirms the diagnosis. MRI may be a good tool for postsurgery follow-up. On the basis of these ideas, we performed histological examination and confirmed the diagnosis of ABC of the metacarpus by radiograph and MRI.
The goals in the treatment of primary ABCs are preservation of function and avoidance of recurrence. Unfortunately, it is impossible to predict the possible aggressive behavior in ABCs. Active or aggressive character in certain localizations of ABC in children requires either curettage, which has a considerable recurrence rate, or radical segmental excision, which raises complex reconstructive challenges. Frassica and colleagues7 reported no recurrences in 3 patients treated by complete excision and bone grafting. Curettage and bone grafting in 7 cases were associated with 4 recurrences.7
Because optimal treatment has not been established,3 current recommendations vary, depending on the condition of the lesion. Several authors recommend more radical treatment modalities, such as en bloc resection, excision diaphysectomy, cryotherapy, and strut bone grafting, and a relatively low rate of recurrence and other complications indicates that those techniques would serve as a good strategy for patients with expansile ABCs in the hand.3,7,10 On the other hand, successful results with less aggressive procedures, such as curettage and autologous bone grafting, have been reported.4,5,8
In pediatric patients, surgery to preserve the growth plate is recommended.5 Ropars and colleagues4 suggested that aggressive treatment approaches, such as cryotherapy and resection with reconstruction, should be used only in cases when the articular surface is involved, when full-bone invasion of the phalanx or metacarpal has occurred, or in cases of more than 1 recurrence.
In conclusion, despite the high risk of recurrence of ABC treated with curettage with bone grafting, the findings of the present case show that ABC of the metacarpal bone in children can be treated successfully with curettage followed by morselized autologous bone grafting without recurrence.
1. Athanasian EA. Aneurysmal bone cyst and giant cell tumor of bone of the hand and distal radius. Hand Clin. 2004;20(3):269-281, vi.
2. Tarazona-Velutini P, Romo-Rodriguez R, Saleme-Cruz J. Aneurysmatic bone cyst in the proximal phalanx of a finger. Case report and literature review. Acta Ortop Mex. 2012;26(4):245-249.
3. Jafari D, Jamshidi K, Najdmazhar F, Shariatzade H, Liaghat O. Expansile aneurysmal bone cyst in the tubular bones of the hand treated with en bloc excision and autograft reconstruction: a report of 12 cases. J Hand Surg Eur Vol. 2011;36(8):648-655.
4. Ropars M, Kaila R, Briggs T, Cannon S. Aneurysmal bone cysts of the metacarpals and phalanges of the hand. A 6 case series and literature review. Chir Main. 2007;26(4-5):214-217.
5. Sproule JA, Salmo E, Mortimer G, O’Sullivan M. Aneursymal bone cyst of the proximal phalanx of the thumb in a child. Hand Surg. 2002;7(1):147-150.
6. Schwartz GB, Hammerman MZ. Aneurysmal bone cyst of the fifth metacarpal. Orthop Rev. 1989;18(12):1309-1314.
7. Frassica FJ, Amadio PC, Wold LE, Beabout JW. Aneurysmal bone cyst: clinicopathologic features and treatment of ten cases involving the hand. J Hand Surg Am. 1988;13(5):676-683.
8. Louahem D, Kouyoumdjian P, Ghanem I, et al. Active aneurysmal bone cysts in children: possible evolution after biopsy. J Child Orthop. 2012;6(4):333-338.
9. Lindfors NC. Treatment of a recurrent aneurysmal bone cyst with bioactive glass in a child allows for good bone remodelling and growth. Bone. 2009;45(2):398-400.
10. Salon A, Rémi J, Brunelle F, Drapé JL, Glorion Ch. Total replacement of a middle phalanx by free non-vascularized chondral graft, after failure of sclerotherapy for treatment of an aneurysmal bone cyst. Chir Main. 2005;24(3-4):187-192.
1. Athanasian EA. Aneurysmal bone cyst and giant cell tumor of bone of the hand and distal radius. Hand Clin. 2004;20(3):269-281, vi.
2. Tarazona-Velutini P, Romo-Rodriguez R, Saleme-Cruz J. Aneurysmatic bone cyst in the proximal phalanx of a finger. Case report and literature review. Acta Ortop Mex. 2012;26(4):245-249.
3. Jafari D, Jamshidi K, Najdmazhar F, Shariatzade H, Liaghat O. Expansile aneurysmal bone cyst in the tubular bones of the hand treated with en bloc excision and autograft reconstruction: a report of 12 cases. J Hand Surg Eur Vol. 2011;36(8):648-655.
4. Ropars M, Kaila R, Briggs T, Cannon S. Aneurysmal bone cysts of the metacarpals and phalanges of the hand. A 6 case series and literature review. Chir Main. 2007;26(4-5):214-217.
5. Sproule JA, Salmo E, Mortimer G, O’Sullivan M. Aneursymal bone cyst of the proximal phalanx of the thumb in a child. Hand Surg. 2002;7(1):147-150.
6. Schwartz GB, Hammerman MZ. Aneurysmal bone cyst of the fifth metacarpal. Orthop Rev. 1989;18(12):1309-1314.
7. Frassica FJ, Amadio PC, Wold LE, Beabout JW. Aneurysmal bone cyst: clinicopathologic features and treatment of ten cases involving the hand. J Hand Surg Am. 1988;13(5):676-683.
8. Louahem D, Kouyoumdjian P, Ghanem I, et al. Active aneurysmal bone cysts in children: possible evolution after biopsy. J Child Orthop. 2012;6(4):333-338.
9. Lindfors NC. Treatment of a recurrent aneurysmal bone cyst with bioactive glass in a child allows for good bone remodelling and growth. Bone. 2009;45(2):398-400.
10. Salon A, Rémi J, Brunelle F, Drapé JL, Glorion Ch. Total replacement of a middle phalanx by free non-vascularized chondral graft, after failure of sclerotherapy for treatment of an aneurysmal bone cyst. Chir Main. 2005;24(3-4):187-192.
Use of Cross-Leg Flap for Wound Complications Resulting From Open Pilon Fracture
Soft-tissue complications are a known problem in the treatment of pilon fractures of the distal end of the tibia. These fractures typically occur as the result of a high-energy mechanism, and axial load and shear forces often lead to a severe soft-tissue injury. In many cases, these injuries may require additional procedures to provide adequate soft-tissue coverage. These procedures can include use of either a rotational muscle flap or a free flap transfer. In some cases, however, these flaps are not possible secondary to vascular compromise.
In this article, we report the case of a pilon fracture combined with severe soft-tissue injury and vascular compromise of the leg. A cross-leg fasciocutaneous flap was performed as a salvage procedure for coverage of the soft-tissue defect. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
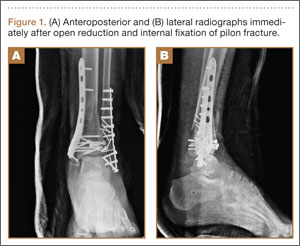
A 23-year-old man sustained a left grade III open pilon fracture after a fall off a cherry picker. He was initially treated with irrigation and débridement of the open anteromedial wound, wound closure, application of external fixation, and open reduction and internal fixation (ORIF) of the concomitant comminuted fibular fracture. Operative fixation of the pilon was performed 3 weeks after injury, once skin and soft tissues were in acceptable condition (Figure 1). Skin closure was performed with 2-0 Vicryl sutures (Ethicon, Inc, Somerville, New Jersey) followed by 3-0 nylon skin sutures and No. 2 nylon retention sutures to reduce tension at the incision.
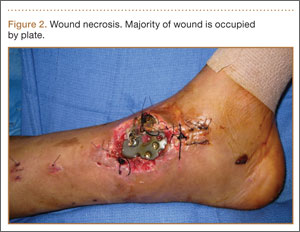
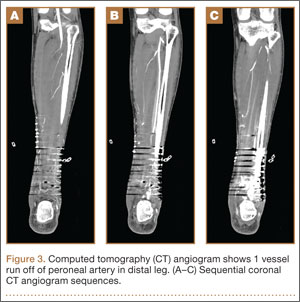
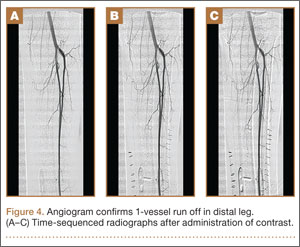
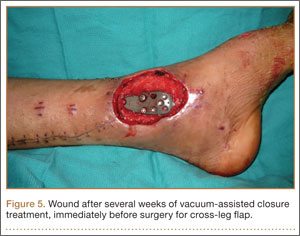
On postoperative day 17, the patient was found to have skin necrosis with exposed hardware over the medial laceration that had resulted from the open fracture (Figure 2). The wound measured 7×6 cm. The plastic surgery team was consulted, and a soft-tissue flap was recommended. Preoperative computed tomography angiogram (Figure 3) revealed 1 vessel runoff in the leg, constituting the peroneal artery, and a conventional angiogram confirmed this finding (Figure 4). Despite these findings, the patient was taken to the operating room 4 weeks after initial injury to try to find a vessel compatible with anastomosis. Intraoperative wound exploration confirmed no patent blood supply for local soft-tissue flap coverage. Therefore, the wound was irrigated and débrided, and a vacuum-assisted closure (VAC) dressing was applied despite exposed hardware and bone. A decision was then made to attempt a cross-leg flap as a salvage procedure, and VAC dressing therapy was continued for several weeks to prepare the recipient site (Figure 5).
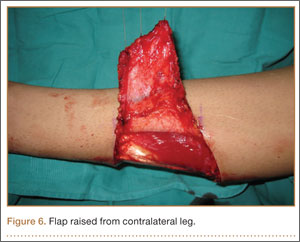
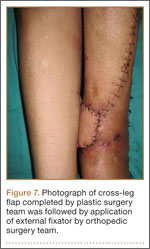
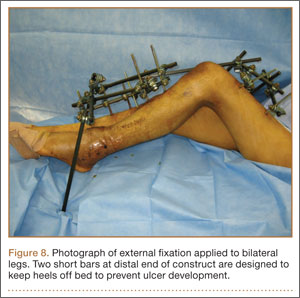
Seven weeks after injury, the patient was taken to the operating room by the orthopedic surgery and plastic surgery teams. After débridement, a fasciocutaneous flap was raised from the middle third of the contralateral leg (Figure 6) based on a posterior tibial artery perforator. The flap, which measured 7×7 cm (sufficient to cover the defect), was raised from lateral to medial from the posterior aspect of the leg with the pedicle located on the medial aspect of the right leg. Flap placement was facilitated by flexing the left knee to 80°. The flap was sutured into place with 4-0 Vicryl deep sutures followed by 4-0 nylon and superficial sutures in an interrupted fashion (Figure 7). Rigid external fixation was then applied to both extremities, bridging them together in optimal position (Figure 8). This construct included 2 short bars that would elevate the patient’s heels off the bed to reduce the chance of heel decubiti. Although including the feet in the external fixator construct may help prevent equinus contracture, we splinted the ankles in neutral position immediately after surgery so that we could begin early range-of-motion (ROM) exercises of the ankles to prevent stiffness. Ankle ROM exercises were started once the flap incorporated, 3 weeks after placement of the external fixator. Lacking medical insurance coverage, the patient could not be admitted to a rehabilitation facility or receive home care. He lived independently and had no help at home, so he had to remain hospitalized after placement of the external fixator. While hospitalized, the surgical site was treated with frequent dressing changes, including use of bacitracin and nonadherent dressing.
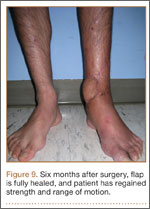
After flap coverage and 4 weeks of bed rest, a base clamping test confirmed the flap was incorporated into the recipient bed. The patient was then returned to the operating room for removal of the external fixator and skin grafting of the donor site. After surgery, he was started on physical therapy, including exercises for bilateral hip, knee, and ankle ROM and strengthening of the lower extremities. Four months after initial injury, the fracture was healed, based on bone consolidation, seen on radiographs, that is consistent with other pilon fractures treated at our institution. Six months after external fixator removal, the patient was able to ambulate independently with minimal discomfort (Figure 9). Passive and active ankle ROM was 20° of dorsiflexion and 25° of plantarflexion, compared with 25° of dorsiflexion and 45° of plantarflexion on the contralateral extremity. Subtalar motion had some stiffness with a 10° arc, compared with a 25° arc on the contralateral extremity. On simple manual testing, the patient had 5/5 motor strength with dorsiflexion, plantarflexion, inversion, and eversion. He returned to full duty as a landscaper about 1 year after initial injury and had no recurrence of wound complications or infection.
Discussion
Fractures of the distal tibia are commonly known as pilon or plafond fractures. They represent up to 10% of all tibial fractures. The injury consists of an intra-articular fracture of the tibiotalar joint with varying degrees of proximal extension into the tibial metaphysis. The etiology is an axial load on the tibia with or without a rotational force.1 Treatment is challenging. The literature includes many reports of wound and soft-tissue complications after ORIF. In 1969, Rüedi and Allgöwer2 published recommendations that have become the standard for treatment of pilon fractures. Twelve percent of the 84 fractures included in their study were associated with wound complications. In 2004, Sirkin and colleagues3 suggested that wound problems associated with ORIF of pilon fractures may be caused by attempts at immediate fixation through swollen soft tissue. They postulated that staging the procedure and waiting for decreased soft-tissue swelling may reduce the incidence of wound complications. In their series, only 2.9% of closed pilon fractures and only 9.1% of open fractures had any wound complications, and none of their patients required skin grafts, rotation flaps, or free tissue transfers.
However, soft-tissue complications still remain a significant threat in the treatment of pilon fracture, and cases that require additional procedures for soft-tissue coverage are common. In some cases, wound necrosis may lead to below-knee amputation.4 There are several coverage options, including local rotational flaps using the soleus muscle5,6 as well as free flaps using the latissimus dorsi, gracilis, or rectus abdominis muscles.7 These options require a sufficient blood supply to the region.
Many high-energy pilon fractures may be associated with vascular injury, and therefore flap survival may be compromised. We have reported such a case in the present article. Our patient’s preoperative angiogram indicated he had 1-vessel runoff to the distal leg—a situation incompatible with free tissue transfer. It is not clear whether this finding is secondary to trauma to the leg or is caused by an anatomical anomaly. Nevertheless, the poor vascularity posed a challenge to providing soft-tissue coverage. Cross-finger8 and cross-foot9 flaps have been described in upper and lower extremity injuries. In 2006, Zhao and colleagues10 reported on 5 patients with tibia and/or hardware exposure after operative fixation of tibia fractures. These patients had poor local soft tissue around the wound and therefore underwent cross-leg flap for coverage. It is not clear where the soft-tissue defects were located and whether any studies were performed to assess the local blood flow.
From our patient’s case, we learned that multiple factors should be considered when assessing such high-energy injuries. First, respecting the soft tissues is of paramount importance. Our initial management on presentation consisted of irrigation and débridement of the wound, fixation of the fibula, and application of an external fixator to allow for soft-tissue healing before definitive fixation of the pilon. Although ultimately the patient required soft-tissue coverage, soft-tissue healing and viability are important in preventing unnecessary soft-tissue procedures, and therefore we would not have handled our initial treatment differently.
Patient selection is also important. The ideal candidate for a cross-leg flap is a young, healthy person who is compliant and has a strong support system to help with activities of daily living. Unfortunately, because of financial issues and lack of home support, our patient remained hospitalized during his treatment course. For a patient who has support, it is possible to be discharged either home or to a rehabilitation facility once flap viability has been confirmed after surgery.
Another consideration is type of immobilization. Immobilization options include casting, use of Kirschner wires (K-wires), and use of rigid external fixation. For cross-leg flaps, external fixation is superior to casting and K-wires, as it provides a more rigid construct and easier access to the flap for serial evaluation. Further, it is easier for the patient to maintain personal hygiene, and it can provide heel rises to avoid pressure ulcers.
Conclusion
To our knowledge, there have been no reports of using a cross-leg flap for wound complications in high-energy pilon fractures. As already mentioned, many of these fractures may be associated with severe soft-tissue injury and may need flap coverage. A cross-leg flap with external fixation of both legs provides a limb salvage option with satisfactory patient outcomes.
1. McCann PA, Jackson M, Mitchell ST, Atkins RM. Complications of definitive open reduction and internal fixation of pilon fractures of the distal tibia. Int Orthop. 2011;35(3):413-418.
2. Rüedi TP, Allgöwer M. Fractures of the lower end of the tibia into the ankle joint. Injury. 1969;1:92-99.
3. Sirkin M, Sanders R, DiPasquale T, Herscovici D Jr. A staged protocol for soft tissue management in the treatment of complex pilon fractures. J Orthop Trauma. 2004;18(8 suppl):S32-S38.
4. Boraiah S, Kemp TJ, Erwteman A, Lucas PA, Asprinio DE. Outcome following open reduction and internal fixation of open pilon fractures. J Bone Joint Surg Am. 2010;92(2):346-352.
5. Cheng C, Li X, Abudu S. Repairing postoperative soft tissue defects of tibia and ankle open fractures with muscle flap pedicled with medial half of soleus [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2009;23(12):1440-1442.
6. Yunus A, Yusuf A, Chen G. Repair of soft tissue defect by reverse soleus muscle flap after pilon fracture fixation [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2007;21(9):925-927.
7. Conroy J, Agarwal M, Giannoudis PV, Matthews SJ. Early internal fixation and soft tissue cover of severe open tibial pilon fractures. Int Orthop. 2003;27(6):343-347.
8. Megerle K, Palm-Bröking K, Germann G. The cross-finger flap [in German]. Oper Orthop Traumatol. 2008;20(2):97-102.
9. Largey A, Faline A, Hebrard W, Hamoui M, Canovas F. Management of massive traumatic compound defects of the foot. Orthop Traumatol Surg Res. 2009;95(4):301-304.
10. Zhao L, Wan L, Wang S. Clinical studies on maintenance of cross-leg position through internal fixation with Kirschner wire after cross-leg flap procedure. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2006;20(12):1211-1213.
Soft-tissue complications are a known problem in the treatment of pilon fractures of the distal end of the tibia. These fractures typically occur as the result of a high-energy mechanism, and axial load and shear forces often lead to a severe soft-tissue injury. In many cases, these injuries may require additional procedures to provide adequate soft-tissue coverage. These procedures can include use of either a rotational muscle flap or a free flap transfer. In some cases, however, these flaps are not possible secondary to vascular compromise.
In this article, we report the case of a pilon fracture combined with severe soft-tissue injury and vascular compromise of the leg. A cross-leg fasciocutaneous flap was performed as a salvage procedure for coverage of the soft-tissue defect. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 23-year-old man sustained a left grade III open pilon fracture after a fall off a cherry picker. He was initially treated with irrigation and débridement of the open anteromedial wound, wound closure, application of external fixation, and open reduction and internal fixation (ORIF) of the concomitant comminuted fibular fracture. Operative fixation of the pilon was performed 3 weeks after injury, once skin and soft tissues were in acceptable condition (Figure 1). Skin closure was performed with 2-0 Vicryl sutures (Ethicon, Inc, Somerville, New Jersey) followed by 3-0 nylon skin sutures and No. 2 nylon retention sutures to reduce tension at the incision.
On postoperative day 17, the patient was found to have skin necrosis with exposed hardware over the medial laceration that had resulted from the open fracture (Figure 2). The wound measured 7×6 cm. The plastic surgery team was consulted, and a soft-tissue flap was recommended. Preoperative computed tomography angiogram (Figure 3) revealed 1 vessel runoff in the leg, constituting the peroneal artery, and a conventional angiogram confirmed this finding (Figure 4). Despite these findings, the patient was taken to the operating room 4 weeks after initial injury to try to find a vessel compatible with anastomosis. Intraoperative wound exploration confirmed no patent blood supply for local soft-tissue flap coverage. Therefore, the wound was irrigated and débrided, and a vacuum-assisted closure (VAC) dressing was applied despite exposed hardware and bone. A decision was then made to attempt a cross-leg flap as a salvage procedure, and VAC dressing therapy was continued for several weeks to prepare the recipient site (Figure 5).
Seven weeks after injury, the patient was taken to the operating room by the orthopedic surgery and plastic surgery teams. After débridement, a fasciocutaneous flap was raised from the middle third of the contralateral leg (Figure 6) based on a posterior tibial artery perforator. The flap, which measured 7×7 cm (sufficient to cover the defect), was raised from lateral to medial from the posterior aspect of the leg with the pedicle located on the medial aspect of the right leg. Flap placement was facilitated by flexing the left knee to 80°. The flap was sutured into place with 4-0 Vicryl deep sutures followed by 4-0 nylon and superficial sutures in an interrupted fashion (Figure 7). Rigid external fixation was then applied to both extremities, bridging them together in optimal position (Figure 8). This construct included 2 short bars that would elevate the patient’s heels off the bed to reduce the chance of heel decubiti. Although including the feet in the external fixator construct may help prevent equinus contracture, we splinted the ankles in neutral position immediately after surgery so that we could begin early range-of-motion (ROM) exercises of the ankles to prevent stiffness. Ankle ROM exercises were started once the flap incorporated, 3 weeks after placement of the external fixator. Lacking medical insurance coverage, the patient could not be admitted to a rehabilitation facility or receive home care. He lived independently and had no help at home, so he had to remain hospitalized after placement of the external fixator. While hospitalized, the surgical site was treated with frequent dressing changes, including use of bacitracin and nonadherent dressing.
After flap coverage and 4 weeks of bed rest, a base clamping test confirmed the flap was incorporated into the recipient bed. The patient was then returned to the operating room for removal of the external fixator and skin grafting of the donor site. After surgery, he was started on physical therapy, including exercises for bilateral hip, knee, and ankle ROM and strengthening of the lower extremities. Four months after initial injury, the fracture was healed, based on bone consolidation, seen on radiographs, that is consistent with other pilon fractures treated at our institution. Six months after external fixator removal, the patient was able to ambulate independently with minimal discomfort (Figure 9). Passive and active ankle ROM was 20° of dorsiflexion and 25° of plantarflexion, compared with 25° of dorsiflexion and 45° of plantarflexion on the contralateral extremity. Subtalar motion had some stiffness with a 10° arc, compared with a 25° arc on the contralateral extremity. On simple manual testing, the patient had 5/5 motor strength with dorsiflexion, plantarflexion, inversion, and eversion. He returned to full duty as a landscaper about 1 year after initial injury and had no recurrence of wound complications or infection.
Discussion
Fractures of the distal tibia are commonly known as pilon or plafond fractures. They represent up to 10% of all tibial fractures. The injury consists of an intra-articular fracture of the tibiotalar joint with varying degrees of proximal extension into the tibial metaphysis. The etiology is an axial load on the tibia with or without a rotational force.1 Treatment is challenging. The literature includes many reports of wound and soft-tissue complications after ORIF. In 1969, Rüedi and Allgöwer2 published recommendations that have become the standard for treatment of pilon fractures. Twelve percent of the 84 fractures included in their study were associated with wound complications. In 2004, Sirkin and colleagues3 suggested that wound problems associated with ORIF of pilon fractures may be caused by attempts at immediate fixation through swollen soft tissue. They postulated that staging the procedure and waiting for decreased soft-tissue swelling may reduce the incidence of wound complications. In their series, only 2.9% of closed pilon fractures and only 9.1% of open fractures had any wound complications, and none of their patients required skin grafts, rotation flaps, or free tissue transfers.
However, soft-tissue complications still remain a significant threat in the treatment of pilon fracture, and cases that require additional procedures for soft-tissue coverage are common. In some cases, wound necrosis may lead to below-knee amputation.4 There are several coverage options, including local rotational flaps using the soleus muscle5,6 as well as free flaps using the latissimus dorsi, gracilis, or rectus abdominis muscles.7 These options require a sufficient blood supply to the region.
Many high-energy pilon fractures may be associated with vascular injury, and therefore flap survival may be compromised. We have reported such a case in the present article. Our patient’s preoperative angiogram indicated he had 1-vessel runoff to the distal leg—a situation incompatible with free tissue transfer. It is not clear whether this finding is secondary to trauma to the leg or is caused by an anatomical anomaly. Nevertheless, the poor vascularity posed a challenge to providing soft-tissue coverage. Cross-finger8 and cross-foot9 flaps have been described in upper and lower extremity injuries. In 2006, Zhao and colleagues10 reported on 5 patients with tibia and/or hardware exposure after operative fixation of tibia fractures. These patients had poor local soft tissue around the wound and therefore underwent cross-leg flap for coverage. It is not clear where the soft-tissue defects were located and whether any studies were performed to assess the local blood flow.
From our patient’s case, we learned that multiple factors should be considered when assessing such high-energy injuries. First, respecting the soft tissues is of paramount importance. Our initial management on presentation consisted of irrigation and débridement of the wound, fixation of the fibula, and application of an external fixator to allow for soft-tissue healing before definitive fixation of the pilon. Although ultimately the patient required soft-tissue coverage, soft-tissue healing and viability are important in preventing unnecessary soft-tissue procedures, and therefore we would not have handled our initial treatment differently.
Patient selection is also important. The ideal candidate for a cross-leg flap is a young, healthy person who is compliant and has a strong support system to help with activities of daily living. Unfortunately, because of financial issues and lack of home support, our patient remained hospitalized during his treatment course. For a patient who has support, it is possible to be discharged either home or to a rehabilitation facility once flap viability has been confirmed after surgery.
Another consideration is type of immobilization. Immobilization options include casting, use of Kirschner wires (K-wires), and use of rigid external fixation. For cross-leg flaps, external fixation is superior to casting and K-wires, as it provides a more rigid construct and easier access to the flap for serial evaluation. Further, it is easier for the patient to maintain personal hygiene, and it can provide heel rises to avoid pressure ulcers.
Conclusion
To our knowledge, there have been no reports of using a cross-leg flap for wound complications in high-energy pilon fractures. As already mentioned, many of these fractures may be associated with severe soft-tissue injury and may need flap coverage. A cross-leg flap with external fixation of both legs provides a limb salvage option with satisfactory patient outcomes.
Soft-tissue complications are a known problem in the treatment of pilon fractures of the distal end of the tibia. These fractures typically occur as the result of a high-energy mechanism, and axial load and shear forces often lead to a severe soft-tissue injury. In many cases, these injuries may require additional procedures to provide adequate soft-tissue coverage. These procedures can include use of either a rotational muscle flap or a free flap transfer. In some cases, however, these flaps are not possible secondary to vascular compromise.
In this article, we report the case of a pilon fracture combined with severe soft-tissue injury and vascular compromise of the leg. A cross-leg fasciocutaneous flap was performed as a salvage procedure for coverage of the soft-tissue defect. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 23-year-old man sustained a left grade III open pilon fracture after a fall off a cherry picker. He was initially treated with irrigation and débridement of the open anteromedial wound, wound closure, application of external fixation, and open reduction and internal fixation (ORIF) of the concomitant comminuted fibular fracture. Operative fixation of the pilon was performed 3 weeks after injury, once skin and soft tissues were in acceptable condition (Figure 1). Skin closure was performed with 2-0 Vicryl sutures (Ethicon, Inc, Somerville, New Jersey) followed by 3-0 nylon skin sutures and No. 2 nylon retention sutures to reduce tension at the incision.
On postoperative day 17, the patient was found to have skin necrosis with exposed hardware over the medial laceration that had resulted from the open fracture (Figure 2). The wound measured 7×6 cm. The plastic surgery team was consulted, and a soft-tissue flap was recommended. Preoperative computed tomography angiogram (Figure 3) revealed 1 vessel runoff in the leg, constituting the peroneal artery, and a conventional angiogram confirmed this finding (Figure 4). Despite these findings, the patient was taken to the operating room 4 weeks after initial injury to try to find a vessel compatible with anastomosis. Intraoperative wound exploration confirmed no patent blood supply for local soft-tissue flap coverage. Therefore, the wound was irrigated and débrided, and a vacuum-assisted closure (VAC) dressing was applied despite exposed hardware and bone. A decision was then made to attempt a cross-leg flap as a salvage procedure, and VAC dressing therapy was continued for several weeks to prepare the recipient site (Figure 5).
Seven weeks after injury, the patient was taken to the operating room by the orthopedic surgery and plastic surgery teams. After débridement, a fasciocutaneous flap was raised from the middle third of the contralateral leg (Figure 6) based on a posterior tibial artery perforator. The flap, which measured 7×7 cm (sufficient to cover the defect), was raised from lateral to medial from the posterior aspect of the leg with the pedicle located on the medial aspect of the right leg. Flap placement was facilitated by flexing the left knee to 80°. The flap was sutured into place with 4-0 Vicryl deep sutures followed by 4-0 nylon and superficial sutures in an interrupted fashion (Figure 7). Rigid external fixation was then applied to both extremities, bridging them together in optimal position (Figure 8). This construct included 2 short bars that would elevate the patient’s heels off the bed to reduce the chance of heel decubiti. Although including the feet in the external fixator construct may help prevent equinus contracture, we splinted the ankles in neutral position immediately after surgery so that we could begin early range-of-motion (ROM) exercises of the ankles to prevent stiffness. Ankle ROM exercises were started once the flap incorporated, 3 weeks after placement of the external fixator. Lacking medical insurance coverage, the patient could not be admitted to a rehabilitation facility or receive home care. He lived independently and had no help at home, so he had to remain hospitalized after placement of the external fixator. While hospitalized, the surgical site was treated with frequent dressing changes, including use of bacitracin and nonadherent dressing.
After flap coverage and 4 weeks of bed rest, a base clamping test confirmed the flap was incorporated into the recipient bed. The patient was then returned to the operating room for removal of the external fixator and skin grafting of the donor site. After surgery, he was started on physical therapy, including exercises for bilateral hip, knee, and ankle ROM and strengthening of the lower extremities. Four months after initial injury, the fracture was healed, based on bone consolidation, seen on radiographs, that is consistent with other pilon fractures treated at our institution. Six months after external fixator removal, the patient was able to ambulate independently with minimal discomfort (Figure 9). Passive and active ankle ROM was 20° of dorsiflexion and 25° of plantarflexion, compared with 25° of dorsiflexion and 45° of plantarflexion on the contralateral extremity. Subtalar motion had some stiffness with a 10° arc, compared with a 25° arc on the contralateral extremity. On simple manual testing, the patient had 5/5 motor strength with dorsiflexion, plantarflexion, inversion, and eversion. He returned to full duty as a landscaper about 1 year after initial injury and had no recurrence of wound complications or infection.
Discussion
Fractures of the distal tibia are commonly known as pilon or plafond fractures. They represent up to 10% of all tibial fractures. The injury consists of an intra-articular fracture of the tibiotalar joint with varying degrees of proximal extension into the tibial metaphysis. The etiology is an axial load on the tibia with or without a rotational force.1 Treatment is challenging. The literature includes many reports of wound and soft-tissue complications after ORIF. In 1969, Rüedi and Allgöwer2 published recommendations that have become the standard for treatment of pilon fractures. Twelve percent of the 84 fractures included in their study were associated with wound complications. In 2004, Sirkin and colleagues3 suggested that wound problems associated with ORIF of pilon fractures may be caused by attempts at immediate fixation through swollen soft tissue. They postulated that staging the procedure and waiting for decreased soft-tissue swelling may reduce the incidence of wound complications. In their series, only 2.9% of closed pilon fractures and only 9.1% of open fractures had any wound complications, and none of their patients required skin grafts, rotation flaps, or free tissue transfers.
However, soft-tissue complications still remain a significant threat in the treatment of pilon fracture, and cases that require additional procedures for soft-tissue coverage are common. In some cases, wound necrosis may lead to below-knee amputation.4 There are several coverage options, including local rotational flaps using the soleus muscle5,6 as well as free flaps using the latissimus dorsi, gracilis, or rectus abdominis muscles.7 These options require a sufficient blood supply to the region.
Many high-energy pilon fractures may be associated with vascular injury, and therefore flap survival may be compromised. We have reported such a case in the present article. Our patient’s preoperative angiogram indicated he had 1-vessel runoff to the distal leg—a situation incompatible with free tissue transfer. It is not clear whether this finding is secondary to trauma to the leg or is caused by an anatomical anomaly. Nevertheless, the poor vascularity posed a challenge to providing soft-tissue coverage. Cross-finger8 and cross-foot9 flaps have been described in upper and lower extremity injuries. In 2006, Zhao and colleagues10 reported on 5 patients with tibia and/or hardware exposure after operative fixation of tibia fractures. These patients had poor local soft tissue around the wound and therefore underwent cross-leg flap for coverage. It is not clear where the soft-tissue defects were located and whether any studies were performed to assess the local blood flow.
From our patient’s case, we learned that multiple factors should be considered when assessing such high-energy injuries. First, respecting the soft tissues is of paramount importance. Our initial management on presentation consisted of irrigation and débridement of the wound, fixation of the fibula, and application of an external fixator to allow for soft-tissue healing before definitive fixation of the pilon. Although ultimately the patient required soft-tissue coverage, soft-tissue healing and viability are important in preventing unnecessary soft-tissue procedures, and therefore we would not have handled our initial treatment differently.
Patient selection is also important. The ideal candidate for a cross-leg flap is a young, healthy person who is compliant and has a strong support system to help with activities of daily living. Unfortunately, because of financial issues and lack of home support, our patient remained hospitalized during his treatment course. For a patient who has support, it is possible to be discharged either home or to a rehabilitation facility once flap viability has been confirmed after surgery.
Another consideration is type of immobilization. Immobilization options include casting, use of Kirschner wires (K-wires), and use of rigid external fixation. For cross-leg flaps, external fixation is superior to casting and K-wires, as it provides a more rigid construct and easier access to the flap for serial evaluation. Further, it is easier for the patient to maintain personal hygiene, and it can provide heel rises to avoid pressure ulcers.
Conclusion
To our knowledge, there have been no reports of using a cross-leg flap for wound complications in high-energy pilon fractures. As already mentioned, many of these fractures may be associated with severe soft-tissue injury and may need flap coverage. A cross-leg flap with external fixation of both legs provides a limb salvage option with satisfactory patient outcomes.
1. McCann PA, Jackson M, Mitchell ST, Atkins RM. Complications of definitive open reduction and internal fixation of pilon fractures of the distal tibia. Int Orthop. 2011;35(3):413-418.
2. Rüedi TP, Allgöwer M. Fractures of the lower end of the tibia into the ankle joint. Injury. 1969;1:92-99.
3. Sirkin M, Sanders R, DiPasquale T, Herscovici D Jr. A staged protocol for soft tissue management in the treatment of complex pilon fractures. J Orthop Trauma. 2004;18(8 suppl):S32-S38.
4. Boraiah S, Kemp TJ, Erwteman A, Lucas PA, Asprinio DE. Outcome following open reduction and internal fixation of open pilon fractures. J Bone Joint Surg Am. 2010;92(2):346-352.
5. Cheng C, Li X, Abudu S. Repairing postoperative soft tissue defects of tibia and ankle open fractures with muscle flap pedicled with medial half of soleus [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2009;23(12):1440-1442.
6. Yunus A, Yusuf A, Chen G. Repair of soft tissue defect by reverse soleus muscle flap after pilon fracture fixation [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2007;21(9):925-927.
7. Conroy J, Agarwal M, Giannoudis PV, Matthews SJ. Early internal fixation and soft tissue cover of severe open tibial pilon fractures. Int Orthop. 2003;27(6):343-347.
8. Megerle K, Palm-Bröking K, Germann G. The cross-finger flap [in German]. Oper Orthop Traumatol. 2008;20(2):97-102.
9. Largey A, Faline A, Hebrard W, Hamoui M, Canovas F. Management of massive traumatic compound defects of the foot. Orthop Traumatol Surg Res. 2009;95(4):301-304.
10. Zhao L, Wan L, Wang S. Clinical studies on maintenance of cross-leg position through internal fixation with Kirschner wire after cross-leg flap procedure. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2006;20(12):1211-1213.
1. McCann PA, Jackson M, Mitchell ST, Atkins RM. Complications of definitive open reduction and internal fixation of pilon fractures of the distal tibia. Int Orthop. 2011;35(3):413-418.
2. Rüedi TP, Allgöwer M. Fractures of the lower end of the tibia into the ankle joint. Injury. 1969;1:92-99.
3. Sirkin M, Sanders R, DiPasquale T, Herscovici D Jr. A staged protocol for soft tissue management in the treatment of complex pilon fractures. J Orthop Trauma. 2004;18(8 suppl):S32-S38.
4. Boraiah S, Kemp TJ, Erwteman A, Lucas PA, Asprinio DE. Outcome following open reduction and internal fixation of open pilon fractures. J Bone Joint Surg Am. 2010;92(2):346-352.
5. Cheng C, Li X, Abudu S. Repairing postoperative soft tissue defects of tibia and ankle open fractures with muscle flap pedicled with medial half of soleus [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2009;23(12):1440-1442.
6. Yunus A, Yusuf A, Chen G. Repair of soft tissue defect by reverse soleus muscle flap after pilon fracture fixation [in Chinese]. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2007;21(9):925-927.
7. Conroy J, Agarwal M, Giannoudis PV, Matthews SJ. Early internal fixation and soft tissue cover of severe open tibial pilon fractures. Int Orthop. 2003;27(6):343-347.
8. Megerle K, Palm-Bröking K, Germann G. The cross-finger flap [in German]. Oper Orthop Traumatol. 2008;20(2):97-102.
9. Largey A, Faline A, Hebrard W, Hamoui M, Canovas F. Management of massive traumatic compound defects of the foot. Orthop Traumatol Surg Res. 2009;95(4):301-304.
10. Zhao L, Wan L, Wang S. Clinical studies on maintenance of cross-leg position through internal fixation with Kirschner wire after cross-leg flap procedure. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2006;20(12):1211-1213.
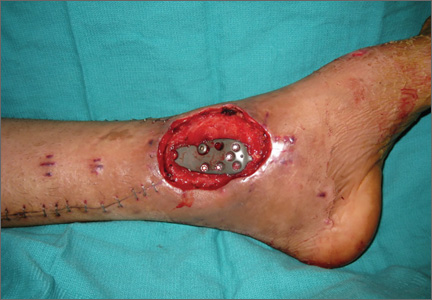
Arthritis, Infectious Tenosynovitis, and Tendon Rupture in a Patient With Rheumatoid Arthritis and Psoriasis
Compared with monoarticular arthritis, polyarticular arthritis may yield an initially narrower differential diagnosis that focuses on systemic inflammatory conditions, such as rheumatoid arthritis (RA). Approximately 15% to 30% of septic arthritis is polyarticular, of which about 45% is associated with underlying RA.1,2 Regardless of the number of joints involved, septic (infectious) arthritis is a valid consideration given the morbidity and mortality.
In a retrospective study in the United Kingdom (UK) between 1982 and 1991, the morbidity and mortality of septic arthritis was 31.6% and 11.5%, respectively, and 16% of the study population had RA.3 A review of the literature by Dubost and colleagues found that polyarticular septic arthritis (PASA) has a mortality of 31% to 42% compared with 4% to 8% for monoarticular septic arthritis, and RA was present in 67% of the PASA fatalities.1
Related: The Golden Era of Treatment in Rheumatology
Rheumatoid arthritis and its treatment predispose patients to septic arthritis. Septic arthritis in the UK general population is 0.42 per 100 patient-years for patients with RA on antitumor necrosis factor therapy.3,4 In a retrospective study in the U.S., the incidence of septic arthritis was 0.40 per 100 patient-years for patients with RA compared with 0.02 per 100 patient-years for patients without RA.5
Other complications of RA include infectious tenosynovitis and tendon rupture. The incidence and prevalence of infectious tenosynovitis and tendon rupture in RA are not firmly established in the literature.
We present a patient with RA and psoriasis who responded initially to acute management for RA but subsequently was diagnosed with culture-negative polyarticular arthritis and infectious tenosynovitis associated with beta hemolytic group G Streptococcus (GGS), a part of Streptococcus milleri (S. milleri). During surgery, he was also found to have bilateral extensor pollicus longus (EPL) tendon rupture. Given the possible morbidity, the authors believe this patient may be of interest to the medical community.
Case Presentation
A 69-year-old African American male presented with 3 to 4 days of swelling and pain of bilateral wrists, bilateral hands, and the left ankle with subjective, but resolved, fevers and chills. His medical history was significant for seropositive erosive RA, psoriasis, hypertension, hyperlipidemia, alcohol abuse, chronic tobacco use, osteoporosis, and glaucoma. He did not have diabetes, reported no IV drug abuse, and except for the immunosuppressive effects of his medications, was not otherwise immunocompromised.
For 2 years in the outpatient setting, the rheumatology clinic had been managing the patient’s rheumatoid factor (RF) positive and anti-cyclic citrullinated peptide (CCP) antibody positive erosive RA with etanercept 25 mg subcutaneously twice a week. The RA affected his hands, wrists, shoulders, and ankles bilaterally but was successfully controlled. The dermatology clinic was managing the patient’s psoriasis with calcipotriene cream 0.005% twice a week and clobetasol ointment 0.05% twice a week. Psoriatic plaques were noted on bilateral elbows, bilateral dorsal hands, and bilateral dorsal feet.
Initial Evaluation
At evaluation, the patient’s vital signs revealed a temperature of 36.3°C (97.3°F), pulse of 102 beats per minute, respiratory rate of 16 breaths per minute, oxygen saturation of 99% on room air, and blood pressure of 102/70 mm Hg. He was found to have edema, tenderness, and erythema of the wrists bilaterally and left metacarpophalangeal joints (MCPs) and edematous right MCPs and left medial ankle.
The patient had been nonadherent with etanercept for 5 monthsand restarted taking the medication only 2 weeks before presentation. He had noticed worsening arthritis for at least 1 month. His last RA flare was approximately 1 year before presentation. Additional symptoms included 4 days of nausea, nonbloody and nonbilious emesis, left lower quadrant pain, and diarrhea without melena or hematochezia.
Initial laboratory studies found 3.2 k/μL white blood cells (WBCs) with a differential of 11.9% lymphocytes, 4.2% monocytes, 83.3% neutrophils, 0.5% eosinophils, and 0.1% basophils; 165 k/μL platelets; 96 mm/h erythrocyte sedimentation rate (ESR); and 45 mg/dL C-reactive protein. The patient was diagnosed with viral gastroenteritis and RA flare and was admitted for inpatient management secondary to limited ability to care for himself.
Related: Infliximab-Induced Complications
The patient was started on prednisone 40 mg orally once a day (for 5 days) for empiric treatment of an RA flare and continued on etanercept. The inpatient rheumatology service was consulted. Further evaluation later that day found involvement of the proximal interphalangeal joints and elbows and tenderness of the tendons of the dorsal hand bilaterally. Over the next 2 days, the patient remained afebrile and WBCs were within normal limits. Edema, erythema, and tenderness of the involved joints somewhat improved, but tenderness along the tendons of the dorsal hand worsened, which concerned the managing teams for infectious tenosynovitis.
By day 4, the patient was afebrile and had a leukocytosis of 12.9 k/μLwith neutrophils 86.7%, but improvement of erythema, pain, and range of motion of involved joints and no tenderness to palpation of tendons was noted. The inpatient orthopedic surgery service evaluated the patient and did not find sufficient evidence necessitating surgical intervention.
Worsening Condition
On day 6, arthrocentesis of the left wrist was performed secondary to worsening of erythema and edema. The patient experienced new edema of the left shoulder and leukocytosis continued to trend upward (15.7 k/μL on day 6). Purulent aspirate (1.5 mL) was obtained from the fluctuance and tenosynovium of the left wrist. Empiric vancomycin 1 g IV twice daily and ceftriaxone 2 g IV daily were started and continued for 3 days. By this point in his hospital course, the patient had received 1 dose of etanercept. Prednisone and etanercept were previously discontinued because of the discovered infection. Blood cultures were drawn and had no growth (Table). Gastroenterology studies were limited to stool cultures and did not include colonoscopy. Leukocytosis began trending down.
On day 8, antibiotics were tailored to penicillin G 4 million units IV every 4 hours following growth of GGS from the sample of the left wrist. Subsequently, synovial fluid (3 mL) from the left shoulder was obtained following initiation of antibiotic therapy and had no growth. Magnetic resonance imaging (MRI) found tenosynovitis of the left ankle and right wrist.
On day 9, transthoracic echocardiography was performed and found no evidence of infectious endocarditis. Later that night, the patient was taken to surgery for incision and drainage/debridement of bilateral wrists and left ankle, synovectomy of right wrist, and aspiration of right shoulder. Findings included abscess in the left wrist and inflammatory synovitis and bilateral EPL tendon rupture consistent with RA. Pus from the left ankle had few gram-positive cocci in chains with no growth, and the specimens from both wrists grew GGS. Aspirate from the left ankle was an opaque yellow fluid with 14,900/mm3 WBC, 30,000/mm3 red blood cells (RBC), 97% neutrophils, 1% macrophages, 2% lymphocytes, and 0% monocytes. Aspirate from the right shoulder was an opaque bloody fluid with 10,100/mm3 WBC, 40,000/mm3 RBC, 95% neutrophils, 2% macrophages, 1% lymphocytes, and 1% monocytes. On day 10, sulfasalazine 500 mg twice a day was initiated for RA.
Following surgery and continued antibiotics, the patient’s leukocytosis resolved, and improvement was seen in all joints with decreased edema, erythema, and pain and increased range of motion. Postoperative recovery was complicated by ileus, urinary retention, and fungal (Candida albicans) urinary tract infection, all of which resolved without significant complications. The inpatient rheumatology service restarted prednisone at a lower dose of 20 mg. The patient became afebrile and sufficiently stable for transfer to a lower level of care with continued physical therapy and IV antibiotics for another 3 weeks.
Discussion
The patient had 2 underlying systemic inflammatory conditions: RA and psoriasis. The underlying chronic arthritis was likely caused by RA, not psoriatic arthritis (PsA). The patient met the 2010 American College of Rheumatology criteria but failed to meet the classification criteria for PsA.6,7 However, the clinical features of RA and PsA overlap. Rheumatoid factor and CCP can be positive laboratory findings in both RA and PsA.8-14 Tenosynovitis is found in about half of RA patients and PsA patients (P > .05).15 In its evaluation of the patient, the inpatient rheumatology service suspected that the patient may have had RA with components of PsA.
Rheumatoid arthritis complicates the diagnosis of septic arthritis. In a study by Nolla and colleagues, a mean of 7.3 days (range 3 to 18 days) elapsed before a diagnosis of septic arthritis was made in 10 patients with RA on corticosteroids.2 Consideration of risk factors such as increasing age, male sex, tobacco use, extra-articular manifestations of RA, positive RF, rheumatoid nodules, poor functional capacity, high ESR, leukopenia, comorbidities (chronic lung disease, alcoholism, organic brain disease, and diabetes), and the use of corticosteroids may expedite the diagnosis of infections in patients with RA.16 In this case, the patient had some of these risk factors: age, male sex, alcoholism, chronic tobacco use, positive RF, high ESR, and leukopenia (at presentation).
Related: Trend Toward Concomitant Supplements and Medications
The history of medication nonadherence of etanercept with progressively worsening arthritis and early clinical improvement (reduction in erythema, edema, and pain and temporary loss of signs of tenosynovitis on examination) while on prednisone suggested that the patient had a RA flare. The prednisone likely alleviated the inflammatory process but created an immunosuppressed state that allowed GGS to invade and possibly disseminate. Alternately, the patient may have been infected before presentation. The lack of a definitive time line for his case prevented the authors from forming conclusions about a possible causal relationship between the infection and medications. The subjective fevers before admission were nonspecific and could have been caused by RA, presumed gastroenteritis, or other undiagnosed infectious processes. The observed leukocytosis may have been initially corticosteroid-induced.17
Septic Arthritis
The suspicion of septic arthritis and infectious tenosynovitis substantially increased on day 6 with worsening symptoms, involvement of additional joints, and spiking fevers. Group G Streptococcus was obtained from the aspirate of the left wrist and from the surgical specimens from the bilateral wrists. The clinical presentation, MRI imaging studies, and surgical and nonsurgical specimens supported a diagnosis of GGS tenosynovitis. However, there was no clear evidence (ie, positive culture with identified organism) of septic arthritis, likely secondary to early septic arthritis and initiation of antibiotics before joint aspirations. The aspirate from the left ankle was yellow and opaque, but the culture was negative.
The pathogenic organism in the patient was GGS. Group G Streptococcus is normal flora of the oral cavity, gastrointestinal (GI) tract, upper respiratory tract, genital tract, and skin, which were all possible sources of seeding.18 Streptococcal species account for about 20% of septic arthritis, and GGS arthritis accounts for 4% to 19% of streptococcal arthritis.19-22 From a review of the literature, 2 cases of GGS tenosynovitis have been published.23,24 However, in an ultrasound study and MRI study, 49% and 43%, respectively, of patients with RA had tenosynovitis of the tendons of the hands.15,25
GGS Demographics
About three-quarters (71%) of patients with GGS arthritis are male.19 The analysis of the literature by Bronze and colleagues found that chronic joint disease and alcoholism are present in 34% and 14% of patients with GGS arthritis, respectively. One-quarter (23% from Dubost and colleagues) to one-third (32% from Schattner and colleagues) of patients with GGS arthritis have RA.19,26
Fever is present in less than half (43%) of patients with GGS arthritis.19 Positive synovial fluid is expected in 90% of patients.19 Leukocytosis and elevated ESR need not be present.27,28 The arthritis is polyarticular in one-quarter of patients (24% from Bronze and colleagues and 26% from Dubost and colleagues).19,26
Positive blood cultures can be expected in one-fourth (26%) of patients with GGS arthritis.19 The patient’s blood cultures were negative. Blood cultures drawn before initiation of antibiotics yielded no growth, so if the spread was hematogenous, the bacteremia was transient or intermittent. Before and after initiation of antibiotics, specimens from the shoulders did not grow colonies, whereas specimens from the wrists did. If the shoulders were truly infected, these findings and the notably later involvement of the shoulders suggest that the shoulders may have been seeded later in the hospital course.
Trenkner and colleagues proposed that GI abnormalities provide a portal of entry for GGS, which is under the umbrella of S. milleri.29S. milleri is associated with abscess formation, usually of the GI tract.30-32 In the study patient, the possible gastroenteritis may have provided such a portal of entry and subsequent seeding to the joints, and an abscess was found in the left wrist.
Tendon Rupture
Additionally, bilateral EPL tendon rupture likely occurred as a consequence of the inflammatory process from RA and infectious tenosynovitis in the patient. According to Zheng and colleagues, tenosynovitis is an inflammatory process of the synovial tendon sheath that may result in degeneration and rupture of the tendons and may contribute to bone erosions, development of joint deformities, and loss of functional capacity.33 In a histologic study of a ruptured EPL tendon from a patient with RA, Harris observed a chronic inflammatory cellular reaction.34 Harris also described a male with RA with unconfirmed bilateral EPL rupture.34 Björkman and colleague identified previous injury, RA, and local or systemic steroids as important etiologic factors for EPL tendon rupture.35
As in the case of this patient, the utilization of both medical and surgical therapy is not uncommon for treating GGS infection. Antibiotic therapy typically consists of penicillin (74%).26 Surgical intervention is necessary in 16% to 37% of patients.19,26 This patient required both penicillin and incision and drainage/debridement before significant clinical improvement was noted. Prognosis of GGS arthritis is favorable with 5% mortality.26
Conclusion
Septic arthritis and infectious tenosynovitis are readily treatable with low mortality if promptly identified. Identification can be masked by other medical conditions, such as RA and psoriasis, and their associated immunosuppressive treatment. Bilateral EPL tendon rupture may be a complication of RA, particularly with an underlying septic arthritis and infectious tenosynovitis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Dubost JJ, Fis I, Denis P, et al. Polyarticular septic arthritis. Medicine (Baltimore). 1993;72(5):296-310.
2. Nolla JM, Gómez-Vaquero C, Fiter J, et al. Pyarthrosis in patients with rheumatoid arthritis: A detailed analysis of 10 cases and literature review. Semin Arthritis Rheum. 2000;30(2):121-126.
3.Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK Health District 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
4. Galloway JB, Hyrich KL, Mercer LK, et al; BSR Biologics Register. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70(10):1810-1814.
5. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Frequency of infection in patients with rheumatoid arthritis compared with controls: A population-based study. Arthritis Rheum. 2002;46(9):2287-2293.
6. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569-2581.
7. Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H; CASPAR Study Group. Classification criteria for psoriatic arthritis: Development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665-2673.
8. Gladman DD, Shuckett R, Russell ML, Thorne JC, Schachter RK. Psoriatic arthritis (PSA)—An analysis of 220 patients. Q J Med. 1987;62(238):127-141.
9. Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol. 2005;32(3):511-515.
10. Vander Cruyssen B, Hoffman IE, Zmierczak H, et al. Anti-citrullinated peptide antibodies may occur in patients with psoriatic arthritis. Ann Rheum Dis. 2005;64(8):1145-1149.
11. Alenius GM, Berglin E, Rantapää Dahlgvist S. Antibodies against cyclic citrullinated peptide (CCP) in psoriatic patients with or without joint inflammation. Ann Rheum Dis. 2006;65(3):398-400.
12. Candia L, Marquez J, Gonzalez C, et al. Low frequency of anticyclic citrullinated peptide antibodies in psoriatic arthritis but not in cutaneous psoriasis. J Clin Rheumatol. 2006;12(5):226-229.
13. Inanc N, Dalkilic E, Kamali S, et al. Anti-CCP antibodies in rheumatoid arthritis and psoriatic arthritis. Clin Rheumatol. 2007;26(1):17-23.
14. Popescu C, Zofota S, Bojinca V, Ionescu R. Anti-cyclic citrullinated peptide antibodies in psoriatic arthritis—Cross-sectional study and literature review. J Med Life. 2013;6(4):376-382.
15. Schoellnast H, Deutschmann HA, Hermann J, et al. Psoriatic arthritis and rheumatoid arthritis: Findings in contrast-enhanced MRI. AJR Am J Roentgenol. 2006;187(2):351-357.
16. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Predictors of infection in rheumatoid arthritis. Arthritis Rheum. 2002;46(9):2294-2300.
17. Shoenfeld Y, Gurewich Y, Gallant LA, Pinkhas J. Prednisone-induced leukocytosis. Influence of dosage, method and duration of administration on the degree of leukocytosis. Am J Med. 1981;71(5):773-778.
18. Gossling J. Occurrence and pathogenicity of the Streptococcus milleri group. Rev Infect Dis. 1988;10(2):257-285.
19. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Sauvezie B. Streptococcal septic arthritis in adults. A study of 55 cases with a literature review. Joint Bone Spine. 2004;71(4):303-311.
20. Ryan MJ, Kavanagh R, Wall PG, Hazleman BL. Bacterial joint infections in England and Wales: Analysis of bacterial isolates over a four year period. Br J Rheumatol. 1997;36(3):370-373.
21. Morgan DS, Fisher D, Merianos A, Currie BJ. An 18 year clinical review of septic arthritis from tropical Australia. Epidemiol Infect. 1996;117(3):423-428.
22. Kaandorp CJ, Dinant HJ, van de Laar MA, Moens HJ, Prins AP, Dijkmans BA. Incidence and sources of native and prosthetic joint infection: A community based prospective survey. Ann Rheum Dis. 1997;56(8):470-475.
23. Bradlow A, Mitchell RG, Mowat AG. Group G streptococcal arthritis. Rheumatol Rehabil. 1982;21(4):206-210.
24. Meier JL, Gerster JC. Bursitis and tenosynovitis caused by group G streptococci. J Rheumatol. 1983;10(5):817-818.
25. Filippucci E, Gabba A, Di Geso L, Girolimetti R, Salaffi F, Grassi W. Hand tendon involvement in rheumatoid arthritis: An ultrasound study. Semin Arthritis Rheum. 2012;41(6):752-760.
26. Bronze MS, Whitby S, Schaberg DR. Group G streptococcal arthritis: Case report and review of the literature. Am J Med Sci. 1997;313(4):239-243.
27. Schattner A, Vosti KL. Bacterial arthritis due to beta-hemolytic streptococci of serogroups A, B, C, F, and G. Analysis of 23 cases and a review of the literature. Medicine (Baltimore). 1998;77(2):122-139.
28. Gaunt PN, Seal DV. Group G streptococcal infection of joints and joint prostheses. J Infect. 1986;13(2):115-123.
29. Trenkner SW, Braunstein EM, Lynn MD, Ike RW. Group G streptococcal arthritis and bowel disease: A rare enteropathic arthropathy. Gastrointest Radiol. 1987;12(3):265-267.
30. Bert F, Bariou-Lancelin M, Lambert-Zechovsky N. Clinical significance of bacteremia involving the “Streptococcus milleri” group: 51 cases and review. Clin Infect Dis. 1998;27(2):385-387.
31. Casariego E, Rodriguez A, Corredoira JC, et al. Prospective study of Streptococcus milleri bacteremia. Eur J Clin Microbiol Infect Dis. 1996;15(3):194-200.
32. Jacobs JA, Pietersen HG, Stobberingh EE, Soeters PB. Bacteremia involving the “Streptococcus milleri” group: Analysis of 19 cases. Clin Infect Dis. 1994;19(4):704-713.
33. Zheng S, Robinson E, Yeoman S, et al. MRI bone oedema predicts eight year tendon function at the wrist but not the requirement for orthopaedic surgery in rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):607-611.
34. Harris R. Spontaneous rupture of the tendon of extensor pollicis longus as a complication of rheumatoid arthritis. Ann Rheum Dis. 1951;10(3):298-306.
35. Björkman A, Jörgsholm P. Rupture of the extensor pollicis longus tendon: A study of aetiological factors. Scand J Plast Reconstr Surg Hand Surg. 2004;38(1):32-35.
Compared with monoarticular arthritis, polyarticular arthritis may yield an initially narrower differential diagnosis that focuses on systemic inflammatory conditions, such as rheumatoid arthritis (RA). Approximately 15% to 30% of septic arthritis is polyarticular, of which about 45% is associated with underlying RA.1,2 Regardless of the number of joints involved, septic (infectious) arthritis is a valid consideration given the morbidity and mortality.
In a retrospective study in the United Kingdom (UK) between 1982 and 1991, the morbidity and mortality of septic arthritis was 31.6% and 11.5%, respectively, and 16% of the study population had RA.3 A review of the literature by Dubost and colleagues found that polyarticular septic arthritis (PASA) has a mortality of 31% to 42% compared with 4% to 8% for monoarticular septic arthritis, and RA was present in 67% of the PASA fatalities.1
Related: The Golden Era of Treatment in Rheumatology
Rheumatoid arthritis and its treatment predispose patients to septic arthritis. Septic arthritis in the UK general population is 0.42 per 100 patient-years for patients with RA on antitumor necrosis factor therapy.3,4 In a retrospective study in the U.S., the incidence of septic arthritis was 0.40 per 100 patient-years for patients with RA compared with 0.02 per 100 patient-years for patients without RA.5
Other complications of RA include infectious tenosynovitis and tendon rupture. The incidence and prevalence of infectious tenosynovitis and tendon rupture in RA are not firmly established in the literature.
We present a patient with RA and psoriasis who responded initially to acute management for RA but subsequently was diagnosed with culture-negative polyarticular arthritis and infectious tenosynovitis associated with beta hemolytic group G Streptococcus (GGS), a part of Streptococcus milleri (S. milleri). During surgery, he was also found to have bilateral extensor pollicus longus (EPL) tendon rupture. Given the possible morbidity, the authors believe this patient may be of interest to the medical community.
Case Presentation
A 69-year-old African American male presented with 3 to 4 days of swelling and pain of bilateral wrists, bilateral hands, and the left ankle with subjective, but resolved, fevers and chills. His medical history was significant for seropositive erosive RA, psoriasis, hypertension, hyperlipidemia, alcohol abuse, chronic tobacco use, osteoporosis, and glaucoma. He did not have diabetes, reported no IV drug abuse, and except for the immunosuppressive effects of his medications, was not otherwise immunocompromised.
For 2 years in the outpatient setting, the rheumatology clinic had been managing the patient’s rheumatoid factor (RF) positive and anti-cyclic citrullinated peptide (CCP) antibody positive erosive RA with etanercept 25 mg subcutaneously twice a week. The RA affected his hands, wrists, shoulders, and ankles bilaterally but was successfully controlled. The dermatology clinic was managing the patient’s psoriasis with calcipotriene cream 0.005% twice a week and clobetasol ointment 0.05% twice a week. Psoriatic plaques were noted on bilateral elbows, bilateral dorsal hands, and bilateral dorsal feet.
Initial Evaluation
At evaluation, the patient’s vital signs revealed a temperature of 36.3°C (97.3°F), pulse of 102 beats per minute, respiratory rate of 16 breaths per minute, oxygen saturation of 99% on room air, and blood pressure of 102/70 mm Hg. He was found to have edema, tenderness, and erythema of the wrists bilaterally and left metacarpophalangeal joints (MCPs) and edematous right MCPs and left medial ankle.
The patient had been nonadherent with etanercept for 5 monthsand restarted taking the medication only 2 weeks before presentation. He had noticed worsening arthritis for at least 1 month. His last RA flare was approximately 1 year before presentation. Additional symptoms included 4 days of nausea, nonbloody and nonbilious emesis, left lower quadrant pain, and diarrhea without melena or hematochezia.
Initial laboratory studies found 3.2 k/μL white blood cells (WBCs) with a differential of 11.9% lymphocytes, 4.2% monocytes, 83.3% neutrophils, 0.5% eosinophils, and 0.1% basophils; 165 k/μL platelets; 96 mm/h erythrocyte sedimentation rate (ESR); and 45 mg/dL C-reactive protein. The patient was diagnosed with viral gastroenteritis and RA flare and was admitted for inpatient management secondary to limited ability to care for himself.
Related: Infliximab-Induced Complications
The patient was started on prednisone 40 mg orally once a day (for 5 days) for empiric treatment of an RA flare and continued on etanercept. The inpatient rheumatology service was consulted. Further evaluation later that day found involvement of the proximal interphalangeal joints and elbows and tenderness of the tendons of the dorsal hand bilaterally. Over the next 2 days, the patient remained afebrile and WBCs were within normal limits. Edema, erythema, and tenderness of the involved joints somewhat improved, but tenderness along the tendons of the dorsal hand worsened, which concerned the managing teams for infectious tenosynovitis.
By day 4, the patient was afebrile and had a leukocytosis of 12.9 k/μLwith neutrophils 86.7%, but improvement of erythema, pain, and range of motion of involved joints and no tenderness to palpation of tendons was noted. The inpatient orthopedic surgery service evaluated the patient and did not find sufficient evidence necessitating surgical intervention.
Worsening Condition
On day 6, arthrocentesis of the left wrist was performed secondary to worsening of erythema and edema. The patient experienced new edema of the left shoulder and leukocytosis continued to trend upward (15.7 k/μL on day 6). Purulent aspirate (1.5 mL) was obtained from the fluctuance and tenosynovium of the left wrist. Empiric vancomycin 1 g IV twice daily and ceftriaxone 2 g IV daily were started and continued for 3 days. By this point in his hospital course, the patient had received 1 dose of etanercept. Prednisone and etanercept were previously discontinued because of the discovered infection. Blood cultures were drawn and had no growth (Table). Gastroenterology studies were limited to stool cultures and did not include colonoscopy. Leukocytosis began trending down.
On day 8, antibiotics were tailored to penicillin G 4 million units IV every 4 hours following growth of GGS from the sample of the left wrist. Subsequently, synovial fluid (3 mL) from the left shoulder was obtained following initiation of antibiotic therapy and had no growth. Magnetic resonance imaging (MRI) found tenosynovitis of the left ankle and right wrist.
On day 9, transthoracic echocardiography was performed and found no evidence of infectious endocarditis. Later that night, the patient was taken to surgery for incision and drainage/debridement of bilateral wrists and left ankle, synovectomy of right wrist, and aspiration of right shoulder. Findings included abscess in the left wrist and inflammatory synovitis and bilateral EPL tendon rupture consistent with RA. Pus from the left ankle had few gram-positive cocci in chains with no growth, and the specimens from both wrists grew GGS. Aspirate from the left ankle was an opaque yellow fluid with 14,900/mm3 WBC, 30,000/mm3 red blood cells (RBC), 97% neutrophils, 1% macrophages, 2% lymphocytes, and 0% monocytes. Aspirate from the right shoulder was an opaque bloody fluid with 10,100/mm3 WBC, 40,000/mm3 RBC, 95% neutrophils, 2% macrophages, 1% lymphocytes, and 1% monocytes. On day 10, sulfasalazine 500 mg twice a day was initiated for RA.
Following surgery and continued antibiotics, the patient’s leukocytosis resolved, and improvement was seen in all joints with decreased edema, erythema, and pain and increased range of motion. Postoperative recovery was complicated by ileus, urinary retention, and fungal (Candida albicans) urinary tract infection, all of which resolved without significant complications. The inpatient rheumatology service restarted prednisone at a lower dose of 20 mg. The patient became afebrile and sufficiently stable for transfer to a lower level of care with continued physical therapy and IV antibiotics for another 3 weeks.
Discussion
The patient had 2 underlying systemic inflammatory conditions: RA and psoriasis. The underlying chronic arthritis was likely caused by RA, not psoriatic arthritis (PsA). The patient met the 2010 American College of Rheumatology criteria but failed to meet the classification criteria for PsA.6,7 However, the clinical features of RA and PsA overlap. Rheumatoid factor and CCP can be positive laboratory findings in both RA and PsA.8-14 Tenosynovitis is found in about half of RA patients and PsA patients (P > .05).15 In its evaluation of the patient, the inpatient rheumatology service suspected that the patient may have had RA with components of PsA.
Rheumatoid arthritis complicates the diagnosis of septic arthritis. In a study by Nolla and colleagues, a mean of 7.3 days (range 3 to 18 days) elapsed before a diagnosis of septic arthritis was made in 10 patients with RA on corticosteroids.2 Consideration of risk factors such as increasing age, male sex, tobacco use, extra-articular manifestations of RA, positive RF, rheumatoid nodules, poor functional capacity, high ESR, leukopenia, comorbidities (chronic lung disease, alcoholism, organic brain disease, and diabetes), and the use of corticosteroids may expedite the diagnosis of infections in patients with RA.16 In this case, the patient had some of these risk factors: age, male sex, alcoholism, chronic tobacco use, positive RF, high ESR, and leukopenia (at presentation).
Related: Trend Toward Concomitant Supplements and Medications
The history of medication nonadherence of etanercept with progressively worsening arthritis and early clinical improvement (reduction in erythema, edema, and pain and temporary loss of signs of tenosynovitis on examination) while on prednisone suggested that the patient had a RA flare. The prednisone likely alleviated the inflammatory process but created an immunosuppressed state that allowed GGS to invade and possibly disseminate. Alternately, the patient may have been infected before presentation. The lack of a definitive time line for his case prevented the authors from forming conclusions about a possible causal relationship between the infection and medications. The subjective fevers before admission were nonspecific and could have been caused by RA, presumed gastroenteritis, or other undiagnosed infectious processes. The observed leukocytosis may have been initially corticosteroid-induced.17
Septic Arthritis
The suspicion of septic arthritis and infectious tenosynovitis substantially increased on day 6 with worsening symptoms, involvement of additional joints, and spiking fevers. Group G Streptococcus was obtained from the aspirate of the left wrist and from the surgical specimens from the bilateral wrists. The clinical presentation, MRI imaging studies, and surgical and nonsurgical specimens supported a diagnosis of GGS tenosynovitis. However, there was no clear evidence (ie, positive culture with identified organism) of septic arthritis, likely secondary to early septic arthritis and initiation of antibiotics before joint aspirations. The aspirate from the left ankle was yellow and opaque, but the culture was negative.
The pathogenic organism in the patient was GGS. Group G Streptococcus is normal flora of the oral cavity, gastrointestinal (GI) tract, upper respiratory tract, genital tract, and skin, which were all possible sources of seeding.18 Streptococcal species account for about 20% of septic arthritis, and GGS arthritis accounts for 4% to 19% of streptococcal arthritis.19-22 From a review of the literature, 2 cases of GGS tenosynovitis have been published.23,24 However, in an ultrasound study and MRI study, 49% and 43%, respectively, of patients with RA had tenosynovitis of the tendons of the hands.15,25
GGS Demographics
About three-quarters (71%) of patients with GGS arthritis are male.19 The analysis of the literature by Bronze and colleagues found that chronic joint disease and alcoholism are present in 34% and 14% of patients with GGS arthritis, respectively. One-quarter (23% from Dubost and colleagues) to one-third (32% from Schattner and colleagues) of patients with GGS arthritis have RA.19,26
Fever is present in less than half (43%) of patients with GGS arthritis.19 Positive synovial fluid is expected in 90% of patients.19 Leukocytosis and elevated ESR need not be present.27,28 The arthritis is polyarticular in one-quarter of patients (24% from Bronze and colleagues and 26% from Dubost and colleagues).19,26
Positive blood cultures can be expected in one-fourth (26%) of patients with GGS arthritis.19 The patient’s blood cultures were negative. Blood cultures drawn before initiation of antibiotics yielded no growth, so if the spread was hematogenous, the bacteremia was transient or intermittent. Before and after initiation of antibiotics, specimens from the shoulders did not grow colonies, whereas specimens from the wrists did. If the shoulders were truly infected, these findings and the notably later involvement of the shoulders suggest that the shoulders may have been seeded later in the hospital course.
Trenkner and colleagues proposed that GI abnormalities provide a portal of entry for GGS, which is under the umbrella of S. milleri.29S. milleri is associated with abscess formation, usually of the GI tract.30-32 In the study patient, the possible gastroenteritis may have provided such a portal of entry and subsequent seeding to the joints, and an abscess was found in the left wrist.
Tendon Rupture
Additionally, bilateral EPL tendon rupture likely occurred as a consequence of the inflammatory process from RA and infectious tenosynovitis in the patient. According to Zheng and colleagues, tenosynovitis is an inflammatory process of the synovial tendon sheath that may result in degeneration and rupture of the tendons and may contribute to bone erosions, development of joint deformities, and loss of functional capacity.33 In a histologic study of a ruptured EPL tendon from a patient with RA, Harris observed a chronic inflammatory cellular reaction.34 Harris also described a male with RA with unconfirmed bilateral EPL rupture.34 Björkman and colleague identified previous injury, RA, and local or systemic steroids as important etiologic factors for EPL tendon rupture.35
As in the case of this patient, the utilization of both medical and surgical therapy is not uncommon for treating GGS infection. Antibiotic therapy typically consists of penicillin (74%).26 Surgical intervention is necessary in 16% to 37% of patients.19,26 This patient required both penicillin and incision and drainage/debridement before significant clinical improvement was noted. Prognosis of GGS arthritis is favorable with 5% mortality.26
Conclusion
Septic arthritis and infectious tenosynovitis are readily treatable with low mortality if promptly identified. Identification can be masked by other medical conditions, such as RA and psoriasis, and their associated immunosuppressive treatment. Bilateral EPL tendon rupture may be a complication of RA, particularly with an underlying septic arthritis and infectious tenosynovitis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Compared with monoarticular arthritis, polyarticular arthritis may yield an initially narrower differential diagnosis that focuses on systemic inflammatory conditions, such as rheumatoid arthritis (RA). Approximately 15% to 30% of septic arthritis is polyarticular, of which about 45% is associated with underlying RA.1,2 Regardless of the number of joints involved, septic (infectious) arthritis is a valid consideration given the morbidity and mortality.
In a retrospective study in the United Kingdom (UK) between 1982 and 1991, the morbidity and mortality of septic arthritis was 31.6% and 11.5%, respectively, and 16% of the study population had RA.3 A review of the literature by Dubost and colleagues found that polyarticular septic arthritis (PASA) has a mortality of 31% to 42% compared with 4% to 8% for monoarticular septic arthritis, and RA was present in 67% of the PASA fatalities.1
Related: The Golden Era of Treatment in Rheumatology
Rheumatoid arthritis and its treatment predispose patients to septic arthritis. Septic arthritis in the UK general population is 0.42 per 100 patient-years for patients with RA on antitumor necrosis factor therapy.3,4 In a retrospective study in the U.S., the incidence of septic arthritis was 0.40 per 100 patient-years for patients with RA compared with 0.02 per 100 patient-years for patients without RA.5
Other complications of RA include infectious tenosynovitis and tendon rupture. The incidence and prevalence of infectious tenosynovitis and tendon rupture in RA are not firmly established in the literature.
We present a patient with RA and psoriasis who responded initially to acute management for RA but subsequently was diagnosed with culture-negative polyarticular arthritis and infectious tenosynovitis associated with beta hemolytic group G Streptococcus (GGS), a part of Streptococcus milleri (S. milleri). During surgery, he was also found to have bilateral extensor pollicus longus (EPL) tendon rupture. Given the possible morbidity, the authors believe this patient may be of interest to the medical community.
Case Presentation
A 69-year-old African American male presented with 3 to 4 days of swelling and pain of bilateral wrists, bilateral hands, and the left ankle with subjective, but resolved, fevers and chills. His medical history was significant for seropositive erosive RA, psoriasis, hypertension, hyperlipidemia, alcohol abuse, chronic tobacco use, osteoporosis, and glaucoma. He did not have diabetes, reported no IV drug abuse, and except for the immunosuppressive effects of his medications, was not otherwise immunocompromised.
For 2 years in the outpatient setting, the rheumatology clinic had been managing the patient’s rheumatoid factor (RF) positive and anti-cyclic citrullinated peptide (CCP) antibody positive erosive RA with etanercept 25 mg subcutaneously twice a week. The RA affected his hands, wrists, shoulders, and ankles bilaterally but was successfully controlled. The dermatology clinic was managing the patient’s psoriasis with calcipotriene cream 0.005% twice a week and clobetasol ointment 0.05% twice a week. Psoriatic plaques were noted on bilateral elbows, bilateral dorsal hands, and bilateral dorsal feet.
Initial Evaluation
At evaluation, the patient’s vital signs revealed a temperature of 36.3°C (97.3°F), pulse of 102 beats per minute, respiratory rate of 16 breaths per minute, oxygen saturation of 99% on room air, and blood pressure of 102/70 mm Hg. He was found to have edema, tenderness, and erythema of the wrists bilaterally and left metacarpophalangeal joints (MCPs) and edematous right MCPs and left medial ankle.
The patient had been nonadherent with etanercept for 5 monthsand restarted taking the medication only 2 weeks before presentation. He had noticed worsening arthritis for at least 1 month. His last RA flare was approximately 1 year before presentation. Additional symptoms included 4 days of nausea, nonbloody and nonbilious emesis, left lower quadrant pain, and diarrhea without melena or hematochezia.
Initial laboratory studies found 3.2 k/μL white blood cells (WBCs) with a differential of 11.9% lymphocytes, 4.2% monocytes, 83.3% neutrophils, 0.5% eosinophils, and 0.1% basophils; 165 k/μL platelets; 96 mm/h erythrocyte sedimentation rate (ESR); and 45 mg/dL C-reactive protein. The patient was diagnosed with viral gastroenteritis and RA flare and was admitted for inpatient management secondary to limited ability to care for himself.
Related: Infliximab-Induced Complications
The patient was started on prednisone 40 mg orally once a day (for 5 days) for empiric treatment of an RA flare and continued on etanercept. The inpatient rheumatology service was consulted. Further evaluation later that day found involvement of the proximal interphalangeal joints and elbows and tenderness of the tendons of the dorsal hand bilaterally. Over the next 2 days, the patient remained afebrile and WBCs were within normal limits. Edema, erythema, and tenderness of the involved joints somewhat improved, but tenderness along the tendons of the dorsal hand worsened, which concerned the managing teams for infectious tenosynovitis.
By day 4, the patient was afebrile and had a leukocytosis of 12.9 k/μLwith neutrophils 86.7%, but improvement of erythema, pain, and range of motion of involved joints and no tenderness to palpation of tendons was noted. The inpatient orthopedic surgery service evaluated the patient and did not find sufficient evidence necessitating surgical intervention.
Worsening Condition
On day 6, arthrocentesis of the left wrist was performed secondary to worsening of erythema and edema. The patient experienced new edema of the left shoulder and leukocytosis continued to trend upward (15.7 k/μL on day 6). Purulent aspirate (1.5 mL) was obtained from the fluctuance and tenosynovium of the left wrist. Empiric vancomycin 1 g IV twice daily and ceftriaxone 2 g IV daily were started and continued for 3 days. By this point in his hospital course, the patient had received 1 dose of etanercept. Prednisone and etanercept were previously discontinued because of the discovered infection. Blood cultures were drawn and had no growth (Table). Gastroenterology studies were limited to stool cultures and did not include colonoscopy. Leukocytosis began trending down.
On day 8, antibiotics were tailored to penicillin G 4 million units IV every 4 hours following growth of GGS from the sample of the left wrist. Subsequently, synovial fluid (3 mL) from the left shoulder was obtained following initiation of antibiotic therapy and had no growth. Magnetic resonance imaging (MRI) found tenosynovitis of the left ankle and right wrist.
On day 9, transthoracic echocardiography was performed and found no evidence of infectious endocarditis. Later that night, the patient was taken to surgery for incision and drainage/debridement of bilateral wrists and left ankle, synovectomy of right wrist, and aspiration of right shoulder. Findings included abscess in the left wrist and inflammatory synovitis and bilateral EPL tendon rupture consistent with RA. Pus from the left ankle had few gram-positive cocci in chains with no growth, and the specimens from both wrists grew GGS. Aspirate from the left ankle was an opaque yellow fluid with 14,900/mm3 WBC, 30,000/mm3 red blood cells (RBC), 97% neutrophils, 1% macrophages, 2% lymphocytes, and 0% monocytes. Aspirate from the right shoulder was an opaque bloody fluid with 10,100/mm3 WBC, 40,000/mm3 RBC, 95% neutrophils, 2% macrophages, 1% lymphocytes, and 1% monocytes. On day 10, sulfasalazine 500 mg twice a day was initiated for RA.
Following surgery and continued antibiotics, the patient’s leukocytosis resolved, and improvement was seen in all joints with decreased edema, erythema, and pain and increased range of motion. Postoperative recovery was complicated by ileus, urinary retention, and fungal (Candida albicans) urinary tract infection, all of which resolved without significant complications. The inpatient rheumatology service restarted prednisone at a lower dose of 20 mg. The patient became afebrile and sufficiently stable for transfer to a lower level of care with continued physical therapy and IV antibiotics for another 3 weeks.
Discussion
The patient had 2 underlying systemic inflammatory conditions: RA and psoriasis. The underlying chronic arthritis was likely caused by RA, not psoriatic arthritis (PsA). The patient met the 2010 American College of Rheumatology criteria but failed to meet the classification criteria for PsA.6,7 However, the clinical features of RA and PsA overlap. Rheumatoid factor and CCP can be positive laboratory findings in both RA and PsA.8-14 Tenosynovitis is found in about half of RA patients and PsA patients (P > .05).15 In its evaluation of the patient, the inpatient rheumatology service suspected that the patient may have had RA with components of PsA.
Rheumatoid arthritis complicates the diagnosis of septic arthritis. In a study by Nolla and colleagues, a mean of 7.3 days (range 3 to 18 days) elapsed before a diagnosis of septic arthritis was made in 10 patients with RA on corticosteroids.2 Consideration of risk factors such as increasing age, male sex, tobacco use, extra-articular manifestations of RA, positive RF, rheumatoid nodules, poor functional capacity, high ESR, leukopenia, comorbidities (chronic lung disease, alcoholism, organic brain disease, and diabetes), and the use of corticosteroids may expedite the diagnosis of infections in patients with RA.16 In this case, the patient had some of these risk factors: age, male sex, alcoholism, chronic tobacco use, positive RF, high ESR, and leukopenia (at presentation).
Related: Trend Toward Concomitant Supplements and Medications
The history of medication nonadherence of etanercept with progressively worsening arthritis and early clinical improvement (reduction in erythema, edema, and pain and temporary loss of signs of tenosynovitis on examination) while on prednisone suggested that the patient had a RA flare. The prednisone likely alleviated the inflammatory process but created an immunosuppressed state that allowed GGS to invade and possibly disseminate. Alternately, the patient may have been infected before presentation. The lack of a definitive time line for his case prevented the authors from forming conclusions about a possible causal relationship between the infection and medications. The subjective fevers before admission were nonspecific and could have been caused by RA, presumed gastroenteritis, or other undiagnosed infectious processes. The observed leukocytosis may have been initially corticosteroid-induced.17
Septic Arthritis
The suspicion of septic arthritis and infectious tenosynovitis substantially increased on day 6 with worsening symptoms, involvement of additional joints, and spiking fevers. Group G Streptococcus was obtained from the aspirate of the left wrist and from the surgical specimens from the bilateral wrists. The clinical presentation, MRI imaging studies, and surgical and nonsurgical specimens supported a diagnosis of GGS tenosynovitis. However, there was no clear evidence (ie, positive culture with identified organism) of septic arthritis, likely secondary to early septic arthritis and initiation of antibiotics before joint aspirations. The aspirate from the left ankle was yellow and opaque, but the culture was negative.
The pathogenic organism in the patient was GGS. Group G Streptococcus is normal flora of the oral cavity, gastrointestinal (GI) tract, upper respiratory tract, genital tract, and skin, which were all possible sources of seeding.18 Streptococcal species account for about 20% of septic arthritis, and GGS arthritis accounts for 4% to 19% of streptococcal arthritis.19-22 From a review of the literature, 2 cases of GGS tenosynovitis have been published.23,24 However, in an ultrasound study and MRI study, 49% and 43%, respectively, of patients with RA had tenosynovitis of the tendons of the hands.15,25
GGS Demographics
About three-quarters (71%) of patients with GGS arthritis are male.19 The analysis of the literature by Bronze and colleagues found that chronic joint disease and alcoholism are present in 34% and 14% of patients with GGS arthritis, respectively. One-quarter (23% from Dubost and colleagues) to one-third (32% from Schattner and colleagues) of patients with GGS arthritis have RA.19,26
Fever is present in less than half (43%) of patients with GGS arthritis.19 Positive synovial fluid is expected in 90% of patients.19 Leukocytosis and elevated ESR need not be present.27,28 The arthritis is polyarticular in one-quarter of patients (24% from Bronze and colleagues and 26% from Dubost and colleagues).19,26
Positive blood cultures can be expected in one-fourth (26%) of patients with GGS arthritis.19 The patient’s blood cultures were negative. Blood cultures drawn before initiation of antibiotics yielded no growth, so if the spread was hematogenous, the bacteremia was transient or intermittent. Before and after initiation of antibiotics, specimens from the shoulders did not grow colonies, whereas specimens from the wrists did. If the shoulders were truly infected, these findings and the notably later involvement of the shoulders suggest that the shoulders may have been seeded later in the hospital course.
Trenkner and colleagues proposed that GI abnormalities provide a portal of entry for GGS, which is under the umbrella of S. milleri.29S. milleri is associated with abscess formation, usually of the GI tract.30-32 In the study patient, the possible gastroenteritis may have provided such a portal of entry and subsequent seeding to the joints, and an abscess was found in the left wrist.
Tendon Rupture
Additionally, bilateral EPL tendon rupture likely occurred as a consequence of the inflammatory process from RA and infectious tenosynovitis in the patient. According to Zheng and colleagues, tenosynovitis is an inflammatory process of the synovial tendon sheath that may result in degeneration and rupture of the tendons and may contribute to bone erosions, development of joint deformities, and loss of functional capacity.33 In a histologic study of a ruptured EPL tendon from a patient with RA, Harris observed a chronic inflammatory cellular reaction.34 Harris also described a male with RA with unconfirmed bilateral EPL rupture.34 Björkman and colleague identified previous injury, RA, and local or systemic steroids as important etiologic factors for EPL tendon rupture.35
As in the case of this patient, the utilization of both medical and surgical therapy is not uncommon for treating GGS infection. Antibiotic therapy typically consists of penicillin (74%).26 Surgical intervention is necessary in 16% to 37% of patients.19,26 This patient required both penicillin and incision and drainage/debridement before significant clinical improvement was noted. Prognosis of GGS arthritis is favorable with 5% mortality.26
Conclusion
Septic arthritis and infectious tenosynovitis are readily treatable with low mortality if promptly identified. Identification can be masked by other medical conditions, such as RA and psoriasis, and their associated immunosuppressive treatment. Bilateral EPL tendon rupture may be a complication of RA, particularly with an underlying septic arthritis and infectious tenosynovitis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Dubost JJ, Fis I, Denis P, et al. Polyarticular septic arthritis. Medicine (Baltimore). 1993;72(5):296-310.
2. Nolla JM, Gómez-Vaquero C, Fiter J, et al. Pyarthrosis in patients with rheumatoid arthritis: A detailed analysis of 10 cases and literature review. Semin Arthritis Rheum. 2000;30(2):121-126.
3.Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK Health District 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
4. Galloway JB, Hyrich KL, Mercer LK, et al; BSR Biologics Register. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70(10):1810-1814.
5. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Frequency of infection in patients with rheumatoid arthritis compared with controls: A population-based study. Arthritis Rheum. 2002;46(9):2287-2293.
6. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569-2581.
7. Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H; CASPAR Study Group. Classification criteria for psoriatic arthritis: Development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665-2673.
8. Gladman DD, Shuckett R, Russell ML, Thorne JC, Schachter RK. Psoriatic arthritis (PSA)—An analysis of 220 patients. Q J Med. 1987;62(238):127-141.
9. Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol. 2005;32(3):511-515.
10. Vander Cruyssen B, Hoffman IE, Zmierczak H, et al. Anti-citrullinated peptide antibodies may occur in patients with psoriatic arthritis. Ann Rheum Dis. 2005;64(8):1145-1149.
11. Alenius GM, Berglin E, Rantapää Dahlgvist S. Antibodies against cyclic citrullinated peptide (CCP) in psoriatic patients with or without joint inflammation. Ann Rheum Dis. 2006;65(3):398-400.
12. Candia L, Marquez J, Gonzalez C, et al. Low frequency of anticyclic citrullinated peptide antibodies in psoriatic arthritis but not in cutaneous psoriasis. J Clin Rheumatol. 2006;12(5):226-229.
13. Inanc N, Dalkilic E, Kamali S, et al. Anti-CCP antibodies in rheumatoid arthritis and psoriatic arthritis. Clin Rheumatol. 2007;26(1):17-23.
14. Popescu C, Zofota S, Bojinca V, Ionescu R. Anti-cyclic citrullinated peptide antibodies in psoriatic arthritis—Cross-sectional study and literature review. J Med Life. 2013;6(4):376-382.
15. Schoellnast H, Deutschmann HA, Hermann J, et al. Psoriatic arthritis and rheumatoid arthritis: Findings in contrast-enhanced MRI. AJR Am J Roentgenol. 2006;187(2):351-357.
16. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Predictors of infection in rheumatoid arthritis. Arthritis Rheum. 2002;46(9):2294-2300.
17. Shoenfeld Y, Gurewich Y, Gallant LA, Pinkhas J. Prednisone-induced leukocytosis. Influence of dosage, method and duration of administration on the degree of leukocytosis. Am J Med. 1981;71(5):773-778.
18. Gossling J. Occurrence and pathogenicity of the Streptococcus milleri group. Rev Infect Dis. 1988;10(2):257-285.
19. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Sauvezie B. Streptococcal septic arthritis in adults. A study of 55 cases with a literature review. Joint Bone Spine. 2004;71(4):303-311.
20. Ryan MJ, Kavanagh R, Wall PG, Hazleman BL. Bacterial joint infections in England and Wales: Analysis of bacterial isolates over a four year period. Br J Rheumatol. 1997;36(3):370-373.
21. Morgan DS, Fisher D, Merianos A, Currie BJ. An 18 year clinical review of septic arthritis from tropical Australia. Epidemiol Infect. 1996;117(3):423-428.
22. Kaandorp CJ, Dinant HJ, van de Laar MA, Moens HJ, Prins AP, Dijkmans BA. Incidence and sources of native and prosthetic joint infection: A community based prospective survey. Ann Rheum Dis. 1997;56(8):470-475.
23. Bradlow A, Mitchell RG, Mowat AG. Group G streptococcal arthritis. Rheumatol Rehabil. 1982;21(4):206-210.
24. Meier JL, Gerster JC. Bursitis and tenosynovitis caused by group G streptococci. J Rheumatol. 1983;10(5):817-818.
25. Filippucci E, Gabba A, Di Geso L, Girolimetti R, Salaffi F, Grassi W. Hand tendon involvement in rheumatoid arthritis: An ultrasound study. Semin Arthritis Rheum. 2012;41(6):752-760.
26. Bronze MS, Whitby S, Schaberg DR. Group G streptococcal arthritis: Case report and review of the literature. Am J Med Sci. 1997;313(4):239-243.
27. Schattner A, Vosti KL. Bacterial arthritis due to beta-hemolytic streptococci of serogroups A, B, C, F, and G. Analysis of 23 cases and a review of the literature. Medicine (Baltimore). 1998;77(2):122-139.
28. Gaunt PN, Seal DV. Group G streptococcal infection of joints and joint prostheses. J Infect. 1986;13(2):115-123.
29. Trenkner SW, Braunstein EM, Lynn MD, Ike RW. Group G streptococcal arthritis and bowel disease: A rare enteropathic arthropathy. Gastrointest Radiol. 1987;12(3):265-267.
30. Bert F, Bariou-Lancelin M, Lambert-Zechovsky N. Clinical significance of bacteremia involving the “Streptococcus milleri” group: 51 cases and review. Clin Infect Dis. 1998;27(2):385-387.
31. Casariego E, Rodriguez A, Corredoira JC, et al. Prospective study of Streptococcus milleri bacteremia. Eur J Clin Microbiol Infect Dis. 1996;15(3):194-200.
32. Jacobs JA, Pietersen HG, Stobberingh EE, Soeters PB. Bacteremia involving the “Streptococcus milleri” group: Analysis of 19 cases. Clin Infect Dis. 1994;19(4):704-713.
33. Zheng S, Robinson E, Yeoman S, et al. MRI bone oedema predicts eight year tendon function at the wrist but not the requirement for orthopaedic surgery in rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):607-611.
34. Harris R. Spontaneous rupture of the tendon of extensor pollicis longus as a complication of rheumatoid arthritis. Ann Rheum Dis. 1951;10(3):298-306.
35. Björkman A, Jörgsholm P. Rupture of the extensor pollicis longus tendon: A study of aetiological factors. Scand J Plast Reconstr Surg Hand Surg. 2004;38(1):32-35.
1. Dubost JJ, Fis I, Denis P, et al. Polyarticular septic arthritis. Medicine (Baltimore). 1993;72(5):296-310.
2. Nolla JM, Gómez-Vaquero C, Fiter J, et al. Pyarthrosis in patients with rheumatoid arthritis: A detailed analysis of 10 cases and literature review. Semin Arthritis Rheum. 2000;30(2):121-126.
3.Weston VC, Jones AC, Bradbury N, Fawthrop F, Doherty M. Clinical features and outcome of septic arthritis in a single UK Health District 1982-1991. Ann Rheum Dis. 1999;58(4):214-219.
4. Galloway JB, Hyrich KL, Mercer LK, et al; BSR Biologics Register. Risk of septic arthritis in patients with rheumatoid arthritis and the effect of anti-TNF therapy: Results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70(10):1810-1814.
5. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Frequency of infection in patients with rheumatoid arthritis compared with controls: A population-based study. Arthritis Rheum. 2002;46(9):2287-2293.
6. Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569-2581.
7. Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H; CASPAR Study Group. Classification criteria for psoriatic arthritis: Development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665-2673.
8. Gladman DD, Shuckett R, Russell ML, Thorne JC, Schachter RK. Psoriatic arthritis (PSA)—An analysis of 220 patients. Q J Med. 1987;62(238):127-141.
9. Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol. 2005;32(3):511-515.
10. Vander Cruyssen B, Hoffman IE, Zmierczak H, et al. Anti-citrullinated peptide antibodies may occur in patients with psoriatic arthritis. Ann Rheum Dis. 2005;64(8):1145-1149.
11. Alenius GM, Berglin E, Rantapää Dahlgvist S. Antibodies against cyclic citrullinated peptide (CCP) in psoriatic patients with or without joint inflammation. Ann Rheum Dis. 2006;65(3):398-400.
12. Candia L, Marquez J, Gonzalez C, et al. Low frequency of anticyclic citrullinated peptide antibodies in psoriatic arthritis but not in cutaneous psoriasis. J Clin Rheumatol. 2006;12(5):226-229.
13. Inanc N, Dalkilic E, Kamali S, et al. Anti-CCP antibodies in rheumatoid arthritis and psoriatic arthritis. Clin Rheumatol. 2007;26(1):17-23.
14. Popescu C, Zofota S, Bojinca V, Ionescu R. Anti-cyclic citrullinated peptide antibodies in psoriatic arthritis—Cross-sectional study and literature review. J Med Life. 2013;6(4):376-382.
15. Schoellnast H, Deutschmann HA, Hermann J, et al. Psoriatic arthritis and rheumatoid arthritis: Findings in contrast-enhanced MRI. AJR Am J Roentgenol. 2006;187(2):351-357.
16. Doran MF, Crowson CS, Pond GR, O’Fallon WM, Gabriel SE. Predictors of infection in rheumatoid arthritis. Arthritis Rheum. 2002;46(9):2294-2300.
17. Shoenfeld Y, Gurewich Y, Gallant LA, Pinkhas J. Prednisone-induced leukocytosis. Influence of dosage, method and duration of administration on the degree of leukocytosis. Am J Med. 1981;71(5):773-778.
18. Gossling J. Occurrence and pathogenicity of the Streptococcus milleri group. Rev Infect Dis. 1988;10(2):257-285.
19. Dubost JJ, Soubrier M, De Champs C, Ristori JM, Sauvezie B. Streptococcal septic arthritis in adults. A study of 55 cases with a literature review. Joint Bone Spine. 2004;71(4):303-311.
20. Ryan MJ, Kavanagh R, Wall PG, Hazleman BL. Bacterial joint infections in England and Wales: Analysis of bacterial isolates over a four year period. Br J Rheumatol. 1997;36(3):370-373.
21. Morgan DS, Fisher D, Merianos A, Currie BJ. An 18 year clinical review of septic arthritis from tropical Australia. Epidemiol Infect. 1996;117(3):423-428.
22. Kaandorp CJ, Dinant HJ, van de Laar MA, Moens HJ, Prins AP, Dijkmans BA. Incidence and sources of native and prosthetic joint infection: A community based prospective survey. Ann Rheum Dis. 1997;56(8):470-475.
23. Bradlow A, Mitchell RG, Mowat AG. Group G streptococcal arthritis. Rheumatol Rehabil. 1982;21(4):206-210.
24. Meier JL, Gerster JC. Bursitis and tenosynovitis caused by group G streptococci. J Rheumatol. 1983;10(5):817-818.
25. Filippucci E, Gabba A, Di Geso L, Girolimetti R, Salaffi F, Grassi W. Hand tendon involvement in rheumatoid arthritis: An ultrasound study. Semin Arthritis Rheum. 2012;41(6):752-760.
26. Bronze MS, Whitby S, Schaberg DR. Group G streptococcal arthritis: Case report and review of the literature. Am J Med Sci. 1997;313(4):239-243.
27. Schattner A, Vosti KL. Bacterial arthritis due to beta-hemolytic streptococci of serogroups A, B, C, F, and G. Analysis of 23 cases and a review of the literature. Medicine (Baltimore). 1998;77(2):122-139.
28. Gaunt PN, Seal DV. Group G streptococcal infection of joints and joint prostheses. J Infect. 1986;13(2):115-123.
29. Trenkner SW, Braunstein EM, Lynn MD, Ike RW. Group G streptococcal arthritis and bowel disease: A rare enteropathic arthropathy. Gastrointest Radiol. 1987;12(3):265-267.
30. Bert F, Bariou-Lancelin M, Lambert-Zechovsky N. Clinical significance of bacteremia involving the “Streptococcus milleri” group: 51 cases and review. Clin Infect Dis. 1998;27(2):385-387.
31. Casariego E, Rodriguez A, Corredoira JC, et al. Prospective study of Streptococcus milleri bacteremia. Eur J Clin Microbiol Infect Dis. 1996;15(3):194-200.
32. Jacobs JA, Pietersen HG, Stobberingh EE, Soeters PB. Bacteremia involving the “Streptococcus milleri” group: Analysis of 19 cases. Clin Infect Dis. 1994;19(4):704-713.
33. Zheng S, Robinson E, Yeoman S, et al. MRI bone oedema predicts eight year tendon function at the wrist but not the requirement for orthopaedic surgery in rheumatoid arthritis. Ann Rheum Dis. 2006;65(5):607-611.
34. Harris R. Spontaneous rupture of the tendon of extensor pollicis longus as a complication of rheumatoid arthritis. Ann Rheum Dis. 1951;10(3):298-306.
35. Björkman A, Jörgsholm P. Rupture of the extensor pollicis longus tendon: A study of aetiological factors. Scand J Plast Reconstr Surg Hand Surg. 2004;38(1):32-35.
Angioimmunoblastic T-cell Lymphoma Presenting as Purpura Fulminans
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
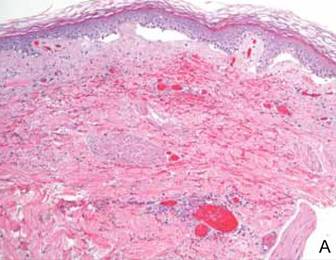
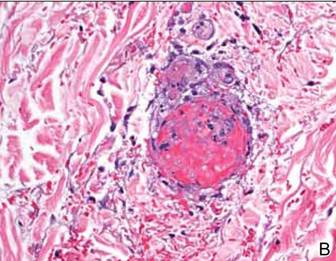
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
|
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
Purpura fulminans is a hematologic emergency, with clinical skin necrosis and laboratory testing showing disseminated intravascular coagulation. The thrombotic occlusion usually affects small and medium-sized blood vessels and may involve any organ. Purpura fulminans has been implicated with sepsis, most commonly meningococcal infections; other infections such as Staphylococcus aureus, groups A and B β-hemolytic streptococci, Streptococcus pneumoniae, and Haemophilus influenzae; and as a sequela to benign childhood infections, such as varicella. Other associations with purpura fulminans include autoimmune disease and heritable or acquired deficiency of anticoagulant proteins, most commonly protein C. We present a rare case of purpura fulminans as the presenting sign of angioimmunoblastic T-cell lymphoma (AITL), an aggressive primary nodal peripheral T-cell lymphoma with a high mortality rate and nonspecific skin manifestations in roughly half of all patients involved.
Case Report
A 56-year-old woman presented with purpuric patches on the left foot (Figure 1A). Seven days after presentation the lesion progressed into ecchymotic geographic plaques and hemorrhagic bullae that spread upward and contralaterally, sparing the digits, trunk, head, neck, and mucous membranes. Ultimately, the involved skin became necrotic and involved 20% of the body surface area (Figure 1B). The lesions were painful with a burning sensation but were not pruritic. The patient also reported intermittent fevers, chills, myalgia, nausea, and shortness of breath. Enlarged lymph nodes were present in the right cervical chain. She denied new medications; stated she had been in good health prior to this episode; and had no history of spontaneous abortion, neurologic symptoms, or other serious illness.
|
|
Computed tomography showed prominent diffuse mediastinal, mesenteric, retroperitoneal, and pelvic lymphadenopathy with involvement of the cervical and inguinal areas. Laboratory values showed thrombocytopenia and increased fibrin degradation products. Blood and tissue cultures were negative; the patient also had a negative viral serology, except for Epstein-Barr virus IgG titers (>1:2560). A skin biopsy of the left thigh demonstrated venules and capillaries in the mid and superficial dermis filled with fibrin thrombi without vasculitis (Figure 2). A lymph node biopsy was consistent with a diagnosis of AITL. The lymph node architecture was largely effaced by a polymorphous lymphoid infiltrate that predominantly expanded into paracortical areas and was associated with a prominent arborizing vascular proliferation. The infiltrate was composed of lymphocytes ranging in size from small to medium, with ample cytoplasm, coarsely clumped chromatin, and mildly irregular nuclear membranes. Large atypical lymphocytes with features of immunoblasts were easily identified. An associated inflammatory background composed of eosinophils, plasma cells, and histiocytes was present (Figure 3). The atypical lymphocytes stained positive for CD3and CD10 on immunohistochemistry. Additionally, a subset of large immunoblastlike lymphocytes was positive for Epstein-Barr–encoded small RNAs by in situ hybridization.
|
The patient was started on rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone. She received 2 cycles with positive response based on subsequent computed tomography and positron emission tomography scans that showed regression of her disease as well as the lack of formation of new skin lesions. She was transferred to a burn unit where she had continuing treatment and skin grafts. Despite 2 cycles of chemotherapy, broad-spectrum antibiotics, and daily wound care management, the patient died secondary to sepsis 6 months after presentation.
Comment
Angioimmunoblastic T-cell lymphoma is a primary nodal lymphoma with occasional cutaneous involvement. Cutaneous manifestations occur in roughly half of all patients with AITL1 and have mainly been described as erythematous macules and papules that can resemble a viral exanthem or a drug reaction.2 However, other skin manifestations include urticaria, papulovesicular lesions, nodules, erythroderma,3 and to a lesser degree purpura.4 The lesions have been noted to occur prior to, concurrent with, or anytime during the disease.3,5,6 This aggressive lymphoma has mortality rates ranging from 50% to 72%, and median survival ranges from 11 to 30 months.6
To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary. Classic findings on histopathology include effacement of normal architecture, marked vascular proliferation, and aggregates of atypical lymphoid cells. CD10 has been shown to be a good objective criterion for the diagnosis of AITL,4 with characteristic tumor cells expressing CD10. Nodal Epstein-Barr virus–positive lymphocytes often are present.2 Other T-cell lymphomas with primarily nodal presentation along with peripheral T-cell lymphoma include peripheral T-cell lymphoma unspecified type and anaplastic large cell lymphoma, according to the World Health Organization classification.7 Anaplastic large cell lymphoma is easily distinguished from AITL based on histopathology, immunostaining, and clinical presentation. Until recently, peripheral T-cell lymphoma unspecified type and reactive lymphoid hyperplasia presented a challenge to differentiate from AITL, especially in the early phases of the disease; however, the introduction of CD10 as a phenotypic marker has been instrumental in distinguishing AITL from other T-cell lymphomas with primary nodal involvement.1,4
The development of purpura fulminans and disseminated intravascular coagulation in a patient with AITL is rare. Although the exact mechanism for the thrombus formation in the skin has not been elucidated, purpura fulminans typically develops secondary to a severe infection. The exact incidence of purpura fulminans in the setting of AITL is unknown, but purpura as a cutaneous eruption has been associated as a clinical finding in AITL.6 Although our case may be a rare presentation of AITL, a prompt and accurate diagnosis can drastically change the prognosis of this aggressive disease.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
1. Ferry JA. Angioimmunoblastic T-cell lymphoma. Adv Anat Pathol. 2002;9:273-279.
2. Brown HA, Macon WR, Kurtin PJ, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma with remarkable heterogeneous Epstein-Barr virus expression. J Cutan Pathol. 2001;28:432-438.
3. Bernstein JE, Soltani K, Lorincz AL. Cutaneous manifestations of angioimmunoblastic lymphadenopathy. J Am Acad Dermatol. 1979;1:227-232.
4. Attygalle A, Al-Jehani R, Diss TC, et al. Neoplastic T cells in angioimmunoblastic T-cell lymphoma express CD10. Blood. 2002;99:627-633.
5. Jayaramna AG, Cassarino D, Advani R, et al. Cutaneous involvement by angioimmunoblastic T-cell lymphoma: a unique histologic presentation, mimicking an infectious etiology. J Cutan Pathol. 2006;33(suppl 2):6-11.
6. Martel P, Laroche L, Courville P, et al. Cutaneous involvement in patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Archives of Dermatology. 2000;136:881-886.
7. Jaffe ES, Harris NL, Stein H, et al, eds. Tumours of Haematopoietic and Lymphoid Tissues. 1st ed. Bethesda, MD: International Agency for Research on Cancer; 2001.
Practice Points
- Angioimmunoblastic T-cell lymphoma (AITL) is a primary nodal lymphoma with occasional nonspecific cutaneous involvement that may be morbilliform, maculopapular, erythrodermic, or rarely purpuric.
- To arrive at the correct diagnosis of AITL, a nodal biopsy with immunochemistry is necessary.
- CD10 positivity is a good objective criterion for the diagnosis of AITL, and Epstein-Barr virus–positive lymphocytes are nearly always present.
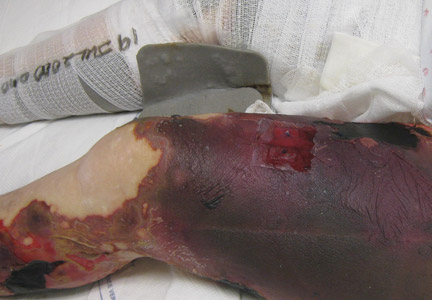
Epithelioid Sarcoma Resembling Benign Fibrous Histiocytoma
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
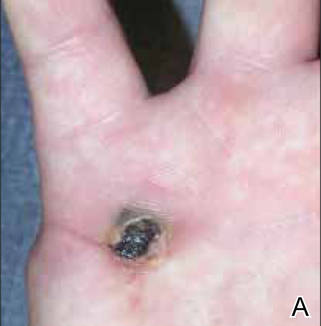
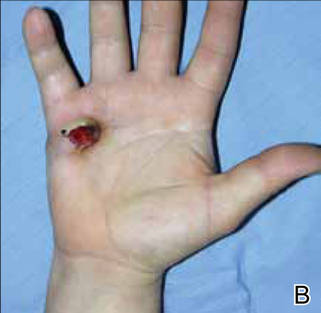
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
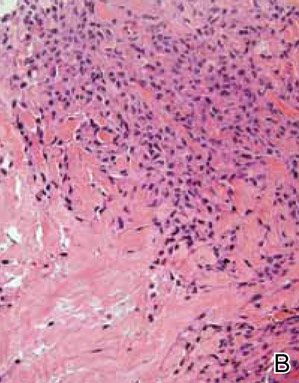
|
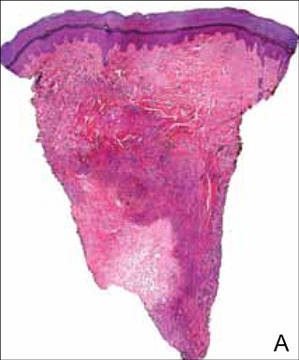
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
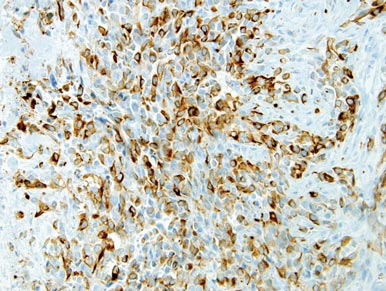
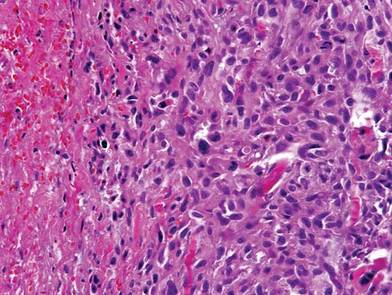
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Practice Points
- Epithelioid sarcoma should be considered in the clinical differential diagnosis of nonhealing recurrent lesions of the distal extremities in a young adult.
- Histological presentation of epithelioid sarcoma can mimic a number of benign granulomatous and fibrohistiocytic processes, including benign fibrous histiocytoma.
- Deeper biopsies may be needed to demonstrate the overtly malignant morphology characteristic of epithelioid sarcoma.
- Inactivation of SMARCB1/INI1 is a common molecular aberration identified in epithelioid sarcoma and can be demonstrated immunohistochemically by absence of nuclear staining in tumor cells.
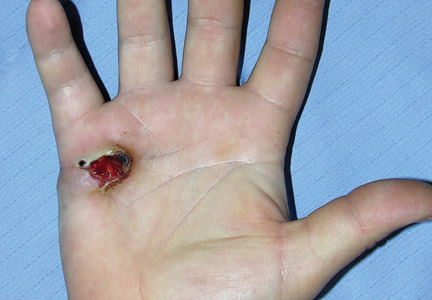
Imiquimod Induces Sustained Remission of Actinic Damage: A Case Report Spanning One Decade of Observation
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
|
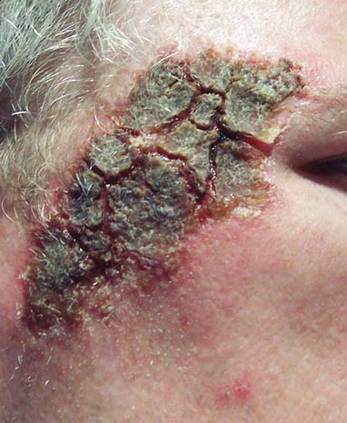
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
|
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
|
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
Practice Points
- Topical immunomodulators such as imiquimod and topical chemotherapeutics such as 5-fluorouracil are effective in the field treatment of actinic keratoses.
- Prior topical immunomodulator use for nonmelanoma skin cancer may induce a sustained remission of actinic damage.
- The field effect of imiquimod treatment in actinically damaged skin may persist for several years.
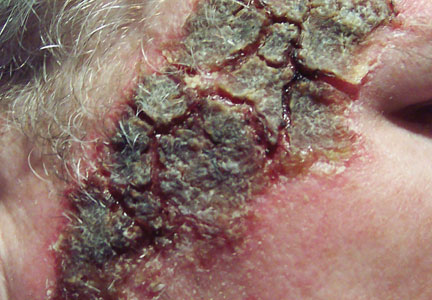
Superficial Acral Fibromyxoma and Other Slow-Growing Tumors in Acral Areas
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
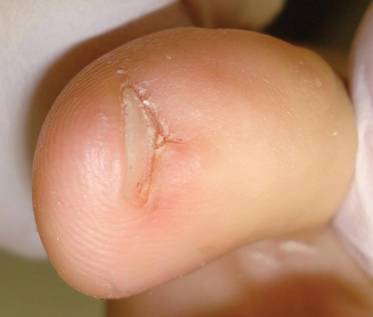
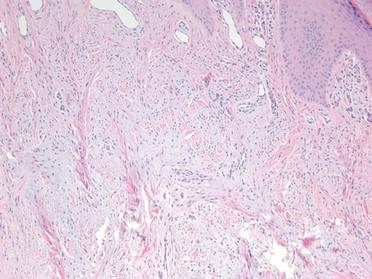

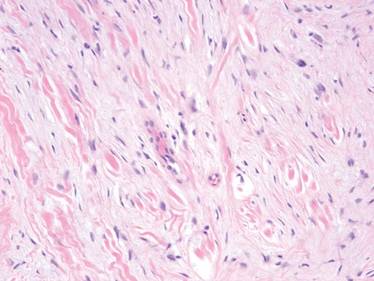
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
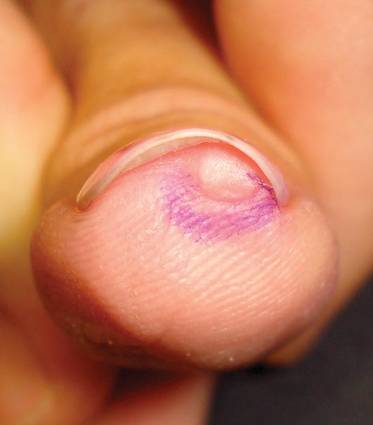
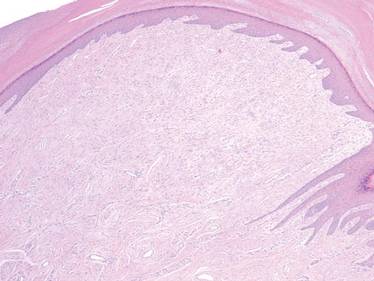
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
Practice Points
- Superficial acral fibromyxoma (SAFM) is a rare but distinct tumor that may affect the nail bed and nail plate, and it may clinically or histopathologically mimic other tumors of the distal extremities.
- Although SAFM is considered a benign tumor, it frequently persists or recurs after incomplete excision, and therefore complete local resection may be recommended, particularly for symptomatic lesions.
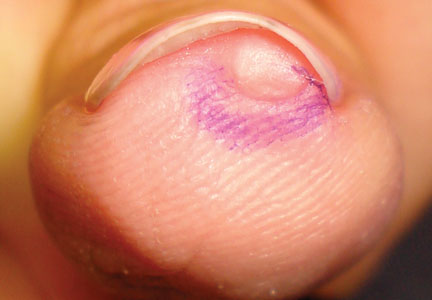
Sharp, left-sided back pain • bilateral leg weakness • degenerative disc disease • Dx?
THE CASE
An 84-year old woman came to the emergency department (ED) with sharp back pain on her left side that she’d had for 4 days. The pain radiated to her posterior hips when standing. She said her whole body felt achy and she was experiencing weakness in both legs.
The patient had a history of hypertension, coronary artery disease, and aortic stenosis; she’d received a bioprosthetic aortic valve 7 years ago. She was not immunocompromised or receiving steroids but was taking docusate, oxybutynin, carvedilol, amlodipine, atorvastatin, furosemide, rivaroxaban, and a multivitamin. Her physical exam, vital signs, and complete blood count (CBC) were normal. An x-ray of the lumbar spine showed degenerative joint/disc disease and spondylosis at L4-L5 and L5-S1. The patient was sent home with oxycodone/acetaminophen 5 mg/325 mg every 6 hours as needed for pain and told to follow up with her family physician (FP).
Six days later, the patient went to see her FP and told her that her symptoms hadn’t improved. She was afebrile and her blood pressure was 150/80 mm Hg. Her muscle strength was 4/5 with hip flexion bilaterally; the rest of her strength was 5/5. There was no lumbar paraspinal tenderness and she had a negative straight leg raise test. No other neurologic deficits were noted. The FP prescribed physical therapy at home with a licensed therapist, which consisted of stretching exercises and active, dynamic exercise to improve the patient’s range of motion. She also ordered outpatient lumbar magnetic resonance imaging (MRI).
THE DIAGNOSIS
Approximately 3 weeks later, the patient’s MRI revealed osteomyelitis/discitis at the L3-L4 level and severe tricompartmental stenosis from L2-L3 through L4-L5. A day after receiving the results—and about a month after having first gone to the ED—the patient was admitted to the hospital. She was afebrile and her blood pressure was 148/75 mm Hg. Her physical exam revealed no leukocytosis or neurologic deficits, but did show a systolic murmur from her aortic valve.
She had an erythrocyte sedimentation rate (ESR) of 77 mm/hr (normal range for women, <30 mm/hr) and her C-reactive protein (CRP) level was 5.88 mg/dL (<.50 mg/dL indicates average risk for cardiovascular disease). A transesophageal echocardiogram was performed and there was no sign of vegetation or thrombi. However, blood cultures were positive for Streptococcus salivarius—a bacterium found on human dental plaque—which we determined was the cause of the osteomyelitis.
To the best of our knowledge, there have been no other case reports that described S. salivarius as having caused osteomyelitis without concurrent endocarditis.
DISCUSSION
Back pain is a common and costly issue among primary care patients. More than two-thirds of adults suffer from low back pain at some point, primarily without underlying malignancy or neurologic deficits.1,2 Acute low back pain is often mechanical (97%); however, other causes, including infection, may be to blame (TABLE).1 Most acute back pain will improve with conservative treatment and patients need only reassurance of a favorable prognosis, but 20% of patients may develop chronic back pain.2
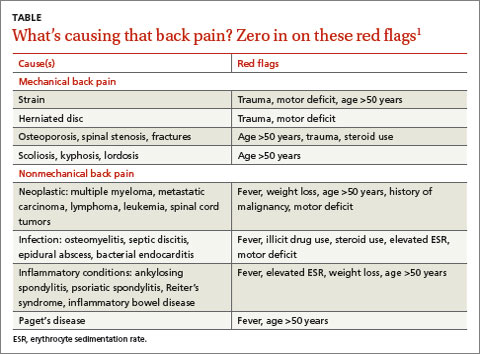
The diagnostic approach to low back pain varies widely.3 Some data indicate that early imaging of back pain can lead to unneeded follow-up testing, radiation exposure, unnecessary surgery, patient “labeling,” and increased health care costs, all of which suggest that routine imaging shouldn’t be pursued in acute low back pain.4
Red flags for acute low back pain that warrant imaging include age >50 years, fever, weight loss, elevated ESR, history of malignancy, trauma, motor deficits, steroid or illicit drug use, and litigation.1 If not already done, it’s also important to order a CBC, ESR, and CRP for patients with any of these red flags.
Imaging studies are important, but clinical correlation is crucial because imaging can reveal disk abnormalities even in healthy, asymptomatic patients.5 Computed tomography scans or MRI is indicated for patients with neurologic deficits or nerve root tension signs, but only if a patient is a potential candidate for surgery or epidural steroid injection.6,7 If you suspect an infection (such as spondylodiscitis or osteomyelitis), diagnosing the condition quickly is key.
Our patient had 2 red flags (age >50 years and elevated ESR) that helped us reach an unlikely diagnosis of lumbar osteomyelitis with S. salivarius as the cause. Degenerative spinal disease seen on x-ray may have delayed our patient’s diagnosis. If our patient had had an ESR or CRP test earlier, or if further imaging had been conducted sooner (given her proximal muscle weakness), the correct diagnosis would have been made more quickly and appropriate treatment provided sooner.
Our patient
The patient was started on a 6-week course of intravenous ceftriaxone 2g/d, which she continued to receive at home via a peripherally inserted central catheter. The patient was instructed at discharge (on Day 8) to follow up with her FP, which she did 12 days later. At that visit, her back pain was improved and her ESR and CRP levels were within normal ranges.
THE TAKEAWAY
When evaluating a patient who presents with low back pain, perform a focused history and be on the lookout for “red flags” that warrant further imaging and testing. Routine imaging is not recommended for patients with nonspecific low back pain, but imaging may be indicated for patients with neurologic deficits or nerve root tension signs.
A patient with low back pain caused by osteomyelitis may present with fever, elevated ESR, and/or motor deficits. Identifying the bacteria underlying the infection will help guide selection of appropriate antibiotics.
1. Deyo RA, Weinstein JN. Low back pain. N Engl J Med. 2001;344:363-370.
2. Deyo RA, Phillips WR. Low back pain. A primary care challenge. Spine (Phila Pa 1976). 1996;21:2826-2832.
3. Cherkin DC, Deyo RA, Wheeler K, et al. Physician variation in diagnostic testing for low back pain. Who you see is what you get. Arthritis Rheum. 1994;37:15-22.
4. Srinivas SV, Deyo RA, Berger ZD. Application of “less is more” to low back pain. Arch Intern Med. 2012;172:1016-1020.
5. Jensen MC, Brant-Zawadzki MN, Obuchowski N, et al. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med. 1994;331:69-73.
6. Wipf JE, Deyo RA. Low back pain. Med Clin North Am. 1995;79:231-246.
7. Chou R, Qaseem A, Snow V, et al; Clinical Efficacy Assessment Committee of the American College of Physicians; American College of Physicians; American Pain Society Low Back Pain Guidelines Panel. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147:478-493.
THE CASE
An 84-year old woman came to the emergency department (ED) with sharp back pain on her left side that she’d had for 4 days. The pain radiated to her posterior hips when standing. She said her whole body felt achy and she was experiencing weakness in both legs.
The patient had a history of hypertension, coronary artery disease, and aortic stenosis; she’d received a bioprosthetic aortic valve 7 years ago. She was not immunocompromised or receiving steroids but was taking docusate, oxybutynin, carvedilol, amlodipine, atorvastatin, furosemide, rivaroxaban, and a multivitamin. Her physical exam, vital signs, and complete blood count (CBC) were normal. An x-ray of the lumbar spine showed degenerative joint/disc disease and spondylosis at L4-L5 and L5-S1. The patient was sent home with oxycodone/acetaminophen 5 mg/325 mg every 6 hours as needed for pain and told to follow up with her family physician (FP).
Six days later, the patient went to see her FP and told her that her symptoms hadn’t improved. She was afebrile and her blood pressure was 150/80 mm Hg. Her muscle strength was 4/5 with hip flexion bilaterally; the rest of her strength was 5/5. There was no lumbar paraspinal tenderness and she had a negative straight leg raise test. No other neurologic deficits were noted. The FP prescribed physical therapy at home with a licensed therapist, which consisted of stretching exercises and active, dynamic exercise to improve the patient’s range of motion. She also ordered outpatient lumbar magnetic resonance imaging (MRI).
THE DIAGNOSIS
Approximately 3 weeks later, the patient’s MRI revealed osteomyelitis/discitis at the L3-L4 level and severe tricompartmental stenosis from L2-L3 through L4-L5. A day after receiving the results—and about a month after having first gone to the ED—the patient was admitted to the hospital. She was afebrile and her blood pressure was 148/75 mm Hg. Her physical exam revealed no leukocytosis or neurologic deficits, but did show a systolic murmur from her aortic valve.
She had an erythrocyte sedimentation rate (ESR) of 77 mm/hr (normal range for women, <30 mm/hr) and her C-reactive protein (CRP) level was 5.88 mg/dL (<.50 mg/dL indicates average risk for cardiovascular disease). A transesophageal echocardiogram was performed and there was no sign of vegetation or thrombi. However, blood cultures were positive for Streptococcus salivarius—a bacterium found on human dental plaque—which we determined was the cause of the osteomyelitis.
To the best of our knowledge, there have been no other case reports that described S. salivarius as having caused osteomyelitis without concurrent endocarditis.
DISCUSSION
Back pain is a common and costly issue among primary care patients. More than two-thirds of adults suffer from low back pain at some point, primarily without underlying malignancy or neurologic deficits.1,2 Acute low back pain is often mechanical (97%); however, other causes, including infection, may be to blame (TABLE).1 Most acute back pain will improve with conservative treatment and patients need only reassurance of a favorable prognosis, but 20% of patients may develop chronic back pain.2
The diagnostic approach to low back pain varies widely.3 Some data indicate that early imaging of back pain can lead to unneeded follow-up testing, radiation exposure, unnecessary surgery, patient “labeling,” and increased health care costs, all of which suggest that routine imaging shouldn’t be pursued in acute low back pain.4
Red flags for acute low back pain that warrant imaging include age >50 years, fever, weight loss, elevated ESR, history of malignancy, trauma, motor deficits, steroid or illicit drug use, and litigation.1 If not already done, it’s also important to order a CBC, ESR, and CRP for patients with any of these red flags.
Imaging studies are important, but clinical correlation is crucial because imaging can reveal disk abnormalities even in healthy, asymptomatic patients.5 Computed tomography scans or MRI is indicated for patients with neurologic deficits or nerve root tension signs, but only if a patient is a potential candidate for surgery or epidural steroid injection.6,7 If you suspect an infection (such as spondylodiscitis or osteomyelitis), diagnosing the condition quickly is key.
Our patient had 2 red flags (age >50 years and elevated ESR) that helped us reach an unlikely diagnosis of lumbar osteomyelitis with S. salivarius as the cause. Degenerative spinal disease seen on x-ray may have delayed our patient’s diagnosis. If our patient had had an ESR or CRP test earlier, or if further imaging had been conducted sooner (given her proximal muscle weakness), the correct diagnosis would have been made more quickly and appropriate treatment provided sooner.
Our patient
The patient was started on a 6-week course of intravenous ceftriaxone 2g/d, which she continued to receive at home via a peripherally inserted central catheter. The patient was instructed at discharge (on Day 8) to follow up with her FP, which she did 12 days later. At that visit, her back pain was improved and her ESR and CRP levels were within normal ranges.
THE TAKEAWAY
When evaluating a patient who presents with low back pain, perform a focused history and be on the lookout for “red flags” that warrant further imaging and testing. Routine imaging is not recommended for patients with nonspecific low back pain, but imaging may be indicated for patients with neurologic deficits or nerve root tension signs.
A patient with low back pain caused by osteomyelitis may present with fever, elevated ESR, and/or motor deficits. Identifying the bacteria underlying the infection will help guide selection of appropriate antibiotics.
THE CASE
An 84-year old woman came to the emergency department (ED) with sharp back pain on her left side that she’d had for 4 days. The pain radiated to her posterior hips when standing. She said her whole body felt achy and she was experiencing weakness in both legs.
The patient had a history of hypertension, coronary artery disease, and aortic stenosis; she’d received a bioprosthetic aortic valve 7 years ago. She was not immunocompromised or receiving steroids but was taking docusate, oxybutynin, carvedilol, amlodipine, atorvastatin, furosemide, rivaroxaban, and a multivitamin. Her physical exam, vital signs, and complete blood count (CBC) were normal. An x-ray of the lumbar spine showed degenerative joint/disc disease and spondylosis at L4-L5 and L5-S1. The patient was sent home with oxycodone/acetaminophen 5 mg/325 mg every 6 hours as needed for pain and told to follow up with her family physician (FP).
Six days later, the patient went to see her FP and told her that her symptoms hadn’t improved. She was afebrile and her blood pressure was 150/80 mm Hg. Her muscle strength was 4/5 with hip flexion bilaterally; the rest of her strength was 5/5. There was no lumbar paraspinal tenderness and she had a negative straight leg raise test. No other neurologic deficits were noted. The FP prescribed physical therapy at home with a licensed therapist, which consisted of stretching exercises and active, dynamic exercise to improve the patient’s range of motion. She also ordered outpatient lumbar magnetic resonance imaging (MRI).
THE DIAGNOSIS
Approximately 3 weeks later, the patient’s MRI revealed osteomyelitis/discitis at the L3-L4 level and severe tricompartmental stenosis from L2-L3 through L4-L5. A day after receiving the results—and about a month after having first gone to the ED—the patient was admitted to the hospital. She was afebrile and her blood pressure was 148/75 mm Hg. Her physical exam revealed no leukocytosis or neurologic deficits, but did show a systolic murmur from her aortic valve.
She had an erythrocyte sedimentation rate (ESR) of 77 mm/hr (normal range for women, <30 mm/hr) and her C-reactive protein (CRP) level was 5.88 mg/dL (<.50 mg/dL indicates average risk for cardiovascular disease). A transesophageal echocardiogram was performed and there was no sign of vegetation or thrombi. However, blood cultures were positive for Streptococcus salivarius—a bacterium found on human dental plaque—which we determined was the cause of the osteomyelitis.
To the best of our knowledge, there have been no other case reports that described S. salivarius as having caused osteomyelitis without concurrent endocarditis.
DISCUSSION
Back pain is a common and costly issue among primary care patients. More than two-thirds of adults suffer from low back pain at some point, primarily without underlying malignancy or neurologic deficits.1,2 Acute low back pain is often mechanical (97%); however, other causes, including infection, may be to blame (TABLE).1 Most acute back pain will improve with conservative treatment and patients need only reassurance of a favorable prognosis, but 20% of patients may develop chronic back pain.2
The diagnostic approach to low back pain varies widely.3 Some data indicate that early imaging of back pain can lead to unneeded follow-up testing, radiation exposure, unnecessary surgery, patient “labeling,” and increased health care costs, all of which suggest that routine imaging shouldn’t be pursued in acute low back pain.4
Red flags for acute low back pain that warrant imaging include age >50 years, fever, weight loss, elevated ESR, history of malignancy, trauma, motor deficits, steroid or illicit drug use, and litigation.1 If not already done, it’s also important to order a CBC, ESR, and CRP for patients with any of these red flags.
Imaging studies are important, but clinical correlation is crucial because imaging can reveal disk abnormalities even in healthy, asymptomatic patients.5 Computed tomography scans or MRI is indicated for patients with neurologic deficits or nerve root tension signs, but only if a patient is a potential candidate for surgery or epidural steroid injection.6,7 If you suspect an infection (such as spondylodiscitis or osteomyelitis), diagnosing the condition quickly is key.
Our patient had 2 red flags (age >50 years and elevated ESR) that helped us reach an unlikely diagnosis of lumbar osteomyelitis with S. salivarius as the cause. Degenerative spinal disease seen on x-ray may have delayed our patient’s diagnosis. If our patient had had an ESR or CRP test earlier, or if further imaging had been conducted sooner (given her proximal muscle weakness), the correct diagnosis would have been made more quickly and appropriate treatment provided sooner.
Our patient
The patient was started on a 6-week course of intravenous ceftriaxone 2g/d, which she continued to receive at home via a peripherally inserted central catheter. The patient was instructed at discharge (on Day 8) to follow up with her FP, which she did 12 days later. At that visit, her back pain was improved and her ESR and CRP levels were within normal ranges.
THE TAKEAWAY
When evaluating a patient who presents with low back pain, perform a focused history and be on the lookout for “red flags” that warrant further imaging and testing. Routine imaging is not recommended for patients with nonspecific low back pain, but imaging may be indicated for patients with neurologic deficits or nerve root tension signs.
A patient with low back pain caused by osteomyelitis may present with fever, elevated ESR, and/or motor deficits. Identifying the bacteria underlying the infection will help guide selection of appropriate antibiotics.
1. Deyo RA, Weinstein JN. Low back pain. N Engl J Med. 2001;344:363-370.
2. Deyo RA, Phillips WR. Low back pain. A primary care challenge. Spine (Phila Pa 1976). 1996;21:2826-2832.
3. Cherkin DC, Deyo RA, Wheeler K, et al. Physician variation in diagnostic testing for low back pain. Who you see is what you get. Arthritis Rheum. 1994;37:15-22.
4. Srinivas SV, Deyo RA, Berger ZD. Application of “less is more” to low back pain. Arch Intern Med. 2012;172:1016-1020.
5. Jensen MC, Brant-Zawadzki MN, Obuchowski N, et al. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med. 1994;331:69-73.
6. Wipf JE, Deyo RA. Low back pain. Med Clin North Am. 1995;79:231-246.
7. Chou R, Qaseem A, Snow V, et al; Clinical Efficacy Assessment Committee of the American College of Physicians; American College of Physicians; American Pain Society Low Back Pain Guidelines Panel. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147:478-493.
1. Deyo RA, Weinstein JN. Low back pain. N Engl J Med. 2001;344:363-370.
2. Deyo RA, Phillips WR. Low back pain. A primary care challenge. Spine (Phila Pa 1976). 1996;21:2826-2832.
3. Cherkin DC, Deyo RA, Wheeler K, et al. Physician variation in diagnostic testing for low back pain. Who you see is what you get. Arthritis Rheum. 1994;37:15-22.
4. Srinivas SV, Deyo RA, Berger ZD. Application of “less is more” to low back pain. Arch Intern Med. 2012;172:1016-1020.
5. Jensen MC, Brant-Zawadzki MN, Obuchowski N, et al. Magnetic resonance imaging of the lumbar spine in people without back pain. N Engl J Med. 1994;331:69-73.
6. Wipf JE, Deyo RA. Low back pain. Med Clin North Am. 1995;79:231-246.
7. Chou R, Qaseem A, Snow V, et al; Clinical Efficacy Assessment Committee of the American College of Physicians; American College of Physicians; American Pain Society Low Back Pain Guidelines Panel. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147:478-493.
Daily episodes of confusion • altered behavior • chronic sleep deprivation • Dx?
THE CASE
A 60-year-old man with hypertension, gout, hyperlipidemia, and chronic sleep deprivation was referred to our neurology department for evaluation because he’d recently developed episodes of confusion and altered behavior that occurred daily. According to the patient’s wife, these episodes had started 4 weeks earlier while the patient was driving. He drove off the road while staring ahead with a “Joker-like” smile on his face. He was unable to utter more than a few words or respond to his wife, who was able to safely bring the car to a stop. The patient had spotty memory of this 40-minute episode.
Since then, he’d had similar but shorter episodes each morning, 20 to 75 minutes after taking his prescribed medications (lisinopril, simvastatin, and allopurinol). According to the patient’s wife, during these episodes, the patient would “act childish.” He would develop a voracious appetite and experience double or distorted vision, an unsteady gait, and poor muscle tone. These episodes were always followed by a long nap.
The man denied drinking, head trauma, acute illness, or taking illicit substances or any medications other than lisinopril, simvastatin, and allopurinol. Computed tomography, magnetic resonance imaging/magnetic resonance angiography, carotid Doppler ultrasound, and routine and 24-hour ambulatory electroencephalography (EEG) were normal.
Before the patient was referred to our neurology department, he had been prescribed a short course of the antiepileptic/mood stabilizer valproate and the wakefulness agent armodafinil, but neither medication had helped. The patient’s episodes continued daily, usually 20 to 75 minutes after taking his regular medications. When he decided to take them at night, the episodes began to occur at night.
His neurologic exam was normal. Family history was positive for a cousin with narcolepsy but negative for seizures and obstructive sleep apnea (OSA). Polysomnography revealed moderate OSA with minimal oxygen desaturation. Inpatient video EEG monitoring captured several of the events that the patient and his wife had described; the patient seemed “uninhibited” in his behavior. His EEG, cardiac telemetry, oxygen saturation, blood pressure, and serum glucose level remained normal.
The episodes’ sudden onset, peculiar symptoms, and duration—and the fact that they occurred after he took his usual medications—made complex partial seizures unlikely. The patient’s chronic sleep deprivation and family history of narcolepsy raised the possibility of “sleep attacks,” but the sudden onset and age of onset of his symptoms made those conditions less likely to explain the complete clinical picture. No particular hormonal disturbance could explain his presentation, and blood work was normal.
THE DIAGNOSIS
Because the patient’s episodes had been occurring shortly after the patient took his lisinopril, simvastatin, and allopurinol, and because his blood pressure and lipid levels were normal and his gout was asymptomatic, we decided to stop these medications. Later that day, the patient reported that he had discovered that his vial of lisinopril, which he had obtained from his regular pharmacy the day before his first episode, contained a different medication. He consulted a pharmacist, who determined that the vial contained extended release zolpidem 12.5 mg, and not his antihypertensive.
DISCUSSION
Although the true incidence of medication errors is difficult to determine, a 2006 Institute of Medicine report estimated that there are at least 1.5 million cases of preventable adverse drug events in the United States each year.1 In light of these statistics, medication errors need to be near the top of our differential diagnosis when patients suddenly develop symptoms for which there is no obvious cause.
Cause to pause? If you observe a temporal association between the onset of a patient’s symptoms and medication administration, consider possible adverse effects of the medication before ordering tests.
In this case … Our patient’s peculiar presentation correlated with regular ingestion of a high dose of zolpidem, a short-acting non-benzodiazepine gamma-aminobutyric acid (GABA) agonist. Zolpidem binds to the same GABAA receptor as benzodiazepines and therefore acts as a hypnotic by increasing GABA transmission.2 Neuropsychiatric adverse events associated with zolpidem include hallucinations, amnesia, parasomnia, psychomotor impairment, and complex behaviors (eg, sleepwalking or sleep-driving).2 Higher doses may cause coma or (rarely) death.2 One case report describes a patient who heard command hallucinations and stabbed himself after ingesting a large dose of zolpidem.3
Our patient
The patient’s episodes stopped after he discontinued the zolpidem. He subsequently received a correct prescription for lisinopril, and did not experience any additional episodes.
THE TAKEAWAY
Consider medication errors and adverse drug events in the differential diagnosis for patients who develop symptoms for which there is no obvious etiology. Educate patients, as well, to question their pharmacist if a recently filled prescription doesn’t look like the pill they usually take or makes them feel different than usual when they take it.
Of course, patients should be reminded that a generic medication may not always look the same as a brand-name drug or a previous generic prescription. But it can’t hurt for the patient to ask whether that medication that “looks different” is just a different generic—or a sign of a more worrisome mix-up.
1. Institute of Medicine. Preventing medication errors. Report Brief. July 2006. Institute of Medicine Web site. Available at: http://iom.edu/~/media/Files/Report%20Files/2006/Preventing-Medication-Errors-Quality-Chasm-Series/medicationerrorsnew.pdf. Accessed January 13, 2015.
2. Gunja N. The clinical and forensic toxicology of Z-drugs. J Med Toxicol. 2013;9:155-162.
3. Manfredi G, Kotzalidis GD, Lazanio S, et al. Command hallucinations with self-stabbing associated with zolpidem overdose. J Clin Psychiatry. 2010;71:92-93.
THE CASE
A 60-year-old man with hypertension, gout, hyperlipidemia, and chronic sleep deprivation was referred to our neurology department for evaluation because he’d recently developed episodes of confusion and altered behavior that occurred daily. According to the patient’s wife, these episodes had started 4 weeks earlier while the patient was driving. He drove off the road while staring ahead with a “Joker-like” smile on his face. He was unable to utter more than a few words or respond to his wife, who was able to safely bring the car to a stop. The patient had spotty memory of this 40-minute episode.
Since then, he’d had similar but shorter episodes each morning, 20 to 75 minutes after taking his prescribed medications (lisinopril, simvastatin, and allopurinol). According to the patient’s wife, during these episodes, the patient would “act childish.” He would develop a voracious appetite and experience double or distorted vision, an unsteady gait, and poor muscle tone. These episodes were always followed by a long nap.
The man denied drinking, head trauma, acute illness, or taking illicit substances or any medications other than lisinopril, simvastatin, and allopurinol. Computed tomography, magnetic resonance imaging/magnetic resonance angiography, carotid Doppler ultrasound, and routine and 24-hour ambulatory electroencephalography (EEG) were normal.
Before the patient was referred to our neurology department, he had been prescribed a short course of the antiepileptic/mood stabilizer valproate and the wakefulness agent armodafinil, but neither medication had helped. The patient’s episodes continued daily, usually 20 to 75 minutes after taking his regular medications. When he decided to take them at night, the episodes began to occur at night.
His neurologic exam was normal. Family history was positive for a cousin with narcolepsy but negative for seizures and obstructive sleep apnea (OSA). Polysomnography revealed moderate OSA with minimal oxygen desaturation. Inpatient video EEG monitoring captured several of the events that the patient and his wife had described; the patient seemed “uninhibited” in his behavior. His EEG, cardiac telemetry, oxygen saturation, blood pressure, and serum glucose level remained normal.
The episodes’ sudden onset, peculiar symptoms, and duration—and the fact that they occurred after he took his usual medications—made complex partial seizures unlikely. The patient’s chronic sleep deprivation and family history of narcolepsy raised the possibility of “sleep attacks,” but the sudden onset and age of onset of his symptoms made those conditions less likely to explain the complete clinical picture. No particular hormonal disturbance could explain his presentation, and blood work was normal.
THE DIAGNOSIS
Because the patient’s episodes had been occurring shortly after the patient took his lisinopril, simvastatin, and allopurinol, and because his blood pressure and lipid levels were normal and his gout was asymptomatic, we decided to stop these medications. Later that day, the patient reported that he had discovered that his vial of lisinopril, which he had obtained from his regular pharmacy the day before his first episode, contained a different medication. He consulted a pharmacist, who determined that the vial contained extended release zolpidem 12.5 mg, and not his antihypertensive.
DISCUSSION
Although the true incidence of medication errors is difficult to determine, a 2006 Institute of Medicine report estimated that there are at least 1.5 million cases of preventable adverse drug events in the United States each year.1 In light of these statistics, medication errors need to be near the top of our differential diagnosis when patients suddenly develop symptoms for which there is no obvious cause.
Cause to pause? If you observe a temporal association between the onset of a patient’s symptoms and medication administration, consider possible adverse effects of the medication before ordering tests.
In this case … Our patient’s peculiar presentation correlated with regular ingestion of a high dose of zolpidem, a short-acting non-benzodiazepine gamma-aminobutyric acid (GABA) agonist. Zolpidem binds to the same GABAA receptor as benzodiazepines and therefore acts as a hypnotic by increasing GABA transmission.2 Neuropsychiatric adverse events associated with zolpidem include hallucinations, amnesia, parasomnia, psychomotor impairment, and complex behaviors (eg, sleepwalking or sleep-driving).2 Higher doses may cause coma or (rarely) death.2 One case report describes a patient who heard command hallucinations and stabbed himself after ingesting a large dose of zolpidem.3
Our patient
The patient’s episodes stopped after he discontinued the zolpidem. He subsequently received a correct prescription for lisinopril, and did not experience any additional episodes.
THE TAKEAWAY
Consider medication errors and adverse drug events in the differential diagnosis for patients who develop symptoms for which there is no obvious etiology. Educate patients, as well, to question their pharmacist if a recently filled prescription doesn’t look like the pill they usually take or makes them feel different than usual when they take it.
Of course, patients should be reminded that a generic medication may not always look the same as a brand-name drug or a previous generic prescription. But it can’t hurt for the patient to ask whether that medication that “looks different” is just a different generic—or a sign of a more worrisome mix-up.
THE CASE
A 60-year-old man with hypertension, gout, hyperlipidemia, and chronic sleep deprivation was referred to our neurology department for evaluation because he’d recently developed episodes of confusion and altered behavior that occurred daily. According to the patient’s wife, these episodes had started 4 weeks earlier while the patient was driving. He drove off the road while staring ahead with a “Joker-like” smile on his face. He was unable to utter more than a few words or respond to his wife, who was able to safely bring the car to a stop. The patient had spotty memory of this 40-minute episode.
Since then, he’d had similar but shorter episodes each morning, 20 to 75 minutes after taking his prescribed medications (lisinopril, simvastatin, and allopurinol). According to the patient’s wife, during these episodes, the patient would “act childish.” He would develop a voracious appetite and experience double or distorted vision, an unsteady gait, and poor muscle tone. These episodes were always followed by a long nap.
The man denied drinking, head trauma, acute illness, or taking illicit substances or any medications other than lisinopril, simvastatin, and allopurinol. Computed tomography, magnetic resonance imaging/magnetic resonance angiography, carotid Doppler ultrasound, and routine and 24-hour ambulatory electroencephalography (EEG) were normal.
Before the patient was referred to our neurology department, he had been prescribed a short course of the antiepileptic/mood stabilizer valproate and the wakefulness agent armodafinil, but neither medication had helped. The patient’s episodes continued daily, usually 20 to 75 minutes after taking his regular medications. When he decided to take them at night, the episodes began to occur at night.
His neurologic exam was normal. Family history was positive for a cousin with narcolepsy but negative for seizures and obstructive sleep apnea (OSA). Polysomnography revealed moderate OSA with minimal oxygen desaturation. Inpatient video EEG monitoring captured several of the events that the patient and his wife had described; the patient seemed “uninhibited” in his behavior. His EEG, cardiac telemetry, oxygen saturation, blood pressure, and serum glucose level remained normal.
The episodes’ sudden onset, peculiar symptoms, and duration—and the fact that they occurred after he took his usual medications—made complex partial seizures unlikely. The patient’s chronic sleep deprivation and family history of narcolepsy raised the possibility of “sleep attacks,” but the sudden onset and age of onset of his symptoms made those conditions less likely to explain the complete clinical picture. No particular hormonal disturbance could explain his presentation, and blood work was normal.
THE DIAGNOSIS
Because the patient’s episodes had been occurring shortly after the patient took his lisinopril, simvastatin, and allopurinol, and because his blood pressure and lipid levels were normal and his gout was asymptomatic, we decided to stop these medications. Later that day, the patient reported that he had discovered that his vial of lisinopril, which he had obtained from his regular pharmacy the day before his first episode, contained a different medication. He consulted a pharmacist, who determined that the vial contained extended release zolpidem 12.5 mg, and not his antihypertensive.
DISCUSSION
Although the true incidence of medication errors is difficult to determine, a 2006 Institute of Medicine report estimated that there are at least 1.5 million cases of preventable adverse drug events in the United States each year.1 In light of these statistics, medication errors need to be near the top of our differential diagnosis when patients suddenly develop symptoms for which there is no obvious cause.
Cause to pause? If you observe a temporal association between the onset of a patient’s symptoms and medication administration, consider possible adverse effects of the medication before ordering tests.
In this case … Our patient’s peculiar presentation correlated with regular ingestion of a high dose of zolpidem, a short-acting non-benzodiazepine gamma-aminobutyric acid (GABA) agonist. Zolpidem binds to the same GABAA receptor as benzodiazepines and therefore acts as a hypnotic by increasing GABA transmission.2 Neuropsychiatric adverse events associated with zolpidem include hallucinations, amnesia, parasomnia, psychomotor impairment, and complex behaviors (eg, sleepwalking or sleep-driving).2 Higher doses may cause coma or (rarely) death.2 One case report describes a patient who heard command hallucinations and stabbed himself after ingesting a large dose of zolpidem.3
Our patient
The patient’s episodes stopped after he discontinued the zolpidem. He subsequently received a correct prescription for lisinopril, and did not experience any additional episodes.
THE TAKEAWAY
Consider medication errors and adverse drug events in the differential diagnosis for patients who develop symptoms for which there is no obvious etiology. Educate patients, as well, to question their pharmacist if a recently filled prescription doesn’t look like the pill they usually take or makes them feel different than usual when they take it.
Of course, patients should be reminded that a generic medication may not always look the same as a brand-name drug or a previous generic prescription. But it can’t hurt for the patient to ask whether that medication that “looks different” is just a different generic—or a sign of a more worrisome mix-up.
1. Institute of Medicine. Preventing medication errors. Report Brief. July 2006. Institute of Medicine Web site. Available at: http://iom.edu/~/media/Files/Report%20Files/2006/Preventing-Medication-Errors-Quality-Chasm-Series/medicationerrorsnew.pdf. Accessed January 13, 2015.
2. Gunja N. The clinical and forensic toxicology of Z-drugs. J Med Toxicol. 2013;9:155-162.
3. Manfredi G, Kotzalidis GD, Lazanio S, et al. Command hallucinations with self-stabbing associated with zolpidem overdose. J Clin Psychiatry. 2010;71:92-93.
1. Institute of Medicine. Preventing medication errors. Report Brief. July 2006. Institute of Medicine Web site. Available at: http://iom.edu/~/media/Files/Report%20Files/2006/Preventing-Medication-Errors-Quality-Chasm-Series/medicationerrorsnew.pdf. Accessed January 13, 2015.
2. Gunja N. The clinical and forensic toxicology of Z-drugs. J Med Toxicol. 2013;9:155-162.
3. Manfredi G, Kotzalidis GD, Lazanio S, et al. Command hallucinations with self-stabbing associated with zolpidem overdose. J Clin Psychiatry. 2010;71:92-93.
