User login
Hot flashes and night sweats • amenorrhea • positive home pregnancy test • Dx?
THE CASE
A 25-year-old G2P2 woman came to our family practice clinic because she had multiple positive home pregnancy test results despite having undergone a sterilization procedure 4 years earlier. She said that 9 months ago, she had begun to experience hot flashes and night sweats that were getting progressively worse. Her menstrual cycles had been regular until 6 months earlier, when her bleeding became very light and irregular (2- to 6-week cycles with only one day of menstruation). Then 3 months ago, she stopped menstruating.
She’d had 2 uncomplicated pregnancies with normal vaginal deliveries 3 and 4 years ago, and had undergone a transcervical sterilization procedure after delivering her second child. Her medical history included hypothyroidism diagnosed at age 15, moderate persistent asthma, and seasonal allergies. She was taking levothyroxine 250 mcg/d, inhaled fluticasone/salmeterol, albuterol, and intranasal mometasone.
Transvaginal ultrasound failed to identify an intrauterine or ectopic pregnancy, and the patient’s ovaries were not visualized (uterine anatomy was normal with an endometrial stripe of 5.7 mm). The result of a serum human chorionic gonadotropin (hCG) test was 6.73 mIU/ mL. (In a nonpregnant, premenopausal woman, hCG is typically undetectable.) Subsequent serial hCG measurements remained low (6.72-7.09 mIU/mL), but persistent. Given these low hCG levels, it was imperative to rule out an intrauterine or ectopic pregnancy. A urine hCG was negative.
THE DIAGNOSIS
Because of our patient’s vasomotor symptoms, we ordered additional laboratory studies, which revealed an elevated follicle-stimulating hormone (FSH) level (66.08 mIU/mL and 42.2 mIU/mL taken one year apart; normal, 1.98-9.58 mIU/mL in a premenopausal female), an elevated luteinizing hormone (LH) level (46.1 mIU/mL; normal, 2.58-15.5 mIU/mL in a premenopausal female), a low thyroid-stimulating hormone (TSH) level (0.445 mIU/mL; normal, 0.465-4.65 mIU/mL), and a normal prolactin level (12.5 mIU/mL). Based on these results, we diagnosed primary ovarian insufficiency (POI).
DISCUSSION
POI, formerly known as premature ovarian failure, is defined as 4 to 6 months of amenorrhea or oligomenorrhea in a woman younger than 40 with an elevated FSH on 2 occasions, at least 4 weeks apart.1-3
The etiology of POI is broad. It can be caused by a failure of the pituitary gland or hypothalamus to secrete regulating hormones to stimulate the ovaries. Possible genetic causes include Turner’s syndrome, fragile X permutation, and other autosomal disorders that cause follicle dysfunction or destruction.1 Infections such as mumps, varicella, and tuberculosis are known to affect ovary function, as well.1,4 In addition, women who are exposed to chemotherapy or radiation are at higher risk for developing POI.1
Because POI and autoimmune disorders tend to occur together, consider screening any patient with POI for disorders such as hypothyroidism and Addison’s disease. A serum analysis to evaluate for autoantibodies against steroid-producing cells may be a potential marker for POI in patients with an autoimmune disease that affects the adrenal glands or thyroid. However, patients with isolated Addison’s disease, autoimmune hypothyroidism, or diabetes mellitus in the absence of POI do not appear to have steroid-specific antibodies.2 In our patient’s case, her hypothyroidism may have placed her at higher risk of having a second organ system adversely affected by her immune system.
What causes a false-positive pregnancy test? This case is unique because our patient reported multiple positive home pregnancy test results and had persistently low serum hCG levels. While she had symptoms that suggested menopause (hot flashes, oligomenorrhea that progressed to amenorrhea), she believed these symptoms were related to pregnancy. In addition to pregnancy, an elevated serum hCG measurement can be due to various malignancies, molar pregnancy, pituitary production of hCG, elevated LH, cross-reactivity with multiple animal exposures (due to the production of human anti-animal antibodies that react with testing), and recent mononucleosis infection.5
Other potential causes for false-positive urine pregnancy test results include tuboovarian abscess,6 adenomyosis,7 and cancers that produce hCG, such as colon, pancreatic, lung, liver, and urothelial bladder carcinoma.8,9 Urine with significant proteinuria can also cause a positive pregnancy test result.10
Our patient likely had a false-positive hCG due to elevated LH, secondary to POI, that demonstrated cross-reactivity on the hCG assay. The similarity in the chemical structure of the beta subunits of hCG and LH have been reported as false-positive tests in the absence of pregnancy.5
Because home pregnancy tests are designed to detect pregnancy as early as possible, they typically feature a high sensitivity by detecting very low levels of hCG, which leads to more frequent false-positive results. It is possible that different assay methods could account for the discrepancy between our patient’s positive home pregnancy tests and our negative laboratory urine pregnancy test.
Our patient and her husband were both counseled regarding her POI diagnosis. We conducted further studies to establish a possible etiology. She was found to have a normal karyotype of 46, XX, which ruled out Turner’s syndrome. Testing for permutations of the FMR1 gene was negative for fragile X syndrome, and antibody testing for thyroid and adrenal glands was negative for autoimmune disease.
Hormone therapy and supplemental calcium and vitamin D are recommended for women with POI to help prevent bone loss and other negative effects of low estrogen.11 We did not take this tack with our patient, however, because she decided she wanted to pursue a tubal ligation reversal in order to get pregnant. So instead, we decreased her dose of levothyroxine to 150 mcg (since her TSH was low) and we referred her to the Reproductive Endocrinology Department.
THE TAKEAWAY
Although many cases of POI have no discernible etiology, it is important to rule out malignancies, failure of pituitary production, genetic causes, infections, and other possible causes. Hormone therapy and prophylactic doses of calcium and vitamin D are recommended for patients diagnosed with POI.
1. Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf). 2008;68:499-509.
2. Betterle C, Rossi A, Dalla Pria S, et al. Premature ovarian failure: autoimmunity and natural history. Clin Endocrinol (Oxf). 1993;39:35-43.
3. Fox H. The pathology of premature ovarian failure. J Pathol. 1992;167:357-363.
4. Panay N, Kalu E. Management of premature ovarian failure. Best Practice & Research Clinical Obstetrics and Gynaecology. 2009;23;129-140.
5. Braunstein GD. False-positive serum human chorionic gonadotropin results: causes, characteristics, and recognition. Am J Obstet Gynecol. 2002;187:217-224.
6. Levsky ME, Handler JA, Suarez RD, et al. False-positive urine beta-HCG in a woman with a tubo-ovarian abscess. J Emerg Med. 2001;21:407-409.
7. Er TK, Chiang CH, Cheng BH, et al. False-positive urine pregnancy test in a woman with adenomysosis. Am J Emerg Med. 2009;27:1019.e5-7.
8. Rajabi B, Khoury J, Brewer C, et al. Urothelial bladder carcinoma with choriocarcinomatous differentiation presenting with a false-positive pregnancy test. Am J Med Sci. 2013;346:256-258.
9. Marcillac I, Troalen F, Bidart JM, et al. Free human chorionic gonadotropin beta subunit in gonadal and nongonadal neoplasms. Cancer Res. 1992;52:3901-3907.
10. Kountz DS, Kolander SA, Rozovsky A. False positive urinary pregnancy test in the nephrotic syndrome. N Engl J Med. 1989;321:1416.
11. National Institute of Health, National Institute of Child Health and Human Development. What are the treatments for POI? National Institute of Child Health and Human Development Web site. Available at: https://www.nichd.nih.gov/health/topics/poi/conditioninfo/Pages/treatments.aspx. Accessed August 5, 2015.
THE CASE
A 25-year-old G2P2 woman came to our family practice clinic because she had multiple positive home pregnancy test results despite having undergone a sterilization procedure 4 years earlier. She said that 9 months ago, she had begun to experience hot flashes and night sweats that were getting progressively worse. Her menstrual cycles had been regular until 6 months earlier, when her bleeding became very light and irregular (2- to 6-week cycles with only one day of menstruation). Then 3 months ago, she stopped menstruating.
She’d had 2 uncomplicated pregnancies with normal vaginal deliveries 3 and 4 years ago, and had undergone a transcervical sterilization procedure after delivering her second child. Her medical history included hypothyroidism diagnosed at age 15, moderate persistent asthma, and seasonal allergies. She was taking levothyroxine 250 mcg/d, inhaled fluticasone/salmeterol, albuterol, and intranasal mometasone.
Transvaginal ultrasound failed to identify an intrauterine or ectopic pregnancy, and the patient’s ovaries were not visualized (uterine anatomy was normal with an endometrial stripe of 5.7 mm). The result of a serum human chorionic gonadotropin (hCG) test was 6.73 mIU/ mL. (In a nonpregnant, premenopausal woman, hCG is typically undetectable.) Subsequent serial hCG measurements remained low (6.72-7.09 mIU/mL), but persistent. Given these low hCG levels, it was imperative to rule out an intrauterine or ectopic pregnancy. A urine hCG was negative.
THE DIAGNOSIS
Because of our patient’s vasomotor symptoms, we ordered additional laboratory studies, which revealed an elevated follicle-stimulating hormone (FSH) level (66.08 mIU/mL and 42.2 mIU/mL taken one year apart; normal, 1.98-9.58 mIU/mL in a premenopausal female), an elevated luteinizing hormone (LH) level (46.1 mIU/mL; normal, 2.58-15.5 mIU/mL in a premenopausal female), a low thyroid-stimulating hormone (TSH) level (0.445 mIU/mL; normal, 0.465-4.65 mIU/mL), and a normal prolactin level (12.5 mIU/mL). Based on these results, we diagnosed primary ovarian insufficiency (POI).
DISCUSSION
POI, formerly known as premature ovarian failure, is defined as 4 to 6 months of amenorrhea or oligomenorrhea in a woman younger than 40 with an elevated FSH on 2 occasions, at least 4 weeks apart.1-3
The etiology of POI is broad. It can be caused by a failure of the pituitary gland or hypothalamus to secrete regulating hormones to stimulate the ovaries. Possible genetic causes include Turner’s syndrome, fragile X permutation, and other autosomal disorders that cause follicle dysfunction or destruction.1 Infections such as mumps, varicella, and tuberculosis are known to affect ovary function, as well.1,4 In addition, women who are exposed to chemotherapy or radiation are at higher risk for developing POI.1
Because POI and autoimmune disorders tend to occur together, consider screening any patient with POI for disorders such as hypothyroidism and Addison’s disease. A serum analysis to evaluate for autoantibodies against steroid-producing cells may be a potential marker for POI in patients with an autoimmune disease that affects the adrenal glands or thyroid. However, patients with isolated Addison’s disease, autoimmune hypothyroidism, or diabetes mellitus in the absence of POI do not appear to have steroid-specific antibodies.2 In our patient’s case, her hypothyroidism may have placed her at higher risk of having a second organ system adversely affected by her immune system.
What causes a false-positive pregnancy test? This case is unique because our patient reported multiple positive home pregnancy test results and had persistently low serum hCG levels. While she had symptoms that suggested menopause (hot flashes, oligomenorrhea that progressed to amenorrhea), she believed these symptoms were related to pregnancy. In addition to pregnancy, an elevated serum hCG measurement can be due to various malignancies, molar pregnancy, pituitary production of hCG, elevated LH, cross-reactivity with multiple animal exposures (due to the production of human anti-animal antibodies that react with testing), and recent mononucleosis infection.5
Other potential causes for false-positive urine pregnancy test results include tuboovarian abscess,6 adenomyosis,7 and cancers that produce hCG, such as colon, pancreatic, lung, liver, and urothelial bladder carcinoma.8,9 Urine with significant proteinuria can also cause a positive pregnancy test result.10
Our patient likely had a false-positive hCG due to elevated LH, secondary to POI, that demonstrated cross-reactivity on the hCG assay. The similarity in the chemical structure of the beta subunits of hCG and LH have been reported as false-positive tests in the absence of pregnancy.5
Because home pregnancy tests are designed to detect pregnancy as early as possible, they typically feature a high sensitivity by detecting very low levels of hCG, which leads to more frequent false-positive results. It is possible that different assay methods could account for the discrepancy between our patient’s positive home pregnancy tests and our negative laboratory urine pregnancy test.
Our patient and her husband were both counseled regarding her POI diagnosis. We conducted further studies to establish a possible etiology. She was found to have a normal karyotype of 46, XX, which ruled out Turner’s syndrome. Testing for permutations of the FMR1 gene was negative for fragile X syndrome, and antibody testing for thyroid and adrenal glands was negative for autoimmune disease.
Hormone therapy and supplemental calcium and vitamin D are recommended for women with POI to help prevent bone loss and other negative effects of low estrogen.11 We did not take this tack with our patient, however, because she decided she wanted to pursue a tubal ligation reversal in order to get pregnant. So instead, we decreased her dose of levothyroxine to 150 mcg (since her TSH was low) and we referred her to the Reproductive Endocrinology Department.
THE TAKEAWAY
Although many cases of POI have no discernible etiology, it is important to rule out malignancies, failure of pituitary production, genetic causes, infections, and other possible causes. Hormone therapy and prophylactic doses of calcium and vitamin D are recommended for patients diagnosed with POI.
THE CASE
A 25-year-old G2P2 woman came to our family practice clinic because she had multiple positive home pregnancy test results despite having undergone a sterilization procedure 4 years earlier. She said that 9 months ago, she had begun to experience hot flashes and night sweats that were getting progressively worse. Her menstrual cycles had been regular until 6 months earlier, when her bleeding became very light and irregular (2- to 6-week cycles with only one day of menstruation). Then 3 months ago, she stopped menstruating.
She’d had 2 uncomplicated pregnancies with normal vaginal deliveries 3 and 4 years ago, and had undergone a transcervical sterilization procedure after delivering her second child. Her medical history included hypothyroidism diagnosed at age 15, moderate persistent asthma, and seasonal allergies. She was taking levothyroxine 250 mcg/d, inhaled fluticasone/salmeterol, albuterol, and intranasal mometasone.
Transvaginal ultrasound failed to identify an intrauterine or ectopic pregnancy, and the patient’s ovaries were not visualized (uterine anatomy was normal with an endometrial stripe of 5.7 mm). The result of a serum human chorionic gonadotropin (hCG) test was 6.73 mIU/ mL. (In a nonpregnant, premenopausal woman, hCG is typically undetectable.) Subsequent serial hCG measurements remained low (6.72-7.09 mIU/mL), but persistent. Given these low hCG levels, it was imperative to rule out an intrauterine or ectopic pregnancy. A urine hCG was negative.
THE DIAGNOSIS
Because of our patient’s vasomotor symptoms, we ordered additional laboratory studies, which revealed an elevated follicle-stimulating hormone (FSH) level (66.08 mIU/mL and 42.2 mIU/mL taken one year apart; normal, 1.98-9.58 mIU/mL in a premenopausal female), an elevated luteinizing hormone (LH) level (46.1 mIU/mL; normal, 2.58-15.5 mIU/mL in a premenopausal female), a low thyroid-stimulating hormone (TSH) level (0.445 mIU/mL; normal, 0.465-4.65 mIU/mL), and a normal prolactin level (12.5 mIU/mL). Based on these results, we diagnosed primary ovarian insufficiency (POI).
DISCUSSION
POI, formerly known as premature ovarian failure, is defined as 4 to 6 months of amenorrhea or oligomenorrhea in a woman younger than 40 with an elevated FSH on 2 occasions, at least 4 weeks apart.1-3
The etiology of POI is broad. It can be caused by a failure of the pituitary gland or hypothalamus to secrete regulating hormones to stimulate the ovaries. Possible genetic causes include Turner’s syndrome, fragile X permutation, and other autosomal disorders that cause follicle dysfunction or destruction.1 Infections such as mumps, varicella, and tuberculosis are known to affect ovary function, as well.1,4 In addition, women who are exposed to chemotherapy or radiation are at higher risk for developing POI.1
Because POI and autoimmune disorders tend to occur together, consider screening any patient with POI for disorders such as hypothyroidism and Addison’s disease. A serum analysis to evaluate for autoantibodies against steroid-producing cells may be a potential marker for POI in patients with an autoimmune disease that affects the adrenal glands or thyroid. However, patients with isolated Addison’s disease, autoimmune hypothyroidism, or diabetes mellitus in the absence of POI do not appear to have steroid-specific antibodies.2 In our patient’s case, her hypothyroidism may have placed her at higher risk of having a second organ system adversely affected by her immune system.
What causes a false-positive pregnancy test? This case is unique because our patient reported multiple positive home pregnancy test results and had persistently low serum hCG levels. While she had symptoms that suggested menopause (hot flashes, oligomenorrhea that progressed to amenorrhea), she believed these symptoms were related to pregnancy. In addition to pregnancy, an elevated serum hCG measurement can be due to various malignancies, molar pregnancy, pituitary production of hCG, elevated LH, cross-reactivity with multiple animal exposures (due to the production of human anti-animal antibodies that react with testing), and recent mononucleosis infection.5
Other potential causes for false-positive urine pregnancy test results include tuboovarian abscess,6 adenomyosis,7 and cancers that produce hCG, such as colon, pancreatic, lung, liver, and urothelial bladder carcinoma.8,9 Urine with significant proteinuria can also cause a positive pregnancy test result.10
Our patient likely had a false-positive hCG due to elevated LH, secondary to POI, that demonstrated cross-reactivity on the hCG assay. The similarity in the chemical structure of the beta subunits of hCG and LH have been reported as false-positive tests in the absence of pregnancy.5
Because home pregnancy tests are designed to detect pregnancy as early as possible, they typically feature a high sensitivity by detecting very low levels of hCG, which leads to more frequent false-positive results. It is possible that different assay methods could account for the discrepancy between our patient’s positive home pregnancy tests and our negative laboratory urine pregnancy test.
Our patient and her husband were both counseled regarding her POI diagnosis. We conducted further studies to establish a possible etiology. She was found to have a normal karyotype of 46, XX, which ruled out Turner’s syndrome. Testing for permutations of the FMR1 gene was negative for fragile X syndrome, and antibody testing for thyroid and adrenal glands was negative for autoimmune disease.
Hormone therapy and supplemental calcium and vitamin D are recommended for women with POI to help prevent bone loss and other negative effects of low estrogen.11 We did not take this tack with our patient, however, because she decided she wanted to pursue a tubal ligation reversal in order to get pregnant. So instead, we decreased her dose of levothyroxine to 150 mcg (since her TSH was low) and we referred her to the Reproductive Endocrinology Department.
THE TAKEAWAY
Although many cases of POI have no discernible etiology, it is important to rule out malignancies, failure of pituitary production, genetic causes, infections, and other possible causes. Hormone therapy and prophylactic doses of calcium and vitamin D are recommended for patients diagnosed with POI.
1. Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf). 2008;68:499-509.
2. Betterle C, Rossi A, Dalla Pria S, et al. Premature ovarian failure: autoimmunity and natural history. Clin Endocrinol (Oxf). 1993;39:35-43.
3. Fox H. The pathology of premature ovarian failure. J Pathol. 1992;167:357-363.
4. Panay N, Kalu E. Management of premature ovarian failure. Best Practice & Research Clinical Obstetrics and Gynaecology. 2009;23;129-140.
5. Braunstein GD. False-positive serum human chorionic gonadotropin results: causes, characteristics, and recognition. Am J Obstet Gynecol. 2002;187:217-224.
6. Levsky ME, Handler JA, Suarez RD, et al. False-positive urine beta-HCG in a woman with a tubo-ovarian abscess. J Emerg Med. 2001;21:407-409.
7. Er TK, Chiang CH, Cheng BH, et al. False-positive urine pregnancy test in a woman with adenomysosis. Am J Emerg Med. 2009;27:1019.e5-7.
8. Rajabi B, Khoury J, Brewer C, et al. Urothelial bladder carcinoma with choriocarcinomatous differentiation presenting with a false-positive pregnancy test. Am J Med Sci. 2013;346:256-258.
9. Marcillac I, Troalen F, Bidart JM, et al. Free human chorionic gonadotropin beta subunit in gonadal and nongonadal neoplasms. Cancer Res. 1992;52:3901-3907.
10. Kountz DS, Kolander SA, Rozovsky A. False positive urinary pregnancy test in the nephrotic syndrome. N Engl J Med. 1989;321:1416.
11. National Institute of Health, National Institute of Child Health and Human Development. What are the treatments for POI? National Institute of Child Health and Human Development Web site. Available at: https://www.nichd.nih.gov/health/topics/poi/conditioninfo/Pages/treatments.aspx. Accessed August 5, 2015.
1. Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol (Oxf). 2008;68:499-509.
2. Betterle C, Rossi A, Dalla Pria S, et al. Premature ovarian failure: autoimmunity and natural history. Clin Endocrinol (Oxf). 1993;39:35-43.
3. Fox H. The pathology of premature ovarian failure. J Pathol. 1992;167:357-363.
4. Panay N, Kalu E. Management of premature ovarian failure. Best Practice & Research Clinical Obstetrics and Gynaecology. 2009;23;129-140.
5. Braunstein GD. False-positive serum human chorionic gonadotropin results: causes, characteristics, and recognition. Am J Obstet Gynecol. 2002;187:217-224.
6. Levsky ME, Handler JA, Suarez RD, et al. False-positive urine beta-HCG in a woman with a tubo-ovarian abscess. J Emerg Med. 2001;21:407-409.
7. Er TK, Chiang CH, Cheng BH, et al. False-positive urine pregnancy test in a woman with adenomysosis. Am J Emerg Med. 2009;27:1019.e5-7.
8. Rajabi B, Khoury J, Brewer C, et al. Urothelial bladder carcinoma with choriocarcinomatous differentiation presenting with a false-positive pregnancy test. Am J Med Sci. 2013;346:256-258.
9. Marcillac I, Troalen F, Bidart JM, et al. Free human chorionic gonadotropin beta subunit in gonadal and nongonadal neoplasms. Cancer Res. 1992;52:3901-3907.
10. Kountz DS, Kolander SA, Rozovsky A. False positive urinary pregnancy test in the nephrotic syndrome. N Engl J Med. 1989;321:1416.
11. National Institute of Health, National Institute of Child Health and Human Development. What are the treatments for POI? National Institute of Child Health and Human Development Web site. Available at: https://www.nichd.nih.gov/health/topics/poi/conditioninfo/Pages/treatments.aspx. Accessed August 5, 2015.

Case Report: A Bittersweet Death
Case
A 32-year-old Hispanic man presented to the ED with complications associated with diabetes mellitus (DM), the symptoms of which started approximately 3 days prior to arrival. The patient reported feelings of fatigue, dry mouth, increased thirst, and frequent urination. He denied sweating, nausea, chest pain, shortness of breath, diarrhea, or blood in his urine; he also denied blurry vision or dizziness.
During history intake, the patient informed the emergency physician (EP) that he had been diagnosed with DM and hyperglycemia earlier that day by his primary care physician, who had immediately referred the patient to the ED for urgent management. The patient’s own medical history was noncontributory; however, his father’s history was notable for DM and chronic renal failure. The patient further stated that he was not on any medications. Regarding his social history, he denied cigarette smoking and noted only occasional alcohol consumption.
The patient’s vital signs on presentation were: blood pressure (BP), 116/74 mm Hg; heart rate, 113 beats/minute; respiratory rate, 26 breaths/minute; and temperature, 97.8°F. Oxygen saturation was 97% on room air. On physical examination, the patient was severely anxious, with tachycardia and respiratory distress. He was obese, with a body mass index of 30.9 kg/m2 (height, 5 feet, 4 inches; weight, 180 lb).
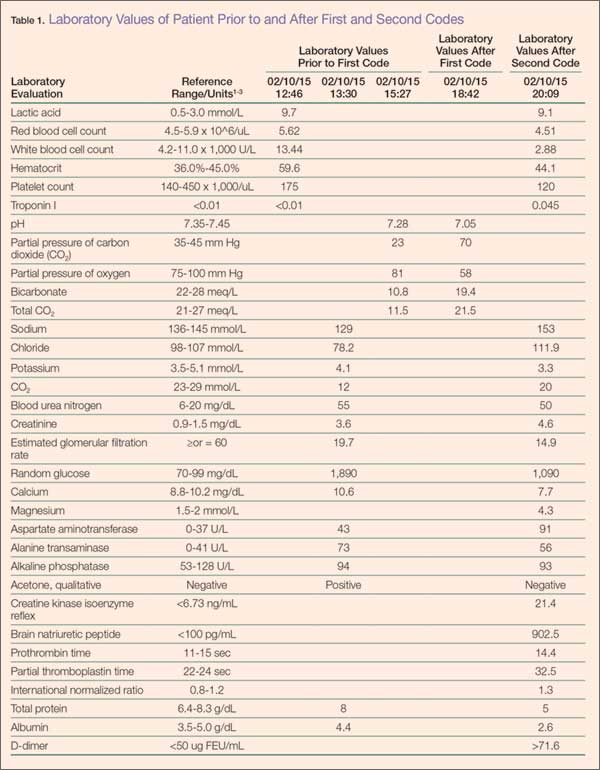
The patient was started on an intravenous (IV) bolus of 0.9% normal saline (2 L at 20 mL/kg). After a consultation with endocrinology, he was then given a maintenance dose of normal saline IV at 250 cc/h and an IV insulin drip at 0.1 U/kg/h following a bolus of 8 units of insulin IV. His glucose levels were carefully monitored via hourly finger-stick glucose testing.
Although the patient’s condition stabilized, he collapsed while walking to the bathroom. He had agonal respirations and no pulse. Resuscitation efforts were started with bag-valve-mask ventilation, along with emergent advanced cardiac life support (ACLS) treatment, the protocol of which included epinephrine administration (x2) IV push 5 minutes apart, 2 ampules of sodium bicarbonate (50 mEq each) IV push, and calcium gluconate 10% (x1) 10 mL (1 g) IV push. A pulse was re-established, and the patient was intubated.
The patient was diagnosed with diabetic ketoacidosis (DKA) and admitted to the intensive care unit where repeat laboratory evaluation was ordered. Additional pharmacological management included IV administration of dopamine, norepinephrine, phenylephrine, vasopressin, antibiotics (azithromycin, meropenem, and vancomycin), pantoprazole, and subcutaneous heparin.
During treatment, the patient coded a second time and was revived according to ACLS protocols. Shortly thereafter, he coded a third time, but resuscitation efforts failed. Pathology reported no biological cause of death, and the coroner closed the case as death due to DM-related complications.
Diabetic Ketoacidosis
Diabetic ketoacidosis is a major complication of DM.4 Although the condition usually occurs in type 1 DM, it can also develop in type 2 DM. Diabetic ketoacidosis may be an inciting event leading to the eventual diagnosis of DM, but can also develop during a concurrent illness such as a urinary tract infection or an eating disorder.5 Risk factors for DKA include patients with type 1 or type 2 DM, a family history of DM, obesity, and nonwhite patients whose ethnic background places them at increased risk.6 Hispanic, black, and African American patients are at a greater risk of developing DKA and are more likely to develop “ketosis-prone” type 2 DM.7
Patients who do not fit into the definitive categories of type 1 or 2 DM can be classified under ketosis-prone DM.7,8 Diabetic ketoacidosis acts as the inciting event for the disease and evolves into severe β-cell dysfunction, hence blurring the lines between the archetypal DM categories. Fifty percent of ketosis-prone DM patients are A-β+ (absent autoantibodies, present β-cell function), which indicates that the dysfunction can be partially reversed. Reversal of the condition is largely based on long-term β-cell reserves, which are dependent on tight glycemic control and insulin dependence. Higher incidences of the A-β+ variant of ketosis-prone diabetes are seen in the male population and are often unprovoked.9-11
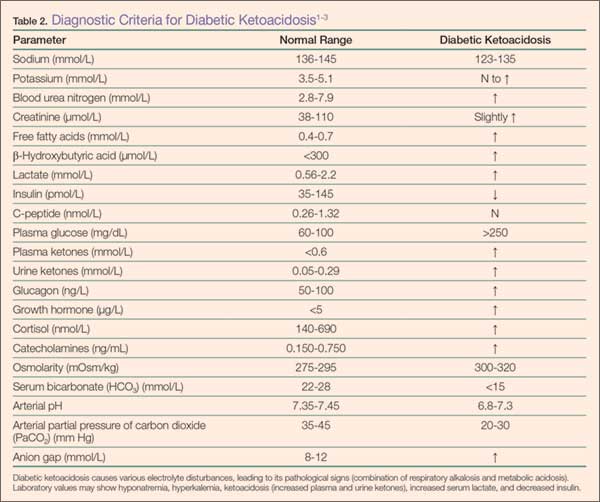
Diabetic ketoacidosis is the result of either a decrease or absence of insulin in the body (Table 2).4 Without insulin modulating exogenous glucose intake and endogenous glucose production (via glucagon, glycogenolysis, and gluconeogenesis), high levels of glucose are found in the circulation, leading to prominent hyperglycemia (>250 mg/dL or >13.8 mmol/L).6 This environment causes the body to switch from carbohydrate metabolism to fatty acid metabolism. As a result, acidic ketone bodies such β-hydroxybutyrate and acetoacetate are produced. These physiological changes in the body cause the signs and symptoms typically found in DKA.
Signs and Symptoms
Over a period of 24 hours, symptoms such as nausea, vomiting, increased thirst, and polyuria develop due to dehydration caused by osmotic diuresis and glucosuria.5 Patients may also present with hypotension and tachycardia. Confusion, deep gasping breaths or Kussmaul respirations, and metabolic acidosis result from hyperventilation and failure to compensate for the increased serum concentration of ketone bodies. Ketone production leads to a fruit-like odor in the patient’s breath and ketonuria in the urinalysis. In DKA, laboratory values will indicate metabolic acidosis and abnormal serum electrolytes. In both DM and DKA, increased urea and creatinine due to dehydration, increased ketones, and the presence of diabetic nephropathy are useful indicators of impaired kidney function.12
Management and Treatment
Diabetic ketoacidosis can be managed and reversed, especially when recognized and treated early.6,13 Dehydration in DKA can be corrected with IV fluid replacement. Normal saline (0.9%) can be started at 15 to 20 mL/kg/h or 1 L/h. As the patient’s vital signs stabilize, IV fluids can be titrated to a lower dose of 250 to 500 mL/h. Monitoring BP and electrolytes are key at this point as alterations in sodium levels and glucose levels may require switching to half-normal saline and/or dextrose.
The hyperglycemic state of patients with DKA is managed by IV insulin. An initial bolus of 0.1 U/kg/h can be given, but should only be administered when potassium levels are greater than 3.3 mmol/L.14 If adequate perfusion can be maintained, then 0.14 U/kg/h can be used instead of a bolus. Glucose levels must be monitored; once the levels decrease to approximately 200 mg/dL, the infusion rate of insulin should be titrated down to 0.05 to 0.1 U/kg/h. Dextrose is then added to maintain glucose levels at approximately 150 to 200 mg/dL.
Electrolytes, especially potassium, must be monitored closely in patients with DKA. Insulin leads to the shift of potassium into cells. The lack of insulin keeps potassium in the extracellular space. Due to osmotic diuresis, potassium is lost in the urine, leading to hypokalemia. Potassium levels in patients with DKA should be maintained at a level between 4 to 5 mmol/L. Patients with potassium levels between 3.3 to 5.2 mmol/L can be started on IV potassium between 20 to 30 mmol/h. If the patient is severely hypokalemic (<3.3 mmol/L), insulin should be withheld, and only IV potassium should be given at a rate of 20 to 30 mmol/h.
Bicarbonate levels can also be managed as acidosis can lead to both neurological and cardiac complications. If the patient’s pH is less than 6.9, the American Diabetes Association recommends starting 100 mmol of sodium bicarbonate in 400 mL sterile water (in addition to potassium chloride at 200 mL/h) for 2 hours. Dosing should be repeated every 2 hours until the patient’s pH is greater than 6.9.
In uncomplicated cases of DKA, the condition is resolved when a patient’s pH is greater than 7.3; glucose level is less than 200 mg/dL; and bicarbonate level is greater than or equal to 18 mmol/L. After patients become hemodynamically stable, they can be discharged and managed at home with a combination of intermediate- or long-acting insulin as well as short- or rapid-acting insulin.
Complications and Mortality
Diabetic ketoacidosis can cause sudden and fluctuating changes in the body. Therefore, it is very important to monitor a patient’s laboratory values very carefully and frequently to avoid any pitfalls. Since patients can present with hyponatremia due to the osmotic draw of glucose in the blood,13 sodium levels may have to be corrected. The corrected serum sodium can be calculated by adding 1.6 mmol/L for every 100 mg/dL of glucose (when finger-stick readings are above 200 mg/dL).15 Patients with DKA can also present with leukocytosis (even in the absence of infection) and hypertriglyceridemia (due to impaired lipoprotein lipase).15 Serum creatine may be elevated due to blood acetoacetate levels.15
Interestingly, there are other acute conditions that can mimic DKA.15 For example, chronic ethanol abuse can lead to ketoacidosis. Unlike DKA, however, alcoholic ketoacidosis does not have profound hyperglycemia, which can help differentiate the two during initial assessment.
Complications due to DKA can arise comprising the patient’s health, including hypoglycemia, hypokalemia, rhabdomyolysis, acute renal failure, pulmonary edema, and shock.16 Cerebral edema is seen in up to 1% of DKA patients,15 the cause of which may be due to the severity of the acidosis, high glucose levels, and rapid hydration. Even when cerebral edema is reduced, patients are often neurologically impaired. Mortality rates from DKA deaths due to cerebral edema can be as high as 24%.13 In the United States, over 100,000 patients with DM per year are admitted to the hospital for DKA, and 9% of patients with DM suffer from DKA-related complications postdischarge.15 With current treatment protocols, mortality rates for DKA-associated deaths are now down to 1%.6,15
Diabetes ketoacidosis-related deaths are usually the result of the following: a triad of DKA symptoms (hyperglycemia, hyperketonemia, and metabolic acidosis), another underlying comorbid condition (eg, myocardial infarction, sepsis, acute respiratory distress syndrome), or the release of biological markers (ie, catecholamines).14,15,17 Thus, as previously stated, the management of potassium levels is important as both hyperkalemia and hypokalemia can lead to fatal arrthythmias.15
Direct mortality from DKA has dropped significantly over the past 20 years, from 8% to less than 1%.6 The US Centers for Disease Control and Prevention has observed a downward trend in death and estimates that 2,417 patients died in 2009 due to DKA,18 and recent postmortem studies have revealed new insights into DKA-related deaths.19 Blood and vitreous acetone concentrations are strong indicators for predeath hyperglycemia and ketosis (if there are no underlying comorbid and/or pharmacological provocations). Blood acetone levels greater than 0.01 g/dL antemortem are suggestive of DKA. It is recommended that these tests should be performed in sudden deaths which have no biological or anatomical cause of death. Postmortem diagnosis of DKA is made with the following criteria: history of DM, increased vitreous glucose concentrations, and elevated blood/vitreous/urine acetone concentrations (>200 mg/dL). If results of the abovementioned parameters are inconclusive, measurement of lactic acid postmortem is thought to further support a diagnosis of DKA.19
Patient Counseling and Education
Approximately 33% of patients whose death was associated with DKA had no personal history of DM.19 This statistic emphasizes the importance of taking a thorough history, physical examination, blood glucose evaluation, and educating patients about the signs and symptoms of DM and DKA.
Patient counseling and education are important, especially in patients whose racial/ethnic background places them at increased risk of developing DM (eg, patients of black or African American, American Indian, Alaskan Native, Asian American, Hispanic, Native Hawaiian, or Pacific Islander descent).20,21 Strategies for preventive management include advocating regular glucose monitoring as well as dietary and lifestyle modifications. In patients with DM, successful management of the condition and its comorbidities can help prevent DKA and associated mortality.
Conclusion
As this case demonstrates, despite prompt diagnosis and management, patients with DKA—especially those with uncontrolled, undiagnosed, or advanced DM—are associated with fatal outcomes. In many cases, however, DKA can be successfully managed and reversed, especially when the condition is recognized early. Management includes not only IV therapy to adjust fluid and insulin levels, but also restoring electrolyte balance (especially potassium and bicarbonate). Frequent and careful evaluation of laboratory values is vital to the successful treatment of DKA, as there are numerous pitfalls and complications that the emergency physician can encounter. Patients who either have or are at an increased risk of developing DM or DKA may benefit from preventive measures, including regular glucose monitoring and appropriate diet and lifestyle modifications.
Mr Hassan-Ali is a fourth-year medical student at Windsor University School of Medicine, St Kitts, West Indies. Dr Raziuddin is an internist and an emergency medicine physician at Weiss Memorial, Thorek Memorial, and Westlake Hospitals, Chicago, Illinois.
- Kitabchi AH, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24(1):131-153.
- Farinda A. Lab values, normal adult: laboratory reference ranges in healthy adults. 2015. Medscape Web site. http://emedicine.medscape.com/article/2172316-overview. Updated May 14, 2014. Accessed August 14, 2015.
- Young D. Implementation of SI units for clinical laboratory data. Ann Intern Med. 1987;106(1):114-129.
- Maitra A. The endocrine system. In: Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th ed. New York, NY: Elsevier Saunders; 2015:1105-1120.
- Powers AC. Diabetes mellitus: management and therapies. In: Kasper DL, Fauci AS, Longo DL, Hauser SL, Jameson JL, Loscalzo J. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY; McGraw-Hill Medical Publishing Division; 2015:2407-2422.
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343.
- Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann Intern Med. 2006;144(5):350-357.
- Umpierrez G, Smiley D, Gosmanov A, Thomason D. Ketosis-prone type 2 diabetes: effect of hyperglycemia on beta-cell function and skeletal muscle insulin signaling. Endocr Pract. 2007;13(3):283-290.
- Mauvais-Jarvis F, Sobngwi E, Porcher R, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes. 2004;53(3):645-653.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes. 1995;44(7):790-795.
- Piñero-Piloña A, Raskin P. Idiopathic type 1 diabetes. J Diabetes Complications. 2001;15(6):328-335.
- Kemperman FA, Weber JA, Gorgels J, van Zanten AP, Krediet RT, Arisz L. The influence of ketoacids on plasma creatinine assays in diabetic ketoacidosis. J Intern Med. 2000;248(6):511-517.
- Westerberg DP. Diabetic ketoacidosis: evaluation and treatment. Am Fam Physician. 2013;87(5):337-346.
- Trachtenbarg DE. Diabetic ketoacidosis. Am Fam Physician. 2005;71(9):1705-1714.
- Umpierrez GE, Murphy MB, Kitabchi AE. Diabetic ketoacidosis and hyperglycemic hyperosmolar ayndrome. Diabetes Spectrum. 2002;15(1):28-36.
- Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: A consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150-1159.
- Rosenbloom AL. Sudden death of a young woman attributed to diabetic ketoacidosis. J Forensic Leg Med. 2013;20(8):1063-1065.
- Centers for Disease Control and Prevention. Number of deaths for hyperglycemic crises as underlying cause, United States, 1980-2009. http://www.cdc.gov/diabetes/statistics/mortalitydka/fnumberofdka.htm. Updated November 19, 2013. Accessed August 14, 2015.
- Ali Z, Levine B, Ripple M, Fowler DR. Diabetic ketoacidosis: a silent death. Am J Forensic Med Pathol. 2012;33(3):189-193.
- US Department of Health and Human Services Office of Minority Health. Diabetes and Hispanic Americans. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=4&lvlid=63. Updated June 15, 2013. Accessed August 14, 2015.
- US Department of Health and Human Services Office of Minority Health. Profile: Native Hawaiian/Other Pacific Islanders. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=3&lvlid=65. Updated January 15, 2015. Accessed August 14, 2015.
Case
A 32-year-old Hispanic man presented to the ED with complications associated with diabetes mellitus (DM), the symptoms of which started approximately 3 days prior to arrival. The patient reported feelings of fatigue, dry mouth, increased thirst, and frequent urination. He denied sweating, nausea, chest pain, shortness of breath, diarrhea, or blood in his urine; he also denied blurry vision or dizziness.
During history intake, the patient informed the emergency physician (EP) that he had been diagnosed with DM and hyperglycemia earlier that day by his primary care physician, who had immediately referred the patient to the ED for urgent management. The patient’s own medical history was noncontributory; however, his father’s history was notable for DM and chronic renal failure. The patient further stated that he was not on any medications. Regarding his social history, he denied cigarette smoking and noted only occasional alcohol consumption.
The patient’s vital signs on presentation were: blood pressure (BP), 116/74 mm Hg; heart rate, 113 beats/minute; respiratory rate, 26 breaths/minute; and temperature, 97.8°F. Oxygen saturation was 97% on room air. On physical examination, the patient was severely anxious, with tachycardia and respiratory distress. He was obese, with a body mass index of 30.9 kg/m2 (height, 5 feet, 4 inches; weight, 180 lb).
The patient was started on an intravenous (IV) bolus of 0.9% normal saline (2 L at 20 mL/kg). After a consultation with endocrinology, he was then given a maintenance dose of normal saline IV at 250 cc/h and an IV insulin drip at 0.1 U/kg/h following a bolus of 8 units of insulin IV. His glucose levels were carefully monitored via hourly finger-stick glucose testing.
Although the patient’s condition stabilized, he collapsed while walking to the bathroom. He had agonal respirations and no pulse. Resuscitation efforts were started with bag-valve-mask ventilation, along with emergent advanced cardiac life support (ACLS) treatment, the protocol of which included epinephrine administration (x2) IV push 5 minutes apart, 2 ampules of sodium bicarbonate (50 mEq each) IV push, and calcium gluconate 10% (x1) 10 mL (1 g) IV push. A pulse was re-established, and the patient was intubated.
The patient was diagnosed with diabetic ketoacidosis (DKA) and admitted to the intensive care unit where repeat laboratory evaluation was ordered. Additional pharmacological management included IV administration of dopamine, norepinephrine, phenylephrine, vasopressin, antibiotics (azithromycin, meropenem, and vancomycin), pantoprazole, and subcutaneous heparin.
During treatment, the patient coded a second time and was revived according to ACLS protocols. Shortly thereafter, he coded a third time, but resuscitation efforts failed. Pathology reported no biological cause of death, and the coroner closed the case as death due to DM-related complications.
Diabetic Ketoacidosis
Diabetic ketoacidosis is a major complication of DM.4 Although the condition usually occurs in type 1 DM, it can also develop in type 2 DM. Diabetic ketoacidosis may be an inciting event leading to the eventual diagnosis of DM, but can also develop during a concurrent illness such as a urinary tract infection or an eating disorder.5 Risk factors for DKA include patients with type 1 or type 2 DM, a family history of DM, obesity, and nonwhite patients whose ethnic background places them at increased risk.6 Hispanic, black, and African American patients are at a greater risk of developing DKA and are more likely to develop “ketosis-prone” type 2 DM.7
Patients who do not fit into the definitive categories of type 1 or 2 DM can be classified under ketosis-prone DM.7,8 Diabetic ketoacidosis acts as the inciting event for the disease and evolves into severe β-cell dysfunction, hence blurring the lines between the archetypal DM categories. Fifty percent of ketosis-prone DM patients are A-β+ (absent autoantibodies, present β-cell function), which indicates that the dysfunction can be partially reversed. Reversal of the condition is largely based on long-term β-cell reserves, which are dependent on tight glycemic control and insulin dependence. Higher incidences of the A-β+ variant of ketosis-prone diabetes are seen in the male population and are often unprovoked.9-11
Diabetic ketoacidosis is the result of either a decrease or absence of insulin in the body (Table 2).4 Without insulin modulating exogenous glucose intake and endogenous glucose production (via glucagon, glycogenolysis, and gluconeogenesis), high levels of glucose are found in the circulation, leading to prominent hyperglycemia (>250 mg/dL or >13.8 mmol/L).6 This environment causes the body to switch from carbohydrate metabolism to fatty acid metabolism. As a result, acidic ketone bodies such β-hydroxybutyrate and acetoacetate are produced. These physiological changes in the body cause the signs and symptoms typically found in DKA.
Signs and Symptoms
Over a period of 24 hours, symptoms such as nausea, vomiting, increased thirst, and polyuria develop due to dehydration caused by osmotic diuresis and glucosuria.5 Patients may also present with hypotension and tachycardia. Confusion, deep gasping breaths or Kussmaul respirations, and metabolic acidosis result from hyperventilation and failure to compensate for the increased serum concentration of ketone bodies. Ketone production leads to a fruit-like odor in the patient’s breath and ketonuria in the urinalysis. In DKA, laboratory values will indicate metabolic acidosis and abnormal serum electrolytes. In both DM and DKA, increased urea and creatinine due to dehydration, increased ketones, and the presence of diabetic nephropathy are useful indicators of impaired kidney function.12
Management and Treatment
Diabetic ketoacidosis can be managed and reversed, especially when recognized and treated early.6,13 Dehydration in DKA can be corrected with IV fluid replacement. Normal saline (0.9%) can be started at 15 to 20 mL/kg/h or 1 L/h. As the patient’s vital signs stabilize, IV fluids can be titrated to a lower dose of 250 to 500 mL/h. Monitoring BP and electrolytes are key at this point as alterations in sodium levels and glucose levels may require switching to half-normal saline and/or dextrose.
The hyperglycemic state of patients with DKA is managed by IV insulin. An initial bolus of 0.1 U/kg/h can be given, but should only be administered when potassium levels are greater than 3.3 mmol/L.14 If adequate perfusion can be maintained, then 0.14 U/kg/h can be used instead of a bolus. Glucose levels must be monitored; once the levels decrease to approximately 200 mg/dL, the infusion rate of insulin should be titrated down to 0.05 to 0.1 U/kg/h. Dextrose is then added to maintain glucose levels at approximately 150 to 200 mg/dL.
Electrolytes, especially potassium, must be monitored closely in patients with DKA. Insulin leads to the shift of potassium into cells. The lack of insulin keeps potassium in the extracellular space. Due to osmotic diuresis, potassium is lost in the urine, leading to hypokalemia. Potassium levels in patients with DKA should be maintained at a level between 4 to 5 mmol/L. Patients with potassium levels between 3.3 to 5.2 mmol/L can be started on IV potassium between 20 to 30 mmol/h. If the patient is severely hypokalemic (<3.3 mmol/L), insulin should be withheld, and only IV potassium should be given at a rate of 20 to 30 mmol/h.
Bicarbonate levels can also be managed as acidosis can lead to both neurological and cardiac complications. If the patient’s pH is less than 6.9, the American Diabetes Association recommends starting 100 mmol of sodium bicarbonate in 400 mL sterile water (in addition to potassium chloride at 200 mL/h) for 2 hours. Dosing should be repeated every 2 hours until the patient’s pH is greater than 6.9.
In uncomplicated cases of DKA, the condition is resolved when a patient’s pH is greater than 7.3; glucose level is less than 200 mg/dL; and bicarbonate level is greater than or equal to 18 mmol/L. After patients become hemodynamically stable, they can be discharged and managed at home with a combination of intermediate- or long-acting insulin as well as short- or rapid-acting insulin.
Complications and Mortality
Diabetic ketoacidosis can cause sudden and fluctuating changes in the body. Therefore, it is very important to monitor a patient’s laboratory values very carefully and frequently to avoid any pitfalls. Since patients can present with hyponatremia due to the osmotic draw of glucose in the blood,13 sodium levels may have to be corrected. The corrected serum sodium can be calculated by adding 1.6 mmol/L for every 100 mg/dL of glucose (when finger-stick readings are above 200 mg/dL).15 Patients with DKA can also present with leukocytosis (even in the absence of infection) and hypertriglyceridemia (due to impaired lipoprotein lipase).15 Serum creatine may be elevated due to blood acetoacetate levels.15
Interestingly, there are other acute conditions that can mimic DKA.15 For example, chronic ethanol abuse can lead to ketoacidosis. Unlike DKA, however, alcoholic ketoacidosis does not have profound hyperglycemia, which can help differentiate the two during initial assessment.
Complications due to DKA can arise comprising the patient’s health, including hypoglycemia, hypokalemia, rhabdomyolysis, acute renal failure, pulmonary edema, and shock.16 Cerebral edema is seen in up to 1% of DKA patients,15 the cause of which may be due to the severity of the acidosis, high glucose levels, and rapid hydration. Even when cerebral edema is reduced, patients are often neurologically impaired. Mortality rates from DKA deaths due to cerebral edema can be as high as 24%.13 In the United States, over 100,000 patients with DM per year are admitted to the hospital for DKA, and 9% of patients with DM suffer from DKA-related complications postdischarge.15 With current treatment protocols, mortality rates for DKA-associated deaths are now down to 1%.6,15
Diabetes ketoacidosis-related deaths are usually the result of the following: a triad of DKA symptoms (hyperglycemia, hyperketonemia, and metabolic acidosis), another underlying comorbid condition (eg, myocardial infarction, sepsis, acute respiratory distress syndrome), or the release of biological markers (ie, catecholamines).14,15,17 Thus, as previously stated, the management of potassium levels is important as both hyperkalemia and hypokalemia can lead to fatal arrthythmias.15
Direct mortality from DKA has dropped significantly over the past 20 years, from 8% to less than 1%.6 The US Centers for Disease Control and Prevention has observed a downward trend in death and estimates that 2,417 patients died in 2009 due to DKA,18 and recent postmortem studies have revealed new insights into DKA-related deaths.19 Blood and vitreous acetone concentrations are strong indicators for predeath hyperglycemia and ketosis (if there are no underlying comorbid and/or pharmacological provocations). Blood acetone levels greater than 0.01 g/dL antemortem are suggestive of DKA. It is recommended that these tests should be performed in sudden deaths which have no biological or anatomical cause of death. Postmortem diagnosis of DKA is made with the following criteria: history of DM, increased vitreous glucose concentrations, and elevated blood/vitreous/urine acetone concentrations (>200 mg/dL). If results of the abovementioned parameters are inconclusive, measurement of lactic acid postmortem is thought to further support a diagnosis of DKA.19
Patient Counseling and Education
Approximately 33% of patients whose death was associated with DKA had no personal history of DM.19 This statistic emphasizes the importance of taking a thorough history, physical examination, blood glucose evaluation, and educating patients about the signs and symptoms of DM and DKA.
Patient counseling and education are important, especially in patients whose racial/ethnic background places them at increased risk of developing DM (eg, patients of black or African American, American Indian, Alaskan Native, Asian American, Hispanic, Native Hawaiian, or Pacific Islander descent).20,21 Strategies for preventive management include advocating regular glucose monitoring as well as dietary and lifestyle modifications. In patients with DM, successful management of the condition and its comorbidities can help prevent DKA and associated mortality.
Conclusion
As this case demonstrates, despite prompt diagnosis and management, patients with DKA—especially those with uncontrolled, undiagnosed, or advanced DM—are associated with fatal outcomes. In many cases, however, DKA can be successfully managed and reversed, especially when the condition is recognized early. Management includes not only IV therapy to adjust fluid and insulin levels, but also restoring electrolyte balance (especially potassium and bicarbonate). Frequent and careful evaluation of laboratory values is vital to the successful treatment of DKA, as there are numerous pitfalls and complications that the emergency physician can encounter. Patients who either have or are at an increased risk of developing DM or DKA may benefit from preventive measures, including regular glucose monitoring and appropriate diet and lifestyle modifications.
Mr Hassan-Ali is a fourth-year medical student at Windsor University School of Medicine, St Kitts, West Indies. Dr Raziuddin is an internist and an emergency medicine physician at Weiss Memorial, Thorek Memorial, and Westlake Hospitals, Chicago, Illinois.
Case
A 32-year-old Hispanic man presented to the ED with complications associated with diabetes mellitus (DM), the symptoms of which started approximately 3 days prior to arrival. The patient reported feelings of fatigue, dry mouth, increased thirst, and frequent urination. He denied sweating, nausea, chest pain, shortness of breath, diarrhea, or blood in his urine; he also denied blurry vision or dizziness.
During history intake, the patient informed the emergency physician (EP) that he had been diagnosed with DM and hyperglycemia earlier that day by his primary care physician, who had immediately referred the patient to the ED for urgent management. The patient’s own medical history was noncontributory; however, his father’s history was notable for DM and chronic renal failure. The patient further stated that he was not on any medications. Regarding his social history, he denied cigarette smoking and noted only occasional alcohol consumption.
The patient’s vital signs on presentation were: blood pressure (BP), 116/74 mm Hg; heart rate, 113 beats/minute; respiratory rate, 26 breaths/minute; and temperature, 97.8°F. Oxygen saturation was 97% on room air. On physical examination, the patient was severely anxious, with tachycardia and respiratory distress. He was obese, with a body mass index of 30.9 kg/m2 (height, 5 feet, 4 inches; weight, 180 lb).
The patient was started on an intravenous (IV) bolus of 0.9% normal saline (2 L at 20 mL/kg). After a consultation with endocrinology, he was then given a maintenance dose of normal saline IV at 250 cc/h and an IV insulin drip at 0.1 U/kg/h following a bolus of 8 units of insulin IV. His glucose levels were carefully monitored via hourly finger-stick glucose testing.
Although the patient’s condition stabilized, he collapsed while walking to the bathroom. He had agonal respirations and no pulse. Resuscitation efforts were started with bag-valve-mask ventilation, along with emergent advanced cardiac life support (ACLS) treatment, the protocol of which included epinephrine administration (x2) IV push 5 minutes apart, 2 ampules of sodium bicarbonate (50 mEq each) IV push, and calcium gluconate 10% (x1) 10 mL (1 g) IV push. A pulse was re-established, and the patient was intubated.
The patient was diagnosed with diabetic ketoacidosis (DKA) and admitted to the intensive care unit where repeat laboratory evaluation was ordered. Additional pharmacological management included IV administration of dopamine, norepinephrine, phenylephrine, vasopressin, antibiotics (azithromycin, meropenem, and vancomycin), pantoprazole, and subcutaneous heparin.
During treatment, the patient coded a second time and was revived according to ACLS protocols. Shortly thereafter, he coded a third time, but resuscitation efforts failed. Pathology reported no biological cause of death, and the coroner closed the case as death due to DM-related complications.
Diabetic Ketoacidosis
Diabetic ketoacidosis is a major complication of DM.4 Although the condition usually occurs in type 1 DM, it can also develop in type 2 DM. Diabetic ketoacidosis may be an inciting event leading to the eventual diagnosis of DM, but can also develop during a concurrent illness such as a urinary tract infection or an eating disorder.5 Risk factors for DKA include patients with type 1 or type 2 DM, a family history of DM, obesity, and nonwhite patients whose ethnic background places them at increased risk.6 Hispanic, black, and African American patients are at a greater risk of developing DKA and are more likely to develop “ketosis-prone” type 2 DM.7
Patients who do not fit into the definitive categories of type 1 or 2 DM can be classified under ketosis-prone DM.7,8 Diabetic ketoacidosis acts as the inciting event for the disease and evolves into severe β-cell dysfunction, hence blurring the lines between the archetypal DM categories. Fifty percent of ketosis-prone DM patients are A-β+ (absent autoantibodies, present β-cell function), which indicates that the dysfunction can be partially reversed. Reversal of the condition is largely based on long-term β-cell reserves, which are dependent on tight glycemic control and insulin dependence. Higher incidences of the A-β+ variant of ketosis-prone diabetes are seen in the male population and are often unprovoked.9-11
Diabetic ketoacidosis is the result of either a decrease or absence of insulin in the body (Table 2).4 Without insulin modulating exogenous glucose intake and endogenous glucose production (via glucagon, glycogenolysis, and gluconeogenesis), high levels of glucose are found in the circulation, leading to prominent hyperglycemia (>250 mg/dL or >13.8 mmol/L).6 This environment causes the body to switch from carbohydrate metabolism to fatty acid metabolism. As a result, acidic ketone bodies such β-hydroxybutyrate and acetoacetate are produced. These physiological changes in the body cause the signs and symptoms typically found in DKA.
Signs and Symptoms
Over a period of 24 hours, symptoms such as nausea, vomiting, increased thirst, and polyuria develop due to dehydration caused by osmotic diuresis and glucosuria.5 Patients may also present with hypotension and tachycardia. Confusion, deep gasping breaths or Kussmaul respirations, and metabolic acidosis result from hyperventilation and failure to compensate for the increased serum concentration of ketone bodies. Ketone production leads to a fruit-like odor in the patient’s breath and ketonuria in the urinalysis. In DKA, laboratory values will indicate metabolic acidosis and abnormal serum electrolytes. In both DM and DKA, increased urea and creatinine due to dehydration, increased ketones, and the presence of diabetic nephropathy are useful indicators of impaired kidney function.12
Management and Treatment
Diabetic ketoacidosis can be managed and reversed, especially when recognized and treated early.6,13 Dehydration in DKA can be corrected with IV fluid replacement. Normal saline (0.9%) can be started at 15 to 20 mL/kg/h or 1 L/h. As the patient’s vital signs stabilize, IV fluids can be titrated to a lower dose of 250 to 500 mL/h. Monitoring BP and electrolytes are key at this point as alterations in sodium levels and glucose levels may require switching to half-normal saline and/or dextrose.
The hyperglycemic state of patients with DKA is managed by IV insulin. An initial bolus of 0.1 U/kg/h can be given, but should only be administered when potassium levels are greater than 3.3 mmol/L.14 If adequate perfusion can be maintained, then 0.14 U/kg/h can be used instead of a bolus. Glucose levels must be monitored; once the levels decrease to approximately 200 mg/dL, the infusion rate of insulin should be titrated down to 0.05 to 0.1 U/kg/h. Dextrose is then added to maintain glucose levels at approximately 150 to 200 mg/dL.
Electrolytes, especially potassium, must be monitored closely in patients with DKA. Insulin leads to the shift of potassium into cells. The lack of insulin keeps potassium in the extracellular space. Due to osmotic diuresis, potassium is lost in the urine, leading to hypokalemia. Potassium levels in patients with DKA should be maintained at a level between 4 to 5 mmol/L. Patients with potassium levels between 3.3 to 5.2 mmol/L can be started on IV potassium between 20 to 30 mmol/h. If the patient is severely hypokalemic (<3.3 mmol/L), insulin should be withheld, and only IV potassium should be given at a rate of 20 to 30 mmol/h.
Bicarbonate levels can also be managed as acidosis can lead to both neurological and cardiac complications. If the patient’s pH is less than 6.9, the American Diabetes Association recommends starting 100 mmol of sodium bicarbonate in 400 mL sterile water (in addition to potassium chloride at 200 mL/h) for 2 hours. Dosing should be repeated every 2 hours until the patient’s pH is greater than 6.9.
In uncomplicated cases of DKA, the condition is resolved when a patient’s pH is greater than 7.3; glucose level is less than 200 mg/dL; and bicarbonate level is greater than or equal to 18 mmol/L. After patients become hemodynamically stable, they can be discharged and managed at home with a combination of intermediate- or long-acting insulin as well as short- or rapid-acting insulin.
Complications and Mortality
Diabetic ketoacidosis can cause sudden and fluctuating changes in the body. Therefore, it is very important to monitor a patient’s laboratory values very carefully and frequently to avoid any pitfalls. Since patients can present with hyponatremia due to the osmotic draw of glucose in the blood,13 sodium levels may have to be corrected. The corrected serum sodium can be calculated by adding 1.6 mmol/L for every 100 mg/dL of glucose (when finger-stick readings are above 200 mg/dL).15 Patients with DKA can also present with leukocytosis (even in the absence of infection) and hypertriglyceridemia (due to impaired lipoprotein lipase).15 Serum creatine may be elevated due to blood acetoacetate levels.15
Interestingly, there are other acute conditions that can mimic DKA.15 For example, chronic ethanol abuse can lead to ketoacidosis. Unlike DKA, however, alcoholic ketoacidosis does not have profound hyperglycemia, which can help differentiate the two during initial assessment.
Complications due to DKA can arise comprising the patient’s health, including hypoglycemia, hypokalemia, rhabdomyolysis, acute renal failure, pulmonary edema, and shock.16 Cerebral edema is seen in up to 1% of DKA patients,15 the cause of which may be due to the severity of the acidosis, high glucose levels, and rapid hydration. Even when cerebral edema is reduced, patients are often neurologically impaired. Mortality rates from DKA deaths due to cerebral edema can be as high as 24%.13 In the United States, over 100,000 patients with DM per year are admitted to the hospital for DKA, and 9% of patients with DM suffer from DKA-related complications postdischarge.15 With current treatment protocols, mortality rates for DKA-associated deaths are now down to 1%.6,15
Diabetes ketoacidosis-related deaths are usually the result of the following: a triad of DKA symptoms (hyperglycemia, hyperketonemia, and metabolic acidosis), another underlying comorbid condition (eg, myocardial infarction, sepsis, acute respiratory distress syndrome), or the release of biological markers (ie, catecholamines).14,15,17 Thus, as previously stated, the management of potassium levels is important as both hyperkalemia and hypokalemia can lead to fatal arrthythmias.15
Direct mortality from DKA has dropped significantly over the past 20 years, from 8% to less than 1%.6 The US Centers for Disease Control and Prevention has observed a downward trend in death and estimates that 2,417 patients died in 2009 due to DKA,18 and recent postmortem studies have revealed new insights into DKA-related deaths.19 Blood and vitreous acetone concentrations are strong indicators for predeath hyperglycemia and ketosis (if there are no underlying comorbid and/or pharmacological provocations). Blood acetone levels greater than 0.01 g/dL antemortem are suggestive of DKA. It is recommended that these tests should be performed in sudden deaths which have no biological or anatomical cause of death. Postmortem diagnosis of DKA is made with the following criteria: history of DM, increased vitreous glucose concentrations, and elevated blood/vitreous/urine acetone concentrations (>200 mg/dL). If results of the abovementioned parameters are inconclusive, measurement of lactic acid postmortem is thought to further support a diagnosis of DKA.19
Patient Counseling and Education
Approximately 33% of patients whose death was associated with DKA had no personal history of DM.19 This statistic emphasizes the importance of taking a thorough history, physical examination, blood glucose evaluation, and educating patients about the signs and symptoms of DM and DKA.
Patient counseling and education are important, especially in patients whose racial/ethnic background places them at increased risk of developing DM (eg, patients of black or African American, American Indian, Alaskan Native, Asian American, Hispanic, Native Hawaiian, or Pacific Islander descent).20,21 Strategies for preventive management include advocating regular glucose monitoring as well as dietary and lifestyle modifications. In patients with DM, successful management of the condition and its comorbidities can help prevent DKA and associated mortality.
Conclusion
As this case demonstrates, despite prompt diagnosis and management, patients with DKA—especially those with uncontrolled, undiagnosed, or advanced DM—are associated with fatal outcomes. In many cases, however, DKA can be successfully managed and reversed, especially when the condition is recognized early. Management includes not only IV therapy to adjust fluid and insulin levels, but also restoring electrolyte balance (especially potassium and bicarbonate). Frequent and careful evaluation of laboratory values is vital to the successful treatment of DKA, as there are numerous pitfalls and complications that the emergency physician can encounter. Patients who either have or are at an increased risk of developing DM or DKA may benefit from preventive measures, including regular glucose monitoring and appropriate diet and lifestyle modifications.
Mr Hassan-Ali is a fourth-year medical student at Windsor University School of Medicine, St Kitts, West Indies. Dr Raziuddin is an internist and an emergency medicine physician at Weiss Memorial, Thorek Memorial, and Westlake Hospitals, Chicago, Illinois.
- Kitabchi AH, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24(1):131-153.
- Farinda A. Lab values, normal adult: laboratory reference ranges in healthy adults. 2015. Medscape Web site. http://emedicine.medscape.com/article/2172316-overview. Updated May 14, 2014. Accessed August 14, 2015.
- Young D. Implementation of SI units for clinical laboratory data. Ann Intern Med. 1987;106(1):114-129.
- Maitra A. The endocrine system. In: Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th ed. New York, NY: Elsevier Saunders; 2015:1105-1120.
- Powers AC. Diabetes mellitus: management and therapies. In: Kasper DL, Fauci AS, Longo DL, Hauser SL, Jameson JL, Loscalzo J. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY; McGraw-Hill Medical Publishing Division; 2015:2407-2422.
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343.
- Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann Intern Med. 2006;144(5):350-357.
- Umpierrez G, Smiley D, Gosmanov A, Thomason D. Ketosis-prone type 2 diabetes: effect of hyperglycemia on beta-cell function and skeletal muscle insulin signaling. Endocr Pract. 2007;13(3):283-290.
- Mauvais-Jarvis F, Sobngwi E, Porcher R, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes. 2004;53(3):645-653.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes. 1995;44(7):790-795.
- Piñero-Piloña A, Raskin P. Idiopathic type 1 diabetes. J Diabetes Complications. 2001;15(6):328-335.
- Kemperman FA, Weber JA, Gorgels J, van Zanten AP, Krediet RT, Arisz L. The influence of ketoacids on plasma creatinine assays in diabetic ketoacidosis. J Intern Med. 2000;248(6):511-517.
- Westerberg DP. Diabetic ketoacidosis: evaluation and treatment. Am Fam Physician. 2013;87(5):337-346.
- Trachtenbarg DE. Diabetic ketoacidosis. Am Fam Physician. 2005;71(9):1705-1714.
- Umpierrez GE, Murphy MB, Kitabchi AE. Diabetic ketoacidosis and hyperglycemic hyperosmolar ayndrome. Diabetes Spectrum. 2002;15(1):28-36.
- Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: A consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150-1159.
- Rosenbloom AL. Sudden death of a young woman attributed to diabetic ketoacidosis. J Forensic Leg Med. 2013;20(8):1063-1065.
- Centers for Disease Control and Prevention. Number of deaths for hyperglycemic crises as underlying cause, United States, 1980-2009. http://www.cdc.gov/diabetes/statistics/mortalitydka/fnumberofdka.htm. Updated November 19, 2013. Accessed August 14, 2015.
- Ali Z, Levine B, Ripple M, Fowler DR. Diabetic ketoacidosis: a silent death. Am J Forensic Med Pathol. 2012;33(3):189-193.
- US Department of Health and Human Services Office of Minority Health. Diabetes and Hispanic Americans. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=4&lvlid=63. Updated June 15, 2013. Accessed August 14, 2015.
- US Department of Health and Human Services Office of Minority Health. Profile: Native Hawaiian/Other Pacific Islanders. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=3&lvlid=65. Updated January 15, 2015. Accessed August 14, 2015.
- Kitabchi AH, Umpierrez GE, Murphy MB, et al. Management of hyperglycemic crises in patients with diabetes. Diabetes Care. 2001;24(1):131-153.
- Farinda A. Lab values, normal adult: laboratory reference ranges in healthy adults. 2015. Medscape Web site. http://emedicine.medscape.com/article/2172316-overview. Updated May 14, 2014. Accessed August 14, 2015.
- Young D. Implementation of SI units for clinical laboratory data. Ann Intern Med. 1987;106(1):114-129.
- Maitra A. The endocrine system. In: Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th ed. New York, NY: Elsevier Saunders; 2015:1105-1120.
- Powers AC. Diabetes mellitus: management and therapies. In: Kasper DL, Fauci AS, Longo DL, Hauser SL, Jameson JL, Loscalzo J. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY; McGraw-Hill Medical Publishing Division; 2015:2407-2422.
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes Care. 2009;32(7):1335-1343.
- Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Ann Intern Med. 2006;144(5):350-357.
- Umpierrez G, Smiley D, Gosmanov A, Thomason D. Ketosis-prone type 2 diabetes: effect of hyperglycemia on beta-cell function and skeletal muscle insulin signaling. Endocr Pract. 2007;13(3):283-290.
- Mauvais-Jarvis F, Sobngwi E, Porcher R, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes. 2004;53(3):645-653.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes. 1995;44(7):790-795.
- Piñero-Piloña A, Raskin P. Idiopathic type 1 diabetes. J Diabetes Complications. 2001;15(6):328-335.
- Kemperman FA, Weber JA, Gorgels J, van Zanten AP, Krediet RT, Arisz L. The influence of ketoacids on plasma creatinine assays in diabetic ketoacidosis. J Intern Med. 2000;248(6):511-517.
- Westerberg DP. Diabetic ketoacidosis: evaluation and treatment. Am Fam Physician. 2013;87(5):337-346.
- Trachtenbarg DE. Diabetic ketoacidosis. Am Fam Physician. 2005;71(9):1705-1714.
- Umpierrez GE, Murphy MB, Kitabchi AE. Diabetic ketoacidosis and hyperglycemic hyperosmolar ayndrome. Diabetes Spectrum. 2002;15(1):28-36.
- Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: A consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150-1159.
- Rosenbloom AL. Sudden death of a young woman attributed to diabetic ketoacidosis. J Forensic Leg Med. 2013;20(8):1063-1065.
- Centers for Disease Control and Prevention. Number of deaths for hyperglycemic crises as underlying cause, United States, 1980-2009. http://www.cdc.gov/diabetes/statistics/mortalitydka/fnumberofdka.htm. Updated November 19, 2013. Accessed August 14, 2015.
- Ali Z, Levine B, Ripple M, Fowler DR. Diabetic ketoacidosis: a silent death. Am J Forensic Med Pathol. 2012;33(3):189-193.
- US Department of Health and Human Services Office of Minority Health. Diabetes and Hispanic Americans. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=4&lvlid=63. Updated June 15, 2013. Accessed August 14, 2015.
- US Department of Health and Human Services Office of Minority Health. Profile: Native Hawaiian/Other Pacific Islanders. http://minorityhealth.hhs.gov/omh/browse.aspx?lvl=3&lvlid=65. Updated January 15, 2015. Accessed August 14, 2015.
Erythema Induratum of Bazin Presenting as Peripheral Neuropathy
Case Report
A 54-year-old Hispanic woman with a history of type 2 diabetes mellitus and hyperlipidemia presented with recurrent painful plaques and nodules on the bilateral lower extremities and a severe burning sensation on the feet of 3 years’ duration. The patient denied experiencing any associated fevers, chills, night sweats, weight loss, joint aches, cough, or shortness of breath. She had a history of pustular psoriasis and reported a positive purified protein derivative (PPD)(tuberculin) skin test approximately 40 years prior.
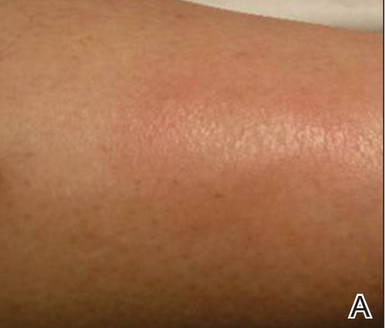
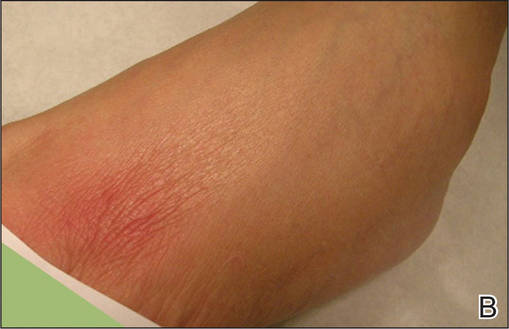
She presented with a 4×4-cm, poorly defined, tender, indurated plaque on the middle of the left shin and a 2×3-cm, red-brown plaque on the dorsal aspect of the left foot (Figure 1). No lymphadenopathy or any other abnormalities were noted. The clinical differential diagnosis included various panniculitides, such as erythema nodosum, erythema induratum of Bazin (EIB), and lupus panniculitis, as well as other conditions, including polyarteritis nodosa, sarcoidosis, Sweet disease, deep fungal and mycobacterial infections, and cutaneous lymphoma.
|
| Figure 1. Erythematous indurated plaques on the middle of the left shin (A) and on the dorsal aspect of the left foot (B). |
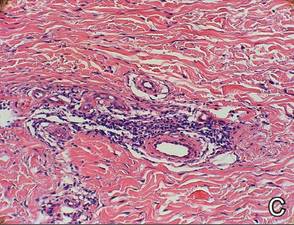
Two skin biopsies taken for histopathologic evaluation revealed primarily granulomatous lobular panniculitis with foci of microthrombi and vasculitis (Figure 2). These findings were consistent with nodular vasculitis. Acid-fast bacillus and Gomori methenamine-silver stains were negative for mycobacterial or fungal organisms. Tissue cultures also were negative. Results from a complete blood cell count, chemistry panel, thyroid and liver function tests, hepatitis panel, and rapid plasma reagin test were unremarkable. Immunologic markers, including antinuclear antibody, antineutrophil cytoplasmic antibody, rheumatoid factor, and cryoglobulins, also revealed no abnormalities. A chest radiograph showed scarring in the suprahilar region of the upper lobe of the left lung consistent with a prior case of pulmonary tuberculosis, and exposure to Mycobacterium tuberculosis was confirmed with an IFN-γ release assay (IGRA) result of 1.49 IU/mL (>0.34 IU/mL indicates positive test). These findings from clinical and histopathologic examination as well as laboratory tests were consistent with a diagnosis of EIB.
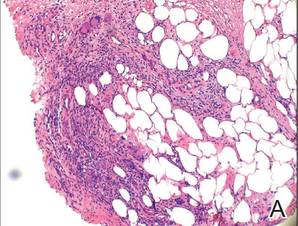

|
Figure 2. Granulomatous lobular panniculitis (A)(H&E, original magnification ×40) with foci of microthrombi and vasculitis (B and C)(both H&E, original magnifications ×200). |
Standard antituberculosis therapy with rifampin, isoniazid, pyrazinamide, and ethambutol (RIPE) was simplified to rifampin and isoniazid due to her inability to tolerate the full regimen because of gastrointestinal tract upset and diarrhea. After 6 months of therapy, a repeat IGRA decreased to 0.43 IU/mL, and the painful plaques and nodules on the lower extremities and burning sensation in the feet completely resolved.
Comment
Our case of EIB associated with peripheral neuropathy is a unique presentation of lesions on the pretibial area of the bilateral legs and dorsal aspect of the feet. We confirmed the presence of latent tuberculosis infection with a chest radiograph and an IGRA. Symptoms of peripheral neuropathy resolved after antituberculosis treatment, which suggests an immune-mediated mechanism of neuronal damage from circulating tuberculosis antigens.
Pathogenesis
Although erythema induratum was first described by Ernest Bazin in 1861, it was not until the early 1900s that the link between tuberculosis and erythema induratum was made by French dermatologists.1,2 Around the same time, similar cases of erythema induratum were discovered in England with no evidence of tuberculosis, which led to the distinct classification of erythema induratum of Whitfield (EIW). This classification described these nontuberculoid cases.1,2 In 1945, Montgomery et al3 in the United States coined the term nodular vasculitis for EIW and categorized its clinical features and histopathology as separate from EIB.3 Today, some authors use EIB, EIW, and nodular vasculitis interchangeably and believe they all are the same entity.2 We use EIB for all cases related to tuberculosis and nodular vasulitis when referring to all other etiologies, including nontuberculoid bacterial infections, chronic hepatitis B and C virus, thrombophlebitis, hypothyroidism, and rheumatoid arthritis.4,5
Erythema induratum of Bazin, lichen scrofulosorum, and papulonecrotic tuberculids are considered tuberculid diseases and are thought to be caused by hypersensitivity reactions to mycobacterial antigens rather than local mycobacterial infections. Tuberculids are believed to be a reaction to an id reaction of circulating mycobacterial antigens in the setting of latent or active tuberculosis infection. The basis for this view is that mycobacteria cannot be cultured or visualized from lesions in tuberculid diseases.6 Cutaneous tuberculosis, such as scrofuloderma, miliary tuberculosis, tuberculosis chancre, lupus vulgaris, and gummatous tuberculosis, differ from EIB and other tuberculid diseases in that mycobacteria can be cultured and visualized on histologic examination with Ziehl-Neelsen staining.6 The pathology of cutaneous tuberculosis results from a mycobacterial infection of the skin, and cutaneous tuberculosis diseases are categorized as multibacillary or paucibacillary based on the number of organisms visualized in biopsies.
The absence of M tuberculosis organisms in skin lesions has led some to doubt the causal relationship between M tuberculosis and EIB.7 However, the advent of polymerase chain reaction (PCR) and specific DNA primers for M tuberculosis has allowed for the detection of M tuberculosis DNA in biopsy specimens, which has further established the relationship between tuberculosis and EIB.5 Some authors have suggested that the absence of mycobacteria in EIB and other tuberculid lesions may be due to small numbers of bacilli in the lesions or early destruction of mycobacteria organisms before biopsy.8,9 These authors consider cutaneous tuberculosis and tuberculids as diseases on the same spectrum, with tuberculids representing one extreme in which there are few mycobacteria organisms present in the lesions.
Presentation
Erythema induratum commonly affects middle-aged women and presents with recurrent crops of tender nodules on the lower extremities.10-13 Nodules often are most commonly found on the lower calves but also can present on the arms, thighs, feet, or buttocks.10 Our patient’s presentation was atypical in that lesions were distributed on the pretibial area of the legs and dorsal aspect of the feet. Obesity and venous insufficiency of the lower extremities are believed to be predisposing factors to the development of EIB nodules.2 The nodules develop over several weeks and heal over several months with possible ulceration and hyperpigmented scarring.10,11 Ulcerated nodules often are irregular and shallow with an overlying crust and a bluish border.11,13 Nodules often are precipitated by cold weather or venostasis.1,11,12
Silva et al14 reported a case of EIB on the lower legs associated with a burning sensation on the feet and paresthesia; all known causes of peripheral neuropathy were excluded by a comprehensive laboratory workup. The burning sensation on the feet resolved after several weeks of antituberculosis therapy. Our patient also presented with a burning sensation on the feet that remarkably improved after 6 months of antituberculosis therapy. Peripheral neuropathy could have been a consequence of diabetes mellitus in our patient, though neuronal damage also could be a consequence of hypersensitivity to tuberculosis antigens. Silva et al14 proposed that macrophages activated by M tuberculosis antigens produce lytic enzymes that can cause tissue necrosis and nerve damage if released into surrounding tissue.
Diagnosis
The diagnosis of EIB is made based on clinical presentation, evidence of prior or current tuberculosis infection, histopathologic findings, and response to antituberculosis therapy.15 Evidence of active or latent tuberculosis infection typically is gathered by patient history, chest radiograph, tuberculin skin tests, interferon-releasing assays, and PCR of skin biopsies. Tuberculin skin tests in patients with EIB result in reactive induration that is typically more than 20 mm.8 In vitro T-lymphocyte proliferation assays in response to PPD have further supported the suggestion that there is a markedly enhanced T-lymphocyte response to M tuberculosis antigens in patients with EIB.16
IFN-γ release assays have provided useful methods for the detection of latent tuberculosis infection.17 The IGRA is effective when the tuberculin skin test yields a suspected false-negative or in the context of prior bacille Calmette-Guérin vaccination.17 IFN-γ release assays also may be preferred to tuberculin skin tests because it provides less discomfort to the patient in the event of a positive hypersensitive reaction to the PPD.
Before the advent of IGRAs, PCR was used to detect M tuberculosis DNA in skin biopsies and to confirm the diagnosis of EIB. Some researchers believe PCR can be an important tool for confirming a diagnosis of EIB, especially in cases and countries where results from the Mantoux test do not have great value.15,18 A PCR assay for detecting M tuberculosis DNA in blood and urine samples also was found helpful in confirming a diagnosis of EIB when skin biopsies were unavailable.8,19 However, PCR has been shown to have low sensitivity for the diagnosis of EIB because of its ability to detect M tuberculosis DNA ranging from 0% to 77% of skin biopsy specimens.20,21 Therefore, a negative PCR for the detection of M tuberculosis DNA in nodules does not exclude a diagnosis of erythema induratum.
Treatment
The mainstay of EIB treatment is a multidrug antituberculosis regimen.5,8,10-12 Our patient was successfully treated with rifampin and isoniazid and a repeat IGRA was used as a laboratory marker of response to therapy. Single-drug therapy with isoniazid has been shown to result in greater likelihood of EIB relapse in comparison to multidrug regimens.22 Other treatments include potassium iodide and gold, but they are not well-studied.23-25 Treatment of venous insufficiency with bed rest and nonsteroidal anti-inflammatory drugs for pain also may be helpful.2 In cases of nodular vasulitis that are not associated with tuberculosis infection, treatment should be targeted at the underlying cause of the immune response. For example, a case of nodular vasculitis associated with hepatitis C virus did not respond to antituberculosis multidrug therapy, but skin lesions did improve with pegylated interferon and ribavirin.4
1. Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
2. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
3. Montgomery H, O’Leary PA, Barker NW. Nodular vascular disease of the legs: erythema induratum and allied conditions. JAMA. 1945;128:335-342.
4. Fernandes SS, Carvalho J, Leite S, et al. Erythema induratum and chronic hepatitis C infection [published online ahead of print February 23, 2009]. J Clin Virol. 2009;44:333-336.
5. Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
6. Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
7. Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin. a clinicopathological study of 20 cases and detection of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356.
8. Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
9. Bravo FG, Gotuzzo E. Cutaneous tuberculosis. Clin Dermatol. 2007;25:173-180.
10. Rademaker M, Lowe DG, Munro DD. Erythema induratum (Bazin’s disease). J Am Acad Dermatol. 1989;21 (4, pt 1):740-745.
11. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
12. Feiwel M, Munro DD. Diagnosis and treatment of erythema induratum (Bazin). Br Med J. 1965;1:1109-1111.
13. Lebel M, Lassonde M. Erythema induratum of Bazin. J Am Acad Dermatol. 1986;14(5, pt 1):738-742.
14. Silva MT, Antunes SL, Rolla VC, et al. Distal painful peripheral neuropathy associated with erythema induratum of Bazin. Eur J Neurol. 2006;13:e5-e6.
15. Jacinto SS, Nograles KB. Erythema induratum of bazin: role of polymerase chain reaction in diagnosis. Int J Dermatol. 2003;42:380-381.
16. Ollert MW, Thomas P, Korting HC, et al. Erythema induratum of Bazin. evidence of T-lymphocyte hyperresponsiveness to purified protein derivative of tuberculin: report of two cases and treatment. Arch Dermatol. 1993;129:469-473.
17. Angus J, Roberts C, Kulkarni K, et al. Usefulness of the QuantiFERON test in the confirmation of latent tuberculosis in association with erythema induratum [published online ahead of print October 10, 2007]. Br J Dermatol. 2007;157:1293-1294.
18. Seckin D, Hízel N, Demirhan B, et al. The diagnostic value of polymerase chain reaction in erythema induratum of Bazin. Br J Dermatol. 1997;137:1011-1012.
19. Cannas A, Goletti D, Girardi E, et al. Mycobacterium tuberculosis DNA detection in soluble fraction of urine from pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2008;12:146-151.
20. Tan SH, Tan BH, Goh CL, et al. Detection of Mycobacterium tuberculosis DNA using polymerase chain reaction in cutaneous tuberculosis and tuberculids. Int J Dermatol. 1999;38:122-127.
21. Baselga E, Margall N, Barnadas MA, et al. Detection of Mycobacterium tuberculosis DNA in lobular granulomatous panniculitis (erythema induratum-nodular vasculitis). Arch Dermatol. 1997;133:457-462.
22. Cho KH, Lee DY, Kim CW. Erythema induratum of Bazin. Int J Dermatol. 1996;35:802-808.
23. Schulz EJ, Whiting DA. Treatment of erythema nodosum and nodular vasculitis with potassium iodide. Br J Dermatol. 1976;94:75-78.
24. Horio T, Imamura S, Danno K, et al. Potassium iodide in the treatment of erythema nodosum and nodular vasculitis. Arch Dermatol. 1981;117:29-31.
25. Shaffer N, Kerdel FA. Nodular vasculitis (erythema induratum): treatment with auranofin. J Am Acad Dermatol. 1991;25(2, pt 2):426-429.
Case Report
A 54-year-old Hispanic woman with a history of type 2 diabetes mellitus and hyperlipidemia presented with recurrent painful plaques and nodules on the bilateral lower extremities and a severe burning sensation on the feet of 3 years’ duration. The patient denied experiencing any associated fevers, chills, night sweats, weight loss, joint aches, cough, or shortness of breath. She had a history of pustular psoriasis and reported a positive purified protein derivative (PPD)(tuberculin) skin test approximately 40 years prior.
She presented with a 4×4-cm, poorly defined, tender, indurated plaque on the middle of the left shin and a 2×3-cm, red-brown plaque on the dorsal aspect of the left foot (Figure 1). No lymphadenopathy or any other abnormalities were noted. The clinical differential diagnosis included various panniculitides, such as erythema nodosum, erythema induratum of Bazin (EIB), and lupus panniculitis, as well as other conditions, including polyarteritis nodosa, sarcoidosis, Sweet disease, deep fungal and mycobacterial infections, and cutaneous lymphoma.
|
|
| Figure 1. Erythematous indurated plaques on the middle of the left shin (A) and on the dorsal aspect of the left foot (B). |
Two skin biopsies taken for histopathologic evaluation revealed primarily granulomatous lobular panniculitis with foci of microthrombi and vasculitis (Figure 2). These findings were consistent with nodular vasculitis. Acid-fast bacillus and Gomori methenamine-silver stains were negative for mycobacterial or fungal organisms. Tissue cultures also were negative. Results from a complete blood cell count, chemistry panel, thyroid and liver function tests, hepatitis panel, and rapid plasma reagin test were unremarkable. Immunologic markers, including antinuclear antibody, antineutrophil cytoplasmic antibody, rheumatoid factor, and cryoglobulins, also revealed no abnormalities. A chest radiograph showed scarring in the suprahilar region of the upper lobe of the left lung consistent with a prior case of pulmonary tuberculosis, and exposure to Mycobacterium tuberculosis was confirmed with an IFN-γ release assay (IGRA) result of 1.49 IU/mL (>0.34 IU/mL indicates positive test). These findings from clinical and histopathologic examination as well as laboratory tests were consistent with a diagnosis of EIB.
|
|
Figure 2. Granulomatous lobular panniculitis (A)(H&E, original magnification ×40) with foci of microthrombi and vasculitis (B and C)(both H&E, original magnifications ×200). |
Standard antituberculosis therapy with rifampin, isoniazid, pyrazinamide, and ethambutol (RIPE) was simplified to rifampin and isoniazid due to her inability to tolerate the full regimen because of gastrointestinal tract upset and diarrhea. After 6 months of therapy, a repeat IGRA decreased to 0.43 IU/mL, and the painful plaques and nodules on the lower extremities and burning sensation in the feet completely resolved.
Comment
Our case of EIB associated with peripheral neuropathy is a unique presentation of lesions on the pretibial area of the bilateral legs and dorsal aspect of the feet. We confirmed the presence of latent tuberculosis infection with a chest radiograph and an IGRA. Symptoms of peripheral neuropathy resolved after antituberculosis treatment, which suggests an immune-mediated mechanism of neuronal damage from circulating tuberculosis antigens.
Pathogenesis
Although erythema induratum was first described by Ernest Bazin in 1861, it was not until the early 1900s that the link between tuberculosis and erythema induratum was made by French dermatologists.1,2 Around the same time, similar cases of erythema induratum were discovered in England with no evidence of tuberculosis, which led to the distinct classification of erythema induratum of Whitfield (EIW). This classification described these nontuberculoid cases.1,2 In 1945, Montgomery et al3 in the United States coined the term nodular vasculitis for EIW and categorized its clinical features and histopathology as separate from EIB.3 Today, some authors use EIB, EIW, and nodular vasculitis interchangeably and believe they all are the same entity.2 We use EIB for all cases related to tuberculosis and nodular vasulitis when referring to all other etiologies, including nontuberculoid bacterial infections, chronic hepatitis B and C virus, thrombophlebitis, hypothyroidism, and rheumatoid arthritis.4,5
Erythema induratum of Bazin, lichen scrofulosorum, and papulonecrotic tuberculids are considered tuberculid diseases and are thought to be caused by hypersensitivity reactions to mycobacterial antigens rather than local mycobacterial infections. Tuberculids are believed to be a reaction to an id reaction of circulating mycobacterial antigens in the setting of latent or active tuberculosis infection. The basis for this view is that mycobacteria cannot be cultured or visualized from lesions in tuberculid diseases.6 Cutaneous tuberculosis, such as scrofuloderma, miliary tuberculosis, tuberculosis chancre, lupus vulgaris, and gummatous tuberculosis, differ from EIB and other tuberculid diseases in that mycobacteria can be cultured and visualized on histologic examination with Ziehl-Neelsen staining.6 The pathology of cutaneous tuberculosis results from a mycobacterial infection of the skin, and cutaneous tuberculosis diseases are categorized as multibacillary or paucibacillary based on the number of organisms visualized in biopsies.
The absence of M tuberculosis organisms in skin lesions has led some to doubt the causal relationship between M tuberculosis and EIB.7 However, the advent of polymerase chain reaction (PCR) and specific DNA primers for M tuberculosis has allowed for the detection of M tuberculosis DNA in biopsy specimens, which has further established the relationship between tuberculosis and EIB.5 Some authors have suggested that the absence of mycobacteria in EIB and other tuberculid lesions may be due to small numbers of bacilli in the lesions or early destruction of mycobacteria organisms before biopsy.8,9 These authors consider cutaneous tuberculosis and tuberculids as diseases on the same spectrum, with tuberculids representing one extreme in which there are few mycobacteria organisms present in the lesions.
Presentation
Erythema induratum commonly affects middle-aged women and presents with recurrent crops of tender nodules on the lower extremities.10-13 Nodules often are most commonly found on the lower calves but also can present on the arms, thighs, feet, or buttocks.10 Our patient’s presentation was atypical in that lesions were distributed on the pretibial area of the legs and dorsal aspect of the feet. Obesity and venous insufficiency of the lower extremities are believed to be predisposing factors to the development of EIB nodules.2 The nodules develop over several weeks and heal over several months with possible ulceration and hyperpigmented scarring.10,11 Ulcerated nodules often are irregular and shallow with an overlying crust and a bluish border.11,13 Nodules often are precipitated by cold weather or venostasis.1,11,12
Silva et al14 reported a case of EIB on the lower legs associated with a burning sensation on the feet and paresthesia; all known causes of peripheral neuropathy were excluded by a comprehensive laboratory workup. The burning sensation on the feet resolved after several weeks of antituberculosis therapy. Our patient also presented with a burning sensation on the feet that remarkably improved after 6 months of antituberculosis therapy. Peripheral neuropathy could have been a consequence of diabetes mellitus in our patient, though neuronal damage also could be a consequence of hypersensitivity to tuberculosis antigens. Silva et al14 proposed that macrophages activated by M tuberculosis antigens produce lytic enzymes that can cause tissue necrosis and nerve damage if released into surrounding tissue.
Diagnosis
The diagnosis of EIB is made based on clinical presentation, evidence of prior or current tuberculosis infection, histopathologic findings, and response to antituberculosis therapy.15 Evidence of active or latent tuberculosis infection typically is gathered by patient history, chest radiograph, tuberculin skin tests, interferon-releasing assays, and PCR of skin biopsies. Tuberculin skin tests in patients with EIB result in reactive induration that is typically more than 20 mm.8 In vitro T-lymphocyte proliferation assays in response to PPD have further supported the suggestion that there is a markedly enhanced T-lymphocyte response to M tuberculosis antigens in patients with EIB.16
IFN-γ release assays have provided useful methods for the detection of latent tuberculosis infection.17 The IGRA is effective when the tuberculin skin test yields a suspected false-negative or in the context of prior bacille Calmette-Guérin vaccination.17 IFN-γ release assays also may be preferred to tuberculin skin tests because it provides less discomfort to the patient in the event of a positive hypersensitive reaction to the PPD.
Before the advent of IGRAs, PCR was used to detect M tuberculosis DNA in skin biopsies and to confirm the diagnosis of EIB. Some researchers believe PCR can be an important tool for confirming a diagnosis of EIB, especially in cases and countries where results from the Mantoux test do not have great value.15,18 A PCR assay for detecting M tuberculosis DNA in blood and urine samples also was found helpful in confirming a diagnosis of EIB when skin biopsies were unavailable.8,19 However, PCR has been shown to have low sensitivity for the diagnosis of EIB because of its ability to detect M tuberculosis DNA ranging from 0% to 77% of skin biopsy specimens.20,21 Therefore, a negative PCR for the detection of M tuberculosis DNA in nodules does not exclude a diagnosis of erythema induratum.
Treatment
The mainstay of EIB treatment is a multidrug antituberculosis regimen.5,8,10-12 Our patient was successfully treated with rifampin and isoniazid and a repeat IGRA was used as a laboratory marker of response to therapy. Single-drug therapy with isoniazid has been shown to result in greater likelihood of EIB relapse in comparison to multidrug regimens.22 Other treatments include potassium iodide and gold, but they are not well-studied.23-25 Treatment of venous insufficiency with bed rest and nonsteroidal anti-inflammatory drugs for pain also may be helpful.2 In cases of nodular vasulitis that are not associated with tuberculosis infection, treatment should be targeted at the underlying cause of the immune response. For example, a case of nodular vasculitis associated with hepatitis C virus did not respond to antituberculosis multidrug therapy, but skin lesions did improve with pegylated interferon and ribavirin.4
Case Report
A 54-year-old Hispanic woman with a history of type 2 diabetes mellitus and hyperlipidemia presented with recurrent painful plaques and nodules on the bilateral lower extremities and a severe burning sensation on the feet of 3 years’ duration. The patient denied experiencing any associated fevers, chills, night sweats, weight loss, joint aches, cough, or shortness of breath. She had a history of pustular psoriasis and reported a positive purified protein derivative (PPD)(tuberculin) skin test approximately 40 years prior.
She presented with a 4×4-cm, poorly defined, tender, indurated plaque on the middle of the left shin and a 2×3-cm, red-brown plaque on the dorsal aspect of the left foot (Figure 1). No lymphadenopathy or any other abnormalities were noted. The clinical differential diagnosis included various panniculitides, such as erythema nodosum, erythema induratum of Bazin (EIB), and lupus panniculitis, as well as other conditions, including polyarteritis nodosa, sarcoidosis, Sweet disease, deep fungal and mycobacterial infections, and cutaneous lymphoma.
|
|
| Figure 1. Erythematous indurated plaques on the middle of the left shin (A) and on the dorsal aspect of the left foot (B). |
Two skin biopsies taken for histopathologic evaluation revealed primarily granulomatous lobular panniculitis with foci of microthrombi and vasculitis (Figure 2). These findings were consistent with nodular vasculitis. Acid-fast bacillus and Gomori methenamine-silver stains were negative for mycobacterial or fungal organisms. Tissue cultures also were negative. Results from a complete blood cell count, chemistry panel, thyroid and liver function tests, hepatitis panel, and rapid plasma reagin test were unremarkable. Immunologic markers, including antinuclear antibody, antineutrophil cytoplasmic antibody, rheumatoid factor, and cryoglobulins, also revealed no abnormalities. A chest radiograph showed scarring in the suprahilar region of the upper lobe of the left lung consistent with a prior case of pulmonary tuberculosis, and exposure to Mycobacterium tuberculosis was confirmed with an IFN-γ release assay (IGRA) result of 1.49 IU/mL (>0.34 IU/mL indicates positive test). These findings from clinical and histopathologic examination as well as laboratory tests were consistent with a diagnosis of EIB.
|
|
Figure 2. Granulomatous lobular panniculitis (A)(H&E, original magnification ×40) with foci of microthrombi and vasculitis (B and C)(both H&E, original magnifications ×200). |
Standard antituberculosis therapy with rifampin, isoniazid, pyrazinamide, and ethambutol (RIPE) was simplified to rifampin and isoniazid due to her inability to tolerate the full regimen because of gastrointestinal tract upset and diarrhea. After 6 months of therapy, a repeat IGRA decreased to 0.43 IU/mL, and the painful plaques and nodules on the lower extremities and burning sensation in the feet completely resolved.
Comment
Our case of EIB associated with peripheral neuropathy is a unique presentation of lesions on the pretibial area of the bilateral legs and dorsal aspect of the feet. We confirmed the presence of latent tuberculosis infection with a chest radiograph and an IGRA. Symptoms of peripheral neuropathy resolved after antituberculosis treatment, which suggests an immune-mediated mechanism of neuronal damage from circulating tuberculosis antigens.
Pathogenesis
Although erythema induratum was first described by Ernest Bazin in 1861, it was not until the early 1900s that the link between tuberculosis and erythema induratum was made by French dermatologists.1,2 Around the same time, similar cases of erythema induratum were discovered in England with no evidence of tuberculosis, which led to the distinct classification of erythema induratum of Whitfield (EIW). This classification described these nontuberculoid cases.1,2 In 1945, Montgomery et al3 in the United States coined the term nodular vasculitis for EIW and categorized its clinical features and histopathology as separate from EIB.3 Today, some authors use EIB, EIW, and nodular vasculitis interchangeably and believe they all are the same entity.2 We use EIB for all cases related to tuberculosis and nodular vasulitis when referring to all other etiologies, including nontuberculoid bacterial infections, chronic hepatitis B and C virus, thrombophlebitis, hypothyroidism, and rheumatoid arthritis.4,5
Erythema induratum of Bazin, lichen scrofulosorum, and papulonecrotic tuberculids are considered tuberculid diseases and are thought to be caused by hypersensitivity reactions to mycobacterial antigens rather than local mycobacterial infections. Tuberculids are believed to be a reaction to an id reaction of circulating mycobacterial antigens in the setting of latent or active tuberculosis infection. The basis for this view is that mycobacteria cannot be cultured or visualized from lesions in tuberculid diseases.6 Cutaneous tuberculosis, such as scrofuloderma, miliary tuberculosis, tuberculosis chancre, lupus vulgaris, and gummatous tuberculosis, differ from EIB and other tuberculid diseases in that mycobacteria can be cultured and visualized on histologic examination with Ziehl-Neelsen staining.6 The pathology of cutaneous tuberculosis results from a mycobacterial infection of the skin, and cutaneous tuberculosis diseases are categorized as multibacillary or paucibacillary based on the number of organisms visualized in biopsies.
The absence of M tuberculosis organisms in skin lesions has led some to doubt the causal relationship between M tuberculosis and EIB.7 However, the advent of polymerase chain reaction (PCR) and specific DNA primers for M tuberculosis has allowed for the detection of M tuberculosis DNA in biopsy specimens, which has further established the relationship between tuberculosis and EIB.5 Some authors have suggested that the absence of mycobacteria in EIB and other tuberculid lesions may be due to small numbers of bacilli in the lesions or early destruction of mycobacteria organisms before biopsy.8,9 These authors consider cutaneous tuberculosis and tuberculids as diseases on the same spectrum, with tuberculids representing one extreme in which there are few mycobacteria organisms present in the lesions.
Presentation
Erythema induratum commonly affects middle-aged women and presents with recurrent crops of tender nodules on the lower extremities.10-13 Nodules often are most commonly found on the lower calves but also can present on the arms, thighs, feet, or buttocks.10 Our patient’s presentation was atypical in that lesions were distributed on the pretibial area of the legs and dorsal aspect of the feet. Obesity and venous insufficiency of the lower extremities are believed to be predisposing factors to the development of EIB nodules.2 The nodules develop over several weeks and heal over several months with possible ulceration and hyperpigmented scarring.10,11 Ulcerated nodules often are irregular and shallow with an overlying crust and a bluish border.11,13 Nodules often are precipitated by cold weather or venostasis.1,11,12
Silva et al14 reported a case of EIB on the lower legs associated with a burning sensation on the feet and paresthesia; all known causes of peripheral neuropathy were excluded by a comprehensive laboratory workup. The burning sensation on the feet resolved after several weeks of antituberculosis therapy. Our patient also presented with a burning sensation on the feet that remarkably improved after 6 months of antituberculosis therapy. Peripheral neuropathy could have been a consequence of diabetes mellitus in our patient, though neuronal damage also could be a consequence of hypersensitivity to tuberculosis antigens. Silva et al14 proposed that macrophages activated by M tuberculosis antigens produce lytic enzymes that can cause tissue necrosis and nerve damage if released into surrounding tissue.
Diagnosis
The diagnosis of EIB is made based on clinical presentation, evidence of prior or current tuberculosis infection, histopathologic findings, and response to antituberculosis therapy.15 Evidence of active or latent tuberculosis infection typically is gathered by patient history, chest radiograph, tuberculin skin tests, interferon-releasing assays, and PCR of skin biopsies. Tuberculin skin tests in patients with EIB result in reactive induration that is typically more than 20 mm.8 In vitro T-lymphocyte proliferation assays in response to PPD have further supported the suggestion that there is a markedly enhanced T-lymphocyte response to M tuberculosis antigens in patients with EIB.16
IFN-γ release assays have provided useful methods for the detection of latent tuberculosis infection.17 The IGRA is effective when the tuberculin skin test yields a suspected false-negative or in the context of prior bacille Calmette-Guérin vaccination.17 IFN-γ release assays also may be preferred to tuberculin skin tests because it provides less discomfort to the patient in the event of a positive hypersensitive reaction to the PPD.
Before the advent of IGRAs, PCR was used to detect M tuberculosis DNA in skin biopsies and to confirm the diagnosis of EIB. Some researchers believe PCR can be an important tool for confirming a diagnosis of EIB, especially in cases and countries where results from the Mantoux test do not have great value.15,18 A PCR assay for detecting M tuberculosis DNA in blood and urine samples also was found helpful in confirming a diagnosis of EIB when skin biopsies were unavailable.8,19 However, PCR has been shown to have low sensitivity for the diagnosis of EIB because of its ability to detect M tuberculosis DNA ranging from 0% to 77% of skin biopsy specimens.20,21 Therefore, a negative PCR for the detection of M tuberculosis DNA in nodules does not exclude a diagnosis of erythema induratum.
Treatment
The mainstay of EIB treatment is a multidrug antituberculosis regimen.5,8,10-12 Our patient was successfully treated with rifampin and isoniazid and a repeat IGRA was used as a laboratory marker of response to therapy. Single-drug therapy with isoniazid has been shown to result in greater likelihood of EIB relapse in comparison to multidrug regimens.22 Other treatments include potassium iodide and gold, but they are not well-studied.23-25 Treatment of venous insufficiency with bed rest and nonsteroidal anti-inflammatory drugs for pain also may be helpful.2 In cases of nodular vasulitis that are not associated with tuberculosis infection, treatment should be targeted at the underlying cause of the immune response. For example, a case of nodular vasculitis associated with hepatitis C virus did not respond to antituberculosis multidrug therapy, but skin lesions did improve with pegylated interferon and ribavirin.4
1. Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
2. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
3. Montgomery H, O’Leary PA, Barker NW. Nodular vascular disease of the legs: erythema induratum and allied conditions. JAMA. 1945;128:335-342.
4. Fernandes SS, Carvalho J, Leite S, et al. Erythema induratum and chronic hepatitis C infection [published online ahead of print February 23, 2009]. J Clin Virol. 2009;44:333-336.
5. Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
6. Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
7. Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin. a clinicopathological study of 20 cases and detection of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356.
8. Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
9. Bravo FG, Gotuzzo E. Cutaneous tuberculosis. Clin Dermatol. 2007;25:173-180.
10. Rademaker M, Lowe DG, Munro DD. Erythema induratum (Bazin’s disease). J Am Acad Dermatol. 1989;21 (4, pt 1):740-745.
11. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
12. Feiwel M, Munro DD. Diagnosis and treatment of erythema induratum (Bazin). Br Med J. 1965;1:1109-1111.
13. Lebel M, Lassonde M. Erythema induratum of Bazin. J Am Acad Dermatol. 1986;14(5, pt 1):738-742.
14. Silva MT, Antunes SL, Rolla VC, et al. Distal painful peripheral neuropathy associated with erythema induratum of Bazin. Eur J Neurol. 2006;13:e5-e6.
15. Jacinto SS, Nograles KB. Erythema induratum of bazin: role of polymerase chain reaction in diagnosis. Int J Dermatol. 2003;42:380-381.
16. Ollert MW, Thomas P, Korting HC, et al. Erythema induratum of Bazin. evidence of T-lymphocyte hyperresponsiveness to purified protein derivative of tuberculin: report of two cases and treatment. Arch Dermatol. 1993;129:469-473.
17. Angus J, Roberts C, Kulkarni K, et al. Usefulness of the QuantiFERON test in the confirmation of latent tuberculosis in association with erythema induratum [published online ahead of print October 10, 2007]. Br J Dermatol. 2007;157:1293-1294.
18. Seckin D, Hízel N, Demirhan B, et al. The diagnostic value of polymerase chain reaction in erythema induratum of Bazin. Br J Dermatol. 1997;137:1011-1012.
19. Cannas A, Goletti D, Girardi E, et al. Mycobacterium tuberculosis DNA detection in soluble fraction of urine from pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2008;12:146-151.
20. Tan SH, Tan BH, Goh CL, et al. Detection of Mycobacterium tuberculosis DNA using polymerase chain reaction in cutaneous tuberculosis and tuberculids. Int J Dermatol. 1999;38:122-127.
21. Baselga E, Margall N, Barnadas MA, et al. Detection of Mycobacterium tuberculosis DNA in lobular granulomatous panniculitis (erythema induratum-nodular vasculitis). Arch Dermatol. 1997;133:457-462.
22. Cho KH, Lee DY, Kim CW. Erythema induratum of Bazin. Int J Dermatol. 1996;35:802-808.
23. Schulz EJ, Whiting DA. Treatment of erythema nodosum and nodular vasculitis with potassium iodide. Br J Dermatol. 1976;94:75-78.
24. Horio T, Imamura S, Danno K, et al. Potassium iodide in the treatment of erythema nodosum and nodular vasculitis. Arch Dermatol. 1981;117:29-31.
25. Shaffer N, Kerdel FA. Nodular vasculitis (erythema induratum): treatment with auranofin. J Am Acad Dermatol. 1991;25(2, pt 2):426-429.
1. Segura S, Pujol RM, Trindade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
2. Requena L, Sánchez Yus E. Panniculitis. part II. mostly lobular panniculitis. J Am Acad Dermatol. 2001;45:325-361.
3. Montgomery H, O’Leary PA, Barker NW. Nodular vascular disease of the legs: erythema induratum and allied conditions. JAMA. 1945;128:335-342.
4. Fernandes SS, Carvalho J, Leite S, et al. Erythema induratum and chronic hepatitis C infection [published online ahead of print February 23, 2009]. J Clin Virol. 2009;44:333-336.
5. Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
6. Frankel A, Penrose C, Emer J. Cutaneous tuberculosis: a practical case report and review for the dermatologist. J Clin Aesthet Dermatol. 2009;2:19-27.
7. Schneider JW, Jordaan HF, Geiger DH, et al. Erythema induratum of Bazin. a clinicopathological study of 20 cases and detection of Mycobacterium tuberculosis DNA in skin lesions by polymerase chain reaction. Am J Dermatopathol. 1995;17:350-356.
8. Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
9. Bravo FG, Gotuzzo E. Cutaneous tuberculosis. Clin Dermatol. 2007;25:173-180.
10. Rademaker M, Lowe DG, Munro DD. Erythema induratum (Bazin’s disease). J Am Acad Dermatol. 1989;21 (4, pt 1):740-745.
11. Sharon V, Goodarzi H, Chambers CJ, et al. Erythema induratum of Bazin. Dermatol Online J. 2010;16:1.
12. Feiwel M, Munro DD. Diagnosis and treatment of erythema induratum (Bazin). Br Med J. 1965;1:1109-1111.
13. Lebel M, Lassonde M. Erythema induratum of Bazin. J Am Acad Dermatol. 1986;14(5, pt 1):738-742.
14. Silva MT, Antunes SL, Rolla VC, et al. Distal painful peripheral neuropathy associated with erythema induratum of Bazin. Eur J Neurol. 2006;13:e5-e6.
15. Jacinto SS, Nograles KB. Erythema induratum of bazin: role of polymerase chain reaction in diagnosis. Int J Dermatol. 2003;42:380-381.
16. Ollert MW, Thomas P, Korting HC, et al. Erythema induratum of Bazin. evidence of T-lymphocyte hyperresponsiveness to purified protein derivative of tuberculin: report of two cases and treatment. Arch Dermatol. 1993;129:469-473.
17. Angus J, Roberts C, Kulkarni K, et al. Usefulness of the QuantiFERON test in the confirmation of latent tuberculosis in association with erythema induratum [published online ahead of print October 10, 2007]. Br J Dermatol. 2007;157:1293-1294.
18. Seckin D, Hízel N, Demirhan B, et al. The diagnostic value of polymerase chain reaction in erythema induratum of Bazin. Br J Dermatol. 1997;137:1011-1012.
19. Cannas A, Goletti D, Girardi E, et al. Mycobacterium tuberculosis DNA detection in soluble fraction of urine from pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2008;12:146-151.
20. Tan SH, Tan BH, Goh CL, et al. Detection of Mycobacterium tuberculosis DNA using polymerase chain reaction in cutaneous tuberculosis and tuberculids. Int J Dermatol. 1999;38:122-127.
21. Baselga E, Margall N, Barnadas MA, et al. Detection of Mycobacterium tuberculosis DNA in lobular granulomatous panniculitis (erythema induratum-nodular vasculitis). Arch Dermatol. 1997;133:457-462.
22. Cho KH, Lee DY, Kim CW. Erythema induratum of Bazin. Int J Dermatol. 1996;35:802-808.
23. Schulz EJ, Whiting DA. Treatment of erythema nodosum and nodular vasculitis with potassium iodide. Br J Dermatol. 1976;94:75-78.
24. Horio T, Imamura S, Danno K, et al. Potassium iodide in the treatment of erythema nodosum and nodular vasculitis. Arch Dermatol. 1981;117:29-31.
25. Shaffer N, Kerdel FA. Nodular vasculitis (erythema induratum): treatment with auranofin. J Am Acad Dermatol. 1991;25(2, pt 2):426-429.
Practice Points
- Erythema induratum of Bazin (EIB) is a type of nodular vasculitis related to tuberculosis and commonly presents with plaques and nodules on the lower extremities.
- Peripheral neuropathy may manifest as a result of a hypersensitivity reaction to tuberculosis antigens causing tissue and nerve damage.
- Treatment of EIB and other cases of nodular vasculitis should be directed at the underlying cause of the immune response.
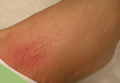
Recorrection Osteotomies and Total Knee Arthroplasties After Failed Bilateral High Tibial Osteotomies
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
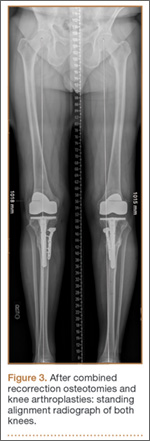
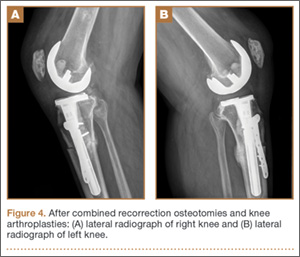
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
High tibial osteotomy has proved successful in treating unicompartmental arthritis in young, active patients.1-3 However, this procedure fails over time because the other compartments deteriorate.4 The next step is conversion of the osteotomy to total knee arthroplasty (TKA). Conversion results vary, with several authors reporting poor outcomes5-9 and others reporting outcomes equal to those of primary TKA.10-14
The long-term success of TKA depends on proper restoration of the mechanical axis and soft-tissue balancing.15 Preexisting extra-articular deformity may adversely affect outcomes. A deformity of more than 15° may make it difficult to obtain intra-articular correction of an extra-articular deformity through soft-tissue balancing alone.16
In this article, we report the unique case of a patient whose bilateral high tibial osteotomies failed because of excessive extra-articular deformity. TKAs were performed consecutively, in 2 separate settings. Each TKA was combined with a recorrection tibial osteotomy in a single operation, allowing for re-creation of normal knee alignment with ligament balance. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 58-year-old man (weight, 250 pounds; body mass index, 30) underwent staged bilateral medial opening wedge osteotomies using distraction osteogenesis. A uniplanar external fixator was used for fixation on each knee. Before surgery, anatomical axis was 2° (right knee) and –1° (left knee) (Figure 1A), and tibial slope was 9° (right) and 8° (left) (Figures 1B, 1C). The procedures were performed 10 months apart. After surgery, anatomical alignment was 17° valgus (right knee) and 12° valgus (left knee) (Figure 2), and tibial slope was 20° (right) and 13° (left).
The patient received mild relief of his arthritis symptoms. Fifty-six months after the index operation, he decided to undergo conversion of the right high tibial osteotomy to TKA because of progressive painful arthritis of the knee. Excessive valgus alignment caused by the initial osteotomy raised concerns about being able to correct the extra-articular deformity intra-articularly while maintaining kinematic ligament balance. For this reason, a recorrection osteotomy was performed concurrently with the TKA. A posterior cruciate ligament–retaining (PCL-retaining) knee design (NexGen, Zimmer) was selected.
The procedure began with bone cuts for the TKA. Initial cuts were made on the femur. The tibial cut was made in valgus corresponding to the preoperative valgus deformity. The tibial recorrection osteotomy was made at the level of the original osteotomy site. A stemmed tibial component was used to cross the osteotomy site, correcting the valgus deformity and providing stability at the osteotomy site. A 3.5-mm locking compression T-plate (Synthes) was medially placed to prevent loss of correction and control rotation of the osteotomy during healing. The patient began range of motion on postoperative day 1. Continuous passive motion was not used. Protective weight-bearing continued for 6 weeks. After 6 weeks, and once there was radiograph evidence of healing at the osteotomy site, full weight-bearing was allowed.
After 4 months, the patient decided to undergo a similar procedure on the left knee. Postoperative rehabilitation was the same. A year after the bilateral TKAs, the patient maintained a Knee Society Score of 95 and a functional score of 90. After surgery, anatomical alignment was 6° (right knee) and 3° (left knee) (Figure 3), and tibial slope was 6° (right) and 7° (left) (Figures 4A, 4B). In each knee, the PCL was preserved with ligament balance.
Discussion
Clinical outcomes of TKA after high tibial osteotomy vary. Windsor and colleagues9 reported that knee arthroplasties after tibial osteotomy were less successful than primary TKAs. In small studies, both Staeheli and colleagues17 and Katz and colleagues5 found that TKA outcomes after osteotomy were satisfactory compared with outcomes of primary TKA without previous osteotomy. A meta-analysis by Ramappa and colleagues18 showed no difference in outcomes between TKAs with and without previous osteotomy. In addition, there were no differences in outcomes between TKAs performed after opening wedge versus closing wedge osteotomies.19
An arthritic knee compartment is unloaded when a high tibial osteotomy produces an extra-articular deformity. Neyret and colleagues7 reported difficulties in correcting angulations of 9° or more through soft-tissue release. Cameron and Welsh16 suggested pre–knee arthroplasty correction of the extra-articular deformity for malalignments of more than 15°. In cases of severe malalignment produced by an osteotomy, Katz and colleagues5 also suggested that a second osteotomy be performed to correct alignment before TKA.
For TKA after high tibial osteotomy, a neutral plateau resection removes more bone medially than laterally, creating medial laxity. Without correction of the tibial deformity, lateral release (or, as Krackow and Holtgrewe20 advocated, medial advancement) is required for ligament stability. Both technically demanding options may not provide complete stability throughout the arc of motion. In addition, neither corrects for rotational or sagittal deformities (the concern with correcting an extra-articular deformity with intra-articular ligament balancing).
Another option is valgus tibial resection, which maintains native ligament balance at the cost of excessive valgus alignment. In the low-demand patient, a condylar constrained implant provides a means of correcting the malalignment with knee stability.8,13,17 The increased restraint produces greater forces at the implant–bone interface and may risk early loosening.
The case presented here represents a unique situation of failed bilateral high tibial osteotomies with excessive valgus malalignment. In a similar situation, Papagelopoulos and colleagues21 suggested correcting fracture deformities before or at time of knee arthroplasty. Yoshina and colleagues22 reported using a stemmed tibial component with TKA in treating nonunion of a high tibial osteotomy. As mentioned, Katz and colleagues5 and Neyret and colleagues7 suggested preoperative correction of the osteotomy in cases of severe malalignment. Others have suggested combining recorrection osteotomy and knee arthroplasty in either consecutive operations or a single operation.23-26 Wolff and colleagues27 and Uchinou and colleagues28 described recorrection osteotomy performed concurrent with TKA. The present article is the first to report a case involving concurrent bilateral recorrection osteotomy and TKA.
In one setting, the recorrection osteotomy is performed after the bony cuts are made for the TKA. The initial tibial plateau resection is performed in valgus at the same degree of malalignment as the osteotomy. This allows the plane of the tibial resection to parallel the floor once the recorrection is finished. With use of a tibial stem crossing the osteotomy site and a derotation plate, adequate fixation of the osteotomy is obtained. The recorrection osteotomy prevents the ligaments from overlengthening, allows the native ligament balance of the knee, and preserves the PCL—which lets the surgeon obtain ligament balance for the TKA throughout the arc of motion, avoiding midstance instabilities and achieving knee alignment rotationally and in the coronal and sagittal planes.
The TKA used in the present case was a PCL-retaining design. Both posterior-stabilized and PCL-retaining designs are reasonable options for use in combination with recorrection osteotomy. A stemmed tibial component is needed to cross the osteotomy site. In our patient’s case, use of a PCL-retaining design was based on surgeon preference and experience.
Patella infera has been noted as a problem in studies on converting high tibial osteotomy to TKA.9,12,29 A postulated cause is scarring of the infrapatellar tendon after high tibial osteotomy. In addition, a higher incidence of lateral retinacular release has been identified.9-11 Patella infera did not occur in either knee in the present case, and lateral release was not required.
Our patient’s lateral radiographs (Figures 4A, 4B) showed persistence of the osteotomy plane anterior to the tibia. The osteotomy healed posteriorly but not completely anteriorly. This raises the issue of risk for nonunion when recorrection osteotomy is performed with TKA. Use of a stemmed tibial implant with a derotation plate provides the benefit of intramedullary fixation for the recorrection osteotomy. If the recorrection osteotomy were performed in a separate setting before TKA, plate fixation would be the primary fixation option. Should nonunion occur at the recorrection osteotomy site, revision of the tibial plateau with a new stemmed implant would be required in combination with plate fixation. Madelaine and colleagues30 reported on a series of 15 severe varus knees treated with both osteotomy and TKA. Two nonunions occurred. Fixation was a staple in one case and a cement wedge in the other. Risk for nonunion may be reduced with the combination of stemmed tibial implant and internal fixation with a derotation plate. Protective weight-bearing is recommended for the first 6 postoperative weeks.
Conclusion
Ligament imbalances produced by high tibial osteotomy and exacerbated by conversion to TKA are difficult to address. In this report, we have described successful single-stage high tibial osteotomy recorrection and TKA performed bilaterally in separate settings. With use of a stemmed tibial component and a derotation plate, solid fixation was obtained with an excellent clinical outcome. The malalignment was corrected while ligament balance was maintained for a PCL-retaining TKA design.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.
1. Billings A, Scott DF, Camargo MP, Hofmann AA. High tibial osteotomy with a calibrated osteotomy guide, rigid internal fixation, and early motion. Long-term follow-up. J Bone Joint Surg Am. 2000;82(1):70-79.
2. Coventry MB, Ilstrup DM, Wallrichs SL. Proximal tibial osteotomy. A critical long-term study of eighty-seven cases. J Bone Joint Surg Am. 1993;75(2):196-201.
3. Rinonapoli E, Mancini GB, Corvaglia A, Musiello S. Tibial osteotomy for varus gonarthrosis. A 10- to 21-year followup study. Clin Orthop Relat Res. 1998;(353):185-193.
4. Ritter MA, Fechtman RA. Proximal tibial osteotomy. A survivorship analysis. J Arthroplasty. 1988;3(4):309-311.
5. Katz MM, Hungerford DS, Krackow KA, Lennox DW. Results of total knee arthroplasty after failed proximal tibial osteotomy for osteoarthritis. J Bone Joint Surg Am. 1987;69(2):225-233.
6. Mont MA, Antonaides S, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. A comparison with a matched group. Clin Orthop Relat Res. 1994;299:125-130.
7. Neyret P, Deroche P, Deschamps G, Dejour H. Total knee replacement after valgus tibial osteotomy. Technical problems [in French]. Rev Chir Orthop Reparatrice Appar Mot. 1992;78(7):438-448.
8. Parvizi J, Hanssen AD, Spangehl MJ. Total knee arthroplasty following proximal tibial osteotomy: risk factors for failure. J Bone Joint Surg Am. 2004;86(3):474-479.
9. Windsor RE, Insall JN, Vince KG. Technical considerations of total knee arthroplasty after proximal tibial osteotomy. J Bone Joint Surg Am. 1988;70(4):547-555.
10. Amendola A, Rorabeck CH, Bourne RB, Apyan PM. Total knee arthroplasty following high tibial osteotomy for osteoarthritis. J Arthroplasty. 1989;(4 suppl):S11-S17.
11. Kazakos KJ, Chatzipapas C, Verettas D, Galanis V, Xarchas KC, Psillakis I. Mid-term results of total knee arthroplasty after high tibial osteotomy. Arch Orthop Trauma Surg. 2008;128(2):167-173.
12. Meding JB, Keating EM, Ritter MA, Faris PM. Total knee arthroplasty after high tibial osteotomy. A comparison study in patients who had bilateral total knee replacement. J Bone Joint Surg Am. 2000;82(9):1252-1259.
13. Niinimaki T, Eskelinen A, Ohtonen P, Puhto AP, Mann BS, Leppilahti J. Total knee arthroplasty after high tibial osteotomy: a registry-based case–control study of 1,036 knees. Arch Orthop Trauma Surg. 2014;134(1):73-77.
14. van Raaij TM, Reijman M, Furlan AD, Verhaar JA. Total knee arthroplasty after high tibial osteotomy. A systematic review. BMC Musculoskelet Disord. 2009;10:88-98.
15. Lotke PA, Ecker ML. Influence of positioning of prosthesis in total knee replacement. J Bone Joint Surg Am. 1977;59(1):77-79.
16. Cameron HU, Welsh RP. Potential complications of total knee replacement following tibial osteotomy. Orthop Rev. 1988;17(1):39-43.
17. Staeheli JW, Cass JR, Morrey BF. Condylar total knee arthroplasty after failed proximal tibial osteotomy. J Bone Joint Surg Am. 1987;69(1):28-31.
18. Ramappa M, Anand S, Jennings A. Total knee replacement following high tibial osteotomy versus total knee replacement without high tibial osteotomy: a systematic review and meta analysis. Arch Orthop Trauma Surg. 2013;133(11):1587-1593.
19. Preston S, Howard J, Naudie D, Somerville L, McAuley J. Total knee arthroplasty after high tibial osteotomy: no differences between medial and lateral osteotomy approaches. Clin Orthop Relat Res. 2014;472(1):105-110.
20. Krackow KA, Holtgrewe JL. Experience with a new technique for managing severely overcorrected valgus high tibial osteotomy at total knee arthroplasty. Clin Orthop Relat Res. 1990;(258):213-224.
21. Papagelopoulos PJ, Karachalios T, Themistocleous GS, Papadopoulos ECh, Savvidou OD, Rand JA. Total knee arthroplasty in patients with pre-existing fracture deformity. Orthopaedics. 2007;30(5):373-378.
22. Yoshina N, Takai S, Watanabe Y, Nakamura S, Kubo T. Total knee arthroplasty with long stem for treatment of nonunion after high tibial osteotomy. J Arthroplasty. 2004;19(4):528-531.
23. Mont MA, Alexander N, Krackow KA, Hungerford DS. Total knee arthroplasty after failed high tibial osteotomy. Orthop Clin North Am. 1994;25(3):515-525.
24. Scott WN. Insall & Scott’s Surgery of the Knee. Vol 1. 4th ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2006.
25. Gill T, Schemitsch EH, Brick GW, Thornhill TS. Revision total knee arthroplasty after failed unicompartmental knee arthroplasty or high tibial osteotomy. Clin Orthop Relat Res. 1995;(321):10-18.
26. Figgie HE 3rd, Goldberg VM, Heiple KG, Moller HS 3rd, Gordon NH. The influence of tibial-patellofemoral location on function of the knee in patients with the posterior stabilized condylar knee prosthesis. J Bone Joint Surg Am. 1986;68(7):1035-1040.
27. Wolff AM, Hungerford DS, Pepe CL. The effect of extraarticular varus and valgus deformity on total knee arthroplasty. Clin Orthop Relat Res. 1994;(271):35-51.
28. Uchinou S, Yano H, Shimizu K, Masumi S. A severely overcorrected high tibial osteotomy: revision by osteotomy and a long stem component. Acta Orthop Scand. 1996;67(2):193-194.
29. Noda T, Yasuda S, Nagano K, Takahara Y, Namba Y, Inoue H. Clinico-radiological study of total knee arthroplasty after high tibial osteotomy. J Orthop Sci. 2000;5(1):25-36.
30. Madelaine A, Villa V, Yela C, et al. Results and complications of single-stage total knee arthroplasty and high tibial osteotomy. Int Orthop. 2014;38(10):2091-2098.

Drug-induced immune hemolytic anemia associated with albumin-bound paclitaxel
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Life-threatening hypoglycemia resulting from a nonislet cell tumor
Nonislet cell tumor-induced hypoglycemia (NICTH), also known as Doege-Potter syndrome, is a rare paraneoplastic syndrome seen in association with various nonpancreatic tumors, benign and malignant, and comprising mesenchymal, vascular, or epithelial cell types. We report a case of recurrent life-threatening hypoglycemia from a large pelvic solitary fibrous tumor.
Click on the PDF icon at the top of this introduction to read the full article.
Nonislet cell tumor-induced hypoglycemia (NICTH), also known as Doege-Potter syndrome, is a rare paraneoplastic syndrome seen in association with various nonpancreatic tumors, benign and malignant, and comprising mesenchymal, vascular, or epithelial cell types. We report a case of recurrent life-threatening hypoglycemia from a large pelvic solitary fibrous tumor.
Click on the PDF icon at the top of this introduction to read the full article.
Nonislet cell tumor-induced hypoglycemia (NICTH), also known as Doege-Potter syndrome, is a rare paraneoplastic syndrome seen in association with various nonpancreatic tumors, benign and malignant, and comprising mesenchymal, vascular, or epithelial cell types. We report a case of recurrent life-threatening hypoglycemia from a large pelvic solitary fibrous tumor.
Click on the PDF icon at the top of this introduction to read the full article.
Colonic Dyspnea and the Morgagni Hernia: A Rare Adult Diagnosis
Congenital diaphragmatic hernias (CDHs) occur from a disruption in the muscular formation of the diaphragm, resulting in herniation of abdominal contents into the thoracic cavity. A rare diagnosis, most cases are identified in the pediatric and neonatal populations with an overall historical 50% mortality related to the diagnosis.1 More recent data published in the U.S. and Japan cite an overall survival rate of 67% to 80% secondary to improved understanding of the pathophysiology and subsequent enhancement of neonatal cardiopulmonary support adjuncts.2,3
Bochladek hernias (posterolateral space) are the most common presentation of CDH, accounting for > 90% of cases. First described by the Giovanni Batista Morgagni in On the Seats and Causes of Disease Investigated by Anatomy, the anteromedial sternocostal location is far less common and accounts for only 2% to 3% of cases.4,5 More commonly found on the right side of the diaphragm, despite protection from the liver, the right-sided space has been traditionally referred to as the Morgagni space. A left-sided defect is occasionally called the Larrey gap or space, after Napoleon’s surgeon who described the space as a potential location for pericardial drainage of tamponade.6,7
Related: Colonoscopy Bowel Preparation Instructions
There are a few congenital conditions, such as trisomy 21, Turner syndrome, Prader Willi syndrome, dextrocardia, and Tetralogy of Fallot, that have been associated with Morgagni hernias.7 Pulmonary hypertension and respiratory distress are the most common symptoms for neonatal patients; chest pain, sensations of tightness/fullness, reflux, and transient obstructive symptoms constitute the typical symptoms of adult patients with CDH. In this case study, the authors present a case of adult-onset Morgagni hernia as well as a review of the relevant literature.
Case Report
The patient was a 48-year-old man on active-duty who presented to the Naval Medical Center Portsmouth General Surgery clinic in Virginia with a 4-year history of gastroesophageal reflux-related symptoms. Specifically, he reported epigastric fullness, pyrosis, and discomfort that radiated toward his bilateral lower ribs for the previous 4 years. This discomfort was typically associated with the intake of solid food and was followed a few hours later by a loose bowel movement.
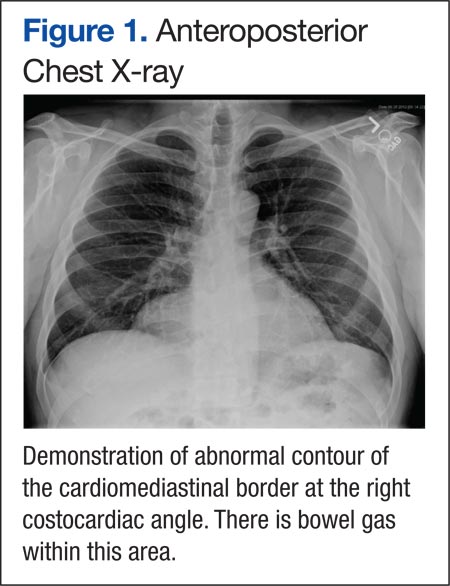
The patient was initially treated with antacids and proton pump inhibitors by his primary care physician, with only minimal relief. He also reported several months of chronic cough as well as intermittent episodes of “gasping air hunger” for about 6 years, which had been incidentally brought up during his separation physical examination. A chest X-ray performed during the workup revealed findings suggesting a right diaphragmatic hernia vs a bronchogenic cyst (Figure 1). A computed tomography (CT) of the thorax demonstrated a 3 x 8-cm hernia through the foramen of Morgagni containing a portion of the transverse colon along with intraperitoneal fat (Figures 2 and 3). 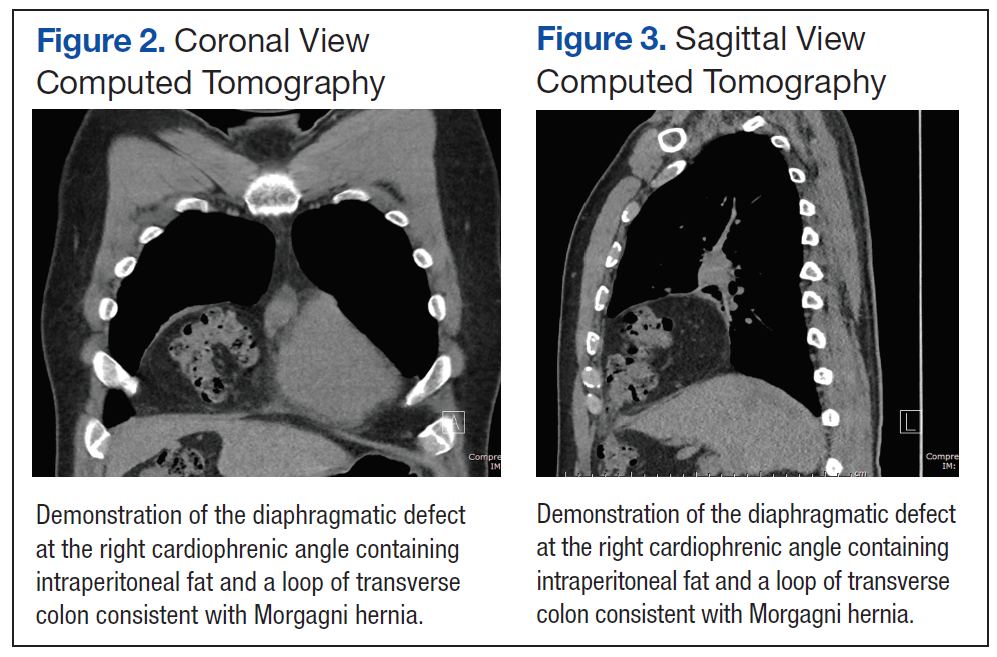
The patient underwent repair of this right Morgagni hernia via a laparoscopic approach. Intraoperative findings confirmed preoperative radiologic studies demonstrating colonic and omental contents within an easily reducible hernia sac (Figures 4 and 5). The hernia sac was left in vivo, and a combined direct hernia repair with mesh reinforcement was performed using Surgimesh XB (BG Medical, Barrington, IL) (Figure 6). The patient remained in the hospital for overnight observation and was discharged on postoperative day 1. The patient has since been seen in follow-up and is doing quite well with complete resolution of his reflux and pulmonary symptoms.

Discussion
A recent review of surgical literature revealed that over a 57-year period, 298 cases of Morgagni hernias have been described in adults.7 Although previous studies have postulated that a majority of adult patients are asymptomatic, more recent retrospective studies have found about a 70% symptomatic rate of patients with Morgagni hernias.7 The natural history of adult presentations lends itself to pulmonary (most common) or chronic upper gastrointestinal symptoms, although an acute presentation with potential volvulus and strangulation of the herniated contents has been described.7
Diagnosis is typically confirmed with a chest X-ray, although the CT scan has become more popular in the era of multimodal imaging.4,7 Multiple methods of repair have been described; however, thoracotomy has been the most widely used approach, and laparoscopy has gained popularity since the early 1990s.7 Mesh has been described in more than 60% of cases, and a laparoscopic repair has proven to have a low (< 5%) complication rate and short hospital stay.8,9 In particular, it has been suggested that a hernia defect larger than 20 to 30 cm2 should be repaired with a prosthetic adjunct, such as polypropylene, polytetrafluoroethylene, and bovine pericardium with a 1.5- to 2.5-cm mesh overlap.7,8
Related: Unusual Congenital Pulmonary Anomaly in an Adult Patient With Dyspnea
There is some controversy about the management of the hernia sac, with about 69% of surgeons choosing not to excise the sac due to concerns of intrathoracic or pericardial injury.7 In a separate study, 36 patients were evaluated retrospectively, and the hernia sac was not resected in any of the patients, with long-term follow-up revealing no evidence of recurrence.6
Conclusion
To allow for early intervention and avoidance of potentially life-threatening volvulus/strangulation, the medical practitioner has to be aware of this rare diagnosis when performing a workup for vague pulmonary and abdominal symptoms as described here. Disagreement exists over the method of repair and management of the hernia sac as well as the need for mesh buttressing of the defect. A well-planned surgical approach individualized to the patient’s anatomy, surgeon’s expertise, and hernia defect size will provide the best possible outcome with a low operative morbidity.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Holcomb GW, Murphy JP. Ashcraft’s Pediatric Surgery. 5th ed. Kansas City: Saunders Elsevier; 2010: 319-320.
2. Haroon, J, Chamberlain RS. An evidence-based review of the current treatment of congenital diaphragmatic hernia. Clin Pediatr (Phila). 2013;52(2):115-124.
3. Nagata K, Usui N, Kanamori Y, et al. The current profile and outcome of congenital diaphragmatic hernia: a nationwide survey in Japan. J Pediatr Surg. 2013;48(4):738-744.
4. Abraham V, Myla Y, Verghese S, Chandran BS. Morgagni-larrey hernia—a review of 20 cases. Indian J Surg. 2012;74(5):391-395.
5. Arora S, Haji A, Ng P. Adult Morgagni hernia: the need for clinical awareness, early diagnosis, and prompt surgical intervention. Ann R Coll Surg Engl. 2008;90(8):694-695.
6. Aghajanzadeh M, Khadem S, Khajeh Jahromi S, Gorabi HE, Ebrahimi H, Maafi AA. Clinical presentation and operative repair of Morgagni hernia. Interact Cardiovasc Thorac Surg. 2012;15(4):608-611.
7. Horton JD, Hofmann LJ, Hetz SP. Presentation and management of Morgagni hernias in adults: a review of 298 cases. Surg Endosc. 2008;22(6):1413-1420.
8. Terrosu G, Brizzolari M, Intini S, Cattin F, Bresadola V, De Anna D. Morgagni hernia: technical variation in the laparoscopic treatment. Ann Ital Chir. 2012;83(5):415-420.
9. Durak E, Gur S, Cokmez A, Atahan K, Zahtz E, Tarcan E. Laparoscopic repair of Morgagni hernia. Hernia. 2007;11(3):265-270.
Congenital diaphragmatic hernias (CDHs) occur from a disruption in the muscular formation of the diaphragm, resulting in herniation of abdominal contents into the thoracic cavity. A rare diagnosis, most cases are identified in the pediatric and neonatal populations with an overall historical 50% mortality related to the diagnosis.1 More recent data published in the U.S. and Japan cite an overall survival rate of 67% to 80% secondary to improved understanding of the pathophysiology and subsequent enhancement of neonatal cardiopulmonary support adjuncts.2,3
Bochladek hernias (posterolateral space) are the most common presentation of CDH, accounting for > 90% of cases. First described by the Giovanni Batista Morgagni in On the Seats and Causes of Disease Investigated by Anatomy, the anteromedial sternocostal location is far less common and accounts for only 2% to 3% of cases.4,5 More commonly found on the right side of the diaphragm, despite protection from the liver, the right-sided space has been traditionally referred to as the Morgagni space. A left-sided defect is occasionally called the Larrey gap or space, after Napoleon’s surgeon who described the space as a potential location for pericardial drainage of tamponade.6,7
Related: Colonoscopy Bowel Preparation Instructions
There are a few congenital conditions, such as trisomy 21, Turner syndrome, Prader Willi syndrome, dextrocardia, and Tetralogy of Fallot, that have been associated with Morgagni hernias.7 Pulmonary hypertension and respiratory distress are the most common symptoms for neonatal patients; chest pain, sensations of tightness/fullness, reflux, and transient obstructive symptoms constitute the typical symptoms of adult patients with CDH. In this case study, the authors present a case of adult-onset Morgagni hernia as well as a review of the relevant literature.
Case Report
The patient was a 48-year-old man on active-duty who presented to the Naval Medical Center Portsmouth General Surgery clinic in Virginia with a 4-year history of gastroesophageal reflux-related symptoms. Specifically, he reported epigastric fullness, pyrosis, and discomfort that radiated toward his bilateral lower ribs for the previous 4 years. This discomfort was typically associated with the intake of solid food and was followed a few hours later by a loose bowel movement.
The patient was initially treated with antacids and proton pump inhibitors by his primary care physician, with only minimal relief. He also reported several months of chronic cough as well as intermittent episodes of “gasping air hunger” for about 6 years, which had been incidentally brought up during his separation physical examination. A chest X-ray performed during the workup revealed findings suggesting a right diaphragmatic hernia vs a bronchogenic cyst (Figure 1). A computed tomography (CT) of the thorax demonstrated a 3 x 8-cm hernia through the foramen of Morgagni containing a portion of the transverse colon along with intraperitoneal fat (Figures 2 and 3).
The patient underwent repair of this right Morgagni hernia via a laparoscopic approach. Intraoperative findings confirmed preoperative radiologic studies demonstrating colonic and omental contents within an easily reducible hernia sac (Figures 4 and 5). The hernia sac was left in vivo, and a combined direct hernia repair with mesh reinforcement was performed using Surgimesh XB (BG Medical, Barrington, IL) (Figure 6). The patient remained in the hospital for overnight observation and was discharged on postoperative day 1. The patient has since been seen in follow-up and is doing quite well with complete resolution of his reflux and pulmonary symptoms.
Discussion
A recent review of surgical literature revealed that over a 57-year period, 298 cases of Morgagni hernias have been described in adults.7 Although previous studies have postulated that a majority of adult patients are asymptomatic, more recent retrospective studies have found about a 70% symptomatic rate of patients with Morgagni hernias.7 The natural history of adult presentations lends itself to pulmonary (most common) or chronic upper gastrointestinal symptoms, although an acute presentation with potential volvulus and strangulation of the herniated contents has been described.7
Diagnosis is typically confirmed with a chest X-ray, although the CT scan has become more popular in the era of multimodal imaging.4,7 Multiple methods of repair have been described; however, thoracotomy has been the most widely used approach, and laparoscopy has gained popularity since the early 1990s.7 Mesh has been described in more than 60% of cases, and a laparoscopic repair has proven to have a low (< 5%) complication rate and short hospital stay.8,9 In particular, it has been suggested that a hernia defect larger than 20 to 30 cm2 should be repaired with a prosthetic adjunct, such as polypropylene, polytetrafluoroethylene, and bovine pericardium with a 1.5- to 2.5-cm mesh overlap.7,8
Related: Unusual Congenital Pulmonary Anomaly in an Adult Patient With Dyspnea
There is some controversy about the management of the hernia sac, with about 69% of surgeons choosing not to excise the sac due to concerns of intrathoracic or pericardial injury.7 In a separate study, 36 patients were evaluated retrospectively, and the hernia sac was not resected in any of the patients, with long-term follow-up revealing no evidence of recurrence.6
Conclusion
To allow for early intervention and avoidance of potentially life-threatening volvulus/strangulation, the medical practitioner has to be aware of this rare diagnosis when performing a workup for vague pulmonary and abdominal symptoms as described here. Disagreement exists over the method of repair and management of the hernia sac as well as the need for mesh buttressing of the defect. A well-planned surgical approach individualized to the patient’s anatomy, surgeon’s expertise, and hernia defect size will provide the best possible outcome with a low operative morbidity.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Congenital diaphragmatic hernias (CDHs) occur from a disruption in the muscular formation of the diaphragm, resulting in herniation of abdominal contents into the thoracic cavity. A rare diagnosis, most cases are identified in the pediatric and neonatal populations with an overall historical 50% mortality related to the diagnosis.1 More recent data published in the U.S. and Japan cite an overall survival rate of 67% to 80% secondary to improved understanding of the pathophysiology and subsequent enhancement of neonatal cardiopulmonary support adjuncts.2,3
Bochladek hernias (posterolateral space) are the most common presentation of CDH, accounting for > 90% of cases. First described by the Giovanni Batista Morgagni in On the Seats and Causes of Disease Investigated by Anatomy, the anteromedial sternocostal location is far less common and accounts for only 2% to 3% of cases.4,5 More commonly found on the right side of the diaphragm, despite protection from the liver, the right-sided space has been traditionally referred to as the Morgagni space. A left-sided defect is occasionally called the Larrey gap or space, after Napoleon’s surgeon who described the space as a potential location for pericardial drainage of tamponade.6,7
Related: Colonoscopy Bowel Preparation Instructions
There are a few congenital conditions, such as trisomy 21, Turner syndrome, Prader Willi syndrome, dextrocardia, and Tetralogy of Fallot, that have been associated with Morgagni hernias.7 Pulmonary hypertension and respiratory distress are the most common symptoms for neonatal patients; chest pain, sensations of tightness/fullness, reflux, and transient obstructive symptoms constitute the typical symptoms of adult patients with CDH. In this case study, the authors present a case of adult-onset Morgagni hernia as well as a review of the relevant literature.
Case Report
The patient was a 48-year-old man on active-duty who presented to the Naval Medical Center Portsmouth General Surgery clinic in Virginia with a 4-year history of gastroesophageal reflux-related symptoms. Specifically, he reported epigastric fullness, pyrosis, and discomfort that radiated toward his bilateral lower ribs for the previous 4 years. This discomfort was typically associated with the intake of solid food and was followed a few hours later by a loose bowel movement.
The patient was initially treated with antacids and proton pump inhibitors by his primary care physician, with only minimal relief. He also reported several months of chronic cough as well as intermittent episodes of “gasping air hunger” for about 6 years, which had been incidentally brought up during his separation physical examination. A chest X-ray performed during the workup revealed findings suggesting a right diaphragmatic hernia vs a bronchogenic cyst (Figure 1). A computed tomography (CT) of the thorax demonstrated a 3 x 8-cm hernia through the foramen of Morgagni containing a portion of the transverse colon along with intraperitoneal fat (Figures 2 and 3).
The patient underwent repair of this right Morgagni hernia via a laparoscopic approach. Intraoperative findings confirmed preoperative radiologic studies demonstrating colonic and omental contents within an easily reducible hernia sac (Figures 4 and 5). The hernia sac was left in vivo, and a combined direct hernia repair with mesh reinforcement was performed using Surgimesh XB (BG Medical, Barrington, IL) (Figure 6). The patient remained in the hospital for overnight observation and was discharged on postoperative day 1. The patient has since been seen in follow-up and is doing quite well with complete resolution of his reflux and pulmonary symptoms.
Discussion
A recent review of surgical literature revealed that over a 57-year period, 298 cases of Morgagni hernias have been described in adults.7 Although previous studies have postulated that a majority of adult patients are asymptomatic, more recent retrospective studies have found about a 70% symptomatic rate of patients with Morgagni hernias.7 The natural history of adult presentations lends itself to pulmonary (most common) or chronic upper gastrointestinal symptoms, although an acute presentation with potential volvulus and strangulation of the herniated contents has been described.7
Diagnosis is typically confirmed with a chest X-ray, although the CT scan has become more popular in the era of multimodal imaging.4,7 Multiple methods of repair have been described; however, thoracotomy has been the most widely used approach, and laparoscopy has gained popularity since the early 1990s.7 Mesh has been described in more than 60% of cases, and a laparoscopic repair has proven to have a low (< 5%) complication rate and short hospital stay.8,9 In particular, it has been suggested that a hernia defect larger than 20 to 30 cm2 should be repaired with a prosthetic adjunct, such as polypropylene, polytetrafluoroethylene, and bovine pericardium with a 1.5- to 2.5-cm mesh overlap.7,8
Related: Unusual Congenital Pulmonary Anomaly in an Adult Patient With Dyspnea
There is some controversy about the management of the hernia sac, with about 69% of surgeons choosing not to excise the sac due to concerns of intrathoracic or pericardial injury.7 In a separate study, 36 patients were evaluated retrospectively, and the hernia sac was not resected in any of the patients, with long-term follow-up revealing no evidence of recurrence.6
Conclusion
To allow for early intervention and avoidance of potentially life-threatening volvulus/strangulation, the medical practitioner has to be aware of this rare diagnosis when performing a workup for vague pulmonary and abdominal symptoms as described here. Disagreement exists over the method of repair and management of the hernia sac as well as the need for mesh buttressing of the defect. A well-planned surgical approach individualized to the patient’s anatomy, surgeon’s expertise, and hernia defect size will provide the best possible outcome with a low operative morbidity.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Holcomb GW, Murphy JP. Ashcraft’s Pediatric Surgery. 5th ed. Kansas City: Saunders Elsevier; 2010: 319-320.
2. Haroon, J, Chamberlain RS. An evidence-based review of the current treatment of congenital diaphragmatic hernia. Clin Pediatr (Phila). 2013;52(2):115-124.
3. Nagata K, Usui N, Kanamori Y, et al. The current profile and outcome of congenital diaphragmatic hernia: a nationwide survey in Japan. J Pediatr Surg. 2013;48(4):738-744.
4. Abraham V, Myla Y, Verghese S, Chandran BS. Morgagni-larrey hernia—a review of 20 cases. Indian J Surg. 2012;74(5):391-395.
5. Arora S, Haji A, Ng P. Adult Morgagni hernia: the need for clinical awareness, early diagnosis, and prompt surgical intervention. Ann R Coll Surg Engl. 2008;90(8):694-695.
6. Aghajanzadeh M, Khadem S, Khajeh Jahromi S, Gorabi HE, Ebrahimi H, Maafi AA. Clinical presentation and operative repair of Morgagni hernia. Interact Cardiovasc Thorac Surg. 2012;15(4):608-611.
7. Horton JD, Hofmann LJ, Hetz SP. Presentation and management of Morgagni hernias in adults: a review of 298 cases. Surg Endosc. 2008;22(6):1413-1420.
8. Terrosu G, Brizzolari M, Intini S, Cattin F, Bresadola V, De Anna D. Morgagni hernia: technical variation in the laparoscopic treatment. Ann Ital Chir. 2012;83(5):415-420.
9. Durak E, Gur S, Cokmez A, Atahan K, Zahtz E, Tarcan E. Laparoscopic repair of Morgagni hernia. Hernia. 2007;11(3):265-270.
1. Holcomb GW, Murphy JP. Ashcraft’s Pediatric Surgery. 5th ed. Kansas City: Saunders Elsevier; 2010: 319-320.
2. Haroon, J, Chamberlain RS. An evidence-based review of the current treatment of congenital diaphragmatic hernia. Clin Pediatr (Phila). 2013;52(2):115-124.
3. Nagata K, Usui N, Kanamori Y, et al. The current profile and outcome of congenital diaphragmatic hernia: a nationwide survey in Japan. J Pediatr Surg. 2013;48(4):738-744.
4. Abraham V, Myla Y, Verghese S, Chandran BS. Morgagni-larrey hernia—a review of 20 cases. Indian J Surg. 2012;74(5):391-395.
5. Arora S, Haji A, Ng P. Adult Morgagni hernia: the need for clinical awareness, early diagnosis, and prompt surgical intervention. Ann R Coll Surg Engl. 2008;90(8):694-695.
6. Aghajanzadeh M, Khadem S, Khajeh Jahromi S, Gorabi HE, Ebrahimi H, Maafi AA. Clinical presentation and operative repair of Morgagni hernia. Interact Cardiovasc Thorac Surg. 2012;15(4):608-611.
7. Horton JD, Hofmann LJ, Hetz SP. Presentation and management of Morgagni hernias in adults: a review of 298 cases. Surg Endosc. 2008;22(6):1413-1420.
8. Terrosu G, Brizzolari M, Intini S, Cattin F, Bresadola V, De Anna D. Morgagni hernia: technical variation in the laparoscopic treatment. Ann Ital Chir. 2012;83(5):415-420.
9. Durak E, Gur S, Cokmez A, Atahan K, Zahtz E, Tarcan E. Laparoscopic repair of Morgagni hernia. Hernia. 2007;11(3):265-270.
Onychomatricoma: An Often Misdiagnosed Tumor of the Nails
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
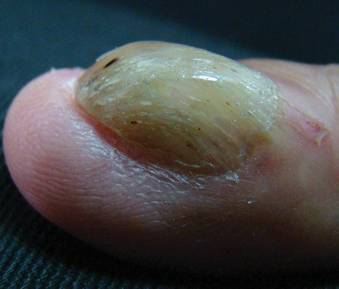
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
| 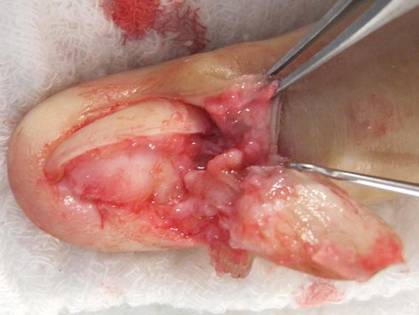
|
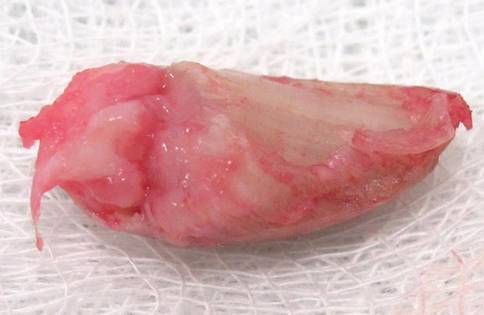
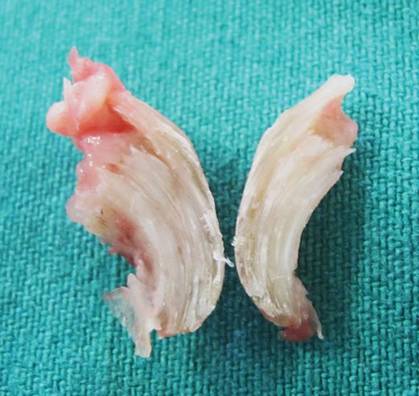
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
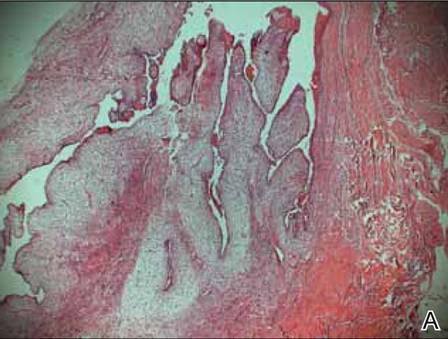
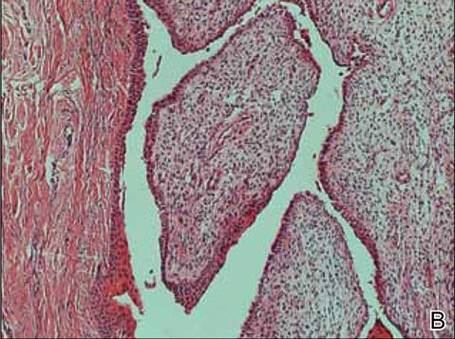
|
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
|
|
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
|
|
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
|
|
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
|
|
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
Practice Points
- Onychomatricoma has been described mostly in white individuals, but it can occur in all races and ethnic groups.
- Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors.
- Treatment of onychomatricoma is complete surgical excision.
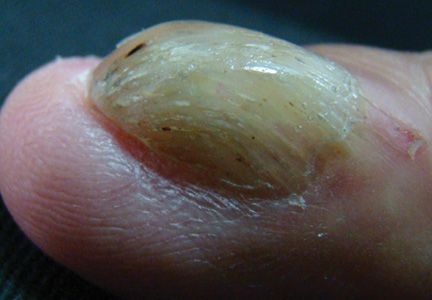
Epidermodysplasia Verruciformis: Successful Treatment With Squaric Acid Dibutylester
Epidermodysplasia verruciformis (EV) is an uncommon autosomal-recessive inherited disorder characterized by disseminated cutaneous warts in predisposed patients who are highly susceptible to genus â-papillomavirus infections. Squaric acid dibutylester (SADBE) is a contact sensitizer agent that has gained general acceptance over the years for the treatment of a variety of dermatologic diseases, including alopecia areata and cutaneous warts. We report the case of a 40-year-old woman with a balanced chromosomal translocation and lymphocytopenia who presented with the sole clinical finding of refractory multiple flat warts that had been present for 25 years. After failed attempts at therapy with oral isotretinoin, cryotherapy with topical trichloroacetic acid, and topical tretinoin, the lesions were successfully eradicated with topical SADBE with prior sensitization.
Case Report
A 40-year-old woman presented with multiple flat warts on the bilateral arms and legs of 25 years’ duration (Figure 1) that had been unsuccessfully treated by an outside physician with imiquimod cream 5% and tazarotene gel 0.1%. Her medical history was remarkable for recurrent upper respiratory tract infections, urinary tract infections, yeast infections, and otitis media. She also reported a history of 6 spontaneous miscarriages that had been attributed to a balanced chromosomal translocation between chromosomes 12 and 14.
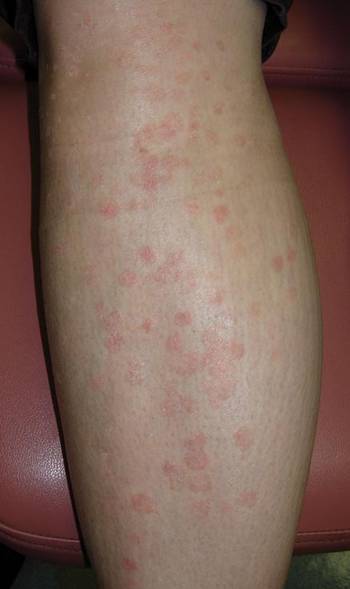
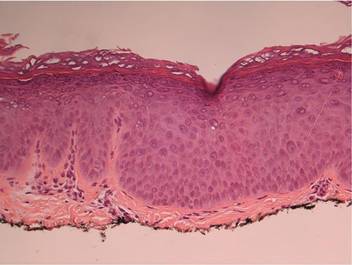
|
Laboratory evaluation revealed leukopenia, lymphopenia, and hypogammaglobulinemia, with a white blood cell count of 3600/μL (reference range, 4500–11,000/mL), a lymphocyte count of 12.1% (20%–45%), absolute CD4 count of 77 cells/μL (490–1740 cells/μL), absolute CD8 count of 56 cells/mL (180–1170 cells/μL), and serum IgM level of 17 mg/dL (48–271 mg/dL). Human immunodeficiency virus (HIV) titers were negative.
On physical examination numerous pink, flat-topped papules were noted on the forehead and bilateral arms and legs. Histologic analysis of a tangential plane biopsy of a lesion on the right leg revealed hyperkeratosis of the stratum corneum and epidermal hyperplasia (Figure 2). The epidermis also showed focal papillomatosis with areas of hypergranulosis and viropathic changes; these findings were consistent with a diagnosis of verruca plana. Human papillomavirus (HPV) DNA typing by polymerase chain reaction from the verrucous lesions showed HPV type 20, which has been associated with EV. Based on the patient’s clinical findings and HPV subtype, she was diagnosed with atypical EV.
Subsequent treatment with liquid nitrogen, tretinoin cream 0.1%, and topical trichloroacetic acid 50% failed. She received oral isotretinoin at a dosage of 80 mg daily for 9 months, but the lesions persisted and she developed alopecia and ankle stiffness; therefore, the isotretinoin was discontinued. Candida antigen testing revealed that the patient was anergic, and SADBE sensitization was subsequently initiated. Squaric acid dibutylester was utilized as a sensitizing agent, and it was formulated as 2% and 0.2% solutions in acetone, supplied in 20-mL tinted glass bottles.
Squaric acid dibutylester solution 2% under occlusion was applied to a test area on the right forearm. Three days later, results indicated prominent erythema and inflammation at the application site. Two weeks later, a chronic dermatitic response was noted at the test site (Figure 3). Squaric acid dibutylester 0.2% was then applied to an affected area on the right shin and was kept under occlusion for 48 hours. One month later, no notable changes in the lesions were observed, and no further treatments were performed. Three months later, the patient returned for evaluation and it was noted that the flat warts on the right shin that had been treated with SADBE 0.2% 4 months prior had resolved (Figure 4). Subsequently, it was noted that all of the lesions had regressed, even those that had not been treated with SADBE.
Comment
Epidermodysplasia verruciformis is a rare genodermatosis caused by a group of phylogenetically related viruses1 belonging to the β-papillomavirus genus.2,3 It is characterized by a combination of pityriasis versicolor–like lesions, reddish verrucalike plaques, and seborrheic keratosis–like plaques,1,4 preferentially on sun-exposed areas.5 The lesions undergo malignant transformation in 30% to 60% of patients,3,6 especially into squamous cell carcinomas.7 The most frequent HPV types found in EV skin lesions are 5, 8, 9, 12, 14, 15, 17, and 19 to 25; types 5 and 8 are found in 90% of cutaneous squamous cell carcinomas in EV patients.2 Human papillomavirus type 20, the type identified in our patient, has been isolated from warts in EV patients,1,2 though it is not the most common type. It has been shown that more than one HPV type could be present concurrently in the same EV patient,1 which necessitates close follow-up for skin cancer evaluation in all EV patients, as oncogenic strains may be present in some lesions.
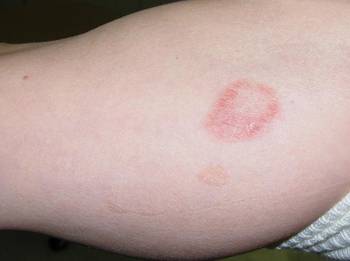
|
Epidermodysplasia verruciformis has no particular predisposition for race or geographic location.1,7 It usually is inherited in an autosomal-recessive fashion1,4,7 and has been linked to mutations in 2 EV genes located on chromosome 17: EVER1/TCM6 and EVER2/TCM8.8 However, approximately 25% of EV cases are not associated with these gene mutations,5,9 as demonstrated in our patient. Autosomal-dominant or X-linked mutations also have been reported.10 In our case, a chromosomal abnormality in the form of a balanced chromosomal translocation was present, which is unique. A connection between EV and balanced chromosomal translocation cannot be excluded and warrants further investigation.
Epidermodysplasia verruciformis has been associated with decreased cell-mediated immunity.1,7 However, nonimmunologic factors likely contribute considering the rarity of EV-like eruptions in immunodeficiency disorders11 as well as its frequent coinfection with HPV type 312 and its association with EVER1/TCM6 and EVER2/TCM8.8 Epidermodysplasia verruciformis–like lesions have been reported in several immunosuppressed states, including HIV infection,13 combined variable immunodeficiency syndrome,14 IgM deficiency,15 and CD4+ T-cell lymphocytopenia.11 Our patient’s findings fit the latter diagnostic criteria, as she had a chronically low CD4 count of 77 cells/μL, negative HIV titers, and absence of alternative explanation to the lymphopenia. Thus, we could consider her as having EV, as a low CD4 count is a known association. Her immunodeficient state could possibly be attributed to the chromosomal translocation; however, the genetic loci surrounding the chromosomal translocation have not been identified to date, leaving this hypothesis unsubstantiated. Nevertheless, in our otherwise healthy patient, no explanation was found as to why a cell-mediated deficiency would selectively favor a cutaneous HPV infection. According to Zavattaro et al,5 a possible cause could be the presence of additional genetic or environmental factors in the patient that predisposed her to this particular infection.
Every patient with EV requires close lifelong observation for skin cancer and education regarding strict sun avoidance and protection.1 Treatment options for the lesions include topical therapies with imiquimod 5%, immunomodulators, and salicylic acid16,17; oral isotretinoin18; and combinations of acitretin and interferon alfa.19 Physical ablative procedures also have been proposed, including cryotherapy with liquid nitrogen, electrosurgery, surgical excision, and laser therapies.20
Topical immunotherapy with SADBE initially was used to treat refractory alopecia areata and also has been described in the treatment of recalcitrant warts.21-24 Historically, 2,4-dinitrochlorobenzene was used for contact immunotherapy in wart management but is now avoided due to its mutagenic potential.25 Squaric acid dibutylester and diphenylcyclopropenone currently are the favored contact sensitizers, with a resolution rate of 60% reported in refractory warts.26
Topical immunotherapy involves sensitization of the patient with high-concentration (2%) SADBE on a small surface area until an eczematous dermatitis appears. The rash indicates sensitization has been achieved, and then a lower-concentration SADBE is applied to the warts. Observation of mild contact dermatitis should not be an indication to stop treatment, as this effect is an integral part of therapeutic response. No serious side effects were reported to SADBE; erythema, desquamation, edema, itching, and burning were described.23
The mechanism of action of SADBE is not clear. The most common proposed theory is the induction of a type IV hypersensitivity reaction in the warts, leading to their destruction. Other authors suggest that wart resolution is caused by a nonspecific inflammatory reaction. An argument in favor of the latter hypothesis is the spontaneous regression of untreated warts in patients treated with SADBE at a remote site, suggesting a mechanism of action beyond a simple cell-mediated process.23
Epidermodysplasia verruciformis should be included in the differential diagnosis for any eruptive, warty, papular, and plaque-type lesions that appear in immunocompromised individuals. Moreover, the diagnosis of idiopathic CD4+ T-cell lymphocytopenia should be considered in any patient with a CD4 count deficit presenting with widespread viral, fungal, or mycobacterial infection with negative HIV test. Appropriate evaluation of the absolute CD4+ counts also should be performed. In our case, it was hypothesized that the patient’s balanced chromosomal translocation was related to her lymphopenia and EV, though this correlation has yet to be confirmed. However, it is notable that her son carried the same translocation and has a normal white blood cell count and no evidence of flat warts. This case demonstrates the success of contact immunotherapy in treating these widespread and often recalcitrant lesions.
1. Vohra S, Sharma NL, Shanker V, et al. Autosomal dominant epidermodysplasia verruciformis: a clinicotherapeutic experience in two cases. Indian J Dermatol Venereol Leprol. 2010;76:557-561.
2. Dell’Oste V, Azzimonti B, De Andrea M, et al. High beta-HPV DNA loads and strong seroreactivity are present in epidermodysplasia verruciformis. J Invest Dermatol. 2009;129:1026-1034.
3. Orth G. Epidermodysplasia verruciformis: a model for understanding the oncogenicity of human papillomavirus. Ciba Found Symp. 1986;120:157-174.
4. Michael KM, Waterboer T, Pfister H, et al. Seroreactivity of 38 human papillomavirus types in epidermodysplasia verruciformis patients, relatives, and controls. J Invest Dermatol. 2010;130:841-848.
5. Zavattaro E, Azzimonti B, Mondini M, et al. Identification of defective Fas function and variation of the perforin gene in an epidermodysplasia verruciformis patient lacking EVER1 and EVER2 mutations. J Invest Dermatol. 2008;128:732-735.
6. Majewski S, Jabłońska S. Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol. 1995;131:1312-1318.
7. Robati RM, Marefat A, Saeedi M, et al. Four familial cases of epidermodysplasia verruciformis: mother and three sons. Dermatol Online J. 2009;15:8.
8. Ramoz N, Rueda LA, Bouadjar B, et al. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579-581.
9. Azzimonti B, Mondini M, De Andrea M, et al. CD8+ T-cell lymphocytopenia and lack of EVER mutations in a patient with clinically and virologically typical epidermodysplasia verruciformis. Arch Dermatol. 2005;141:1323-1325.
10. Androphy EJ, Dvoretzky I, Lowy DR. X-linked inheritance of epidermodysplasia verruciformis. genetic and virologic studies of a kindred. Arch Dermatol. 1985;121:864-868.
11. Tobin E, Rohwedder A, Holland SM, et al. Recurrent ‘sterile’ verrucous cyst abscesses and epidermodysplasia verruciformis-like eruption associated with idiopathic CD4 lymphopenia. Br J Dermatol. 2003;149:627-633.
12. Obalek S, Favre M, Szymanczyk J, et al. Human papillomavirus (HPV) types specific of epidermodysplasia verruciformis detected in warts induced by HPV3 or HPV3-related types in immunosuppressed patients. J Invest Dermatol. 1992;98:936-941.
13. Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
14. Vu J, Wallace GR, Singh R, et al. Common variable immunodeficiency syndrome associated with epidermodysplasia verruciformis. Am J Clin Dermatol. 2007;8:307-310.
15. Gul U, Soylu S, Yavuzer R. Epidermodysplasia verruciformis associated with IgM deficiency. Indian J Dermatol Venereol Leprol. 2007;73:420-422.
16. De Oliveira WR, Festa Neto C, Rady PL, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
17. Jablonska S, Majewski S. Epidermodysplasia verruciformis: what’s new? J Eur Acad Dermatol Venereol. 2003;17:381-382.
18. Rallis E, Papatheodorou G, Bimpakis E, et al. Systemic low-dose isotretinoin maintains remission status in epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2008;22:523-525.
19. Anadolu R, Oskay T, Erdem C, et al. Treatment of epidermodysplasia verruciformis with a combination of acitretin and interferon alfa-2a. J Am Acad Dermatol. 2001;45:296-299.
20. Fang F, Zhao L, Jiang MJ, et al. Epidermodysplasia verruciformis with severe hand and foot deformity successfully treated with surgical excision. J Plast Reconstr Aesthet Surg. 2008;61:338-341.
21. Huang W, Morrell D. Successful treatment of recalcitrant warts with topical squaric acid in immunosuppressed child. Pediatr Dermatol. 2008;25:275-276.
22. Hama N, Hatamochi A, Hayashi S, et al. Usefulness of topical immunotherapy with squaric acid dibutylester for refractory common warts on the face and neck. J Dermatol. 2009;36:660-662.
23. Micali G, Nasca MR, Tedeschi A, et al. Use of squaric acid dibutylester (SADBE) for cutaneous warts in children. Pediatr Dermatol. 2000;17:315-318.
24. Mastrolonardo M, Lopalco PL, Diaferio A. Topical immunotherapy with contact sensitizers: a model to study the natural history of delayedhypersensitivity. Contact Dermatitis. 2002;47:210-214.
25. Lewis HM. Topical immunotherapy of refractory warts. Cutis. 1973;12:863-869.
26. Weisshaar E, Neumann HJ, Gollnick H. Successful treatment of disseminated facial verrucae with contact immunotherapy. Eur J Dermatol. 1998;8:488-491.
Epidermodysplasia verruciformis (EV) is an uncommon autosomal-recessive inherited disorder characterized by disseminated cutaneous warts in predisposed patients who are highly susceptible to genus â-papillomavirus infections. Squaric acid dibutylester (SADBE) is a contact sensitizer agent that has gained general acceptance over the years for the treatment of a variety of dermatologic diseases, including alopecia areata and cutaneous warts. We report the case of a 40-year-old woman with a balanced chromosomal translocation and lymphocytopenia who presented with the sole clinical finding of refractory multiple flat warts that had been present for 25 years. After failed attempts at therapy with oral isotretinoin, cryotherapy with topical trichloroacetic acid, and topical tretinoin, the lesions were successfully eradicated with topical SADBE with prior sensitization.
Case Report
A 40-year-old woman presented with multiple flat warts on the bilateral arms and legs of 25 years’ duration (Figure 1) that had been unsuccessfully treated by an outside physician with imiquimod cream 5% and tazarotene gel 0.1%. Her medical history was remarkable for recurrent upper respiratory tract infections, urinary tract infections, yeast infections, and otitis media. She also reported a history of 6 spontaneous miscarriages that had been attributed to a balanced chromosomal translocation between chromosomes 12 and 14.
|
|
Laboratory evaluation revealed leukopenia, lymphopenia, and hypogammaglobulinemia, with a white blood cell count of 3600/μL (reference range, 4500–11,000/mL), a lymphocyte count of 12.1% (20%–45%), absolute CD4 count of 77 cells/μL (490–1740 cells/μL), absolute CD8 count of 56 cells/mL (180–1170 cells/μL), and serum IgM level of 17 mg/dL (48–271 mg/dL). Human immunodeficiency virus (HIV) titers were negative.
On physical examination numerous pink, flat-topped papules were noted on the forehead and bilateral arms and legs. Histologic analysis of a tangential plane biopsy of a lesion on the right leg revealed hyperkeratosis of the stratum corneum and epidermal hyperplasia (Figure 2). The epidermis also showed focal papillomatosis with areas of hypergranulosis and viropathic changes; these findings were consistent with a diagnosis of verruca plana. Human papillomavirus (HPV) DNA typing by polymerase chain reaction from the verrucous lesions showed HPV type 20, which has been associated with EV. Based on the patient’s clinical findings and HPV subtype, she was diagnosed with atypical EV.
Subsequent treatment with liquid nitrogen, tretinoin cream 0.1%, and topical trichloroacetic acid 50% failed. She received oral isotretinoin at a dosage of 80 mg daily for 9 months, but the lesions persisted and she developed alopecia and ankle stiffness; therefore, the isotretinoin was discontinued. Candida antigen testing revealed that the patient was anergic, and SADBE sensitization was subsequently initiated. Squaric acid dibutylester was utilized as a sensitizing agent, and it was formulated as 2% and 0.2% solutions in acetone, supplied in 20-mL tinted glass bottles.
Squaric acid dibutylester solution 2% under occlusion was applied to a test area on the right forearm. Three days later, results indicated prominent erythema and inflammation at the application site. Two weeks later, a chronic dermatitic response was noted at the test site (Figure 3). Squaric acid dibutylester 0.2% was then applied to an affected area on the right shin and was kept under occlusion for 48 hours. One month later, no notable changes in the lesions were observed, and no further treatments were performed. Three months later, the patient returned for evaluation and it was noted that the flat warts on the right shin that had been treated with SADBE 0.2% 4 months prior had resolved (Figure 4). Subsequently, it was noted that all of the lesions had regressed, even those that had not been treated with SADBE.
Comment
Epidermodysplasia verruciformis is a rare genodermatosis caused by a group of phylogenetically related viruses1 belonging to the β-papillomavirus genus.2,3 It is characterized by a combination of pityriasis versicolor–like lesions, reddish verrucalike plaques, and seborrheic keratosis–like plaques,1,4 preferentially on sun-exposed areas.5 The lesions undergo malignant transformation in 30% to 60% of patients,3,6 especially into squamous cell carcinomas.7 The most frequent HPV types found in EV skin lesions are 5, 8, 9, 12, 14, 15, 17, and 19 to 25; types 5 and 8 are found in 90% of cutaneous squamous cell carcinomas in EV patients.2 Human papillomavirus type 20, the type identified in our patient, has been isolated from warts in EV patients,1,2 though it is not the most common type. It has been shown that more than one HPV type could be present concurrently in the same EV patient,1 which necessitates close follow-up for skin cancer evaluation in all EV patients, as oncogenic strains may be present in some lesions.
|
|
Epidermodysplasia verruciformis has no particular predisposition for race or geographic location.1,7 It usually is inherited in an autosomal-recessive fashion1,4,7 and has been linked to mutations in 2 EV genes located on chromosome 17: EVER1/TCM6 and EVER2/TCM8.8 However, approximately 25% of EV cases are not associated with these gene mutations,5,9 as demonstrated in our patient. Autosomal-dominant or X-linked mutations also have been reported.10 In our case, a chromosomal abnormality in the form of a balanced chromosomal translocation was present, which is unique. A connection between EV and balanced chromosomal translocation cannot be excluded and warrants further investigation.
Epidermodysplasia verruciformis has been associated with decreased cell-mediated immunity.1,7 However, nonimmunologic factors likely contribute considering the rarity of EV-like eruptions in immunodeficiency disorders11 as well as its frequent coinfection with HPV type 312 and its association with EVER1/TCM6 and EVER2/TCM8.8 Epidermodysplasia verruciformis–like lesions have been reported in several immunosuppressed states, including HIV infection,13 combined variable immunodeficiency syndrome,14 IgM deficiency,15 and CD4+ T-cell lymphocytopenia.11 Our patient’s findings fit the latter diagnostic criteria, as she had a chronically low CD4 count of 77 cells/μL, negative HIV titers, and absence of alternative explanation to the lymphopenia. Thus, we could consider her as having EV, as a low CD4 count is a known association. Her immunodeficient state could possibly be attributed to the chromosomal translocation; however, the genetic loci surrounding the chromosomal translocation have not been identified to date, leaving this hypothesis unsubstantiated. Nevertheless, in our otherwise healthy patient, no explanation was found as to why a cell-mediated deficiency would selectively favor a cutaneous HPV infection. According to Zavattaro et al,5 a possible cause could be the presence of additional genetic or environmental factors in the patient that predisposed her to this particular infection.
Every patient with EV requires close lifelong observation for skin cancer and education regarding strict sun avoidance and protection.1 Treatment options for the lesions include topical therapies with imiquimod 5%, immunomodulators, and salicylic acid16,17; oral isotretinoin18; and combinations of acitretin and interferon alfa.19 Physical ablative procedures also have been proposed, including cryotherapy with liquid nitrogen, electrosurgery, surgical excision, and laser therapies.20
Topical immunotherapy with SADBE initially was used to treat refractory alopecia areata and also has been described in the treatment of recalcitrant warts.21-24 Historically, 2,4-dinitrochlorobenzene was used for contact immunotherapy in wart management but is now avoided due to its mutagenic potential.25 Squaric acid dibutylester and diphenylcyclopropenone currently are the favored contact sensitizers, with a resolution rate of 60% reported in refractory warts.26
Topical immunotherapy involves sensitization of the patient with high-concentration (2%) SADBE on a small surface area until an eczematous dermatitis appears. The rash indicates sensitization has been achieved, and then a lower-concentration SADBE is applied to the warts. Observation of mild contact dermatitis should not be an indication to stop treatment, as this effect is an integral part of therapeutic response. No serious side effects were reported to SADBE; erythema, desquamation, edema, itching, and burning were described.23
The mechanism of action of SADBE is not clear. The most common proposed theory is the induction of a type IV hypersensitivity reaction in the warts, leading to their destruction. Other authors suggest that wart resolution is caused by a nonspecific inflammatory reaction. An argument in favor of the latter hypothesis is the spontaneous regression of untreated warts in patients treated with SADBE at a remote site, suggesting a mechanism of action beyond a simple cell-mediated process.23
Epidermodysplasia verruciformis should be included in the differential diagnosis for any eruptive, warty, papular, and plaque-type lesions that appear in immunocompromised individuals. Moreover, the diagnosis of idiopathic CD4+ T-cell lymphocytopenia should be considered in any patient with a CD4 count deficit presenting with widespread viral, fungal, or mycobacterial infection with negative HIV test. Appropriate evaluation of the absolute CD4+ counts also should be performed. In our case, it was hypothesized that the patient’s balanced chromosomal translocation was related to her lymphopenia and EV, though this correlation has yet to be confirmed. However, it is notable that her son carried the same translocation and has a normal white blood cell count and no evidence of flat warts. This case demonstrates the success of contact immunotherapy in treating these widespread and often recalcitrant lesions.
Epidermodysplasia verruciformis (EV) is an uncommon autosomal-recessive inherited disorder characterized by disseminated cutaneous warts in predisposed patients who are highly susceptible to genus â-papillomavirus infections. Squaric acid dibutylester (SADBE) is a contact sensitizer agent that has gained general acceptance over the years for the treatment of a variety of dermatologic diseases, including alopecia areata and cutaneous warts. We report the case of a 40-year-old woman with a balanced chromosomal translocation and lymphocytopenia who presented with the sole clinical finding of refractory multiple flat warts that had been present for 25 years. After failed attempts at therapy with oral isotretinoin, cryotherapy with topical trichloroacetic acid, and topical tretinoin, the lesions were successfully eradicated with topical SADBE with prior sensitization.
Case Report
A 40-year-old woman presented with multiple flat warts on the bilateral arms and legs of 25 years’ duration (Figure 1) that had been unsuccessfully treated by an outside physician with imiquimod cream 5% and tazarotene gel 0.1%. Her medical history was remarkable for recurrent upper respiratory tract infections, urinary tract infections, yeast infections, and otitis media. She also reported a history of 6 spontaneous miscarriages that had been attributed to a balanced chromosomal translocation between chromosomes 12 and 14.
|
|
Laboratory evaluation revealed leukopenia, lymphopenia, and hypogammaglobulinemia, with a white blood cell count of 3600/μL (reference range, 4500–11,000/mL), a lymphocyte count of 12.1% (20%–45%), absolute CD4 count of 77 cells/μL (490–1740 cells/μL), absolute CD8 count of 56 cells/mL (180–1170 cells/μL), and serum IgM level of 17 mg/dL (48–271 mg/dL). Human immunodeficiency virus (HIV) titers were negative.
On physical examination numerous pink, flat-topped papules were noted on the forehead and bilateral arms and legs. Histologic analysis of a tangential plane biopsy of a lesion on the right leg revealed hyperkeratosis of the stratum corneum and epidermal hyperplasia (Figure 2). The epidermis also showed focal papillomatosis with areas of hypergranulosis and viropathic changes; these findings were consistent with a diagnosis of verruca plana. Human papillomavirus (HPV) DNA typing by polymerase chain reaction from the verrucous lesions showed HPV type 20, which has been associated with EV. Based on the patient’s clinical findings and HPV subtype, she was diagnosed with atypical EV.
Subsequent treatment with liquid nitrogen, tretinoin cream 0.1%, and topical trichloroacetic acid 50% failed. She received oral isotretinoin at a dosage of 80 mg daily for 9 months, but the lesions persisted and she developed alopecia and ankle stiffness; therefore, the isotretinoin was discontinued. Candida antigen testing revealed that the patient was anergic, and SADBE sensitization was subsequently initiated. Squaric acid dibutylester was utilized as a sensitizing agent, and it was formulated as 2% and 0.2% solutions in acetone, supplied in 20-mL tinted glass bottles.
Squaric acid dibutylester solution 2% under occlusion was applied to a test area on the right forearm. Three days later, results indicated prominent erythema and inflammation at the application site. Two weeks later, a chronic dermatitic response was noted at the test site (Figure 3). Squaric acid dibutylester 0.2% was then applied to an affected area on the right shin and was kept under occlusion for 48 hours. One month later, no notable changes in the lesions were observed, and no further treatments were performed. Three months later, the patient returned for evaluation and it was noted that the flat warts on the right shin that had been treated with SADBE 0.2% 4 months prior had resolved (Figure 4). Subsequently, it was noted that all of the lesions had regressed, even those that had not been treated with SADBE.
Comment
Epidermodysplasia verruciformis is a rare genodermatosis caused by a group of phylogenetically related viruses1 belonging to the β-papillomavirus genus.2,3 It is characterized by a combination of pityriasis versicolor–like lesions, reddish verrucalike plaques, and seborrheic keratosis–like plaques,1,4 preferentially on sun-exposed areas.5 The lesions undergo malignant transformation in 30% to 60% of patients,3,6 especially into squamous cell carcinomas.7 The most frequent HPV types found in EV skin lesions are 5, 8, 9, 12, 14, 15, 17, and 19 to 25; types 5 and 8 are found in 90% of cutaneous squamous cell carcinomas in EV patients.2 Human papillomavirus type 20, the type identified in our patient, has been isolated from warts in EV patients,1,2 though it is not the most common type. It has been shown that more than one HPV type could be present concurrently in the same EV patient,1 which necessitates close follow-up for skin cancer evaluation in all EV patients, as oncogenic strains may be present in some lesions.
|
|
Epidermodysplasia verruciformis has no particular predisposition for race or geographic location.1,7 It usually is inherited in an autosomal-recessive fashion1,4,7 and has been linked to mutations in 2 EV genes located on chromosome 17: EVER1/TCM6 and EVER2/TCM8.8 However, approximately 25% of EV cases are not associated with these gene mutations,5,9 as demonstrated in our patient. Autosomal-dominant or X-linked mutations also have been reported.10 In our case, a chromosomal abnormality in the form of a balanced chromosomal translocation was present, which is unique. A connection between EV and balanced chromosomal translocation cannot be excluded and warrants further investigation.
Epidermodysplasia verruciformis has been associated with decreased cell-mediated immunity.1,7 However, nonimmunologic factors likely contribute considering the rarity of EV-like eruptions in immunodeficiency disorders11 as well as its frequent coinfection with HPV type 312 and its association with EVER1/TCM6 and EVER2/TCM8.8 Epidermodysplasia verruciformis–like lesions have been reported in several immunosuppressed states, including HIV infection,13 combined variable immunodeficiency syndrome,14 IgM deficiency,15 and CD4+ T-cell lymphocytopenia.11 Our patient’s findings fit the latter diagnostic criteria, as she had a chronically low CD4 count of 77 cells/μL, negative HIV titers, and absence of alternative explanation to the lymphopenia. Thus, we could consider her as having EV, as a low CD4 count is a known association. Her immunodeficient state could possibly be attributed to the chromosomal translocation; however, the genetic loci surrounding the chromosomal translocation have not been identified to date, leaving this hypothesis unsubstantiated. Nevertheless, in our otherwise healthy patient, no explanation was found as to why a cell-mediated deficiency would selectively favor a cutaneous HPV infection. According to Zavattaro et al,5 a possible cause could be the presence of additional genetic or environmental factors in the patient that predisposed her to this particular infection.
Every patient with EV requires close lifelong observation for skin cancer and education regarding strict sun avoidance and protection.1 Treatment options for the lesions include topical therapies with imiquimod 5%, immunomodulators, and salicylic acid16,17; oral isotretinoin18; and combinations of acitretin and interferon alfa.19 Physical ablative procedures also have been proposed, including cryotherapy with liquid nitrogen, electrosurgery, surgical excision, and laser therapies.20
Topical immunotherapy with SADBE initially was used to treat refractory alopecia areata and also has been described in the treatment of recalcitrant warts.21-24 Historically, 2,4-dinitrochlorobenzene was used for contact immunotherapy in wart management but is now avoided due to its mutagenic potential.25 Squaric acid dibutylester and diphenylcyclopropenone currently are the favored contact sensitizers, with a resolution rate of 60% reported in refractory warts.26
Topical immunotherapy involves sensitization of the patient with high-concentration (2%) SADBE on a small surface area until an eczematous dermatitis appears. The rash indicates sensitization has been achieved, and then a lower-concentration SADBE is applied to the warts. Observation of mild contact dermatitis should not be an indication to stop treatment, as this effect is an integral part of therapeutic response. No serious side effects were reported to SADBE; erythema, desquamation, edema, itching, and burning were described.23
The mechanism of action of SADBE is not clear. The most common proposed theory is the induction of a type IV hypersensitivity reaction in the warts, leading to their destruction. Other authors suggest that wart resolution is caused by a nonspecific inflammatory reaction. An argument in favor of the latter hypothesis is the spontaneous regression of untreated warts in patients treated with SADBE at a remote site, suggesting a mechanism of action beyond a simple cell-mediated process.23
Epidermodysplasia verruciformis should be included in the differential diagnosis for any eruptive, warty, papular, and plaque-type lesions that appear in immunocompromised individuals. Moreover, the diagnosis of idiopathic CD4+ T-cell lymphocytopenia should be considered in any patient with a CD4 count deficit presenting with widespread viral, fungal, or mycobacterial infection with negative HIV test. Appropriate evaluation of the absolute CD4+ counts also should be performed. In our case, it was hypothesized that the patient’s balanced chromosomal translocation was related to her lymphopenia and EV, though this correlation has yet to be confirmed. However, it is notable that her son carried the same translocation and has a normal white blood cell count and no evidence of flat warts. This case demonstrates the success of contact immunotherapy in treating these widespread and often recalcitrant lesions.
1. Vohra S, Sharma NL, Shanker V, et al. Autosomal dominant epidermodysplasia verruciformis: a clinicotherapeutic experience in two cases. Indian J Dermatol Venereol Leprol. 2010;76:557-561.
2. Dell’Oste V, Azzimonti B, De Andrea M, et al. High beta-HPV DNA loads and strong seroreactivity are present in epidermodysplasia verruciformis. J Invest Dermatol. 2009;129:1026-1034.
3. Orth G. Epidermodysplasia verruciformis: a model for understanding the oncogenicity of human papillomavirus. Ciba Found Symp. 1986;120:157-174.
4. Michael KM, Waterboer T, Pfister H, et al. Seroreactivity of 38 human papillomavirus types in epidermodysplasia verruciformis patients, relatives, and controls. J Invest Dermatol. 2010;130:841-848.
5. Zavattaro E, Azzimonti B, Mondini M, et al. Identification of defective Fas function and variation of the perforin gene in an epidermodysplasia verruciformis patient lacking EVER1 and EVER2 mutations. J Invest Dermatol. 2008;128:732-735.
6. Majewski S, Jabłońska S. Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol. 1995;131:1312-1318.
7. Robati RM, Marefat A, Saeedi M, et al. Four familial cases of epidermodysplasia verruciformis: mother and three sons. Dermatol Online J. 2009;15:8.
8. Ramoz N, Rueda LA, Bouadjar B, et al. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579-581.
9. Azzimonti B, Mondini M, De Andrea M, et al. CD8+ T-cell lymphocytopenia and lack of EVER mutations in a patient with clinically and virologically typical epidermodysplasia verruciformis. Arch Dermatol. 2005;141:1323-1325.
10. Androphy EJ, Dvoretzky I, Lowy DR. X-linked inheritance of epidermodysplasia verruciformis. genetic and virologic studies of a kindred. Arch Dermatol. 1985;121:864-868.
11. Tobin E, Rohwedder A, Holland SM, et al. Recurrent ‘sterile’ verrucous cyst abscesses and epidermodysplasia verruciformis-like eruption associated with idiopathic CD4 lymphopenia. Br J Dermatol. 2003;149:627-633.
12. Obalek S, Favre M, Szymanczyk J, et al. Human papillomavirus (HPV) types specific of epidermodysplasia verruciformis detected in warts induced by HPV3 or HPV3-related types in immunosuppressed patients. J Invest Dermatol. 1992;98:936-941.
13. Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
14. Vu J, Wallace GR, Singh R, et al. Common variable immunodeficiency syndrome associated with epidermodysplasia verruciformis. Am J Clin Dermatol. 2007;8:307-310.
15. Gul U, Soylu S, Yavuzer R. Epidermodysplasia verruciformis associated with IgM deficiency. Indian J Dermatol Venereol Leprol. 2007;73:420-422.
16. De Oliveira WR, Festa Neto C, Rady PL, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
17. Jablonska S, Majewski S. Epidermodysplasia verruciformis: what’s new? J Eur Acad Dermatol Venereol. 2003;17:381-382.
18. Rallis E, Papatheodorou G, Bimpakis E, et al. Systemic low-dose isotretinoin maintains remission status in epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2008;22:523-525.
19. Anadolu R, Oskay T, Erdem C, et al. Treatment of epidermodysplasia verruciformis with a combination of acitretin and interferon alfa-2a. J Am Acad Dermatol. 2001;45:296-299.
20. Fang F, Zhao L, Jiang MJ, et al. Epidermodysplasia verruciformis with severe hand and foot deformity successfully treated with surgical excision. J Plast Reconstr Aesthet Surg. 2008;61:338-341.
21. Huang W, Morrell D. Successful treatment of recalcitrant warts with topical squaric acid in immunosuppressed child. Pediatr Dermatol. 2008;25:275-276.
22. Hama N, Hatamochi A, Hayashi S, et al. Usefulness of topical immunotherapy with squaric acid dibutylester for refractory common warts on the face and neck. J Dermatol. 2009;36:660-662.
23. Micali G, Nasca MR, Tedeschi A, et al. Use of squaric acid dibutylester (SADBE) for cutaneous warts in children. Pediatr Dermatol. 2000;17:315-318.
24. Mastrolonardo M, Lopalco PL, Diaferio A. Topical immunotherapy with contact sensitizers: a model to study the natural history of delayedhypersensitivity. Contact Dermatitis. 2002;47:210-214.
25. Lewis HM. Topical immunotherapy of refractory warts. Cutis. 1973;12:863-869.
26. Weisshaar E, Neumann HJ, Gollnick H. Successful treatment of disseminated facial verrucae with contact immunotherapy. Eur J Dermatol. 1998;8:488-491.
1. Vohra S, Sharma NL, Shanker V, et al. Autosomal dominant epidermodysplasia verruciformis: a clinicotherapeutic experience in two cases. Indian J Dermatol Venereol Leprol. 2010;76:557-561.
2. Dell’Oste V, Azzimonti B, De Andrea M, et al. High beta-HPV DNA loads and strong seroreactivity are present in epidermodysplasia verruciformis. J Invest Dermatol. 2009;129:1026-1034.
3. Orth G. Epidermodysplasia verruciformis: a model for understanding the oncogenicity of human papillomavirus. Ciba Found Symp. 1986;120:157-174.
4. Michael KM, Waterboer T, Pfister H, et al. Seroreactivity of 38 human papillomavirus types in epidermodysplasia verruciformis patients, relatives, and controls. J Invest Dermatol. 2010;130:841-848.
5. Zavattaro E, Azzimonti B, Mondini M, et al. Identification of defective Fas function and variation of the perforin gene in an epidermodysplasia verruciformis patient lacking EVER1 and EVER2 mutations. J Invest Dermatol. 2008;128:732-735.
6. Majewski S, Jabłońska S. Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol. 1995;131:1312-1318.
7. Robati RM, Marefat A, Saeedi M, et al. Four familial cases of epidermodysplasia verruciformis: mother and three sons. Dermatol Online J. 2009;15:8.
8. Ramoz N, Rueda LA, Bouadjar B, et al. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579-581.
9. Azzimonti B, Mondini M, De Andrea M, et al. CD8+ T-cell lymphocytopenia and lack of EVER mutations in a patient with clinically and virologically typical epidermodysplasia verruciformis. Arch Dermatol. 2005;141:1323-1325.
10. Androphy EJ, Dvoretzky I, Lowy DR. X-linked inheritance of epidermodysplasia verruciformis. genetic and virologic studies of a kindred. Arch Dermatol. 1985;121:864-868.
11. Tobin E, Rohwedder A, Holland SM, et al. Recurrent ‘sterile’ verrucous cyst abscesses and epidermodysplasia verruciformis-like eruption associated with idiopathic CD4 lymphopenia. Br J Dermatol. 2003;149:627-633.
12. Obalek S, Favre M, Szymanczyk J, et al. Human papillomavirus (HPV) types specific of epidermodysplasia verruciformis detected in warts induced by HPV3 or HPV3-related types in immunosuppressed patients. J Invest Dermatol. 1992;98:936-941.
13. Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
14. Vu J, Wallace GR, Singh R, et al. Common variable immunodeficiency syndrome associated with epidermodysplasia verruciformis. Am J Clin Dermatol. 2007;8:307-310.
15. Gul U, Soylu S, Yavuzer R. Epidermodysplasia verruciformis associated with IgM deficiency. Indian J Dermatol Venereol Leprol. 2007;73:420-422.
16. De Oliveira WR, Festa Neto C, Rady PL, et al. Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394-398.
17. Jablonska S, Majewski S. Epidermodysplasia verruciformis: what’s new? J Eur Acad Dermatol Venereol. 2003;17:381-382.
18. Rallis E, Papatheodorou G, Bimpakis E, et al. Systemic low-dose isotretinoin maintains remission status in epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2008;22:523-525.
19. Anadolu R, Oskay T, Erdem C, et al. Treatment of epidermodysplasia verruciformis with a combination of acitretin and interferon alfa-2a. J Am Acad Dermatol. 2001;45:296-299.
20. Fang F, Zhao L, Jiang MJ, et al. Epidermodysplasia verruciformis with severe hand and foot deformity successfully treated with surgical excision. J Plast Reconstr Aesthet Surg. 2008;61:338-341.
21. Huang W, Morrell D. Successful treatment of recalcitrant warts with topical squaric acid in immunosuppressed child. Pediatr Dermatol. 2008;25:275-276.
22. Hama N, Hatamochi A, Hayashi S, et al. Usefulness of topical immunotherapy with squaric acid dibutylester for refractory common warts on the face and neck. J Dermatol. 2009;36:660-662.
23. Micali G, Nasca MR, Tedeschi A, et al. Use of squaric acid dibutylester (SADBE) for cutaneous warts in children. Pediatr Dermatol. 2000;17:315-318.
24. Mastrolonardo M, Lopalco PL, Diaferio A. Topical immunotherapy with contact sensitizers: a model to study the natural history of delayedhypersensitivity. Contact Dermatitis. 2002;47:210-214.
25. Lewis HM. Topical immunotherapy of refractory warts. Cutis. 1973;12:863-869.
26. Weisshaar E, Neumann HJ, Gollnick H. Successful treatment of disseminated facial verrucae with contact immunotherapy. Eur J Dermatol. 1998;8:488-491.
Practice Points
- Epidermodysplasia verruciformis (EV) is a rare immune deficiency. Associated warts are difficult to treat.
- Topical immunotherapy with squaric acid dibutylester (SADBE) has successfully treated long-standing warts in an EV patient.
- Consider immunotherapy with a contact sensitizer such as SADBE to treat resistant warts, even in immune deficiency patients.
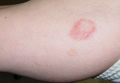
Evaluating Endoleaks in the Dermatology Office
Endoleaks are common complications following endovascular aneurysm repairs (EVARs) that may occur any time after surgery. There are 5 types of endoleaks with various etiologies. A type V endoleak (also known as endotension) is not considered a true endoleak but instead is characterized by continued aneurysm expansion without a leak, which is demonstrated via imaging tests.1 Type V endoleaks typically require open aneurysm repair.2 We report the case of a 69-year-old woman who presented to our dermatology office for treatment of a suspected lipoma overlying the right mid sternum that was confirmed to be a type V endoleak via computed tomography angiography.
Case Report
A 69-year-old woman was referred to our dermatology office by her primary care physician for evaluation of a subcutaneous mass overlying the right mid sternum, which was a suspected lipoma. The patient reported that the mass had been present for approximately 2 weeks and was enlarging but otherwise asymptomatic. Her medical history was remarkable for hypertension, an ascending aortic aneurysm, and a subsequent aortic valve replacement approximately 2.5 years prior. Her current medications included amlodipine, lisinopril, nebivolol, ibuprofen, and aspirin. She denied use of alcohol, tobacco, or illicit drugs. A review of systems was noncontributory.
Physical examination revealed a single 3.5×4.5-cm, soft, nonmobile subcutaneous mass located at the site of the thoracotomy scar (Figure 1). The mass appeared to have a central attachment to the sternum. No erythema, swelling, or exudate was noted, and the patient denied tenderness on palpation. The diagnosis of lipoma was questioned, and the patient was referred for ultrasonography and computed tomography angiography. Ultrasonography showed a nonspecific chest wall mass with internal blood flow, and computed tomography angiography showed a large, low-attenuation collection of blood around the entire circumference of the ascending aorta, extending from the aortic root to the arch of the aorta. There was extension of the collection of blood through either the sternocostal junction or the sternotomy defect into the subcutaneous tissue anterior to the sternum (Figure 2). Findings were most consistent with a type V endoleak, and the patient was referred to a cardiothoracic surgeon for treatment. We later learned that our patient died during surgery attempting to repair the aneurysm approximately 2 weeks after her presentation to our office.

Comment
An endoleak is a common complication following an EVAR that is characterized by persistent blood flow within the aneurysm sac. Endoleaks have been described as the Achilles’ heel of EVARs.1 The goal of an EVAR is to create a complete seal so that the flow of blood completely excludes the aneurysm, thus ultimately preventing an aneurysm rupture. An endoleak results when there is failure to obtain a complete seal due to a variety of different mechanisms. White et al3 first described and classified endoleaks in 1997. The initial terminology used to classify endoleaks was based on timing (primary or secondary/late) and location (graft related/perigraft or non–graft related/retrograde). Today, endoleaks are classified into 5 types, 3 of which are considered true endoleaks and 2 of which are not.4 Type I endoleaks result from a failure to create an adequate seal at one of the attachments of the graft to the vessel wall. Type II endoleaks are due to retrograde flow through collateral vessels into the aneurysm sac. They are much more common than type I, occurring in 10% to 25% of abdominal endograft cases. The last true endoleak, type III, occurs due to device failure in the form of disjunction of the components of the graft system (type IIIa) or a defect in the graft fabric (type IIIb). Type IV and type V endoleaks are not considered to be true endoleaks. Type IV endoleaks are due to the porosity of the graft material and have virtually been eliminated by changes in graft materials to decrease porosity. Type V endoleaks are characterized by continued blood flow into the aneurysm without any evidence of a leak on any imaging modality. Type V endoleaks are poorly understood but are believed to be due to pulsation of the graft wall, which is transmitted through the perivascular space to the aneurysm wall.4

|
Treatment of type V endoleaks is controversial. It is important to characterize the endoleak by various imaging modalities, and if a type V endoleak is confirmed, an open aneurysm repair often is required.2 A case of nonsurgical management of a type V endoleak has been described but is rare.5 In this case, the patient was referred to our dermatology office by her primary care physician for what appeared to be a benign lipoma, but it proved to be a type V endoleak on further examination. It is imperative that dermatologists are aware of endoleaks as common complications of EVARs, as they can be life threatening and usually require surgical intervention.
Conclusion
Endoleaks are common complications of EVARs. Dermatologists may encounter endoleaks that have been misdiagnosed as benign subcutaneous masses such as lipomas. It is imperative that dermatologists are aware of endoleaks, and patients who present with subcutaneous thoracic masses with a history of aneurysm repair require imaging, including computed tomography angiography, and referral to a cardiothoracic surgeon if appropriate.
1. Rosen RJ, Green RM. Endoleak management following endovascular aneurysm repair. J Vasc Interv Radiol. 2008;19(suppl 6):S37-S43.
2. Stavropoulos SW, Charagundla SR. Imaging techniques for detection and management of endoleaks after endovascular aortic aneurysm repair. Radiology. 2007;243:641-655.
3. White GH, Yu W, May J, et al. Endoleak as a complication of endoluminal grafting of abdominal aortic aneurysms: classification, incidence, diagnosis and management. J Endovascular Surg. 1997;4:152-168.
4. Veith FJ, Baum BA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg. 2002;35:1029-1035.
5. Mennander A, Pimenoff G, Heikkinen M, et al. Nonoperative approach to endotension. J Vasc Surg. 2005;42:194-198.
Endoleaks are common complications following endovascular aneurysm repairs (EVARs) that may occur any time after surgery. There are 5 types of endoleaks with various etiologies. A type V endoleak (also known as endotension) is not considered a true endoleak but instead is characterized by continued aneurysm expansion without a leak, which is demonstrated via imaging tests.1 Type V endoleaks typically require open aneurysm repair.2 We report the case of a 69-year-old woman who presented to our dermatology office for treatment of a suspected lipoma overlying the right mid sternum that was confirmed to be a type V endoleak via computed tomography angiography.
Case Report
A 69-year-old woman was referred to our dermatology office by her primary care physician for evaluation of a subcutaneous mass overlying the right mid sternum, which was a suspected lipoma. The patient reported that the mass had been present for approximately 2 weeks and was enlarging but otherwise asymptomatic. Her medical history was remarkable for hypertension, an ascending aortic aneurysm, and a subsequent aortic valve replacement approximately 2.5 years prior. Her current medications included amlodipine, lisinopril, nebivolol, ibuprofen, and aspirin. She denied use of alcohol, tobacco, or illicit drugs. A review of systems was noncontributory.
Physical examination revealed a single 3.5×4.5-cm, soft, nonmobile subcutaneous mass located at the site of the thoracotomy scar (Figure 1). The mass appeared to have a central attachment to the sternum. No erythema, swelling, or exudate was noted, and the patient denied tenderness on palpation. The diagnosis of lipoma was questioned, and the patient was referred for ultrasonography and computed tomography angiography. Ultrasonography showed a nonspecific chest wall mass with internal blood flow, and computed tomography angiography showed a large, low-attenuation collection of blood around the entire circumference of the ascending aorta, extending from the aortic root to the arch of the aorta. There was extension of the collection of blood through either the sternocostal junction or the sternotomy defect into the subcutaneous tissue anterior to the sternum (Figure 2). Findings were most consistent with a type V endoleak, and the patient was referred to a cardiothoracic surgeon for treatment. We later learned that our patient died during surgery attempting to repair the aneurysm approximately 2 weeks after her presentation to our office.
Comment
An endoleak is a common complication following an EVAR that is characterized by persistent blood flow within the aneurysm sac. Endoleaks have been described as the Achilles’ heel of EVARs.1 The goal of an EVAR is to create a complete seal so that the flow of blood completely excludes the aneurysm, thus ultimately preventing an aneurysm rupture. An endoleak results when there is failure to obtain a complete seal due to a variety of different mechanisms. White et al3 first described and classified endoleaks in 1997. The initial terminology used to classify endoleaks was based on timing (primary or secondary/late) and location (graft related/perigraft or non–graft related/retrograde). Today, endoleaks are classified into 5 types, 3 of which are considered true endoleaks and 2 of which are not.4 Type I endoleaks result from a failure to create an adequate seal at one of the attachments of the graft to the vessel wall. Type II endoleaks are due to retrograde flow through collateral vessels into the aneurysm sac. They are much more common than type I, occurring in 10% to 25% of abdominal endograft cases. The last true endoleak, type III, occurs due to device failure in the form of disjunction of the components of the graft system (type IIIa) or a defect in the graft fabric (type IIIb). Type IV and type V endoleaks are not considered to be true endoleaks. Type IV endoleaks are due to the porosity of the graft material and have virtually been eliminated by changes in graft materials to decrease porosity. Type V endoleaks are characterized by continued blood flow into the aneurysm without any evidence of a leak on any imaging modality. Type V endoleaks are poorly understood but are believed to be due to pulsation of the graft wall, which is transmitted through the perivascular space to the aneurysm wall.4
|
|
Treatment of type V endoleaks is controversial. It is important to characterize the endoleak by various imaging modalities, and if a type V endoleak is confirmed, an open aneurysm repair often is required.2 A case of nonsurgical management of a type V endoleak has been described but is rare.5 In this case, the patient was referred to our dermatology office by her primary care physician for what appeared to be a benign lipoma, but it proved to be a type V endoleak on further examination. It is imperative that dermatologists are aware of endoleaks as common complications of EVARs, as they can be life threatening and usually require surgical intervention.
Conclusion
Endoleaks are common complications of EVARs. Dermatologists may encounter endoleaks that have been misdiagnosed as benign subcutaneous masses such as lipomas. It is imperative that dermatologists are aware of endoleaks, and patients who present with subcutaneous thoracic masses with a history of aneurysm repair require imaging, including computed tomography angiography, and referral to a cardiothoracic surgeon if appropriate.
Endoleaks are common complications following endovascular aneurysm repairs (EVARs) that may occur any time after surgery. There are 5 types of endoleaks with various etiologies. A type V endoleak (also known as endotension) is not considered a true endoleak but instead is characterized by continued aneurysm expansion without a leak, which is demonstrated via imaging tests.1 Type V endoleaks typically require open aneurysm repair.2 We report the case of a 69-year-old woman who presented to our dermatology office for treatment of a suspected lipoma overlying the right mid sternum that was confirmed to be a type V endoleak via computed tomography angiography.
Case Report
A 69-year-old woman was referred to our dermatology office by her primary care physician for evaluation of a subcutaneous mass overlying the right mid sternum, which was a suspected lipoma. The patient reported that the mass had been present for approximately 2 weeks and was enlarging but otherwise asymptomatic. Her medical history was remarkable for hypertension, an ascending aortic aneurysm, and a subsequent aortic valve replacement approximately 2.5 years prior. Her current medications included amlodipine, lisinopril, nebivolol, ibuprofen, and aspirin. She denied use of alcohol, tobacco, or illicit drugs. A review of systems was noncontributory.
Physical examination revealed a single 3.5×4.5-cm, soft, nonmobile subcutaneous mass located at the site of the thoracotomy scar (Figure 1). The mass appeared to have a central attachment to the sternum. No erythema, swelling, or exudate was noted, and the patient denied tenderness on palpation. The diagnosis of lipoma was questioned, and the patient was referred for ultrasonography and computed tomography angiography. Ultrasonography showed a nonspecific chest wall mass with internal blood flow, and computed tomography angiography showed a large, low-attenuation collection of blood around the entire circumference of the ascending aorta, extending from the aortic root to the arch of the aorta. There was extension of the collection of blood through either the sternocostal junction or the sternotomy defect into the subcutaneous tissue anterior to the sternum (Figure 2). Findings were most consistent with a type V endoleak, and the patient was referred to a cardiothoracic surgeon for treatment. We later learned that our patient died during surgery attempting to repair the aneurysm approximately 2 weeks after her presentation to our office.
Comment
An endoleak is a common complication following an EVAR that is characterized by persistent blood flow within the aneurysm sac. Endoleaks have been described as the Achilles’ heel of EVARs.1 The goal of an EVAR is to create a complete seal so that the flow of blood completely excludes the aneurysm, thus ultimately preventing an aneurysm rupture. An endoleak results when there is failure to obtain a complete seal due to a variety of different mechanisms. White et al3 first described and classified endoleaks in 1997. The initial terminology used to classify endoleaks was based on timing (primary or secondary/late) and location (graft related/perigraft or non–graft related/retrograde). Today, endoleaks are classified into 5 types, 3 of which are considered true endoleaks and 2 of which are not.4 Type I endoleaks result from a failure to create an adequate seal at one of the attachments of the graft to the vessel wall. Type II endoleaks are due to retrograde flow through collateral vessels into the aneurysm sac. They are much more common than type I, occurring in 10% to 25% of abdominal endograft cases. The last true endoleak, type III, occurs due to device failure in the form of disjunction of the components of the graft system (type IIIa) or a defect in the graft fabric (type IIIb). Type IV and type V endoleaks are not considered to be true endoleaks. Type IV endoleaks are due to the porosity of the graft material and have virtually been eliminated by changes in graft materials to decrease porosity. Type V endoleaks are characterized by continued blood flow into the aneurysm without any evidence of a leak on any imaging modality. Type V endoleaks are poorly understood but are believed to be due to pulsation of the graft wall, which is transmitted through the perivascular space to the aneurysm wall.4
|
|
Treatment of type V endoleaks is controversial. It is important to characterize the endoleak by various imaging modalities, and if a type V endoleak is confirmed, an open aneurysm repair often is required.2 A case of nonsurgical management of a type V endoleak has been described but is rare.5 In this case, the patient was referred to our dermatology office by her primary care physician for what appeared to be a benign lipoma, but it proved to be a type V endoleak on further examination. It is imperative that dermatologists are aware of endoleaks as common complications of EVARs, as they can be life threatening and usually require surgical intervention.
Conclusion
Endoleaks are common complications of EVARs. Dermatologists may encounter endoleaks that have been misdiagnosed as benign subcutaneous masses such as lipomas. It is imperative that dermatologists are aware of endoleaks, and patients who present with subcutaneous thoracic masses with a history of aneurysm repair require imaging, including computed tomography angiography, and referral to a cardiothoracic surgeon if appropriate.
1. Rosen RJ, Green RM. Endoleak management following endovascular aneurysm repair. J Vasc Interv Radiol. 2008;19(suppl 6):S37-S43.
2. Stavropoulos SW, Charagundla SR. Imaging techniques for detection and management of endoleaks after endovascular aortic aneurysm repair. Radiology. 2007;243:641-655.
3. White GH, Yu W, May J, et al. Endoleak as a complication of endoluminal grafting of abdominal aortic aneurysms: classification, incidence, diagnosis and management. J Endovascular Surg. 1997;4:152-168.
4. Veith FJ, Baum BA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg. 2002;35:1029-1035.
5. Mennander A, Pimenoff G, Heikkinen M, et al. Nonoperative approach to endotension. J Vasc Surg. 2005;42:194-198.
1. Rosen RJ, Green RM. Endoleak management following endovascular aneurysm repair. J Vasc Interv Radiol. 2008;19(suppl 6):S37-S43.
2. Stavropoulos SW, Charagundla SR. Imaging techniques for detection and management of endoleaks after endovascular aortic aneurysm repair. Radiology. 2007;243:641-655.
3. White GH, Yu W, May J, et al. Endoleak as a complication of endoluminal grafting of abdominal aortic aneurysms: classification, incidence, diagnosis and management. J Endovascular Surg. 1997;4:152-168.
4. Veith FJ, Baum BA, Ohki T, et al. Nature and significance of endoleaks and endotension: summary of opinions expressed at an international conference. J Vasc Surg. 2002;35:1029-1035.
5. Mennander A, Pimenoff G, Heikkinen M, et al. Nonoperative approach to endotension. J Vasc Surg. 2005;42:194-198.
Practice Points
- An endoleak should be considered in any patient with a thoracic subcutaneous mass and history of aneurysm repair.
- Order imaging when an endoleak is suspected, including computed tomography angiography. Endoleaks can result in substantial morbidity and mortality if they are not recognized and treated appropriately.
- Dermatologists should be familiar with and able to recognize endoleaks, as patients may present to a dermatologist for evaluation of a subcutaneous mass that proves to be an endoleak.