User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
A brief scale IDs sleep-disordered breathing in pregnancy
CHICAGO – A four-item pregnancy-specific sleep disturbance scale proved valid as a screening tool for sleep-disordered breathing in pregnancy, and was associated with preeclampsia in a study of more than 1,100 women.
After adjustment for potential confounders – including sociodemographics, body mass index, and high blood pressure – a higher score on the short-form pregnancy-specific questionnaire (SF-SPQ) was significantly associated with an increase in the risk of preeclampsia (adjusted risk ratio, 1.54) Alpna Agrawal, Ph.D., of the University of Texas Health Science Center, Houston, reported in a poster at the annual meeting of the American Congress of Obstetricians and Gynecologists.
"In a clinical setting, the short format and validity of SF-SPQ shown in this study suggests it may be a quick and effective method to screen women at risk for sleep-disordered breathing," Dr. Agrawal wrote.
The SF-SPQ was developed based on data collected from 1,153 pregnant women seen in three outpatient clinics between 2010 and 2013. Sleep patterns were assessed by the Berlin Questionnaire, Epworth Sleepiness Scale, and by questions regarding napping behavior. A confirmatory factor analysis (CFA) was performed to develop a sensitive and specific sleep scale derived from the findings. The scale ultimately included "snoring frequently," "bothersome snoring," "stopped breathing while sleeping," and "falling asleep while driving."
"These items were conceptually related to sleep-disordered breathing during pregnancy, statistically intercorrelated, and/or associated with adverse outcomes. CFA factor loadings were significant and model fit was good," Dr. Agrawal wrote.
In addition to higher score on the SF-SPQ, adjusted relative risks of adverse outcomes were associated with BMI greater than 30 (adjusted relative risk, 1.55), hypertension (adjusted RR, 5.07), and screening positive on the Berlin Questionnaire (adjusted RR, 2.45).
"These data suggest that comorbid conditions such as obesity and hypertension (which are themselves a part of the Berlin Questionnaire) drive association with pregnancy outcomes. However, preeclampsia was independently associated with the SF-SPQ," Dr. Agrawal noted.
Prior studies have demonstrated that sleep-disordered breathing during pregnancy is associated with adverse pregnancy outcomes, but an efficient and efficacious screening tool for sleep disorders has been lacking.
The findings are important, because in the United States, preeclampsia affects up to 6% of pregnancies and is linked with other morbidities such as intrauterine growth restriction, and because studies suggest that the use of continuous positive airway pressure (CPAP) can reduce the risk of preeclampsia in pregnant patients with sleep-disordered breathing.
Additional research is needed to evaluate the efficacy of the SF-SPQ for detecting women at risk, Dr. Agrawal concluded.
Dr. Agrawal reported having no disclosures.
CHICAGO – A four-item pregnancy-specific sleep disturbance scale proved valid as a screening tool for sleep-disordered breathing in pregnancy, and was associated with preeclampsia in a study of more than 1,100 women.
After adjustment for potential confounders – including sociodemographics, body mass index, and high blood pressure – a higher score on the short-form pregnancy-specific questionnaire (SF-SPQ) was significantly associated with an increase in the risk of preeclampsia (adjusted risk ratio, 1.54) Alpna Agrawal, Ph.D., of the University of Texas Health Science Center, Houston, reported in a poster at the annual meeting of the American Congress of Obstetricians and Gynecologists.
"In a clinical setting, the short format and validity of SF-SPQ shown in this study suggests it may be a quick and effective method to screen women at risk for sleep-disordered breathing," Dr. Agrawal wrote.
The SF-SPQ was developed based on data collected from 1,153 pregnant women seen in three outpatient clinics between 2010 and 2013. Sleep patterns were assessed by the Berlin Questionnaire, Epworth Sleepiness Scale, and by questions regarding napping behavior. A confirmatory factor analysis (CFA) was performed to develop a sensitive and specific sleep scale derived from the findings. The scale ultimately included "snoring frequently," "bothersome snoring," "stopped breathing while sleeping," and "falling asleep while driving."
"These items were conceptually related to sleep-disordered breathing during pregnancy, statistically intercorrelated, and/or associated with adverse outcomes. CFA factor loadings were significant and model fit was good," Dr. Agrawal wrote.
In addition to higher score on the SF-SPQ, adjusted relative risks of adverse outcomes were associated with BMI greater than 30 (adjusted relative risk, 1.55), hypertension (adjusted RR, 5.07), and screening positive on the Berlin Questionnaire (adjusted RR, 2.45).
"These data suggest that comorbid conditions such as obesity and hypertension (which are themselves a part of the Berlin Questionnaire) drive association with pregnancy outcomes. However, preeclampsia was independently associated with the SF-SPQ," Dr. Agrawal noted.
Prior studies have demonstrated that sleep-disordered breathing during pregnancy is associated with adverse pregnancy outcomes, but an efficient and efficacious screening tool for sleep disorders has been lacking.
The findings are important, because in the United States, preeclampsia affects up to 6% of pregnancies and is linked with other morbidities such as intrauterine growth restriction, and because studies suggest that the use of continuous positive airway pressure (CPAP) can reduce the risk of preeclampsia in pregnant patients with sleep-disordered breathing.
Additional research is needed to evaluate the efficacy of the SF-SPQ for detecting women at risk, Dr. Agrawal concluded.
Dr. Agrawal reported having no disclosures.
CHICAGO – A four-item pregnancy-specific sleep disturbance scale proved valid as a screening tool for sleep-disordered breathing in pregnancy, and was associated with preeclampsia in a study of more than 1,100 women.
After adjustment for potential confounders – including sociodemographics, body mass index, and high blood pressure – a higher score on the short-form pregnancy-specific questionnaire (SF-SPQ) was significantly associated with an increase in the risk of preeclampsia (adjusted risk ratio, 1.54) Alpna Agrawal, Ph.D., of the University of Texas Health Science Center, Houston, reported in a poster at the annual meeting of the American Congress of Obstetricians and Gynecologists.
"In a clinical setting, the short format and validity of SF-SPQ shown in this study suggests it may be a quick and effective method to screen women at risk for sleep-disordered breathing," Dr. Agrawal wrote.
The SF-SPQ was developed based on data collected from 1,153 pregnant women seen in three outpatient clinics between 2010 and 2013. Sleep patterns were assessed by the Berlin Questionnaire, Epworth Sleepiness Scale, and by questions regarding napping behavior. A confirmatory factor analysis (CFA) was performed to develop a sensitive and specific sleep scale derived from the findings. The scale ultimately included "snoring frequently," "bothersome snoring," "stopped breathing while sleeping," and "falling asleep while driving."
"These items were conceptually related to sleep-disordered breathing during pregnancy, statistically intercorrelated, and/or associated with adverse outcomes. CFA factor loadings were significant and model fit was good," Dr. Agrawal wrote.
In addition to higher score on the SF-SPQ, adjusted relative risks of adverse outcomes were associated with BMI greater than 30 (adjusted relative risk, 1.55), hypertension (adjusted RR, 5.07), and screening positive on the Berlin Questionnaire (adjusted RR, 2.45).
"These data suggest that comorbid conditions such as obesity and hypertension (which are themselves a part of the Berlin Questionnaire) drive association with pregnancy outcomes. However, preeclampsia was independently associated with the SF-SPQ," Dr. Agrawal noted.
Prior studies have demonstrated that sleep-disordered breathing during pregnancy is associated with adverse pregnancy outcomes, but an efficient and efficacious screening tool for sleep disorders has been lacking.
The findings are important, because in the United States, preeclampsia affects up to 6% of pregnancies and is linked with other morbidities such as intrauterine growth restriction, and because studies suggest that the use of continuous positive airway pressure (CPAP) can reduce the risk of preeclampsia in pregnant patients with sleep-disordered breathing.
Additional research is needed to evaluate the efficacy of the SF-SPQ for detecting women at risk, Dr. Agrawal concluded.
Dr. Agrawal reported having no disclosures.
AT THE ACOG ANNUAL CLINICAL MEETING
Key clinical point: A short pregnancy-specific sleep disturbance scale may be useful as a screening tool for sleep-disordered breathing in pregnancy, which is associated with preeclampsia.
Major finding: Higher SF-SPQ score was significantly associated with an increase in the risk of preeclampsia (adjusted risk ratio, 1.54).
Data source: Confirmatory factor analysis of data from 1,153 pregnant women.
Disclosures: Dr. Agrawal reported having no disclosures
CDC: Illinois man cleared of MERS-CoV; not infected by Indiana patient
An Indiana man infected with Middle East Respiratory Syndrome Coronavirus had not transmitted the virus to a colleague in Illinois, as was previously suspected, Centers for Disease Control and Prevention officials said May 28.
On May 17, the CDC disclosed that the Illinois resident, who had attended an extended business meeting with the confirmed Indiana MERS-CoV patient prior to that patient’s hospitalization, had tested positive for MERS-CoV antibodies on serological assays. The case was particularly worrisome because it suggested transmission through a more casual level of contact than has been seen in most MERS cases to date.
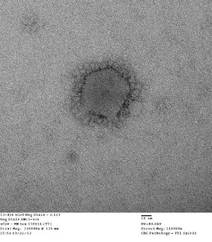
However, CDC officials now say that additional testing with a neutralizing antibody assay, a more definitive blood test that takes 5 or more days to produce results, revealed that the Illinois man had not been infected with MERS-CoV.
Thus far only two U.S. residents have been confirmed with MERS-CoV, the Indiana patient and a Florida patient. Both became ill after travel to Saudi Arabia. None of their stateside contacts has yet tested positive, though voluntary testing of family members, colleagues, fellow passengers on flights, and attending health care workers continues.
In a news conference May 28, Mark Pallansch, Ph.D., director of CDC’s Division of Viral Diseases at the National Center for Immunization and Respiratory Diseases, acknowledged that the two serological assays used in preliminary testing – the enzyme-linked immunosorbent assay (ELISA) and the immunofluorescent assay (IFA), have high specificity in excluding infection, but can result in false positives.
In the case of the Illinois resident, "there are several potential explanations for our preliminary results – that is still something not completely understood," Dr. Pallansch said. The most common and likely explanation, he said, is some cross-reactivity with another type of coronavirus.
Dr. Pallansch said that CDC would continue its protocol of using ELISA and IFA, with any positive results followed by the neutralizing antibody assay.
The neutralizing antibody assay, he said, uses live MERS-CoV and must be conducted in a high-containment lab, making it difficult to test hundreds of samples; thus initial screening with the other assays would continue.
Various serology assays are being developed around the world, including by the CDC, that may supplant the assays currently being used in the initial serological detection of MERS-CoV, Dr. Pallansch reported.
Dr. David Swerdlow, who is leading CDC’s MERS-CoV response, said during the press conference that the CDC would continue to maintain its high level of vigilance, which includes disclosing to the public any initial positive results pending confirmation, and asking individuals who test positive on first assays to take significant measures to avoid contacts that could result in transmission.
Before definitive serological results were returned, the Illinois man was asked to wear a mask, to avoid close contact with other people, and to not come in contact with crowds. He was fully compliant with those requests, Dr. Swerdlow said.
"We can’t have all the tests back in order to take public health action," Dr. Swerdlow said, noting that MERS-CoV has been seen associated with a 30% fatality rate.
"It’s our priority to make sure the public is protected," he added. While there have been only two imported cases to date in the United States, and no infections yet found to have occurred as a result of local transmission, MERS-CoV "can and likely will enter our country again."
An Indiana man infected with Middle East Respiratory Syndrome Coronavirus had not transmitted the virus to a colleague in Illinois, as was previously suspected, Centers for Disease Control and Prevention officials said May 28.
On May 17, the CDC disclosed that the Illinois resident, who had attended an extended business meeting with the confirmed Indiana MERS-CoV patient prior to that patient’s hospitalization, had tested positive for MERS-CoV antibodies on serological assays. The case was particularly worrisome because it suggested transmission through a more casual level of contact than has been seen in most MERS cases to date.
However, CDC officials now say that additional testing with a neutralizing antibody assay, a more definitive blood test that takes 5 or more days to produce results, revealed that the Illinois man had not been infected with MERS-CoV.
Thus far only two U.S. residents have been confirmed with MERS-CoV, the Indiana patient and a Florida patient. Both became ill after travel to Saudi Arabia. None of their stateside contacts has yet tested positive, though voluntary testing of family members, colleagues, fellow passengers on flights, and attending health care workers continues.
In a news conference May 28, Mark Pallansch, Ph.D., director of CDC’s Division of Viral Diseases at the National Center for Immunization and Respiratory Diseases, acknowledged that the two serological assays used in preliminary testing – the enzyme-linked immunosorbent assay (ELISA) and the immunofluorescent assay (IFA), have high specificity in excluding infection, but can result in false positives.
In the case of the Illinois resident, "there are several potential explanations for our preliminary results – that is still something not completely understood," Dr. Pallansch said. The most common and likely explanation, he said, is some cross-reactivity with another type of coronavirus.
Dr. Pallansch said that CDC would continue its protocol of using ELISA and IFA, with any positive results followed by the neutralizing antibody assay.
The neutralizing antibody assay, he said, uses live MERS-CoV and must be conducted in a high-containment lab, making it difficult to test hundreds of samples; thus initial screening with the other assays would continue.
Various serology assays are being developed around the world, including by the CDC, that may supplant the assays currently being used in the initial serological detection of MERS-CoV, Dr. Pallansch reported.
Dr. David Swerdlow, who is leading CDC’s MERS-CoV response, said during the press conference that the CDC would continue to maintain its high level of vigilance, which includes disclosing to the public any initial positive results pending confirmation, and asking individuals who test positive on first assays to take significant measures to avoid contacts that could result in transmission.
Before definitive serological results were returned, the Illinois man was asked to wear a mask, to avoid close contact with other people, and to not come in contact with crowds. He was fully compliant with those requests, Dr. Swerdlow said.
"We can’t have all the tests back in order to take public health action," Dr. Swerdlow said, noting that MERS-CoV has been seen associated with a 30% fatality rate.
"It’s our priority to make sure the public is protected," he added. While there have been only two imported cases to date in the United States, and no infections yet found to have occurred as a result of local transmission, MERS-CoV "can and likely will enter our country again."
An Indiana man infected with Middle East Respiratory Syndrome Coronavirus had not transmitted the virus to a colleague in Illinois, as was previously suspected, Centers for Disease Control and Prevention officials said May 28.
On May 17, the CDC disclosed that the Illinois resident, who had attended an extended business meeting with the confirmed Indiana MERS-CoV patient prior to that patient’s hospitalization, had tested positive for MERS-CoV antibodies on serological assays. The case was particularly worrisome because it suggested transmission through a more casual level of contact than has been seen in most MERS cases to date.
However, CDC officials now say that additional testing with a neutralizing antibody assay, a more definitive blood test that takes 5 or more days to produce results, revealed that the Illinois man had not been infected with MERS-CoV.
Thus far only two U.S. residents have been confirmed with MERS-CoV, the Indiana patient and a Florida patient. Both became ill after travel to Saudi Arabia. None of their stateside contacts has yet tested positive, though voluntary testing of family members, colleagues, fellow passengers on flights, and attending health care workers continues.
In a news conference May 28, Mark Pallansch, Ph.D., director of CDC’s Division of Viral Diseases at the National Center for Immunization and Respiratory Diseases, acknowledged that the two serological assays used in preliminary testing – the enzyme-linked immunosorbent assay (ELISA) and the immunofluorescent assay (IFA), have high specificity in excluding infection, but can result in false positives.
In the case of the Illinois resident, "there are several potential explanations for our preliminary results – that is still something not completely understood," Dr. Pallansch said. The most common and likely explanation, he said, is some cross-reactivity with another type of coronavirus.
Dr. Pallansch said that CDC would continue its protocol of using ELISA and IFA, with any positive results followed by the neutralizing antibody assay.
The neutralizing antibody assay, he said, uses live MERS-CoV and must be conducted in a high-containment lab, making it difficult to test hundreds of samples; thus initial screening with the other assays would continue.
Various serology assays are being developed around the world, including by the CDC, that may supplant the assays currently being used in the initial serological detection of MERS-CoV, Dr. Pallansch reported.
Dr. David Swerdlow, who is leading CDC’s MERS-CoV response, said during the press conference that the CDC would continue to maintain its high level of vigilance, which includes disclosing to the public any initial positive results pending confirmation, and asking individuals who test positive on first assays to take significant measures to avoid contacts that could result in transmission.
Before definitive serological results were returned, the Illinois man was asked to wear a mask, to avoid close contact with other people, and to not come in contact with crowds. He was fully compliant with those requests, Dr. Swerdlow said.
"We can’t have all the tests back in order to take public health action," Dr. Swerdlow said, noting that MERS-CoV has been seen associated with a 30% fatality rate.
"It’s our priority to make sure the public is protected," he added. While there have been only two imported cases to date in the United States, and no infections yet found to have occurred as a result of local transmission, MERS-CoV "can and likely will enter our country again."
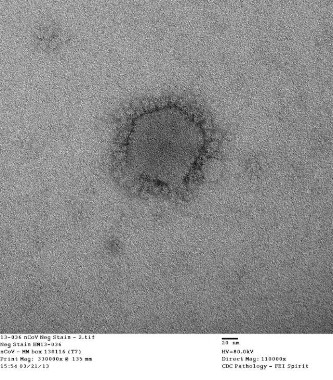
Implantable device approved for remotely monitoring PA in heart failure patients
An implantable device that provides measurements of pulmonary arterial pressure in patients with class III heart failure has been approved by the Food and Drug Administration, based on a study that showed the use of the device to remotely monitor patients reduced heart failure hospitalization rates.
The CardioMEMS HF System "is the first permanently implantable wireless system intended to provide PA pressure measurements, including systolic, diastolic, and mean PA pressures," according to the FDA statement announcing the approval on May 28. This information is remotely reviewed by the patient’s physician, who "can make decisions regarding the status of the patient and, if necessary, initiate changes in medical therapy, with the goal of reducing hospitalization due to heart failure," the statement said.

It is specifically approved for patients with New York Heart Association (NYHA) class III heart failure (HF) who have been hospitalized for heart failure in the previous year.
The three components of the system are the battery-free sensor/monitor that is permanently implanted in the pulmonary artery, a transvenous catheter that deploys the sensor in the distal PA, and an electronic system that receives and processes the signals from the sensor/monitor and transfers the PA pressure measurements to a secure database, the statement said. Patients can be monitored from their home or another remote location.
Approval was based on a study of 550 patients with NYHA class III HF and a recent hospitalization for HF, who had the device implanted. Physicians had access to daily PA measurements only for the patients randomized to the treatment group, and adjusted HF medications based on the values provided. At 6 months, the HF hospitalization rate was significantly lower among those in the treatment group. The FDA statement noted that at 6 months, almost 99% of the patients who had the device implanted or in whom implantation was attempted had no complications related to the device or system, and all of the devices that were implanted were operating normally.
However, concerns about the study held up approval of the device for several years, and at a meeting in December 2011, the majority of the FDA’s Circulatory System Devices Panel agreed that the risks of the device outweighed the benefits. The manufacturer, CardioMEMS, provided follow-up data and further analyses of the data that were provided at another meeting of the panel, in October 2013. At that meeting, the majority of the panel agreed that the benefits of the device outweighed its risks for monitoring patients who met the criteria specified in the indication that has been approved, patients with NYHA class III heart failure who have been hospitalized for HF in the previous year.
In the May 28 statement, the FDA said that the company is required to conduct a postmarketing study to evaluate the performance of the device when used outside of a clinical trial. One concern of the panelists who supported approval at the 2013 meeting was that the benefit in terms of HF hospitalizations was not evident in women in the study, which they said could have been due to the low number of women enrolled in the trial, and they recommended that the device be studied in more women.
An implantable device that provides measurements of pulmonary arterial pressure in patients with class III heart failure has been approved by the Food and Drug Administration, based on a study that showed the use of the device to remotely monitor patients reduced heart failure hospitalization rates.
The CardioMEMS HF System "is the first permanently implantable wireless system intended to provide PA pressure measurements, including systolic, diastolic, and mean PA pressures," according to the FDA statement announcing the approval on May 28. This information is remotely reviewed by the patient’s physician, who "can make decisions regarding the status of the patient and, if necessary, initiate changes in medical therapy, with the goal of reducing hospitalization due to heart failure," the statement said.
It is specifically approved for patients with New York Heart Association (NYHA) class III heart failure (HF) who have been hospitalized for heart failure in the previous year.
The three components of the system are the battery-free sensor/monitor that is permanently implanted in the pulmonary artery, a transvenous catheter that deploys the sensor in the distal PA, and an electronic system that receives and processes the signals from the sensor/monitor and transfers the PA pressure measurements to a secure database, the statement said. Patients can be monitored from their home or another remote location.
Approval was based on a study of 550 patients with NYHA class III HF and a recent hospitalization for HF, who had the device implanted. Physicians had access to daily PA measurements only for the patients randomized to the treatment group, and adjusted HF medications based on the values provided. At 6 months, the HF hospitalization rate was significantly lower among those in the treatment group. The FDA statement noted that at 6 months, almost 99% of the patients who had the device implanted or in whom implantation was attempted had no complications related to the device or system, and all of the devices that were implanted were operating normally.
However, concerns about the study held up approval of the device for several years, and at a meeting in December 2011, the majority of the FDA’s Circulatory System Devices Panel agreed that the risks of the device outweighed the benefits. The manufacturer, CardioMEMS, provided follow-up data and further analyses of the data that were provided at another meeting of the panel, in October 2013. At that meeting, the majority of the panel agreed that the benefits of the device outweighed its risks for monitoring patients who met the criteria specified in the indication that has been approved, patients with NYHA class III heart failure who have been hospitalized for HF in the previous year.
In the May 28 statement, the FDA said that the company is required to conduct a postmarketing study to evaluate the performance of the device when used outside of a clinical trial. One concern of the panelists who supported approval at the 2013 meeting was that the benefit in terms of HF hospitalizations was not evident in women in the study, which they said could have been due to the low number of women enrolled in the trial, and they recommended that the device be studied in more women.
An implantable device that provides measurements of pulmonary arterial pressure in patients with class III heart failure has been approved by the Food and Drug Administration, based on a study that showed the use of the device to remotely monitor patients reduced heart failure hospitalization rates.
The CardioMEMS HF System "is the first permanently implantable wireless system intended to provide PA pressure measurements, including systolic, diastolic, and mean PA pressures," according to the FDA statement announcing the approval on May 28. This information is remotely reviewed by the patient’s physician, who "can make decisions regarding the status of the patient and, if necessary, initiate changes in medical therapy, with the goal of reducing hospitalization due to heart failure," the statement said.
It is specifically approved for patients with New York Heart Association (NYHA) class III heart failure (HF) who have been hospitalized for heart failure in the previous year.
The three components of the system are the battery-free sensor/monitor that is permanently implanted in the pulmonary artery, a transvenous catheter that deploys the sensor in the distal PA, and an electronic system that receives and processes the signals from the sensor/monitor and transfers the PA pressure measurements to a secure database, the statement said. Patients can be monitored from their home or another remote location.
Approval was based on a study of 550 patients with NYHA class III HF and a recent hospitalization for HF, who had the device implanted. Physicians had access to daily PA measurements only for the patients randomized to the treatment group, and adjusted HF medications based on the values provided. At 6 months, the HF hospitalization rate was significantly lower among those in the treatment group. The FDA statement noted that at 6 months, almost 99% of the patients who had the device implanted or in whom implantation was attempted had no complications related to the device or system, and all of the devices that were implanted were operating normally.
However, concerns about the study held up approval of the device for several years, and at a meeting in December 2011, the majority of the FDA’s Circulatory System Devices Panel agreed that the risks of the device outweighed the benefits. The manufacturer, CardioMEMS, provided follow-up data and further analyses of the data that were provided at another meeting of the panel, in October 2013. At that meeting, the majority of the panel agreed that the benefits of the device outweighed its risks for monitoring patients who met the criteria specified in the indication that has been approved, patients with NYHA class III heart failure who have been hospitalized for HF in the previous year.
In the May 28 statement, the FDA said that the company is required to conduct a postmarketing study to evaluate the performance of the device when used outside of a clinical trial. One concern of the panelists who supported approval at the 2013 meeting was that the benefit in terms of HF hospitalizations was not evident in women in the study, which they said could have been due to the low number of women enrolled in the trial, and they recommended that the device be studied in more women.

ADX-N05: Favorable results in narcolepsy patients
A novel agent for symptoms of excessive daytime sleepiness in narcolepsy patients has shown evidence of efficacy, according to researchers who will report their results as a late-breaking abstract at Sleep 2014.
ADX-N05, a wake-promoting agent with dopaminergic and noradrenergic activity, reduced symptoms in a 12-week, placebo-controlled, double-blind study of nearly 100 patients with narcolepsy, according to Dr. Jed Black of Jazz Pharmaceuticals and Stanford (Calif.) Sleep Medicine Center, and his colleagues.
Efficacy was based on improvements at 4 and 12 weeks from baseline in average sleep-onset latency on the Maintenance of Wakefulness Test and Clinical Global Impression-Change scale. A secondary endpoint was change on the Epworth Sleepiness Scale.
For the study, 49 patients were randomized to placebo and 44 to the active drug. The active drug was initiated at 150 mg/day for 4 weeks and increased to 300 mg/day for weeks 5-12.
At week 4, average sleep-onset latency on the Maintenance of Wakefulness Test was 9.5 minutes in those on the active drug and 1.4 minutes in those on placebo, a difference that was statistically significant (P less than .0001). Improvements on the Clinical Global Impression-Change scale were 80% vs. 51%; (P = .0066), and Epworth Sleepiness Scale scores had decreased (5.6 points vs. 2.4 points; P = 0.0038).
Further improvements were noted at 12 weeks, with average sleep-onset latency on the Maintenance of Wakefulness Test of 12.8 minutes vs. 2.1 minutes; (P less than .0001). Epworth Sleepiness Scale scores were 8.5 points vs. 2.5 points (P less than .0001), and the proportion of patients with Clinical Global Impression-Change scale improvements was 86% vs. 38% (P less than .0001), according to the researchers, who will present their complete study results at the annual meeting of the Associated Professional Sleep Societies.
Adverse events were more common with the active drug and included headache, nausea, diarrhea, insomnia, decreased appetite, and anxiety. Three study subjects halted active therapy because of adverse events. Two serious events—conversion disorder and acute cholecystitis—occurred in the active treatment group and were probably unrelated to drug therapy, according to the researchers.
The study was supported by Aerial BioPharma. Dr. Black is with Jazz Pharmaceuticals, which has acquired ADX-N05 from Aerial BioPharma.
A novel agent for symptoms of excessive daytime sleepiness in narcolepsy patients has shown evidence of efficacy, according to researchers who will report their results as a late-breaking abstract at Sleep 2014.
ADX-N05, a wake-promoting agent with dopaminergic and noradrenergic activity, reduced symptoms in a 12-week, placebo-controlled, double-blind study of nearly 100 patients with narcolepsy, according to Dr. Jed Black of Jazz Pharmaceuticals and Stanford (Calif.) Sleep Medicine Center, and his colleagues.
Efficacy was based on improvements at 4 and 12 weeks from baseline in average sleep-onset latency on the Maintenance of Wakefulness Test and Clinical Global Impression-Change scale. A secondary endpoint was change on the Epworth Sleepiness Scale.
For the study, 49 patients were randomized to placebo and 44 to the active drug. The active drug was initiated at 150 mg/day for 4 weeks and increased to 300 mg/day for weeks 5-12.
At week 4, average sleep-onset latency on the Maintenance of Wakefulness Test was 9.5 minutes in those on the active drug and 1.4 minutes in those on placebo, a difference that was statistically significant (P less than .0001). Improvements on the Clinical Global Impression-Change scale were 80% vs. 51%; (P = .0066), and Epworth Sleepiness Scale scores had decreased (5.6 points vs. 2.4 points; P = 0.0038).
Further improvements were noted at 12 weeks, with average sleep-onset latency on the Maintenance of Wakefulness Test of 12.8 minutes vs. 2.1 minutes; (P less than .0001). Epworth Sleepiness Scale scores were 8.5 points vs. 2.5 points (P less than .0001), and the proportion of patients with Clinical Global Impression-Change scale improvements was 86% vs. 38% (P less than .0001), according to the researchers, who will present their complete study results at the annual meeting of the Associated Professional Sleep Societies.
Adverse events were more common with the active drug and included headache, nausea, diarrhea, insomnia, decreased appetite, and anxiety. Three study subjects halted active therapy because of adverse events. Two serious events—conversion disorder and acute cholecystitis—occurred in the active treatment group and were probably unrelated to drug therapy, according to the researchers.
The study was supported by Aerial BioPharma. Dr. Black is with Jazz Pharmaceuticals, which has acquired ADX-N05 from Aerial BioPharma.
A novel agent for symptoms of excessive daytime sleepiness in narcolepsy patients has shown evidence of efficacy, according to researchers who will report their results as a late-breaking abstract at Sleep 2014.
ADX-N05, a wake-promoting agent with dopaminergic and noradrenergic activity, reduced symptoms in a 12-week, placebo-controlled, double-blind study of nearly 100 patients with narcolepsy, according to Dr. Jed Black of Jazz Pharmaceuticals and Stanford (Calif.) Sleep Medicine Center, and his colleagues.
Efficacy was based on improvements at 4 and 12 weeks from baseline in average sleep-onset latency on the Maintenance of Wakefulness Test and Clinical Global Impression-Change scale. A secondary endpoint was change on the Epworth Sleepiness Scale.
For the study, 49 patients were randomized to placebo and 44 to the active drug. The active drug was initiated at 150 mg/day for 4 weeks and increased to 300 mg/day for weeks 5-12.
At week 4, average sleep-onset latency on the Maintenance of Wakefulness Test was 9.5 minutes in those on the active drug and 1.4 minutes in those on placebo, a difference that was statistically significant (P less than .0001). Improvements on the Clinical Global Impression-Change scale were 80% vs. 51%; (P = .0066), and Epworth Sleepiness Scale scores had decreased (5.6 points vs. 2.4 points; P = 0.0038).
Further improvements were noted at 12 weeks, with average sleep-onset latency on the Maintenance of Wakefulness Test of 12.8 minutes vs. 2.1 minutes; (P less than .0001). Epworth Sleepiness Scale scores were 8.5 points vs. 2.5 points (P less than .0001), and the proportion of patients with Clinical Global Impression-Change scale improvements was 86% vs. 38% (P less than .0001), according to the researchers, who will present their complete study results at the annual meeting of the Associated Professional Sleep Societies.
Adverse events were more common with the active drug and included headache, nausea, diarrhea, insomnia, decreased appetite, and anxiety. Three study subjects halted active therapy because of adverse events. Two serious events—conversion disorder and acute cholecystitis—occurred in the active treatment group and were probably unrelated to drug therapy, according to the researchers.
The study was supported by Aerial BioPharma. Dr. Black is with Jazz Pharmaceuticals, which has acquired ADX-N05 from Aerial BioPharma.
FROM SLEEP 2014
Key clinical point: ADX-N05, a wake-promoting agent with dopaminergic and noradrenergic activity, may prove to limit daytime sleepiness in narcolepsy.
Major finding: After 12 weeks, average sleep-onset latency on the Maintenance of Wakefulness Test was 12.8 minutes for those on the active drug and 2.1 minutes for those on placebo (P less than .0001).
Data source: A 12-week, placebo-controlled, double-blind study of nearly 100 patients with narcolepsy.
Disclosures: The study was supported by Aerial BioPharma. Dr. Black is with Jazz Pharmaceuticals, which has acquired ADX-N05 from Aerial BioPharma.
In seasonal affective disorder, the eyes have it
Pupil response to light differs in people with seasonal affective disorder and is affected by their total exposure to light, researchers will report at Sleep 2014 in Minneapolis.
"We speculate that low light levels in SAD [seasonal affective disorder] trigger downstream changes in mood and behavior, and that the link between light and SAD may be mediated by the PIPR [postillumination pupil response]," Kathryn A. Roecklein, Ph.D., of the University of Pittsburgh and her colleagues wrote in a late-breaker abstract to be presented at the annual meeting of the Associated Professional Sleep Societies.
For their study, the researchers examined postillumination pupil response during summer and winter in 33 people with SAD and 17 controls. One-second light exposures to red light and to blue light were used for testing. Actigraphy was used to measure light exposure in the days before the tests.
In the study subjects with SAD, most of the variance in postillumination pupil response was associated with total photon exposures on the day of testing. In the controls, however, postillumination pupil response was independent of total photon exposures on the day of testing. Blue total photon exposures accounted for the greatest proportion of variance in postillumination pupil response (P = .013) and were predictive of SAD independent of the subject’s gender, time since waking, and whether they were early risers or night owls (P = .013).
The postillumination pupil response was lower in subjects with SAD than in controls (P less than .05), especially among those with SAD who were night owls (P less than .001).
The data, which are the first to link light exposure and the postillumination pupil response in SAD, will be presented in their entirety at the meeting.
The study was funded by a grant from the National Institutes of Health. The authors reported having no financial disclosures.
Pupil response to light differs in people with seasonal affective disorder and is affected by their total exposure to light, researchers will report at Sleep 2014 in Minneapolis.
"We speculate that low light levels in SAD [seasonal affective disorder] trigger downstream changes in mood and behavior, and that the link between light and SAD may be mediated by the PIPR [postillumination pupil response]," Kathryn A. Roecklein, Ph.D., of the University of Pittsburgh and her colleagues wrote in a late-breaker abstract to be presented at the annual meeting of the Associated Professional Sleep Societies.
For their study, the researchers examined postillumination pupil response during summer and winter in 33 people with SAD and 17 controls. One-second light exposures to red light and to blue light were used for testing. Actigraphy was used to measure light exposure in the days before the tests.
In the study subjects with SAD, most of the variance in postillumination pupil response was associated with total photon exposures on the day of testing. In the controls, however, postillumination pupil response was independent of total photon exposures on the day of testing. Blue total photon exposures accounted for the greatest proportion of variance in postillumination pupil response (P = .013) and were predictive of SAD independent of the subject’s gender, time since waking, and whether they were early risers or night owls (P = .013).
The postillumination pupil response was lower in subjects with SAD than in controls (P less than .05), especially among those with SAD who were night owls (P less than .001).
The data, which are the first to link light exposure and the postillumination pupil response in SAD, will be presented in their entirety at the meeting.
The study was funded by a grant from the National Institutes of Health. The authors reported having no financial disclosures.
Pupil response to light differs in people with seasonal affective disorder and is affected by their total exposure to light, researchers will report at Sleep 2014 in Minneapolis.
"We speculate that low light levels in SAD [seasonal affective disorder] trigger downstream changes in mood and behavior, and that the link between light and SAD may be mediated by the PIPR [postillumination pupil response]," Kathryn A. Roecklein, Ph.D., of the University of Pittsburgh and her colleagues wrote in a late-breaker abstract to be presented at the annual meeting of the Associated Professional Sleep Societies.
For their study, the researchers examined postillumination pupil response during summer and winter in 33 people with SAD and 17 controls. One-second light exposures to red light and to blue light were used for testing. Actigraphy was used to measure light exposure in the days before the tests.
In the study subjects with SAD, most of the variance in postillumination pupil response was associated with total photon exposures on the day of testing. In the controls, however, postillumination pupil response was independent of total photon exposures on the day of testing. Blue total photon exposures accounted for the greatest proportion of variance in postillumination pupil response (P = .013) and were predictive of SAD independent of the subject’s gender, time since waking, and whether they were early risers or night owls (P = .013).
The postillumination pupil response was lower in subjects with SAD than in controls (P less than .05), especially among those with SAD who were night owls (P less than .001).
The data, which are the first to link light exposure and the postillumination pupil response in SAD, will be presented in their entirety at the meeting.
The study was funded by a grant from the National Institutes of Health. The authors reported having no financial disclosures.
FROM SLEEP 2014
Key clinical point: Those with seasonal affective disorder, especially night owls, might benefit from more light exposure.
Major finding: The postillumination pupil response was lower in subjects with SAD than in controls (P less than .05) and especially those who were night owls (P less than .001).
Data source: Postillumination pupil response during summer and winter in 33 people with SAD and 17 controls.
Disclosures: The study was funded by a grant from the National Institutes of Health. The authors reported having no financial disclosures.
Teens who skip sleep risk insulin resistance
Teens who miss sleep are more likely to set their metabolism in motion for insulin resistance.
Based on a pilot study of 10 lean and obese adolescents, sleep duration was the primary predictor of abnormal 90-minute glucose values on oral glucose tolerance tests, Dr. Dorit Koren and her colleagues will report at the annual meeting of the Associated Professional Sleep Societies in Minneapolis. The finding was independent of the teens’ body weights.
The University of Chicago researchers wrote in their late-breaking abstract that the pilot study is "the first to our knowledge to examine potential interrelationships between home sleep duration and dynamic insulin and glucose homeostasis in adolescents." Previous studies in children have associated short sleep with insulin resistance, but have not examined the relationship between home sleep and postprandial glucose metabolism. Studies in adults have linked type 2 diabetes risks and experimental sleep restriction to acute insulin resistance and glucose intolerance.
For the study, the 13- to 18-year-olds had oral glucose tolerance tests, evaluations of body weight, an overnight polysomnogram, and home sleep assessments based on actigraphy and sleep diaries. Sleep duration was linearly correlated with 90-minute oral glucose tolerance test results (r = –0.66, P = .036). There were trends toward negative associations between home sleep duration, obesity, and insulin resistance.
The study was supported by a grant from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health.
Teens who miss sleep are more likely to set their metabolism in motion for insulin resistance.
Based on a pilot study of 10 lean and obese adolescents, sleep duration was the primary predictor of abnormal 90-minute glucose values on oral glucose tolerance tests, Dr. Dorit Koren and her colleagues will report at the annual meeting of the Associated Professional Sleep Societies in Minneapolis. The finding was independent of the teens’ body weights.
The University of Chicago researchers wrote in their late-breaking abstract that the pilot study is "the first to our knowledge to examine potential interrelationships between home sleep duration and dynamic insulin and glucose homeostasis in adolescents." Previous studies in children have associated short sleep with insulin resistance, but have not examined the relationship between home sleep and postprandial glucose metabolism. Studies in adults have linked type 2 diabetes risks and experimental sleep restriction to acute insulin resistance and glucose intolerance.
For the study, the 13- to 18-year-olds had oral glucose tolerance tests, evaluations of body weight, an overnight polysomnogram, and home sleep assessments based on actigraphy and sleep diaries. Sleep duration was linearly correlated with 90-minute oral glucose tolerance test results (r = –0.66, P = .036). There were trends toward negative associations between home sleep duration, obesity, and insulin resistance.
The study was supported by a grant from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health.
Teens who miss sleep are more likely to set their metabolism in motion for insulin resistance.
Based on a pilot study of 10 lean and obese adolescents, sleep duration was the primary predictor of abnormal 90-minute glucose values on oral glucose tolerance tests, Dr. Dorit Koren and her colleagues will report at the annual meeting of the Associated Professional Sleep Societies in Minneapolis. The finding was independent of the teens’ body weights.
The University of Chicago researchers wrote in their late-breaking abstract that the pilot study is "the first to our knowledge to examine potential interrelationships between home sleep duration and dynamic insulin and glucose homeostasis in adolescents." Previous studies in children have associated short sleep with insulin resistance, but have not examined the relationship between home sleep and postprandial glucose metabolism. Studies in adults have linked type 2 diabetes risks and experimental sleep restriction to acute insulin resistance and glucose intolerance.
For the study, the 13- to 18-year-olds had oral glucose tolerance tests, evaluations of body weight, an overnight polysomnogram, and home sleep assessments based on actigraphy and sleep diaries. Sleep duration was linearly correlated with 90-minute oral glucose tolerance test results (r = –0.66, P = .036). There were trends toward negative associations between home sleep duration, obesity, and insulin resistance.
The study was supported by a grant from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health.
FROM SLEEP 2014
Key clinical point: Sleep deprivation in teens affects insulin metabolism.
Major finding: Independent of weight, sleep duration was linearly associated with 90-minute results on oral glucose tolerance tests (r = –0.66, P = .036).
Data source: A pilot study of 10 lean and obese adolescents.
Disclosures: The study was supported by a grant from the National Institutes of Health.
Hypertonic saline indications for bronchiolitis lack evidence for clear guidance
The therapeutic value of hypertonic saline in treating bronchiolitis in young children remains unclear, based on the findings of two randomized controlled trials with conflicting results.
While one found no significant improvements in outcomes between hypertonic and normal saline, the other found a lower risk of hospitalization in children receiving hypertonic saline.
"Based on the results of this and other studies, the administration of a single dose of 3% hypertonic saline in the acute care setting does not appear to be more effective than normal saline in improving short-term respiratory distress in bronchiolitis," reported Dr. Todd Florin of Cincinnati Children’s Hospital Medical Center and his associates in the first study (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2013.5306]).
Dr. Florin’s team equally randomized 62 children under age 24 months to receive 4 mL of either 3% hypertonic saline or normal saline within 90 minutes after receiving standard therapy for bronchiolitis, which included nasal suctioning and a single dose of nebulized albuterol. The children had all presented to the emergency department of Children’s Hospital of Philadelphia during one of two consecutive bronchiolitis seasons, from November to April in 2010 and 2011.
All children were assessed using the Respiratory Distress Assessment Instrument (RDAI) 1 hour after treatment and then 2 hours after treatment for those being discharged or still in the ED at that time. Based on the Respiratory Assessment Change Score (RACS) – which uses the RDAI score and a standardized change in respiratory rate to assess respiratory status changes – the normal saline group showed clinically significant improvement (a RACS of –3) after an hour, whereas the hypertonic saline group did not.
There was no significant difference in the median RDAI scores, heart rate, oxygen saturation, hospitalization rate, or child’s breathing or feeding status (based on parental perception) between the two groups.
Yet the hospitalization rate was lower in the hypertonic saline group of the other study, which used similar protocols in a larger population, reported Dr. Susan Wu of Children’s Hospital Los Angeles and her associates, also in JAMA Pediatrics (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2014.301].
Their participants included 408 children under age 24 months presenting with bronchiolitis at two different children’s hospital EDs between March 2008 and April 2011. All patients received 2.5 mg of nebulized albuterol, after which 197 children received 4 mL of normal saline and 211 children received 4 mL of 3% hypertonic saline, each inhaled up to three times. Children admitted received their assigned saline (always premedicated with albuterol sulfate) every 8 hours until discharge.
While 42.6% of the normal saline patients were admitted to the hospital, only 28.9% of the hypertonic saline patients were admitted (P = .01), for an adjusted odds ratio of 0.49 and a number needed to treat of 8 to prevent one hospitalization. "Other statistically significant predictors of admission included site, male sex, patient weight, baseline respiratory rate, and baseline oxygen saturation," the authors reported.
Among those admitted, the length of stay was 3.16 days for the hypertonic saline participants and 3.92 days for the normal saline participants, but the difference was not significant (P = .24). No significant difference was found in the RDAI score, which decreased in both groups. The RACS was calculated using the same methods as in the Florin study for 366 cases, but no significant differences in the mean RACS scores existed between the two groups after adjustment for RDAI baseline scores.
Dr. Wu and her team recommended that future research "investigate the optimal dosing and administration regimen and the patient-level factors that may affect response to hypertonic saline."
The Florin et al. study was funded by an Academic Pediatric Association Young Investigator Award. The Wu et al. study was funded by the Thrasher Research Fund and the department of pediatrics, University of Southern California Keck School of Medicine, Los Angeles. The authors of both studies declared that they had no relevant financial disclosures.
For pediatricians looking for answers on how best to provide treatment for their patients, nothing can be more frustrating than two randomized clinical trials (RCTs) with contradictory results. When we consider study design and risk of bias, these two RCTs appear to be well designed and well implemented. Differences in populations, interventions, controls, and outcome measures may also result in study results being different; however, examination of these two studies shows no obvious reasons for their results to differ because of these factors. It remains to be seen whether the way in which albuterol was used in reference to hypertonic saline has an effect on the outcome.
Evaluating the efficacy of hypertonic saline in the treatment of bronchiolitis is not an easy task. Evidently, it is difficult to base treatment decisions on the result of a single study. The summary of a systematic review from 2013 in the Cochrane Database is that there is probably not a role for hypertonic saline for patients with bronchiolitis treated in the emergency department, but it may be helpful for inpatients by decreasing their length of hospital stay by 1.15 days.
These two trials point us to the importance of using scientifically developed systematic reviews and meta-analyses to get the best sense of optimal treatment for children. From our read of the current systematic review (which now will need to be updated) and our read of these two individual trials, we would not start using hypertonic saline in the emergency department on a routine basis. However, nebulized hypertonic saline may have a role to play for children hospitalized with bronchiolitis.
Dr. Sim Grewal is in the division of pediatric emergency medicine at the University of Alberta in Edmonton. Dr. Terry P. Klassen is part of Translating Emergency Knowledge for Kids at the Manitoba Institute of Child Health, and is in the department of pediatrics at the University of Manitoba, both in Winnipeg. Neither physician reported any relevant financial disclosures. These comments have been adapted from an editorial accompanying the studies in JAMA Pediatrics (JAMA Pediatr. 2014 26 May [doi: 10.1001/jamapediatrics.2014.423]).
For pediatricians looking for answers on how best to provide treatment for their patients, nothing can be more frustrating than two randomized clinical trials (RCTs) with contradictory results. When we consider study design and risk of bias, these two RCTs appear to be well designed and well implemented. Differences in populations, interventions, controls, and outcome measures may also result in study results being different; however, examination of these two studies shows no obvious reasons for their results to differ because of these factors. It remains to be seen whether the way in which albuterol was used in reference to hypertonic saline has an effect on the outcome.
Evaluating the efficacy of hypertonic saline in the treatment of bronchiolitis is not an easy task. Evidently, it is difficult to base treatment decisions on the result of a single study. The summary of a systematic review from 2013 in the Cochrane Database is that there is probably not a role for hypertonic saline for patients with bronchiolitis treated in the emergency department, but it may be helpful for inpatients by decreasing their length of hospital stay by 1.15 days.
These two trials point us to the importance of using scientifically developed systematic reviews and meta-analyses to get the best sense of optimal treatment for children. From our read of the current systematic review (which now will need to be updated) and our read of these two individual trials, we would not start using hypertonic saline in the emergency department on a routine basis. However, nebulized hypertonic saline may have a role to play for children hospitalized with bronchiolitis.
Dr. Sim Grewal is in the division of pediatric emergency medicine at the University of Alberta in Edmonton. Dr. Terry P. Klassen is part of Translating Emergency Knowledge for Kids at the Manitoba Institute of Child Health, and is in the department of pediatrics at the University of Manitoba, both in Winnipeg. Neither physician reported any relevant financial disclosures. These comments have been adapted from an editorial accompanying the studies in JAMA Pediatrics (JAMA Pediatr. 2014 26 May [doi: 10.1001/jamapediatrics.2014.423]).
For pediatricians looking for answers on how best to provide treatment for their patients, nothing can be more frustrating than two randomized clinical trials (RCTs) with contradictory results. When we consider study design and risk of bias, these two RCTs appear to be well designed and well implemented. Differences in populations, interventions, controls, and outcome measures may also result in study results being different; however, examination of these two studies shows no obvious reasons for their results to differ because of these factors. It remains to be seen whether the way in which albuterol was used in reference to hypertonic saline has an effect on the outcome.
Evaluating the efficacy of hypertonic saline in the treatment of bronchiolitis is not an easy task. Evidently, it is difficult to base treatment decisions on the result of a single study. The summary of a systematic review from 2013 in the Cochrane Database is that there is probably not a role for hypertonic saline for patients with bronchiolitis treated in the emergency department, but it may be helpful for inpatients by decreasing their length of hospital stay by 1.15 days.
These two trials point us to the importance of using scientifically developed systematic reviews and meta-analyses to get the best sense of optimal treatment for children. From our read of the current systematic review (which now will need to be updated) and our read of these two individual trials, we would not start using hypertonic saline in the emergency department on a routine basis. However, nebulized hypertonic saline may have a role to play for children hospitalized with bronchiolitis.
Dr. Sim Grewal is in the division of pediatric emergency medicine at the University of Alberta in Edmonton. Dr. Terry P. Klassen is part of Translating Emergency Knowledge for Kids at the Manitoba Institute of Child Health, and is in the department of pediatrics at the University of Manitoba, both in Winnipeg. Neither physician reported any relevant financial disclosures. These comments have been adapted from an editorial accompanying the studies in JAMA Pediatrics (JAMA Pediatr. 2014 26 May [doi: 10.1001/jamapediatrics.2014.423]).
The therapeutic value of hypertonic saline in treating bronchiolitis in young children remains unclear, based on the findings of two randomized controlled trials with conflicting results.
While one found no significant improvements in outcomes between hypertonic and normal saline, the other found a lower risk of hospitalization in children receiving hypertonic saline.
"Based on the results of this and other studies, the administration of a single dose of 3% hypertonic saline in the acute care setting does not appear to be more effective than normal saline in improving short-term respiratory distress in bronchiolitis," reported Dr. Todd Florin of Cincinnati Children’s Hospital Medical Center and his associates in the first study (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2013.5306]).
Dr. Florin’s team equally randomized 62 children under age 24 months to receive 4 mL of either 3% hypertonic saline or normal saline within 90 minutes after receiving standard therapy for bronchiolitis, which included nasal suctioning and a single dose of nebulized albuterol. The children had all presented to the emergency department of Children’s Hospital of Philadelphia during one of two consecutive bronchiolitis seasons, from November to April in 2010 and 2011.
All children were assessed using the Respiratory Distress Assessment Instrument (RDAI) 1 hour after treatment and then 2 hours after treatment for those being discharged or still in the ED at that time. Based on the Respiratory Assessment Change Score (RACS) – which uses the RDAI score and a standardized change in respiratory rate to assess respiratory status changes – the normal saline group showed clinically significant improvement (a RACS of –3) after an hour, whereas the hypertonic saline group did not.
There was no significant difference in the median RDAI scores, heart rate, oxygen saturation, hospitalization rate, or child’s breathing or feeding status (based on parental perception) between the two groups.
Yet the hospitalization rate was lower in the hypertonic saline group of the other study, which used similar protocols in a larger population, reported Dr. Susan Wu of Children’s Hospital Los Angeles and her associates, also in JAMA Pediatrics (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2014.301].
Their participants included 408 children under age 24 months presenting with bronchiolitis at two different children’s hospital EDs between March 2008 and April 2011. All patients received 2.5 mg of nebulized albuterol, after which 197 children received 4 mL of normal saline and 211 children received 4 mL of 3% hypertonic saline, each inhaled up to three times. Children admitted received their assigned saline (always premedicated with albuterol sulfate) every 8 hours until discharge.
While 42.6% of the normal saline patients were admitted to the hospital, only 28.9% of the hypertonic saline patients were admitted (P = .01), for an adjusted odds ratio of 0.49 and a number needed to treat of 8 to prevent one hospitalization. "Other statistically significant predictors of admission included site, male sex, patient weight, baseline respiratory rate, and baseline oxygen saturation," the authors reported.
Among those admitted, the length of stay was 3.16 days for the hypertonic saline participants and 3.92 days for the normal saline participants, but the difference was not significant (P = .24). No significant difference was found in the RDAI score, which decreased in both groups. The RACS was calculated using the same methods as in the Florin study for 366 cases, but no significant differences in the mean RACS scores existed between the two groups after adjustment for RDAI baseline scores.
Dr. Wu and her team recommended that future research "investigate the optimal dosing and administration regimen and the patient-level factors that may affect response to hypertonic saline."
The Florin et al. study was funded by an Academic Pediatric Association Young Investigator Award. The Wu et al. study was funded by the Thrasher Research Fund and the department of pediatrics, University of Southern California Keck School of Medicine, Los Angeles. The authors of both studies declared that they had no relevant financial disclosures.
The therapeutic value of hypertonic saline in treating bronchiolitis in young children remains unclear, based on the findings of two randomized controlled trials with conflicting results.
While one found no significant improvements in outcomes between hypertonic and normal saline, the other found a lower risk of hospitalization in children receiving hypertonic saline.
"Based on the results of this and other studies, the administration of a single dose of 3% hypertonic saline in the acute care setting does not appear to be more effective than normal saline in improving short-term respiratory distress in bronchiolitis," reported Dr. Todd Florin of Cincinnati Children’s Hospital Medical Center and his associates in the first study (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2013.5306]).
Dr. Florin’s team equally randomized 62 children under age 24 months to receive 4 mL of either 3% hypertonic saline or normal saline within 90 minutes after receiving standard therapy for bronchiolitis, which included nasal suctioning and a single dose of nebulized albuterol. The children had all presented to the emergency department of Children’s Hospital of Philadelphia during one of two consecutive bronchiolitis seasons, from November to April in 2010 and 2011.
All children were assessed using the Respiratory Distress Assessment Instrument (RDAI) 1 hour after treatment and then 2 hours after treatment for those being discharged or still in the ED at that time. Based on the Respiratory Assessment Change Score (RACS) – which uses the RDAI score and a standardized change in respiratory rate to assess respiratory status changes – the normal saline group showed clinically significant improvement (a RACS of –3) after an hour, whereas the hypertonic saline group did not.
There was no significant difference in the median RDAI scores, heart rate, oxygen saturation, hospitalization rate, or child’s breathing or feeding status (based on parental perception) between the two groups.
Yet the hospitalization rate was lower in the hypertonic saline group of the other study, which used similar protocols in a larger population, reported Dr. Susan Wu of Children’s Hospital Los Angeles and her associates, also in JAMA Pediatrics (JAMA Pediatr. 2014 May 26 [doi: 10.1001/jamapediatrics.2014.301].
Their participants included 408 children under age 24 months presenting with bronchiolitis at two different children’s hospital EDs between March 2008 and April 2011. All patients received 2.5 mg of nebulized albuterol, after which 197 children received 4 mL of normal saline and 211 children received 4 mL of 3% hypertonic saline, each inhaled up to three times. Children admitted received their assigned saline (always premedicated with albuterol sulfate) every 8 hours until discharge.
While 42.6% of the normal saline patients were admitted to the hospital, only 28.9% of the hypertonic saline patients were admitted (P = .01), for an adjusted odds ratio of 0.49 and a number needed to treat of 8 to prevent one hospitalization. "Other statistically significant predictors of admission included site, male sex, patient weight, baseline respiratory rate, and baseline oxygen saturation," the authors reported.
Among those admitted, the length of stay was 3.16 days for the hypertonic saline participants and 3.92 days for the normal saline participants, but the difference was not significant (P = .24). No significant difference was found in the RDAI score, which decreased in both groups. The RACS was calculated using the same methods as in the Florin study for 366 cases, but no significant differences in the mean RACS scores existed between the two groups after adjustment for RDAI baseline scores.
Dr. Wu and her team recommended that future research "investigate the optimal dosing and administration regimen and the patient-level factors that may affect response to hypertonic saline."
The Florin et al. study was funded by an Academic Pediatric Association Young Investigator Award. The Wu et al. study was funded by the Thrasher Research Fund and the department of pediatrics, University of Southern California Keck School of Medicine, Los Angeles. The authors of both studies declared that they had no relevant financial disclosures.
FROM JAMA PEDIATRICS
Key clinical point: Hypertonic saline indications for bronchiolitis lack evidence for clear guidance.
Major finding: Compared with normal saline, administration of 3% hypertonic saline for bronchiolitis resulted in less improvement in the median RDAI score and no significant differences in heart rate, oxygen saturation, hospitalization rate, or other outcomes an hour after intervention in one study (Florin et al.). Another study showed a lower hospital admission rate for those receiving 3% hypertonic saline (28.9%, vs. 42.6% for normal saline; P = .01; adjusted odds ratio, 0.49) and no significant differences in RDAI score (Wu et al.).
Data source: The findings are based on two randomized controlled studies, one (Florin et al.) involving 62 children under age 24 months during November to April of 2010 and 2011, and another (Wu et al.) involving 408 children under age 24 months presenting at two emergency departments between March 2008 and April 2011.
Disclosures: The Florin et al. study was funded by an Academic Pediatric Association Young Investigator Award. The Wu et al. study was funded by the Thrasher Research Fund and the department of pediatrics, University of Southern California Keck School of Medicine, Los Angeles. The authors of both studies declared that they had no relevant financial disclosures.
Subsyndromal delirium common in critically ill patients
SAN DIEGO – Subsyndromal delirium was present in 86% of critically ill patients, results from a large observational study demonstrated. In addition, the duration of delirium was associated with increased odds of institutionalization, an association that was modified by the duration of delirium.
"In patients with less delirium, the effect of subsyndromal delirium on institutionalization was actually stronger," lead author Dr. Nathan E. Brummel said in an interview at an international conference of the American Thoracic Society, where the research was presented. "This identifies a cohort of people who previously were considered to have normal brain function, but it appears that this has long-term implications for their lives.

"Screening for delirium should occur not only in the ICU but on the wards as well. Patients who have delirium or delirium symptoms may benefit from measures used to prevent and treat this syndrome, such as the Hospital Elder Life Program, routine mobilization, and frequent reorientation through the use of nursing staff or even family members," he later added. This study data may help clinicians discuss long-term outcomes of critical illness with patients and their family members," he said.
For the study, Dr. Brummel, an instructor in medicine in the division of allergy, pulmonary and critical care medicine at Vanderbilt University Medical Center, Nashville, Tenn., and his associates evaluated 821 medical or surgical ICU patients with respiratory failure and/or shock who were enrolled in the BRAIN-ICU observational cohort study (N. Engl. J. Med. 2013;369:1306-16).
They used the Confusion Assessment Method for the ICU (CAM-ICU) to screen for delirium symptoms twice daily in the ICU and daily thereafter. The researchers considered delirium to be present if the CAM-ICU was positive. If the CAM-ICU was negative, they considered subsyndromal delirium (SSD) to be present if any delirium features were present or if inattention was present with or without other features of delirium.
SSD "is said to be present when a patient exhibits some delirium symptoms but does not meet the full delirium diagnostic criteria," the researchers wrote in their poster. "In patients without critical illness, SSD is associated with institutionalization, mortality, and cognitive decline, but these associations remain unclear in the critically ill."
The researchers tracked discharge location, mortality after hospital discharge and assessed for cognitive impairment at 3 and 12 months follow-up and used multivariate regression to determine the relationship between days of SSD and outcomes.
The mean age of the 821 patients was 61 years and their mean APACHE II score was 25. In all, 702 patients (86%) had SSD that lasted an average of 3 days. The most common SSU pattern based on the CAM-ICU was fluctuation of mental status (which occurred in 50% of assessments) and fluctuation in mental status plus altered level of consciousness (which occurred in 22% of assessments).
Dr. Brummel and his associates also found that the duration of SSD was independently associated with increased odds of institutionalization (odds ratio, 1.90), but SSD did not predict mortality or long-term cognitive impairment at 3 or 12 months. "We don’t yet understand the mechanism behind why subsyndromal delirium and institutionalization are associated," Dr. Brummel said. "It probably relates to an association between SSD and factors that drive institutionalization, such as physical disability and cognitive impairment. Once patients survived the hospital stay, subsyndromal delirium wasn’t associated with an increased risk of mortality. It may be the fact that this less severe form of brain dysfunction in the ICU does not have the same effect as the full syndrome of delirium."
He acknowledged certain limitations of the study, including the fact that the CAM-ICU only measures four features of delirium and that no assessments of cognitive or physical function were conducted at hospital discharge.
The study was supported by the National Institutes of Health, the Vanderbilt Clinical and Translational Scholars Program, and the Veterans Affairs Tennessee Valley Healthcare System Geriatric Research Education and Clinical Centers. Dr. Brummel said that he had no relevant financial conflicts to disclose.
SAN DIEGO – Subsyndromal delirium was present in 86% of critically ill patients, results from a large observational study demonstrated. In addition, the duration of delirium was associated with increased odds of institutionalization, an association that was modified by the duration of delirium.
"In patients with less delirium, the effect of subsyndromal delirium on institutionalization was actually stronger," lead author Dr. Nathan E. Brummel said in an interview at an international conference of the American Thoracic Society, where the research was presented. "This identifies a cohort of people who previously were considered to have normal brain function, but it appears that this has long-term implications for their lives.
"Screening for delirium should occur not only in the ICU but on the wards as well. Patients who have delirium or delirium symptoms may benefit from measures used to prevent and treat this syndrome, such as the Hospital Elder Life Program, routine mobilization, and frequent reorientation through the use of nursing staff or even family members," he later added. This study data may help clinicians discuss long-term outcomes of critical illness with patients and their family members," he said.
For the study, Dr. Brummel, an instructor in medicine in the division of allergy, pulmonary and critical care medicine at Vanderbilt University Medical Center, Nashville, Tenn., and his associates evaluated 821 medical or surgical ICU patients with respiratory failure and/or shock who were enrolled in the BRAIN-ICU observational cohort study (N. Engl. J. Med. 2013;369:1306-16).
They used the Confusion Assessment Method for the ICU (CAM-ICU) to screen for delirium symptoms twice daily in the ICU and daily thereafter. The researchers considered delirium to be present if the CAM-ICU was positive. If the CAM-ICU was negative, they considered subsyndromal delirium (SSD) to be present if any delirium features were present or if inattention was present with or without other features of delirium.
SSD "is said to be present when a patient exhibits some delirium symptoms but does not meet the full delirium diagnostic criteria," the researchers wrote in their poster. "In patients without critical illness, SSD is associated with institutionalization, mortality, and cognitive decline, but these associations remain unclear in the critically ill."
The researchers tracked discharge location, mortality after hospital discharge and assessed for cognitive impairment at 3 and 12 months follow-up and used multivariate regression to determine the relationship between days of SSD and outcomes.
The mean age of the 821 patients was 61 years and their mean APACHE II score was 25. In all, 702 patients (86%) had SSD that lasted an average of 3 days. The most common SSU pattern based on the CAM-ICU was fluctuation of mental status (which occurred in 50% of assessments) and fluctuation in mental status plus altered level of consciousness (which occurred in 22% of assessments).
Dr. Brummel and his associates also found that the duration of SSD was independently associated with increased odds of institutionalization (odds ratio, 1.90), but SSD did not predict mortality or long-term cognitive impairment at 3 or 12 months. "We don’t yet understand the mechanism behind why subsyndromal delirium and institutionalization are associated," Dr. Brummel said. "It probably relates to an association between SSD and factors that drive institutionalization, such as physical disability and cognitive impairment. Once patients survived the hospital stay, subsyndromal delirium wasn’t associated with an increased risk of mortality. It may be the fact that this less severe form of brain dysfunction in the ICU does not have the same effect as the full syndrome of delirium."
He acknowledged certain limitations of the study, including the fact that the CAM-ICU only measures four features of delirium and that no assessments of cognitive or physical function were conducted at hospital discharge.
The study was supported by the National Institutes of Health, the Vanderbilt Clinical and Translational Scholars Program, and the Veterans Affairs Tennessee Valley Healthcare System Geriatric Research Education and Clinical Centers. Dr. Brummel said that he had no relevant financial conflicts to disclose.
SAN DIEGO – Subsyndromal delirium was present in 86% of critically ill patients, results from a large observational study demonstrated. In addition, the duration of delirium was associated with increased odds of institutionalization, an association that was modified by the duration of delirium.
"In patients with less delirium, the effect of subsyndromal delirium on institutionalization was actually stronger," lead author Dr. Nathan E. Brummel said in an interview at an international conference of the American Thoracic Society, where the research was presented. "This identifies a cohort of people who previously were considered to have normal brain function, but it appears that this has long-term implications for their lives.
"Screening for delirium should occur not only in the ICU but on the wards as well. Patients who have delirium or delirium symptoms may benefit from measures used to prevent and treat this syndrome, such as the Hospital Elder Life Program, routine mobilization, and frequent reorientation through the use of nursing staff or even family members," he later added. This study data may help clinicians discuss long-term outcomes of critical illness with patients and their family members," he said.
For the study, Dr. Brummel, an instructor in medicine in the division of allergy, pulmonary and critical care medicine at Vanderbilt University Medical Center, Nashville, Tenn., and his associates evaluated 821 medical or surgical ICU patients with respiratory failure and/or shock who were enrolled in the BRAIN-ICU observational cohort study (N. Engl. J. Med. 2013;369:1306-16).
They used the Confusion Assessment Method for the ICU (CAM-ICU) to screen for delirium symptoms twice daily in the ICU and daily thereafter. The researchers considered delirium to be present if the CAM-ICU was positive. If the CAM-ICU was negative, they considered subsyndromal delirium (SSD) to be present if any delirium features were present or if inattention was present with or without other features of delirium.
SSD "is said to be present when a patient exhibits some delirium symptoms but does not meet the full delirium diagnostic criteria," the researchers wrote in their poster. "In patients without critical illness, SSD is associated with institutionalization, mortality, and cognitive decline, but these associations remain unclear in the critically ill."
The researchers tracked discharge location, mortality after hospital discharge and assessed for cognitive impairment at 3 and 12 months follow-up and used multivariate regression to determine the relationship between days of SSD and outcomes.
The mean age of the 821 patients was 61 years and their mean APACHE II score was 25. In all, 702 patients (86%) had SSD that lasted an average of 3 days. The most common SSU pattern based on the CAM-ICU was fluctuation of mental status (which occurred in 50% of assessments) and fluctuation in mental status plus altered level of consciousness (which occurred in 22% of assessments).
Dr. Brummel and his associates also found that the duration of SSD was independently associated with increased odds of institutionalization (odds ratio, 1.90), but SSD did not predict mortality or long-term cognitive impairment at 3 or 12 months. "We don’t yet understand the mechanism behind why subsyndromal delirium and institutionalization are associated," Dr. Brummel said. "It probably relates to an association between SSD and factors that drive institutionalization, such as physical disability and cognitive impairment. Once patients survived the hospital stay, subsyndromal delirium wasn’t associated with an increased risk of mortality. It may be the fact that this less severe form of brain dysfunction in the ICU does not have the same effect as the full syndrome of delirium."
He acknowledged certain limitations of the study, including the fact that the CAM-ICU only measures four features of delirium and that no assessments of cognitive or physical function were conducted at hospital discharge.
The study was supported by the National Institutes of Health, the Vanderbilt Clinical and Translational Scholars Program, and the Veterans Affairs Tennessee Valley Healthcare System Geriatric Research Education and Clinical Centers. Dr. Brummel said that he had no relevant financial conflicts to disclose.
AT ATS 2014
Key clinical point: Patients who have even a few delirium symptoms may benefit from prevention and treatment measures.
Major finding: Among 821 critically ill patients, 702 (86%) had subsyndromal delirium that lasted an average of 3 days.
Data source: An evaluation of 821 medical or surgical ICU patients with respiratory failure and/or shock who were enrolled in the BRAIN-ICU observational cohort study.
Disclosures: The study was supported by the National Institutes of Health, the Vanderbilt Clinical and Translational Scholars Program, and the VA Tennessee Valley Healthcare System Geriatric Research Education and Clinical Centers. Dr. Brummel had no financial conflicts.

A CPAP alternative, implantable tongue stimulator is ahead for apnea
An implantable device that stimulates the hypoglossal nerve during inspiration has been approved for treating a subgroup of adults with moderate to severe obstructive sleep apnea, the Food and Drug Administration has announced.
In a statement reviewing the approval, posted on the agency’s website on May 21, the FDA says that the device is indicated for adults, age 22 years and older, with moderate to severe OSA, "who have been confirmed to fail or cannot tolerate positive airway pressure (PAP) treatments," such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) machines, "and who do not have a complete concentric collapse (as seen during drug induced sleep endoscopy) at the soft palate level."
The components of the device include a stimulation lead placed around the distal hypoglossal nerve, a pulse generator implanted subcutaneously below the clavicle in the upper chest, and a respiration-sensing lead inserted between the fourth and fifth ribs – implanted under general anesthesia. The pulse generator "detects the patient’s breathing pattern and maintains an open airway with mild stimulation of the hypoglossal nerve, which controls tongue movement, during inhaled breathing," according to the FDA statement. The stimulation settings are adjusted by physicians with an external programmer, and patients use a remote to turn the device on before they go to sleep and to turn it off when they wake up.
The device is manufactured by Inspire Medical Systems.
At a meeting in February 2014, the FDA’s Anesthesiology and Respiratory Therapy Devices Panel supported approval of the Inspire Upper Airway Stimulation System, based on the results of the pivotal study, the STAR (Stimulation Therapy for Apnea Reduction) trial, of 126 patients with moderate to severe OSA, which was published in January. Treatment with the device, which delivered unilateral stimulation of the hypoglossal nerve, "synchronous with ventilation, resulted in significant and clinically meaningful reductions in the severity of sleep apnea and self-reported sleepiness and improvement in quality of life measures at 1 year," the authors concluded (N. Engl. J. Med. 2014;370:139-49).
The device was approved on May 1. It will be commercially available during the second half of 2014, according to the manufacturer’s statement announcing the approval.
An implantable device that stimulates the hypoglossal nerve during inspiration has been approved for treating a subgroup of adults with moderate to severe obstructive sleep apnea, the Food and Drug Administration has announced.
In a statement reviewing the approval, posted on the agency’s website on May 21, the FDA says that the device is indicated for adults, age 22 years and older, with moderate to severe OSA, "who have been confirmed to fail or cannot tolerate positive airway pressure (PAP) treatments," such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) machines, "and who do not have a complete concentric collapse (as seen during drug induced sleep endoscopy) at the soft palate level."
The components of the device include a stimulation lead placed around the distal hypoglossal nerve, a pulse generator implanted subcutaneously below the clavicle in the upper chest, and a respiration-sensing lead inserted between the fourth and fifth ribs – implanted under general anesthesia. The pulse generator "detects the patient’s breathing pattern and maintains an open airway with mild stimulation of the hypoglossal nerve, which controls tongue movement, during inhaled breathing," according to the FDA statement. The stimulation settings are adjusted by physicians with an external programmer, and patients use a remote to turn the device on before they go to sleep and to turn it off when they wake up.
The device is manufactured by Inspire Medical Systems.
At a meeting in February 2014, the FDA’s Anesthesiology and Respiratory Therapy Devices Panel supported approval of the Inspire Upper Airway Stimulation System, based on the results of the pivotal study, the STAR (Stimulation Therapy for Apnea Reduction) trial, of 126 patients with moderate to severe OSA, which was published in January. Treatment with the device, which delivered unilateral stimulation of the hypoglossal nerve, "synchronous with ventilation, resulted in significant and clinically meaningful reductions in the severity of sleep apnea and self-reported sleepiness and improvement in quality of life measures at 1 year," the authors concluded (N. Engl. J. Med. 2014;370:139-49).
The device was approved on May 1. It will be commercially available during the second half of 2014, according to the manufacturer’s statement announcing the approval.
An implantable device that stimulates the hypoglossal nerve during inspiration has been approved for treating a subgroup of adults with moderate to severe obstructive sleep apnea, the Food and Drug Administration has announced.
In a statement reviewing the approval, posted on the agency’s website on May 21, the FDA says that the device is indicated for adults, age 22 years and older, with moderate to severe OSA, "who have been confirmed to fail or cannot tolerate positive airway pressure (PAP) treatments," such as continuous positive airway pressure (CPAP) or bilevel positive airway pressure (BPAP) machines, "and who do not have a complete concentric collapse (as seen during drug induced sleep endoscopy) at the soft palate level."
The components of the device include a stimulation lead placed around the distal hypoglossal nerve, a pulse generator implanted subcutaneously below the clavicle in the upper chest, and a respiration-sensing lead inserted between the fourth and fifth ribs – implanted under general anesthesia. The pulse generator "detects the patient’s breathing pattern and maintains an open airway with mild stimulation of the hypoglossal nerve, which controls tongue movement, during inhaled breathing," according to the FDA statement. The stimulation settings are adjusted by physicians with an external programmer, and patients use a remote to turn the device on before they go to sleep and to turn it off when they wake up.
The device is manufactured by Inspire Medical Systems.
At a meeting in February 2014, the FDA’s Anesthesiology and Respiratory Therapy Devices Panel supported approval of the Inspire Upper Airway Stimulation System, based on the results of the pivotal study, the STAR (Stimulation Therapy for Apnea Reduction) trial, of 126 patients with moderate to severe OSA, which was published in January. Treatment with the device, which delivered unilateral stimulation of the hypoglossal nerve, "synchronous with ventilation, resulted in significant and clinically meaningful reductions in the severity of sleep apnea and self-reported sleepiness and improvement in quality of life measures at 1 year," the authors concluded (N. Engl. J. Med. 2014;370:139-49).
The device was approved on May 1. It will be commercially available during the second half of 2014, according to the manufacturer’s statement announcing the approval.
Housing code violations are marker for asthma risk
VANCOUVER, B.C. – Pediatricians and public health professionals can estimate children’s asthma risk based on the density of housing code violations in their neighborhood, according to a retrospective cohort study reported at the annual meeting of the Pediatric Academic Societies.
Investigators led by Dr. Andrew F. Beck, of the pediatrics department at Cincinnati Children’s Hospital Medical Center, calculated the density of asthma-relevant housing code violations – those for cockroaches, rodents, mold, and water damage – for 113 neighborhoods (census tracts) in the Greater Cincinnati area.

Analyses showed that this density was significantly and positively associated with the neighborhood’s rate of pediatric asthma-related emergency department (ED) visits and hospital admissions.
Also, among the subset of patients who were admitted for asthma, the odds of another ED visit or admission in the next year increased with the density of violations. It was elevated by more than 50% for those living in neighborhoods with a medium-high or high density of violations relative to those living in neighborhoods with a low density.
"A measure of housing code violation density ... was found to be significantly correlated with asthma utilization rates at the population level, and significantly and independently associated with reutilization at the patient level," Dr. Beck commented.
"In an era where home visits are rare, local geographic data that highlight the impact of context on health provide a virtual home or neighborhood visit. Such information could help to identify upstream areas where disparity reduction can be targeted within care delivery, a focus especially relevant for diseases like asthma that are heavily impacted by the quality of one’s surroundings," he maintained.
"We expect that local risk-specific data can influence delivery of public health services more proactively. For example, the pairing of housing data with outcome data could facilitate identification of geographic areas likely to benefit from targeted housing assessments and actions," Dr. Beck added. "We also believe that contextual housing data can be integrated into clinical care. Such information could rapidly inform and drive focused interventions for connections to housing experts; for example, patients could be more efficiently connected to social workers, home health providers, and community partners such as legal advocates and community health workers, all of whom may be more suited to handling underlying housing risks than the medical team."
Study results showed that across neighborhoods, the median density of asthma-relevant housing code violations was 11 per 1,000 homes and apartments, with a range from 0 to 120, according to Dr. Beck.
The investigators identified 8,736 asthma-related ED visits and admissions to Cincinnati Children’s Hospital Medical Center by children aged 1-16 years during the study period. As the neighborhood density of housing code violations increased, so did this rate (r = 0.59; P less than .0001).
Moreover, the association remained significant after adjustment for neighborhood poverty level. Overall, the density of violations accounted for 25% of the variation across neighborhoods in such asthma-related health care use.
Among 1,531 pediatric patients with an index hospital admission for asthma, 37% had another ED visit or admission during a year of follow-up.
In a multivariate analysis, the adjusted odds of such repeat use increased with the neighborhood housing code violation density. Relative to peers living in neighborhoods with low density, children living in ones with medium-high density or high density had a significantly elevated risk (odds ratios, 1.54 and 1.84, respectively).
"In our region, housing assessments are complaint-driven; thus, the true breadth of violations is unknown," Dr. Beck noted, addressing study limitations. Also, generalizability is unclear; "we expect that many jurisdictions, however, collect similar data and use that data in similar ways."
Dr. Beck disclosed no relevant conflicts of interest.
VANCOUVER, B.C. – Pediatricians and public health professionals can estimate children’s asthma risk based on the density of housing code violations in their neighborhood, according to a retrospective cohort study reported at the annual meeting of the Pediatric Academic Societies.
Investigators led by Dr. Andrew F. Beck, of the pediatrics department at Cincinnati Children’s Hospital Medical Center, calculated the density of asthma-relevant housing code violations – those for cockroaches, rodents, mold, and water damage – for 113 neighborhoods (census tracts) in the Greater Cincinnati area.
Analyses showed that this density was significantly and positively associated with the neighborhood’s rate of pediatric asthma-related emergency department (ED) visits and hospital admissions.
Also, among the subset of patients who were admitted for asthma, the odds of another ED visit or admission in the next year increased with the density of violations. It was elevated by more than 50% for those living in neighborhoods with a medium-high or high density of violations relative to those living in neighborhoods with a low density.
"A measure of housing code violation density ... was found to be significantly correlated with asthma utilization rates at the population level, and significantly and independently associated with reutilization at the patient level," Dr. Beck commented.
"In an era where home visits are rare, local geographic data that highlight the impact of context on health provide a virtual home or neighborhood visit. Such information could help to identify upstream areas where disparity reduction can be targeted within care delivery, a focus especially relevant for diseases like asthma that are heavily impacted by the quality of one’s surroundings," he maintained.
"We expect that local risk-specific data can influence delivery of public health services more proactively. For example, the pairing of housing data with outcome data could facilitate identification of geographic areas likely to benefit from targeted housing assessments and actions," Dr. Beck added. "We also believe that contextual housing data can be integrated into clinical care. Such information could rapidly inform and drive focused interventions for connections to housing experts; for example, patients could be more efficiently connected to social workers, home health providers, and community partners such as legal advocates and community health workers, all of whom may be more suited to handling underlying housing risks than the medical team."
Study results showed that across neighborhoods, the median density of asthma-relevant housing code violations was 11 per 1,000 homes and apartments, with a range from 0 to 120, according to Dr. Beck.
The investigators identified 8,736 asthma-related ED visits and admissions to Cincinnati Children’s Hospital Medical Center by children aged 1-16 years during the study period. As the neighborhood density of housing code violations increased, so did this rate (r = 0.59; P less than .0001).
Moreover, the association remained significant after adjustment for neighborhood poverty level. Overall, the density of violations accounted for 25% of the variation across neighborhoods in such asthma-related health care use.
Among 1,531 pediatric patients with an index hospital admission for asthma, 37% had another ED visit or admission during a year of follow-up.
In a multivariate analysis, the adjusted odds of such repeat use increased with the neighborhood housing code violation density. Relative to peers living in neighborhoods with low density, children living in ones with medium-high density or high density had a significantly elevated risk (odds ratios, 1.54 and 1.84, respectively).
"In our region, housing assessments are complaint-driven; thus, the true breadth of violations is unknown," Dr. Beck noted, addressing study limitations. Also, generalizability is unclear; "we expect that many jurisdictions, however, collect similar data and use that data in similar ways."
Dr. Beck disclosed no relevant conflicts of interest.
VANCOUVER, B.C. – Pediatricians and public health professionals can estimate children’s asthma risk based on the density of housing code violations in their neighborhood, according to a retrospective cohort study reported at the annual meeting of the Pediatric Academic Societies.
Investigators led by Dr. Andrew F. Beck, of the pediatrics department at Cincinnati Children’s Hospital Medical Center, calculated the density of asthma-relevant housing code violations – those for cockroaches, rodents, mold, and water damage – for 113 neighborhoods (census tracts) in the Greater Cincinnati area.
Analyses showed that this density was significantly and positively associated with the neighborhood’s rate of pediatric asthma-related emergency department (ED) visits and hospital admissions.
Also, among the subset of patients who were admitted for asthma, the odds of another ED visit or admission in the next year increased with the density of violations. It was elevated by more than 50% for those living in neighborhoods with a medium-high or high density of violations relative to those living in neighborhoods with a low density.
"A measure of housing code violation density ... was found to be significantly correlated with asthma utilization rates at the population level, and significantly and independently associated with reutilization at the patient level," Dr. Beck commented.
"In an era where home visits are rare, local geographic data that highlight the impact of context on health provide a virtual home or neighborhood visit. Such information could help to identify upstream areas where disparity reduction can be targeted within care delivery, a focus especially relevant for diseases like asthma that are heavily impacted by the quality of one’s surroundings," he maintained.
"We expect that local risk-specific data can influence delivery of public health services more proactively. For example, the pairing of housing data with outcome data could facilitate identification of geographic areas likely to benefit from targeted housing assessments and actions," Dr. Beck added. "We also believe that contextual housing data can be integrated into clinical care. Such information could rapidly inform and drive focused interventions for connections to housing experts; for example, patients could be more efficiently connected to social workers, home health providers, and community partners such as legal advocates and community health workers, all of whom may be more suited to handling underlying housing risks than the medical team."
Study results showed that across neighborhoods, the median density of asthma-relevant housing code violations was 11 per 1,000 homes and apartments, with a range from 0 to 120, according to Dr. Beck.
The investigators identified 8,736 asthma-related ED visits and admissions to Cincinnati Children’s Hospital Medical Center by children aged 1-16 years during the study period. As the neighborhood density of housing code violations increased, so did this rate (r = 0.59; P less than .0001).
Moreover, the association remained significant after adjustment for neighborhood poverty level. Overall, the density of violations accounted for 25% of the variation across neighborhoods in such asthma-related health care use.
Among 1,531 pediatric patients with an index hospital admission for asthma, 37% had another ED visit or admission during a year of follow-up.
In a multivariate analysis, the adjusted odds of such repeat use increased with the neighborhood housing code violation density. Relative to peers living in neighborhoods with low density, children living in ones with medium-high density or high density had a significantly elevated risk (odds ratios, 1.54 and 1.84, respectively).
"In our region, housing assessments are complaint-driven; thus, the true breadth of violations is unknown," Dr. Beck noted, addressing study limitations. Also, generalizability is unclear; "we expect that many jurisdictions, however, collect similar data and use that data in similar ways."
Dr. Beck disclosed no relevant conflicts of interest.
AT THE PAS ANNUAL MEETING
Key clinical point: Children’s asthma risk can be estimated based on the density of housing code violations in their neighborhood.
Major finding: The density of housing code violations explained 25% of the variation in pediatric asthma-related health care use across neighborhoods, and hospitalized children from higher-density neighborhoods had increased odds of repeat use.
Data source: A retrospective cohort study of 113 neighborhoods, 8,736 pediatric asthma-related ED visits and admissions, and 1,531 children admitted for asthma.
Disclosures: Dr. Beck disclosed no relevant conflicts of interest.
