User login
Autoantibody profile may affect the number and size of RA bone erosions
Rheumatoid arthritis patients who tested positive for both rheumatoid factor and anticitrullinated protein antibodies showed a greater prevalence and size of bone erosions than did patients who came up negative for both in a prospective cohort study.
High-resolution peripheral quantitative CT revealed a prevalence of around 5.35 bone erosions per patient among 112 who were positive for both rheumatoid factor (RF) and anticitrullinated protein antibodies (ACPAs), compared with 2.49 in 69 who were negative for both of the autoantibodies. Another 29 patients who were ACPA positive and RH negative had 2.41 erosions, while 28 patients who were ACPA negative and RH positive had 2.00 erosions. The size of the bone erosion had a similar pattern, with the greatest volume observed in patients positive for both autoantibodies (7.66 mm3), followed by ACPA-positive patients (6.20 mm3), patients negative for both autoantibodies (3.32 mm3), and RH-positive patients (2.76 mm3).
The size of erosions was also associated with the presence and titer of rheumatoid factor in ACPA-positive but not ACPA-negative patients (Ann. Rheum. Dis. 2014 Aug. 12 [doi:10.1136/annrheumdis-2014-205428]).
"These observations suggest that RF may act as an enhancer of bone loss in patients with RA, and acts as an additive to ACPAs," wrote Dr. Carolin Hecht of the University of Erlangen-Nuremberg (Germany) and colleagues.
The study was supported by the Deutsche Forschungsgemeinschaft, the Bundesministerium fu¨r Bildung und Forschung, the Marie Curie project OSTEOIMMUNE, the TEAM and MASTERSWITCH projects of the European Union, and the IMI-funded project BTCure. There were no other conflicts of interest declared.
Rheumatoid arthritis patients who tested positive for both rheumatoid factor and anticitrullinated protein antibodies showed a greater prevalence and size of bone erosions than did patients who came up negative for both in a prospective cohort study.
High-resolution peripheral quantitative CT revealed a prevalence of around 5.35 bone erosions per patient among 112 who were positive for both rheumatoid factor (RF) and anticitrullinated protein antibodies (ACPAs), compared with 2.49 in 69 who were negative for both of the autoantibodies. Another 29 patients who were ACPA positive and RH negative had 2.41 erosions, while 28 patients who were ACPA negative and RH positive had 2.00 erosions. The size of the bone erosion had a similar pattern, with the greatest volume observed in patients positive for both autoantibodies (7.66 mm3), followed by ACPA-positive patients (6.20 mm3), patients negative for both autoantibodies (3.32 mm3), and RH-positive patients (2.76 mm3).
The size of erosions was also associated with the presence and titer of rheumatoid factor in ACPA-positive but not ACPA-negative patients (Ann. Rheum. Dis. 2014 Aug. 12 [doi:10.1136/annrheumdis-2014-205428]).
"These observations suggest that RF may act as an enhancer of bone loss in patients with RA, and acts as an additive to ACPAs," wrote Dr. Carolin Hecht of the University of Erlangen-Nuremberg (Germany) and colleagues.
The study was supported by the Deutsche Forschungsgemeinschaft, the Bundesministerium fu¨r Bildung und Forschung, the Marie Curie project OSTEOIMMUNE, the TEAM and MASTERSWITCH projects of the European Union, and the IMI-funded project BTCure. There were no other conflicts of interest declared.
Rheumatoid arthritis patients who tested positive for both rheumatoid factor and anticitrullinated protein antibodies showed a greater prevalence and size of bone erosions than did patients who came up negative for both in a prospective cohort study.
High-resolution peripheral quantitative CT revealed a prevalence of around 5.35 bone erosions per patient among 112 who were positive for both rheumatoid factor (RF) and anticitrullinated protein antibodies (ACPAs), compared with 2.49 in 69 who were negative for both of the autoantibodies. Another 29 patients who were ACPA positive and RH negative had 2.41 erosions, while 28 patients who were ACPA negative and RH positive had 2.00 erosions. The size of the bone erosion had a similar pattern, with the greatest volume observed in patients positive for both autoantibodies (7.66 mm3), followed by ACPA-positive patients (6.20 mm3), patients negative for both autoantibodies (3.32 mm3), and RH-positive patients (2.76 mm3).
The size of erosions was also associated with the presence and titer of rheumatoid factor in ACPA-positive but not ACPA-negative patients (Ann. Rheum. Dis. 2014 Aug. 12 [doi:10.1136/annrheumdis-2014-205428]).
"These observations suggest that RF may act as an enhancer of bone loss in patients with RA, and acts as an additive to ACPAs," wrote Dr. Carolin Hecht of the University of Erlangen-Nuremberg (Germany) and colleagues.
The study was supported by the Deutsche Forschungsgemeinschaft, the Bundesministerium fu¨r Bildung und Forschung, the Marie Curie project OSTEOIMMUNE, the TEAM and MASTERSWITCH projects of the European Union, and the IMI-funded project BTCure. There were no other conflicts of interest declared.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: RF may enhance the loss of bone in patients who are positive for ACPAs.
Major finding: There was a prevalence of around 5.35 bone erosions per patient among 112 who were positive for both rheumatoid factor and anticitrullinated protein antibodies, compared with 2.49 in 69 who were negative for both of the autoantibodies.
Data source: A prospective cohort study in 238 patients with rheumatoid arthritis.
Disclosures: The study was supported by the Deutsche Forschungsgemeinschaft, the Bundesministerium fu¨r Bildung und Forschung, the Marie Curie project OSTEOIMMUNE, the TEAM and MASTERSWITCH projects of the European Union, and the IMI-funded project BTCure.
2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis
The levels of evidence supporting the recommendations (A-C) are defined at the end of the "Major Recommendations" field.
Recommendations for the Use of Disease-Modifying Antirheumatic Drugs (DMARDs) and Biologic Agents in Patients Who Qualify for Treatment of Rheumatoid Arthritis (RA)
This 2012 American College of Rheumatology (ACR) recommendations update incorporates the evidence from systematic literature review synthesis and recommendations from 2008 and rates updated and new clinical scenarios regarding the use of DMARDs and biologic agents for the treatment of RA. Terms used in the recommendations are defined in Table 2 of the original guideline document. The 2012 recommendations are listed in the 4 sections below and in the following order:
- Indications for and switching DMARDs and biologic agents: early RA (indications, see Figure 1 in the original guideline document) followed by established RA (indications and switching, see Figure 2 in the original guideline document), along with details of the level of evidence supporting these recommendations (see Supplementary Appendix 7, available in the online version at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
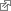
- Use of biologic agents in patients with hepatitis, malignancy, or congestive heart failure (CHF) who qualify for RA management (see Table 4 in the original guideline document)
- Screening for tuberculosis (TB) in patients starting or currently receiving biologic agents as part of their RA therapy (see Figure 3 in the original guideline document)
- Vaccination in patients starting or currently receiving DMARDs or biologic agents as part of their RA therapy (see Table 5 in the original guideline document)
The recommendations in the text below and in Tables 4 and 5 in the original guideline document represent the results of the 2012 update only, whereas Figures 1–3 in the original guideline document also incorporate some of the 2008 ACR RA recommendations that did not change. Areas of uncertainty by the panel (that did not lead to recommendations) are noted in Supplementary Appendix 8 (available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658 ![]()
- Indications for Starting, Resuming, Adding, or Switching DMARDs or Biologic Agents
The panel first describes a recommendation targeting remission or low disease activity in RA (section 1A). This is followed by recommendations for DMARD or biologic agent use in early RA (section 1B). Next, the panel provides recommendations for initiating and switching between DMARDs and biologic agents in established RA (section 1C).
1A. Target Low Disease Activity or Remission
The panel recommends targeting either low disease activity (see Table 3 in the original guideline document) or remission (see Table 2 in the original guideline document) in all patients with early RA (see Figure 1 in the original guideline document; level of evidence C) and established RA (see Figure 2 in the original guideline document; level of evidence C) receiving any DMARD or biologic agent.
1B. Early RA (Disease Duration <6 Months)
In patients with early RA, the panel recommends the use of DMARD monotherapy both for low disease activity and for moderate or high disease activity with the absence of poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C) (details are shown in Supplementary Appendix 7, available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
In patients with early RA, the panel recommends the use of DMARD combination therapy (including double and triple therapy) in patients with moderate or high disease activity plus poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C).
In patients with early RA, the panel also recommends the use of an anti-tumor necrosis factor (anti-TNF) biologic with or without methotrexate in patients who have high disease activity with poor prognostic features (see Figure 1 in the original guideline document; level of evidence A and B). Infliximab is the only exception and the recommendation is to use it in combination with methotrexate, but not as monotherapy.
1C. Established RA (Disease Duration ≥6 Months or Meeting the 1987 ACR RA Classification Criteria)
The remainder of panel recommendations regarding indications for DMARDs and biologic agents are for patients with established RA. The 3 subsections below define recommendations for initiating and switching therapies in established RA (see Figure 2 in the original guideline document). Where the prognosis is not mentioned, the recommendation to use/switch to a DMARD or a biologic agent applies to all patients, regardless of prognostic features.
Initiating and Switching Among DMARDs
If after 3 months of DMARD monotherapy (in patients without poor prognostic features), a patient deteriorates from low to moderate/high disease activity, then methotrexate, hydroxychloroquine, or leflunomide should be added (see rectangle A of Figure 2 in the original guideline document; level of evidence A and B).
If after 3 months of methotrexate or methotrexate/DMARD combination, a patient still has moderate or high disease activity, then add another non-methotrexate DMARD or switch to a different non-methotrexate DMARD (see rectangle B of Figure 2 in the original guideline document; level of evidence B and C).
Switching from DMARDs to Biologic Agents
If a patient has moderate or high disease activity after 3 months of methotrexate monotherapy or DMARD combination therapy, as an alternative to the DMARD recommendation just noted above, the panel recommends adding or switching to an anti-TNF biologic, abatacept, or rituximab (see rectangles C and D of Figure 2 in the original guideline document; level of evidence A–C).
If after 3 months of intensified DMARD combination therapy or after a second DMARD, a patient still has moderate or high disease activity, add or switch to an anti-TNF biologic (see rectangle C of Figure 2 in the original guideline document; level of evidence C).
Switching Among Biologic Agents Due to Lack of Benefit or Loss of Benefit
If a patient still has moderate or high disease activity after 3 months of anti-TNF biologic therapy and this is due to a lack or loss of benefit, switching to another anti-TNF biologic or a non-TNF biologic is recommended (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C).
If a patient still has moderate or high disease activity after 6 months of a non-TNF biologic and the failure is due to a lack or loss of benefit, switch to another non-TNF biologic or an anti-TNF biologic (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C). An assessment period of 6 months was chosen rather than 3 months, due to the anticipation that a longer time may be required for efficacy of a non-TNF biologic.
Switching Among Biologic Agents Due to Harms/Adverse Events
If a patient has high disease activity after failing an anti-TNF biologic because of a serious adverse event, switch to a non-TNF biologic (see rectangle E of Figure 2 in the original guideline document; level of evidence C).
If a patient has moderate or high disease activity after failing an anti-TNF biologic because of a nonserious adverse event, switch to another anti-TNF biologic or a non-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence B and C).
If a patient has moderate or high disease activity after failing a non-TNF biologic because of an adverse event (serious or nonserious), switch to another non-TNF biologic or an anti-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence C).
- Use of Biologic Agents in RA Patients With Hepatitis, Malignancy, or Chronic Heart Failure (CHF), Qualifying for More Aggressive Treatment (level of evidence C for all recommendations)
Hepatitis B or C
The panel recommends that etanercept could potentially be used in RA patients with hepatitis C requiring RA treatment (see Table 4 in the original guideline document).
The panel also recommends not using biologic agents in RA patients with untreated chronic hepatitis B (disease not treated due to contraindications to treatment or intolerable adverse events) and in RA patients with treated chronic hepatitis B with Child-Pugh class B and higher (see Table 4 in the original guideline document; for details of Child-Pugh classification, see Table 2 in the original guideline document). The panel did not make recommendations regarding the use of any biologic agent for treatment in RA patients with a history of hepatitis B and a positive hepatitis B core antibody.
Malignancies
For patients who have been treated for solid malignancies more than 5 years ago or who have been treated for nonmelanoma skin cancer more than 5 years ago, the panel recommends starting or resuming any biologic agent if those patients would otherwise qualify for this RA management strategy (see Table 4 in the original guideline document).
The panel only recommends starting or resuming rituximab in RA patients with: 1) a previously treated solid malignancy within the last 5 years, 2) a previously treated nonmelanoma skin cancer within the last 5 years, 3) a previously treated melanoma skin cancer, or 4) a previously treated lymphoproliferative malignancy. Little is known about the effects of biologic therapy in patients with a history of a solid cancer within the past 5 years owing to the exclusion of such patients from participation in clinical trials and the lack of studies examining the risk of recurrent cancer in this subgroup of patients.
CHF
The panel recommends not using an anti-TNF biologic in RA patients with CHF that is New York Heart Association (NYHA) class III or IV and who have an ejection fraction of 50% or less (see Table 4 in the original guideline document).
- TB Screening for Biologic Agents (level of evidence C for all recommendations except for initiation of biologic agents in patients being treated for latent TB infection [LTBI], where the level of evidence is B)
The panel recommends screening to identify LTBI in all RA patients being considered for therapy with biologic agents, regardless of the presence of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends that clinicians assess the patient's medical history to identify risk factors for TB (specified by the Centers for Disease Control and Prevention [CDC]) (see Table 2 in the original guideline document).
The panel recommends the tuberculin skin test (TST) or interferon-gamma–release assays (IGRAs) as the initial test in all RA patients starting biologic agents, regardless of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends the use of the IGRA over the TST in patients who had previously received a bacillus Calmette-Guerin (BCG) vaccination, due to the high false-positive test rates for TST (see Figure 3 in the original guideline document).
The panel recommends that RA patients with a positive initial or repeat TST or IGRA should have a chest radiograph and, if suggestive of active TB, a subsequent sputum examination to check for the presence of active TB (see diamonds B and C of Figure 3 in the original guideline document). RA patients with a negative screening TST or IGRA may not need further evaluation in the absence of risk factors and/or clinical suspicion for TB. Since patients with RA may have false-negative TST or IGRA results due to immunosuppression, a negative TST or IGRA should not be interpreted as excluding the possibility that a patient has LTBI. Accordingly, in immunosuppressed RA patients with risk factors for LTBI and negative initial screening tests, the panel recommends that a repeat TST or IGRA could be considered 1–3 weeks after the initial negative screening (see diamond A of Figure 3 in the original guideline document).
If the RA patient has active or latent TB based on the test results, the panel recommends appropriate antitubercular treatment and consideration of referral to a specialist. Treatment with biologic agents can be initiated or resumed after 1 month of latent TB treatment with antitubercular medications and after completion of the treatment of active TB, as applicable (see Figure 3 in the original guideline document).
The panel recommends annual testing in RA patients who live, travel, or work in situations where TB exposure is likely while they continue treatment with biologic agents (see diamond D of Figure 3 in the original guideline document). Patients who test positive for TST or IGRA at baseline can remain positive for these tests even after successful treatment of TB. These patients need monitoring for clinical signs and symptoms of recurrent TB, since repeating tests will not help in the diagnosis of recurrent TB.
- Vaccination in Patients Starting or Currently Receiving DMARDs or Biologic Agents as Part of Their RA Therapy (level of evidence C for all recommendations)
The panel recommends that all killed (pneumococcal, influenza intramuscular, and hepatitis B), recombinant (human papillomavirus [HPV] vaccine for cervical cancer), and live attenuated (herpes zoster) vaccinations should be undertaken before starting a DMARD or a biologic agent (see Table 5 in the original guideline document).
It also recommends that, if not previously done, vaccination with indicated pneumococcal (killed), influenza intramuscular (killed), hepatitis B (killed), and HPV vaccine (recombinant) should be undertaken in RA patients already taking a DMARD or a biologic agent (see Table 5 in the original guideline document).
The panel recommends vaccination with herpes zoster vaccine in RA patients already taking a DMARD. All vaccines should be given based on age and risk, and physicians should refer to vaccine instructions and CDC recommendations for details about dosing and timing issues related to vaccinations.
Definitions:
Level of Evidence
- Level of Evidence A: Data derived from multiple randomized clinical trials.
- Level of Evidence B: Data derived from a single randomized trial, or nonrandomized studies.
- Level of Evidence C: Only consensus opinion of experts, case studies, or standard-of-care.
Note: Level C evidence often denoted a circumstance where medical literature addressed the general topic under discussion but it did not address the specific clinical situations or scenarios reviewed by the panel.
- 2012 American College of Rheumatology (ACR) recommendations update for the treatment of early rheumatoid arthritis (RA), defined as a disease duration <6 months
- 2012 ACR recommendations update for the treatment of established RA, defined as a disease duration ≥6 months or meeting the 1987 ACR classification criteria
- 2012 ACR recommendations update for tuberculosis (TB) screening with biologic agent use
The levels of evidence supporting the recommendations (A-C) are defined at the end of the "Major Recommendations" field.
Recommendations for the Use of Disease-Modifying Antirheumatic Drugs (DMARDs) and Biologic Agents in Patients Who Qualify for Treatment of Rheumatoid Arthritis (RA)
This 2012 American College of Rheumatology (ACR) recommendations update incorporates the evidence from systematic literature review synthesis and recommendations from 2008 and rates updated and new clinical scenarios regarding the use of DMARDs and biologic agents for the treatment of RA. Terms used in the recommendations are defined in Table 2 of the original guideline document. The 2012 recommendations are listed in the 4 sections below and in the following order:
- Indications for and switching DMARDs and biologic agents: early RA (indications, see Figure 1 in the original guideline document) followed by established RA (indications and switching, see Figure 2 in the original guideline document), along with details of the level of evidence supporting these recommendations (see Supplementary Appendix 7, available in the online version at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
- Use of biologic agents in patients with hepatitis, malignancy, or congestive heart failure (CHF) who qualify for RA management (see Table 4 in the original guideline document)
- Screening for tuberculosis (TB) in patients starting or currently receiving biologic agents as part of their RA therapy (see Figure 3 in the original guideline document)
- Vaccination in patients starting or currently receiving DMARDs or biologic agents as part of their RA therapy (see Table 5 in the original guideline document)
The recommendations in the text below and in Tables 4 and 5 in the original guideline document represent the results of the 2012 update only, whereas Figures 1–3 in the original guideline document also incorporate some of the 2008 ACR RA recommendations that did not change. Areas of uncertainty by the panel (that did not lead to recommendations) are noted in Supplementary Appendix 8 (available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658 ![]()
- Indications for Starting, Resuming, Adding, or Switching DMARDs or Biologic Agents
The panel first describes a recommendation targeting remission or low disease activity in RA (section 1A). This is followed by recommendations for DMARD or biologic agent use in early RA (section 1B). Next, the panel provides recommendations for initiating and switching between DMARDs and biologic agents in established RA (section 1C).
1A. Target Low Disease Activity or Remission
The panel recommends targeting either low disease activity (see Table 3 in the original guideline document) or remission (see Table 2 in the original guideline document) in all patients with early RA (see Figure 1 in the original guideline document; level of evidence C) and established RA (see Figure 2 in the original guideline document; level of evidence C) receiving any DMARD or biologic agent.
1B. Early RA (Disease Duration <6 Months)
In patients with early RA, the panel recommends the use of DMARD monotherapy both for low disease activity and for moderate or high disease activity with the absence of poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C) (details are shown in Supplementary Appendix 7, available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
In patients with early RA, the panel recommends the use of DMARD combination therapy (including double and triple therapy) in patients with moderate or high disease activity plus poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C).
In patients with early RA, the panel also recommends the use of an anti-tumor necrosis factor (anti-TNF) biologic with or without methotrexate in patients who have high disease activity with poor prognostic features (see Figure 1 in the original guideline document; level of evidence A and B). Infliximab is the only exception and the recommendation is to use it in combination with methotrexate, but not as monotherapy.
1C. Established RA (Disease Duration ≥6 Months or Meeting the 1987 ACR RA Classification Criteria)
The remainder of panel recommendations regarding indications for DMARDs and biologic agents are for patients with established RA. The 3 subsections below define recommendations for initiating and switching therapies in established RA (see Figure 2 in the original guideline document). Where the prognosis is not mentioned, the recommendation to use/switch to a DMARD or a biologic agent applies to all patients, regardless of prognostic features.
Initiating and Switching Among DMARDs
If after 3 months of DMARD monotherapy (in patients without poor prognostic features), a patient deteriorates from low to moderate/high disease activity, then methotrexate, hydroxychloroquine, or leflunomide should be added (see rectangle A of Figure 2 in the original guideline document; level of evidence A and B).
If after 3 months of methotrexate or methotrexate/DMARD combination, a patient still has moderate or high disease activity, then add another non-methotrexate DMARD or switch to a different non-methotrexate DMARD (see rectangle B of Figure 2 in the original guideline document; level of evidence B and C).
Switching from DMARDs to Biologic Agents
If a patient has moderate or high disease activity after 3 months of methotrexate monotherapy or DMARD combination therapy, as an alternative to the DMARD recommendation just noted above, the panel recommends adding or switching to an anti-TNF biologic, abatacept, or rituximab (see rectangles C and D of Figure 2 in the original guideline document; level of evidence A–C).
If after 3 months of intensified DMARD combination therapy or after a second DMARD, a patient still has moderate or high disease activity, add or switch to an anti-TNF biologic (see rectangle C of Figure 2 in the original guideline document; level of evidence C).
Switching Among Biologic Agents Due to Lack of Benefit or Loss of Benefit
If a patient still has moderate or high disease activity after 3 months of anti-TNF biologic therapy and this is due to a lack or loss of benefit, switching to another anti-TNF biologic or a non-TNF biologic is recommended (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C).
If a patient still has moderate or high disease activity after 6 months of a non-TNF biologic and the failure is due to a lack or loss of benefit, switch to another non-TNF biologic or an anti-TNF biologic (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C). An assessment period of 6 months was chosen rather than 3 months, due to the anticipation that a longer time may be required for efficacy of a non-TNF biologic.
Switching Among Biologic Agents Due to Harms/Adverse Events
If a patient has high disease activity after failing an anti-TNF biologic because of a serious adverse event, switch to a non-TNF biologic (see rectangle E of Figure 2 in the original guideline document; level of evidence C).
If a patient has moderate or high disease activity after failing an anti-TNF biologic because of a nonserious adverse event, switch to another anti-TNF biologic or a non-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence B and C).
If a patient has moderate or high disease activity after failing a non-TNF biologic because of an adverse event (serious or nonserious), switch to another non-TNF biologic or an anti-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence C).
- Use of Biologic Agents in RA Patients With Hepatitis, Malignancy, or Chronic Heart Failure (CHF), Qualifying for More Aggressive Treatment (level of evidence C for all recommendations)
Hepatitis B or C
The panel recommends that etanercept could potentially be used in RA patients with hepatitis C requiring RA treatment (see Table 4 in the original guideline document).
The panel also recommends not using biologic agents in RA patients with untreated chronic hepatitis B (disease not treated due to contraindications to treatment or intolerable adverse events) and in RA patients with treated chronic hepatitis B with Child-Pugh class B and higher (see Table 4 in the original guideline document; for details of Child-Pugh classification, see Table 2 in the original guideline document). The panel did not make recommendations regarding the use of any biologic agent for treatment in RA patients with a history of hepatitis B and a positive hepatitis B core antibody.
Malignancies
For patients who have been treated for solid malignancies more than 5 years ago or who have been treated for nonmelanoma skin cancer more than 5 years ago, the panel recommends starting or resuming any biologic agent if those patients would otherwise qualify for this RA management strategy (see Table 4 in the original guideline document).
The panel only recommends starting or resuming rituximab in RA patients with: 1) a previously treated solid malignancy within the last 5 years, 2) a previously treated nonmelanoma skin cancer within the last 5 years, 3) a previously treated melanoma skin cancer, or 4) a previously treated lymphoproliferative malignancy. Little is known about the effects of biologic therapy in patients with a history of a solid cancer within the past 5 years owing to the exclusion of such patients from participation in clinical trials and the lack of studies examining the risk of recurrent cancer in this subgroup of patients.
CHF
The panel recommends not using an anti-TNF biologic in RA patients with CHF that is New York Heart Association (NYHA) class III or IV and who have an ejection fraction of 50% or less (see Table 4 in the original guideline document).
- TB Screening for Biologic Agents (level of evidence C for all recommendations except for initiation of biologic agents in patients being treated for latent TB infection [LTBI], where the level of evidence is B)
The panel recommends screening to identify LTBI in all RA patients being considered for therapy with biologic agents, regardless of the presence of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends that clinicians assess the patient's medical history to identify risk factors for TB (specified by the Centers for Disease Control and Prevention [CDC]) (see Table 2 in the original guideline document).
The panel recommends the tuberculin skin test (TST) or interferon-gamma–release assays (IGRAs) as the initial test in all RA patients starting biologic agents, regardless of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends the use of the IGRA over the TST in patients who had previously received a bacillus Calmette-Guerin (BCG) vaccination, due to the high false-positive test rates for TST (see Figure 3 in the original guideline document).
The panel recommends that RA patients with a positive initial or repeat TST or IGRA should have a chest radiograph and, if suggestive of active TB, a subsequent sputum examination to check for the presence of active TB (see diamonds B and C of Figure 3 in the original guideline document). RA patients with a negative screening TST or IGRA may not need further evaluation in the absence of risk factors and/or clinical suspicion for TB. Since patients with RA may have false-negative TST or IGRA results due to immunosuppression, a negative TST or IGRA should not be interpreted as excluding the possibility that a patient has LTBI. Accordingly, in immunosuppressed RA patients with risk factors for LTBI and negative initial screening tests, the panel recommends that a repeat TST or IGRA could be considered 1–3 weeks after the initial negative screening (see diamond A of Figure 3 in the original guideline document).
If the RA patient has active or latent TB based on the test results, the panel recommends appropriate antitubercular treatment and consideration of referral to a specialist. Treatment with biologic agents can be initiated or resumed after 1 month of latent TB treatment with antitubercular medications and after completion of the treatment of active TB, as applicable (see Figure 3 in the original guideline document).
The panel recommends annual testing in RA patients who live, travel, or work in situations where TB exposure is likely while they continue treatment with biologic agents (see diamond D of Figure 3 in the original guideline document). Patients who test positive for TST or IGRA at baseline can remain positive for these tests even after successful treatment of TB. These patients need monitoring for clinical signs and symptoms of recurrent TB, since repeating tests will not help in the diagnosis of recurrent TB.
- Vaccination in Patients Starting or Currently Receiving DMARDs or Biologic Agents as Part of Their RA Therapy (level of evidence C for all recommendations)
The panel recommends that all killed (pneumococcal, influenza intramuscular, and hepatitis B), recombinant (human papillomavirus [HPV] vaccine for cervical cancer), and live attenuated (herpes zoster) vaccinations should be undertaken before starting a DMARD or a biologic agent (see Table 5 in the original guideline document).
It also recommends that, if not previously done, vaccination with indicated pneumococcal (killed), influenza intramuscular (killed), hepatitis B (killed), and HPV vaccine (recombinant) should be undertaken in RA patients already taking a DMARD or a biologic agent (see Table 5 in the original guideline document).
The panel recommends vaccination with herpes zoster vaccine in RA patients already taking a DMARD. All vaccines should be given based on age and risk, and physicians should refer to vaccine instructions and CDC recommendations for details about dosing and timing issues related to vaccinations.
Definitions:
Level of Evidence
- Level of Evidence A: Data derived from multiple randomized clinical trials.
- Level of Evidence B: Data derived from a single randomized trial, or nonrandomized studies.
- Level of Evidence C: Only consensus opinion of experts, case studies, or standard-of-care.
Note: Level C evidence often denoted a circumstance where medical literature addressed the general topic under discussion but it did not address the specific clinical situations or scenarios reviewed by the panel.
- 2012 American College of Rheumatology (ACR) recommendations update for the treatment of early rheumatoid arthritis (RA), defined as a disease duration <6 months
- 2012 ACR recommendations update for the treatment of established RA, defined as a disease duration ≥6 months or meeting the 1987 ACR classification criteria
- 2012 ACR recommendations update for tuberculosis (TB) screening with biologic agent use
The levels of evidence supporting the recommendations (A-C) are defined at the end of the "Major Recommendations" field.
Recommendations for the Use of Disease-Modifying Antirheumatic Drugs (DMARDs) and Biologic Agents in Patients Who Qualify for Treatment of Rheumatoid Arthritis (RA)
This 2012 American College of Rheumatology (ACR) recommendations update incorporates the evidence from systematic literature review synthesis and recommendations from 2008 and rates updated and new clinical scenarios regarding the use of DMARDs and biologic agents for the treatment of RA. Terms used in the recommendations are defined in Table 2 of the original guideline document. The 2012 recommendations are listed in the 4 sections below and in the following order:
- Indications for and switching DMARDs and biologic agents: early RA (indications, see Figure 1 in the original guideline document) followed by established RA (indications and switching, see Figure 2 in the original guideline document), along with details of the level of evidence supporting these recommendations (see Supplementary Appendix 7, available in the online version at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
- Use of biologic agents in patients with hepatitis, malignancy, or congestive heart failure (CHF) who qualify for RA management (see Table 4 in the original guideline document)
- Screening for tuberculosis (TB) in patients starting or currently receiving biologic agents as part of their RA therapy (see Figure 3 in the original guideline document)
- Vaccination in patients starting or currently receiving DMARDs or biologic agents as part of their RA therapy (see Table 5 in the original guideline document)
The recommendations in the text below and in Tables 4 and 5 in the original guideline document represent the results of the 2012 update only, whereas Figures 1–3 in the original guideline document also incorporate some of the 2008 ACR RA recommendations that did not change. Areas of uncertainty by the panel (that did not lead to recommendations) are noted in Supplementary Appendix 8 (available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658 ![]()
- Indications for Starting, Resuming, Adding, or Switching DMARDs or Biologic Agents
The panel first describes a recommendation targeting remission or low disease activity in RA (section 1A). This is followed by recommendations for DMARD or biologic agent use in early RA (section 1B). Next, the panel provides recommendations for initiating and switching between DMARDs and biologic agents in established RA (section 1C).
1A. Target Low Disease Activity or Remission
The panel recommends targeting either low disease activity (see Table 3 in the original guideline document) or remission (see Table 2 in the original guideline document) in all patients with early RA (see Figure 1 in the original guideline document; level of evidence C) and established RA (see Figure 2 in the original guideline document; level of evidence C) receiving any DMARD or biologic agent.
1B. Early RA (Disease Duration <6 Months)
In patients with early RA, the panel recommends the use of DMARD monotherapy both for low disease activity and for moderate or high disease activity with the absence of poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C) (details are shown in Supplementary Appendix 7, available in the online version of this article at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2151-4658
In patients with early RA, the panel recommends the use of DMARD combination therapy (including double and triple therapy) in patients with moderate or high disease activity plus poor prognostic features (see Figure 1 in the original guideline document; level of evidence A–C).
In patients with early RA, the panel also recommends the use of an anti-tumor necrosis factor (anti-TNF) biologic with or without methotrexate in patients who have high disease activity with poor prognostic features (see Figure 1 in the original guideline document; level of evidence A and B). Infliximab is the only exception and the recommendation is to use it in combination with methotrexate, but not as monotherapy.
1C. Established RA (Disease Duration ≥6 Months or Meeting the 1987 ACR RA Classification Criteria)
The remainder of panel recommendations regarding indications for DMARDs and biologic agents are for patients with established RA. The 3 subsections below define recommendations for initiating and switching therapies in established RA (see Figure 2 in the original guideline document). Where the prognosis is not mentioned, the recommendation to use/switch to a DMARD or a biologic agent applies to all patients, regardless of prognostic features.
Initiating and Switching Among DMARDs
If after 3 months of DMARD monotherapy (in patients without poor prognostic features), a patient deteriorates from low to moderate/high disease activity, then methotrexate, hydroxychloroquine, or leflunomide should be added (see rectangle A of Figure 2 in the original guideline document; level of evidence A and B).
If after 3 months of methotrexate or methotrexate/DMARD combination, a patient still has moderate or high disease activity, then add another non-methotrexate DMARD or switch to a different non-methotrexate DMARD (see rectangle B of Figure 2 in the original guideline document; level of evidence B and C).
Switching from DMARDs to Biologic Agents
If a patient has moderate or high disease activity after 3 months of methotrexate monotherapy or DMARD combination therapy, as an alternative to the DMARD recommendation just noted above, the panel recommends adding or switching to an anti-TNF biologic, abatacept, or rituximab (see rectangles C and D of Figure 2 in the original guideline document; level of evidence A–C).
If after 3 months of intensified DMARD combination therapy or after a second DMARD, a patient still has moderate or high disease activity, add or switch to an anti-TNF biologic (see rectangle C of Figure 2 in the original guideline document; level of evidence C).
Switching Among Biologic Agents Due to Lack of Benefit or Loss of Benefit
If a patient still has moderate or high disease activity after 3 months of anti-TNF biologic therapy and this is due to a lack or loss of benefit, switching to another anti-TNF biologic or a non-TNF biologic is recommended (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C).
If a patient still has moderate or high disease activity after 6 months of a non-TNF biologic and the failure is due to a lack or loss of benefit, switch to another non-TNF biologic or an anti-TNF biologic (see rectangles F and G of Figure 2 in the original guideline document; level of evidence B and C). An assessment period of 6 months was chosen rather than 3 months, due to the anticipation that a longer time may be required for efficacy of a non-TNF biologic.
Switching Among Biologic Agents Due to Harms/Adverse Events
If a patient has high disease activity after failing an anti-TNF biologic because of a serious adverse event, switch to a non-TNF biologic (see rectangle E of Figure 2 in the original guideline document; level of evidence C).
If a patient has moderate or high disease activity after failing an anti-TNF biologic because of a nonserious adverse event, switch to another anti-TNF biologic or a non-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence B and C).
If a patient has moderate or high disease activity after failing a non-TNF biologic because of an adverse event (serious or nonserious), switch to another non-TNF biologic or an anti-TNF biologic (see rectangle F of Figure 2 in the original guideline document; level of evidence C).
- Use of Biologic Agents in RA Patients With Hepatitis, Malignancy, or Chronic Heart Failure (CHF), Qualifying for More Aggressive Treatment (level of evidence C for all recommendations)
Hepatitis B or C
The panel recommends that etanercept could potentially be used in RA patients with hepatitis C requiring RA treatment (see Table 4 in the original guideline document).
The panel also recommends not using biologic agents in RA patients with untreated chronic hepatitis B (disease not treated due to contraindications to treatment or intolerable adverse events) and in RA patients with treated chronic hepatitis B with Child-Pugh class B and higher (see Table 4 in the original guideline document; for details of Child-Pugh classification, see Table 2 in the original guideline document). The panel did not make recommendations regarding the use of any biologic agent for treatment in RA patients with a history of hepatitis B and a positive hepatitis B core antibody.
Malignancies
For patients who have been treated for solid malignancies more than 5 years ago or who have been treated for nonmelanoma skin cancer more than 5 years ago, the panel recommends starting or resuming any biologic agent if those patients would otherwise qualify for this RA management strategy (see Table 4 in the original guideline document).
The panel only recommends starting or resuming rituximab in RA patients with: 1) a previously treated solid malignancy within the last 5 years, 2) a previously treated nonmelanoma skin cancer within the last 5 years, 3) a previously treated melanoma skin cancer, or 4) a previously treated lymphoproliferative malignancy. Little is known about the effects of biologic therapy in patients with a history of a solid cancer within the past 5 years owing to the exclusion of such patients from participation in clinical trials and the lack of studies examining the risk of recurrent cancer in this subgroup of patients.
CHF
The panel recommends not using an anti-TNF biologic in RA patients with CHF that is New York Heart Association (NYHA) class III or IV and who have an ejection fraction of 50% or less (see Table 4 in the original guideline document).
- TB Screening for Biologic Agents (level of evidence C for all recommendations except for initiation of biologic agents in patients being treated for latent TB infection [LTBI], where the level of evidence is B)
The panel recommends screening to identify LTBI in all RA patients being considered for therapy with biologic agents, regardless of the presence of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends that clinicians assess the patient's medical history to identify risk factors for TB (specified by the Centers for Disease Control and Prevention [CDC]) (see Table 2 in the original guideline document).
The panel recommends the tuberculin skin test (TST) or interferon-gamma–release assays (IGRAs) as the initial test in all RA patients starting biologic agents, regardless of risk factors for LTBI (see diamond A of Figure 3 in the original guideline document). It recommends the use of the IGRA over the TST in patients who had previously received a bacillus Calmette-Guerin (BCG) vaccination, due to the high false-positive test rates for TST (see Figure 3 in the original guideline document).
The panel recommends that RA patients with a positive initial or repeat TST or IGRA should have a chest radiograph and, if suggestive of active TB, a subsequent sputum examination to check for the presence of active TB (see diamonds B and C of Figure 3 in the original guideline document). RA patients with a negative screening TST or IGRA may not need further evaluation in the absence of risk factors and/or clinical suspicion for TB. Since patients with RA may have false-negative TST or IGRA results due to immunosuppression, a negative TST or IGRA should not be interpreted as excluding the possibility that a patient has LTBI. Accordingly, in immunosuppressed RA patients with risk factors for LTBI and negative initial screening tests, the panel recommends that a repeat TST or IGRA could be considered 1–3 weeks after the initial negative screening (see diamond A of Figure 3 in the original guideline document).
If the RA patient has active or latent TB based on the test results, the panel recommends appropriate antitubercular treatment and consideration of referral to a specialist. Treatment with biologic agents can be initiated or resumed after 1 month of latent TB treatment with antitubercular medications and after completion of the treatment of active TB, as applicable (see Figure 3 in the original guideline document).
The panel recommends annual testing in RA patients who live, travel, or work in situations where TB exposure is likely while they continue treatment with biologic agents (see diamond D of Figure 3 in the original guideline document). Patients who test positive for TST or IGRA at baseline can remain positive for these tests even after successful treatment of TB. These patients need monitoring for clinical signs and symptoms of recurrent TB, since repeating tests will not help in the diagnosis of recurrent TB.
- Vaccination in Patients Starting or Currently Receiving DMARDs or Biologic Agents as Part of Their RA Therapy (level of evidence C for all recommendations)
The panel recommends that all killed (pneumococcal, influenza intramuscular, and hepatitis B), recombinant (human papillomavirus [HPV] vaccine for cervical cancer), and live attenuated (herpes zoster) vaccinations should be undertaken before starting a DMARD or a biologic agent (see Table 5 in the original guideline document).
It also recommends that, if not previously done, vaccination with indicated pneumococcal (killed), influenza intramuscular (killed), hepatitis B (killed), and HPV vaccine (recombinant) should be undertaken in RA patients already taking a DMARD or a biologic agent (see Table 5 in the original guideline document).
The panel recommends vaccination with herpes zoster vaccine in RA patients already taking a DMARD. All vaccines should be given based on age and risk, and physicians should refer to vaccine instructions and CDC recommendations for details about dosing and timing issues related to vaccinations.
Definitions:
Level of Evidence
- Level of Evidence A: Data derived from multiple randomized clinical trials.
- Level of Evidence B: Data derived from a single randomized trial, or nonrandomized studies.
- Level of Evidence C: Only consensus opinion of experts, case studies, or standard-of-care.
Note: Level C evidence often denoted a circumstance where medical literature addressed the general topic under discussion but it did not address the specific clinical situations or scenarios reviewed by the panel.
- 2012 American College of Rheumatology (ACR) recommendations update for the treatment of early rheumatoid arthritis (RA), defined as a disease duration <6 months
- 2012 ACR recommendations update for the treatment of established RA, defined as a disease duration ≥6 months or meeting the 1987 ACR classification criteria
- 2012 ACR recommendations update for tuberculosis (TB) screening with biologic agent use
OBJECTIVE:
-To simplify the treatment algorithms for patients with rheumatoid arthritis (RA) and providers
-To provide clinicians with choices for treatments of patients with active RA, both in early and established disease phases
-To provide guidance regarding treatment choices in RA patients with comorbidities such as hepatitis, congestive heart failure (CHF), and malignancy
Guidelines are copyright © 2012 American College of Rheumatology. All rights reserved. The summary is provided by the Agency for Healthcare Research and Quality.
Provider Resilience

Part of self-care is avoiding compassion fatigue, which means avoiding negative feelings toward the ability to complete a job effectively or handle work-related stress. Compassion fatigue has 2 components, according to T2: burnout and secondary traumatic stress. To avoid or combat compassion fatigue, the app gives frontline providers tools to keep themselves productive and emotionally healthy as they help our nation’s service members, veterans, and their families.
DASHBOARD
The app’s home screen is a dashboard, which tracks wellbeing across 4 parameters, all based on self-assessments: vacation time, burnout scale, builders and killers checklist, and professional quality-of-life (ProQOL) measures. Each of these 4 assessments calculates the user’s resilience rating, displayed by a colored meter measuring from 0 (low) to 100 (high).
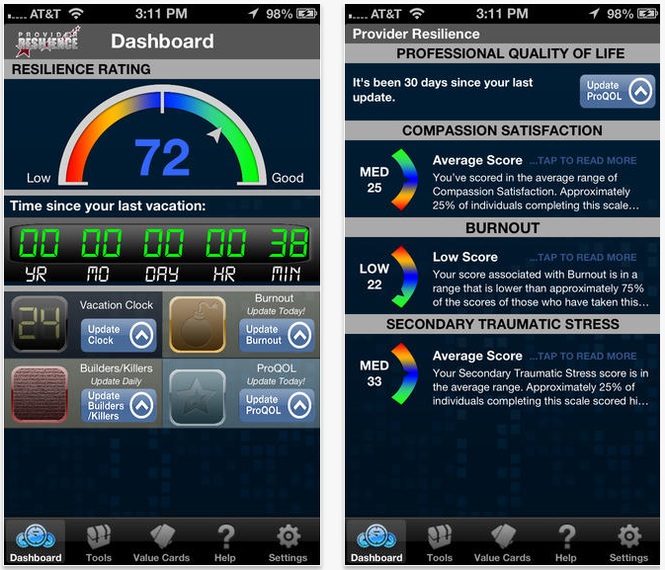
Beneath the resilience rating meter is a prominent vacation clock, running time down to the minute since the user’s last recorded vacation day. To measure burnout, the app offers a survey for 10 measures that should be updated by the user at least once a week. The question asked of users is, “How would you describe yourself as you approach your workday today?” All measures, including happy, trapped, satisfied, and preoccupied, can be rated from “Not at All” to “Very Much So.”
The professional quality-of-life (ProQOL) quiz is the longest of all—30 questions—but the resilience rating relies on an update only every 30 days. This quiz measures 3 critical components of ProQOL: compassion satisfaction, burnout, and secondary traumatic stress. Average scores provide the following output for each section:
1. Compassion Satisfaction: “You’ve scored in the average range of Compassion Satisfaction. … This suggests that you don’t find your work to be consistently satisfying, but that you do derive some degree of satisfaction from your daily work activities."
2. Burnout: “Your Burnout score is in the average range. … This may indicate that you are currently experiencing frustration in your work and may be feeling discouraged or ineffective. It may be worth reviewing your answers to see if you can pin down the source of these feelings and determine how you might improve your experience at work.”
3. Secondary Traumatic Stress: “If you are working with clients or patients who are describing highly traumatic experiences, be sure to focus on self-care, which includes maintaining physical, emotional, social, and spiritual supports.”
The dashboard also tracks how many days until the user needs to update each section, and daily reminders (push notifications) to do so may be modified in Settings.
TOOLS
There are 7 videos currently loaded into the app. Two informational videos (both running under 10 minutes) cover burnout and secondary traumatic stress so that users can gain a richer understanding either of what they are experiencing or what they are trying to avoid. The remaining 5 videos are shorter and can be found in the “Remind Me Why I Do This” menu. These videos cover important patient characteristics that have been found to lead to compassion fatigue, including alcohol, anger, depression, depression support, and stigma.
Scrolling flashcards can be found in “Physical Exercise” and include an assortment of stretching exercises the user can perform at his or her work desk. And if stretching doesn’t release enough endorphins, the user can raise his or her spirit browsing the cartoons found in “I Need A Laugh.” All of the cartoons are office-related, updated daily, and archived for the user’s pleasure.

Nearly 100 inspirational flashcards are loaded onto the app, each one defining a valuable character trait, associated quote, and affirmations. To scroll through the cards, which are listed alphabetically from “Acceptance” through “Unity,” simply swipe left or right. To flip the card over, swipe up or down on the screen.
FINAL THOUGHTS
Certain luxuries we have grown accustomed to, such as rotating a device to change display orientation, don’t perform on this app, a feature missed greatly when viewing videos and a good reason to use the app on a tablet, not a phone.
And although everyone needs the occasional short break or multiday vacation, tolerance levels differ from person to person. Hence, it is important to use the app over a period of time so that the user can learn what his or her “normal” is and track readings not necessarily against the general population, but against him- or herself on the plotted ProQOL and burnout graphs.
Part of self-care is avoiding compassion fatigue, which means avoiding negative feelings toward the ability to complete a job effectively or handle work-related stress. Compassion fatigue has 2 components, according to T2: burnout and secondary traumatic stress. To avoid or combat compassion fatigue, the app gives frontline providers tools to keep themselves productive and emotionally healthy as they help our nation’s service members, veterans, and their families.
DASHBOARD
The app’s home screen is a dashboard, which tracks wellbeing across 4 parameters, all based on self-assessments: vacation time, burnout scale, builders and killers checklist, and professional quality-of-life (ProQOL) measures. Each of these 4 assessments calculates the user’s resilience rating, displayed by a colored meter measuring from 0 (low) to 100 (high).
Beneath the resilience rating meter is a prominent vacation clock, running time down to the minute since the user’s last recorded vacation day. To measure burnout, the app offers a survey for 10 measures that should be updated by the user at least once a week. The question asked of users is, “How would you describe yourself as you approach your workday today?” All measures, including happy, trapped, satisfied, and preoccupied, can be rated from “Not at All” to “Very Much So.”
The professional quality-of-life (ProQOL) quiz is the longest of all—30 questions—but the resilience rating relies on an update only every 30 days. This quiz measures 3 critical components of ProQOL: compassion satisfaction, burnout, and secondary traumatic stress. Average scores provide the following output for each section:
1. Compassion Satisfaction: “You’ve scored in the average range of Compassion Satisfaction. … This suggests that you don’t find your work to be consistently satisfying, but that you do derive some degree of satisfaction from your daily work activities."
2. Burnout: “Your Burnout score is in the average range. … This may indicate that you are currently experiencing frustration in your work and may be feeling discouraged or ineffective. It may be worth reviewing your answers to see if you can pin down the source of these feelings and determine how you might improve your experience at work.”
3. Secondary Traumatic Stress: “If you are working with clients or patients who are describing highly traumatic experiences, be sure to focus on self-care, which includes maintaining physical, emotional, social, and spiritual supports.”
The dashboard also tracks how many days until the user needs to update each section, and daily reminders (push notifications) to do so may be modified in Settings.
TOOLS
There are 7 videos currently loaded into the app. Two informational videos (both running under 10 minutes) cover burnout and secondary traumatic stress so that users can gain a richer understanding either of what they are experiencing or what they are trying to avoid. The remaining 5 videos are shorter and can be found in the “Remind Me Why I Do This” menu. These videos cover important patient characteristics that have been found to lead to compassion fatigue, including alcohol, anger, depression, depression support, and stigma.
Scrolling flashcards can be found in “Physical Exercise” and include an assortment of stretching exercises the user can perform at his or her work desk. And if stretching doesn’t release enough endorphins, the user can raise his or her spirit browsing the cartoons found in “I Need A Laugh.” All of the cartoons are office-related, updated daily, and archived for the user’s pleasure.
Nearly 100 inspirational flashcards are loaded onto the app, each one defining a valuable character trait, associated quote, and affirmations. To scroll through the cards, which are listed alphabetically from “Acceptance” through “Unity,” simply swipe left or right. To flip the card over, swipe up or down on the screen.
FINAL THOUGHTS
Certain luxuries we have grown accustomed to, such as rotating a device to change display orientation, don’t perform on this app, a feature missed greatly when viewing videos and a good reason to use the app on a tablet, not a phone.
And although everyone needs the occasional short break or multiday vacation, tolerance levels differ from person to person. Hence, it is important to use the app over a period of time so that the user can learn what his or her “normal” is and track readings not necessarily against the general population, but against him- or herself on the plotted ProQOL and burnout graphs.
Part of self-care is avoiding compassion fatigue, which means avoiding negative feelings toward the ability to complete a job effectively or handle work-related stress. Compassion fatigue has 2 components, according to T2: burnout and secondary traumatic stress. To avoid or combat compassion fatigue, the app gives frontline providers tools to keep themselves productive and emotionally healthy as they help our nation’s service members, veterans, and their families.
DASHBOARD
The app’s home screen is a dashboard, which tracks wellbeing across 4 parameters, all based on self-assessments: vacation time, burnout scale, builders and killers checklist, and professional quality-of-life (ProQOL) measures. Each of these 4 assessments calculates the user’s resilience rating, displayed by a colored meter measuring from 0 (low) to 100 (high).
Beneath the resilience rating meter is a prominent vacation clock, running time down to the minute since the user’s last recorded vacation day. To measure burnout, the app offers a survey for 10 measures that should be updated by the user at least once a week. The question asked of users is, “How would you describe yourself as you approach your workday today?” All measures, including happy, trapped, satisfied, and preoccupied, can be rated from “Not at All” to “Very Much So.”
The professional quality-of-life (ProQOL) quiz is the longest of all—30 questions—but the resilience rating relies on an update only every 30 days. This quiz measures 3 critical components of ProQOL: compassion satisfaction, burnout, and secondary traumatic stress. Average scores provide the following output for each section:
1. Compassion Satisfaction: “You’ve scored in the average range of Compassion Satisfaction. … This suggests that you don’t find your work to be consistently satisfying, but that you do derive some degree of satisfaction from your daily work activities."
2. Burnout: “Your Burnout score is in the average range. … This may indicate that you are currently experiencing frustration in your work and may be feeling discouraged or ineffective. It may be worth reviewing your answers to see if you can pin down the source of these feelings and determine how you might improve your experience at work.”
3. Secondary Traumatic Stress: “If you are working with clients or patients who are describing highly traumatic experiences, be sure to focus on self-care, which includes maintaining physical, emotional, social, and spiritual supports.”
The dashboard also tracks how many days until the user needs to update each section, and daily reminders (push notifications) to do so may be modified in Settings.
TOOLS
There are 7 videos currently loaded into the app. Two informational videos (both running under 10 minutes) cover burnout and secondary traumatic stress so that users can gain a richer understanding either of what they are experiencing or what they are trying to avoid. The remaining 5 videos are shorter and can be found in the “Remind Me Why I Do This” menu. These videos cover important patient characteristics that have been found to lead to compassion fatigue, including alcohol, anger, depression, depression support, and stigma.
Scrolling flashcards can be found in “Physical Exercise” and include an assortment of stretching exercises the user can perform at his or her work desk. And if stretching doesn’t release enough endorphins, the user can raise his or her spirit browsing the cartoons found in “I Need A Laugh.” All of the cartoons are office-related, updated daily, and archived for the user’s pleasure.
Nearly 100 inspirational flashcards are loaded onto the app, each one defining a valuable character trait, associated quote, and affirmations. To scroll through the cards, which are listed alphabetically from “Acceptance” through “Unity,” simply swipe left or right. To flip the card over, swipe up or down on the screen.
FINAL THOUGHTS
Certain luxuries we have grown accustomed to, such as rotating a device to change display orientation, don’t perform on this app, a feature missed greatly when viewing videos and a good reason to use the app on a tablet, not a phone.
And although everyone needs the occasional short break or multiday vacation, tolerance levels differ from person to person. Hence, it is important to use the app over a period of time so that the user can learn what his or her “normal” is and track readings not necessarily against the general population, but against him- or herself on the plotted ProQOL and burnout graphs.
Risk factors for premature menopause in HL survivors
Credit: NCI
The risk of premature menopause among survivors of Hodgkin lymphoma (HL) may vary greatly, according to a study of more than 2000 women.
The results suggest that ovarian radiotherapy and certain chemotherapeutic regimens increase a woman’s risk of premature menopause.
And the radiation dose, number of treatment cycles, and patient age at treatment all influence that risk.
The findings appear in the Journal of the National Cancer Institute.
Previous research suggested that women with HL who receive certain types of chemotherapy or radiotherapy are at an increased risk of early menopause, but there was insufficient information to provide patients with detailed advice.
To gain more insight, Anthony Swerdlow, DSc, of The Institute of Cancer Research in London, UK, and his colleagues studied 2127 women who were treated for HL in England or Wales between 1960 and 2004.
All of the women were younger than 36 at the time of treatment, and all had received chest radiotherapy, sometimes alongside other treatments.
Some 605 of the women underwent non-surgical menopause before the age of 40. The researchers said this was a large enough number for them to estimate accurate risks of menopause, depending on the type and dose of treatments patients received and the age they received them.
The team found that several treatments caused a sharp increase in premature menopause risk, and menopause was more like among women treated at older ages.
The risk of premature menopause increased more than 20-fold after ovarian radiotherapy, alkylating chemotherapy other than dacarbazine, or BEAM (bis-chloroethylnitrosourea, etoposide, cytarabine, melphalan) chemotherapy given prior to stem cell transplant.
However, there was no significant increase in risk after receiving adriamycin, bleomycin, vinblastine, dacarbazine (ABVD).
Within 5 years of treatment, menopause had occurred in 62.5% of patients who received ≥5 Gy of ovarian radiotherapy, 50.9% of patients who received BEAM, and 24.2% of patients who received ≥6 cycles of alkylating chemotherapy.
The cumulative risk of menopause by age 40 was 81.3% after ≥ 5 Gy of ovarian radiotherapy, 75.3% after BEAM, 49.1% after ≥ 6 cycles of alkylating chemotherapy, 3.0% after solely supradiaphragmatic radiotherapy, and 1.4% after ABVD.
“We hope our study will help women to understand better, in consultation with their doctors, their risks of future infertility following treatment for this malignancy,” Dr Swerdlow said.
“By looking in a much larger group of women than previous studies of this type, we were able to produce age- and treatment-specific risk estimates that we hope will be of practical use to individual women.” ![]()
Credit: NCI
The risk of premature menopause among survivors of Hodgkin lymphoma (HL) may vary greatly, according to a study of more than 2000 women.
The results suggest that ovarian radiotherapy and certain chemotherapeutic regimens increase a woman’s risk of premature menopause.
And the radiation dose, number of treatment cycles, and patient age at treatment all influence that risk.
The findings appear in the Journal of the National Cancer Institute.
Previous research suggested that women with HL who receive certain types of chemotherapy or radiotherapy are at an increased risk of early menopause, but there was insufficient information to provide patients with detailed advice.
To gain more insight, Anthony Swerdlow, DSc, of The Institute of Cancer Research in London, UK, and his colleagues studied 2127 women who were treated for HL in England or Wales between 1960 and 2004.
All of the women were younger than 36 at the time of treatment, and all had received chest radiotherapy, sometimes alongside other treatments.
Some 605 of the women underwent non-surgical menopause before the age of 40. The researchers said this was a large enough number for them to estimate accurate risks of menopause, depending on the type and dose of treatments patients received and the age they received them.
The team found that several treatments caused a sharp increase in premature menopause risk, and menopause was more like among women treated at older ages.
The risk of premature menopause increased more than 20-fold after ovarian radiotherapy, alkylating chemotherapy other than dacarbazine, or BEAM (bis-chloroethylnitrosourea, etoposide, cytarabine, melphalan) chemotherapy given prior to stem cell transplant.
However, there was no significant increase in risk after receiving adriamycin, bleomycin, vinblastine, dacarbazine (ABVD).
Within 5 years of treatment, menopause had occurred in 62.5% of patients who received ≥5 Gy of ovarian radiotherapy, 50.9% of patients who received BEAM, and 24.2% of patients who received ≥6 cycles of alkylating chemotherapy.
The cumulative risk of menopause by age 40 was 81.3% after ≥ 5 Gy of ovarian radiotherapy, 75.3% after BEAM, 49.1% after ≥ 6 cycles of alkylating chemotherapy, 3.0% after solely supradiaphragmatic radiotherapy, and 1.4% after ABVD.
“We hope our study will help women to understand better, in consultation with their doctors, their risks of future infertility following treatment for this malignancy,” Dr Swerdlow said.
“By looking in a much larger group of women than previous studies of this type, we were able to produce age- and treatment-specific risk estimates that we hope will be of practical use to individual women.” ![]()
Credit: NCI
The risk of premature menopause among survivors of Hodgkin lymphoma (HL) may vary greatly, according to a study of more than 2000 women.
The results suggest that ovarian radiotherapy and certain chemotherapeutic regimens increase a woman’s risk of premature menopause.
And the radiation dose, number of treatment cycles, and patient age at treatment all influence that risk.
The findings appear in the Journal of the National Cancer Institute.
Previous research suggested that women with HL who receive certain types of chemotherapy or radiotherapy are at an increased risk of early menopause, but there was insufficient information to provide patients with detailed advice.
To gain more insight, Anthony Swerdlow, DSc, of The Institute of Cancer Research in London, UK, and his colleagues studied 2127 women who were treated for HL in England or Wales between 1960 and 2004.
All of the women were younger than 36 at the time of treatment, and all had received chest radiotherapy, sometimes alongside other treatments.
Some 605 of the women underwent non-surgical menopause before the age of 40. The researchers said this was a large enough number for them to estimate accurate risks of menopause, depending on the type and dose of treatments patients received and the age they received them.
The team found that several treatments caused a sharp increase in premature menopause risk, and menopause was more like among women treated at older ages.
The risk of premature menopause increased more than 20-fold after ovarian radiotherapy, alkylating chemotherapy other than dacarbazine, or BEAM (bis-chloroethylnitrosourea, etoposide, cytarabine, melphalan) chemotherapy given prior to stem cell transplant.
However, there was no significant increase in risk after receiving adriamycin, bleomycin, vinblastine, dacarbazine (ABVD).
Within 5 years of treatment, menopause had occurred in 62.5% of patients who received ≥5 Gy of ovarian radiotherapy, 50.9% of patients who received BEAM, and 24.2% of patients who received ≥6 cycles of alkylating chemotherapy.
The cumulative risk of menopause by age 40 was 81.3% after ≥ 5 Gy of ovarian radiotherapy, 75.3% after BEAM, 49.1% after ≥ 6 cycles of alkylating chemotherapy, 3.0% after solely supradiaphragmatic radiotherapy, and 1.4% after ABVD.
“We hope our study will help women to understand better, in consultation with their doctors, their risks of future infertility following treatment for this malignancy,” Dr Swerdlow said.
“By looking in a much larger group of women than previous studies of this type, we were able to produce age- and treatment-specific risk estimates that we hope will be of practical use to individual women.” ![]()
A new system for malaria diagnosis
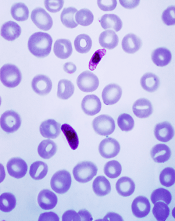
Plasmodium falciparum
Credit: CDC/Mae Melvin
A semi-automated system may allow healthcare professionals to diagnose malaria infection with more than 90% accuracy.
With this system, a computer algorithm analyzes red blood cells from a digitized slide, ranks them according to the likelihood of infection, and presents the user with more than 100 images from which to make a diagnosis.
Johan Lundin, MD, PhD, of the Institute for Molecular Medicine Finland, and his colleagues described the system in PLOS ONE.
The researchers noted that high-quality microscopy is still the most accurate method for detecting malaria infection. However, as microscopy can be time-consuming, the team wanted to streamline the process by developing a semi-automated system.
“We are not suggesting that the whole malaria diagnostic process could or should be automated,” Dr Lundin said. “Rather, our aim is to develop methods that are significantly less labor-intensive than the traditional ones and have a potential to considerably increase the throughput in malaria diagnostics.”
The group’s method is based on computer vision algorithms similar to those used in facial recognition software. First, a thin layer of blood smeared on a microscope slide is digitized. Then, a computer algorithm analyzes more than 50,000 red blood cells per sample and ranks them according to the probability of malaria infection.
Next, the program creates a panel containing images of more than a hundred cells that are the most likely to be infected and presents that panel to the user. The final diagnosis is made by a healthcare professional based on the images.
To test this system, Dr Lundin and his colleagues used a set of samples from 19 patients already diagnosed with malaria and 12 control subjects. From each sample, the researchers created a digitized slide, and the system generated 128 images of the most probable parasite candidate regions.
Two expert microscopists viewed the images on a tablet computer to determine whether a subject was infected with Plasmodium falciparum.
The diagnostic sensitivity was 90% with one viewer and 95% for the other. The specificity was 100% for both viewers.
Based on these results, the researchers said this system has the potential to increase the throughput in malaria diagnostics. However, it does require some tweaking, and the team would like to expand its capabilities.
“The equipment needed for digitization of the samples is a challenge in developed countries,” said study author Nina Linder, MD, PhD, also of the Institute for Molecular Medicine Finland. “In the next phase of our project, we will test the system in combination with inexpensive mobile microscopy devices that our group has also developed.”
“There is also a strong need for fast and accurate methods for measuring the malaria parasite load in a sample,” she added. “Various malaria drug screening programs are underway, and the parasite load in a large number of samples needs to be quantified for determining the efficacy of potential drugs. We are further developing the computer algorithms used in this study to meet this need as well.”
Lastly, the researchers said this system could be applied in various other fields of medicine. In addition to other infectious diseases such as tuberculosis, the group is planning to test the system’s utility in cancer diagnosis. ![]()
Plasmodium falciparum
Credit: CDC/Mae Melvin
A semi-automated system may allow healthcare professionals to diagnose malaria infection with more than 90% accuracy.
With this system, a computer algorithm analyzes red blood cells from a digitized slide, ranks them according to the likelihood of infection, and presents the user with more than 100 images from which to make a diagnosis.
Johan Lundin, MD, PhD, of the Institute for Molecular Medicine Finland, and his colleagues described the system in PLOS ONE.
The researchers noted that high-quality microscopy is still the most accurate method for detecting malaria infection. However, as microscopy can be time-consuming, the team wanted to streamline the process by developing a semi-automated system.
“We are not suggesting that the whole malaria diagnostic process could or should be automated,” Dr Lundin said. “Rather, our aim is to develop methods that are significantly less labor-intensive than the traditional ones and have a potential to considerably increase the throughput in malaria diagnostics.”
The group’s method is based on computer vision algorithms similar to those used in facial recognition software. First, a thin layer of blood smeared on a microscope slide is digitized. Then, a computer algorithm analyzes more than 50,000 red blood cells per sample and ranks them according to the probability of malaria infection.
Next, the program creates a panel containing images of more than a hundred cells that are the most likely to be infected and presents that panel to the user. The final diagnosis is made by a healthcare professional based on the images.
To test this system, Dr Lundin and his colleagues used a set of samples from 19 patients already diagnosed with malaria and 12 control subjects. From each sample, the researchers created a digitized slide, and the system generated 128 images of the most probable parasite candidate regions.
Two expert microscopists viewed the images on a tablet computer to determine whether a subject was infected with Plasmodium falciparum.
The diagnostic sensitivity was 90% with one viewer and 95% for the other. The specificity was 100% for both viewers.
Based on these results, the researchers said this system has the potential to increase the throughput in malaria diagnostics. However, it does require some tweaking, and the team would like to expand its capabilities.
“The equipment needed for digitization of the samples is a challenge in developed countries,” said study author Nina Linder, MD, PhD, also of the Institute for Molecular Medicine Finland. “In the next phase of our project, we will test the system in combination with inexpensive mobile microscopy devices that our group has also developed.”
“There is also a strong need for fast and accurate methods for measuring the malaria parasite load in a sample,” she added. “Various malaria drug screening programs are underway, and the parasite load in a large number of samples needs to be quantified for determining the efficacy of potential drugs. We are further developing the computer algorithms used in this study to meet this need as well.”
Lastly, the researchers said this system could be applied in various other fields of medicine. In addition to other infectious diseases such as tuberculosis, the group is planning to test the system’s utility in cancer diagnosis. ![]()
Plasmodium falciparum
Credit: CDC/Mae Melvin
A semi-automated system may allow healthcare professionals to diagnose malaria infection with more than 90% accuracy.
With this system, a computer algorithm analyzes red blood cells from a digitized slide, ranks them according to the likelihood of infection, and presents the user with more than 100 images from which to make a diagnosis.
Johan Lundin, MD, PhD, of the Institute for Molecular Medicine Finland, and his colleagues described the system in PLOS ONE.
The researchers noted that high-quality microscopy is still the most accurate method for detecting malaria infection. However, as microscopy can be time-consuming, the team wanted to streamline the process by developing a semi-automated system.
“We are not suggesting that the whole malaria diagnostic process could or should be automated,” Dr Lundin said. “Rather, our aim is to develop methods that are significantly less labor-intensive than the traditional ones and have a potential to considerably increase the throughput in malaria diagnostics.”
The group’s method is based on computer vision algorithms similar to those used in facial recognition software. First, a thin layer of blood smeared on a microscope slide is digitized. Then, a computer algorithm analyzes more than 50,000 red blood cells per sample and ranks them according to the probability of malaria infection.
Next, the program creates a panel containing images of more than a hundred cells that are the most likely to be infected and presents that panel to the user. The final diagnosis is made by a healthcare professional based on the images.
To test this system, Dr Lundin and his colleagues used a set of samples from 19 patients already diagnosed with malaria and 12 control subjects. From each sample, the researchers created a digitized slide, and the system generated 128 images of the most probable parasite candidate regions.
Two expert microscopists viewed the images on a tablet computer to determine whether a subject was infected with Plasmodium falciparum.
The diagnostic sensitivity was 90% with one viewer and 95% for the other. The specificity was 100% for both viewers.
Based on these results, the researchers said this system has the potential to increase the throughput in malaria diagnostics. However, it does require some tweaking, and the team would like to expand its capabilities.
“The equipment needed for digitization of the samples is a challenge in developed countries,” said study author Nina Linder, MD, PhD, also of the Institute for Molecular Medicine Finland. “In the next phase of our project, we will test the system in combination with inexpensive mobile microscopy devices that our group has also developed.”
“There is also a strong need for fast and accurate methods for measuring the malaria parasite load in a sample,” she added. “Various malaria drug screening programs are underway, and the parasite load in a large number of samples needs to be quantified for determining the efficacy of potential drugs. We are further developing the computer algorithms used in this study to meet this need as well.”
Lastly, the researchers said this system could be applied in various other fields of medicine. In addition to other infectious diseases such as tuberculosis, the group is planning to test the system’s utility in cancer diagnosis. ![]()
Product prevents bleeding in hemophilia A, company says
A recombinant factor VIII (rFVIII) product known as BAX 855 can prevent bleeding in hemophilia A patients, according to Baxter International, the company developing the product.
BAX 855 is a pegylated version of ADVATE, a full-length rFVIII product already approved to treat hemophilia A.
In a phase 3 study, BAX 855 met the primary endpoint of reducing annualized bleed rates (ABRs) among hemophilia A patients when used as prophylaxis rather than on-demand treatment.
The trial included 138 patients age 12 and older with previously treated hemophilia A. The patients received BAX 855 either twice weekly (45 IU/kg) or on-demand and were followed for 6 months.
The primary objective was to show that BAX 855 prophylaxis can reduce ABRs compared to on-demand treatment. The researchers’ other objectives were to evaluate the safety and immunogenicity of the compound when administered as prophylaxis or on-demand.
BAX 855 met its primary endpoint in the control and prevention of bleeding, routine prophylaxis, and perioperative management.
Patients in the twice-weekly prophylaxis arm experienced a 95% reduction in median ABR compared to those in the on-demand arm (1.9% vs 41.5%, respectively). BAX 855 was also effective in treating bleeding episodes, 96% of which were controlled with 1 or 2 infusions.
The half-life of BAX 855 was 1.4 to 1.5 times that of ADVATE, which is consistent with the findings from a phase 1 study.
No patients developed inhibitors to BAX 855 and no treatment-related serious adverse events, including hypersensitivity, were reported. The most common treatment-related adverse event was headache, which occurred in 3 patients.
Baxter said it expects to submit a biologics license application for BAX 855 to the US Food and Drug Administration before the end of 2014 and to present additional trial data in the coming months.
In addition to an ongoing continuation study for patients who have completed the pivotal trial, the company is starting a phase 3 study to evaluate the safety and efficacy of BAX 855 among 60 previously treated hemophilia A patients younger than 12 years of age.
Consistent with guidelines published by the European Medicines Agency that require a study in children less than 12 years of age prior to filing, Baxter expects to file a marketing authorization application with the agency upon the completing the pediatric study. ![]()
A recombinant factor VIII (rFVIII) product known as BAX 855 can prevent bleeding in hemophilia A patients, according to Baxter International, the company developing the product.
BAX 855 is a pegylated version of ADVATE, a full-length rFVIII product already approved to treat hemophilia A.
In a phase 3 study, BAX 855 met the primary endpoint of reducing annualized bleed rates (ABRs) among hemophilia A patients when used as prophylaxis rather than on-demand treatment.
The trial included 138 patients age 12 and older with previously treated hemophilia A. The patients received BAX 855 either twice weekly (45 IU/kg) or on-demand and were followed for 6 months.
The primary objective was to show that BAX 855 prophylaxis can reduce ABRs compared to on-demand treatment. The researchers’ other objectives were to evaluate the safety and immunogenicity of the compound when administered as prophylaxis or on-demand.
BAX 855 met its primary endpoint in the control and prevention of bleeding, routine prophylaxis, and perioperative management.
Patients in the twice-weekly prophylaxis arm experienced a 95% reduction in median ABR compared to those in the on-demand arm (1.9% vs 41.5%, respectively). BAX 855 was also effective in treating bleeding episodes, 96% of which were controlled with 1 or 2 infusions.
The half-life of BAX 855 was 1.4 to 1.5 times that of ADVATE, which is consistent with the findings from a phase 1 study.
No patients developed inhibitors to BAX 855 and no treatment-related serious adverse events, including hypersensitivity, were reported. The most common treatment-related adverse event was headache, which occurred in 3 patients.
Baxter said it expects to submit a biologics license application for BAX 855 to the US Food and Drug Administration before the end of 2014 and to present additional trial data in the coming months.
In addition to an ongoing continuation study for patients who have completed the pivotal trial, the company is starting a phase 3 study to evaluate the safety and efficacy of BAX 855 among 60 previously treated hemophilia A patients younger than 12 years of age.
Consistent with guidelines published by the European Medicines Agency that require a study in children less than 12 years of age prior to filing, Baxter expects to file a marketing authorization application with the agency upon the completing the pediatric study. ![]()
A recombinant factor VIII (rFVIII) product known as BAX 855 can prevent bleeding in hemophilia A patients, according to Baxter International, the company developing the product.
BAX 855 is a pegylated version of ADVATE, a full-length rFVIII product already approved to treat hemophilia A.
In a phase 3 study, BAX 855 met the primary endpoint of reducing annualized bleed rates (ABRs) among hemophilia A patients when used as prophylaxis rather than on-demand treatment.
The trial included 138 patients age 12 and older with previously treated hemophilia A. The patients received BAX 855 either twice weekly (45 IU/kg) or on-demand and were followed for 6 months.
The primary objective was to show that BAX 855 prophylaxis can reduce ABRs compared to on-demand treatment. The researchers’ other objectives were to evaluate the safety and immunogenicity of the compound when administered as prophylaxis or on-demand.
BAX 855 met its primary endpoint in the control and prevention of bleeding, routine prophylaxis, and perioperative management.
Patients in the twice-weekly prophylaxis arm experienced a 95% reduction in median ABR compared to those in the on-demand arm (1.9% vs 41.5%, respectively). BAX 855 was also effective in treating bleeding episodes, 96% of which were controlled with 1 or 2 infusions.
The half-life of BAX 855 was 1.4 to 1.5 times that of ADVATE, which is consistent with the findings from a phase 1 study.
No patients developed inhibitors to BAX 855 and no treatment-related serious adverse events, including hypersensitivity, were reported. The most common treatment-related adverse event was headache, which occurred in 3 patients.
Baxter said it expects to submit a biologics license application for BAX 855 to the US Food and Drug Administration before the end of 2014 and to present additional trial data in the coming months.
In addition to an ongoing continuation study for patients who have completed the pivotal trial, the company is starting a phase 3 study to evaluate the safety and efficacy of BAX 855 among 60 previously treated hemophilia A patients younger than 12 years of age.
Consistent with guidelines published by the European Medicines Agency that require a study in children less than 12 years of age prior to filing, Baxter expects to file a marketing authorization application with the agency upon the completing the pediatric study. ![]()
New technology enables malaria discovery
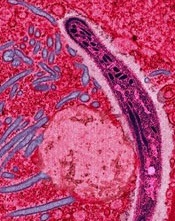
Credit: Ute Frevert
and Margaret Shear
Researchers say they’ve developed a new computational method to predict the function of disease-causing genes and proteins, and this has enabled an important discovery.
The team used the method to study EXP1, a protein known to be essential to the malaria parasite Plasmodium falciparum, although its exact mechanism has been unclear.
The research revealed that EXP1 enables the parasite to detoxify the main metabolic byproducts it creates in red blood cells.
The researchers also found that EXP1 has a direct role in drug action and susceptibility to artesunate, an important member of the artemisinin drug family.
“Through this multiyear collaborative effort, we now have an improved understanding of the protective molecular mechanisms of the malaria parasite and its drug susceptibility to artesunate,” said study author Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas.
“As we are witnessing a rise of resistance to artemisinins, these results may help [us in] finding new pathways to successor drugs.”
Dr Lichtarge and his colleagues described this research in Cell.
The team devised a computational method that allows biological information to flow from gene to gene across the supergenomic network.
“The network connects millions of genes from hundreds of species based on their interactions within the organism or based on their ancestral relations between different species,” said study author Andreas Martin Lisewski, PhD, also of the Baylor College of Medicine.
“Normally, computing the flow of functional information would be costly and slow, but we developed a compression method that reduces this gigantic network into one that is much smaller and now computationally tractable. The surprise is that these biological networks are compressible, much like digital data in today’s computers.”
To test their method, the researchers looked at functional predictions of P falciparum. It’s been more than 10 years since this parasite’s genome was fully sequenced, but little is known about the function for most of its genes.
“To better understand this disease, we need to identify more functions of the parasite’s genes,” Dr Lisewski said. “This understanding may eventually help us to stem the rise of drug-resistant malaria, such as the emerging resistance to artemisinins.”
With that in mind, the researchers used their computational method to study EXP1. And they discovered the protein is a membrane glutathione S-transferase that degrades cytotoxic hematin but is inhibited by artesunate. ![]()
Credit: Ute Frevert
and Margaret Shear
Researchers say they’ve developed a new computational method to predict the function of disease-causing genes and proteins, and this has enabled an important discovery.
The team used the method to study EXP1, a protein known to be essential to the malaria parasite Plasmodium falciparum, although its exact mechanism has been unclear.
The research revealed that EXP1 enables the parasite to detoxify the main metabolic byproducts it creates in red blood cells.
The researchers also found that EXP1 has a direct role in drug action and susceptibility to artesunate, an important member of the artemisinin drug family.
“Through this multiyear collaborative effort, we now have an improved understanding of the protective molecular mechanisms of the malaria parasite and its drug susceptibility to artesunate,” said study author Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas.
“As we are witnessing a rise of resistance to artemisinins, these results may help [us in] finding new pathways to successor drugs.”
Dr Lichtarge and his colleagues described this research in Cell.
The team devised a computational method that allows biological information to flow from gene to gene across the supergenomic network.
“The network connects millions of genes from hundreds of species based on their interactions within the organism or based on their ancestral relations between different species,” said study author Andreas Martin Lisewski, PhD, also of the Baylor College of Medicine.
“Normally, computing the flow of functional information would be costly and slow, but we developed a compression method that reduces this gigantic network into one that is much smaller and now computationally tractable. The surprise is that these biological networks are compressible, much like digital data in today’s computers.”
To test their method, the researchers looked at functional predictions of P falciparum. It’s been more than 10 years since this parasite’s genome was fully sequenced, but little is known about the function for most of its genes.
“To better understand this disease, we need to identify more functions of the parasite’s genes,” Dr Lisewski said. “This understanding may eventually help us to stem the rise of drug-resistant malaria, such as the emerging resistance to artemisinins.”
With that in mind, the researchers used their computational method to study EXP1. And they discovered the protein is a membrane glutathione S-transferase that degrades cytotoxic hematin but is inhibited by artesunate. ![]()
Credit: Ute Frevert
and Margaret Shear
Researchers say they’ve developed a new computational method to predict the function of disease-causing genes and proteins, and this has enabled an important discovery.
The team used the method to study EXP1, a protein known to be essential to the malaria parasite Plasmodium falciparum, although its exact mechanism has been unclear.
The research revealed that EXP1 enables the parasite to detoxify the main metabolic byproducts it creates in red blood cells.
The researchers also found that EXP1 has a direct role in drug action and susceptibility to artesunate, an important member of the artemisinin drug family.
“Through this multiyear collaborative effort, we now have an improved understanding of the protective molecular mechanisms of the malaria parasite and its drug susceptibility to artesunate,” said study author Olivier Lichtarge, MD, PhD, of the Baylor College of Medicine in Houston, Texas.
“As we are witnessing a rise of resistance to artemisinins, these results may help [us in] finding new pathways to successor drugs.”
Dr Lichtarge and his colleagues described this research in Cell.
The team devised a computational method that allows biological information to flow from gene to gene across the supergenomic network.
“The network connects millions of genes from hundreds of species based on their interactions within the organism or based on their ancestral relations between different species,” said study author Andreas Martin Lisewski, PhD, also of the Baylor College of Medicine.
“Normally, computing the flow of functional information would be costly and slow, but we developed a compression method that reduces this gigantic network into one that is much smaller and now computationally tractable. The surprise is that these biological networks are compressible, much like digital data in today’s computers.”
To test their method, the researchers looked at functional predictions of P falciparum. It’s been more than 10 years since this parasite’s genome was fully sequenced, but little is known about the function for most of its genes.
“To better understand this disease, we need to identify more functions of the parasite’s genes,” Dr Lisewski said. “This understanding may eventually help us to stem the rise of drug-resistant malaria, such as the emerging resistance to artemisinins.”
With that in mind, the researchers used their computational method to study EXP1. And they discovered the protein is a membrane glutathione S-transferase that degrades cytotoxic hematin but is inhibited by artesunate. ![]()
Toward better treatment of CML
Credit: UC San Diego
In vitro experiments have revealed new insight into tyrosine kinase inhibitor (TKI) resistance among patients with chronic myeloid leukemia (CML).
Though it’s now possible to overcome TKI resistance resulting from single BCR-ABL1 mutants, targeting compound mutants remains a challenge.
So researchers tested several TKIs on various BCR-ABL1 compound mutants to determine which drug, if any, would be most effective for each combination.
The results appear in Cancer Cell.
Thomas O’Hare, PhD, of the Huntsman Cancer Institute at the University of Utah, and his colleagues first took an inventory of clinical BCR-ABL1 compound mutations associated with TKI resistance that had been reported in the literature.
The team identified 12 kinase domain positions that account for most clinical BCR-ABL1 TKI resistance—M244, G250, Q252, Y253, E255, V299, F311, T315, F317, M351, F359, and H396.
All of the clinically reported compound mutations include at least 1 of the 12 key positions, and most (65%) include 2. Each position has been implicated in resistance to 1 or more TKIs, including imatinib, nilotinib, dasatinib, bosutinib, rebastinib, and ponatinib.
The researchers found that some of the compound mutations they studied conferred resistance several-fold higher than that of either contributing mutation alone.
“We were able to sequence about 100 clinical samples, which gave us a very large body of data to shed light on the number of compound mutations and how they develop,” said Michael Deininger, MD, PhD, also of the Huntsman Cancer Institute.
“One key finding was that compound mutations containing an already known mutation called T315I tend to confer complete resistance to all available TKIs.”
The researchers had focused their testing on ponatinib, as the drug has proven effective against resistant CML, particularly cases with the T315I mutation. Unfortunately, ponatinib was often no match for compound mutations including T315I.
Tests did suggest that a 30 mg/day dose of ponatinib would maintain efficacy against 7 of the 8 non-T315I compound mutants tested, though the Y253H/E255V mutant proved resistant.
The researchers also found that a 15 mg/day dose of ponatinib could pre-empt outgrowth of 5 of the 8 non-T315I compound mutants, though Y253H/E255V, E255V/V299L, and F317L/F359V might be problematic.
But ponatinib proved substantially less effective against T315I-inclusive compound mutants. Nine of 10 T315I-inclusive compound mutants showed little or no sensitivity to ponatinib or any of the other TKIs tested. M244V/T315I was the only compound mutant not resistant to ponatinib.
The researchers noted that because ponatinib has proven effective against the T315I mutant in isolation, many patients treated with ponatinib are likely to have this mutation.
So it may be necessary to perform more sensitive screening on these patients to determine whether they might have T315I-inclusive compound mutants that could confer resistance.
“Fortunately, the problems we are studying affect a minority of CML patients,” Dr O’Hare said. “[S]till, this leaves some patients with no good treatment option at all. Our goal is to have a TKI option for every patient.”
According to Dr O’Hare, it’s only a matter of time until analogous compound mutations emerge in many other cancers, including acute myeloid leukemia and non-small cell lung cancer.
“Our findings in CML will provide a blueprint for contending with resistance in these highly aggressive diseases as well,” he concluded. ![]()
Credit: UC San Diego
In vitro experiments have revealed new insight into tyrosine kinase inhibitor (TKI) resistance among patients with chronic myeloid leukemia (CML).
Though it’s now possible to overcome TKI resistance resulting from single BCR-ABL1 mutants, targeting compound mutants remains a challenge.
So researchers tested several TKIs on various BCR-ABL1 compound mutants to determine which drug, if any, would be most effective for each combination.
The results appear in Cancer Cell.
Thomas O’Hare, PhD, of the Huntsman Cancer Institute at the University of Utah, and his colleagues first took an inventory of clinical BCR-ABL1 compound mutations associated with TKI resistance that had been reported in the literature.
The team identified 12 kinase domain positions that account for most clinical BCR-ABL1 TKI resistance—M244, G250, Q252, Y253, E255, V299, F311, T315, F317, M351, F359, and H396.
All of the clinically reported compound mutations include at least 1 of the 12 key positions, and most (65%) include 2. Each position has been implicated in resistance to 1 or more TKIs, including imatinib, nilotinib, dasatinib, bosutinib, rebastinib, and ponatinib.
The researchers found that some of the compound mutations they studied conferred resistance several-fold higher than that of either contributing mutation alone.
“We were able to sequence about 100 clinical samples, which gave us a very large body of data to shed light on the number of compound mutations and how they develop,” said Michael Deininger, MD, PhD, also of the Huntsman Cancer Institute.
“One key finding was that compound mutations containing an already known mutation called T315I tend to confer complete resistance to all available TKIs.”
The researchers had focused their testing on ponatinib, as the drug has proven effective against resistant CML, particularly cases with the T315I mutation. Unfortunately, ponatinib was often no match for compound mutations including T315I.
Tests did suggest that a 30 mg/day dose of ponatinib would maintain efficacy against 7 of the 8 non-T315I compound mutants tested, though the Y253H/E255V mutant proved resistant.
The researchers also found that a 15 mg/day dose of ponatinib could pre-empt outgrowth of 5 of the 8 non-T315I compound mutants, though Y253H/E255V, E255V/V299L, and F317L/F359V might be problematic.
But ponatinib proved substantially less effective against T315I-inclusive compound mutants. Nine of 10 T315I-inclusive compound mutants showed little or no sensitivity to ponatinib or any of the other TKIs tested. M244V/T315I was the only compound mutant not resistant to ponatinib.
The researchers noted that because ponatinib has proven effective against the T315I mutant in isolation, many patients treated with ponatinib are likely to have this mutation.
So it may be necessary to perform more sensitive screening on these patients to determine whether they might have T315I-inclusive compound mutants that could confer resistance.
“Fortunately, the problems we are studying affect a minority of CML patients,” Dr O’Hare said. “[S]till, this leaves some patients with no good treatment option at all. Our goal is to have a TKI option for every patient.”
According to Dr O’Hare, it’s only a matter of time until analogous compound mutations emerge in many other cancers, including acute myeloid leukemia and non-small cell lung cancer.
“Our findings in CML will provide a blueprint for contending with resistance in these highly aggressive diseases as well,” he concluded. ![]()
Credit: UC San Diego
In vitro experiments have revealed new insight into tyrosine kinase inhibitor (TKI) resistance among patients with chronic myeloid leukemia (CML).
Though it’s now possible to overcome TKI resistance resulting from single BCR-ABL1 mutants, targeting compound mutants remains a challenge.
So researchers tested several TKIs on various BCR-ABL1 compound mutants to determine which drug, if any, would be most effective for each combination.
The results appear in Cancer Cell.
Thomas O’Hare, PhD, of the Huntsman Cancer Institute at the University of Utah, and his colleagues first took an inventory of clinical BCR-ABL1 compound mutations associated with TKI resistance that had been reported in the literature.
The team identified 12 kinase domain positions that account for most clinical BCR-ABL1 TKI resistance—M244, G250, Q252, Y253, E255, V299, F311, T315, F317, M351, F359, and H396.
All of the clinically reported compound mutations include at least 1 of the 12 key positions, and most (65%) include 2. Each position has been implicated in resistance to 1 or more TKIs, including imatinib, nilotinib, dasatinib, bosutinib, rebastinib, and ponatinib.
The researchers found that some of the compound mutations they studied conferred resistance several-fold higher than that of either contributing mutation alone.
“We were able to sequence about 100 clinical samples, which gave us a very large body of data to shed light on the number of compound mutations and how they develop,” said Michael Deininger, MD, PhD, also of the Huntsman Cancer Institute.
“One key finding was that compound mutations containing an already known mutation called T315I tend to confer complete resistance to all available TKIs.”
The researchers had focused their testing on ponatinib, as the drug has proven effective against resistant CML, particularly cases with the T315I mutation. Unfortunately, ponatinib was often no match for compound mutations including T315I.
Tests did suggest that a 30 mg/day dose of ponatinib would maintain efficacy against 7 of the 8 non-T315I compound mutants tested, though the Y253H/E255V mutant proved resistant.
The researchers also found that a 15 mg/day dose of ponatinib could pre-empt outgrowth of 5 of the 8 non-T315I compound mutants, though Y253H/E255V, E255V/V299L, and F317L/F359V might be problematic.
But ponatinib proved substantially less effective against T315I-inclusive compound mutants. Nine of 10 T315I-inclusive compound mutants showed little or no sensitivity to ponatinib or any of the other TKIs tested. M244V/T315I was the only compound mutant not resistant to ponatinib.
The researchers noted that because ponatinib has proven effective against the T315I mutant in isolation, many patients treated with ponatinib are likely to have this mutation.
So it may be necessary to perform more sensitive screening on these patients to determine whether they might have T315I-inclusive compound mutants that could confer resistance.
“Fortunately, the problems we are studying affect a minority of CML patients,” Dr O’Hare said. “[S]till, this leaves some patients with no good treatment option at all. Our goal is to have a TKI option for every patient.”
According to Dr O’Hare, it’s only a matter of time until analogous compound mutations emerge in many other cancers, including acute myeloid leukemia and non-small cell lung cancer.
“Our findings in CML will provide a blueprint for contending with resistance in these highly aggressive diseases as well,” he concluded. ![]()
Path to drug development often not straightforward, study shows

Credit: Rhoda Baer
An analysis of university discoveries licensed to biotechnology firms has revealed early bottlenecks in the drug development process.
Typically, universities do most of the basic research and then license a discovery to a small biotech firm that advances the research. The small firm will then sublicense the discovery to a large firm that can run clinical trials.
But an analysis published in Science Translational Medicine suggests the process rarely follows this straightforward path.
Instead, it often zigzags across biotech firms and between research areas before a drug is finally commercialized.
“The timeline for commercialization is much longer than most people think,” said study author Jerry Thursby, PhD, of the Georgia Institute of Technology in Atlanta.
To study the path of drug development, Dr Thursby and his colleagues built a database of 835 patents in 342 university licenses with biotech firms.
They then traced the path of patents to document whether the inventions were sublicensed to another firm for testing in a new disease category or whether the sublicense was to a large firm for clinical trials or marketing.
In all, 27% of inventions appeared in a second license (sublicense). The average time between invention and first license was 5.5 years, and the average time between first and second license was 3.5 years.
This time span is substantial, the researchers said, given that the average time from discovery to drug approval in the US is 13 years.
The team also found that sublicensing often resets the development timeline because a drug must be tested for an entirely new indication or several new indications.
The disease categories in the licenses analyzed spanned 20 distinct indications, and individual licenses included up to 5 indications. But the categories were very broad, such as “cancer” or “infectious diseases.”
Nevertheless, the researchers saw substantial changes in disease indications from the first license to the second. Only 19% of the inventions remained completely unchanged between the first and second license.
For 44% of inventions, none of the first-license indications remained in the second license. Twenty-eight percent of inventions had indications added between the first and second license, and 9% had indications subtracted.
The researchers said these results suggest a need for policies and initiatives that enhance early translation by more efficiently driving more inventions into multiple disease pipelines.
One option might be the formation of an open-source translational research database that complements clinicaltrials.gov, where patents and licenses for biomedical research thought to be destined for eventual therapeutic use would be logged and shared. ![]()
Credit: Rhoda Baer
An analysis of university discoveries licensed to biotechnology firms has revealed early bottlenecks in the drug development process.
Typically, universities do most of the basic research and then license a discovery to a small biotech firm that advances the research. The small firm will then sublicense the discovery to a large firm that can run clinical trials.
But an analysis published in Science Translational Medicine suggests the process rarely follows this straightforward path.
Instead, it often zigzags across biotech firms and between research areas before a drug is finally commercialized.
“The timeline for commercialization is much longer than most people think,” said study author Jerry Thursby, PhD, of the Georgia Institute of Technology in Atlanta.
To study the path of drug development, Dr Thursby and his colleagues built a database of 835 patents in 342 university licenses with biotech firms.
They then traced the path of patents to document whether the inventions were sublicensed to another firm for testing in a new disease category or whether the sublicense was to a large firm for clinical trials or marketing.
In all, 27% of inventions appeared in a second license (sublicense). The average time between invention and first license was 5.5 years, and the average time between first and second license was 3.5 years.
This time span is substantial, the researchers said, given that the average time from discovery to drug approval in the US is 13 years.
The team also found that sublicensing often resets the development timeline because a drug must be tested for an entirely new indication or several new indications.
The disease categories in the licenses analyzed spanned 20 distinct indications, and individual licenses included up to 5 indications. But the categories were very broad, such as “cancer” or “infectious diseases.”
Nevertheless, the researchers saw substantial changes in disease indications from the first license to the second. Only 19% of the inventions remained completely unchanged between the first and second license.
For 44% of inventions, none of the first-license indications remained in the second license. Twenty-eight percent of inventions had indications added between the first and second license, and 9% had indications subtracted.
The researchers said these results suggest a need for policies and initiatives that enhance early translation by more efficiently driving more inventions into multiple disease pipelines.
One option might be the formation of an open-source translational research database that complements clinicaltrials.gov, where patents and licenses for biomedical research thought to be destined for eventual therapeutic use would be logged and shared. ![]()
Credit: Rhoda Baer
An analysis of university discoveries licensed to biotechnology firms has revealed early bottlenecks in the drug development process.
Typically, universities do most of the basic research and then license a discovery to a small biotech firm that advances the research. The small firm will then sublicense the discovery to a large firm that can run clinical trials.
But an analysis published in Science Translational Medicine suggests the process rarely follows this straightforward path.
Instead, it often zigzags across biotech firms and between research areas before a drug is finally commercialized.
“The timeline for commercialization is much longer than most people think,” said study author Jerry Thursby, PhD, of the Georgia Institute of Technology in Atlanta.
To study the path of drug development, Dr Thursby and his colleagues built a database of 835 patents in 342 university licenses with biotech firms.
They then traced the path of patents to document whether the inventions were sublicensed to another firm for testing in a new disease category or whether the sublicense was to a large firm for clinical trials or marketing.
In all, 27% of inventions appeared in a second license (sublicense). The average time between invention and first license was 5.5 years, and the average time between first and second license was 3.5 years.
This time span is substantial, the researchers said, given that the average time from discovery to drug approval in the US is 13 years.
The team also found that sublicensing often resets the development timeline because a drug must be tested for an entirely new indication or several new indications.
The disease categories in the licenses analyzed spanned 20 distinct indications, and individual licenses included up to 5 indications. But the categories were very broad, such as “cancer” or “infectious diseases.”
Nevertheless, the researchers saw substantial changes in disease indications from the first license to the second. Only 19% of the inventions remained completely unchanged between the first and second license.
For 44% of inventions, none of the first-license indications remained in the second license. Twenty-eight percent of inventions had indications added between the first and second license, and 9% had indications subtracted.
The researchers said these results suggest a need for policies and initiatives that enhance early translation by more efficiently driving more inventions into multiple disease pipelines.
One option might be the formation of an open-source translational research database that complements clinicaltrials.gov, where patents and licenses for biomedical research thought to be destined for eventual therapeutic use would be logged and shared. ![]()
New pipette can move single cells

Credit: Rhoda Baer
Researchers say they have developed a pipette that can transfer a single cell at a time.
The device, called the handheld single-cell pipette (hSCP), has 2 plungers. The first is used to wash and capture a single cell, and the second can release the cell in the desired location.
Lidong Qin, PhD, of Houston Methodist Research Institute in Texas, and his colleagues described the hSCP and reported preliminary results with the device in the Journal of the American Chemical Society.
“Studying single cells and their unique functions has become a frontier in current biomedical research,” Dr Qin said. “One of the biggest challenges for single-cell research is picking out only one cell from a collection of millions of cells.”
He noted that current techniques for withdrawing single cells from a tube or Petri dish can be cumbersome, expensive, and time-consuming.
“Some old and clumsy methods are used to capture single cells,” he explained. “Some researchers use their mouths at one end of the pipette, driven by their own mouth force, to try to ensure only a minimum amount of cell suspension collected. The sample is then checked with a microscope to find out the number of cells captured. The opportunity to get only one cell is hit-or-miss and a bit troublesome.”
“One company provides a million-dollar machine that can help biologists transfer single cells to 96-well plates. Each run costs an additional $1000 to purchase the plate. Such technology will not be widely accessible to biologists.”
With that in mind, Dr Qin and his colleagues developed their 2-plunger hSCP. The first plunger withdraws fluid from a suspension of cells.
Fluid travels through canals on either side of a nanoscopic, laser-sculpted “hook” that is just big enough to trap a single cell. This hook can be altered depending on the size and type of cells a researcher is using.
The first plunger is also used to wash and separate the captured cell from other cells that may have been extracted. The second plunger pushes the captured cell out of the pipette into growth medium or onto a slide or welled plate for study.
Dr Qin said one of his goals is to make the technology cost $10 or less per run. And future designs of the hSCP will be developed with mass production in mind.
Dr Qin said his group can also produce hSCPs that pick up virtually any small number of cells, depending on a scientist’s needs, by etching more hooks during the pipette’s construction. ![]()
Credit: Rhoda Baer
Researchers say they have developed a pipette that can transfer a single cell at a time.
The device, called the handheld single-cell pipette (hSCP), has 2 plungers. The first is used to wash and capture a single cell, and the second can release the cell in the desired location.
Lidong Qin, PhD, of Houston Methodist Research Institute in Texas, and his colleagues described the hSCP and reported preliminary results with the device in the Journal of the American Chemical Society.
“Studying single cells and their unique functions has become a frontier in current biomedical research,” Dr Qin said. “One of the biggest challenges for single-cell research is picking out only one cell from a collection of millions of cells.”
He noted that current techniques for withdrawing single cells from a tube or Petri dish can be cumbersome, expensive, and time-consuming.
“Some old and clumsy methods are used to capture single cells,” he explained. “Some researchers use their mouths at one end of the pipette, driven by their own mouth force, to try to ensure only a minimum amount of cell suspension collected. The sample is then checked with a microscope to find out the number of cells captured. The opportunity to get only one cell is hit-or-miss and a bit troublesome.”
“One company provides a million-dollar machine that can help biologists transfer single cells to 96-well plates. Each run costs an additional $1000 to purchase the plate. Such technology will not be widely accessible to biologists.”
With that in mind, Dr Qin and his colleagues developed their 2-plunger hSCP. The first plunger withdraws fluid from a suspension of cells.
Fluid travels through canals on either side of a nanoscopic, laser-sculpted “hook” that is just big enough to trap a single cell. This hook can be altered depending on the size and type of cells a researcher is using.
The first plunger is also used to wash and separate the captured cell from other cells that may have been extracted. The second plunger pushes the captured cell out of the pipette into growth medium or onto a slide or welled plate for study.
Dr Qin said one of his goals is to make the technology cost $10 or less per run. And future designs of the hSCP will be developed with mass production in mind.
Dr Qin said his group can also produce hSCPs that pick up virtually any small number of cells, depending on a scientist’s needs, by etching more hooks during the pipette’s construction. ![]()
Credit: Rhoda Baer
Researchers say they have developed a pipette that can transfer a single cell at a time.
The device, called the handheld single-cell pipette (hSCP), has 2 plungers. The first is used to wash and capture a single cell, and the second can release the cell in the desired location.
Lidong Qin, PhD, of Houston Methodist Research Institute in Texas, and his colleagues described the hSCP and reported preliminary results with the device in the Journal of the American Chemical Society.
“Studying single cells and their unique functions has become a frontier in current biomedical research,” Dr Qin said. “One of the biggest challenges for single-cell research is picking out only one cell from a collection of millions of cells.”
He noted that current techniques for withdrawing single cells from a tube or Petri dish can be cumbersome, expensive, and time-consuming.
“Some old and clumsy methods are used to capture single cells,” he explained. “Some researchers use their mouths at one end of the pipette, driven by their own mouth force, to try to ensure only a minimum amount of cell suspension collected. The sample is then checked with a microscope to find out the number of cells captured. The opportunity to get only one cell is hit-or-miss and a bit troublesome.”
“One company provides a million-dollar machine that can help biologists transfer single cells to 96-well plates. Each run costs an additional $1000 to purchase the plate. Such technology will not be widely accessible to biologists.”
With that in mind, Dr Qin and his colleagues developed their 2-plunger hSCP. The first plunger withdraws fluid from a suspension of cells.
Fluid travels through canals on either side of a nanoscopic, laser-sculpted “hook” that is just big enough to trap a single cell. This hook can be altered depending on the size and type of cells a researcher is using.
The first plunger is also used to wash and separate the captured cell from other cells that may have been extracted. The second plunger pushes the captured cell out of the pipette into growth medium or onto a slide or welled plate for study.
Dr Qin said one of his goals is to make the technology cost $10 or less per run. And future designs of the hSCP will be developed with mass production in mind.
Dr Qin said his group can also produce hSCPs that pick up virtually any small number of cells, depending on a scientist’s needs, by etching more hooks during the pipette’s construction.