User login
Novel anticoagulant system no better than existing drug

SAN DIEGO—In a now-terminated phase 3 trial, a novel anticoagulant system proved about as effective as an established drug in patients undergoing percutaneous coronary intervention (PCI).
The system also conferred higher rates of bleeding and prompted more allergic reactions.
The trial, known as REGULATE-PCI, was designed to compare the Revolixys Kit (also known as the REG-1 Anticoagulation System) to bivalirudin (Angiomax).
The study was officially halted in August due to an excess of severe allergic reactions associated with the Revolixys Kit. Given the early termination, investigators said the data should be considered exploratory.
Roxana Mehran, MD, of Mount Sinai Hospital in New York, New York, presented the study’s results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 402-12).
The goal of the REGULATE-PCI trial was to compare the reversible thrombin inhibitor bivalirudin to the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing PCI.
Before the trial was stopped, 3232 patients were enrolled. They underwent PCI at 225 hospitals in 17 countries. Patients were equally randomized to the bivalirudin or Revolixys arms, and investigators collected data at 3 days and 30 days.
Efficacy and safety results
There were no differences between the treatment arms in terms of the study’s primary efficacy endpoint—a composite of all-cause death, heart attack, stroke, or urgent revascularization.
The endpoint occurred in 6.7% of patients in the Revolixys arm and 6.4% of patients receiving bivalirudin 3 days after PCI (P=0.72). Efficacy was still comparable at 30 days.
In addition, the Revolixys system failed to show a benefit over bivalirudin with regard to the primary safety endpoint of bleeding.
The rate of BARC 3 or 5 bleeding was 0.4% in the Revolixys arm and 0.1% in the bivalirudin arm (P=0.09). And the rates of BARC 2, 3, or 5 bleeding were 6.5% and 4.1%, respectively (P=0.002).
Serious allergic reactions occurred in 10 of 1605 patients in the Revolixys arm. One of these reactions was fatal, and the others were anaphylactic reactions. Only one patient in the bivalirudin group had a serious allergic event.
“This anticoagulant system is associated with infrequent but an unacceptably high rate of severe allergic reactions,” Dr Mehran said.
Research is ongoing to determine the exact cause of the allergic reactions, and Dr Mehran said she hopes this does not deter the search for novel anticoagulants for use in this patient population.
More about REGULATE-PCI
Investigators started recruiting patients to the trial in September 2013. In April, because there were a handful of allergic reactions seen in the earlier phase 2 trial, a data safety and monitoring board reviewed all of the safety endpoints for the first 1000 patients enrolled in REGULATE-PCI.
In June, both the executive committee and the sponsor decided to suspend the trial, and the US Food and Drug Administration announced a clinical hold in July. The trial was permanently halted in August.
This study was funded by Regado Biosciences Inc., the company developing the Revolixys kit. Dr Mehran has served on the scientific advisory board for Regado Biosciences. ![]()
SAN DIEGO—In a now-terminated phase 3 trial, a novel anticoagulant system proved about as effective as an established drug in patients undergoing percutaneous coronary intervention (PCI).
The system also conferred higher rates of bleeding and prompted more allergic reactions.
The trial, known as REGULATE-PCI, was designed to compare the Revolixys Kit (also known as the REG-1 Anticoagulation System) to bivalirudin (Angiomax).
The study was officially halted in August due to an excess of severe allergic reactions associated with the Revolixys Kit. Given the early termination, investigators said the data should be considered exploratory.
Roxana Mehran, MD, of Mount Sinai Hospital in New York, New York, presented the study’s results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 402-12).
The goal of the REGULATE-PCI trial was to compare the reversible thrombin inhibitor bivalirudin to the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing PCI.
Before the trial was stopped, 3232 patients were enrolled. They underwent PCI at 225 hospitals in 17 countries. Patients were equally randomized to the bivalirudin or Revolixys arms, and investigators collected data at 3 days and 30 days.
Efficacy and safety results
There were no differences between the treatment arms in terms of the study’s primary efficacy endpoint—a composite of all-cause death, heart attack, stroke, or urgent revascularization.
The endpoint occurred in 6.7% of patients in the Revolixys arm and 6.4% of patients receiving bivalirudin 3 days after PCI (P=0.72). Efficacy was still comparable at 30 days.
In addition, the Revolixys system failed to show a benefit over bivalirudin with regard to the primary safety endpoint of bleeding.
The rate of BARC 3 or 5 bleeding was 0.4% in the Revolixys arm and 0.1% in the bivalirudin arm (P=0.09). And the rates of BARC 2, 3, or 5 bleeding were 6.5% and 4.1%, respectively (P=0.002).
Serious allergic reactions occurred in 10 of 1605 patients in the Revolixys arm. One of these reactions was fatal, and the others were anaphylactic reactions. Only one patient in the bivalirudin group had a serious allergic event.
“This anticoagulant system is associated with infrequent but an unacceptably high rate of severe allergic reactions,” Dr Mehran said.
Research is ongoing to determine the exact cause of the allergic reactions, and Dr Mehran said she hopes this does not deter the search for novel anticoagulants for use in this patient population.
More about REGULATE-PCI
Investigators started recruiting patients to the trial in September 2013. In April, because there were a handful of allergic reactions seen in the earlier phase 2 trial, a data safety and monitoring board reviewed all of the safety endpoints for the first 1000 patients enrolled in REGULATE-PCI.
In June, both the executive committee and the sponsor decided to suspend the trial, and the US Food and Drug Administration announced a clinical hold in July. The trial was permanently halted in August.
This study was funded by Regado Biosciences Inc., the company developing the Revolixys kit. Dr Mehran has served on the scientific advisory board for Regado Biosciences. ![]()
SAN DIEGO—In a now-terminated phase 3 trial, a novel anticoagulant system proved about as effective as an established drug in patients undergoing percutaneous coronary intervention (PCI).
The system also conferred higher rates of bleeding and prompted more allergic reactions.
The trial, known as REGULATE-PCI, was designed to compare the Revolixys Kit (also known as the REG-1 Anticoagulation System) to bivalirudin (Angiomax).
The study was officially halted in August due to an excess of severe allergic reactions associated with the Revolixys Kit. Given the early termination, investigators said the data should be considered exploratory.
Roxana Mehran, MD, of Mount Sinai Hospital in New York, New York, presented the study’s results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 402-12).
The goal of the REGULATE-PCI trial was to compare the reversible thrombin inhibitor bivalirudin to the Revolixys Kit—a 2-component system consisting of pegnivacogin, an anticoagulant aptamer targeting coagulation factor IXa, and its complementary oligonucleotide active control agent, anivamersen—in patients undergoing PCI.
Before the trial was stopped, 3232 patients were enrolled. They underwent PCI at 225 hospitals in 17 countries. Patients were equally randomized to the bivalirudin or Revolixys arms, and investigators collected data at 3 days and 30 days.
Efficacy and safety results
There were no differences between the treatment arms in terms of the study’s primary efficacy endpoint—a composite of all-cause death, heart attack, stroke, or urgent revascularization.
The endpoint occurred in 6.7% of patients in the Revolixys arm and 6.4% of patients receiving bivalirudin 3 days after PCI (P=0.72). Efficacy was still comparable at 30 days.
In addition, the Revolixys system failed to show a benefit over bivalirudin with regard to the primary safety endpoint of bleeding.
The rate of BARC 3 or 5 bleeding was 0.4% in the Revolixys arm and 0.1% in the bivalirudin arm (P=0.09). And the rates of BARC 2, 3, or 5 bleeding were 6.5% and 4.1%, respectively (P=0.002).
Serious allergic reactions occurred in 10 of 1605 patients in the Revolixys arm. One of these reactions was fatal, and the others were anaphylactic reactions. Only one patient in the bivalirudin group had a serious allergic event.
“This anticoagulant system is associated with infrequent but an unacceptably high rate of severe allergic reactions,” Dr Mehran said.
Research is ongoing to determine the exact cause of the allergic reactions, and Dr Mehran said she hopes this does not deter the search for novel anticoagulants for use in this patient population.
More about REGULATE-PCI
Investigators started recruiting patients to the trial in September 2013. In April, because there were a handful of allergic reactions seen in the earlier phase 2 trial, a data safety and monitoring board reviewed all of the safety endpoints for the first 1000 patients enrolled in REGULATE-PCI.
In June, both the executive committee and the sponsor decided to suspend the trial, and the US Food and Drug Administration announced a clinical hold in July. The trial was permanently halted in August.
This study was funded by Regado Biosciences Inc., the company developing the Revolixys kit. Dr Mehran has served on the scientific advisory board for Regado Biosciences. ![]()
Group reprograms B-ALL cells into macrophage-like cells
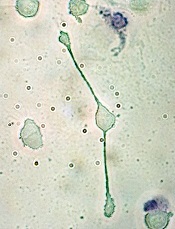
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
Lowering the cost of cancer drugs in the US

Photo by Petr Kratochvil
Increasingly high prices for cancer drugs are affecting patient care and the overall healthcare system in the US, according to authors of an article in Mayo Clinic Proceedings.
The authors noted that the average price of cancer drugs for about a year of therapy increased from $5000 to $10,000 before 2000, and to more than $100,000 by 2012.
Over nearly the same period, the average household income in the US decreased by about 8%.
“Americans with cancer pay 50% to 100% more for the same patented drug than patients in other countries,” said author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“As oncologists, we have a moral obligation to advocate for affordable cancer drugs for our patients.”
Dr Rajkumar and co-author Hagop Kantarjian, MD, of MD Anderson Cancer Center in Houston, Texas, rebutted the major arguments the pharmaceutical industry uses to justify the high price of cancer drugs; namely, the expense of conducting research and drug development, the comparative benefits to patients, that market forces will settle prices to reasonable levels, and that price controls on cancer drugs will stifle innovation.
“One of the facts that people do not realize is that cancer drugs, for the most part, are not operating under a free market economy,” Dr Rajkumar said. “The fact that there are 5 approved drugs to treat an incurable cancer does not mean there is competition.”
“Typically, the standard of care is that each drug is used sequentially or in combination, so that each new drug represents a monopoly with exclusivity granted by patent protection for many years.”
Drs Rajkumar and Kantarjian said other reasons for the high cost of cancer drugs include legislation that prevents Medicare from being able to negotiate drug prices and a lack of value-based pricing, which ties the cost of a drug to its relative effectiveness compared to other drugs.
The authors recommended a set of potential solutions to help control and reduce the high cost of cancer drugs in the US. Some of their recommendations are already in practice in other developed countries. Their recommendations include:
- Allow Medicare to negotiate drug prices
- Develop cancer treatment pathways/guidelines that incorporate the cost and benefit of cancer drugs
- Allow the Food and Drug Administration or physician panels to recommend target prices based on a drug’s magnitude of benefit (value-based pricing)
- Eliminate “pay-for-delay” strategies in which a pharmaceutical company with a brand name drug shares profits on that drug with a generic drug manufacturer for the remainder of a patent period, effectively eliminating a patent challenge and competition
- Allow the importation of drugs from abroad for personal use
- Allow the Patient-Centered Outcomes Research Institute and other cancer advocacy groups to consider cost in their recommendations
- Create patient-driven grassroots movements and organizations to advocate effectively for the interests of patients with cancer to balance advocacy efforts of pharmaceutical companies, insurance companies, pharmacy outlets, and hospitals.
Dr Kantarjian has organized a petition, which is available on change.org, asking the federal government to implement these changes. ![]()
Photo by Petr Kratochvil
Increasingly high prices for cancer drugs are affecting patient care and the overall healthcare system in the US, according to authors of an article in Mayo Clinic Proceedings.
The authors noted that the average price of cancer drugs for about a year of therapy increased from $5000 to $10,000 before 2000, and to more than $100,000 by 2012.
Over nearly the same period, the average household income in the US decreased by about 8%.
“Americans with cancer pay 50% to 100% more for the same patented drug than patients in other countries,” said author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“As oncologists, we have a moral obligation to advocate for affordable cancer drugs for our patients.”
Dr Rajkumar and co-author Hagop Kantarjian, MD, of MD Anderson Cancer Center in Houston, Texas, rebutted the major arguments the pharmaceutical industry uses to justify the high price of cancer drugs; namely, the expense of conducting research and drug development, the comparative benefits to patients, that market forces will settle prices to reasonable levels, and that price controls on cancer drugs will stifle innovation.
“One of the facts that people do not realize is that cancer drugs, for the most part, are not operating under a free market economy,” Dr Rajkumar said. “The fact that there are 5 approved drugs to treat an incurable cancer does not mean there is competition.”
“Typically, the standard of care is that each drug is used sequentially or in combination, so that each new drug represents a monopoly with exclusivity granted by patent protection for many years.”
Drs Rajkumar and Kantarjian said other reasons for the high cost of cancer drugs include legislation that prevents Medicare from being able to negotiate drug prices and a lack of value-based pricing, which ties the cost of a drug to its relative effectiveness compared to other drugs.
The authors recommended a set of potential solutions to help control and reduce the high cost of cancer drugs in the US. Some of their recommendations are already in practice in other developed countries. Their recommendations include:
- Allow Medicare to negotiate drug prices
- Develop cancer treatment pathways/guidelines that incorporate the cost and benefit of cancer drugs
- Allow the Food and Drug Administration or physician panels to recommend target prices based on a drug’s magnitude of benefit (value-based pricing)
- Eliminate “pay-for-delay” strategies in which a pharmaceutical company with a brand name drug shares profits on that drug with a generic drug manufacturer for the remainder of a patent period, effectively eliminating a patent challenge and competition
- Allow the importation of drugs from abroad for personal use
- Allow the Patient-Centered Outcomes Research Institute and other cancer advocacy groups to consider cost in their recommendations
- Create patient-driven grassroots movements and organizations to advocate effectively for the interests of patients with cancer to balance advocacy efforts of pharmaceutical companies, insurance companies, pharmacy outlets, and hospitals.
Dr Kantarjian has organized a petition, which is available on change.org, asking the federal government to implement these changes. ![]()
Photo by Petr Kratochvil
Increasingly high prices for cancer drugs are affecting patient care and the overall healthcare system in the US, according to authors of an article in Mayo Clinic Proceedings.
The authors noted that the average price of cancer drugs for about a year of therapy increased from $5000 to $10,000 before 2000, and to more than $100,000 by 2012.
Over nearly the same period, the average household income in the US decreased by about 8%.
“Americans with cancer pay 50% to 100% more for the same patented drug than patients in other countries,” said author S. Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
“As oncologists, we have a moral obligation to advocate for affordable cancer drugs for our patients.”
Dr Rajkumar and co-author Hagop Kantarjian, MD, of MD Anderson Cancer Center in Houston, Texas, rebutted the major arguments the pharmaceutical industry uses to justify the high price of cancer drugs; namely, the expense of conducting research and drug development, the comparative benefits to patients, that market forces will settle prices to reasonable levels, and that price controls on cancer drugs will stifle innovation.
“One of the facts that people do not realize is that cancer drugs, for the most part, are not operating under a free market economy,” Dr Rajkumar said. “The fact that there are 5 approved drugs to treat an incurable cancer does not mean there is competition.”
“Typically, the standard of care is that each drug is used sequentially or in combination, so that each new drug represents a monopoly with exclusivity granted by patent protection for many years.”
Drs Rajkumar and Kantarjian said other reasons for the high cost of cancer drugs include legislation that prevents Medicare from being able to negotiate drug prices and a lack of value-based pricing, which ties the cost of a drug to its relative effectiveness compared to other drugs.
The authors recommended a set of potential solutions to help control and reduce the high cost of cancer drugs in the US. Some of their recommendations are already in practice in other developed countries. Their recommendations include:
- Allow Medicare to negotiate drug prices
- Develop cancer treatment pathways/guidelines that incorporate the cost and benefit of cancer drugs
- Allow the Food and Drug Administration or physician panels to recommend target prices based on a drug’s magnitude of benefit (value-based pricing)
- Eliminate “pay-for-delay” strategies in which a pharmaceutical company with a brand name drug shares profits on that drug with a generic drug manufacturer for the remainder of a patent period, effectively eliminating a patent challenge and competition
- Allow the importation of drugs from abroad for personal use
- Allow the Patient-Centered Outcomes Research Institute and other cancer advocacy groups to consider cost in their recommendations
- Create patient-driven grassroots movements and organizations to advocate effectively for the interests of patients with cancer to balance advocacy efforts of pharmaceutical companies, insurance companies, pharmacy outlets, and hospitals.
Dr Kantarjian has organized a petition, which is available on change.org, asking the federal government to implement these changes. ![]()
EMA grants vaccine orphan status for MM

showing MM
The European Medicines Agency (EMA) has given a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is also being evaluated in a phase 1/2 study to treat patients with metastatic breast cancer who are receiving first-line hormone therapy.
ImMucin is under development by Vaxil Biotherapeutics Ltd.
About orphan designation
The EMA grants orphan designation to promote the clinical development of drugs that treat rare, life-threatening, or debilitating conditions and are expected to provide significant therapeutic advantage over existing treatments.
Orphan designation provides the company developing a drug with significant benefits, including 10 years of market exclusivity following approval, reductions in the fees and costs of the regulatory process, and scientific assistance from the EMA in clinical development. ![]()
showing MM
The European Medicines Agency (EMA) has given a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is also being evaluated in a phase 1/2 study to treat patients with metastatic breast cancer who are receiving first-line hormone therapy.
ImMucin is under development by Vaxil Biotherapeutics Ltd.
About orphan designation
The EMA grants orphan designation to promote the clinical development of drugs that treat rare, life-threatening, or debilitating conditions and are expected to provide significant therapeutic advantage over existing treatments.
Orphan designation provides the company developing a drug with significant benefits, including 10 years of market exclusivity following approval, reductions in the fees and costs of the regulatory process, and scientific assistance from the EMA in clinical development. ![]()
showing MM
The European Medicines Agency (EMA) has given a novel vaccine orphan designation as a treatment for multiple myeloma (MM).
The vaccine, known as ImMucin, targets the signal peptide domain of the MUC1 tumor antigen.
ImMucin works by “teaching” the immune system to identify and destroy cells that display a short, specific, 21-mer portion from MUC1, which appears on 90% of all cancer cells but not in patients’ blood.
Results of a phase 1/2 trial suggested that ImMucin was safe and active in MM patients. The trial included 15 MUC1-positive patients who had residual or biochemically progressive disease after autologous stem cell transplant.
The patients received 6 or 12 bi-weekly intradermal doses of ImMucin co-administered with human granulocyte-macrophage colony-stimulating factor.
The researchers said the vaccine was well-tolerated, as all adverse events were temporary, grade 1-2 in nature, and resolved spontaneously.
There was a significant decrease in soluble MUC1 levels in 9 patients, and 11 patients had stable disease or clinical improvement that persisted for 17.5 months to more than 41.3 months.
A follow-on study (which is ongoing) in patients who responded to ImMucin has shown that some patients can go more than 4 years without requiring any further treatment for their disease.
ImMucin is also being evaluated in a phase 1/2 study to treat patients with metastatic breast cancer who are receiving first-line hormone therapy.
ImMucin is under development by Vaxil Biotherapeutics Ltd.
About orphan designation
The EMA grants orphan designation to promote the clinical development of drugs that treat rare, life-threatening, or debilitating conditions and are expected to provide significant therapeutic advantage over existing treatments.
Orphan designation provides the company developing a drug with significant benefits, including 10 years of market exclusivity following approval, reductions in the fees and costs of the regulatory process, and scientific assistance from the EMA in clinical development. ![]()
New approvals, genetic testing, maintenance therapy, and DFS in ovarian cancer
Recent study findings have indicated that women with ovarian cancer may have BRCA1 or BRCA2 mutations despite a negative family history, and current NCCN (National Comprehensive Cancer Network) guidelines endorse genetic testing for all women with epithelial cancer of the ovary. Despite this, recent reports indicate that most women with ovarian cancer are not being tested, particularly those who are elderly or without a family history. In this paper by Daniels and colleagues, the investigators examined targeted versus universal genetic testing to see if the use of a well-regarded risk model (BRCAPRO) based on personal and family history could discriminate among patients with high-grade serous ovarian cancer. Targeted genetic testing in this group might help lower costs and encourage testing for those women who actually have a significant chance of carrying a deleterious gene mutation.
Click on the PDF icon at the top of this introduction to read the full article.
Recent study findings have indicated that women with ovarian cancer may have BRCA1 or BRCA2 mutations despite a negative family history, and current NCCN (National Comprehensive Cancer Network) guidelines endorse genetic testing for all women with epithelial cancer of the ovary. Despite this, recent reports indicate that most women with ovarian cancer are not being tested, particularly those who are elderly or without a family history. In this paper by Daniels and colleagues, the investigators examined targeted versus universal genetic testing to see if the use of a well-regarded risk model (BRCAPRO) based on personal and family history could discriminate among patients with high-grade serous ovarian cancer. Targeted genetic testing in this group might help lower costs and encourage testing for those women who actually have a significant chance of carrying a deleterious gene mutation.
Click on the PDF icon at the top of this introduction to read the full article.
Recent study findings have indicated that women with ovarian cancer may have BRCA1 or BRCA2 mutations despite a negative family history, and current NCCN (National Comprehensive Cancer Network) guidelines endorse genetic testing for all women with epithelial cancer of the ovary. Despite this, recent reports indicate that most women with ovarian cancer are not being tested, particularly those who are elderly or without a family history. In this paper by Daniels and colleagues, the investigators examined targeted versus universal genetic testing to see if the use of a well-regarded risk model (BRCAPRO) based on personal and family history could discriminate among patients with high-grade serous ovarian cancer. Targeted genetic testing in this group might help lower costs and encourage testing for those women who actually have a significant chance of carrying a deleterious gene mutation.
Click on the PDF icon at the top of this introduction to read the full article.
Pelvic pleomorphic rhabdomyosarcoma presenting as oliguria in a 61-year-old woman
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Guideline recommends combination therapy for smoking cessation in cancer patients
The National Comprehensive Cancer Network has published a new guideline on smoking cessation for cancer patients that recommends combining pharmacologic therapy with counseling as the most effective approach, along with rigorous review and close follow-ups to prevent relapses.
“Although the medical community recognizes the importance of smoking cessation, supporting patients in ceasing to smoke is generally not done well. Our hope is that by addressing smoking cessation in a cancer patient population, we can make it easier for oncologists to effectively support their patients in achieving their smoking cessation goals,” Dr. Peter Shields, deputy director of the Ohio State University Comprehensive Cancer Center, said in a written statement. Of the estimated 590,000 cancer deaths in 2015, about 170,000, or nearly 30%, will be caused by tobacco smoking. Quitting tobacco improves cancer treatment effectiveness and reduces cancer recurrence, according to the NCCN.
Read the full statement on the NCCN website.
The National Comprehensive Cancer Network has published a new guideline on smoking cessation for cancer patients that recommends combining pharmacologic therapy with counseling as the most effective approach, along with rigorous review and close follow-ups to prevent relapses.
“Although the medical community recognizes the importance of smoking cessation, supporting patients in ceasing to smoke is generally not done well. Our hope is that by addressing smoking cessation in a cancer patient population, we can make it easier for oncologists to effectively support their patients in achieving their smoking cessation goals,” Dr. Peter Shields, deputy director of the Ohio State University Comprehensive Cancer Center, said in a written statement. Of the estimated 590,000 cancer deaths in 2015, about 170,000, or nearly 30%, will be caused by tobacco smoking. Quitting tobacco improves cancer treatment effectiveness and reduces cancer recurrence, according to the NCCN.
Read the full statement on the NCCN website.
The National Comprehensive Cancer Network has published a new guideline on smoking cessation for cancer patients that recommends combining pharmacologic therapy with counseling as the most effective approach, along with rigorous review and close follow-ups to prevent relapses.
“Although the medical community recognizes the importance of smoking cessation, supporting patients in ceasing to smoke is generally not done well. Our hope is that by addressing smoking cessation in a cancer patient population, we can make it easier for oncologists to effectively support their patients in achieving their smoking cessation goals,” Dr. Peter Shields, deputy director of the Ohio State University Comprehensive Cancer Center, said in a written statement. Of the estimated 590,000 cancer deaths in 2015, about 170,000, or nearly 30%, will be caused by tobacco smoking. Quitting tobacco improves cancer treatment effectiveness and reduces cancer recurrence, according to the NCCN.
Read the full statement on the NCCN website.
Stem cells from sickle cell disease patients used to generate gene-corrected cells
Cells from patients with sickle cell disease have been used to generate mature red blood cells without the genetic mutation that causes the disease, investigators at Johns Hopkins University reported.
This advance may lead to improved treatment of sickle cell disease patients, who frequently need blood transfusions from healthy donors but often reject foreign blood.
The corrected cells were created through genome editing of human induced pluripotent stem cells (iPSCs), which can make any cell in the body and grow indefinitely in the laboratory.
“Our results represent a significant step towards the clinical applications of genome editing using patient-derived iPSCs to generate disease-free cells for cell and gene therapy,” according to the study’s lead researcher, Xiaosong Huang, and his associates.
The edited iPSCs, which were derived from the patients’ blood cells, produced blood cells as “efficiently” as stem cells that had not been edited, according to a written statement from Johns Hopkins. Still, “to become medically useful, the technique … will have to be made even more efficient and scaled up significantly,” study investigator Linzhao Cheng, Ph.D., noted in the statement.
Find the full study in Stem Cells (2015 Feb. 20 [doi:10.1002/stem.1969]).
Cells from patients with sickle cell disease have been used to generate mature red blood cells without the genetic mutation that causes the disease, investigators at Johns Hopkins University reported.
This advance may lead to improved treatment of sickle cell disease patients, who frequently need blood transfusions from healthy donors but often reject foreign blood.
The corrected cells were created through genome editing of human induced pluripotent stem cells (iPSCs), which can make any cell in the body and grow indefinitely in the laboratory.
“Our results represent a significant step towards the clinical applications of genome editing using patient-derived iPSCs to generate disease-free cells for cell and gene therapy,” according to the study’s lead researcher, Xiaosong Huang, and his associates.
The edited iPSCs, which were derived from the patients’ blood cells, produced blood cells as “efficiently” as stem cells that had not been edited, according to a written statement from Johns Hopkins. Still, “to become medically useful, the technique … will have to be made even more efficient and scaled up significantly,” study investigator Linzhao Cheng, Ph.D., noted in the statement.
Find the full study in Stem Cells (2015 Feb. 20 [doi:10.1002/stem.1969]).
Cells from patients with sickle cell disease have been used to generate mature red blood cells without the genetic mutation that causes the disease, investigators at Johns Hopkins University reported.
This advance may lead to improved treatment of sickle cell disease patients, who frequently need blood transfusions from healthy donors but often reject foreign blood.
The corrected cells were created through genome editing of human induced pluripotent stem cells (iPSCs), which can make any cell in the body and grow indefinitely in the laboratory.
“Our results represent a significant step towards the clinical applications of genome editing using patient-derived iPSCs to generate disease-free cells for cell and gene therapy,” according to the study’s lead researcher, Xiaosong Huang, and his associates.
The edited iPSCs, which were derived from the patients’ blood cells, produced blood cells as “efficiently” as stem cells that had not been edited, according to a written statement from Johns Hopkins. Still, “to become medically useful, the technique … will have to be made even more efficient and scaled up significantly,” study investigator Linzhao Cheng, Ph.D., noted in the statement.
Find the full study in Stem Cells (2015 Feb. 20 [doi:10.1002/stem.1969]).
Immunotoxin could treat B-cell malignancies, team says

Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Risks and benefits of long-term ticagrelor

Photo courtesy of AstraZeneca
SAN DIEGO—New research suggests that adding the antiplatelet drug ticagrelor to aspirin as long-term therapy after a heart attack can significantly reduce the risk of cardiovascular death, heart attack, or stroke, but it can also increase the risk of major bleeding.
Marc S. Sabatine, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, presented these results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 400-18). The findings were published in NEJM as well.
The research was sponsored by AstraZeneca, the company developing ticagrelor.
The trial, known as PEGASUS-TIMI 54, included 21,162 patients who had experienced a heart attack in the previous 1 to 3 years. Each had another factor, such as age or diabetes, that put them at risk for a second heart attack.
The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
The twice-daily 90 mg dose of ticagrelor is already approved for patients with acute coronary syndrome. The researchers included a lower dose in this study to investigate whether platelet inhibition needed 2 years after a heart attack might be different from what is needed 2 hours after a heart attack.
The team found that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke. At 3 years, the rate was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
“The benefit we saw was remarkably consistent across the individual components of the endpoint and in all the major subgroups of patients,” Dr Sabatine said. “Moreover, we followed patients for an average of just under 3 years, and our event curves continue to spread out over time, suggesting that the benefit continues to accrue over time.”
“Efficacy was virtually identical with both ticagrelor doses,” he added. “Risk of bleeding and dyspnea tended to be, as predicted, a bit more with the 90 mg than the 60 mg dose, but the trial wasn’t designed to compare those two dose levels.”
The rates of TIMI major bleeding were 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea were 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea leading to treatment discontinuation were 6.5% in the 90 mg group, 4.55% in the 60 mg group, and 0.79% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
Although the differences in dyspnea and TIMI major bleeding between the 2 ticagrelor dose groups were not statistically significant, Dr Sabatine and his colleagues said the 60 mg dose may offer a more attractive benefit-risk profile.
“Now that we have the evidence, when faced with a patient who has had a heart attack, based on these data, I would continue treatment with ticagrelor as long as the patient tolerated it,” Dr Sabatine concluded. ![]()
Photo courtesy of AstraZeneca
SAN DIEGO—New research suggests that adding the antiplatelet drug ticagrelor to aspirin as long-term therapy after a heart attack can significantly reduce the risk of cardiovascular death, heart attack, or stroke, but it can also increase the risk of major bleeding.
Marc S. Sabatine, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, presented these results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 400-18). The findings were published in NEJM as well.
The research was sponsored by AstraZeneca, the company developing ticagrelor.
The trial, known as PEGASUS-TIMI 54, included 21,162 patients who had experienced a heart attack in the previous 1 to 3 years. Each had another factor, such as age or diabetes, that put them at risk for a second heart attack.
The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
The twice-daily 90 mg dose of ticagrelor is already approved for patients with acute coronary syndrome. The researchers included a lower dose in this study to investigate whether platelet inhibition needed 2 years after a heart attack might be different from what is needed 2 hours after a heart attack.
The team found that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke. At 3 years, the rate was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
“The benefit we saw was remarkably consistent across the individual components of the endpoint and in all the major subgroups of patients,” Dr Sabatine said. “Moreover, we followed patients for an average of just under 3 years, and our event curves continue to spread out over time, suggesting that the benefit continues to accrue over time.”
“Efficacy was virtually identical with both ticagrelor doses,” he added. “Risk of bleeding and dyspnea tended to be, as predicted, a bit more with the 90 mg than the 60 mg dose, but the trial wasn’t designed to compare those two dose levels.”
The rates of TIMI major bleeding were 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea were 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea leading to treatment discontinuation were 6.5% in the 90 mg group, 4.55% in the 60 mg group, and 0.79% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
Although the differences in dyspnea and TIMI major bleeding between the 2 ticagrelor dose groups were not statistically significant, Dr Sabatine and his colleagues said the 60 mg dose may offer a more attractive benefit-risk profile.
“Now that we have the evidence, when faced with a patient who has had a heart attack, based on these data, I would continue treatment with ticagrelor as long as the patient tolerated it,” Dr Sabatine concluded. ![]()
Photo courtesy of AstraZeneca
SAN DIEGO—New research suggests that adding the antiplatelet drug ticagrelor to aspirin as long-term therapy after a heart attack can significantly reduce the risk of cardiovascular death, heart attack, or stroke, but it can also increase the risk of major bleeding.
Marc S. Sabatine, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, presented these results at the American College of Cardiology’s 64th Annual Scientific Session (abstract 400-18). The findings were published in NEJM as well.
The research was sponsored by AstraZeneca, the company developing ticagrelor.
The trial, known as PEGASUS-TIMI 54, included 21,162 patients who had experienced a heart attack in the previous 1 to 3 years. Each had another factor, such as age or diabetes, that put them at risk for a second heart attack.
The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
The twice-daily 90 mg dose of ticagrelor is already approved for patients with acute coronary syndrome. The researchers included a lower dose in this study to investigate whether platelet inhibition needed 2 years after a heart attack might be different from what is needed 2 hours after a heart attack.
The team found that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke. At 3 years, the rate was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
“The benefit we saw was remarkably consistent across the individual components of the endpoint and in all the major subgroups of patients,” Dr Sabatine said. “Moreover, we followed patients for an average of just under 3 years, and our event curves continue to spread out over time, suggesting that the benefit continues to accrue over time.”
“Efficacy was virtually identical with both ticagrelor doses,” he added. “Risk of bleeding and dyspnea tended to be, as predicted, a bit more with the 90 mg than the 60 mg dose, but the trial wasn’t designed to compare those two dose levels.”
The rates of TIMI major bleeding were 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea were 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rates of dyspnea leading to treatment discontinuation were 6.5% in the 90 mg group, 4.55% in the 60 mg group, and 0.79% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
Although the differences in dyspnea and TIMI major bleeding between the 2 ticagrelor dose groups were not statistically significant, Dr Sabatine and his colleagues said the 60 mg dose may offer a more attractive benefit-risk profile.
“Now that we have the evidence, when faced with a patient who has had a heart attack, based on these data, I would continue treatment with ticagrelor as long as the patient tolerated it,” Dr Sabatine concluded. ![]()