User login
Brentuximab doubles PFS in Hodgkin’s lymphoma patients
Brentuximab vedotin (Adcetris) increased progression-free survival to 43 months when given to adults with hard-to-treat Hodgkin’s lymphoma immediately after stem cell transplant, compared to 24 months for placebo, according to research published online March 18 in The Lancet.
As part of the AETHERA phase III trial, 329 patients with Hodgkin’s lymphoma who were at high risk of relapse or progression after autologous stem cell transplant were given brentuximab vedotin infusions or placebo every 3 weeks for up to 16 cycles. After a 2-year follow-up, the cancer had not progressed in 65% of the patients in the treatment group, compared with 45% in the placebo group.
The most common side effects were peripheral neuropathy (67% vs. 13% placebo) and neutropenia (35% vs. 12% placebo), noted Dr. Craig Moskowitz of Memorial Sloan Kettering Cancer Center, New York, and his associates.
Read the full article here.
Brentuximab vedotin (Adcetris) increased progression-free survival to 43 months when given to adults with hard-to-treat Hodgkin’s lymphoma immediately after stem cell transplant, compared to 24 months for placebo, according to research published online March 18 in The Lancet.
As part of the AETHERA phase III trial, 329 patients with Hodgkin’s lymphoma who were at high risk of relapse or progression after autologous stem cell transplant were given brentuximab vedotin infusions or placebo every 3 weeks for up to 16 cycles. After a 2-year follow-up, the cancer had not progressed in 65% of the patients in the treatment group, compared with 45% in the placebo group.
The most common side effects were peripheral neuropathy (67% vs. 13% placebo) and neutropenia (35% vs. 12% placebo), noted Dr. Craig Moskowitz of Memorial Sloan Kettering Cancer Center, New York, and his associates.
Read the full article here.
Brentuximab vedotin (Adcetris) increased progression-free survival to 43 months when given to adults with hard-to-treat Hodgkin’s lymphoma immediately after stem cell transplant, compared to 24 months for placebo, according to research published online March 18 in The Lancet.
As part of the AETHERA phase III trial, 329 patients with Hodgkin’s lymphoma who were at high risk of relapse or progression after autologous stem cell transplant were given brentuximab vedotin infusions or placebo every 3 weeks for up to 16 cycles. After a 2-year follow-up, the cancer had not progressed in 65% of the patients in the treatment group, compared with 45% in the placebo group.
The most common side effects were peripheral neuropathy (67% vs. 13% placebo) and neutropenia (35% vs. 12% placebo), noted Dr. Craig Moskowitz of Memorial Sloan Kettering Cancer Center, New York, and his associates.
Read the full article here.
ACP: Avoid ECG, MPI cardiac screening in low-risk patients
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.

The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.
The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
Clinicians should not screen for cardiac disease in asymptomatic, low-risk adults using resting or stress electrocardiography, stress echocardiography, or stress myocardial perfusion imaging , according to new guidelines from the American College of Physicians.
“There is no evidence that cardiac screening of low-risk adults with resting or stress ECG, stress echocardiography, or stress MPI improves outcomes, but it is associated with increased costs and potential harms,” wrote the guideline’s author, Dr. Roger Chou, associate professor of medicine at Oregon Health & Science University, Portland.
The recommendation is based on a systematic literature review, recommendations from the U.S. Preventive Services Task Force, and American College of Cardiology guidelines. The new ACP clinical guideline was published March 17 in Annals of Internal Medicine (doi: 10.7326/M14-1225).
“What we are saying here is that, as physicians, we have responsibility to understand what the pretest probability is, and what the likelihood is that someone actually has disease – and if it’s low enough, then doing the screening test is going to cause a lot more false positives than true positives,” Dr. Robert Centor, regional dean of the Huntsville Medical Campus of the University of Alabama at Birmingham, explained in an interview.
“Even if it is a true positive, there is no evidence that we can find that finding that heart disease will do anything other than lead someone to do a procedure that we have no evidence will improve their outcomes,” added Dr. Centor, chair of the ACP Board of Regents.
Despite existing recommendations to the contrary, physicians are increasingly performing these tests on low-risk patients, the ACP cautioned.
For example, a Consumer Reports survey found that “39% of asymptomatic adults without high blood pressure or a high cholesterol level reported having ECG within the past 5 years, and 12% reported undergoing exercise ECG,” Dr. Chou wrote in his report on behalf of the ACP High Value Care Task Force. More than half of those patients said their physicians recommended the tests as part of their routine health care.
The rise in the use of such tests is likely the result of a combination of factors, Dr. Centor said. Those factors include money (patients see no out-of-pocket cost and thus don’t consider the cost of tests in their decision making), direct-to-consumer advertising, fear on behalf of physicians that they might miss a diagnosis, and a lack of understanding by patients on the adverse effects of screening if they are at low risk for heart disease.
Dr. Chou identified a number of potential harms related to unnecessary screenings, including sudden death or hospitalization during stress tests; adverse events from pharmacologics used to induce stress; radiation exposure from myocardial perfusion imaging; false positive results that, in turn, lead to anxiety by the patient and additional unnecessary tests and treatments; disease labeling; and downstream harms from follow-up testing and interventions.
“To be most effective, efforts to reduce the use of imaging should be multifocal and should address clinician behavior, patient expectations, direct-to-consumer screening programs, and financial incentives,” Dr. Chou explained.
In low-risk patients, physicians instead should “focus on treating modifiable risk factors (such as smoking, diabetes, hypertension, hyperlipidemia, and overweight) and encouraging healthy levels of exercise,” according to the guideline.
FROM ANNALS OF INTERNAL MEDICINE
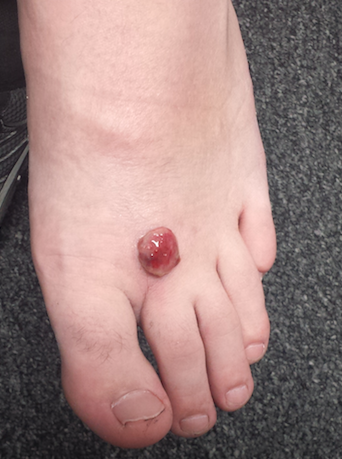
Attempt at “Wart” Removal Backfires
Two months ago, a tender lesion manifested on the dorsum of a 14-year-old boy’s right foot. Before it appeared, there was a tiny papule in the same spot; the boy’s mother says they thought it was a wart and attempted to remove it. As a result, the papule bled and became inflamed before quickly growing to its present form.
Besides being tender to touch, the lesion bleeds with minimal trauma. Attempts to remove it—using silver nitrate, liquid nitrogen, and triple-antibiotic cream—have all failed to have any positive impact.
The patient is reportedly otherwise healthy. He takes no medications of any kind.
EXAMINATION
The lesion is a 2-cm, domelike, red shiny nodule located on the mid-dorsum of the right foot. It is attached to the underlying skin by a thick sessile base. There is no surrounding erythema or other skin changes.
The site is anesthetized with local infiltrate of 1% lidocaine with epinephrine before deep shave biopsy is used to remove the lesion. The base is curetted, then cauterized for hemostasis.
PATHOLOGY
Microscopic examination shows a tangle of capillaries arranged in lobules separated by septae of connective tissue. A brisk inflammatory reaction and faint erosions are seen on the lesion’s surface.
What is the diagnosis?
DISCUSSION
First described by Ponce and Dor in 1897, pyogenic granuloma (PG) was thought to represent a kind of infection but turned out to be neither infectious nor granulomatous. It is now understood to represent the body’s attempt to heal a wound or other trauma (albeit this process goes awry for unknown reasons).
PG has a range of morphologic presentations; this case, with a berry-like friable papule, is fairly typical. PG is common on fingertips, lips, and faces (in children) and can even appear on gingival mucosae (particularly in first-trimester pregnancy). Other common areas of manifestation include ingrown toenails (lesion forms where the distal edge of the nail cuts into the lateral aspect of the nail bed) and the umbilical stump of newborns. An extremely common scenario is the child who cannot leave a wart or skin tag alone, picking and squeezing the lesion repeatedly until a PG forms.
PG is reactive in nature and not neoplastic. It is essentially vascular (other names for it include lobular capillary hemangioma and eruptive hemangioma), and the amount of blood that can flow from these lesions tends to frighten patients and parents.
While PG is completely benign, it is part of a differential of “look-alikes” that include nodular melanoma and metastatic cancer. For this reason, once removed, PGs are always sent for pathologic examination.
Removal should be done by deep shave, followed by electrodessication and curettage to prevent recurrence. Cryosurgery or silver nitrate application, though frequently done, is seldom effective. Significantly, neither yields a definitive diagnosis.
Certain drugs are associated with PG formation. These include isotretinoin, some chemotherapy drugs, and retroviral medications.
TAKE-HOME LEARNING POINTS
• Pyogenic granuloma (PG) is neither pyogenic nor granulomatous and is instead a reactive process.
• PG represents the body’s incomplete attempt to heal a wound.
• The trauma is often repetitive, particularly in PG associated with ingrown toenails or digitally manipulated by the patient.
• A berry-like appearance, sudden growth, and friability are all characteristic of PG.
• PG, though extremely common, needs to be removed to provide relief and to establish the lesion’s benign nature by pathologic examination.
Two months ago, a tender lesion manifested on the dorsum of a 14-year-old boy’s right foot. Before it appeared, there was a tiny papule in the same spot; the boy’s mother says they thought it was a wart and attempted to remove it. As a result, the papule bled and became inflamed before quickly growing to its present form.
Besides being tender to touch, the lesion bleeds with minimal trauma. Attempts to remove it—using silver nitrate, liquid nitrogen, and triple-antibiotic cream—have all failed to have any positive impact.
The patient is reportedly otherwise healthy. He takes no medications of any kind.
EXAMINATION
The lesion is a 2-cm, domelike, red shiny nodule located on the mid-dorsum of the right foot. It is attached to the underlying skin by a thick sessile base. There is no surrounding erythema or other skin changes.
The site is anesthetized with local infiltrate of 1% lidocaine with epinephrine before deep shave biopsy is used to remove the lesion. The base is curetted, then cauterized for hemostasis.
PATHOLOGY
Microscopic examination shows a tangle of capillaries arranged in lobules separated by septae of connective tissue. A brisk inflammatory reaction and faint erosions are seen on the lesion’s surface.
What is the diagnosis?
DISCUSSION
First described by Ponce and Dor in 1897, pyogenic granuloma (PG) was thought to represent a kind of infection but turned out to be neither infectious nor granulomatous. It is now understood to represent the body’s attempt to heal a wound or other trauma (albeit this process goes awry for unknown reasons).
PG has a range of morphologic presentations; this case, with a berry-like friable papule, is fairly typical. PG is common on fingertips, lips, and faces (in children) and can even appear on gingival mucosae (particularly in first-trimester pregnancy). Other common areas of manifestation include ingrown toenails (lesion forms where the distal edge of the nail cuts into the lateral aspect of the nail bed) and the umbilical stump of newborns. An extremely common scenario is the child who cannot leave a wart or skin tag alone, picking and squeezing the lesion repeatedly until a PG forms.
PG is reactive in nature and not neoplastic. It is essentially vascular (other names for it include lobular capillary hemangioma and eruptive hemangioma), and the amount of blood that can flow from these lesions tends to frighten patients and parents.
While PG is completely benign, it is part of a differential of “look-alikes” that include nodular melanoma and metastatic cancer. For this reason, once removed, PGs are always sent for pathologic examination.
Removal should be done by deep shave, followed by electrodessication and curettage to prevent recurrence. Cryosurgery or silver nitrate application, though frequently done, is seldom effective. Significantly, neither yields a definitive diagnosis.
Certain drugs are associated with PG formation. These include isotretinoin, some chemotherapy drugs, and retroviral medications.
TAKE-HOME LEARNING POINTS
• Pyogenic granuloma (PG) is neither pyogenic nor granulomatous and is instead a reactive process.
• PG represents the body’s incomplete attempt to heal a wound.
• The trauma is often repetitive, particularly in PG associated with ingrown toenails or digitally manipulated by the patient.
• A berry-like appearance, sudden growth, and friability are all characteristic of PG.
• PG, though extremely common, needs to be removed to provide relief and to establish the lesion’s benign nature by pathologic examination.
Two months ago, a tender lesion manifested on the dorsum of a 14-year-old boy’s right foot. Before it appeared, there was a tiny papule in the same spot; the boy’s mother says they thought it was a wart and attempted to remove it. As a result, the papule bled and became inflamed before quickly growing to its present form.
Besides being tender to touch, the lesion bleeds with minimal trauma. Attempts to remove it—using silver nitrate, liquid nitrogen, and triple-antibiotic cream—have all failed to have any positive impact.
The patient is reportedly otherwise healthy. He takes no medications of any kind.
EXAMINATION
The lesion is a 2-cm, domelike, red shiny nodule located on the mid-dorsum of the right foot. It is attached to the underlying skin by a thick sessile base. There is no surrounding erythema or other skin changes.
The site is anesthetized with local infiltrate of 1% lidocaine with epinephrine before deep shave biopsy is used to remove the lesion. The base is curetted, then cauterized for hemostasis.
PATHOLOGY
Microscopic examination shows a tangle of capillaries arranged in lobules separated by septae of connective tissue. A brisk inflammatory reaction and faint erosions are seen on the lesion’s surface.
What is the diagnosis?
DISCUSSION
First described by Ponce and Dor in 1897, pyogenic granuloma (PG) was thought to represent a kind of infection but turned out to be neither infectious nor granulomatous. It is now understood to represent the body’s attempt to heal a wound or other trauma (albeit this process goes awry for unknown reasons).
PG has a range of morphologic presentations; this case, with a berry-like friable papule, is fairly typical. PG is common on fingertips, lips, and faces (in children) and can even appear on gingival mucosae (particularly in first-trimester pregnancy). Other common areas of manifestation include ingrown toenails (lesion forms where the distal edge of the nail cuts into the lateral aspect of the nail bed) and the umbilical stump of newborns. An extremely common scenario is the child who cannot leave a wart or skin tag alone, picking and squeezing the lesion repeatedly until a PG forms.
PG is reactive in nature and not neoplastic. It is essentially vascular (other names for it include lobular capillary hemangioma and eruptive hemangioma), and the amount of blood that can flow from these lesions tends to frighten patients and parents.
While PG is completely benign, it is part of a differential of “look-alikes” that include nodular melanoma and metastatic cancer. For this reason, once removed, PGs are always sent for pathologic examination.
Removal should be done by deep shave, followed by electrodessication and curettage to prevent recurrence. Cryosurgery or silver nitrate application, though frequently done, is seldom effective. Significantly, neither yields a definitive diagnosis.
Certain drugs are associated with PG formation. These include isotretinoin, some chemotherapy drugs, and retroviral medications.
TAKE-HOME LEARNING POINTS
• Pyogenic granuloma (PG) is neither pyogenic nor granulomatous and is instead a reactive process.
• PG represents the body’s incomplete attempt to heal a wound.
• The trauma is often repetitive, particularly in PG associated with ingrown toenails or digitally manipulated by the patient.
• A berry-like appearance, sudden growth, and friability are all characteristic of PG.
• PG, though extremely common, needs to be removed to provide relief and to establish the lesion’s benign nature by pathologic examination.
FDA guidance focuses on infections with reusable devices, including duodenoscopes
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
Keeping your religious belief outside the office
Religion isn’t an uncommon topic in a doctor’s office, and mine is no exception. Patients often express their personal beliefs in difficult situations, and part of my job is to listen and support.
But when they ask me about my own, I don’t answer. I simply tell them that I don’t discuss such things with patients.
People can have pretty strong feelings about religion, and whether I agree or disagree with them doesn’t have a place in my office. Religion, like politics, opens a can of personal opinion worms that disrupts the doctor-patient relationship. It can make things unworkable.
The last thing I want, or need, during an appointment is a debate over evolution, the perennial Middle East crisis, or belief (or lack thereof) in a deity. There are plenty of good forums to argue such subjects, but my office isn’t one of them.
On rare occasions, someone calling for an appointment will ask about my religious orientation. My secretary has been told to say “I don’t know.” If that matters to you when you’re looking for a doctor, you’re probably better off going elsewhere.
I have nothing against social pleasantries. They’re part of the ordinary patter in my office, and help maintain a degree of doctor-patient comfort to let us talk openly. But religious beliefs are a topic that, with some people, can rapidly spiral out of control. On the rare occasions where they become acrimonious, it pretty much destroys the fabric of the professional relationship. So, my belief is that it’s best not to start in the first place.
Some find religion to be an important part of who they are, and I’m willing to listen to that and not be judgmental. But don’t expect me to share my own thoughts at an appointment. It’s not why either of us is there.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Religion isn’t an uncommon topic in a doctor’s office, and mine is no exception. Patients often express their personal beliefs in difficult situations, and part of my job is to listen and support.
But when they ask me about my own, I don’t answer. I simply tell them that I don’t discuss such things with patients.
People can have pretty strong feelings about religion, and whether I agree or disagree with them doesn’t have a place in my office. Religion, like politics, opens a can of personal opinion worms that disrupts the doctor-patient relationship. It can make things unworkable.
The last thing I want, or need, during an appointment is a debate over evolution, the perennial Middle East crisis, or belief (or lack thereof) in a deity. There are plenty of good forums to argue such subjects, but my office isn’t one of them.
On rare occasions, someone calling for an appointment will ask about my religious orientation. My secretary has been told to say “I don’t know.” If that matters to you when you’re looking for a doctor, you’re probably better off going elsewhere.
I have nothing against social pleasantries. They’re part of the ordinary patter in my office, and help maintain a degree of doctor-patient comfort to let us talk openly. But religious beliefs are a topic that, with some people, can rapidly spiral out of control. On the rare occasions where they become acrimonious, it pretty much destroys the fabric of the professional relationship. So, my belief is that it’s best not to start in the first place.
Some find religion to be an important part of who they are, and I’m willing to listen to that and not be judgmental. But don’t expect me to share my own thoughts at an appointment. It’s not why either of us is there.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Religion isn’t an uncommon topic in a doctor’s office, and mine is no exception. Patients often express their personal beliefs in difficult situations, and part of my job is to listen and support.
But when they ask me about my own, I don’t answer. I simply tell them that I don’t discuss such things with patients.
People can have pretty strong feelings about religion, and whether I agree or disagree with them doesn’t have a place in my office. Religion, like politics, opens a can of personal opinion worms that disrupts the doctor-patient relationship. It can make things unworkable.
The last thing I want, or need, during an appointment is a debate over evolution, the perennial Middle East crisis, or belief (or lack thereof) in a deity. There are plenty of good forums to argue such subjects, but my office isn’t one of them.
On rare occasions, someone calling for an appointment will ask about my religious orientation. My secretary has been told to say “I don’t know.” If that matters to you when you’re looking for a doctor, you’re probably better off going elsewhere.
I have nothing against social pleasantries. They’re part of the ordinary patter in my office, and help maintain a degree of doctor-patient comfort to let us talk openly. But religious beliefs are a topic that, with some people, can rapidly spiral out of control. On the rare occasions where they become acrimonious, it pretty much destroys the fabric of the professional relationship. So, my belief is that it’s best not to start in the first place.
Some find religion to be an important part of who they are, and I’m willing to listen to that and not be judgmental. But don’t expect me to share my own thoughts at an appointment. It’s not why either of us is there.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.

Storage time doesn’t affect blood quality, study shows

Photo by Elise Amendola
The length of time red blood cells (RBCs) are stored does not affect transfusion outcomes in critically ill patients, results of the ABLE study suggest.
Researchers compared critically ill patients who received RBCs stored for an average of about 3 weeks to those who received RBCs stored for less than a week.
And there were no significant differences between the groups with regard to mortality, transfusion reactions, and other outcomes.
“Previous observational and laboratory studies have suggested that fresh blood may be better because of the breakdown of red blood cells and accumulation of toxins during storage,” said Alan Tinmouth, MD, of the University of Ottawa in Ontario, Canada.
“But this definitive clinical trial clearly shows that these changes do not affect the quality of blood.”
Dr Tinmouth and his colleagues reported these results in NEJM.
The researchers enrolled 2430 adult intensive care patients in the trial, comparing patients who received fresh RBCs (n=1211) to those who received older RBCs (n=1219). Fresh RBCs were stored for a median of 6.1±4.9 days, and older RBCs were stored for a median of 22.0±8.4 days.
There was no significant difference between the arms with regard to the primary outcome, 90-day mortality. This endpoint was met by 37% of patients who received fresh blood and 35.3% of those who received older blood.
There were no significant differences between the fresh and older RBC arms for other mortality outcomes, either. This included death in the intensive care unit (26.7% vs 24.2%), in-hospital death (33.3% vs 31.9%), and death by day 28 (30.6% vs 28.8%).
Similarly, there were no significant differences between the fresh and older blood arms with regard to major illnesses, including multiple organ dysfunction syndrome (13.4% vs 13%), acute respiratory distress syndrome (5.7% vs 6.6%), cardiovascular failure (5.1% vs 4.2%), cardiac ischemia or infarction (4.5% vs 3.6%), venous thromboembolism (3.6% for both), nosocomial infection (34.1% vs 31.3%), and acute transfusion reaction (0.3% vs 0.5%).
In addition, there was no significant difference between the fresh RBC arm and the older RBC arm in the length of time patients required mechanical ventilation (15.0±18.0 days vs 14.7±14.9 days), cardiac or vasoactive drugs (7.1±10.2 days vs 7.5±11.2 days), or extrarenal epuration (2.5±10.1 days vs 2.5±8.3 days).
And there was no significant difference between the fresh and older blood arms in patients’ length of stay in the hospital (34.4±39.5 days vs 33.9±38.8 days) or the intensive care unit (15.3±15.4 days vs 15.3±14.8 days).
Based on these results, the researchers said there is no need to worry about the age of blood routinely used in hospitals. The team is now conducting a trial to determine if the same can be said for transfusions in pediatric patients. ![]()
Photo by Elise Amendola
The length of time red blood cells (RBCs) are stored does not affect transfusion outcomes in critically ill patients, results of the ABLE study suggest.
Researchers compared critically ill patients who received RBCs stored for an average of about 3 weeks to those who received RBCs stored for less than a week.
And there were no significant differences between the groups with regard to mortality, transfusion reactions, and other outcomes.
“Previous observational and laboratory studies have suggested that fresh blood may be better because of the breakdown of red blood cells and accumulation of toxins during storage,” said Alan Tinmouth, MD, of the University of Ottawa in Ontario, Canada.
“But this definitive clinical trial clearly shows that these changes do not affect the quality of blood.”
Dr Tinmouth and his colleagues reported these results in NEJM.
The researchers enrolled 2430 adult intensive care patients in the trial, comparing patients who received fresh RBCs (n=1211) to those who received older RBCs (n=1219). Fresh RBCs were stored for a median of 6.1±4.9 days, and older RBCs were stored for a median of 22.0±8.4 days.
There was no significant difference between the arms with regard to the primary outcome, 90-day mortality. This endpoint was met by 37% of patients who received fresh blood and 35.3% of those who received older blood.
There were no significant differences between the fresh and older RBC arms for other mortality outcomes, either. This included death in the intensive care unit (26.7% vs 24.2%), in-hospital death (33.3% vs 31.9%), and death by day 28 (30.6% vs 28.8%).
Similarly, there were no significant differences between the fresh and older blood arms with regard to major illnesses, including multiple organ dysfunction syndrome (13.4% vs 13%), acute respiratory distress syndrome (5.7% vs 6.6%), cardiovascular failure (5.1% vs 4.2%), cardiac ischemia or infarction (4.5% vs 3.6%), venous thromboembolism (3.6% for both), nosocomial infection (34.1% vs 31.3%), and acute transfusion reaction (0.3% vs 0.5%).
In addition, there was no significant difference between the fresh RBC arm and the older RBC arm in the length of time patients required mechanical ventilation (15.0±18.0 days vs 14.7±14.9 days), cardiac or vasoactive drugs (7.1±10.2 days vs 7.5±11.2 days), or extrarenal epuration (2.5±10.1 days vs 2.5±8.3 days).
And there was no significant difference between the fresh and older blood arms in patients’ length of stay in the hospital (34.4±39.5 days vs 33.9±38.8 days) or the intensive care unit (15.3±15.4 days vs 15.3±14.8 days).
Based on these results, the researchers said there is no need to worry about the age of blood routinely used in hospitals. The team is now conducting a trial to determine if the same can be said for transfusions in pediatric patients. ![]()
Photo by Elise Amendola
The length of time red blood cells (RBCs) are stored does not affect transfusion outcomes in critically ill patients, results of the ABLE study suggest.
Researchers compared critically ill patients who received RBCs stored for an average of about 3 weeks to those who received RBCs stored for less than a week.
And there were no significant differences between the groups with regard to mortality, transfusion reactions, and other outcomes.
“Previous observational and laboratory studies have suggested that fresh blood may be better because of the breakdown of red blood cells and accumulation of toxins during storage,” said Alan Tinmouth, MD, of the University of Ottawa in Ontario, Canada.
“But this definitive clinical trial clearly shows that these changes do not affect the quality of blood.”
Dr Tinmouth and his colleagues reported these results in NEJM.
The researchers enrolled 2430 adult intensive care patients in the trial, comparing patients who received fresh RBCs (n=1211) to those who received older RBCs (n=1219). Fresh RBCs were stored for a median of 6.1±4.9 days, and older RBCs were stored for a median of 22.0±8.4 days.
There was no significant difference between the arms with regard to the primary outcome, 90-day mortality. This endpoint was met by 37% of patients who received fresh blood and 35.3% of those who received older blood.
There were no significant differences between the fresh and older RBC arms for other mortality outcomes, either. This included death in the intensive care unit (26.7% vs 24.2%), in-hospital death (33.3% vs 31.9%), and death by day 28 (30.6% vs 28.8%).
Similarly, there were no significant differences between the fresh and older blood arms with regard to major illnesses, including multiple organ dysfunction syndrome (13.4% vs 13%), acute respiratory distress syndrome (5.7% vs 6.6%), cardiovascular failure (5.1% vs 4.2%), cardiac ischemia or infarction (4.5% vs 3.6%), venous thromboembolism (3.6% for both), nosocomial infection (34.1% vs 31.3%), and acute transfusion reaction (0.3% vs 0.5%).
In addition, there was no significant difference between the fresh RBC arm and the older RBC arm in the length of time patients required mechanical ventilation (15.0±18.0 days vs 14.7±14.9 days), cardiac or vasoactive drugs (7.1±10.2 days vs 7.5±11.2 days), or extrarenal epuration (2.5±10.1 days vs 2.5±8.3 days).
And there was no significant difference between the fresh and older blood arms in patients’ length of stay in the hospital (34.4±39.5 days vs 33.9±38.8 days) or the intensive care unit (15.3±15.4 days vs 15.3±14.8 days).
Based on these results, the researchers said there is no need to worry about the age of blood routinely used in hospitals. The team is now conducting a trial to determine if the same can be said for transfusions in pediatric patients. ![]()
Bivalirudin bests heparin in patients with AMI
Data from the BRIGHT trial suggest bivalirudin may be more suitable than heparin alone or heparin plus tirofiban as anticoagulant therapy for patients with acute myocardial infarction (AMI) who are undergoing percutaneous coronary intervention (PCI).
At 30 days and 1 year after PCI, patients who received bivalirudin had a lower rate of net adverse clinical events (NACE)—death, reinfarction, bleeding, and
other events—than patients who received heparin.
Complete results from this study were published in JAMA alongside a related editorial.
Research has yet to reveal the optimal anticoagulant strategy for patients with AMI. Previous multicenter trials, such as HORIZONS-AMI and EUROMAX, have suggested that bivalirudin is superior to heparin plus glycoprotein IIb/IIIa inhibitors. But a recent single-center trial, HEAT-PPCI, indicated that heparin monotherapy was superior to bivalirudin alone.
So Gregg W. Stone, MD, of Columbia University Medical Center in New York, New York, and his colleagues conducted the BRIGHT trial to gain some insight into the issue.
The team analyzed 2194 patients with AMI who underwent emergency PCI at 82 Chinese sites. The patients were randomized to receive bivalirudin with a post-PCI infusion (n=735), heparin alone (n=729), or heparin plus tirofiban with a post-PCI infusion (n=730).
The primary endpoint was 30-day NACE, a composite of major adverse cardiac and cerebral events (MACCE) and bleeding. The secondary endpoints were NACE at 1 year, as well as MACCE and bleeding at 30 days and 1 year.
MACCE includes all-cause death, reinfarction, ischemia-driven target vessel revascularization, and stroke. Bleeding was defined by the Bleeding Academic Research Consortium (BARC) definition.
At 30 days, NACE had occurred in 8.8% of bivalirudin-treated patients, 13.2% of heparin-treated patients, and 17.0% of patients who received heparin plus tirofiban. The relative risk (RR) for bivalirudin vs heparin was 0.67 (P=0.008), and the RR for bivalirudin vs heparin plus tirofiban was 0.52 (P<0.001).
Patients who received bivalirudin had a lower rate of bleeding at 30 days than patients who received heparin or heparin plus tirofiban—4.1%, 7.5%, and 12.3% respectively (P<0.001).
There were no significant differences between treatments in the 30-day rates of MACCE (5.0%, 5.8%, and 4.9% respectively, P=0.74) and stent thrombosis (0.6%, 0.9%, and 0.7%, respectively, P=0.77). And there was no significant difference in acute (<24 hour) stent thrombosis (0.3% in each group).
At 1 year, patients in the bivalirudin arm still had a lower rate of NACE compared to patients in the heparin arm (12.8% vs 16.5%, RR=0.78, P=0.048) or patients who received heparin plus tirofiban (12.8% vs 20.5%, RR=0.62, P<0.001), due to lower rates of bleeding.
Rates of MACCE and stent thrombosis at 1 year were not significantly different between the treatment arms.
“By reducing bleeding with comparable rates of MACCE and stent thrombosis, bivalirudin significantly improved overall 30-day and 1-year outcomes, compared with both heparin alone and heparin plus tirofiban in patients with AMI undergoing primary PCI,” Dr Stone concluded.
The BRIGHT trial was funded, in part, by Salubris Pharmaceutical Co., makers of bivalirudin. Other funding came from the General Hospital of Shenyang Military Region and the Chinese Government National Key R&D project for the 12th five-year plan. ![]()
Data from the BRIGHT trial suggest bivalirudin may be more suitable than heparin alone or heparin plus tirofiban as anticoagulant therapy for patients with acute myocardial infarction (AMI) who are undergoing percutaneous coronary intervention (PCI).
At 30 days and 1 year after PCI, patients who received bivalirudin had a lower rate of net adverse clinical events (NACE)—death, reinfarction, bleeding, and
other events—than patients who received heparin.
Complete results from this study were published in JAMA alongside a related editorial.
Research has yet to reveal the optimal anticoagulant strategy for patients with AMI. Previous multicenter trials, such as HORIZONS-AMI and EUROMAX, have suggested that bivalirudin is superior to heparin plus glycoprotein IIb/IIIa inhibitors. But a recent single-center trial, HEAT-PPCI, indicated that heparin monotherapy was superior to bivalirudin alone.
So Gregg W. Stone, MD, of Columbia University Medical Center in New York, New York, and his colleagues conducted the BRIGHT trial to gain some insight into the issue.
The team analyzed 2194 patients with AMI who underwent emergency PCI at 82 Chinese sites. The patients were randomized to receive bivalirudin with a post-PCI infusion (n=735), heparin alone (n=729), or heparin plus tirofiban with a post-PCI infusion (n=730).
The primary endpoint was 30-day NACE, a composite of major adverse cardiac and cerebral events (MACCE) and bleeding. The secondary endpoints were NACE at 1 year, as well as MACCE and bleeding at 30 days and 1 year.
MACCE includes all-cause death, reinfarction, ischemia-driven target vessel revascularization, and stroke. Bleeding was defined by the Bleeding Academic Research Consortium (BARC) definition.
At 30 days, NACE had occurred in 8.8% of bivalirudin-treated patients, 13.2% of heparin-treated patients, and 17.0% of patients who received heparin plus tirofiban. The relative risk (RR) for bivalirudin vs heparin was 0.67 (P=0.008), and the RR for bivalirudin vs heparin plus tirofiban was 0.52 (P<0.001).
Patients who received bivalirudin had a lower rate of bleeding at 30 days than patients who received heparin or heparin plus tirofiban—4.1%, 7.5%, and 12.3% respectively (P<0.001).
There were no significant differences between treatments in the 30-day rates of MACCE (5.0%, 5.8%, and 4.9% respectively, P=0.74) and stent thrombosis (0.6%, 0.9%, and 0.7%, respectively, P=0.77). And there was no significant difference in acute (<24 hour) stent thrombosis (0.3% in each group).
At 1 year, patients in the bivalirudin arm still had a lower rate of NACE compared to patients in the heparin arm (12.8% vs 16.5%, RR=0.78, P=0.048) or patients who received heparin plus tirofiban (12.8% vs 20.5%, RR=0.62, P<0.001), due to lower rates of bleeding.
Rates of MACCE and stent thrombosis at 1 year were not significantly different between the treatment arms.
“By reducing bleeding with comparable rates of MACCE and stent thrombosis, bivalirudin significantly improved overall 30-day and 1-year outcomes, compared with both heparin alone and heparin plus tirofiban in patients with AMI undergoing primary PCI,” Dr Stone concluded.
The BRIGHT trial was funded, in part, by Salubris Pharmaceutical Co., makers of bivalirudin. Other funding came from the General Hospital of Shenyang Military Region and the Chinese Government National Key R&D project for the 12th five-year plan. ![]()
Data from the BRIGHT trial suggest bivalirudin may be more suitable than heparin alone or heparin plus tirofiban as anticoagulant therapy for patients with acute myocardial infarction (AMI) who are undergoing percutaneous coronary intervention (PCI).
At 30 days and 1 year after PCI, patients who received bivalirudin had a lower rate of net adverse clinical events (NACE)—death, reinfarction, bleeding, and
other events—than patients who received heparin.
Complete results from this study were published in JAMA alongside a related editorial.
Research has yet to reveal the optimal anticoagulant strategy for patients with AMI. Previous multicenter trials, such as HORIZONS-AMI and EUROMAX, have suggested that bivalirudin is superior to heparin plus glycoprotein IIb/IIIa inhibitors. But a recent single-center trial, HEAT-PPCI, indicated that heparin monotherapy was superior to bivalirudin alone.
So Gregg W. Stone, MD, of Columbia University Medical Center in New York, New York, and his colleagues conducted the BRIGHT trial to gain some insight into the issue.
The team analyzed 2194 patients with AMI who underwent emergency PCI at 82 Chinese sites. The patients were randomized to receive bivalirudin with a post-PCI infusion (n=735), heparin alone (n=729), or heparin plus tirofiban with a post-PCI infusion (n=730).
The primary endpoint was 30-day NACE, a composite of major adverse cardiac and cerebral events (MACCE) and bleeding. The secondary endpoints were NACE at 1 year, as well as MACCE and bleeding at 30 days and 1 year.
MACCE includes all-cause death, reinfarction, ischemia-driven target vessel revascularization, and stroke. Bleeding was defined by the Bleeding Academic Research Consortium (BARC) definition.
At 30 days, NACE had occurred in 8.8% of bivalirudin-treated patients, 13.2% of heparin-treated patients, and 17.0% of patients who received heparin plus tirofiban. The relative risk (RR) for bivalirudin vs heparin was 0.67 (P=0.008), and the RR for bivalirudin vs heparin plus tirofiban was 0.52 (P<0.001).
Patients who received bivalirudin had a lower rate of bleeding at 30 days than patients who received heparin or heparin plus tirofiban—4.1%, 7.5%, and 12.3% respectively (P<0.001).
There were no significant differences between treatments in the 30-day rates of MACCE (5.0%, 5.8%, and 4.9% respectively, P=0.74) and stent thrombosis (0.6%, 0.9%, and 0.7%, respectively, P=0.77). And there was no significant difference in acute (<24 hour) stent thrombosis (0.3% in each group).
At 1 year, patients in the bivalirudin arm still had a lower rate of NACE compared to patients in the heparin arm (12.8% vs 16.5%, RR=0.78, P=0.048) or patients who received heparin plus tirofiban (12.8% vs 20.5%, RR=0.62, P<0.001), due to lower rates of bleeding.
Rates of MACCE and stent thrombosis at 1 year were not significantly different between the treatment arms.
“By reducing bleeding with comparable rates of MACCE and stent thrombosis, bivalirudin significantly improved overall 30-day and 1-year outcomes, compared with both heparin alone and heparin plus tirofiban in patients with AMI undergoing primary PCI,” Dr Stone concluded.
The BRIGHT trial was funded, in part, by Salubris Pharmaceutical Co., makers of bivalirudin. Other funding came from the General Hospital of Shenyang Military Region and the Chinese Government National Key R&D project for the 12th five-year plan. ![]()
EC approves drug for polycythemia vera

Image courtesy of AFIP
The European Commission (EC) has approved ruxolitinib (Jakavi) to treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
This is the first targeted treatment the EC has approved for these patients.
The approval applies to all 28 member states of the European Union (EU), plus Iceland, Norway, and Liechtenstein.
Ruxolitinib is already approved to treat PV in the US, and additional regulatory applications for ruxolitinib in PV are ongoing worldwide.
The drug is also approved to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF in more than 70 countries, including EU member states and the US.
“The European Commission’s approval of Jakavi [for PV] is encouraging news for patients,” said Claire Harrison, MD, a consultant hematologist at Guy’s and St Thomas’ NHS Foundation Trust in London, England.
“Jakavi will fill an unmet need as the first treatment shown to significantly improve hematocrit, as well as symptom control and reduce spleen size in patients with polycythemia vera resistant to or intolerant of hydroxyurea.”
RESPONSE trial
The EC’s approval is based on data from the phase 3 RESPONSE trial. The study included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had splenomegaly.
Patients were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study’s primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
Twenty-one percent of ruxolitinib-treated patients met this endpoint (achieving hematocrit control and spleen reduction), compared to 1% of patients who received BAT (P<0.001).
And the researchers said ruxolitinib was well-tolerated. Common adverse events included headache, diarrhea, and fatigue.
Grade 3/4 anemia, grade 3/4 thrombocytopenia, and herpes zoster infections of all grades were more common in the ruxolitinib arm than the BAT arm. But thromboembolic events were more common with BAT than ruxolitinib.
This trial was funded by the Incyte Corporation, which markets ruxolitinib in the US. Novartis licensed ruxolitinib from Incyte for development and commercialization outside the US.
For more details on ruxolitinib, see the full prescribing information, available at www.jakavi.com. ![]()
Image courtesy of AFIP
The European Commission (EC) has approved ruxolitinib (Jakavi) to treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
This is the first targeted treatment the EC has approved for these patients.
The approval applies to all 28 member states of the European Union (EU), plus Iceland, Norway, and Liechtenstein.
Ruxolitinib is already approved to treat PV in the US, and additional regulatory applications for ruxolitinib in PV are ongoing worldwide.
The drug is also approved to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF in more than 70 countries, including EU member states and the US.
“The European Commission’s approval of Jakavi [for PV] is encouraging news for patients,” said Claire Harrison, MD, a consultant hematologist at Guy’s and St Thomas’ NHS Foundation Trust in London, England.
“Jakavi will fill an unmet need as the first treatment shown to significantly improve hematocrit, as well as symptom control and reduce spleen size in patients with polycythemia vera resistant to or intolerant of hydroxyurea.”
RESPONSE trial
The EC’s approval is based on data from the phase 3 RESPONSE trial. The study included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had splenomegaly.
Patients were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study’s primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
Twenty-one percent of ruxolitinib-treated patients met this endpoint (achieving hematocrit control and spleen reduction), compared to 1% of patients who received BAT (P<0.001).
And the researchers said ruxolitinib was well-tolerated. Common adverse events included headache, diarrhea, and fatigue.
Grade 3/4 anemia, grade 3/4 thrombocytopenia, and herpes zoster infections of all grades were more common in the ruxolitinib arm than the BAT arm. But thromboembolic events were more common with BAT than ruxolitinib.
This trial was funded by the Incyte Corporation, which markets ruxolitinib in the US. Novartis licensed ruxolitinib from Incyte for development and commercialization outside the US.
For more details on ruxolitinib, see the full prescribing information, available at www.jakavi.com. ![]()
Image courtesy of AFIP
The European Commission (EC) has approved ruxolitinib (Jakavi) to treat adults with polycythemia vera (PV) who are resistant to or cannot tolerate hydroxyurea.
This is the first targeted treatment the EC has approved for these patients.
The approval applies to all 28 member states of the European Union (EU), plus Iceland, Norway, and Liechtenstein.
Ruxolitinib is already approved to treat PV in the US, and additional regulatory applications for ruxolitinib in PV are ongoing worldwide.
The drug is also approved to treat adults with primary myelofibrosis (MF), post-PV MF, or post-essential thrombocythemia MF in more than 70 countries, including EU member states and the US.
“The European Commission’s approval of Jakavi [for PV] is encouraging news for patients,” said Claire Harrison, MD, a consultant hematologist at Guy’s and St Thomas’ NHS Foundation Trust in London, England.
“Jakavi will fill an unmet need as the first treatment shown to significantly improve hematocrit, as well as symptom control and reduce spleen size in patients with polycythemia vera resistant to or intolerant of hydroxyurea.”
RESPONSE trial
The EC’s approval is based on data from the phase 3 RESPONSE trial. The study included 222 patients who had PV for at least 24 weeks. All patients had an inadequate response to or could not tolerate hydroxyurea, had undergone a phlebotomy, and had splenomegaly.
Patients were randomized to receive ruxolitinib starting at 10 mg twice daily or best available therapy (BAT) as determined by the investigator. The ruxolitinib dose was adjusted as needed.
The study’s primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
Twenty-one percent of ruxolitinib-treated patients met this endpoint (achieving hematocrit control and spleen reduction), compared to 1% of patients who received BAT (P<0.001).
And the researchers said ruxolitinib was well-tolerated. Common adverse events included headache, diarrhea, and fatigue.
Grade 3/4 anemia, grade 3/4 thrombocytopenia, and herpes zoster infections of all grades were more common in the ruxolitinib arm than the BAT arm. But thromboembolic events were more common with BAT than ruxolitinib.
This trial was funded by the Incyte Corporation, which markets ruxolitinib in the US. Novartis licensed ruxolitinib from Incyte for development and commercialization outside the US.
For more details on ruxolitinib, see the full prescribing information, available at www.jakavi.com. ![]()
4F-PCC proves more effective than plasma

Photo by Cristina Granados
Results of a phase 3 trial indicate that a 4-factor prothrombin complex concentrate (4F-PCC) is more effective than plasma for reversing acquired coagulation factor deficiency induced by vitamin K antagonist therapy in adults who require urgent surgery or an invasive procedure.
4F-PCC induced hemostasis in more patients and reduced international normalized ratios (INRs) more quickly than plasma.
And the rates of adverse events (AEs) were similar in the 2 groups.
“[4F-PCC] is more effective than plasma for INR reduction and periprocedural hemostasis in adults who are taking warfarin and require an urgent procedure,” said Joshua N. Goldstein, MD, PhD, of Massachusetts General Hospital in Boston.
He and his colleagues reported these findings in The Lancet.
The study included 181 patients, but only 168 were evaluable for efficacy. Eighty-seven of these patients received 4F-PCC, and 81 received plasma.
Ninety percent of patients treated with 4F-PCC achieved effective hemostasis, compared to 75% of patients treated with plasma (P=0.0142).
Fifty-five percent of patients who received 4F-PCC achieved rapid INR reduction (to ≤ 1.3 at 30 minutes after the end of infusion), compared to 10% of patients treated with plasma (P<0.0001).
In post-hoc analysis, the median time from the start of infusion to the start of the urgent surgical procedure was shorter in the 4F-PCC group than in the plasma group—3.6 hours and 8.5 hours, respectively (P=0.0098).
Eighty-eight patients in each group were evaluable for safety. And the incidence of AEs was similar in the 4F-PCC and plasma groups, at 56% and 60%, respectively.
Treatment-related AEs occurred in 9% of 4F-PCC-treated patients and 17% of plasma-treated patients. In both groups, 3% of these AEs were serious.
The rate of death at day 45 was 3% in the 4F-PCC group and 9% in the plasma group. The rates of thromboembolic AEs were 7% and 8%, respectively.
The rates of fluid overload or similar cardiac events were 3% and 13%, respectively. And the rates of bleeding after the primary outcome assessment were 3% and 5%, respectively.
This study was funded by CSL Behring, makers of 4F-PCC, which is marketed as Kcentra, Beriplex, or Confidex. ![]()
Photo by Cristina Granados
Results of a phase 3 trial indicate that a 4-factor prothrombin complex concentrate (4F-PCC) is more effective than plasma for reversing acquired coagulation factor deficiency induced by vitamin K antagonist therapy in adults who require urgent surgery or an invasive procedure.
4F-PCC induced hemostasis in more patients and reduced international normalized ratios (INRs) more quickly than plasma.
And the rates of adverse events (AEs) were similar in the 2 groups.
“[4F-PCC] is more effective than plasma for INR reduction and periprocedural hemostasis in adults who are taking warfarin and require an urgent procedure,” said Joshua N. Goldstein, MD, PhD, of Massachusetts General Hospital in Boston.
He and his colleagues reported these findings in The Lancet.
The study included 181 patients, but only 168 were evaluable for efficacy. Eighty-seven of these patients received 4F-PCC, and 81 received plasma.
Ninety percent of patients treated with 4F-PCC achieved effective hemostasis, compared to 75% of patients treated with plasma (P=0.0142).
Fifty-five percent of patients who received 4F-PCC achieved rapid INR reduction (to ≤ 1.3 at 30 minutes after the end of infusion), compared to 10% of patients treated with plasma (P<0.0001).
In post-hoc analysis, the median time from the start of infusion to the start of the urgent surgical procedure was shorter in the 4F-PCC group than in the plasma group—3.6 hours and 8.5 hours, respectively (P=0.0098).
Eighty-eight patients in each group were evaluable for safety. And the incidence of AEs was similar in the 4F-PCC and plasma groups, at 56% and 60%, respectively.
Treatment-related AEs occurred in 9% of 4F-PCC-treated patients and 17% of plasma-treated patients. In both groups, 3% of these AEs were serious.
The rate of death at day 45 was 3% in the 4F-PCC group and 9% in the plasma group. The rates of thromboembolic AEs were 7% and 8%, respectively.
The rates of fluid overload or similar cardiac events were 3% and 13%, respectively. And the rates of bleeding after the primary outcome assessment were 3% and 5%, respectively.
This study was funded by CSL Behring, makers of 4F-PCC, which is marketed as Kcentra, Beriplex, or Confidex. ![]()
Photo by Cristina Granados
Results of a phase 3 trial indicate that a 4-factor prothrombin complex concentrate (4F-PCC) is more effective than plasma for reversing acquired coagulation factor deficiency induced by vitamin K antagonist therapy in adults who require urgent surgery or an invasive procedure.
4F-PCC induced hemostasis in more patients and reduced international normalized ratios (INRs) more quickly than plasma.
And the rates of adverse events (AEs) were similar in the 2 groups.
“[4F-PCC] is more effective than plasma for INR reduction and periprocedural hemostasis in adults who are taking warfarin and require an urgent procedure,” said Joshua N. Goldstein, MD, PhD, of Massachusetts General Hospital in Boston.
He and his colleagues reported these findings in The Lancet.
The study included 181 patients, but only 168 were evaluable for efficacy. Eighty-seven of these patients received 4F-PCC, and 81 received plasma.
Ninety percent of patients treated with 4F-PCC achieved effective hemostasis, compared to 75% of patients treated with plasma (P=0.0142).
Fifty-five percent of patients who received 4F-PCC achieved rapid INR reduction (to ≤ 1.3 at 30 minutes after the end of infusion), compared to 10% of patients treated with plasma (P<0.0001).
In post-hoc analysis, the median time from the start of infusion to the start of the urgent surgical procedure was shorter in the 4F-PCC group than in the plasma group—3.6 hours and 8.5 hours, respectively (P=0.0098).
Eighty-eight patients in each group were evaluable for safety. And the incidence of AEs was similar in the 4F-PCC and plasma groups, at 56% and 60%, respectively.
Treatment-related AEs occurred in 9% of 4F-PCC-treated patients and 17% of plasma-treated patients. In both groups, 3% of these AEs were serious.
The rate of death at day 45 was 3% in the 4F-PCC group and 9% in the plasma group. The rates of thromboembolic AEs were 7% and 8%, respectively.
The rates of fluid overload or similar cardiac events were 3% and 13%, respectively. And the rates of bleeding after the primary outcome assessment were 3% and 5%, respectively.
This study was funded by CSL Behring, makers of 4F-PCC, which is marketed as Kcentra, Beriplex, or Confidex. ![]()
LISTEN NOW: My iPad Went to Medical School
{kind=link}
Mobile devices put information in the palm of your hand. For hospitalists, this presents real opportunities to engage patients, improve care, and streamline hospital workflows. Two hospitalists who were early adopters of mobile tech in their practices, Dr. Henry Feldman of Beth Israel Deaconess and Dr. Richard Pittman of Emory University/Grady share their lessons learned, and their advice for other hospital clinicians and informaticists on using mobile tech in their practices.
Mobile devices put information in the palm of your hand. For hospitalists, this presents real opportunities to engage patients, improve care, and streamline hospital workflows. Two hospitalists who were early adopters of mobile tech in their practices, Dr. Henry Feldman of Beth Israel Deaconess and Dr. Richard Pittman of Emory University/Grady share their lessons learned, and their advice for other hospital clinicians and informaticists on using mobile tech in their practices.
Mobile devices put information in the palm of your hand. For hospitalists, this presents real opportunities to engage patients, improve care, and streamline hospital workflows. Two hospitalists who were early adopters of mobile tech in their practices, Dr. Henry Feldman of Beth Israel Deaconess and Dr. Richard Pittman of Emory University/Grady share their lessons learned, and their advice for other hospital clinicians and informaticists on using mobile tech in their practices.