User login
EHA: Venetoclax-rituxumab combo highly active in relapsed/refractory CLL
VIENNA – A daily dose of the investigational BCL-2 inhibitor venetoclax plus rituximab induced responses in 84% of patients with relapsed or refractory chronic lymphocytic leukemia in a phase Ib study.
Of the 49 patients, 20 (41%) achieved a complete response by standard assessment and 13 (27%) achieved a complete response with no evidence of residual disease on flow cytometry.
Moreover, six patients elected to stop venetoclax after achieving a complete response and, to date, only one has had recurrence of disease after 24 months without therapy, lead investigator Dr. Andrew W. Roberts reported at the annual congress of the European Hematology Association.

Not only were patients able to come off treatment and continue to remain in complete response, but responses were seen at the same frequencies across all classes of cytogenetic and molecular abnormalities, he noted.
“The greatest advance that this drug brings is for those patients who currently have a terrible prognosis with all other drugs that we now have,” Dr. Roberts of Royal Melbourne Hospital said in a press briefing.
“This is an important step forward in finding chemotherapy-free regimens in these vulnerable, elderly patients,” said press briefing moderator Dr. Anton Hagenbeek of the University Medical Center, Utrecht, the Netherlands.
Patients in the open-label, dose-escalation study had received a median of two prior lines of therapy (range, one to five) for chronic lymphocytic leukemia (CLL); their median age was 68 years (50-88 years). They began treatment with 20 mg or 50 mg venetoclax daily, increasing weekly to final cohort doses of 200 mg to 600 mg. Six cycles of monthly standard rituximab were added after the weekly lead-in phase.
CLL depends on high levels of B-cell lymphoma-2 (BCL-2) to stay alive. Venetoclax binds to and switches off the BCL-2 protein function, triggering the death of the CLL cell.
Grade 3 or 4 adverse events occurring in more than 10% of patients were neutropenia (51%), thrombocytopenia (16%), and anemia (14%). There was one treatment-emergent case of tumor lysis syndrome leading to death early in the trial. This phenomenon can occur when the CLL breaks down very quickly and, as a consequence, the study was redesigned and a lower starting dose is now used, Dr. Roberts said.
“That problem has been eliminated, but we still see a very large improvement in patients in a few weeks,” he said. “Other than that, there is a little bit of neutropenia, but that is very manageable.”
“So do you think you are curing patients with this approach?” Dr. Hagenbeek asked, to which Dr. Roberts replied, “Too early to say.”
Venetoclax is currently being evaluated in less heavily pretreated patients and a phase III trial is comparing the combination of venetoclax and rituximab with standard bendamustine chemotherapy plus rituximab, he said.
In May, the Food and Drug Administration granted venetoclax breakthrough therapy designation for use in relapsed or refractory chronic lymphocytic leukemia with a 17p deletion mutation.
AbbVie, which is developing venetoclax in partnership with Roche and Genentech, plans to submit regulatory applications for venetoclax to the FDA and the European Medicines Agency before the end of 2015.
On Twitter @pwendl
VIENNA – A daily dose of the investigational BCL-2 inhibitor venetoclax plus rituximab induced responses in 84% of patients with relapsed or refractory chronic lymphocytic leukemia in a phase Ib study.
Of the 49 patients, 20 (41%) achieved a complete response by standard assessment and 13 (27%) achieved a complete response with no evidence of residual disease on flow cytometry.
Moreover, six patients elected to stop venetoclax after achieving a complete response and, to date, only one has had recurrence of disease after 24 months without therapy, lead investigator Dr. Andrew W. Roberts reported at the annual congress of the European Hematology Association.
Not only were patients able to come off treatment and continue to remain in complete response, but responses were seen at the same frequencies across all classes of cytogenetic and molecular abnormalities, he noted.
“The greatest advance that this drug brings is for those patients who currently have a terrible prognosis with all other drugs that we now have,” Dr. Roberts of Royal Melbourne Hospital said in a press briefing.
“This is an important step forward in finding chemotherapy-free regimens in these vulnerable, elderly patients,” said press briefing moderator Dr. Anton Hagenbeek of the University Medical Center, Utrecht, the Netherlands.
Patients in the open-label, dose-escalation study had received a median of two prior lines of therapy (range, one to five) for chronic lymphocytic leukemia (CLL); their median age was 68 years (50-88 years). They began treatment with 20 mg or 50 mg venetoclax daily, increasing weekly to final cohort doses of 200 mg to 600 mg. Six cycles of monthly standard rituximab were added after the weekly lead-in phase.
CLL depends on high levels of B-cell lymphoma-2 (BCL-2) to stay alive. Venetoclax binds to and switches off the BCL-2 protein function, triggering the death of the CLL cell.
Grade 3 or 4 adverse events occurring in more than 10% of patients were neutropenia (51%), thrombocytopenia (16%), and anemia (14%). There was one treatment-emergent case of tumor lysis syndrome leading to death early in the trial. This phenomenon can occur when the CLL breaks down very quickly and, as a consequence, the study was redesigned and a lower starting dose is now used, Dr. Roberts said.
“That problem has been eliminated, but we still see a very large improvement in patients in a few weeks,” he said. “Other than that, there is a little bit of neutropenia, but that is very manageable.”
“So do you think you are curing patients with this approach?” Dr. Hagenbeek asked, to which Dr. Roberts replied, “Too early to say.”
Venetoclax is currently being evaluated in less heavily pretreated patients and a phase III trial is comparing the combination of venetoclax and rituximab with standard bendamustine chemotherapy plus rituximab, he said.
In May, the Food and Drug Administration granted venetoclax breakthrough therapy designation for use in relapsed or refractory chronic lymphocytic leukemia with a 17p deletion mutation.
AbbVie, which is developing venetoclax in partnership with Roche and Genentech, plans to submit regulatory applications for venetoclax to the FDA and the European Medicines Agency before the end of 2015.
On Twitter @pwendl
VIENNA – A daily dose of the investigational BCL-2 inhibitor venetoclax plus rituximab induced responses in 84% of patients with relapsed or refractory chronic lymphocytic leukemia in a phase Ib study.
Of the 49 patients, 20 (41%) achieved a complete response by standard assessment and 13 (27%) achieved a complete response with no evidence of residual disease on flow cytometry.
Moreover, six patients elected to stop venetoclax after achieving a complete response and, to date, only one has had recurrence of disease after 24 months without therapy, lead investigator Dr. Andrew W. Roberts reported at the annual congress of the European Hematology Association.
Not only were patients able to come off treatment and continue to remain in complete response, but responses were seen at the same frequencies across all classes of cytogenetic and molecular abnormalities, he noted.
“The greatest advance that this drug brings is for those patients who currently have a terrible prognosis with all other drugs that we now have,” Dr. Roberts of Royal Melbourne Hospital said in a press briefing.
“This is an important step forward in finding chemotherapy-free regimens in these vulnerable, elderly patients,” said press briefing moderator Dr. Anton Hagenbeek of the University Medical Center, Utrecht, the Netherlands.
Patients in the open-label, dose-escalation study had received a median of two prior lines of therapy (range, one to five) for chronic lymphocytic leukemia (CLL); their median age was 68 years (50-88 years). They began treatment with 20 mg or 50 mg venetoclax daily, increasing weekly to final cohort doses of 200 mg to 600 mg. Six cycles of monthly standard rituximab were added after the weekly lead-in phase.
CLL depends on high levels of B-cell lymphoma-2 (BCL-2) to stay alive. Venetoclax binds to and switches off the BCL-2 protein function, triggering the death of the CLL cell.
Grade 3 or 4 adverse events occurring in more than 10% of patients were neutropenia (51%), thrombocytopenia (16%), and anemia (14%). There was one treatment-emergent case of tumor lysis syndrome leading to death early in the trial. This phenomenon can occur when the CLL breaks down very quickly and, as a consequence, the study was redesigned and a lower starting dose is now used, Dr. Roberts said.
“That problem has been eliminated, but we still see a very large improvement in patients in a few weeks,” he said. “Other than that, there is a little bit of neutropenia, but that is very manageable.”
“So do you think you are curing patients with this approach?” Dr. Hagenbeek asked, to which Dr. Roberts replied, “Too early to say.”
Venetoclax is currently being evaluated in less heavily pretreated patients and a phase III trial is comparing the combination of venetoclax and rituximab with standard bendamustine chemotherapy plus rituximab, he said.
In May, the Food and Drug Administration granted venetoclax breakthrough therapy designation for use in relapsed or refractory chronic lymphocytic leukemia with a 17p deletion mutation.
AbbVie, which is developing venetoclax in partnership with Roche and Genentech, plans to submit regulatory applications for venetoclax to the FDA and the European Medicines Agency before the end of 2015.
On Twitter @pwendl
AT THE EHA CONGRESS
Key clinical point: Venetoclax plus rituximab is a highly active nonchemotherapy combination for patients with relapsed or refractory chronic lymphocytic leukemia.
Major finding: Overall, 84% of patients responded to venetoclax plus rituximab.
Data source: Phase Ib trial in 49 patients with relapsed or refractory chronic lymphocytic leukemia.
Disclosures: AbbVie sponsored the study. Dr. Roberts’ financial disclosures were not available at press time.

Pruritic Urticarial Papules and Plaques of Pregnancy Occurring Postpartum
The cutaneous effects of pregnancy are variable and numerous. We all have likely seen the pigmentary changes induced by pregnancy as well as both exacerbation and complete resolution of preexisting skin conditions. The dermatoses of pregnancy are classified as a group of inflammatory skin conditions exclusively seen in pregnant women, the most common being pruritic urticarial papules and plaques of pregnancy (PUPPP).1 Also known as polymorphic eruption of pregnancy in Europe, PUPPP was first recognized in 1979 as a distinct entity that manifested as an intense pruritic eruption unique to women in the third trimester of pregnancy.2 The condition usually is self-limited, with the majority of cases spontaneously resolving within 4 to 6 weeks after delivery.3,4 Presentation of PUPPP in the postpartum period is rare.1-4 We report a biopsy-proven case of PUPPP in a 30-year-old woman who presented 2 weeks postpartum with an intensely pruritic generalized eruption. A PubMed search of articles indexed for MEDLINE using the search terms pruritic urticarial papules and plaques of pregnancy or polymorphic eruption of pregnancy and postpartum revealed only 5 reports of PUPPP or polymorphic eruption of pregnancy occurring in the postpartum period, 2 occurring in the United States.5-9
Case Report
A 30-year-old woman who was 2 weeks postpartum presented to our dermatology clinic with an intensely pruritic generalized rash. Within 24 hours of delivery of her first child, the patient developed an itchy rash on the abdomen and was started on oral corticosteroids and antihistamines in the hospital. On discharge, she was instructed to follow up with the dermatology department if the rash did not resolve. After leaving the hospital, she reported that the eruption had progressively spread to the buttocks, legs, and arms, and the itching seemed to be worse despite finishing the course of oral corticosteroids and antihistamines.
The patient’s prenatal course was uneventful. She gained 16 kg during pregnancy, with a prepregnancy weight of 50 kg. A healthy male neonate was delivered at 38 weeks’ gestation without complication. The patient’s medical history was unremarkable. Her current medications included prenatal vitamins, oral prednisone, and loratadine, and she reported no known drug allergies.
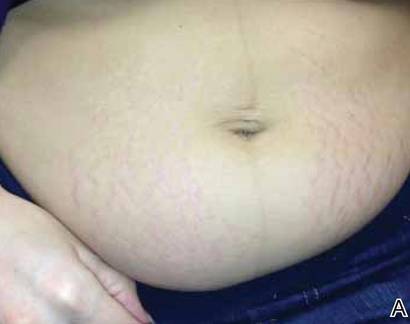
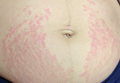
On physical examination, the patient was afebrile and her blood pressure was normal. Examination of the skin revealed erythematous papules and urticarial plaques involving the abdominal striae with periumbilical sparing (Figure 1A). Similar lesions were noted on the legs, buttocks, and arms (Figure 1B). The face, palms, and soles were uninvolved. No vesicles or pustules were noted. The oral mucosa was pink, moist, and unremarkable.

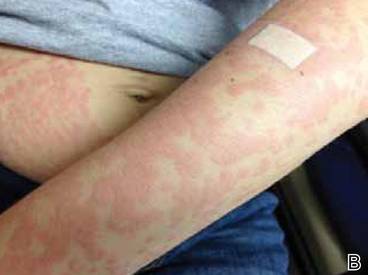
Figure 1. Initial presentation of urticarial plaques involving the abdominal striae with periumbilical sparing (A) and the left arm (B). |
Based on the patient’s clinical presentation, the differential diagnosis included pemphigoid gestationis, a hypersensitivity reaction, cutaneous lupus, cholestasis of pregnancy, and PUPPP. Pruritic urticarial papules and plaques of pregnancy was considered to be unlikely because of the uncharacteristic postpartum presentation of the eruption.
Two 4-mm punch biopsies were performed on the left upper arm and were sent for histopathologic examination and direct immunofluorescence. Laboratory studies including complete blood cell count with differential, complete metabolic panel, antinuclear antibodies, and IgE levels were conducted. The patient was started on triamcinolone cream 0.1% twice daily and her antihistamine was switched from loratadine to cetirizine.
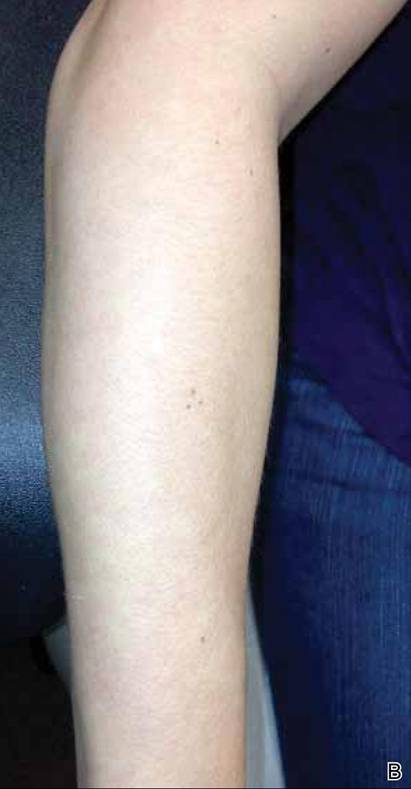
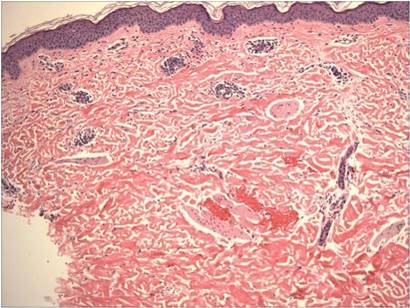
Histopathologic examination revealed a mixed perivascular infiltrate in the superficial dermis consisting of lymphocytes, mast cells, and eosinophils (Figures 2 and 3), which was consistent with a diagnosis of PUPPP. Direct immunofluorescence was negative. Laboratory studies were within reference range and antinuclear antibodies and IgE levels were negative. A diagnosis of postpartum PUPPP was made. Complete resolution of the eruption was experienced by 2-week follow-up (Figures 4A and 4B). The patient noted that her symptoms improved within 2 days of starting topical therapy.
|
|

Comment
Pruritic urticarial papules and plaques of pregnancy complicates 1 of 160 to 1 of 300 pregnancies.1 As seen in our case, the majority of cases of PUPPP are diagnosed in women who are nulliparous or primigravida.10 A study by Aronson et al10 reported that of 57 cases of PUPPP, 24 (42%) patients were primigravida, 16 (28%) were gravida 2, 9 (16%) were gravida 3, 4 (7%) were gravida 4, 3 (5%) were gravida 6, and 1 (2%) was gravida 7. Thirty-nine (68%) patients were nulliparous.10 The average onset of symptoms is approximately 35 weeks’ gestation.9
Classical presentation of PUPPP starts with erythematous papules within the abdominal striae, sparing the periumbilical skin.1 The abdominal striae are most commonly affected, and in some women, it may be the only site affected.10 The lesions then may pro-gress to urticarial plaques involving the extremities, while the face, palms, and soles usually are spared.11 However, clinical manifestations of PUPPP can vary, with reports of targetlike lesions with a surrounding halo resembling erythema multiforme as well as involvement of the face and palmoplantar skin.10-13 Histologic findings are not diagnostic but can help distinguish PUPPP from other pregnancy-associated dermatoses.14 Histologically, PUPPP demonstrates variable epidermal spongiosis and a nonspecific superficial perivascular infiltrate in the dermis composed of lymphocytes with eosinophils or neutrophils, and there may be dermal edema.10,15 Direct immunofluorescence usually is negative in PUPPP; however, 31% of cases have demonstrated deposition of C3 and IgM or IgA, either perivascularly or at the dermoepidermal junction.1,10,15
There are no systemic alterations seen in PUPPP; however, all patients report severe pruritus.12 Pruritic urticarial papules and plaques of pregnancy typically affects women in the third trimester, and delivery is curative in most patients.13 Recurrence of PUPPP usually is not seen with subsequent pregnancies, and the long-term prognosis is excellent.15
The pathogenesis of PUPPP is not well understood and likely is multifactorial. Ohel et al12 found PUPPP to be strongly associated with hypertensive disorders, multiple gestation pregnancies, excessive maternal weight gain, excessive stretching of the abdominal skin, and nulliparity.13 One theory suggests that abdominal skin stretching, if drastic, can damage underlying connective tissue, resulting in the release of antigens that can trigger a reactive inflammatory response.16 The majority of maternal weight gain occurs during the third trimester, which may explain why most cases of PUPPP present in the third trimester.17 Alternative theories have suggested that PUPPP may represent an immunologic response to circulating fetal antigens.18 It is possible, as in our case, that certain nulliparous women who have a healthy weight prior to pregnancy (as determined by a body mass index of 18.5 to 24.9) in combination with excessive weight gain during the third trimester and drastic hormone fluctuations associated with labor and delivery may be at greater risk for developing PUPPP. Another theory may be related to the degree of skin stretching during the third trimester and the abrupt decrease in the stretching of the skin that occurs with delivery.16
Conclusion
Pruritic urticarial papules and plaques of pregnancy can present in a variety of ways, most commonly in the third trimester but also in the postpartum period. When a patient presents in the postpartum period with a pruritic eruption, PUPPP should be included in the differential diagnosis. The pathogenesis of PUPPP is multifactorial and not well understood, and additional research in the field may lead to improved prediction of who may be at risk and what we can do to prevent it.
1. Pomeranz MK. Dermatoses of pregnancy. UpToDate Web site. http://www.uptodate.com/contents/dermatoses-of-pregnancy. Updated December 22, 2014. Accessed May 5, 2015.
2. Lawley TJ, Hertz KC, Wade TR, et al. Pruritic urticarial papules and plaques of pregnancy. JAMA. 1979;241:1696-1699.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
4. Callen JP, Hanno R. Pruritic urticarial papules and plaques of pregnancy (PUPPP): a clinicopathologic study. J Am Acad Dermatol.1981;5:401-405.
5. Ozcan D, Ozcakmak B, Aydogan FC. J Obstet Gynaecol Res. 2011;37:1158-1161.
6. Journet-Tollhupp J, Tchen T, Remy-Leroux V, et al. Polymorphic eruption of pregnancy and acquired hemophilia A [in French]. Ann Dermatol Venereol. 2010;137:713-717.
7. Buccolo LS, Viera AJ. Pruritic urticarial papules and plaques of pregnancy presenting in the postpartum period: a case report. J Reprod Med. 2005;50:61-63.
8. Kirkup ME, Dunnill MG. Polymorphic eruption of pregnancy developing in the puerrperium. Clin Exp Dermatol. 2002;27:657-660.
9. Yancy KB, Hall RP, Lawley TJ. Pruritic urticarial papules and plaques of pregnancy: clinical experience in twenty-five patients. J Am Acad Dermatol. 1984;10:473-480.
10. Aronson IK, Bond S, Fiedler VC, et al. Pruritic urticarial papules and plaques of pregnancy: clinical and immunopathologic observations in 57 patients. J Am Acad Dermatol. 1998;39:933-939.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic dermatoses of pregnancy: a prospective study of 3129 women. Arch Dermatol. 1994;130:734-739.
12. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Matern Fetal Neonatal Med. 2006;19:305-308.
13. Elling SV, McKenna P, Pawell FC. Pruritic urticarial papules and plaques of pregnancy in twin and triplet pregnancies. J Eur Acad Dermatol Venereol. 2000;14:378-381.
14. Scheinfeld N. Pruritic urticarial papules and plaques of pregnancy wholly abated with one week twice daily application of fluticasone propionate lotion: a case report and review of the literature. Dermatol Online J. 2008;14:4.
15. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
16. Cohen LM, Capeless EL, Krusinski PA, et al. Pruritic urticarial papules and plaques of pregnancy and its relationship to maternal-fetal weight gain and twin pregnancy. Arch Dermatol. 1989;125:1534-1536.
17. Drehmer M, Duncan BB, Kac G, et al. Association of second and third trimester weight gain in pregnancy with maternal and fetal outcomes. PLoS One. 2013;8:e54704.
18. Aractingi S, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
The cutaneous effects of pregnancy are variable and numerous. We all have likely seen the pigmentary changes induced by pregnancy as well as both exacerbation and complete resolution of preexisting skin conditions. The dermatoses of pregnancy are classified as a group of inflammatory skin conditions exclusively seen in pregnant women, the most common being pruritic urticarial papules and plaques of pregnancy (PUPPP).1 Also known as polymorphic eruption of pregnancy in Europe, PUPPP was first recognized in 1979 as a distinct entity that manifested as an intense pruritic eruption unique to women in the third trimester of pregnancy.2 The condition usually is self-limited, with the majority of cases spontaneously resolving within 4 to 6 weeks after delivery.3,4 Presentation of PUPPP in the postpartum period is rare.1-4 We report a biopsy-proven case of PUPPP in a 30-year-old woman who presented 2 weeks postpartum with an intensely pruritic generalized eruption. A PubMed search of articles indexed for MEDLINE using the search terms pruritic urticarial papules and plaques of pregnancy or polymorphic eruption of pregnancy and postpartum revealed only 5 reports of PUPPP or polymorphic eruption of pregnancy occurring in the postpartum period, 2 occurring in the United States.5-9
Case Report
A 30-year-old woman who was 2 weeks postpartum presented to our dermatology clinic with an intensely pruritic generalized rash. Within 24 hours of delivery of her first child, the patient developed an itchy rash on the abdomen and was started on oral corticosteroids and antihistamines in the hospital. On discharge, she was instructed to follow up with the dermatology department if the rash did not resolve. After leaving the hospital, she reported that the eruption had progressively spread to the buttocks, legs, and arms, and the itching seemed to be worse despite finishing the course of oral corticosteroids and antihistamines.
The patient’s prenatal course was uneventful. She gained 16 kg during pregnancy, with a prepregnancy weight of 50 kg. A healthy male neonate was delivered at 38 weeks’ gestation without complication. The patient’s medical history was unremarkable. Her current medications included prenatal vitamins, oral prednisone, and loratadine, and she reported no known drug allergies.
On physical examination, the patient was afebrile and her blood pressure was normal. Examination of the skin revealed erythematous papules and urticarial plaques involving the abdominal striae with periumbilical sparing (Figure 1A). Similar lesions were noted on the legs, buttocks, and arms (Figure 1B). The face, palms, and soles were uninvolved. No vesicles or pustules were noted. The oral mucosa was pink, moist, and unremarkable.
|
Figure 1. Initial presentation of urticarial plaques involving the abdominal striae with periumbilical sparing (A) and the left arm (B). |
Based on the patient’s clinical presentation, the differential diagnosis included pemphigoid gestationis, a hypersensitivity reaction, cutaneous lupus, cholestasis of pregnancy, and PUPPP. Pruritic urticarial papules and plaques of pregnancy was considered to be unlikely because of the uncharacteristic postpartum presentation of the eruption.
Two 4-mm punch biopsies were performed on the left upper arm and were sent for histopathologic examination and direct immunofluorescence. Laboratory studies including complete blood cell count with differential, complete metabolic panel, antinuclear antibodies, and IgE levels were conducted. The patient was started on triamcinolone cream 0.1% twice daily and her antihistamine was switched from loratadine to cetirizine.
Histopathologic examination revealed a mixed perivascular infiltrate in the superficial dermis consisting of lymphocytes, mast cells, and eosinophils (Figures 2 and 3), which was consistent with a diagnosis of PUPPP. Direct immunofluorescence was negative. Laboratory studies were within reference range and antinuclear antibodies and IgE levels were negative. A diagnosis of postpartum PUPPP was made. Complete resolution of the eruption was experienced by 2-week follow-up (Figures 4A and 4B). The patient noted that her symptoms improved within 2 days of starting topical therapy.
|
|
Comment
Pruritic urticarial papules and plaques of pregnancy complicates 1 of 160 to 1 of 300 pregnancies.1 As seen in our case, the majority of cases of PUPPP are diagnosed in women who are nulliparous or primigravida.10 A study by Aronson et al10 reported that of 57 cases of PUPPP, 24 (42%) patients were primigravida, 16 (28%) were gravida 2, 9 (16%) were gravida 3, 4 (7%) were gravida 4, 3 (5%) were gravida 6, and 1 (2%) was gravida 7. Thirty-nine (68%) patients were nulliparous.10 The average onset of symptoms is approximately 35 weeks’ gestation.9
Classical presentation of PUPPP starts with erythematous papules within the abdominal striae, sparing the periumbilical skin.1 The abdominal striae are most commonly affected, and in some women, it may be the only site affected.10 The lesions then may pro-gress to urticarial plaques involving the extremities, while the face, palms, and soles usually are spared.11 However, clinical manifestations of PUPPP can vary, with reports of targetlike lesions with a surrounding halo resembling erythema multiforme as well as involvement of the face and palmoplantar skin.10-13 Histologic findings are not diagnostic but can help distinguish PUPPP from other pregnancy-associated dermatoses.14 Histologically, PUPPP demonstrates variable epidermal spongiosis and a nonspecific superficial perivascular infiltrate in the dermis composed of lymphocytes with eosinophils or neutrophils, and there may be dermal edema.10,15 Direct immunofluorescence usually is negative in PUPPP; however, 31% of cases have demonstrated deposition of C3 and IgM or IgA, either perivascularly or at the dermoepidermal junction.1,10,15
There are no systemic alterations seen in PUPPP; however, all patients report severe pruritus.12 Pruritic urticarial papules and plaques of pregnancy typically affects women in the third trimester, and delivery is curative in most patients.13 Recurrence of PUPPP usually is not seen with subsequent pregnancies, and the long-term prognosis is excellent.15
The pathogenesis of PUPPP is not well understood and likely is multifactorial. Ohel et al12 found PUPPP to be strongly associated with hypertensive disorders, multiple gestation pregnancies, excessive maternal weight gain, excessive stretching of the abdominal skin, and nulliparity.13 One theory suggests that abdominal skin stretching, if drastic, can damage underlying connective tissue, resulting in the release of antigens that can trigger a reactive inflammatory response.16 The majority of maternal weight gain occurs during the third trimester, which may explain why most cases of PUPPP present in the third trimester.17 Alternative theories have suggested that PUPPP may represent an immunologic response to circulating fetal antigens.18 It is possible, as in our case, that certain nulliparous women who have a healthy weight prior to pregnancy (as determined by a body mass index of 18.5 to 24.9) in combination with excessive weight gain during the third trimester and drastic hormone fluctuations associated with labor and delivery may be at greater risk for developing PUPPP. Another theory may be related to the degree of skin stretching during the third trimester and the abrupt decrease in the stretching of the skin that occurs with delivery.16
Conclusion
Pruritic urticarial papules and plaques of pregnancy can present in a variety of ways, most commonly in the third trimester but also in the postpartum period. When a patient presents in the postpartum period with a pruritic eruption, PUPPP should be included in the differential diagnosis. The pathogenesis of PUPPP is multifactorial and not well understood, and additional research in the field may lead to improved prediction of who may be at risk and what we can do to prevent it.
The cutaneous effects of pregnancy are variable and numerous. We all have likely seen the pigmentary changes induced by pregnancy as well as both exacerbation and complete resolution of preexisting skin conditions. The dermatoses of pregnancy are classified as a group of inflammatory skin conditions exclusively seen in pregnant women, the most common being pruritic urticarial papules and plaques of pregnancy (PUPPP).1 Also known as polymorphic eruption of pregnancy in Europe, PUPPP was first recognized in 1979 as a distinct entity that manifested as an intense pruritic eruption unique to women in the third trimester of pregnancy.2 The condition usually is self-limited, with the majority of cases spontaneously resolving within 4 to 6 weeks after delivery.3,4 Presentation of PUPPP in the postpartum period is rare.1-4 We report a biopsy-proven case of PUPPP in a 30-year-old woman who presented 2 weeks postpartum with an intensely pruritic generalized eruption. A PubMed search of articles indexed for MEDLINE using the search terms pruritic urticarial papules and plaques of pregnancy or polymorphic eruption of pregnancy and postpartum revealed only 5 reports of PUPPP or polymorphic eruption of pregnancy occurring in the postpartum period, 2 occurring in the United States.5-9
Case Report
A 30-year-old woman who was 2 weeks postpartum presented to our dermatology clinic with an intensely pruritic generalized rash. Within 24 hours of delivery of her first child, the patient developed an itchy rash on the abdomen and was started on oral corticosteroids and antihistamines in the hospital. On discharge, she was instructed to follow up with the dermatology department if the rash did not resolve. After leaving the hospital, she reported that the eruption had progressively spread to the buttocks, legs, and arms, and the itching seemed to be worse despite finishing the course of oral corticosteroids and antihistamines.
The patient’s prenatal course was uneventful. She gained 16 kg during pregnancy, with a prepregnancy weight of 50 kg. A healthy male neonate was delivered at 38 weeks’ gestation without complication. The patient’s medical history was unremarkable. Her current medications included prenatal vitamins, oral prednisone, and loratadine, and she reported no known drug allergies.
On physical examination, the patient was afebrile and her blood pressure was normal. Examination of the skin revealed erythematous papules and urticarial plaques involving the abdominal striae with periumbilical sparing (Figure 1A). Similar lesions were noted on the legs, buttocks, and arms (Figure 1B). The face, palms, and soles were uninvolved. No vesicles or pustules were noted. The oral mucosa was pink, moist, and unremarkable.
|
Figure 1. Initial presentation of urticarial plaques involving the abdominal striae with periumbilical sparing (A) and the left arm (B). |
Based on the patient’s clinical presentation, the differential diagnosis included pemphigoid gestationis, a hypersensitivity reaction, cutaneous lupus, cholestasis of pregnancy, and PUPPP. Pruritic urticarial papules and plaques of pregnancy was considered to be unlikely because of the uncharacteristic postpartum presentation of the eruption.
Two 4-mm punch biopsies were performed on the left upper arm and were sent for histopathologic examination and direct immunofluorescence. Laboratory studies including complete blood cell count with differential, complete metabolic panel, antinuclear antibodies, and IgE levels were conducted. The patient was started on triamcinolone cream 0.1% twice daily and her antihistamine was switched from loratadine to cetirizine.
Histopathologic examination revealed a mixed perivascular infiltrate in the superficial dermis consisting of lymphocytes, mast cells, and eosinophils (Figures 2 and 3), which was consistent with a diagnosis of PUPPP. Direct immunofluorescence was negative. Laboratory studies were within reference range and antinuclear antibodies and IgE levels were negative. A diagnosis of postpartum PUPPP was made. Complete resolution of the eruption was experienced by 2-week follow-up (Figures 4A and 4B). The patient noted that her symptoms improved within 2 days of starting topical therapy.
|
|
Comment
Pruritic urticarial papules and plaques of pregnancy complicates 1 of 160 to 1 of 300 pregnancies.1 As seen in our case, the majority of cases of PUPPP are diagnosed in women who are nulliparous or primigravida.10 A study by Aronson et al10 reported that of 57 cases of PUPPP, 24 (42%) patients were primigravida, 16 (28%) were gravida 2, 9 (16%) were gravida 3, 4 (7%) were gravida 4, 3 (5%) were gravida 6, and 1 (2%) was gravida 7. Thirty-nine (68%) patients were nulliparous.10 The average onset of symptoms is approximately 35 weeks’ gestation.9
Classical presentation of PUPPP starts with erythematous papules within the abdominal striae, sparing the periumbilical skin.1 The abdominal striae are most commonly affected, and in some women, it may be the only site affected.10 The lesions then may pro-gress to urticarial plaques involving the extremities, while the face, palms, and soles usually are spared.11 However, clinical manifestations of PUPPP can vary, with reports of targetlike lesions with a surrounding halo resembling erythema multiforme as well as involvement of the face and palmoplantar skin.10-13 Histologic findings are not diagnostic but can help distinguish PUPPP from other pregnancy-associated dermatoses.14 Histologically, PUPPP demonstrates variable epidermal spongiosis and a nonspecific superficial perivascular infiltrate in the dermis composed of lymphocytes with eosinophils or neutrophils, and there may be dermal edema.10,15 Direct immunofluorescence usually is negative in PUPPP; however, 31% of cases have demonstrated deposition of C3 and IgM or IgA, either perivascularly or at the dermoepidermal junction.1,10,15
There are no systemic alterations seen in PUPPP; however, all patients report severe pruritus.12 Pruritic urticarial papules and plaques of pregnancy typically affects women in the third trimester, and delivery is curative in most patients.13 Recurrence of PUPPP usually is not seen with subsequent pregnancies, and the long-term prognosis is excellent.15
The pathogenesis of PUPPP is not well understood and likely is multifactorial. Ohel et al12 found PUPPP to be strongly associated with hypertensive disorders, multiple gestation pregnancies, excessive maternal weight gain, excessive stretching of the abdominal skin, and nulliparity.13 One theory suggests that abdominal skin stretching, if drastic, can damage underlying connective tissue, resulting in the release of antigens that can trigger a reactive inflammatory response.16 The majority of maternal weight gain occurs during the third trimester, which may explain why most cases of PUPPP present in the third trimester.17 Alternative theories have suggested that PUPPP may represent an immunologic response to circulating fetal antigens.18 It is possible, as in our case, that certain nulliparous women who have a healthy weight prior to pregnancy (as determined by a body mass index of 18.5 to 24.9) in combination with excessive weight gain during the third trimester and drastic hormone fluctuations associated with labor and delivery may be at greater risk for developing PUPPP. Another theory may be related to the degree of skin stretching during the third trimester and the abrupt decrease in the stretching of the skin that occurs with delivery.16
Conclusion
Pruritic urticarial papules and plaques of pregnancy can present in a variety of ways, most commonly in the third trimester but also in the postpartum period. When a patient presents in the postpartum period with a pruritic eruption, PUPPP should be included in the differential diagnosis. The pathogenesis of PUPPP is multifactorial and not well understood, and additional research in the field may lead to improved prediction of who may be at risk and what we can do to prevent it.
1. Pomeranz MK. Dermatoses of pregnancy. UpToDate Web site. http://www.uptodate.com/contents/dermatoses-of-pregnancy. Updated December 22, 2014. Accessed May 5, 2015.
2. Lawley TJ, Hertz KC, Wade TR, et al. Pruritic urticarial papules and plaques of pregnancy. JAMA. 1979;241:1696-1699.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
4. Callen JP, Hanno R. Pruritic urticarial papules and plaques of pregnancy (PUPPP): a clinicopathologic study. J Am Acad Dermatol.1981;5:401-405.
5. Ozcan D, Ozcakmak B, Aydogan FC. J Obstet Gynaecol Res. 2011;37:1158-1161.
6. Journet-Tollhupp J, Tchen T, Remy-Leroux V, et al. Polymorphic eruption of pregnancy and acquired hemophilia A [in French]. Ann Dermatol Venereol. 2010;137:713-717.
7. Buccolo LS, Viera AJ. Pruritic urticarial papules and plaques of pregnancy presenting in the postpartum period: a case report. J Reprod Med. 2005;50:61-63.
8. Kirkup ME, Dunnill MG. Polymorphic eruption of pregnancy developing in the puerrperium. Clin Exp Dermatol. 2002;27:657-660.
9. Yancy KB, Hall RP, Lawley TJ. Pruritic urticarial papules and plaques of pregnancy: clinical experience in twenty-five patients. J Am Acad Dermatol. 1984;10:473-480.
10. Aronson IK, Bond S, Fiedler VC, et al. Pruritic urticarial papules and plaques of pregnancy: clinical and immunopathologic observations in 57 patients. J Am Acad Dermatol. 1998;39:933-939.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic dermatoses of pregnancy: a prospective study of 3129 women. Arch Dermatol. 1994;130:734-739.
12. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Matern Fetal Neonatal Med. 2006;19:305-308.
13. Elling SV, McKenna P, Pawell FC. Pruritic urticarial papules and plaques of pregnancy in twin and triplet pregnancies. J Eur Acad Dermatol Venereol. 2000;14:378-381.
14. Scheinfeld N. Pruritic urticarial papules and plaques of pregnancy wholly abated with one week twice daily application of fluticasone propionate lotion: a case report and review of the literature. Dermatol Online J. 2008;14:4.
15. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
16. Cohen LM, Capeless EL, Krusinski PA, et al. Pruritic urticarial papules and plaques of pregnancy and its relationship to maternal-fetal weight gain and twin pregnancy. Arch Dermatol. 1989;125:1534-1536.
17. Drehmer M, Duncan BB, Kac G, et al. Association of second and third trimester weight gain in pregnancy with maternal and fetal outcomes. PLoS One. 2013;8:e54704.
18. Aractingi S, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
1. Pomeranz MK. Dermatoses of pregnancy. UpToDate Web site. http://www.uptodate.com/contents/dermatoses-of-pregnancy. Updated December 22, 2014. Accessed May 5, 2015.
2. Lawley TJ, Hertz KC, Wade TR, et al. Pruritic urticarial papules and plaques of pregnancy. JAMA. 1979;241:1696-1699.
3. Kroumpouzos G, Cohen LM. Specific dermatoses of pregnancy: an evidence-based systematic review. Am J Obstet Gynecol. 2003;188:1083-1092.
4. Callen JP, Hanno R. Pruritic urticarial papules and plaques of pregnancy (PUPPP): a clinicopathologic study. J Am Acad Dermatol.1981;5:401-405.
5. Ozcan D, Ozcakmak B, Aydogan FC. J Obstet Gynaecol Res. 2011;37:1158-1161.
6. Journet-Tollhupp J, Tchen T, Remy-Leroux V, et al. Polymorphic eruption of pregnancy and acquired hemophilia A [in French]. Ann Dermatol Venereol. 2010;137:713-717.
7. Buccolo LS, Viera AJ. Pruritic urticarial papules and plaques of pregnancy presenting in the postpartum period: a case report. J Reprod Med. 2005;50:61-63.
8. Kirkup ME, Dunnill MG. Polymorphic eruption of pregnancy developing in the puerrperium. Clin Exp Dermatol. 2002;27:657-660.
9. Yancy KB, Hall RP, Lawley TJ. Pruritic urticarial papules and plaques of pregnancy: clinical experience in twenty-five patients. J Am Acad Dermatol. 1984;10:473-480.
10. Aronson IK, Bond S, Fiedler VC, et al. Pruritic urticarial papules and plaques of pregnancy: clinical and immunopathologic observations in 57 patients. J Am Acad Dermatol. 1998;39:933-939.
11. Roger D, Vaillant L, Fignon A, et al. Specific pruritic dermatoses of pregnancy: a prospective study of 3129 women. Arch Dermatol. 1994;130:734-739.
12. Ohel I, Levy A, Silberstein T, et al. Pregnancy outcome of patients with pruritic urticarial papules and plaques of pregnancy. J Matern Fetal Neonatal Med. 2006;19:305-308.
13. Elling SV, McKenna P, Pawell FC. Pruritic urticarial papules and plaques of pregnancy in twin and triplet pregnancies. J Eur Acad Dermatol Venereol. 2000;14:378-381.
14. Scheinfeld N. Pruritic urticarial papules and plaques of pregnancy wholly abated with one week twice daily application of fluticasone propionate lotion: a case report and review of the literature. Dermatol Online J. 2008;14:4.
15. Shornick JK. Dermatoses of pregnancy. Semin Cutan Med Surg. 1998;17:172-181.
16. Cohen LM, Capeless EL, Krusinski PA, et al. Pruritic urticarial papules and plaques of pregnancy and its relationship to maternal-fetal weight gain and twin pregnancy. Arch Dermatol. 1989;125:1534-1536.
17. Drehmer M, Duncan BB, Kac G, et al. Association of second and third trimester weight gain in pregnancy with maternal and fetal outcomes. PLoS One. 2013;8:e54704.
18. Aractingi S, Berkane N, Bertheau P, et al. Fetal DNA in skin of polymorphic eruptions of pregnancy. Lancet. 1998;352:1898-1901.
Practice Points
- Pruritic urticarial papules and plaques of pregnancy (PUPPP) is an intensely pruritic eruption that typically affects women during the third trimester of pregnancy.
- Because clinical manifestations can vary, PUPPP should be considered in the differential diagnosis when patients present in the postpartum period with a pruritic eruption.
- Histologic findings are not diagnostic but can help distinguish PUPPP from other pregnancy-associated dermatoses.
Identifying melasma triggers
Melasma can be a very frustrating, remitting, and relapsing condition, particularly in the summer months. Often patients get good results with at-home and in-office treatments and return frustrated as the melasma frequently recurs. A thorough history can help identify melasma triggers.

Ask about exposure to:
1. Any heat source. You will be surprised by the answers. Examples include overhead work lights, overhead desk lamps, extensive cooking over an oven or a grill, lamps used to treat seasonal affective disorder, heating lamps, and hair dryers. Heat is a very common trigger for melasma as it increases vasodilation. Melasma is typically thought of as solely hyperpigmentation; however, vascular dilatation often occurs in the affected area. In addition, heat may lead to more inflammation, also stimulating melanocyte pigment production.
2. UV sources. These include computer screens, car side windows, sunroofs (even if the roof glass is closed, UV can penetrate the glass, so the sunroof shade also should be closed), and a window near an office desk or a window near a bed (UVA penetrates window glass).
3. Visible light sources. Examples are overhead lights at home and in office buildings. These lights increase pigmentation. Iron oxide in sunscreens helps block visible light.

4. Hormonal triggers. These include birth control pills, hormone-releasing intrauterine devices, hormone therapy, and vitamin supplements such as those used for pregnancy, nursing, and perimenopausal symptoms (such as black cohosh and dong quai).
5. Other triggers:• Scented or deodorant soaps, toiletries, cosmetics, or fragrances that may cause phototoxic reactions. These reactions may in turn trigger melasma, which may then persist.
• Sunglasses. This is the most common avoidable trigger. Aviator sunglasses or sunglasses with metal rims, or metal attached to the inside handle or rim absorb the heat when in the sun and/or when left in the car. The metal gets warm, and the heat transfers to the skin when the sunglasses are placed on the face. I ask every melasma patient to bring in all their sunglasses so I can check for metal on the rim or handles. This is a very common trigger, and patients are shocked after they observe that streaks of melasma can often follow the pattern of their sunglasses.
• Autoimmune thyroid disorders, chronic stress, or adrenal dysfunction.
• Triggers of melanocyte-stimulating hormone.
The history is crucial to long-term clearance of melasma. Asking questions to get to the source of the trigger often can help isolate the cause and help eliminate significant recurrences of melasma in skin of color patients.
Dr. Wesley and Dr. Talakoub are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month's column is by Dr. Talakoub.
Melasma can be a very frustrating, remitting, and relapsing condition, particularly in the summer months. Often patients get good results with at-home and in-office treatments and return frustrated as the melasma frequently recurs. A thorough history can help identify melasma triggers.
Ask about exposure to:
1. Any heat source. You will be surprised by the answers. Examples include overhead work lights, overhead desk lamps, extensive cooking over an oven or a grill, lamps used to treat seasonal affective disorder, heating lamps, and hair dryers. Heat is a very common trigger for melasma as it increases vasodilation. Melasma is typically thought of as solely hyperpigmentation; however, vascular dilatation often occurs in the affected area. In addition, heat may lead to more inflammation, also stimulating melanocyte pigment production.
2. UV sources. These include computer screens, car side windows, sunroofs (even if the roof glass is closed, UV can penetrate the glass, so the sunroof shade also should be closed), and a window near an office desk or a window near a bed (UVA penetrates window glass).
3. Visible light sources. Examples are overhead lights at home and in office buildings. These lights increase pigmentation. Iron oxide in sunscreens helps block visible light.
4. Hormonal triggers. These include birth control pills, hormone-releasing intrauterine devices, hormone therapy, and vitamin supplements such as those used for pregnancy, nursing, and perimenopausal symptoms (such as black cohosh and dong quai).
5. Other triggers:• Scented or deodorant soaps, toiletries, cosmetics, or fragrances that may cause phototoxic reactions. These reactions may in turn trigger melasma, which may then persist.
• Sunglasses. This is the most common avoidable trigger. Aviator sunglasses or sunglasses with metal rims, or metal attached to the inside handle or rim absorb the heat when in the sun and/or when left in the car. The metal gets warm, and the heat transfers to the skin when the sunglasses are placed on the face. I ask every melasma patient to bring in all their sunglasses so I can check for metal on the rim or handles. This is a very common trigger, and patients are shocked after they observe that streaks of melasma can often follow the pattern of their sunglasses.
• Autoimmune thyroid disorders, chronic stress, or adrenal dysfunction.
• Triggers of melanocyte-stimulating hormone.
The history is crucial to long-term clearance of melasma. Asking questions to get to the source of the trigger often can help isolate the cause and help eliminate significant recurrences of melasma in skin of color patients.
Dr. Wesley and Dr. Talakoub are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month's column is by Dr. Talakoub.
Melasma can be a very frustrating, remitting, and relapsing condition, particularly in the summer months. Often patients get good results with at-home and in-office treatments and return frustrated as the melasma frequently recurs. A thorough history can help identify melasma triggers.
Ask about exposure to:
1. Any heat source. You will be surprised by the answers. Examples include overhead work lights, overhead desk lamps, extensive cooking over an oven or a grill, lamps used to treat seasonal affective disorder, heating lamps, and hair dryers. Heat is a very common trigger for melasma as it increases vasodilation. Melasma is typically thought of as solely hyperpigmentation; however, vascular dilatation often occurs in the affected area. In addition, heat may lead to more inflammation, also stimulating melanocyte pigment production.
2. UV sources. These include computer screens, car side windows, sunroofs (even if the roof glass is closed, UV can penetrate the glass, so the sunroof shade also should be closed), and a window near an office desk or a window near a bed (UVA penetrates window glass).
3. Visible light sources. Examples are overhead lights at home and in office buildings. These lights increase pigmentation. Iron oxide in sunscreens helps block visible light.
4. Hormonal triggers. These include birth control pills, hormone-releasing intrauterine devices, hormone therapy, and vitamin supplements such as those used for pregnancy, nursing, and perimenopausal symptoms (such as black cohosh and dong quai).
5. Other triggers:• Scented or deodorant soaps, toiletries, cosmetics, or fragrances that may cause phototoxic reactions. These reactions may in turn trigger melasma, which may then persist.
• Sunglasses. This is the most common avoidable trigger. Aviator sunglasses or sunglasses with metal rims, or metal attached to the inside handle or rim absorb the heat when in the sun and/or when left in the car. The metal gets warm, and the heat transfers to the skin when the sunglasses are placed on the face. I ask every melasma patient to bring in all their sunglasses so I can check for metal on the rim or handles. This is a very common trigger, and patients are shocked after they observe that streaks of melasma can often follow the pattern of their sunglasses.
• Autoimmune thyroid disorders, chronic stress, or adrenal dysfunction.
• Triggers of melanocyte-stimulating hormone.
The history is crucial to long-term clearance of melasma. Asking questions to get to the source of the trigger often can help isolate the cause and help eliminate significant recurrences of melasma in skin of color patients.
Dr. Wesley and Dr. Talakoub are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month's column is by Dr. Talakoub.

Ingenol mebutate for AKs gets thumbs-up from patients
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.

Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.
Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.
Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
AT WCD 2015
Key clinical point: Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes.
Major finding: Mean scores on the Skindex-16, which reflects the quality of life impact of a patient’s skin disease, improved significantly from 24.3 pretreatment to 12.1 after 8 weeks.
Data source: A prospective observational study of patient-reported outcomes of ingenol mebutate therapy for actinic keratoses in 826 patients treated in 292 German dermatologists’ offices.
Disclosures: The study was sponsored by Leo Pharma, which markets ingenol mebutate. The presenter reported having received research grants and speakers’ fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.

Simulated daylight PDT advantageous for AKs
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
AT WCD 2015
Key clinical point: A field containing multiple actinic keratoses can be treated virtually painlessly using lamps that simulate daylight to activate photodynamic therapy.
Major finding: 3 months after simulated daylight PDT, the mean number of AKs in treated patients was reduced from 5.3 at baseline to 0.4.
Data source: This was a retrospective study including 32 patients whose actinic keratoses was treated using simulated daylight PDT.
Disclosures: The study was supported by Biofrontera. The presenter reported serving as a consultant to and receiving speaking fees from the company.
FDA extends approval of ITP drug to kids

Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) has approved eltrombopag (Promacta) to treat children age 6 and older with chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Eltrombopag is an oral thrombopoietin receptor agonist that works by inducing the stimulation and differentiation of megakaryocytes to increase platelet production.
The drug is already FDA-approved to treat adults with ITP.
The FDA’s latest approval of eltrombopag was based on data from the phase 2 PETIT trial and the phase 3 PETIT2 trial.
“Young patients with chronic ITP who have either an insufficient response to or side effects from standard therapies have limited treatment options, making this FDA approval of eltrombopag for children 6 years and older particularly important,” said James B. Bussel, MD, a professor at Weill Cornell Medical College in New York and lead investigator of the PETIT study.
“Through the eltrombopag studies, one of which is the largest randomized trial ever performed in children with chronic ITP, we discovered that Promacta—a treatment that can be taken once daily by mouth and shown to be well tolerated—can manage this disorder and help these young patients.”
PETIT trials: Efficacy
The PETIT trial included 67 ITP patients stratified by age cohort (12-17 years, 6-11 years, and 1-5 years). They were randomized (2:1) to receive eltrombopag or placebo for 7 weeks. Eltrombopag dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects achieving platelet counts of 50 x109/L or higher at least once between days 8 and 43 of the randomized period of the study.
Significantly more patients in the eltrombopag arm met this endpoint—62.2%—compared to 31.8% in the placebo arm (P=0.011).
The PETIT2 trial enrolled 92 patients with chronic ITP who were randomized (2:1) to receive eltrombopag or placebo for 13 weeks. The eltrombopag
dose was titrated to a target platelet count of 50-200 x109/L.
The primary efficacy endpoint was the proportion of subjects who achieve platelet counts of 50 x109/L or higher for at least 6 out of 8 weeks, between weeks 5 and 12 of the randomized period.
Significantly more patients in the eltrombopag arm met this endpoint—41.3%—compared to 3.4% of patients in the placebo arm (P<0.001).
PETIT trials: Safety
For both trials, there were 107 eltrombopag-treated patients evaluable for safety.
The most common adverse events occurring more frequently in the eltrombopag arms than the placebo arms were upper respiratory tract infection,
nasopharyngitis, cough, diarrhea, pyrexia, rhinitis, abdominal pain, oropharyngeal pain, toothache, increased ALT or AST, rash, and rhinorrhea.
Serious adverse events were reported in 8% of patients during the randomized part of both trials, although no serious adverse event occurred in more than 1 patient (1%).
An ALT elevation of at least 3 times the upper limit of normal occurred in 5% of eltrombopag-treated patients. Of those patients, 2% had ALT increases
of at least 5 times the upper limit of normal.
There were no deaths or thromboembolic events during either study.
Eltrombopag is marketed as Promacta in the US and Revolade in most countries outside the US. For more information on the drug, see the full prescribing information. ![]()
IDH1 inhibitor gets orphan designation for AML

The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for AG-120 to treat patients with acute myeloid leukemia (AML) who harbor an isocitrate dehydrogenase-1 (IDH1) mutation.
AG-120 is an oral, selective inhibitor of the mutated IDH1 protein that is under investigation in two phase 1 clinical trials, one in hematologic malignancies and one in advanced solid tumors.
The FDA granted AG-120 fast track designation last month.
“Receiving orphan drug designation for AG-120 is an important milestone as we continue to move this program to late-stage development,” said Chris Bowden, MD, chief medical officer of Agios Pharmaceuticals, Inc., the company developing AG-120.
“We are pleased with the progress we are making in the clinic and look forward to presenting new data from our ongoing phase 1 study of AG-120 at the Congress of the European Hematology Association later this week.”
Phase 1 trial
Results from the phase 1 study of AG-120 in patients with hematologic malignancies were previously presented at the EORTC-NCI-AACR symposium in November 2014.
The data included 17 patients with relapsed and/or refractory AML who had received a median of 2 prior treatments. The patients were scheduled to receive AG-120 in 1 of 4 dose groups: 100 mg twice a day, 300 mg once a day, 500 mg once a day, and 800 mg once a day over continuous, 28-day cycles.
Of the 14 patients evaluable for response, 7 responded. Four patients achieved a complete response, 2 had a complete response in the marrow, and 1 had a partial response.
Responses occurred at all the dose levels tested. The maximum-tolerated dose was not reached. All responding patients were still on AG-120 at the time of presentation, and 1 patient with stable disease remained on the drug.
Researchers said AG-120 was generally well-tolerated. The majority of adverse events were grade 1 and 2. The most common of these were nausea, fatigue, and dyspnea.
Eight patients experienced serious adverse events, but these were primarily related to disease progression.
One patient experienced a dose-limiting toxicity of asymptomatic grade 3 QT prolongation at the highest dose tested, which improved to grade 1 with dose reduction. The patient was in complete remission and remained on AG-120 at the time of presentation.
There were 6 patient deaths, all unrelated to AG-120. Five deaths occurred after patients discontinued treatment due to progressive disease, and 1 patient died due to disease-related intracranial hemorrhage while on treatment.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If the FDA approves AG-120 to treat patients with AML, orphan designation will provide Agios with 7 years of marketing exclusivity in the US. ![]()
There may be room for improvement with VTE prophylaxis, team says

Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Photo courtesy of the CDC
Results of a large, retrospective study suggest a need for more frequent use of post-discharge thromboprophylaxis in colorectal surgery patients.
Although the overall rate of venous thromboembolism (VTE) in this study was low, nearly 40% of the VTEs occurred after hospital discharge.
And discharge prophylaxis was used in a small percentage of patients. So researchers believe this may be an area for improvement in patient care.
The team described this research in JAMA Surgery alongside a related commentary.
The study was conducted by the Colorectal Writing Group for the Surgical Care and Outcomes Assessment Program-Comparative Effectiveness Research Translation Network (SCOAP-CERTAIN) Collaborative.
The group analyzed data from 16,120 patients who underwent colorectal surgery between 2006 and 2011 at 52 hospitals in Washington. The goal was to determine whether the incidence of VTE had changed with evolving prophylaxis patterns.
The researchers found the use of VTE prophylaxis increased significantly during the study period, but there was no significant change in VTE incidence.
The use of perioperative prophylaxis increased from 31.6% (323/1021) to 86.4% (3007/3480). The use of postoperative, in-hospital prophylaxis increased from 59.6% (603/1012) to 91.4% (3223/3527). And the use of discharge prophylaxis increased from 8.6% to 11.7%. Overall, 10.6% of patients (1399/13,230) were discharged on VTE prophylaxis.
The incidence of any VTE up to 90 days after surgery was 2.2% (360/16,120), and 60.6% of these events (218/360) occurred during a patient’s hospital stay.
The unadjusted, 90-day VTE rate increased during the study period, from 1.2% in 2006 to 3.0% in 2011 (P<0.01 for trend). However, there were no significant differences in VTE incidence over time after the researchers adjusted for patient and operative variables (P=0.09).
The researchers also found that patients who underwent abdominal operations had higher rates of 90-day VTE than patients who had pelvic operations—2.5% vs 1.8%. And patients undergoing cancer-related operations had a similar incidence of VTE as patients having operations not related to malignancy—2.1% vs 2.3%.
These results were surprising because previous research suggested that VTE rates tend to be higher among cancer patients and those who undergo pelvic surgery, said study author Scott R. Steele, MD, of Madigan Army Medical Center in Tacoma, Washington.
Dr Steele also noted that this study suggests a low overall rate of VTE in patients who undergo colorectal surgery, but discharge prophylaxis may be an area for quality improvement. Nearly 40% of VTEs occurred after hospital discharge, and only about 11% of patients received discharge prophylaxis.
Still, he said researchers would need to conduct a large-scale, randomized trial to confirm a benefit for discharge prophylaxis in these patients. ![]()
Drug granted orphan status for hemophilia A

The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to the investigational agent ATX-F8-117 for use in patients with hemophilia A.
The drug is designed to prevent the development of factor VIII (FVIII) inhibitors in patients receiving FVIII therapy or treat patients who already have FVIII inhibitors.
ATX-F8-117 received orphan designation from the European Medicines Agency in November 2014.
“These designations emphasize the need for an effective treatment for hemophilia A patients developing factor VIII inhibitors that occurs in approximately 30% of patients,” said Keith Martin, CEO of Apitope, the company developing ATX-F8-117.
“This results in poor clotting of the blood, leading to severe health issues. This orphan drug designation follows extensive preclinical evaluation, and we look forward to advancing a clinical development program for this important medical condition.”
ATX-F8-117 consists of 2 peptides derived from FVIII. Research conducted by Apitope investigators has shown that ATX-F8-117 induces T-cell tolerance toward FVIII in HLA-DRB1*1501 transgenic mice and decreases FVIII inhibitor formation in mice with FVIII neutralizing antibodies.
About orphan designation
The FDA grants orphan designation to encourage companies to develop therapies for diseases that affect fewer than 200,000 individuals in the US.
Orphan designation provides a company with research and development tax credits, an opportunity to obtain grant funding, exemption from FDA application fees, and other benefits.
If ATX-F8-117 is approved to treat patients with hemophilia A, orphan designation will provide Apitope with 7 years of marketing exclusivity in the US. ![]()
The Rural Surgeon: Surgical practice in the Indian Health Service
Only last week I thought to myself: an almost perfect surgery day. A few endoscopy cases, a breast case, a parathyroid adenoma, and a gastrectomy. I remind myself from time to time how fortunate I am to have the diversity of cases that I am afforded by my unique rural location and employment in the Indian Health Service (IHS).
Over 2 decades ago with what seemed to be an upheaval in health care, I decided to either leave surgery altogether or find some alternative to the business side of medicine that I was experiencing in the world of my private surgical practice. It was 1993 and the Health Security Act was being formulated with a task force with a paucity of physician input. It looked like medicine was headed to a period of increasing bureaucracy and decreased autonomy.

While thumbing through one of the recruiting magazines, I noticed an article about an internal medicine physician and his wife, an obstetrician/ gynecologist, who together joined the Indian Health Service. I made some inquiries.
I knew nothing about the Indian Health Service. I had a picture in my mind of a remote barren reservation working with doctors who couldn’t get a job in the real world. What I found was the best career I could have imagined.
I landed in a rural community in Oklahoma. The colleagues that I have come to know have been some of the best I have seen anywhere. I have had the distinct privilege of taking care of patients who are for the most part very grateful for the care I can give them.
I can recall during the interview process I was concerned that as a non-Native, I might not be accepted by the patients in this part of the country. My concerns were dispelled. I have felt accepted and appreciated.
What I found was that in many ways, my Native American patients are similar to the rural patients I have had in private practice. In the Native American culture, elders are respected. Family is very important – not just the nuclear family but the extended family, cousins, and multiple generations. Patients are proud of their heritage. There is a sense of interdependence and connection. I have been blessed to be a part of healing ceremonies that have left a lasting influence on my approach to disease, health, and spirituality.
Many of these patients have limited resources and astounding health burdens. Native Americans are disproportionately afflicted with diabetes, cardiovascular disease, and obesity. Because of these health problems, programs that address these specific issues have been developed within our system. We have a diabetic clinic that includes foot care, eye care, nutritional counseling, general medicine, and pharmacy needs as well as extensive education about prevention and disease control. We have also developed a Healthy Eating for Life Program (HELP) involving a multidisciplinary approach to weight loss that includes a cognitive behavioral health program, one-on-one education with a certified bariatric nurse, support groups, nutritional instruction, and for some patients, surgical intervention.
Patients may access the Indian and Tribal Health Systems regardless of insurance status. While our practice is not totally devoid of the business aspects of medicine, most of the time we are unaware of the patients’ insurance status. Procedures or diagnostic studies that cannot be done onsite are sometimes covered through contract health services.
Our facility was built with the intent of providing health care for the adjacent counties, but by the time it was completed the need had already outstripped the resources. While funding has improved over the years, the rising costs of medical care and increases in the volume of the service population have continued to translate into unmet needs, especially for services not directly provided in our facility.
There are many physicians who have come and gone during my tenure. Some have Indian Health Service scholarship paybacks that they fulfill and move on, and others may be in transition from one greener pasture to another. The surgical service has grown from two surgeons to six. We have a good mix of youth and seasoned doctors, with half the group over 40 and half younger. The gender mix is also balanced with three females and three males.
There is a plethora of pathology. Most of us have carved out niches of surgical interest. We average 150 referrals per week, which translates into plenty to do. There are no turf battles. We have not adopted the hospitalist model. The surgeons here round and follow their own patients, which is great for continuity. Our patients appreciate seeing the same doctor. We are not, however, tethered to the facility. The surgeon on call will graciously cover any patients if needed, and we are fortunate in that we all have similar practice styles. Thus, we have cross coverage by surgeons who think and operate similarly.
We have had the pleasure of hosting both fellowship trained surgeons (vascular and trauma) and general surgeons interested in a rural lifestyle and Native medicine. The facility is also a teaching hospital. An array of students including surgical technicians, Certified Registered Nurse Anesthetist (CRNA) students, residents, medical students, and U.S. Army Special Forces all rotate through our operating rooms.
One of the benefits of the Indian Health Service is that you are part of a system. Sometimes one can forget that point amid the daily work. Going to meetings specifically geared toward IHS issues is often very rewarding. You are a part of something much bigger than your own practice. Progress is defined over time – a decrease in amputation rates as hemoglobin A1C’s improve, a system-wide approach to colorectal cancer screening, the development of a tumor registry that specifically tracks cancer for Native Americans (no longer grouping them under “white” or lost under “other”).
Although we are rural surgeons, we do not work in isolation. All of our providers are board certified. We are currently the only facility in Oklahoma participating in the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP).
In training, we were encouraged to be aggressive and work independently. It was the era of the pyramid system. No one cared how much sleep you got or when you last ate, and yet there was a team approach among the residents. That same independence is fostered here but in a much more conducive environment.
Many of us sought surgical careers because we truly enjoyed being in the operating room, the haven. We liked the technical aspects, the challenges. We joked that we liked our patients asleep, either on early morning rounds or in the surgical suite.
Where once clinic was a necessary evil, I now enjoy the interaction and find I can often do as much for the patient or family in the clinic as I can in the operating room.
I have also found that with the support of administration, we can have an impact on care beyond the individual level. We can affect the health status of an entire population.
There has been much progress in this system over the last 2 decades and there continues to be room for even more. The key may be in the name: Indian Health Service. I would encourage those who think they might find this type of practice intriguing to explore www.ihs.gov and look under career opportunities.
Dr. Hope Baluh is an ACS fellow. She serves as chief of surgery at Cherokee Nation W.W. Hastings Hospital in Oklahoma and is a recent graduate from Johns Hopkins School of Public Health.
Only last week I thought to myself: an almost perfect surgery day. A few endoscopy cases, a breast case, a parathyroid adenoma, and a gastrectomy. I remind myself from time to time how fortunate I am to have the diversity of cases that I am afforded by my unique rural location and employment in the Indian Health Service (IHS).
Over 2 decades ago with what seemed to be an upheaval in health care, I decided to either leave surgery altogether or find some alternative to the business side of medicine that I was experiencing in the world of my private surgical practice. It was 1993 and the Health Security Act was being formulated with a task force with a paucity of physician input. It looked like medicine was headed to a period of increasing bureaucracy and decreased autonomy.
While thumbing through one of the recruiting magazines, I noticed an article about an internal medicine physician and his wife, an obstetrician/ gynecologist, who together joined the Indian Health Service. I made some inquiries.
I knew nothing about the Indian Health Service. I had a picture in my mind of a remote barren reservation working with doctors who couldn’t get a job in the real world. What I found was the best career I could have imagined.
I landed in a rural community in Oklahoma. The colleagues that I have come to know have been some of the best I have seen anywhere. I have had the distinct privilege of taking care of patients who are for the most part very grateful for the care I can give them.
I can recall during the interview process I was concerned that as a non-Native, I might not be accepted by the patients in this part of the country. My concerns were dispelled. I have felt accepted and appreciated.
What I found was that in many ways, my Native American patients are similar to the rural patients I have had in private practice. In the Native American culture, elders are respected. Family is very important – not just the nuclear family but the extended family, cousins, and multiple generations. Patients are proud of their heritage. There is a sense of interdependence and connection. I have been blessed to be a part of healing ceremonies that have left a lasting influence on my approach to disease, health, and spirituality.
Many of these patients have limited resources and astounding health burdens. Native Americans are disproportionately afflicted with diabetes, cardiovascular disease, and obesity. Because of these health problems, programs that address these specific issues have been developed within our system. We have a diabetic clinic that includes foot care, eye care, nutritional counseling, general medicine, and pharmacy needs as well as extensive education about prevention and disease control. We have also developed a Healthy Eating for Life Program (HELP) involving a multidisciplinary approach to weight loss that includes a cognitive behavioral health program, one-on-one education with a certified bariatric nurse, support groups, nutritional instruction, and for some patients, surgical intervention.
Patients may access the Indian and Tribal Health Systems regardless of insurance status. While our practice is not totally devoid of the business aspects of medicine, most of the time we are unaware of the patients’ insurance status. Procedures or diagnostic studies that cannot be done onsite are sometimes covered through contract health services.
Our facility was built with the intent of providing health care for the adjacent counties, but by the time it was completed the need had already outstripped the resources. While funding has improved over the years, the rising costs of medical care and increases in the volume of the service population have continued to translate into unmet needs, especially for services not directly provided in our facility.
There are many physicians who have come and gone during my tenure. Some have Indian Health Service scholarship paybacks that they fulfill and move on, and others may be in transition from one greener pasture to another. The surgical service has grown from two surgeons to six. We have a good mix of youth and seasoned doctors, with half the group over 40 and half younger. The gender mix is also balanced with three females and three males.
There is a plethora of pathology. Most of us have carved out niches of surgical interest. We average 150 referrals per week, which translates into plenty to do. There are no turf battles. We have not adopted the hospitalist model. The surgeons here round and follow their own patients, which is great for continuity. Our patients appreciate seeing the same doctor. We are not, however, tethered to the facility. The surgeon on call will graciously cover any patients if needed, and we are fortunate in that we all have similar practice styles. Thus, we have cross coverage by surgeons who think and operate similarly.
We have had the pleasure of hosting both fellowship trained surgeons (vascular and trauma) and general surgeons interested in a rural lifestyle and Native medicine. The facility is also a teaching hospital. An array of students including surgical technicians, Certified Registered Nurse Anesthetist (CRNA) students, residents, medical students, and U.S. Army Special Forces all rotate through our operating rooms.
One of the benefits of the Indian Health Service is that you are part of a system. Sometimes one can forget that point amid the daily work. Going to meetings specifically geared toward IHS issues is often very rewarding. You are a part of something much bigger than your own practice. Progress is defined over time – a decrease in amputation rates as hemoglobin A1C’s improve, a system-wide approach to colorectal cancer screening, the development of a tumor registry that specifically tracks cancer for Native Americans (no longer grouping them under “white” or lost under “other”).
Although we are rural surgeons, we do not work in isolation. All of our providers are board certified. We are currently the only facility in Oklahoma participating in the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP).
In training, we were encouraged to be aggressive and work independently. It was the era of the pyramid system. No one cared how much sleep you got or when you last ate, and yet there was a team approach among the residents. That same independence is fostered here but in a much more conducive environment.
Many of us sought surgical careers because we truly enjoyed being in the operating room, the haven. We liked the technical aspects, the challenges. We joked that we liked our patients asleep, either on early morning rounds or in the surgical suite.
Where once clinic was a necessary evil, I now enjoy the interaction and find I can often do as much for the patient or family in the clinic as I can in the operating room.
I have also found that with the support of administration, we can have an impact on care beyond the individual level. We can affect the health status of an entire population.
There has been much progress in this system over the last 2 decades and there continues to be room for even more. The key may be in the name: Indian Health Service. I would encourage those who think they might find this type of practice intriguing to explore www.ihs.gov and look under career opportunities.
Dr. Hope Baluh is an ACS fellow. She serves as chief of surgery at Cherokee Nation W.W. Hastings Hospital in Oklahoma and is a recent graduate from Johns Hopkins School of Public Health.
Only last week I thought to myself: an almost perfect surgery day. A few endoscopy cases, a breast case, a parathyroid adenoma, and a gastrectomy. I remind myself from time to time how fortunate I am to have the diversity of cases that I am afforded by my unique rural location and employment in the Indian Health Service (IHS).
Over 2 decades ago with what seemed to be an upheaval in health care, I decided to either leave surgery altogether or find some alternative to the business side of medicine that I was experiencing in the world of my private surgical practice. It was 1993 and the Health Security Act was being formulated with a task force with a paucity of physician input. It looked like medicine was headed to a period of increasing bureaucracy and decreased autonomy.
While thumbing through one of the recruiting magazines, I noticed an article about an internal medicine physician and his wife, an obstetrician/ gynecologist, who together joined the Indian Health Service. I made some inquiries.
I knew nothing about the Indian Health Service. I had a picture in my mind of a remote barren reservation working with doctors who couldn’t get a job in the real world. What I found was the best career I could have imagined.
I landed in a rural community in Oklahoma. The colleagues that I have come to know have been some of the best I have seen anywhere. I have had the distinct privilege of taking care of patients who are for the most part very grateful for the care I can give them.
I can recall during the interview process I was concerned that as a non-Native, I might not be accepted by the patients in this part of the country. My concerns were dispelled. I have felt accepted and appreciated.
What I found was that in many ways, my Native American patients are similar to the rural patients I have had in private practice. In the Native American culture, elders are respected. Family is very important – not just the nuclear family but the extended family, cousins, and multiple generations. Patients are proud of their heritage. There is a sense of interdependence and connection. I have been blessed to be a part of healing ceremonies that have left a lasting influence on my approach to disease, health, and spirituality.
Many of these patients have limited resources and astounding health burdens. Native Americans are disproportionately afflicted with diabetes, cardiovascular disease, and obesity. Because of these health problems, programs that address these specific issues have been developed within our system. We have a diabetic clinic that includes foot care, eye care, nutritional counseling, general medicine, and pharmacy needs as well as extensive education about prevention and disease control. We have also developed a Healthy Eating for Life Program (HELP) involving a multidisciplinary approach to weight loss that includes a cognitive behavioral health program, one-on-one education with a certified bariatric nurse, support groups, nutritional instruction, and for some patients, surgical intervention.
Patients may access the Indian and Tribal Health Systems regardless of insurance status. While our practice is not totally devoid of the business aspects of medicine, most of the time we are unaware of the patients’ insurance status. Procedures or diagnostic studies that cannot be done onsite are sometimes covered through contract health services.
Our facility was built with the intent of providing health care for the adjacent counties, but by the time it was completed the need had already outstripped the resources. While funding has improved over the years, the rising costs of medical care and increases in the volume of the service population have continued to translate into unmet needs, especially for services not directly provided in our facility.
There are many physicians who have come and gone during my tenure. Some have Indian Health Service scholarship paybacks that they fulfill and move on, and others may be in transition from one greener pasture to another. The surgical service has grown from two surgeons to six. We have a good mix of youth and seasoned doctors, with half the group over 40 and half younger. The gender mix is also balanced with three females and three males.
There is a plethora of pathology. Most of us have carved out niches of surgical interest. We average 150 referrals per week, which translates into plenty to do. There are no turf battles. We have not adopted the hospitalist model. The surgeons here round and follow their own patients, which is great for continuity. Our patients appreciate seeing the same doctor. We are not, however, tethered to the facility. The surgeon on call will graciously cover any patients if needed, and we are fortunate in that we all have similar practice styles. Thus, we have cross coverage by surgeons who think and operate similarly.
We have had the pleasure of hosting both fellowship trained surgeons (vascular and trauma) and general surgeons interested in a rural lifestyle and Native medicine. The facility is also a teaching hospital. An array of students including surgical technicians, Certified Registered Nurse Anesthetist (CRNA) students, residents, medical students, and U.S. Army Special Forces all rotate through our operating rooms.
One of the benefits of the Indian Health Service is that you are part of a system. Sometimes one can forget that point amid the daily work. Going to meetings specifically geared toward IHS issues is often very rewarding. You are a part of something much bigger than your own practice. Progress is defined over time – a decrease in amputation rates as hemoglobin A1C’s improve, a system-wide approach to colorectal cancer screening, the development of a tumor registry that specifically tracks cancer for Native Americans (no longer grouping them under “white” or lost under “other”).
Although we are rural surgeons, we do not work in isolation. All of our providers are board certified. We are currently the only facility in Oklahoma participating in the American College of Surgeons National Surgical Quality Improvement Program (ACS-NSQIP).
In training, we were encouraged to be aggressive and work independently. It was the era of the pyramid system. No one cared how much sleep you got or when you last ate, and yet there was a team approach among the residents. That same independence is fostered here but in a much more conducive environment.
Many of us sought surgical careers because we truly enjoyed being in the operating room, the haven. We liked the technical aspects, the challenges. We joked that we liked our patients asleep, either on early morning rounds or in the surgical suite.
Where once clinic was a necessary evil, I now enjoy the interaction and find I can often do as much for the patient or family in the clinic as I can in the operating room.
I have also found that with the support of administration, we can have an impact on care beyond the individual level. We can affect the health status of an entire population.
There has been much progress in this system over the last 2 decades and there continues to be room for even more. The key may be in the name: Indian Health Service. I would encourage those who think they might find this type of practice intriguing to explore www.ihs.gov and look under career opportunities.
Dr. Hope Baluh is an ACS fellow. She serves as chief of surgery at Cherokee Nation W.W. Hastings Hospital in Oklahoma and is a recent graduate from Johns Hopkins School of Public Health.