User login
FDA updates warning about Treanda

Photo by Rhoda Baer
Last March, the US Food and Drug Administration (FDA) issued a statement warning healthcare professionals not to use the chemotherapy drug Treanda Injection (bendamustine hydrochloride) with closed system transfer devices (CSTDs), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Now, the FDA is providing a list of devices that were tested and deemed compatible with the drug (see the tables below).
The devices were tested by Treanda’s manufacturer, Teva Pharmaceuticals.
Treanda is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available in 2 formulations: a solution, Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution), and a lyophilized powder, Treanda for Injection (25 mg/vial or 100 mg/vial lyophilized powder). The information discussed here is referring to compatibility with the solution, Treanda Injection.
Treanda Injection contains N, N-dimethylacetamide (DMA), which is incompatible with devices that contain polycarbonate or ABS. Devices including CSTDs, adapters, and syringes that contain polycarbonate or ABS have been shown to dissolve when they come in contact with DMA in the drug.
This incompatibility leads to device failure, such as leaking, breaking, or operational failure of CSTD components; possible product contamination; and potential serious adverse health consequences to practitioners, such as skin reactions, or to patients, including the risk of small blood vessel blockage if the product is contaminated with dissolved ABS or polycarbonate.
Users should contact device manufacturers prior to using the specific devices listed below to ensure there have been no changes made to the material composition of the devices and that the devices are compatible with Treanda use.
Table 1. The compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, and gloves (based on testing conducted by Teva from February 2015 through June 2015).
| Component tested | Component brand name (part number) |
| Closed system transfer devices (CSTDs) | BD Phaseal System consisting of:
BD Phaseal Protector P14 (REF 515100), BD Phaseal Injector Luer Lock N35 (REF 515003), BD Phaseal Infusion Adapter C100 (REF 515306), BD syringe 5 mL (REF 309646 and 309657) |
| Vial adapters | Baxter CHEMO-AIDE Dispensing Pin (REF 2N9106)
Medimop Swabable Vial Adapter (REF 8070101) Alaris Smartsite (REF 2202E and 2203E) |
| Polypropylene syringes | BD (Becton Dickinson), 5 mL (REF 309646) and 3 mL (REF 309657)
Covidien Monoject, 5 mL (REF 1180600777) and 3 mL (REF 1180300777) B. Braun, 5 mL (REF 4617053V-02) and 3 mL (REF 4610303-02) Air-Tite Norm Jet, 5 mL (REF 4050.X00V0) and 3 mL (REF 4020.X00V0) Medline, 5 mL (REF SYR105010) and 3 mL (REF SYR103010) Terumo, 5 mL (REF SS-05L) |
| Disposable gloves* | ChemoPlus (REF CT0194-1)
EP-Blue (REF 181350) Jackson Safety G29 (REF 49824) NeoPro (REF NPG-888) NitriDerm (REF 182350) Purple (REF 50604) Purple KC 500 (REF 55084) UltraSense EC (REF USE-880) |
*Part numbers reflect a specific size glove used in the compatibility tests.
Table 2. The IV administration set found to be compatible with Treanda Injection after dilution in a 500 mL 0.9% sodium chloride IV infusion bags (based on testing conducted by Teva from February 2015 through June 2015*).
| Component tested | Brand name (part number) |
| IV administration sets | B. Braun Safeline (REF NF3482) and AdditIV (REF V1921)
Baxter DuoVent Spike (REF 2C7575) and Clearlink System (2H8480) BD Phaseal Secondary set (REF 515301) ICU Medical Clave (REF CH3011) |
*Compatibility studies did not include testing with 2.5% dextrose/0.45% sodium chloride injection. However, the results of these studies are not expected to change. So either diluent, 0.9% sodium chloride or 2.5% dextrose/0.45% sodium chloride injection, can be used with Treanda injection.
The FDA required label changes for both the solution and the powder formulations of Treanda to include information for safe preparation and handling for IV administration. See the full prescribing information for details.
For more details on the compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, gloves, and IV administration sets, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo by Rhoda Baer
Last March, the US Food and Drug Administration (FDA) issued a statement warning healthcare professionals not to use the chemotherapy drug Treanda Injection (bendamustine hydrochloride) with closed system transfer devices (CSTDs), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Now, the FDA is providing a list of devices that were tested and deemed compatible with the drug (see the tables below).
The devices were tested by Treanda’s manufacturer, Teva Pharmaceuticals.
Treanda is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available in 2 formulations: a solution, Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution), and a lyophilized powder, Treanda for Injection (25 mg/vial or 100 mg/vial lyophilized powder). The information discussed here is referring to compatibility with the solution, Treanda Injection.
Treanda Injection contains N, N-dimethylacetamide (DMA), which is incompatible with devices that contain polycarbonate or ABS. Devices including CSTDs, adapters, and syringes that contain polycarbonate or ABS have been shown to dissolve when they come in contact with DMA in the drug.
This incompatibility leads to device failure, such as leaking, breaking, or operational failure of CSTD components; possible product contamination; and potential serious adverse health consequences to practitioners, such as skin reactions, or to patients, including the risk of small blood vessel blockage if the product is contaminated with dissolved ABS or polycarbonate.
Users should contact device manufacturers prior to using the specific devices listed below to ensure there have been no changes made to the material composition of the devices and that the devices are compatible with Treanda use.
Table 1. The compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, and gloves (based on testing conducted by Teva from February 2015 through June 2015).
| Component tested | Component brand name (part number) |
| Closed system transfer devices (CSTDs) | BD Phaseal System consisting of:
BD Phaseal Protector P14 (REF 515100), BD Phaseal Injector Luer Lock N35 (REF 515003), BD Phaseal Infusion Adapter C100 (REF 515306), BD syringe 5 mL (REF 309646 and 309657) |
| Vial adapters | Baxter CHEMO-AIDE Dispensing Pin (REF 2N9106)
Medimop Swabable Vial Adapter (REF 8070101) Alaris Smartsite (REF 2202E and 2203E) |
| Polypropylene syringes | BD (Becton Dickinson), 5 mL (REF 309646) and 3 mL (REF 309657)
Covidien Monoject, 5 mL (REF 1180600777) and 3 mL (REF 1180300777) B. Braun, 5 mL (REF 4617053V-02) and 3 mL (REF 4610303-02) Air-Tite Norm Jet, 5 mL (REF 4050.X00V0) and 3 mL (REF 4020.X00V0) Medline, 5 mL (REF SYR105010) and 3 mL (REF SYR103010) Terumo, 5 mL (REF SS-05L) |
| Disposable gloves* | ChemoPlus (REF CT0194-1)
EP-Blue (REF 181350) Jackson Safety G29 (REF 49824) NeoPro (REF NPG-888) NitriDerm (REF 182350) Purple (REF 50604) Purple KC 500 (REF 55084) UltraSense EC (REF USE-880) |
*Part numbers reflect a specific size glove used in the compatibility tests.
Table 2. The IV administration set found to be compatible with Treanda Injection after dilution in a 500 mL 0.9% sodium chloride IV infusion bags (based on testing conducted by Teva from February 2015 through June 2015*).
| Component tested | Brand name (part number) |
| IV administration sets | B. Braun Safeline (REF NF3482) and AdditIV (REF V1921)
Baxter DuoVent Spike (REF 2C7575) and Clearlink System (2H8480) BD Phaseal Secondary set (REF 515301) ICU Medical Clave (REF CH3011) |
*Compatibility studies did not include testing with 2.5% dextrose/0.45% sodium chloride injection. However, the results of these studies are not expected to change. So either diluent, 0.9% sodium chloride or 2.5% dextrose/0.45% sodium chloride injection, can be used with Treanda injection.
The FDA required label changes for both the solution and the powder formulations of Treanda to include information for safe preparation and handling for IV administration. See the full prescribing information for details.
For more details on the compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, gloves, and IV administration sets, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo by Rhoda Baer
Last March, the US Food and Drug Administration (FDA) issued a statement warning healthcare professionals not to use the chemotherapy drug Treanda Injection (bendamustine hydrochloride) with closed system transfer devices (CSTDs), adapters, and syringes containing polycarbonate or acrylonitrile-butadiene-styrene (ABS).
Now, the FDA is providing a list of devices that were tested and deemed compatible with the drug (see the tables below).
The devices were tested by Treanda’s manufacturer, Teva Pharmaceuticals.
Treanda is used to treat patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin lymphoma that has progressed during or within 6 months of treatment with rituximab or a rituximab-containing regimen.
Treanda is available in 2 formulations: a solution, Treanda Injection (45 mg/0.5 mL or 180 mg/2 mL solution), and a lyophilized powder, Treanda for Injection (25 mg/vial or 100 mg/vial lyophilized powder). The information discussed here is referring to compatibility with the solution, Treanda Injection.
Treanda Injection contains N, N-dimethylacetamide (DMA), which is incompatible with devices that contain polycarbonate or ABS. Devices including CSTDs, adapters, and syringes that contain polycarbonate or ABS have been shown to dissolve when they come in contact with DMA in the drug.
This incompatibility leads to device failure, such as leaking, breaking, or operational failure of CSTD components; possible product contamination; and potential serious adverse health consequences to practitioners, such as skin reactions, or to patients, including the risk of small blood vessel blockage if the product is contaminated with dissolved ABS or polycarbonate.
Users should contact device manufacturers prior to using the specific devices listed below to ensure there have been no changes made to the material composition of the devices and that the devices are compatible with Treanda use.
Table 1. The compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, and gloves (based on testing conducted by Teva from February 2015 through June 2015).
| Component tested | Component brand name (part number) |
| Closed system transfer devices (CSTDs) | BD Phaseal System consisting of:
BD Phaseal Protector P14 (REF 515100), BD Phaseal Injector Luer Lock N35 (REF 515003), BD Phaseal Infusion Adapter C100 (REF 515306), BD syringe 5 mL (REF 309646 and 309657) |
| Vial adapters | Baxter CHEMO-AIDE Dispensing Pin (REF 2N9106)
Medimop Swabable Vial Adapter (REF 8070101) Alaris Smartsite (REF 2202E and 2203E) |
| Polypropylene syringes | BD (Becton Dickinson), 5 mL (REF 309646) and 3 mL (REF 309657)
Covidien Monoject, 5 mL (REF 1180600777) and 3 mL (REF 1180300777) B. Braun, 5 mL (REF 4617053V-02) and 3 mL (REF 4610303-02) Air-Tite Norm Jet, 5 mL (REF 4050.X00V0) and 3 mL (REF 4020.X00V0) Medline, 5 mL (REF SYR105010) and 3 mL (REF SYR103010) Terumo, 5 mL (REF SS-05L) |
| Disposable gloves* | ChemoPlus (REF CT0194-1)
EP-Blue (REF 181350) Jackson Safety G29 (REF 49824) NeoPro (REF NPG-888) NitriDerm (REF 182350) Purple (REF 50604) Purple KC 500 (REF 55084) UltraSense EC (REF USE-880) |
*Part numbers reflect a specific size glove used in the compatibility tests.
Table 2. The IV administration set found to be compatible with Treanda Injection after dilution in a 500 mL 0.9% sodium chloride IV infusion bags (based on testing conducted by Teva from February 2015 through June 2015*).
| Component tested | Brand name (part number) |
| IV administration sets | B. Braun Safeline (REF NF3482) and AdditIV (REF V1921)
Baxter DuoVent Spike (REF 2C7575) and Clearlink System (2H8480) BD Phaseal Secondary set (REF 515301) ICU Medical Clave (REF CH3011) |
*Compatibility studies did not include testing with 2.5% dextrose/0.45% sodium chloride injection. However, the results of these studies are not expected to change. So either diluent, 0.9% sodium chloride or 2.5% dextrose/0.45% sodium chloride injection, can be used with Treanda injection.
The FDA required label changes for both the solution and the powder formulations of Treanda to include information for safe preparation and handling for IV administration. See the full prescribing information for details.
For more details on the compatibility of Treanda Injection with specific CSTDs, syringes, vial adapters, gloves, and IV administration sets, see Teva’s Dear Health Care Provider letter.
Adverse events or quality problems associated with the use of Treanda products can be reported to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Drug deemed ‘breakthrough’ for hemophilia A with inhibitors

Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ACE910 to prevent bleeding in hemophilia A patients age 12 and older who have factor VIII inhibitors.
ACE910 is the first factor VIIIa-mimetic bispecific antibody to be investigated for the prophylactic treatment of hemophilia A.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The breakthrough therapy designation for ACE910 was granted based on results of a phase 1 study of ACE910 in patients with severe hemophilia A.
About ACE910
ACE910 is an investigational, humanized, bispecific monoclonal antibody engineered to simultaneously bind factors IXa and X. ACE910 thereby mimics the cofactor function of factor VIII and is designed to promote blood coagulation in hemophilia A patients, regardless of whether they have developed inhibitors to factor VIII.
ACE910 is administered subcutaneously once weekly. As it is distinct in structure from factor VIII, it is not expected to lead to the formation of factor VIII inhibitors.
ACE910 was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Genentech.
ACE910 research
Results of the phase 1 trial suggested that once-weekly, subcutaneous administration of ACE910 can reduce annualized bleeding rates (ABRs) in adults and adolescents with severe hemophilia A, with or without factor VIII inhibitors.
At ISTH 2015 (abstract AS017), researchers presented data on 18 Japanese patients with severe hemophilia A (factor VIII: C<1%, ages 12 to 58 years).
Patients received once-weekly subcutaneous ACE910 at one of the following dose levels: 0.3 mg/kg (cohort 1), 1 mg/kg (cohort 2), or 3 mg/kg (cohort 3). There were 6 patients in each cohort.
The patients were followed for 5.6 months to 18.5 months.
Efficacy
Whether or not they had inhibitors, patients experienced a decrease in ABR with ACE910. The changes in ABR per treatment cohort and according to inhibitor status are as follows:
| Treatment/patient type | N | ABR reduction | Median ABR change |
| Cohort 1 (0.3 mg/kg) without inhibitors | 2/6 | 22.8%-82.7% | 32.5→1.7 |
| Cohort 1 (0.3 mg/kg) with inhibitors | 4/6 | 49.3%-100% | |
| Cohort 2 (1 mg/kg) without inhibitors | 2/6 | 79.6%-100% | 18.3→0 |
| Cohort 2 (1 mg/kg) with inhibitors | 4/6 | 87.0%-100% | |
| Cohort 3 (3 mg/kg) without inhibitors | 3/6 | 0%*-100% | 15.2→0 |
| Cohort 3 (3 mg/kg) with inhibitors | 3/6 | 93.0%-100% |
*One patient did not report bleeding episodes at baseline or during the study.
Safety
There were 93 adverse events. The researchers said all events were of mild or moderate intensity. One patient discontinued ACE910 due to mild injection-site redness.
There were no thromboembolic events, even when ACE910 was given concomitantly with factor VIII products or bypassing agents as episodic treatment for breakthrough bleeds.
Three patients developed anti-ACE910 antibodies, but they did not affect ACE910 pharmacokinetics or pharmacodynamics.
Genentech is planning to initiate a phase 3 trial of ACE910 in patients with hemophilia A and factor VIII inhibitors by the end of 2015, a phase 3 trial in patients without inhibitors in 2016, and a trial in pediatric patients with hemophilia A in 2016. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ACE910 to prevent bleeding in hemophilia A patients age 12 and older who have factor VIII inhibitors.
ACE910 is the first factor VIIIa-mimetic bispecific antibody to be investigated for the prophylactic treatment of hemophilia A.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The breakthrough therapy designation for ACE910 was granted based on results of a phase 1 study of ACE910 in patients with severe hemophilia A.
About ACE910
ACE910 is an investigational, humanized, bispecific monoclonal antibody engineered to simultaneously bind factors IXa and X. ACE910 thereby mimics the cofactor function of factor VIII and is designed to promote blood coagulation in hemophilia A patients, regardless of whether they have developed inhibitors to factor VIII.
ACE910 is administered subcutaneously once weekly. As it is distinct in structure from factor VIII, it is not expected to lead to the formation of factor VIII inhibitors.
ACE910 was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Genentech.
ACE910 research
Results of the phase 1 trial suggested that once-weekly, subcutaneous administration of ACE910 can reduce annualized bleeding rates (ABRs) in adults and adolescents with severe hemophilia A, with or without factor VIII inhibitors.
At ISTH 2015 (abstract AS017), researchers presented data on 18 Japanese patients with severe hemophilia A (factor VIII: C<1%, ages 12 to 58 years).
Patients received once-weekly subcutaneous ACE910 at one of the following dose levels: 0.3 mg/kg (cohort 1), 1 mg/kg (cohort 2), or 3 mg/kg (cohort 3). There were 6 patients in each cohort.
The patients were followed for 5.6 months to 18.5 months.
Efficacy
Whether or not they had inhibitors, patients experienced a decrease in ABR with ACE910. The changes in ABR per treatment cohort and according to inhibitor status are as follows:
| Treatment/patient type | N | ABR reduction | Median ABR change |
| Cohort 1 (0.3 mg/kg) without inhibitors | 2/6 | 22.8%-82.7% | 32.5→1.7 |
| Cohort 1 (0.3 mg/kg) with inhibitors | 4/6 | 49.3%-100% | |
| Cohort 2 (1 mg/kg) without inhibitors | 2/6 | 79.6%-100% | 18.3→0 |
| Cohort 2 (1 mg/kg) with inhibitors | 4/6 | 87.0%-100% | |
| Cohort 3 (3 mg/kg) without inhibitors | 3/6 | 0%*-100% | 15.2→0 |
| Cohort 3 (3 mg/kg) with inhibitors | 3/6 | 93.0%-100% |
*One patient did not report bleeding episodes at baseline or during the study.
Safety
There were 93 adverse events. The researchers said all events were of mild or moderate intensity. One patient discontinued ACE910 due to mild injection-site redness.
There were no thromboembolic events, even when ACE910 was given concomitantly with factor VIII products or bypassing agents as episodic treatment for breakthrough bleeds.
Three patients developed anti-ACE910 antibodies, but they did not affect ACE910 pharmacokinetics or pharmacodynamics.
Genentech is planning to initiate a phase 3 trial of ACE910 in patients with hemophilia A and factor VIII inhibitors by the end of 2015, a phase 3 trial in patients without inhibitors in 2016, and a trial in pediatric patients with hemophilia A in 2016. ![]()
Photo by Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ACE910 to prevent bleeding in hemophilia A patients age 12 and older who have factor VIII inhibitors.
ACE910 is the first factor VIIIa-mimetic bispecific antibody to be investigated for the prophylactic treatment of hemophilia A.
Breakthrough therapy designation is designed to accelerate the development and review of medicines that demonstrate early clinical evidence of a substantial improvement over current treatment options for serious diseases.
The breakthrough therapy designation for ACE910 was granted based on results of a phase 1 study of ACE910 in patients with severe hemophilia A.
About ACE910
ACE910 is an investigational, humanized, bispecific monoclonal antibody engineered to simultaneously bind factors IXa and X. ACE910 thereby mimics the cofactor function of factor VIII and is designed to promote blood coagulation in hemophilia A patients, regardless of whether they have developed inhibitors to factor VIII.
ACE910 is administered subcutaneously once weekly. As it is distinct in structure from factor VIII, it is not expected to lead to the formation of factor VIII inhibitors.
ACE910 was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Genentech.
ACE910 research
Results of the phase 1 trial suggested that once-weekly, subcutaneous administration of ACE910 can reduce annualized bleeding rates (ABRs) in adults and adolescents with severe hemophilia A, with or without factor VIII inhibitors.
At ISTH 2015 (abstract AS017), researchers presented data on 18 Japanese patients with severe hemophilia A (factor VIII: C<1%, ages 12 to 58 years).
Patients received once-weekly subcutaneous ACE910 at one of the following dose levels: 0.3 mg/kg (cohort 1), 1 mg/kg (cohort 2), or 3 mg/kg (cohort 3). There were 6 patients in each cohort.
The patients were followed for 5.6 months to 18.5 months.
Efficacy
Whether or not they had inhibitors, patients experienced a decrease in ABR with ACE910. The changes in ABR per treatment cohort and according to inhibitor status are as follows:
| Treatment/patient type | N | ABR reduction | Median ABR change |
| Cohort 1 (0.3 mg/kg) without inhibitors | 2/6 | 22.8%-82.7% | 32.5→1.7 |
| Cohort 1 (0.3 mg/kg) with inhibitors | 4/6 | 49.3%-100% | |
| Cohort 2 (1 mg/kg) without inhibitors | 2/6 | 79.6%-100% | 18.3→0 |
| Cohort 2 (1 mg/kg) with inhibitors | 4/6 | 87.0%-100% | |
| Cohort 3 (3 mg/kg) without inhibitors | 3/6 | 0%*-100% | 15.2→0 |
| Cohort 3 (3 mg/kg) with inhibitors | 3/6 | 93.0%-100% |
*One patient did not report bleeding episodes at baseline or during the study.
Safety
There were 93 adverse events. The researchers said all events were of mild or moderate intensity. One patient discontinued ACE910 due to mild injection-site redness.
There were no thromboembolic events, even when ACE910 was given concomitantly with factor VIII products or bypassing agents as episodic treatment for breakthrough bleeds.
Three patients developed anti-ACE910 antibodies, but they did not affect ACE910 pharmacokinetics or pharmacodynamics.
Genentech is planning to initiate a phase 3 trial of ACE910 in patients with hemophilia A and factor VIII inhibitors by the end of 2015, a phase 3 trial in patients without inhibitors in 2016, and a trial in pediatric patients with hemophilia A in 2016. ![]()
ESC: Statins reduce postoperative noncardiac surgery event rates
LONDON – Statin therapy, given the week before a host of noncardiac surgical procedures, reduced the postoperative risk for death and cardiac complications at 30 days by 17% when compared with no statin use in a large, international observational study.
Results of the prospective VISION (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) study showed that the primary composite endpoint of all-cause mortality, myocardial injury after noncardiac surgery (MINS), or stroke at 30 days was 11.8% in a propensity-matched cohort of patients, 2,845 of whom were treated with a statin and 4,492 who were not. The relative risk (RR) for this composite endpoint was 0.83 favoring the use of preoperative statins, with a 95% confidence interval (CI) of 0.73-0.95 and a P value of .007.

Perioperative statin vs. no statin use also cut all-cause mortality by 42% (RR, 0.58; 95% CI, 0.40-0.83; P = .003), cardiovascular mortality by 58% (RR, 0.42; 95% CI, 0.23-0.76; P = .004), and MINS by 14% (RR, 0.86; 95% CI, 0.73-0.98; P = .002).
“These study results are hypothesis generating at most,” emphasized Dr. Otavio Berwanger, who presented the findings at the annual congress of the European Society of Cardiology while they were simultaneously published online (Eur Heart J. 2015 Sept. 1. doi: 10.1093/eurheartj/ehv456).
“It is true that, in this large representative cohort of contemporary patients, statins were associated with lower event rates,” added Dr. Berwanger of Hospital do Coração in São Paulo, Brazil, and “together with the previous body of evidence, statins appear to be an interesting and attractive intervention to reduce postoperative events.” A large-scale, randomized trial is needed, however, to answer the question of whether statins should be used preoperatively to prevent postoperative events in noncardiac surgery patients.
Over a 4-year period that started in August 2007, more than 15,000 individuals aged 45 years or older who were undergoing a variety of noncardiac surgical procedures that required regional or a general anesthetic and at least an overnight stay in the hospital were recruited at 12 centers in eight countries in North and South America, Africa, Asia, Australia, and Europe.
One of the objectives of the study was to examine the use of perioperative statins on cardiovascular events at 30 days, and to do this, the VISION investigators identified the patients who had received a statin in the 7 days prior to surgery and then used propensity matching to form a control group of patients that had not received a statin in the week before surgery.
Just under half of the propensity-matched population was male, with an average age of nearly 69 years. Around 70% of the population had hypertension, 9%-10% had a prior stroke, and 13% had active cancer. Surgeries were urgent in about 2% and emergent in 8%. Around a quarter had undergone orthopedic procedures, and 4% were vascular surgeries. Overall, 36% of surgeries were classified as low risk and 39% as other.
One of the limitations of the study, however, is that, despite the propensity matching, there were some variables that remained different between the two groups, with higher rates of coronary artery disease (20% vs. 14%), peripheral vascular disease (8% vs. 5%), diabetes (30% vs. 25%), and preoperative use of aspirin (25% vs. 19%) and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (53% vs. 48%) in the statin- versus nonstatin-treated patients.
Other limitations are that these data are observational and the use of statins could be just a surrogate for unmeasured confounders that relate to prognosis. Information on the type and dosing of statins also was not obtained, and there was no information collected on potential liver or muscle function side effects.
Nevertheless, the VISION investigators noted in the published paper that the results are consistent with those from other observational studies and prior small-scale, randomized trials and so do add important information. They noted that these data were collected prospectively in a broader range of patients and types of surgeries than has been reported previously. In addition, patients were recruited from several countries and were actively monitored for outcomes and events were centrally adjudicated. VISION is also the only study to report on the effects of statins on MINS.
“Another message from our results is that the use of long-term statins is sub-optimal in high cardiovascular risk patients, who should be on long-term lipid-lowering therapy independently of surgery,” the VISION investigators wrote in their report.
The study was funded by Hamilton Health Sciences Corp. at McMaster University. Dr. Berwanger disclosed receiving research contracts from AstraZeneca, Bayer Healthcare, Amgen, Boehringer-Ingelheim, Pfizer, and Roche Diagnostics.
LONDON – Statin therapy, given the week before a host of noncardiac surgical procedures, reduced the postoperative risk for death and cardiac complications at 30 days by 17% when compared with no statin use in a large, international observational study.
Results of the prospective VISION (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) study showed that the primary composite endpoint of all-cause mortality, myocardial injury after noncardiac surgery (MINS), or stroke at 30 days was 11.8% in a propensity-matched cohort of patients, 2,845 of whom were treated with a statin and 4,492 who were not. The relative risk (RR) for this composite endpoint was 0.83 favoring the use of preoperative statins, with a 95% confidence interval (CI) of 0.73-0.95 and a P value of .007.
Perioperative statin vs. no statin use also cut all-cause mortality by 42% (RR, 0.58; 95% CI, 0.40-0.83; P = .003), cardiovascular mortality by 58% (RR, 0.42; 95% CI, 0.23-0.76; P = .004), and MINS by 14% (RR, 0.86; 95% CI, 0.73-0.98; P = .002).
“These study results are hypothesis generating at most,” emphasized Dr. Otavio Berwanger, who presented the findings at the annual congress of the European Society of Cardiology while they were simultaneously published online (Eur Heart J. 2015 Sept. 1. doi: 10.1093/eurheartj/ehv456).
“It is true that, in this large representative cohort of contemporary patients, statins were associated with lower event rates,” added Dr. Berwanger of Hospital do Coração in São Paulo, Brazil, and “together with the previous body of evidence, statins appear to be an interesting and attractive intervention to reduce postoperative events.” A large-scale, randomized trial is needed, however, to answer the question of whether statins should be used preoperatively to prevent postoperative events in noncardiac surgery patients.
Over a 4-year period that started in August 2007, more than 15,000 individuals aged 45 years or older who were undergoing a variety of noncardiac surgical procedures that required regional or a general anesthetic and at least an overnight stay in the hospital were recruited at 12 centers in eight countries in North and South America, Africa, Asia, Australia, and Europe.
One of the objectives of the study was to examine the use of perioperative statins on cardiovascular events at 30 days, and to do this, the VISION investigators identified the patients who had received a statin in the 7 days prior to surgery and then used propensity matching to form a control group of patients that had not received a statin in the week before surgery.
Just under half of the propensity-matched population was male, with an average age of nearly 69 years. Around 70% of the population had hypertension, 9%-10% had a prior stroke, and 13% had active cancer. Surgeries were urgent in about 2% and emergent in 8%. Around a quarter had undergone orthopedic procedures, and 4% were vascular surgeries. Overall, 36% of surgeries were classified as low risk and 39% as other.
One of the limitations of the study, however, is that, despite the propensity matching, there were some variables that remained different between the two groups, with higher rates of coronary artery disease (20% vs. 14%), peripheral vascular disease (8% vs. 5%), diabetes (30% vs. 25%), and preoperative use of aspirin (25% vs. 19%) and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (53% vs. 48%) in the statin- versus nonstatin-treated patients.
Other limitations are that these data are observational and the use of statins could be just a surrogate for unmeasured confounders that relate to prognosis. Information on the type and dosing of statins also was not obtained, and there was no information collected on potential liver or muscle function side effects.
Nevertheless, the VISION investigators noted in the published paper that the results are consistent with those from other observational studies and prior small-scale, randomized trials and so do add important information. They noted that these data were collected prospectively in a broader range of patients and types of surgeries than has been reported previously. In addition, patients were recruited from several countries and were actively monitored for outcomes and events were centrally adjudicated. VISION is also the only study to report on the effects of statins on MINS.
“Another message from our results is that the use of long-term statins is sub-optimal in high cardiovascular risk patients, who should be on long-term lipid-lowering therapy independently of surgery,” the VISION investigators wrote in their report.
The study was funded by Hamilton Health Sciences Corp. at McMaster University. Dr. Berwanger disclosed receiving research contracts from AstraZeneca, Bayer Healthcare, Amgen, Boehringer-Ingelheim, Pfizer, and Roche Diagnostics.
LONDON – Statin therapy, given the week before a host of noncardiac surgical procedures, reduced the postoperative risk for death and cardiac complications at 30 days by 17% when compared with no statin use in a large, international observational study.
Results of the prospective VISION (Vascular Events in Noncardiac Surgery Patients Cohort Evaluation) study showed that the primary composite endpoint of all-cause mortality, myocardial injury after noncardiac surgery (MINS), or stroke at 30 days was 11.8% in a propensity-matched cohort of patients, 2,845 of whom were treated with a statin and 4,492 who were not. The relative risk (RR) for this composite endpoint was 0.83 favoring the use of preoperative statins, with a 95% confidence interval (CI) of 0.73-0.95 and a P value of .007.
Perioperative statin vs. no statin use also cut all-cause mortality by 42% (RR, 0.58; 95% CI, 0.40-0.83; P = .003), cardiovascular mortality by 58% (RR, 0.42; 95% CI, 0.23-0.76; P = .004), and MINS by 14% (RR, 0.86; 95% CI, 0.73-0.98; P = .002).
“These study results are hypothesis generating at most,” emphasized Dr. Otavio Berwanger, who presented the findings at the annual congress of the European Society of Cardiology while they were simultaneously published online (Eur Heart J. 2015 Sept. 1. doi: 10.1093/eurheartj/ehv456).
“It is true that, in this large representative cohort of contemporary patients, statins were associated with lower event rates,” added Dr. Berwanger of Hospital do Coração in São Paulo, Brazil, and “together with the previous body of evidence, statins appear to be an interesting and attractive intervention to reduce postoperative events.” A large-scale, randomized trial is needed, however, to answer the question of whether statins should be used preoperatively to prevent postoperative events in noncardiac surgery patients.
Over a 4-year period that started in August 2007, more than 15,000 individuals aged 45 years or older who were undergoing a variety of noncardiac surgical procedures that required regional or a general anesthetic and at least an overnight stay in the hospital were recruited at 12 centers in eight countries in North and South America, Africa, Asia, Australia, and Europe.
One of the objectives of the study was to examine the use of perioperative statins on cardiovascular events at 30 days, and to do this, the VISION investigators identified the patients who had received a statin in the 7 days prior to surgery and then used propensity matching to form a control group of patients that had not received a statin in the week before surgery.
Just under half of the propensity-matched population was male, with an average age of nearly 69 years. Around 70% of the population had hypertension, 9%-10% had a prior stroke, and 13% had active cancer. Surgeries were urgent in about 2% and emergent in 8%. Around a quarter had undergone orthopedic procedures, and 4% were vascular surgeries. Overall, 36% of surgeries were classified as low risk and 39% as other.
One of the limitations of the study, however, is that, despite the propensity matching, there were some variables that remained different between the two groups, with higher rates of coronary artery disease (20% vs. 14%), peripheral vascular disease (8% vs. 5%), diabetes (30% vs. 25%), and preoperative use of aspirin (25% vs. 19%) and angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (53% vs. 48%) in the statin- versus nonstatin-treated patients.
Other limitations are that these data are observational and the use of statins could be just a surrogate for unmeasured confounders that relate to prognosis. Information on the type and dosing of statins also was not obtained, and there was no information collected on potential liver or muscle function side effects.
Nevertheless, the VISION investigators noted in the published paper that the results are consistent with those from other observational studies and prior small-scale, randomized trials and so do add important information. They noted that these data were collected prospectively in a broader range of patients and types of surgeries than has been reported previously. In addition, patients were recruited from several countries and were actively monitored for outcomes and events were centrally adjudicated. VISION is also the only study to report on the effects of statins on MINS.
“Another message from our results is that the use of long-term statins is sub-optimal in high cardiovascular risk patients, who should be on long-term lipid-lowering therapy independently of surgery,” the VISION investigators wrote in their report.
The study was funded by Hamilton Health Sciences Corp. at McMaster University. Dr. Berwanger disclosed receiving research contracts from AstraZeneca, Bayer Healthcare, Amgen, Boehringer-Ingelheim, Pfizer, and Roche Diagnostics.
AT THE ESC CONGRESS 2015
Key clinical point: Statin therapy, given the week before a host of noncardiac surgical procedures, reduced the postoperative risk for death and cardiac complications at 30 days, but the findings are hypothesis generating and need validation in a randomized controlled clinical trial.
Major finding: The primary composite outcome (all-cause mortality, myocardial injury after noncardiac surgery, or stroke at 30 days) was 11.8% overall in the propensity-matched cohort, with a 17% relative risk reduction favoring the use of statin versus no statin (P = .007).
Data source: The VISION study is an international, prospective, observational study of more than 15,000 patients who underwent noncardiac surgery between 2007 and 2011.
Disclosures: The study was funded by Hamilton Health Sciences Corp. at McMaster University. Dr. Berwanger disclosed receiving research contracts from AstraZeneca, Bayer Healthcare, Amgen, Boehringer-Ingelheim, Pfizer, and Roche Diagnostics.

Appearances can be deceiving
One of the best parts of my job is meeting people. You can tell a lot by a patient’s name, age, ethnicity, speech, dress, or number and nature of his or her tattoos. But there is a lot more that you won’t know until you have a conversation, and that’s the part that always surprises me. Every interaction offers an opportunity to learn something unexpected about the patient or about oneself.
I recently met a lovely young patient with chronic pain. She had some challenges, including being morbidly obese and on welfare. She had a scar across her left forearm – a deep, well-executed, self-inflicted wound requiring 16 stitches. She’d done it as a teenager and readily admitted that it was a tough time in her life.
But when I got to asking her social history, she lit up with pride. When she was down on her luck some years ago, she decided to learn sign language. She then started a business to incorporate sign language into programs for children with learning disabilities. That left me in awe but also surprised at myself for being so surprised.
And how about a nun who, in addition to having rheumatoid arthritis, also had complex regional pain syndrome after foot surgery. For a year and a half, all she could talk about was how painful her foot was, how miserable she was – so much so that I dreaded each visit, knowing it would make me feel inadequate. I discovered later on that my one-dimensional view of this person as patient was quite limited. “Nun” is not her job description. Her job is with the Social Justice Advocacy arm of her congregation, and most recently her work has focused on interpreting Pope Francis’s encyclical on climate change to make it accessible to congregants. Again, a pleasant surprise.
I was raised Catholic: Heaven and hell, good and evil, Immanuel Kant’s moral imperative. But to be totally postmodern about it, I have a great appreciation for how, unless I walk in another person’s shoes, I will never fully understand them and therefore cannot be the judge of them. In fact, those judgments speak more about me than they do of the patient. What a treat, then, that with each patient interaction I shine a light on my own spirit, and get to know myself a little bit better.
Let me end with a little tribute to Oliver Sacks, who devoted his life to shining a light on the complexities of his patients’ minds: “People will make a life in their own terms, whether they are deaf or colorblind or autistic or whatever. And their world will be quite as rich and interesting and full as our world.”
Dr. Chan practices rheumatology in Pawtucket, R.I.
One of the best parts of my job is meeting people. You can tell a lot by a patient’s name, age, ethnicity, speech, dress, or number and nature of his or her tattoos. But there is a lot more that you won’t know until you have a conversation, and that’s the part that always surprises me. Every interaction offers an opportunity to learn something unexpected about the patient or about oneself.
I recently met a lovely young patient with chronic pain. She had some challenges, including being morbidly obese and on welfare. She had a scar across her left forearm – a deep, well-executed, self-inflicted wound requiring 16 stitches. She’d done it as a teenager and readily admitted that it was a tough time in her life.
But when I got to asking her social history, she lit up with pride. When she was down on her luck some years ago, she decided to learn sign language. She then started a business to incorporate sign language into programs for children with learning disabilities. That left me in awe but also surprised at myself for being so surprised.
And how about a nun who, in addition to having rheumatoid arthritis, also had complex regional pain syndrome after foot surgery. For a year and a half, all she could talk about was how painful her foot was, how miserable she was – so much so that I dreaded each visit, knowing it would make me feel inadequate. I discovered later on that my one-dimensional view of this person as patient was quite limited. “Nun” is not her job description. Her job is with the Social Justice Advocacy arm of her congregation, and most recently her work has focused on interpreting Pope Francis’s encyclical on climate change to make it accessible to congregants. Again, a pleasant surprise.
I was raised Catholic: Heaven and hell, good and evil, Immanuel Kant’s moral imperative. But to be totally postmodern about it, I have a great appreciation for how, unless I walk in another person’s shoes, I will never fully understand them and therefore cannot be the judge of them. In fact, those judgments speak more about me than they do of the patient. What a treat, then, that with each patient interaction I shine a light on my own spirit, and get to know myself a little bit better.
Let me end with a little tribute to Oliver Sacks, who devoted his life to shining a light on the complexities of his patients’ minds: “People will make a life in their own terms, whether they are deaf or colorblind or autistic or whatever. And their world will be quite as rich and interesting and full as our world.”
Dr. Chan practices rheumatology in Pawtucket, R.I.
One of the best parts of my job is meeting people. You can tell a lot by a patient’s name, age, ethnicity, speech, dress, or number and nature of his or her tattoos. But there is a lot more that you won’t know until you have a conversation, and that’s the part that always surprises me. Every interaction offers an opportunity to learn something unexpected about the patient or about oneself.
I recently met a lovely young patient with chronic pain. She had some challenges, including being morbidly obese and on welfare. She had a scar across her left forearm – a deep, well-executed, self-inflicted wound requiring 16 stitches. She’d done it as a teenager and readily admitted that it was a tough time in her life.
But when I got to asking her social history, she lit up with pride. When she was down on her luck some years ago, she decided to learn sign language. She then started a business to incorporate sign language into programs for children with learning disabilities. That left me in awe but also surprised at myself for being so surprised.
And how about a nun who, in addition to having rheumatoid arthritis, also had complex regional pain syndrome after foot surgery. For a year and a half, all she could talk about was how painful her foot was, how miserable she was – so much so that I dreaded each visit, knowing it would make me feel inadequate. I discovered later on that my one-dimensional view of this person as patient was quite limited. “Nun” is not her job description. Her job is with the Social Justice Advocacy arm of her congregation, and most recently her work has focused on interpreting Pope Francis’s encyclical on climate change to make it accessible to congregants. Again, a pleasant surprise.
I was raised Catholic: Heaven and hell, good and evil, Immanuel Kant’s moral imperative. But to be totally postmodern about it, I have a great appreciation for how, unless I walk in another person’s shoes, I will never fully understand them and therefore cannot be the judge of them. In fact, those judgments speak more about me than they do of the patient. What a treat, then, that with each patient interaction I shine a light on my own spirit, and get to know myself a little bit better.
Let me end with a little tribute to Oliver Sacks, who devoted his life to shining a light on the complexities of his patients’ minds: “People will make a life in their own terms, whether they are deaf or colorblind or autistic or whatever. And their world will be quite as rich and interesting and full as our world.”
Dr. Chan practices rheumatology in Pawtucket, R.I.

Dermatologists should be central to wound care, expert says
The way Dr. Adam Friedman sees it, dermatologists deserve a prominent place at the table when it comes to the treatment of acute and chronic wounds.
“As masters of the integument, we should be central to wound care, whether it be for research, in terms of developing better technologies, medications, approaches, diagnostics, but also in terms of managing these wounds, given the rich breadth of pathophysiology and biology we learn during our residency and maintain during our continuing education as practicing dermatologists,” said Dr. Friedman of the department of dermatology at George Washington University, Washington.

When the Journal of Drugs in Dermatology invited Dr. Friedman to serve as guest editor for a special feature section on wound care for its July 2015 issue, he jumped at the chance “to give the dermatology community a small taste of what’s going on in the wound healing world.”
Currently, he said, there is wide variability in the types of clinicians leading wound care centers in the United States, with dermatologists often sitting on the sidelines. “At one institution, it may be the vascular surgery service, at others it may be the family medicine service or even the emergency medicine department,” said Dr. Friedman, who is an editorial advisor to Dermatology News.
“That’s a big problem, in that there’s no uniformity from one center to the next in terms of who is expected to and should be taking responsibility for the wound healing service at their institutions. The reality is, it should be an interdisciplinary team, which not only involves dermatology but vascular surgery, nutrition, internal medicine, subspecialties of medicine like rheumatology, and rehab medicine. However, what is happening more often than not is that you’re getting just one or two of these elements, which cannot be as effective because you miss out on a broader, holistic view.”

There are two chief reasons why dermatologists aren’t more involved in wound care management, he continued. One stems from a lack of training on the topic. In one of the abstracts from the special JDD wound care section, researchers led by Dr. Emily Stamell Ruiz conducted an online survey of dermatology residents in the United States, to ask them about their preparedness to care for wounds and to assess the amount and type of training devoted to wound care during residency. Of the 175 respondents, 78% and 85% did not feel prepared to manage acute and chronic wounds, respectively, while 77% felt that more education is needed during their residency (J Drugs Dermatol. 2015;14[7]:716-20). “Residents felt that there was a clinical as well as a didactical gap, so they felt that they needed more training both through lectures as well as in clinics,” said Dr. Ruiz of the department of dermatology at Brigham and Women’s Hospital, Boston. “It’s not just a focal problem, it really is a universal curriculum problem. Future reforms to the current dermatology curriculum to include wound care training could help close the gap in wound care training.”
Another reason why dermatologists aren’t more involved in wound care management is the time commitment, said Dr. Friedman, who is also director of translational research at George Washington. The treatment of chronic wounds is “physically and financially burdensome,” he said. “It takes not only yourself being comfortable with managing the whole patient which includes the wound[s] with a side order of comorbidities, but your support staff as well – having nurses who know how to use the different wound dressings and how to help you with debridement. You need the right infrastructure. It also costs a lot on the provider side to manage wounds. You need a setup where you can get these patients in, have support staff to help with the wound dressings once you’ve identified what’s necessary, and be able to move on to the next patient.”
In another manuscript contained in the JDD special section, Dr. Friedman and his associates retrospectively reviewed the characteristics of 51 patients with burn injuries who were seen by seven different dermatologists at the Einstein-Montefiore division of dermatology from April 2010 to July 2014 (J Drugs Dermatol. 2015;14[7]:721-4). It found that the main mechanism of injury was burn from hot metal (22%), followed by contact with hot liquids (18%). It also found that silver sulfadiazine was the most commonly prescribed treatment, “even though there are considerable data illustrating that its use will delay wound closure and healing (J Invest Dermatol. 2015 May;135[5]:1459-62),” Dr. Friedman said. He went on to note that for patients who suffer an acute burn, “the ability to access a dermatologist is somewhat limited because their schedules are heavily booked well in advance, and the format doesn’t allow for these types of emergencies. More often than not they go to the ED or to primary care. That might not necessarily be the right decision because these are physicians who may not have the necessary training in terms of not only proper burn care, but skin care overall.”
Another manuscript in the special section describes a method in which partial thickness wounds were induced by cryosurgery to create wounds that could facilitate wound healing research and development. For the study, researchers led by Dr. Robert Kirsner, interim chairman of the department of dermatology and cutaneous surgery at the University of Miami, used liquid nitrogen spray to induce freeze injuries on the forearms of eight healthy adult volunteers (J. Drugs Dermatol. 2015;14[7]: 734-8). They delivered the spray onto a target area of a 1-cm circular opening at a distance from the cryodevice to the skin of 0.5-1 cm and implemented several freeze-thaw time cycles by administering pulses that ranged from 3-12 seconds.
After a 24-hour follow-up, Dr. Kirsner and his associates observed that freeze times exceeding 5 seconds caused a majority of study participants to develop blisters, while freeze times exceeding 8 seconds caused uniform blister formation. Time to healing among subjects in the 8-second freeze time group was 12-13 days, while time to healing among those in the 12-second time freeze group was 21 days.
“Cryo-induced wound healing is a little bit slower than you’d expect with a scalpel, but that wasn’t really surprising,” Dr. Kirsner said. “The fact that it healed a little bit slower was a pretty good thing because if everything healed too fast then it couldn’t serve as a model to speed or slow epithelialization. We were quite pleased.” He noted that the model “could be used as a safety test for chronic wound treatment and as an efficacy test for acute wound treatment. It’s relatively inexpensive and a relatively simple technique. If you’re developing a product for widespread use, it’s probably a minor cost in the whole development process.”
Other manuscripts in the JDD special section include a preclinical study using a murine multithermal burn model which found that N-acetylcysteine S-nitrosothiol nanoparticles prevent wound expansion and accelerate burn closure, and a practical, systematic approach to using wound dressings for the wound care novice. Dr. Friedman hopes that the special section not only stimulates further interest in wound care, but that it serves as “a call for action. We really need to be more involved in wound care from the acute and chronic perspective,” he said. “Wound centers around the country should be involving dermatologists. We have so much to offer from bench to bedside because the skin is our thing. I hope this is a reminder that we should be part of this picture.”
Dr. Friedman disclosed that he serves as a consultant for Galderma, Biogen, Aveeno, Intraderm, Puracore, La Roche-Posay, Amgen, Pfizer, PHD Skin Care. He also serves as an advisory board member for Nerium International, Valeant, Nano BioMed, MicroCures, and Novartis, and has received research grants from Valeant. Dr. Ruiz and Dr. Kirsner reported no financial disclosures.
The way Dr. Adam Friedman sees it, dermatologists deserve a prominent place at the table when it comes to the treatment of acute and chronic wounds.
“As masters of the integument, we should be central to wound care, whether it be for research, in terms of developing better technologies, medications, approaches, diagnostics, but also in terms of managing these wounds, given the rich breadth of pathophysiology and biology we learn during our residency and maintain during our continuing education as practicing dermatologists,” said Dr. Friedman of the department of dermatology at George Washington University, Washington.
When the Journal of Drugs in Dermatology invited Dr. Friedman to serve as guest editor for a special feature section on wound care for its July 2015 issue, he jumped at the chance “to give the dermatology community a small taste of what’s going on in the wound healing world.”
Currently, he said, there is wide variability in the types of clinicians leading wound care centers in the United States, with dermatologists often sitting on the sidelines. “At one institution, it may be the vascular surgery service, at others it may be the family medicine service or even the emergency medicine department,” said Dr. Friedman, who is an editorial advisor to Dermatology News.
“That’s a big problem, in that there’s no uniformity from one center to the next in terms of who is expected to and should be taking responsibility for the wound healing service at their institutions. The reality is, it should be an interdisciplinary team, which not only involves dermatology but vascular surgery, nutrition, internal medicine, subspecialties of medicine like rheumatology, and rehab medicine. However, what is happening more often than not is that you’re getting just one or two of these elements, which cannot be as effective because you miss out on a broader, holistic view.”
There are two chief reasons why dermatologists aren’t more involved in wound care management, he continued. One stems from a lack of training on the topic. In one of the abstracts from the special JDD wound care section, researchers led by Dr. Emily Stamell Ruiz conducted an online survey of dermatology residents in the United States, to ask them about their preparedness to care for wounds and to assess the amount and type of training devoted to wound care during residency. Of the 175 respondents, 78% and 85% did not feel prepared to manage acute and chronic wounds, respectively, while 77% felt that more education is needed during their residency (J Drugs Dermatol. 2015;14[7]:716-20). “Residents felt that there was a clinical as well as a didactical gap, so they felt that they needed more training both through lectures as well as in clinics,” said Dr. Ruiz of the department of dermatology at Brigham and Women’s Hospital, Boston. “It’s not just a focal problem, it really is a universal curriculum problem. Future reforms to the current dermatology curriculum to include wound care training could help close the gap in wound care training.”
Another reason why dermatologists aren’t more involved in wound care management is the time commitment, said Dr. Friedman, who is also director of translational research at George Washington. The treatment of chronic wounds is “physically and financially burdensome,” he said. “It takes not only yourself being comfortable with managing the whole patient which includes the wound[s] with a side order of comorbidities, but your support staff as well – having nurses who know how to use the different wound dressings and how to help you with debridement. You need the right infrastructure. It also costs a lot on the provider side to manage wounds. You need a setup where you can get these patients in, have support staff to help with the wound dressings once you’ve identified what’s necessary, and be able to move on to the next patient.”
In another manuscript contained in the JDD special section, Dr. Friedman and his associates retrospectively reviewed the characteristics of 51 patients with burn injuries who were seen by seven different dermatologists at the Einstein-Montefiore division of dermatology from April 2010 to July 2014 (J Drugs Dermatol. 2015;14[7]:721-4). It found that the main mechanism of injury was burn from hot metal (22%), followed by contact with hot liquids (18%). It also found that silver sulfadiazine was the most commonly prescribed treatment, “even though there are considerable data illustrating that its use will delay wound closure and healing (J Invest Dermatol. 2015 May;135[5]:1459-62),” Dr. Friedman said. He went on to note that for patients who suffer an acute burn, “the ability to access a dermatologist is somewhat limited because their schedules are heavily booked well in advance, and the format doesn’t allow for these types of emergencies. More often than not they go to the ED or to primary care. That might not necessarily be the right decision because these are physicians who may not have the necessary training in terms of not only proper burn care, but skin care overall.”
Another manuscript in the special section describes a method in which partial thickness wounds were induced by cryosurgery to create wounds that could facilitate wound healing research and development. For the study, researchers led by Dr. Robert Kirsner, interim chairman of the department of dermatology and cutaneous surgery at the University of Miami, used liquid nitrogen spray to induce freeze injuries on the forearms of eight healthy adult volunteers (J. Drugs Dermatol. 2015;14[7]: 734-8). They delivered the spray onto a target area of a 1-cm circular opening at a distance from the cryodevice to the skin of 0.5-1 cm and implemented several freeze-thaw time cycles by administering pulses that ranged from 3-12 seconds.
After a 24-hour follow-up, Dr. Kirsner and his associates observed that freeze times exceeding 5 seconds caused a majority of study participants to develop blisters, while freeze times exceeding 8 seconds caused uniform blister formation. Time to healing among subjects in the 8-second freeze time group was 12-13 days, while time to healing among those in the 12-second time freeze group was 21 days.
“Cryo-induced wound healing is a little bit slower than you’d expect with a scalpel, but that wasn’t really surprising,” Dr. Kirsner said. “The fact that it healed a little bit slower was a pretty good thing because if everything healed too fast then it couldn’t serve as a model to speed or slow epithelialization. We were quite pleased.” He noted that the model “could be used as a safety test for chronic wound treatment and as an efficacy test for acute wound treatment. It’s relatively inexpensive and a relatively simple technique. If you’re developing a product for widespread use, it’s probably a minor cost in the whole development process.”
Other manuscripts in the JDD special section include a preclinical study using a murine multithermal burn model which found that N-acetylcysteine S-nitrosothiol nanoparticles prevent wound expansion and accelerate burn closure, and a practical, systematic approach to using wound dressings for the wound care novice. Dr. Friedman hopes that the special section not only stimulates further interest in wound care, but that it serves as “a call for action. We really need to be more involved in wound care from the acute and chronic perspective,” he said. “Wound centers around the country should be involving dermatologists. We have so much to offer from bench to bedside because the skin is our thing. I hope this is a reminder that we should be part of this picture.”
Dr. Friedman disclosed that he serves as a consultant for Galderma, Biogen, Aveeno, Intraderm, Puracore, La Roche-Posay, Amgen, Pfizer, PHD Skin Care. He also serves as an advisory board member for Nerium International, Valeant, Nano BioMed, MicroCures, and Novartis, and has received research grants from Valeant. Dr. Ruiz and Dr. Kirsner reported no financial disclosures.
The way Dr. Adam Friedman sees it, dermatologists deserve a prominent place at the table when it comes to the treatment of acute and chronic wounds.
“As masters of the integument, we should be central to wound care, whether it be for research, in terms of developing better technologies, medications, approaches, diagnostics, but also in terms of managing these wounds, given the rich breadth of pathophysiology and biology we learn during our residency and maintain during our continuing education as practicing dermatologists,” said Dr. Friedman of the department of dermatology at George Washington University, Washington.
When the Journal of Drugs in Dermatology invited Dr. Friedman to serve as guest editor for a special feature section on wound care for its July 2015 issue, he jumped at the chance “to give the dermatology community a small taste of what’s going on in the wound healing world.”
Currently, he said, there is wide variability in the types of clinicians leading wound care centers in the United States, with dermatologists often sitting on the sidelines. “At one institution, it may be the vascular surgery service, at others it may be the family medicine service or even the emergency medicine department,” said Dr. Friedman, who is an editorial advisor to Dermatology News.
“That’s a big problem, in that there’s no uniformity from one center to the next in terms of who is expected to and should be taking responsibility for the wound healing service at their institutions. The reality is, it should be an interdisciplinary team, which not only involves dermatology but vascular surgery, nutrition, internal medicine, subspecialties of medicine like rheumatology, and rehab medicine. However, what is happening more often than not is that you’re getting just one or two of these elements, which cannot be as effective because you miss out on a broader, holistic view.”
There are two chief reasons why dermatologists aren’t more involved in wound care management, he continued. One stems from a lack of training on the topic. In one of the abstracts from the special JDD wound care section, researchers led by Dr. Emily Stamell Ruiz conducted an online survey of dermatology residents in the United States, to ask them about their preparedness to care for wounds and to assess the amount and type of training devoted to wound care during residency. Of the 175 respondents, 78% and 85% did not feel prepared to manage acute and chronic wounds, respectively, while 77% felt that more education is needed during their residency (J Drugs Dermatol. 2015;14[7]:716-20). “Residents felt that there was a clinical as well as a didactical gap, so they felt that they needed more training both through lectures as well as in clinics,” said Dr. Ruiz of the department of dermatology at Brigham and Women’s Hospital, Boston. “It’s not just a focal problem, it really is a universal curriculum problem. Future reforms to the current dermatology curriculum to include wound care training could help close the gap in wound care training.”
Another reason why dermatologists aren’t more involved in wound care management is the time commitment, said Dr. Friedman, who is also director of translational research at George Washington. The treatment of chronic wounds is “physically and financially burdensome,” he said. “It takes not only yourself being comfortable with managing the whole patient which includes the wound[s] with a side order of comorbidities, but your support staff as well – having nurses who know how to use the different wound dressings and how to help you with debridement. You need the right infrastructure. It also costs a lot on the provider side to manage wounds. You need a setup where you can get these patients in, have support staff to help with the wound dressings once you’ve identified what’s necessary, and be able to move on to the next patient.”
In another manuscript contained in the JDD special section, Dr. Friedman and his associates retrospectively reviewed the characteristics of 51 patients with burn injuries who were seen by seven different dermatologists at the Einstein-Montefiore division of dermatology from April 2010 to July 2014 (J Drugs Dermatol. 2015;14[7]:721-4). It found that the main mechanism of injury was burn from hot metal (22%), followed by contact with hot liquids (18%). It also found that silver sulfadiazine was the most commonly prescribed treatment, “even though there are considerable data illustrating that its use will delay wound closure and healing (J Invest Dermatol. 2015 May;135[5]:1459-62),” Dr. Friedman said. He went on to note that for patients who suffer an acute burn, “the ability to access a dermatologist is somewhat limited because their schedules are heavily booked well in advance, and the format doesn’t allow for these types of emergencies. More often than not they go to the ED or to primary care. That might not necessarily be the right decision because these are physicians who may not have the necessary training in terms of not only proper burn care, but skin care overall.”
Another manuscript in the special section describes a method in which partial thickness wounds were induced by cryosurgery to create wounds that could facilitate wound healing research and development. For the study, researchers led by Dr. Robert Kirsner, interim chairman of the department of dermatology and cutaneous surgery at the University of Miami, used liquid nitrogen spray to induce freeze injuries on the forearms of eight healthy adult volunteers (J. Drugs Dermatol. 2015;14[7]: 734-8). They delivered the spray onto a target area of a 1-cm circular opening at a distance from the cryodevice to the skin of 0.5-1 cm and implemented several freeze-thaw time cycles by administering pulses that ranged from 3-12 seconds.
After a 24-hour follow-up, Dr. Kirsner and his associates observed that freeze times exceeding 5 seconds caused a majority of study participants to develop blisters, while freeze times exceeding 8 seconds caused uniform blister formation. Time to healing among subjects in the 8-second freeze time group was 12-13 days, while time to healing among those in the 12-second time freeze group was 21 days.
“Cryo-induced wound healing is a little bit slower than you’d expect with a scalpel, but that wasn’t really surprising,” Dr. Kirsner said. “The fact that it healed a little bit slower was a pretty good thing because if everything healed too fast then it couldn’t serve as a model to speed or slow epithelialization. We were quite pleased.” He noted that the model “could be used as a safety test for chronic wound treatment and as an efficacy test for acute wound treatment. It’s relatively inexpensive and a relatively simple technique. If you’re developing a product for widespread use, it’s probably a minor cost in the whole development process.”
Other manuscripts in the JDD special section include a preclinical study using a murine multithermal burn model which found that N-acetylcysteine S-nitrosothiol nanoparticles prevent wound expansion and accelerate burn closure, and a practical, systematic approach to using wound dressings for the wound care novice. Dr. Friedman hopes that the special section not only stimulates further interest in wound care, but that it serves as “a call for action. We really need to be more involved in wound care from the acute and chronic perspective,” he said. “Wound centers around the country should be involving dermatologists. We have so much to offer from bench to bedside because the skin is our thing. I hope this is a reminder that we should be part of this picture.”
Dr. Friedman disclosed that he serves as a consultant for Galderma, Biogen, Aveeno, Intraderm, Puracore, La Roche-Posay, Amgen, Pfizer, PHD Skin Care. He also serves as an advisory board member for Nerium International, Valeant, Nano BioMed, MicroCures, and Novartis, and has received research grants from Valeant. Dr. Ruiz and Dr. Kirsner reported no financial disclosures.
Agent Orange linked to increased risk of MGUS

Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Photo by Graham Colm
Researchers studying stored blood samples from Vietnam War veterans found that exposure to the herbicide Agent Orange was associated with a more than 2-fold increased risk of monoclonal gammopathy of undetermined significance (MGUS).
The team studied samples from US Air Force personnel who conducted aerial herbicide spray missions of Agent Orange during the war and compared them to blood samples from other Air Force vets.
The incidence of MGUS among the vets exposed to Agent Orange was low, at about 7%. But they still had twice the rate of MGUS as the other vets.
The researchers said this finding supports the previously discovered link between pesticides and myelomagenesis.
While the cause of MGUS and multiple myeloma (MM) remains largely unclear, studies have reported an elevated risk of MM among farmers and other agricultural workers. And pesticides have been thought to be the basis for these associations.
To further investigate the link, Ola Landgren, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York, New York, and his colleagues conducted their study of Vietnam vets. The team reported the results in JAMA Oncology.
The researchers studied store blood samples from 958 male vets—479 Operation Ranch Hand vets who were involved in aerial herbicide spray missions and 479 Air Force vets who had similar duties in Southeast Asia during the same time period (1962 to 1971) but were not involved in herbicide spray missions.
The overall prevalence of MGUS was 7.1% in the Operation Ranch Hand vets and 3.1% in the comparison vets, which translates to a 2.4-fold increased risk for MGUS in Operation Ranch Hand vets.
The odds ratio—after the researchers adjusted for confounding factors such as race, age, and body mass index—was 2.37 (P=0.007).
Dr Landgren and his colleagues conceded that this study has limitations, including a lack of women and the potential for unknown confounding factors such as family medical history and civilian occupation.
Still, the researchers said their findings support an association between Agent Orange exposure and myelomagenesis.
In a related editorial, Niklhil C. Munshi, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, said this study has implications beyond MGUS and MM.
“It also highlights the importance of tissue banking that allows investigation of a number of unanswered questions using modern methods,” Dr Munshi wrote. “The emphasis now is to store samples from almost every major study with correlative science in mind, and this is essential if we are to understand disease biology, mechanism of response, and resistance to therapy in the era of targeted therapy and precision medicine.” ![]()
Despite responses, imetelstat won’t move forward in ET
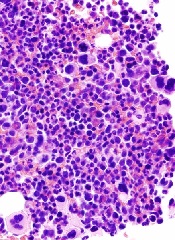
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Although trial results suggest imetelstat is effective against essential thrombocythemia (ET), the telomerase inhibitor is only being developed to treat myelofibrosis (MF).
Imetelstat produced “rapid and durable” responses in a phase 2 trial of ET patients, but the drug also produced side effects that caused the US Food and Drug Administration (FDA) to place a full clinical hold on the drug.
The hold was lifted last November, but the company developing imetelstat decided not to pursue the drug as a treatment for ET or polycythemia vera.
Development continues for MF, however, and results of the pilot study of imetelstat in MF appear in NEJM.
Results from the trial of imetelstat in ET have been published in NEJM as well. Both studies were funded by Geron Corporation, the company developing imetelstat.
The ET trial included 18 patients with a median age of 59.5 (range, 21-83). All patients had received one or more prior treatments, including hydroxyurea (n=17, 94%), anagrelide (n=13, 72%), and interferon (n=4, 22%). Half of patients (n=9) were resistant to at least 1 prior therapy, and 78% (n=14) had experienced unacceptable side effects from a previous therapy.
The patients received imetelstat at an initial dose of 7.5 or 9.4 mg per kilogram of body weight intravenously once a week. Treatment continued until patients attained a platelet count of approximately 250,000 to 300,000 per cubic millimeter.
Responses
All 18 patients experienced hematologic responses, and 16 (89%) had a complete hematologic response. At a median follow-up of 17 months, 10 patients were still receiving treatment, and the median duration of response had not been reached (range, 5 to 30 months).
“[I]metelstat had a clinically significant effect on disease burden in ET patients,” said study investigator David Snyder, MD, of City of Hope in Duarte, California.
“This study was a first look at what happens when you treat ET patients with a drug that has a totally novel mechanism of action.”
Seven of the 8 patients (88%) who were positive for the JAK2 V617F mutation had a molecular response. All of the patients with CALR (n=5) or MPL (n=2) mutations saw a reduction in mutant allele burden, ranging from 15% to 66%.
“The molecular responses suggest that imetelstat may have broad activity across hematologic myeloid malignancies, which warrants further clinical study in other myeloproliferative neoplasms,” said investigator Gabriela M. Baerlocher, MD, of the University of Bern in Switzerland.
Toxicity, clinical hold, and discontinuation
The most common adverse events (≥50%) in this trial were fatigue (83%), diarrhea (78%), nausea (72%), dizziness (61%), increased alanine aminotransferase (ALT, 56%), increased aspartate aminotransferase (AST, 56%), constipation (50%), cough (50%), epistaxis (50%), and headache (50%).
Grade 3/4 adverse events included decreased neutrophil count (22%), neutropenia (22%), anemia (11%), syncope (11%), headache (11%), upper respiratory tract infection (6%), decreased white cell count (6%), myalgia (6%), hypokalemia (6%), fatigue (6%), cellulitis (6%), increased ALT (6%), increased AST (6%), and epistaxis (6%).
All 18 patients had at least one increase in grade, from baseline, in a liver-function value. Seventeen patients had an elevation in ALT, 17 in AST, 15 in alkaline phosphatase, and 8 in total bilirubin. Fourteen patients had persistent abnormalities (≥ 6 weeks), all of which were grade 1 or 2 in severity.
It was these liver-function abnormalities and the potential risk of chronic liver injury that prompted the FDA to place a clinical hold on imetelstat. The agency was concerned about whether the effects were reversible.
It turned out that, in many cases, abnormalities resolved after patients permanently discontinued treatment (as a result of the clinical hold). Sixteen of 17 patients with elevated ALT experienced a resolution, as did 12 of 17 patients with elevated AST, 9 of 15 with elevated alkaline phosphatase, and 7 of 8 with elevated bilirubin.
Of the 14 patients who had persistent abnormalities, 11 had their values reversed to normal or baseline values after stopping imetelstat. The median time to resolution after treatment discontinuation was 12 weeks.
Still, Geron Corporation decided not to develop imetelstat for patients with ET or polycythemia vera. And the FDA said the proposed clinical development plan for the drug, which is focused on MF and other high-risk myeloid disorders, was acceptable. So the clinical hold was lifted. ![]()
Imetelstat in MF: More research needed
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
The telomerase inhibitor imetelstat has exhibited unique activity in a pilot study of patients with intermediate- or high-risk myelofibrosis (MF), but more research is needed, according to investigators.
Imetelstat produced complete and partial responses in a minority of patients and reversed bone marrow fibrosis in complete responders.
However, imetelstat also prompted severe myelosuppression and liver-test abnormalities. And most patients ultimately discontinued treatment.
The investigators therefore concluded that additional research is needed to establish the most effective dosing of the drug, clarify its mechanism of action, and address concerns about toxicity.
Ayalew Tefferi, MD, of the Mayo Clinic in Rochester, Minnesota, and his colleagues reported the results of this trial in NEJM.
A phase 2 trial of imetelstat in patients with essential thrombocythemia was published in NEJM simultaneously. Both trials were sponsored by Geron Corporation, the company developing imetelstat.
The MF study included 33 patients, 18 with primary MF, 10 with post-polycythemia vera MF, and 5 with post-essential thrombocythemia MF. About 52% had high-risk disease, and the rest had intermediate-2-risk MF.
The patients had a median age of 67. About 79% of patients had received prior therapy, and 48% had received a JAK inhibitor. Thirty-nine percent of patients were dependent on red cell transfusions, 64% had constitutional symptoms, 70% had palpable splenomegaly, and 55% had an abnormal karyotype, including 18% with an unfavorable karyotype.
Imetelstat was administered as a 2-hour intravenous infusion, at a starting dose of 9.4 mg per kilogram of body weight. There were 2 dosing schedules: (1) once every 3 weeks or (2) weekly for 4 weeks, followed by once every 3 weeks.
Responses
“We observed that imetelstat was active and induced morphologic and molecular remissions in some patients with myelofibrosis,” Dr Tefferi said. “We also observed that imetelstat demonstrated selective anticlonal activity, inhibiting the growth of cancer cells, which we had not previously documented with other drugs.”
Overall, 21% of patients (7/33) experienced a complete response (n=4) or partial response (n=3) to treatment. The median duration of complete response was 18 months (range, 13-20+), and the median duration of partial response was 10 months (range, 7-10+).
“Some patients treated with imetelstat have reverted back to normal bone marrow,” Dr Tefferi noted. “Typically, myelofibrosis is characterized by marrow scarring, and, although patients may derive symptomatic relief from other treatments, such as ruxolitinib, they usually do not revert back to normal bone marrow.”
Bone marrow fibrosis was reversed in all 4 patients with a complete response. A molecular response was documented in 3 of these patients.
Mutations and telomere length
“We noted a difference in response rates, especially in complete remission rates, in patients with and without certain specific gene mutations, such as ASXL1, SF3B1, and U2AF1,” Dr Tefferi noted. “This underscores the need for laboratory correlative studies in future clinical trials.”
Responses occurred in 27% of patients with a JAK2 mutation and 0% of patients without a JAK2 mutation (P=0.30). Alternatively, responses occurred in 32% of patients without an ASXL1 mutation and 0% of patients with an ASXL1 mutation (P=0.07).
The rate of complete response was 38% among patients with a mutation in SF3B1 or U2AF1, compared to 4% among patients without either mutation (P=0.04).
The investigators also found that responses were not correlated with baseline telomere length.
Toxicity
Dr Tefferi and his colleagues said the most clinically significant side effect of imetelstat was myelosuppression. It was the primary reason for a protocol-mandated dose reduction that occurred in 22 patients (67%).
Another “notable” side effect was the elevation of liver-enzyme levels. The investigators observed treatment-emergent (though not necessarily related) increases from baseline in total bilirubin (49%), alkaline phosphatase (58%), aspartate aminotransferase (58%), and alanine aminotransferase (27%).
None of these abnormalities were linked to clinically overt liver damage, and most patients ultimately saw their values return to baseline levels.
Adverse events that were considered at least possibly related to treatment and occurred in 3 or more patients included thrombocytopenia (45% grade 3/4), anemia (39% overall, 30% grade 3), neutropenia (27% grade 3/4), aspartate aminotransferase elevation (27% grade 1), alkaline phosphatase elevation (21% grade 1/2), elevation in total bilirubin (12% grade 1/2), infusion-related reactions (12% grade 1/2), diarrhea (9% grade 1/2), and epistaxis (9% grade 1/2).
Treatment discontinuation
At the data-cutoff date (December 5, 2014), 76% of patients had discontinued imetelstat (n=25). For all patients, the median duration of treatment was 8.6 months (range, 1.4 to 21.7).
Patients stopped treatment due to insufficient response (n=16), disease progression or relapse after response (n=3), death during the treatment period (n=2), adverse events (n=2), financial constraints (n=1), and pre-existing atrial fibrillation (n=1).
Both patients who discontinued imetelstat due to adverse events had persistent thrombocytopenia. Of the 2 deaths, 1 was considered treatment-related. That patient died of intracranial hemorrhage that was attributed to drug-induced, grade 4 thrombocytopenia after weekly dosing. The non-treatment-related death was the result of an upper gastrointestinal hemorrhage. ![]()
First biosimilar launched in US
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
© Sandoz Inc. 2015
The leukocyte growth factor Zarxio (filgrastim-sndz), the first biosimilar product to gain approval from the US Food and Drug Administration (FDA), is now available in the US.
Zarxio was approved by the FDA on March 6. The product, made by Sandoz, Inc., is biosimilar to Amgen Inc.’s Neupogen, which was originally licensed in 1991.
Zarxio is marketed as Zarzio outside the US. The biosimilar is available in more than 60 countries worldwide.
In the US, Zarxio is approved for the same indications as Neupogen. So Zarxio can be prescribed for the following 5 indications.
Patients with cancer receiving myelosuppressive chemotherapy: to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with nonmyeloid malignancies receiving myelosuppressive anticancer drugs associated with a significant incidence of severe neutropenia with fever.
Patients with acute myeloid leukemia receiving induction or consolidation chemotherapy: to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy.
Patients with cancer undergoing bone marrow transplant: to reduce the duration of neutropenia and neutropenia-related clinical sequelae—eg, febrile neutropenia—in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplant.
Patients undergoing autologous peripheral blood progenitor cell collection and therapy: for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis.
Patients with severe chronic neutropenia: for chronic administration to reduce the incidence and duration of sequelae of neutropenia—eg, fever, infections, oropharyngeal ulcers—in symptomatic patients with congenital neutropenia, cyclic neutropenia, or idiopathic neutropenia.
PIONEER trial
The FDA’s approval of Zarxio was based on data showing that Zarxio is highly similar to Neupogen, with no clinically meaningful differences between the products.
The head-to-head PIONEER study was the final piece of evidence the FDA used to approve Zarxio as biosimilar to Neupogen. Results of the trial were presented at ASH 2014.
Zarxio and Neupogen both produced the expected reduction in the duration of severe neutropenia in breast cancer patients undergoing myelosuppressive chemotherapy—1.17 ± 1.11 and 1.20 ±1.02 days, respectively.
The mean time to absolute neutrophil count recovery in cycle 1 was also similar—1.8 ± 0.97 days in the Zarxio arm and 1.7 ± 0.81 days in the Neupogen arm. No immunogenicity or antibodies against rhG-CSF were detected throughout the study.
The researchers said there were no obvious differences between Zarxio and Neupogen with regard to treatment-emergent adverse events.
The most common side effects observed with Zarxio are aching bones/muscles and redness, swelling, or itching at the injection site. Serious side effects may include spleen rupture; serious allergic reactions that may cause rash, shortness of breath, wheezing and/or swelling around the mouth and eyes; fast pulse and sweating; and acute respiratory distress syndrome.
For more details on Zarxio, see the full prescribing information or visit www.zarxio.com. ![]()
Letter to the Editor
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
We greatly appreciate the thoughtful points made by Dr. Kerman regarding our recently published study evaluating the association of hospitalist continuity on adverse events (AEs).[1] We agree that a 7‐on/7‐off staffing model may limit discontinuity relative to models using shorter rotations lengths. Many hospital medicine programs use a 7‐on/7‐off model to optimize continuity. Longer rotation lengths are uncommon, as they may lead to fatigue and negatively affect physician work‐life balance. Shorter rotation lengths do exist, and we acknowledge that a study in a setting with greater fragmentation may have detected an effect.
We respectfully disagree with Dr. Kerman's concern that our methods for AE detection and confirmation may have been insensitive. We did not rely on incident reports, as these systems suffer from under‐reporting and often represent only a fraction of true AEs. We used a modified version of the classic 2‐stage method to identify and confirm AEs.[2] In the first stage, we used computerized screens, based on criteria from the Harvard Medical Practice Study and Institute for Healthcare Improvement global trigger tool, to identify potential AEs.[3, 4, 5] A research nurse created narrative summaries of potential AEs. A physician researcher then reviewed the narrative summaries to confirm whether an AE was truly present. This time‐consuming method is much more sensitive and specific than other options for patient safety measurement, including administrative data analyses and incident reporting systems.[6, 7]
With respect to other outcomes that may be affected by hospitalist continuity, we recently published a separate study showing that lower inpatient physician continuity was significantly associated with modest increases in hospital costs.[8] We found no association between continuity and patient satisfaction, but were likely underpowered to detect one. Interestingly, some of the models in our study suggested a slightly reduced risk of readmission with lower continuity. We were surprised by this finding and hypothesized that countervailing forces may be at play during handoffs of care from 1 hospitalist to another. Transitions of care introduce the opportunity for critical information to be lost, but they also introduce the potential for patient reassessment. A hospitalist newly taking over care from another may not be anchored to the initial diagnostic impressions and management plan established by the first. Of course, the potential benefit of a reassessment could only occur if the new hospitalist has time to perform one. At extremely high patient volumes, this theoretical benefit is unlikely to exist.
We did not include length of stay (LOS) as an outcome because hospitalist continuity and LOS are interdependent. Although discontinuity may lead to longer LOS, longer LOS definitely increases the probability of discontinuity. Thus, we controlled for LOS in our statistical models to isolate the effect of continuity. The study by Epstein and colleagues did not take into account the interdependence between LOS and hospitalist continuity.[9] Observational studies are not ideal for determining the effect of continuity on LOS. The Combing Incentives and Continuity Leading to Efficiency (CICLE) study by Chandra and colleagues was a pre‐post evaluation of a hospitalist staffing model specifically designed to improve continuity.[10] In the CICLE model, physicians work in a 4‐day rotation. On day 1, physicians exclusively admit patients. On day 2, physicians care for patients admitted on day 1 and accept patients admitted overnight. On days 3 and 4, physicians continue to care for patients received on days 1 and 2, but receive no additional patients. The remaining patients are transitioned to the next physician entering the cycle at the end of day 4. Chandra and colleagues found a 7.5% reduction in LOS and an 8.5% reduction in charges. Interestingly, they also found a 13.5% increase in readmissions that did not achieve statistical significance (P=0.08). The CICLE study suggests continuity does affect LOS, but is limited in that it did not account for a potential preexisting trend toward lower LOS.
Dr. Kerman presents data showing that it takes longer for a physician to care for a patient who is new to him or her than for a patient who is previously known. This finding has face validity. However, as we have suggested, the extra time spent by the oncoming physician may have both advantages and disadvantages. The disadvantages include time‐consuming cognitive work for the physician and the potential for information loss affecting patient care. The potential advantage is a second physician reassessing the diagnosis and management decisions established by the first, potentially correcting errors and optimizing care.
Ultimately, more research is needed to illuminate the effect of hospitalist continuity on patient outcomes. For now, we feel that hospital medicine group leaders need not institute lengthy rotations or staffing models that prioritize continuity above all other factors, as continuity appears to have little impact on patient outcomes.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.
- , , , et al. The effect of hospitalist discontinuity on adverse events. J Hosp Med. 2015;10(3):147–151.
- , , , et al. Comparison of manual abstraction to data warehouse facilitated abstraction to identify hosptial adverse events. BMJ Qual Saf. 2013;22(2):130–138.
- , . IHI global trigger tool for measuring adverse events: IHI innovation series white paper. Cambridge, MA: Institute for Healthcare Improvement; 2007.
- , BA, , et al. A study of medical injury and medical malpractice. N Engl J Med. 1989;321(7):480–484.
- , , , et al. Incidence and types of adverse events and negligent care in Utah and Colorado. Med Care. 2000;38(3):261–271.
- . The elephant of patient safety: what you see depends on how you look. Jt Comm J Qual Patient Saf. 2010;36(9):399–401.
- , . Measuring errors and adverse events in health care. J Gen Intern Med. 2003;18(1):61–67.
- , , , et al. The impact of hospitalist discontinuity on hospital cost, readmissions, and patient satisfaction. J Gen Intern Med. 2014;29(7):1004–1008.
- , , , , . The impact of fragmentation of hospitalist care on length of stay. J Hosp Med. 2010;5(6):335–338.
- , , . The Creating Incentives and Continuity Leading to Efficiency staffing model: a quality improvement initiative in hospital medicine. Mayo Clin Proc. 2012;87(4):364–371.