User login
Combo granted orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
Distress linked to higher risk of death from leukemia, other cancers

patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
patient and her father
Photo by Rhoda Baer
A study published in The BMJ suggests that higher levels of psychological distress (anxiety and depression) may be associated with an increased risk of death from leukemia and other cancers.
The findings are observational, so no firm conclusions about cause and effect can be drawn.
However, the researchers said the findings add to the growing evidence that psychological distress could have some predictive capacity for certain physical conditions.
There is some evidence that psychological distress (anxiety and depression) is related to increased rates of cardiovascular disease, but links with different types of cancer are either unclear or untested.
So David Batty, PhD, of University College London in the UK, and his colleagues set out to examine if psychological distress is a potential predictor of site-specific cancer mortality.
The researchers analyzed data from 16 studies (13 from England and 3 from Scotland), which started between 1994 and 2008. The data included 163,363 men and women age 16 or over who were free from cancer at the start of the study.
Psychological distress scores were measured using the general health questionnaire (GHQ-12), and participants were monitored for an average of 9.5 years. During this time, there were 4353 deaths from cancer.
Several factors that could have influenced the results were taken into account, including age, sex, education, socioeconomic status, body mass index, smoking, and alcohol intake.
“After statistical control for these factors, the results show that, compared with people in the least distressed group, death rates in the most distressed group were consistently higher for cancer of the bowel, prostate, pancreas, and esophagus and for leukemia,” Dr Batty said.
He and his colleagues pointed out that this association may also be affected by reverse causality, where undiagnosed (early) cancer might have had an underlying impact on mood.
In a bid to correct for this, the team conducted a further analysis excluding study participants who died in the first 5 years of follow-up, but this made no difference to the findings. The links between distress and cancer remained.
Specifically, compared to people in the least distressed group (GHQ-12 score 0-6), death rates in the most distressed group (score 7-12) were consistently increased for cancer of all sites combined, with a multivariable adjusted hazard ratio (HR) of 1.32.
Death rates were also increased for colorectal (HR=1.84), prostate (HR=2.42), pancreatic (HR=2.76), and esophageal cancers (HR=2.59), as well as for leukemia (HR=3.86).
“Our findings contribute to the evidence that poor mental health might have some predictive capacity for certain physical diseases,” Dr Batty said, “but we are a long way off from knowing if these relationships are truly causal.” ![]()
Recognize, treat, and teach others to spot hidradenitis suppurativa
MIAMI – Clinicians have many options to treat and help people manage hidradenitis suppurativa, but for most patients, an early and accurate diagnosis remains elusive.
“The problem here is because it has so many mimickers, the diagnosis is often delayed, patients can be [treated for an incorrect diagnosis] and in many ways that treatment can be harmful,” said Adam Friedman, MD, of the George Washington University in Washington. Missed or ignored diagnoses can lead to more pain, impaired function, and wasted time and money.
“There are – no question – gaps in clinical care. Patients seek care outside of dermatology and go to urgent care centers, emergency rooms, [and other settings]. That’s why it’s not only important for us to recognize this, but to teach everyone else as well,” Dr. Friedman said at the Orlando Dermatology Aesthetic and Clinical Conference.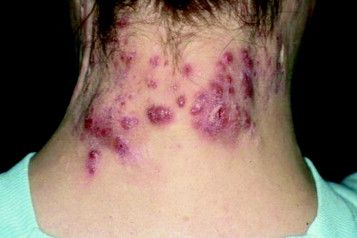
Tips for early detection
Look for chronicity in the disease’s presentation, Dr. Friedman said. “Chronicity is key, but the morphology will change, and lesions will look different over time.” Therefore, “the clinical presentation can be challenging, depending on when you catch the patient.”
Hidradenitis suppurativa is characterized by very purulent, indurated, abscesslike structures often on the underarms and groin. Ask patients where and how often they see lesions. Lesions in certain locations can be very disabling for patients not only because of pain, but also from a psychosocial impact. The groin and chest are prime examples. Also, there is a genetic predisposition so it is important to ask patients about family history as well.
Use combination therapy to hit disease ‘from all angles’
It is imperative to treat patients even when they present at a time of mild disease, Dr. Friedman said. “This is a chronic, snowballing disease that will get worse over time, because inflammation begets inflammation. Even if it’s mild disease, you still want to treat. Combinations are king, and we dermatologists are the synergy masters.”
Effective treatment strategies include medications that curtail inflammation and block hormonal influences; dietary changes (a minimal-dairy, low-carbohydrate diet helps some patients, for example); environmental changes and/or eliminating the invasive proliferative gelatinous mass (IPGM). “This is not step therapy,” Dr. Friedman emphasized. “You want to hit all these angles at once.”
In terms of nutritional support, “I usually put my patients on a combination of zinc and vitamin C, both anti-inflammatories, but also good for wound healing,” Dr. Friedman said. “I also get them on board with V-8, which can be a tough sell sometimes.” He recommends patients drink three small cans per week, adding that he has no financial disclosure related to the vegetable juice.
Patient education, smoking cessation, and keeping affected areas dry and cool are other important management strategies. Instruct patients that stress, friction, and obesity can each worsen the condition “I think obesity is an independent risk factor here, like it is in psoriasis, where obesity alone has been shown to increase the risk of psoriasis later on,” he said.
Ease the inflammation
Hidradenitis suppurativa is a disease of “inappropriate inflammation,” Dr. Friedman said, which explains why anti-inflammatory agents remain the mainstay of treatment. These include antibiotics, classic corticosteroids, and biologics, which all can have a role in therapy. He highlighted the potential role of the cutaneous microbiota and possible dysbiosis associated with disease activity. “I also use a lot chlorhexidine washes to wipe the microbial slate clean in high-risk areas; just be careful to avoid the face.”
Intralesional Kenalog (triamcinolone acetonide), in particular, is useful for its rapid results. “This is such a great and easy trick, and it really works quickly,” Dr. Friedman said. He added that a recent case series provides evidence for its efficacy as well (J Amer Acad Dermatol. 2016 Dec;75:1151-5).
Three take-home treatment pointers
Dr. Friedman shared these three take-aways for treatment of hidradenitis suppurativa with antibiotics:
- The combination of oral clindamycin 300 mg twice daily and oral rifampin 300 mg twice daily carries the most evidence for efficacy and safety, with no evidence of resistance.
- Rifampin also acts against Clostridium difficile infections, which decreases the risk of associated colitis.
- Do not give a tetracycline antibiotic as monotherapy.
In terms of retinoids, “I’ve been pretty disappointed. I find them effective [in other conditions], but for hidradenitis suppurativa, I’m just not impressed, unfortunately,” Dr. Friedman said. “Antihormonal therapies such as oral contraceptives and spironolactone for women and finasteride for men have been a useful adjuncts in my practice, with evidence supporting their use in the literature, he added. Biologics are among the new treatment options, but there are cost and insurance coverage issues, Dr. Friedman said. There are small case series evaluating biologics such as infliximab, which are very supportive – and he himself has had good responses – although the Food and Drug Administration indication has been the hurdle.
One exception is the recent FDA approval of adalimumab (Humira) for hidradenitis suppurativa. “Make sure you realize that the dosing is different, more like a ‘whopping’ Crohn’s disease dose. This is a very important medication in our armamentarium – the issue is when you start it,” Dr. Friedman said. He also cautioned, “if a patient has sinus tracts and scarring, it’s not going to be enough. You still need to address that. Adalimumab is only going to get rid of the inflammation. This is why early diagnosis is so important!”
When it comes to surgery, “my philosophy is it’s all or none. If you’re going to do surgery, do surgery. You better cut these things out. Do a wide global excision,” Dr. Friedman emphasized.
Dr. Friedman is a member of the Dermatology News editorial advisory board.
Dr. Friedman had no relevant financial disclosures.
MIAMI – Clinicians have many options to treat and help people manage hidradenitis suppurativa, but for most patients, an early and accurate diagnosis remains elusive.
“The problem here is because it has so many mimickers, the diagnosis is often delayed, patients can be [treated for an incorrect diagnosis] and in many ways that treatment can be harmful,” said Adam Friedman, MD, of the George Washington University in Washington. Missed or ignored diagnoses can lead to more pain, impaired function, and wasted time and money.
“There are – no question – gaps in clinical care. Patients seek care outside of dermatology and go to urgent care centers, emergency rooms, [and other settings]. That’s why it’s not only important for us to recognize this, but to teach everyone else as well,” Dr. Friedman said at the Orlando Dermatology Aesthetic and Clinical Conference.
Tips for early detection
Look for chronicity in the disease’s presentation, Dr. Friedman said. “Chronicity is key, but the morphology will change, and lesions will look different over time.” Therefore, “the clinical presentation can be challenging, depending on when you catch the patient.”
Hidradenitis suppurativa is characterized by very purulent, indurated, abscesslike structures often on the underarms and groin. Ask patients where and how often they see lesions. Lesions in certain locations can be very disabling for patients not only because of pain, but also from a psychosocial impact. The groin and chest are prime examples. Also, there is a genetic predisposition so it is important to ask patients about family history as well.
Use combination therapy to hit disease ‘from all angles’
It is imperative to treat patients even when they present at a time of mild disease, Dr. Friedman said. “This is a chronic, snowballing disease that will get worse over time, because inflammation begets inflammation. Even if it’s mild disease, you still want to treat. Combinations are king, and we dermatologists are the synergy masters.”
Effective treatment strategies include medications that curtail inflammation and block hormonal influences; dietary changes (a minimal-dairy, low-carbohydrate diet helps some patients, for example); environmental changes and/or eliminating the invasive proliferative gelatinous mass (IPGM). “This is not step therapy,” Dr. Friedman emphasized. “You want to hit all these angles at once.”
In terms of nutritional support, “I usually put my patients on a combination of zinc and vitamin C, both anti-inflammatories, but also good for wound healing,” Dr. Friedman said. “I also get them on board with V-8, which can be a tough sell sometimes.” He recommends patients drink three small cans per week, adding that he has no financial disclosure related to the vegetable juice.
Patient education, smoking cessation, and keeping affected areas dry and cool are other important management strategies. Instruct patients that stress, friction, and obesity can each worsen the condition “I think obesity is an independent risk factor here, like it is in psoriasis, where obesity alone has been shown to increase the risk of psoriasis later on,” he said.
Ease the inflammation
Hidradenitis suppurativa is a disease of “inappropriate inflammation,” Dr. Friedman said, which explains why anti-inflammatory agents remain the mainstay of treatment. These include antibiotics, classic corticosteroids, and biologics, which all can have a role in therapy. He highlighted the potential role of the cutaneous microbiota and possible dysbiosis associated with disease activity. “I also use a lot chlorhexidine washes to wipe the microbial slate clean in high-risk areas; just be careful to avoid the face.”
Intralesional Kenalog (triamcinolone acetonide), in particular, is useful for its rapid results. “This is such a great and easy trick, and it really works quickly,” Dr. Friedman said. He added that a recent case series provides evidence for its efficacy as well (J Amer Acad Dermatol. 2016 Dec;75:1151-5).
Three take-home treatment pointers
Dr. Friedman shared these three take-aways for treatment of hidradenitis suppurativa with antibiotics:
- The combination of oral clindamycin 300 mg twice daily and oral rifampin 300 mg twice daily carries the most evidence for efficacy and safety, with no evidence of resistance.
- Rifampin also acts against Clostridium difficile infections, which decreases the risk of associated colitis.
- Do not give a tetracycline antibiotic as monotherapy.
In terms of retinoids, “I’ve been pretty disappointed. I find them effective [in other conditions], but for hidradenitis suppurativa, I’m just not impressed, unfortunately,” Dr. Friedman said. “Antihormonal therapies such as oral contraceptives and spironolactone for women and finasteride for men have been a useful adjuncts in my practice, with evidence supporting their use in the literature, he added. Biologics are among the new treatment options, but there are cost and insurance coverage issues, Dr. Friedman said. There are small case series evaluating biologics such as infliximab, which are very supportive – and he himself has had good responses – although the Food and Drug Administration indication has been the hurdle.
One exception is the recent FDA approval of adalimumab (Humira) for hidradenitis suppurativa. “Make sure you realize that the dosing is different, more like a ‘whopping’ Crohn’s disease dose. This is a very important medication in our armamentarium – the issue is when you start it,” Dr. Friedman said. He also cautioned, “if a patient has sinus tracts and scarring, it’s not going to be enough. You still need to address that. Adalimumab is only going to get rid of the inflammation. This is why early diagnosis is so important!”
When it comes to surgery, “my philosophy is it’s all or none. If you’re going to do surgery, do surgery. You better cut these things out. Do a wide global excision,” Dr. Friedman emphasized.
Dr. Friedman is a member of the Dermatology News editorial advisory board.
Dr. Friedman had no relevant financial disclosures.
MIAMI – Clinicians have many options to treat and help people manage hidradenitis suppurativa, but for most patients, an early and accurate diagnosis remains elusive.
“The problem here is because it has so many mimickers, the diagnosis is often delayed, patients can be [treated for an incorrect diagnosis] and in many ways that treatment can be harmful,” said Adam Friedman, MD, of the George Washington University in Washington. Missed or ignored diagnoses can lead to more pain, impaired function, and wasted time and money.
“There are – no question – gaps in clinical care. Patients seek care outside of dermatology and go to urgent care centers, emergency rooms, [and other settings]. That’s why it’s not only important for us to recognize this, but to teach everyone else as well,” Dr. Friedman said at the Orlando Dermatology Aesthetic and Clinical Conference.
Tips for early detection
Look for chronicity in the disease’s presentation, Dr. Friedman said. “Chronicity is key, but the morphology will change, and lesions will look different over time.” Therefore, “the clinical presentation can be challenging, depending on when you catch the patient.”
Hidradenitis suppurativa is characterized by very purulent, indurated, abscesslike structures often on the underarms and groin. Ask patients where and how often they see lesions. Lesions in certain locations can be very disabling for patients not only because of pain, but also from a psychosocial impact. The groin and chest are prime examples. Also, there is a genetic predisposition so it is important to ask patients about family history as well.
Use combination therapy to hit disease ‘from all angles’
It is imperative to treat patients even when they present at a time of mild disease, Dr. Friedman said. “This is a chronic, snowballing disease that will get worse over time, because inflammation begets inflammation. Even if it’s mild disease, you still want to treat. Combinations are king, and we dermatologists are the synergy masters.”
Effective treatment strategies include medications that curtail inflammation and block hormonal influences; dietary changes (a minimal-dairy, low-carbohydrate diet helps some patients, for example); environmental changes and/or eliminating the invasive proliferative gelatinous mass (IPGM). “This is not step therapy,” Dr. Friedman emphasized. “You want to hit all these angles at once.”
In terms of nutritional support, “I usually put my patients on a combination of zinc and vitamin C, both anti-inflammatories, but also good for wound healing,” Dr. Friedman said. “I also get them on board with V-8, which can be a tough sell sometimes.” He recommends patients drink three small cans per week, adding that he has no financial disclosure related to the vegetable juice.
Patient education, smoking cessation, and keeping affected areas dry and cool are other important management strategies. Instruct patients that stress, friction, and obesity can each worsen the condition “I think obesity is an independent risk factor here, like it is in psoriasis, where obesity alone has been shown to increase the risk of psoriasis later on,” he said.
Ease the inflammation
Hidradenitis suppurativa is a disease of “inappropriate inflammation,” Dr. Friedman said, which explains why anti-inflammatory agents remain the mainstay of treatment. These include antibiotics, classic corticosteroids, and biologics, which all can have a role in therapy. He highlighted the potential role of the cutaneous microbiota and possible dysbiosis associated with disease activity. “I also use a lot chlorhexidine washes to wipe the microbial slate clean in high-risk areas; just be careful to avoid the face.”
Intralesional Kenalog (triamcinolone acetonide), in particular, is useful for its rapid results. “This is such a great and easy trick, and it really works quickly,” Dr. Friedman said. He added that a recent case series provides evidence for its efficacy as well (J Amer Acad Dermatol. 2016 Dec;75:1151-5).
Three take-home treatment pointers
Dr. Friedman shared these three take-aways for treatment of hidradenitis suppurativa with antibiotics:
- The combination of oral clindamycin 300 mg twice daily and oral rifampin 300 mg twice daily carries the most evidence for efficacy and safety, with no evidence of resistance.
- Rifampin also acts against Clostridium difficile infections, which decreases the risk of associated colitis.
- Do not give a tetracycline antibiotic as monotherapy.
In terms of retinoids, “I’ve been pretty disappointed. I find them effective [in other conditions], but for hidradenitis suppurativa, I’m just not impressed, unfortunately,” Dr. Friedman said. “Antihormonal therapies such as oral contraceptives and spironolactone for women and finasteride for men have been a useful adjuncts in my practice, with evidence supporting their use in the literature, he added. Biologics are among the new treatment options, but there are cost and insurance coverage issues, Dr. Friedman said. There are small case series evaluating biologics such as infliximab, which are very supportive – and he himself has had good responses – although the Food and Drug Administration indication has been the hurdle.
One exception is the recent FDA approval of adalimumab (Humira) for hidradenitis suppurativa. “Make sure you realize that the dosing is different, more like a ‘whopping’ Crohn’s disease dose. This is a very important medication in our armamentarium – the issue is when you start it,” Dr. Friedman said. He also cautioned, “if a patient has sinus tracts and scarring, it’s not going to be enough. You still need to address that. Adalimumab is only going to get rid of the inflammation. This is why early diagnosis is so important!”
When it comes to surgery, “my philosophy is it’s all or none. If you’re going to do surgery, do surgery. You better cut these things out. Do a wide global excision,” Dr. Friedman emphasized.
Dr. Friedman is a member of the Dermatology News editorial advisory board.
Dr. Friedman had no relevant financial disclosures.
EXPERT ANALYSIS FROM ODAC 2017
Endoscopic resection alone sufficed in many T1 colorectal cancers
Patients with T1 colorectal cancer might not benefit from additional surgery after endoscopic resection unless they have positive or indeterminate resection margins or high-risk histology, according to a retrospective, population-based study of 1,315 patients.
After a median follow-up of 6.6 years, the rates of colorectal cancer (CRC) recurrence were 6.2% in patients who underwent endoscopic resection only and 6.4% in patients who also had additional surgery (P = .9), reported Tim D.G. Belderbos, MD, of University Medical Center Utrecht (the Netherlands). Rates of local recurrence also were similar between these groups (4.1% and 3.7%, P = .3), he and his associates reported in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.08.041).
Among high-risk patients, however, the rates of overall recurrence were 14% with endoscopic resection only and 7% with endoscopic resection plus additional surgery (P = .06), and the rates of local recurrence were 12% and 1%, respectively (P = .004). “Based on our study, we recommend performing additional surgery after initial endoscopic resection in cases of high-risk T1 CRC, determined by high-risk histology and/or positive resection margins,” the researchers concluded. Invasive CRCs confined to the colonic submucosa (T1 CRC) present a treatment dilemma – they are usually cured by complete endoscopic resection, but up to 13% involve lymph node metastases and need additional surgery, the investigators noted. To identify predictors of recurrence and metastasis, they studied all patients diagnosed with T1 CRC in the Southeast Netherlands from 1995 through 2011. A total of 370 patients (28%) underwent endoscopic resection only, 220 (17%) underwent endoscopic resection with additional surgery, and 725 (55%) had an initial surgical resection.
Surgery after endoscopic resection was more likely when patients had positive or doubtful resection margins (P less than .001), and this link remained significant after high-risk histology, tumor location, time period, age, sex, and comorbidities were controlled for. Endoscopic resection plus surgery did not reduce the risk of recurrence, compared with endoscopic resection only (P = .3), after the investigators accounted for age, sex, year of procedure, tumor location, and margin characteristics. Initial surgery was associated with significantly lower rates of overall and local recurrence, compared with endoscopic resection only, but the differences also lost significance in the multivariable analysis (P = .2).
Only the presence of positive resection margins significantly predicted recurrence among patients undergoing endoscopic resection (hazard ratio, 6.9; 95% confidence interval, 2.3-20.9). Positive or doubtful resection margins also predicted recurrence after initial surgery, with hazard ratios of 13.2 and 3.4, respectively. High-risk histology – that is, poor differentiation, deep submucosal invasion, or lymphangioinvasion – was significantly associated with lymph node metastasis (OR, 2.2; 95% CI, 1.3-3.7; P less than .002), but not with recurrence after resection margins were accounted for. This might result from missing histology data or the fact that patients with high-risk histology tended to undergo surgical rather than endoscopic resection, the researchers said.
They noted several other study limitations, including a lack of details about lesions and procedures. Also, endoscopic submucosal resection was not practiced in the Netherlands during the study period, they said.
The investigators did not report funding sources and had no disclosures.
Patients with T1 colorectal cancer might not benefit from additional surgery after endoscopic resection unless they have positive or indeterminate resection margins or high-risk histology, according to a retrospective, population-based study of 1,315 patients.
After a median follow-up of 6.6 years, the rates of colorectal cancer (CRC) recurrence were 6.2% in patients who underwent endoscopic resection only and 6.4% in patients who also had additional surgery (P = .9), reported Tim D.G. Belderbos, MD, of University Medical Center Utrecht (the Netherlands). Rates of local recurrence also were similar between these groups (4.1% and 3.7%, P = .3), he and his associates reported in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.08.041).
Among high-risk patients, however, the rates of overall recurrence were 14% with endoscopic resection only and 7% with endoscopic resection plus additional surgery (P = .06), and the rates of local recurrence were 12% and 1%, respectively (P = .004). “Based on our study, we recommend performing additional surgery after initial endoscopic resection in cases of high-risk T1 CRC, determined by high-risk histology and/or positive resection margins,” the researchers concluded. Invasive CRCs confined to the colonic submucosa (T1 CRC) present a treatment dilemma – they are usually cured by complete endoscopic resection, but up to 13% involve lymph node metastases and need additional surgery, the investigators noted. To identify predictors of recurrence and metastasis, they studied all patients diagnosed with T1 CRC in the Southeast Netherlands from 1995 through 2011. A total of 370 patients (28%) underwent endoscopic resection only, 220 (17%) underwent endoscopic resection with additional surgery, and 725 (55%) had an initial surgical resection.
Surgery after endoscopic resection was more likely when patients had positive or doubtful resection margins (P less than .001), and this link remained significant after high-risk histology, tumor location, time period, age, sex, and comorbidities were controlled for. Endoscopic resection plus surgery did not reduce the risk of recurrence, compared with endoscopic resection only (P = .3), after the investigators accounted for age, sex, year of procedure, tumor location, and margin characteristics. Initial surgery was associated with significantly lower rates of overall and local recurrence, compared with endoscopic resection only, but the differences also lost significance in the multivariable analysis (P = .2).
Only the presence of positive resection margins significantly predicted recurrence among patients undergoing endoscopic resection (hazard ratio, 6.9; 95% confidence interval, 2.3-20.9). Positive or doubtful resection margins also predicted recurrence after initial surgery, with hazard ratios of 13.2 and 3.4, respectively. High-risk histology – that is, poor differentiation, deep submucosal invasion, or lymphangioinvasion – was significantly associated with lymph node metastasis (OR, 2.2; 95% CI, 1.3-3.7; P less than .002), but not with recurrence after resection margins were accounted for. This might result from missing histology data or the fact that patients with high-risk histology tended to undergo surgical rather than endoscopic resection, the researchers said.
They noted several other study limitations, including a lack of details about lesions and procedures. Also, endoscopic submucosal resection was not practiced in the Netherlands during the study period, they said.
The investigators did not report funding sources and had no disclosures.
Patients with T1 colorectal cancer might not benefit from additional surgery after endoscopic resection unless they have positive or indeterminate resection margins or high-risk histology, according to a retrospective, population-based study of 1,315 patients.
After a median follow-up of 6.6 years, the rates of colorectal cancer (CRC) recurrence were 6.2% in patients who underwent endoscopic resection only and 6.4% in patients who also had additional surgery (P = .9), reported Tim D.G. Belderbos, MD, of University Medical Center Utrecht (the Netherlands). Rates of local recurrence also were similar between these groups (4.1% and 3.7%, P = .3), he and his associates reported in the March issue of Clinical Gastroenterology and Hepatology (doi: 10.1016/j.cgh.2016.08.041).
Among high-risk patients, however, the rates of overall recurrence were 14% with endoscopic resection only and 7% with endoscopic resection plus additional surgery (P = .06), and the rates of local recurrence were 12% and 1%, respectively (P = .004). “Based on our study, we recommend performing additional surgery after initial endoscopic resection in cases of high-risk T1 CRC, determined by high-risk histology and/or positive resection margins,” the researchers concluded. Invasive CRCs confined to the colonic submucosa (T1 CRC) present a treatment dilemma – they are usually cured by complete endoscopic resection, but up to 13% involve lymph node metastases and need additional surgery, the investigators noted. To identify predictors of recurrence and metastasis, they studied all patients diagnosed with T1 CRC in the Southeast Netherlands from 1995 through 2011. A total of 370 patients (28%) underwent endoscopic resection only, 220 (17%) underwent endoscopic resection with additional surgery, and 725 (55%) had an initial surgical resection.
Surgery after endoscopic resection was more likely when patients had positive or doubtful resection margins (P less than .001), and this link remained significant after high-risk histology, tumor location, time period, age, sex, and comorbidities were controlled for. Endoscopic resection plus surgery did not reduce the risk of recurrence, compared with endoscopic resection only (P = .3), after the investigators accounted for age, sex, year of procedure, tumor location, and margin characteristics. Initial surgery was associated with significantly lower rates of overall and local recurrence, compared with endoscopic resection only, but the differences also lost significance in the multivariable analysis (P = .2).
Only the presence of positive resection margins significantly predicted recurrence among patients undergoing endoscopic resection (hazard ratio, 6.9; 95% confidence interval, 2.3-20.9). Positive or doubtful resection margins also predicted recurrence after initial surgery, with hazard ratios of 13.2 and 3.4, respectively. High-risk histology – that is, poor differentiation, deep submucosal invasion, or lymphangioinvasion – was significantly associated with lymph node metastasis (OR, 2.2; 95% CI, 1.3-3.7; P less than .002), but not with recurrence after resection margins were accounted for. This might result from missing histology data or the fact that patients with high-risk histology tended to undergo surgical rather than endoscopic resection, the researchers said.
They noted several other study limitations, including a lack of details about lesions and procedures. Also, endoscopic submucosal resection was not practiced in the Netherlands during the study period, they said.
The investigators did not report funding sources and had no disclosures.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point. Patients with T1 colorectal cancer might not benefit from additional surgery after endoscopic resection unless they have positive or indeterminate resection margins or high-risk histology.
Major finding: After a median follow-up of 6.6 years, rates of CRC recurrence were 6.2% in patients who underwent endoscopic resection only, and 6.4% in patients who also had additional surgery (P = .9). Among high-risk patients, these rates were 14% and 7%, respectively (P = .06).
Data source: A retrospective population-based study of 1,315 patients who underwent endoscopic or surgical resection of T1 colorectal cancer.
Disclosures: The investigators did not report funding sources and had no disclosures.
COBRA trial takes the long view of absorbable biosynthetic mesh outcomes
Absorbable, biosynthetic surgical mesh used to repair ventral hernia defects may be a good alternative to biologic and permanent synthetic mesh products both in terms of long-term durability and cost, according to a longitudinal cohort study.
The results of the COBRA (Complex Open Bioabsorbable Reconstruction of the Abdominal Wall) study published in the January issue of Annals of Surgery represent the longest follow-up of patients in whom this product was used. Lead author of the study, Michael J. Rosen, MD, professor of surgery at Case Western Reserve University, Cleveland, and his colleagues wrote: “Absorbable synthetic mesh has the prospective advantages of a reduced cost, minimal constraints in manufacturing alternative sizes (lengths, widths, and thicknesses), informed consent in certain religious or cultural groups, and ability to be iterative in generational improvements in mesh constructs based on outcome studies, compared with allogeneic or xenogenic mesh.”
Contaminated wounds were present in 77% of participants. About one-fourth of patients had concomitant procedures for fistula takedown; a quarter of the cohort also required the removal of infected, previously placed mesh. More than a fifth of patients required a concomitant repair of both a midline and parastomal hernia; the mean size for the hernia defects was 137 cm2, and the average width was 9 cm.
Placement of the biosynthetic mesh was left to the discretion of the surgeon, but 90% chose retrorectus placement. Primary fascial closure using a single unit of the material was successful in all patients, 68 of whom required concomitant component separation; 21 of these had an external oblique release. Another 50 of these had transversus abdominis release.
At 24 months, when 84% of patients completed follow-up, 17% were found to have a hernia recurrence. Intraperitoneal placement of the material was found to significantly increase the risk of hernia recurrence (P less than or equal to .04). Infections at the surgical site were associated with a higher risk of recurrence (P less than .01). Patient-reported physical and mental quality-of-life scores at 24 months improved significantly from baseline (P less than .05), showing sustained improvement at 6 and 12 months post procedure.
While more studies are needed, the COBRA findings suggest absorbable, biosynthetic mesh compares favorably with biologic mesh when it comes to recurrence. “The Repair of Infected and Contaminated Hernias (RICH) trial is the only long-term, multicentered, prospective trial to evaluate biologic mesh in CDC [Centers for Disease Control and Prevention] class II to IV wounds. The RICH trial reported 66% surgical site occurrence and 28% hernia recurrence after 2 years’ follow-up in patients who underwent ventral hernia repair with a non–cross-linked porcine dermis,” the investigators noted.
Cost is another area in which bioabsorbable synthetic mesh compares favorably with the biologics, not only in terms of savings from fewer recurrences but also the cost of the mesh itself. Biologic mesh can cost $10,000 or more while synthetics run about a quarter of that (Clin Colon Rectal Surg. 2014 Dec; 27[4]:140-8).
In conclusion, the investigators commented that despite the lack of a control group and random assignment in the study, the results “should not be underestimated” when considering alternatives to biologic and costlier, permanent synthetic meshes.
The study was funded by W.L. Gore. The authors had no relevant financial disclosures.
Absorbable, biosynthetic surgical mesh used to repair ventral hernia defects may be a good alternative to biologic and permanent synthetic mesh products both in terms of long-term durability and cost, according to a longitudinal cohort study.
The results of the COBRA (Complex Open Bioabsorbable Reconstruction of the Abdominal Wall) study published in the January issue of Annals of Surgery represent the longest follow-up of patients in whom this product was used. Lead author of the study, Michael J. Rosen, MD, professor of surgery at Case Western Reserve University, Cleveland, and his colleagues wrote: “Absorbable synthetic mesh has the prospective advantages of a reduced cost, minimal constraints in manufacturing alternative sizes (lengths, widths, and thicknesses), informed consent in certain religious or cultural groups, and ability to be iterative in generational improvements in mesh constructs based on outcome studies, compared with allogeneic or xenogenic mesh.”
Contaminated wounds were present in 77% of participants. About one-fourth of patients had concomitant procedures for fistula takedown; a quarter of the cohort also required the removal of infected, previously placed mesh. More than a fifth of patients required a concomitant repair of both a midline and parastomal hernia; the mean size for the hernia defects was 137 cm2, and the average width was 9 cm.
Placement of the biosynthetic mesh was left to the discretion of the surgeon, but 90% chose retrorectus placement. Primary fascial closure using a single unit of the material was successful in all patients, 68 of whom required concomitant component separation; 21 of these had an external oblique release. Another 50 of these had transversus abdominis release.
At 24 months, when 84% of patients completed follow-up, 17% were found to have a hernia recurrence. Intraperitoneal placement of the material was found to significantly increase the risk of hernia recurrence (P less than or equal to .04). Infections at the surgical site were associated with a higher risk of recurrence (P less than .01). Patient-reported physical and mental quality-of-life scores at 24 months improved significantly from baseline (P less than .05), showing sustained improvement at 6 and 12 months post procedure.
While more studies are needed, the COBRA findings suggest absorbable, biosynthetic mesh compares favorably with biologic mesh when it comes to recurrence. “The Repair of Infected and Contaminated Hernias (RICH) trial is the only long-term, multicentered, prospective trial to evaluate biologic mesh in CDC [Centers for Disease Control and Prevention] class II to IV wounds. The RICH trial reported 66% surgical site occurrence and 28% hernia recurrence after 2 years’ follow-up in patients who underwent ventral hernia repair with a non–cross-linked porcine dermis,” the investigators noted.
Cost is another area in which bioabsorbable synthetic mesh compares favorably with the biologics, not only in terms of savings from fewer recurrences but also the cost of the mesh itself. Biologic mesh can cost $10,000 or more while synthetics run about a quarter of that (Clin Colon Rectal Surg. 2014 Dec; 27[4]:140-8).
In conclusion, the investigators commented that despite the lack of a control group and random assignment in the study, the results “should not be underestimated” when considering alternatives to biologic and costlier, permanent synthetic meshes.
The study was funded by W.L. Gore. The authors had no relevant financial disclosures.
Absorbable, biosynthetic surgical mesh used to repair ventral hernia defects may be a good alternative to biologic and permanent synthetic mesh products both in terms of long-term durability and cost, according to a longitudinal cohort study.
The results of the COBRA (Complex Open Bioabsorbable Reconstruction of the Abdominal Wall) study published in the January issue of Annals of Surgery represent the longest follow-up of patients in whom this product was used. Lead author of the study, Michael J. Rosen, MD, professor of surgery at Case Western Reserve University, Cleveland, and his colleagues wrote: “Absorbable synthetic mesh has the prospective advantages of a reduced cost, minimal constraints in manufacturing alternative sizes (lengths, widths, and thicknesses), informed consent in certain religious or cultural groups, and ability to be iterative in generational improvements in mesh constructs based on outcome studies, compared with allogeneic or xenogenic mesh.”
Contaminated wounds were present in 77% of participants. About one-fourth of patients had concomitant procedures for fistula takedown; a quarter of the cohort also required the removal of infected, previously placed mesh. More than a fifth of patients required a concomitant repair of both a midline and parastomal hernia; the mean size for the hernia defects was 137 cm2, and the average width was 9 cm.
Placement of the biosynthetic mesh was left to the discretion of the surgeon, but 90% chose retrorectus placement. Primary fascial closure using a single unit of the material was successful in all patients, 68 of whom required concomitant component separation; 21 of these had an external oblique release. Another 50 of these had transversus abdominis release.
At 24 months, when 84% of patients completed follow-up, 17% were found to have a hernia recurrence. Intraperitoneal placement of the material was found to significantly increase the risk of hernia recurrence (P less than or equal to .04). Infections at the surgical site were associated with a higher risk of recurrence (P less than .01). Patient-reported physical and mental quality-of-life scores at 24 months improved significantly from baseline (P less than .05), showing sustained improvement at 6 and 12 months post procedure.
While more studies are needed, the COBRA findings suggest absorbable, biosynthetic mesh compares favorably with biologic mesh when it comes to recurrence. “The Repair of Infected and Contaminated Hernias (RICH) trial is the only long-term, multicentered, prospective trial to evaluate biologic mesh in CDC [Centers for Disease Control and Prevention] class II to IV wounds. The RICH trial reported 66% surgical site occurrence and 28% hernia recurrence after 2 years’ follow-up in patients who underwent ventral hernia repair with a non–cross-linked porcine dermis,” the investigators noted.
Cost is another area in which bioabsorbable synthetic mesh compares favorably with the biologics, not only in terms of savings from fewer recurrences but also the cost of the mesh itself. Biologic mesh can cost $10,000 or more while synthetics run about a quarter of that (Clin Colon Rectal Surg. 2014 Dec; 27[4]:140-8).
In conclusion, the investigators commented that despite the lack of a control group and random assignment in the study, the results “should not be underestimated” when considering alternatives to biologic and costlier, permanent synthetic meshes.
The study was funded by W.L. Gore. The authors had no relevant financial disclosures.
Key clinical point:
Major finding: At 24 months, 17% of patients were found to have a hernia recurrence.
Data source: An international, multisite, prospective, intention-to-treat cohort analysis of 104 patients with contaminated or noncontaminated hernia defects of at least 9 cm2 in size.
Disclosures: The study was funded by W.L. Gore. The authors had no relevant financial disclosures.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.

“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).

“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Everolimus fails in pretreated gastric cancer
SAN FRANCISCO – Adding everolimus to paclitaxel failed to significantly improve outcomes in pretreated patients with gastric or esophagogastric junction adenocarcinoma in a German randomized phase III study.
Median overall survival in the double-blind multicenter study (RADPAC) was 6.12 months among 150 patients randomized to receive treatment with paclitaxel plus everolimus as 2nd, 3rd, or 4th line therapy, and 5.03 months among those who received paclitaxel and placebo (hazard ratio, 0.93), Salah-Eddin Al-Batran, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
Median progression-free survival was 2.20 vs. 2.07 months in the treatment and placebo groups, respectively (hazard ratio, 0.88), said Dr. Al-Batran of Krankenhaus Nordwest, University Cancer Center, Frankfurt, Germany.
Study subjects had a mean age of 62 years and had progressed after treatment with a fluoropyrimidine/platinum-containing regimen. All had at least one, and a maximum of three prior lines of therapy (median of two in both groups).
Of note, accrual was slow and was stopped early, largely because of the very-high rate of taxane use for first-line treatment, but also because combination ramucirumab/paclitaxel was approved during the course of the study, Dr. Al-Batran said.
The treatment and placebo groups were well balanced. Treatment included 80 mg/m2 of paclitaxel on days 1, 8 and 15, plus placebo or 10 mg of everolimus daily on days 1-28, repeated every 28 days. Dose adjustment was more common in the treatment group (26% vs. 13%) but cumulative doses were similar in the groups.
Also, more patients in the everolimus group discontinued treatment for toxicity (11% vs. 5%). However, the only toxicity that was significantly increased was grade 3-5 oral mucositis in the treatment group (13% vs. 1% in the placebo group).
Gastric cancer is aggressive and difficult to treat, with median survival of only 9-11 months, Dr. Al-Batran said, adding that at the time the RADPAC study was initiated, no treatments had been approved for patients who failed first-line therapy, although agents like paclitaxel and irinotecan were in use.
He and his colleagues sought to evaluate everolimus, because 50%-60% of gastric cancers are driven by dysregulation in the P13k/Akt/mTOR pathway – a key regulator of cell proliferation, growth, survival, metabolism, and angiogenesis, and because everolimus – an oral mTOR inhibitor – showed efficacy in preclinical models of gastric cancer.
In the phase III GRANITE-1 trial, it was associated with trends toward improved progression-free survival and overall survival, compared with best supportive care, he noted.
Subgroup analyses in the current trial suggested that patients with prior taxane use derived greater benefit from everolimus. Overall survival in those patients, who comprised about half of the study population, was 6 months vs. 4 months with placebo; the difference did not reach statistical significance, but showed a strong trend in that direction (P = .072). Progression-free survival was, however, significantly greater with everolimus than with placebo (2.66 vs. 1.81 months; HR, 0.50) in those with prior taxane use.
“Interestingly, the very few patients having ECOG performance status of 2 really performed very poorly,” Dr. Al-Batran said, explaining that those patients had better outcomes with paclitaxel monotherapy.
“So, in conclusion, everolimus combined with paclitaxel improved outcomes as compared with paclitaxel alone in the intention to treat population. However, activity was seen in the taxane pretreated subgroup. Biomarker studies could attempt to identify a subgroup with more benefit, as we see some activity, but this activity is not enough,” he concluded.
Dr. Al-Batran reported receiving honoraria, serving as a consultant or advisor, receiving research funding from, and/or being on the speakers’ bureau for Celgene, Hospira, Lilly, Medac, Merck, Roche, Sanofi, Vifor, and Nordic Bioscience.
SAN FRANCISCO – Adding everolimus to paclitaxel failed to significantly improve outcomes in pretreated patients with gastric or esophagogastric junction adenocarcinoma in a German randomized phase III study.
Median overall survival in the double-blind multicenter study (RADPAC) was 6.12 months among 150 patients randomized to receive treatment with paclitaxel plus everolimus as 2nd, 3rd, or 4th line therapy, and 5.03 months among those who received paclitaxel and placebo (hazard ratio, 0.93), Salah-Eddin Al-Batran, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
Median progression-free survival was 2.20 vs. 2.07 months in the treatment and placebo groups, respectively (hazard ratio, 0.88), said Dr. Al-Batran of Krankenhaus Nordwest, University Cancer Center, Frankfurt, Germany.
Study subjects had a mean age of 62 years and had progressed after treatment with a fluoropyrimidine/platinum-containing regimen. All had at least one, and a maximum of three prior lines of therapy (median of two in both groups).
Of note, accrual was slow and was stopped early, largely because of the very-high rate of taxane use for first-line treatment, but also because combination ramucirumab/paclitaxel was approved during the course of the study, Dr. Al-Batran said.
The treatment and placebo groups were well balanced. Treatment included 80 mg/m2 of paclitaxel on days 1, 8 and 15, plus placebo or 10 mg of everolimus daily on days 1-28, repeated every 28 days. Dose adjustment was more common in the treatment group (26% vs. 13%) but cumulative doses were similar in the groups.
Also, more patients in the everolimus group discontinued treatment for toxicity (11% vs. 5%). However, the only toxicity that was significantly increased was grade 3-5 oral mucositis in the treatment group (13% vs. 1% in the placebo group).
Gastric cancer is aggressive and difficult to treat, with median survival of only 9-11 months, Dr. Al-Batran said, adding that at the time the RADPAC study was initiated, no treatments had been approved for patients who failed first-line therapy, although agents like paclitaxel and irinotecan were in use.
He and his colleagues sought to evaluate everolimus, because 50%-60% of gastric cancers are driven by dysregulation in the P13k/Akt/mTOR pathway – a key regulator of cell proliferation, growth, survival, metabolism, and angiogenesis, and because everolimus – an oral mTOR inhibitor – showed efficacy in preclinical models of gastric cancer.
In the phase III GRANITE-1 trial, it was associated with trends toward improved progression-free survival and overall survival, compared with best supportive care, he noted.
Subgroup analyses in the current trial suggested that patients with prior taxane use derived greater benefit from everolimus. Overall survival in those patients, who comprised about half of the study population, was 6 months vs. 4 months with placebo; the difference did not reach statistical significance, but showed a strong trend in that direction (P = .072). Progression-free survival was, however, significantly greater with everolimus than with placebo (2.66 vs. 1.81 months; HR, 0.50) in those with prior taxane use.
“Interestingly, the very few patients having ECOG performance status of 2 really performed very poorly,” Dr. Al-Batran said, explaining that those patients had better outcomes with paclitaxel monotherapy.
“So, in conclusion, everolimus combined with paclitaxel improved outcomes as compared with paclitaxel alone in the intention to treat population. However, activity was seen in the taxane pretreated subgroup. Biomarker studies could attempt to identify a subgroup with more benefit, as we see some activity, but this activity is not enough,” he concluded.
Dr. Al-Batran reported receiving honoraria, serving as a consultant or advisor, receiving research funding from, and/or being on the speakers’ bureau for Celgene, Hospira, Lilly, Medac, Merck, Roche, Sanofi, Vifor, and Nordic Bioscience.
SAN FRANCISCO – Adding everolimus to paclitaxel failed to significantly improve outcomes in pretreated patients with gastric or esophagogastric junction adenocarcinoma in a German randomized phase III study.
Median overall survival in the double-blind multicenter study (RADPAC) was 6.12 months among 150 patients randomized to receive treatment with paclitaxel plus everolimus as 2nd, 3rd, or 4th line therapy, and 5.03 months among those who received paclitaxel and placebo (hazard ratio, 0.93), Salah-Eddin Al-Batran, MD, reported at the symposium sponsored by ASCO, ASTRO, the American Gastroenterological Association, and the Society of Surgical Oncology.
Median progression-free survival was 2.20 vs. 2.07 months in the treatment and placebo groups, respectively (hazard ratio, 0.88), said Dr. Al-Batran of Krankenhaus Nordwest, University Cancer Center, Frankfurt, Germany.
Study subjects had a mean age of 62 years and had progressed after treatment with a fluoropyrimidine/platinum-containing regimen. All had at least one, and a maximum of three prior lines of therapy (median of two in both groups).
Of note, accrual was slow and was stopped early, largely because of the very-high rate of taxane use for first-line treatment, but also because combination ramucirumab/paclitaxel was approved during the course of the study, Dr. Al-Batran said.
The treatment and placebo groups were well balanced. Treatment included 80 mg/m2 of paclitaxel on days 1, 8 and 15, plus placebo or 10 mg of everolimus daily on days 1-28, repeated every 28 days. Dose adjustment was more common in the treatment group (26% vs. 13%) but cumulative doses were similar in the groups.
Also, more patients in the everolimus group discontinued treatment for toxicity (11% vs. 5%). However, the only toxicity that was significantly increased was grade 3-5 oral mucositis in the treatment group (13% vs. 1% in the placebo group).
Gastric cancer is aggressive and difficult to treat, with median survival of only 9-11 months, Dr. Al-Batran said, adding that at the time the RADPAC study was initiated, no treatments had been approved for patients who failed first-line therapy, although agents like paclitaxel and irinotecan were in use.
He and his colleagues sought to evaluate everolimus, because 50%-60% of gastric cancers are driven by dysregulation in the P13k/Akt/mTOR pathway – a key regulator of cell proliferation, growth, survival, metabolism, and angiogenesis, and because everolimus – an oral mTOR inhibitor – showed efficacy in preclinical models of gastric cancer.
In the phase III GRANITE-1 trial, it was associated with trends toward improved progression-free survival and overall survival, compared with best supportive care, he noted.
Subgroup analyses in the current trial suggested that patients with prior taxane use derived greater benefit from everolimus. Overall survival in those patients, who comprised about half of the study population, was 6 months vs. 4 months with placebo; the difference did not reach statistical significance, but showed a strong trend in that direction (P = .072). Progression-free survival was, however, significantly greater with everolimus than with placebo (2.66 vs. 1.81 months; HR, 0.50) in those with prior taxane use.
“Interestingly, the very few patients having ECOG performance status of 2 really performed very poorly,” Dr. Al-Batran said, explaining that those patients had better outcomes with paclitaxel monotherapy.
“So, in conclusion, everolimus combined with paclitaxel improved outcomes as compared with paclitaxel alone in the intention to treat population. However, activity was seen in the taxane pretreated subgroup. Biomarker studies could attempt to identify a subgroup with more benefit, as we see some activity, but this activity is not enough,” he concluded.
Dr. Al-Batran reported receiving honoraria, serving as a consultant or advisor, receiving research funding from, and/or being on the speakers’ bureau for Celgene, Hospira, Lilly, Medac, Merck, Roche, Sanofi, Vifor, and Nordic Bioscience.
AT THE 2017 GASTROINTESTINAL CANCERS SYMPOSIUM
Key clinical point:
Major finding: Median overall survival was 6.12 vs. 5.03 months with paclitaxel plus everolimus vs. placebo (hazard ratio, 0.93).
Data source: The randomized phase III RADPAC study of 300 patients.
Disclosures: Dr. Al-Batran reported receiving honoraria, serving as a consultant or advisor, receiving research funding from, and/or being on the speakers’ bureau for Celgene, Hospira, Lilly, Medac, Merck, Roche, Sanofi, Vifor, and Nordic Bioscience.
Fresh Press: ACS Surgery News January issue now online
The January issue of ACS Surgery News is available on the website. This month’s issue features a special report on burnout. A new paradigm of burnout is emerging: The roots of the problem may be institutional. Addressing physician burnout must begin with recognition of the challenge and a commitment to change from the top levels of management, according to a study by Tait D. Shanafelt, MD, and John Noseworthy, MD, of the Mayo Clinic.

Don’t miss our annual Meet the Editorial Advisory Board feature. This year, we welcome seven new members: Joshua A. Broghammer, MD, FACS; Samer G. Mattar, MD, FACS; Arden M. Morris, MD, FACS; Rudolfo J. Oviedo, MD, FACS; Kevin M. Reavis, MD, FACS; Michael D. Sarap, MD, FACS; and Gary Timmerman, MD, FACS. On behalf of the editors and our readers, we sincerely thank our members who have finished their term. These colleagues have given of their time and expertise for the benefit of their fellow surgeons. They have earned our admiration and gratitude.
The January issue of ACS Surgery News is available on the website. This month’s issue features a special report on burnout. A new paradigm of burnout is emerging: The roots of the problem may be institutional. Addressing physician burnout must begin with recognition of the challenge and a commitment to change from the top levels of management, according to a study by Tait D. Shanafelt, MD, and John Noseworthy, MD, of the Mayo Clinic.
Don’t miss our annual Meet the Editorial Advisory Board feature. This year, we welcome seven new members: Joshua A. Broghammer, MD, FACS; Samer G. Mattar, MD, FACS; Arden M. Morris, MD, FACS; Rudolfo J. Oviedo, MD, FACS; Kevin M. Reavis, MD, FACS; Michael D. Sarap, MD, FACS; and Gary Timmerman, MD, FACS. On behalf of the editors and our readers, we sincerely thank our members who have finished their term. These colleagues have given of their time and expertise for the benefit of their fellow surgeons. They have earned our admiration and gratitude.
The January issue of ACS Surgery News is available on the website. This month’s issue features a special report on burnout. A new paradigm of burnout is emerging: The roots of the problem may be institutional. Addressing physician burnout must begin with recognition of the challenge and a commitment to change from the top levels of management, according to a study by Tait D. Shanafelt, MD, and John Noseworthy, MD, of the Mayo Clinic.
Don’t miss our annual Meet the Editorial Advisory Board feature. This year, we welcome seven new members: Joshua A. Broghammer, MD, FACS; Samer G. Mattar, MD, FACS; Arden M. Morris, MD, FACS; Rudolfo J. Oviedo, MD, FACS; Kevin M. Reavis, MD, FACS; Michael D. Sarap, MD, FACS; and Gary Timmerman, MD, FACS. On behalf of the editors and our readers, we sincerely thank our members who have finished their term. These colleagues have given of their time and expertise for the benefit of their fellow surgeons. They have earned our admiration and gratitude.
Elements for Success in Managing Type 2 Diabetes With SGLT-2 Inhibitors

Faculty/Faculty Disclosure
Eden M. Miller, DO
Executive Director and Co-founder, Diabetes Nation
High Lakes Health Care
St. Charles Hospital
Bend, Oregon
Competing Interest and Financial Disclosures: Dr. Miller discloses that she is on the advisory boards and speakers’ bureaus for AstraZeneca, Eli Lilly and Company, and Janssen Pharmaceuticals, Inc.
Faculty/Faculty Disclosure
Eden M. Miller, DO
Executive Director and Co-founder, Diabetes Nation
High Lakes Health Care
St. Charles Hospital
Bend, Oregon
Competing Interest and Financial Disclosures: Dr. Miller discloses that she is on the advisory boards and speakers’ bureaus for AstraZeneca, Eli Lilly and Company, and Janssen Pharmaceuticals, Inc.
Faculty/Faculty Disclosure
Eden M. Miller, DO
Executive Director and Co-founder, Diabetes Nation
High Lakes Health Care
St. Charles Hospital
Bend, Oregon
Competing Interest and Financial Disclosures: Dr. Miller discloses that she is on the advisory boards and speakers’ bureaus for AstraZeneca, Eli Lilly and Company, and Janssen Pharmaceuticals, Inc.
Semaglutide compares well with sitagliptin
NEW ORLEANS – Semaglutide, a GLP-1 agonist for type 2 diabetes that’s dosed weekly, was superior to daily sitagliptin in improving glycemic control and reducing body weight in people who are also on metformin and/or thiazolidinediones (TZDs), based on results from a phase III trial. But while the serious adverse event profile was similar for both treatments, far more patients on semaglutide discontinued treatment because of adverse events.
The SUSTAIN study includes more than 8,000 patients with type 2 diabetes. The results are the basis for a new drug application filed in December with the Food and Drug Administration by the investigational drug’s manufacturer, Novo Nordisk, which made the announcement in a press release.

“The SUSTAIN 2 trial has shown that semaglutide at both doses, 0.5 and 1 mg, is superior at improving glycemic control in subjects with type 2 diabetes, compared with sitagliptin, and showed a reduction of 1.3% and 1.6%, respectively, from the baseline HbA1c of 8.1%,” Dr. Ahrén said. For comparison, the sitagliptin group showed an average HbA1c reduction of 0.5%, he said.
The treatments were well tolerated with no new safety concerns, Dr. Ahrén said. “As expected, semaglutide caused more gastrointestinal adverse events, but the frequency was similar to those reported with other GLP-1 receptor agonists,” he said.
The study’s investigators also looked at a composite endpoint of HbA1c less than 7% without symptomatic hypoglycemia and no weight gain, Dr. Ahrén said, achieved by 63% on 0.5 mg and 74% on 1 mg of semaglutide vs. 27% of the sitagliptin group.
The serious adverse event (AE) profile was similar in all three groups: 7.3% in both semaglutide groups and 7.1% in the sitagliptin group. However, far more patients on semaglutide discontinued treatment because of AEs: 8.1% and 9.5% on 0.5 and 1 mg, respectively, vs. 2.9% on sitagliptin. Gastrointestinal AEs in all groups were 43.5% and 39.9% in the 0.5- and 1-mg semaglutide groups, respectively, and 23.6% in the sitagliptin group.
Six deaths were reported in the study population, Dr. Ahrén said: two on 0.5-mg semaglutide dosing, one on the 1-mg dosing, and three on sitagliptin.
Hypoglycemia rates were also “very low,” he said, with 14 patients overall having reported it; seven on 0.5-mg semaglutide therapy and two in the 1-mg group, and five on sitagliptin, “So there were no increased risks for hypoglycemia with semaglutide.”
Dr. Ahrén disclosed relationships with Novo Nordisk and several other drug companies.
NEW ORLEANS – Semaglutide, a GLP-1 agonist for type 2 diabetes that’s dosed weekly, was superior to daily sitagliptin in improving glycemic control and reducing body weight in people who are also on metformin and/or thiazolidinediones (TZDs), based on results from a phase III trial. But while the serious adverse event profile was similar for both treatments, far more patients on semaglutide discontinued treatment because of adverse events.
The SUSTAIN study includes more than 8,000 patients with type 2 diabetes. The results are the basis for a new drug application filed in December with the Food and Drug Administration by the investigational drug’s manufacturer, Novo Nordisk, which made the announcement in a press release.
“The SUSTAIN 2 trial has shown that semaglutide at both doses, 0.5 and 1 mg, is superior at improving glycemic control in subjects with type 2 diabetes, compared with sitagliptin, and showed a reduction of 1.3% and 1.6%, respectively, from the baseline HbA1c of 8.1%,” Dr. Ahrén said. For comparison, the sitagliptin group showed an average HbA1c reduction of 0.5%, he said.
The treatments were well tolerated with no new safety concerns, Dr. Ahrén said. “As expected, semaglutide caused more gastrointestinal adverse events, but the frequency was similar to those reported with other GLP-1 receptor agonists,” he said.
The study’s investigators also looked at a composite endpoint of HbA1c less than 7% without symptomatic hypoglycemia and no weight gain, Dr. Ahrén said, achieved by 63% on 0.5 mg and 74% on 1 mg of semaglutide vs. 27% of the sitagliptin group.
The serious adverse event (AE) profile was similar in all three groups: 7.3% in both semaglutide groups and 7.1% in the sitagliptin group. However, far more patients on semaglutide discontinued treatment because of AEs: 8.1% and 9.5% on 0.5 and 1 mg, respectively, vs. 2.9% on sitagliptin. Gastrointestinal AEs in all groups were 43.5% and 39.9% in the 0.5- and 1-mg semaglutide groups, respectively, and 23.6% in the sitagliptin group.
Six deaths were reported in the study population, Dr. Ahrén said: two on 0.5-mg semaglutide dosing, one on the 1-mg dosing, and three on sitagliptin.
Hypoglycemia rates were also “very low,” he said, with 14 patients overall having reported it; seven on 0.5-mg semaglutide therapy and two in the 1-mg group, and five on sitagliptin, “So there were no increased risks for hypoglycemia with semaglutide.”
Dr. Ahrén disclosed relationships with Novo Nordisk and several other drug companies.
NEW ORLEANS – Semaglutide, a GLP-1 agonist for type 2 diabetes that’s dosed weekly, was superior to daily sitagliptin in improving glycemic control and reducing body weight in people who are also on metformin and/or thiazolidinediones (TZDs), based on results from a phase III trial. But while the serious adverse event profile was similar for both treatments, far more patients on semaglutide discontinued treatment because of adverse events.
The SUSTAIN study includes more than 8,000 patients with type 2 diabetes. The results are the basis for a new drug application filed in December with the Food and Drug Administration by the investigational drug’s manufacturer, Novo Nordisk, which made the announcement in a press release.
“The SUSTAIN 2 trial has shown that semaglutide at both doses, 0.5 and 1 mg, is superior at improving glycemic control in subjects with type 2 diabetes, compared with sitagliptin, and showed a reduction of 1.3% and 1.6%, respectively, from the baseline HbA1c of 8.1%,” Dr. Ahrén said. For comparison, the sitagliptin group showed an average HbA1c reduction of 0.5%, he said.
The treatments were well tolerated with no new safety concerns, Dr. Ahrén said. “As expected, semaglutide caused more gastrointestinal adverse events, but the frequency was similar to those reported with other GLP-1 receptor agonists,” he said.
The study’s investigators also looked at a composite endpoint of HbA1c less than 7% without symptomatic hypoglycemia and no weight gain, Dr. Ahrén said, achieved by 63% on 0.5 mg and 74% on 1 mg of semaglutide vs. 27% of the sitagliptin group.
The serious adverse event (AE) profile was similar in all three groups: 7.3% in both semaglutide groups and 7.1% in the sitagliptin group. However, far more patients on semaglutide discontinued treatment because of AEs: 8.1% and 9.5% on 0.5 and 1 mg, respectively, vs. 2.9% on sitagliptin. Gastrointestinal AEs in all groups were 43.5% and 39.9% in the 0.5- and 1-mg semaglutide groups, respectively, and 23.6% in the sitagliptin group.
Six deaths were reported in the study population, Dr. Ahrén said: two on 0.5-mg semaglutide dosing, one on the 1-mg dosing, and three on sitagliptin.
Hypoglycemia rates were also “very low,” he said, with 14 patients overall having reported it; seven on 0.5-mg semaglutide therapy and two in the 1-mg group, and five on sitagliptin, “So there were no increased risks for hypoglycemia with semaglutide.”
Dr. Ahrén disclosed relationships with Novo Nordisk and several other drug companies.
AT THE ADA ANNUAL SCIENTIFIC SESSIONS
Key clinical point: Investigators for a phase III trial have found weekly semaglutide superior to daily sitagliptin as add-on therapy for improving glycemic control and reducing body weight in type 2 diabetes.
Major finding: Semaglutide 0.5 and 1 mg showed a reduction of 1.3% and 1.6%, respectively, from the baseline HbA1c, compared with an average reduction of 0.5% for sitagliptin.
Data source: SUSTAIN 2 double-blind, randomized trial of 1,231 patients with type 2 diabetes taking either metformin or thiazolidinediones.
Disclosures: Dr. Ahrén disclosed relationships with Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Merck, Novartis, Novo Nordisk, and Sanofi-Aventis Deutschland.