User login
Single dose of ZA pre-transplant prevents bone loss
ORLANDO, FL—A single, 4 mg-dose of the bisphosphonate zoledronic acid (ZA) prior to allogeneic hematopoietic stem cell transplant (HSCT) prevents femoral neck (FN) bone loss at day 100 in patients with lymphoid or myeloid malignancies, according to new research.
The study also suggests that patients who receive risk-adapted ZA therapy after transplant can experience significant reductions in bone loss between days 100 and 365.
However, patients with acute and chronic graft-versus-host disease (GVHD) continue to be at risk of bone loss.
Eric Wong, of the Royal Melbourne Hospital in Parkville, Australia, presented these findings at the 2017 BMT Tandem Meetings (abstract 53) on behalf of the Australasian Leukaemia and Lymphoma Group.
“Previous studies have demonstrated that efforts to prevent bone loss through calcium and vitamin D supplementation as well as hormone-replacement therapy alone have been ineffective in preventing bone loss,” Wong explained.
And monthly pamidronate begun prior to HSCT reduces, but does not prevent, FN bone density loss.
So Wong and his colleagues began a trial of ZA, which is approximately 100-fold more potent than pamidronate.
Study design
The researchers enrolled 82 patients into the phase 2 ALLG BM07 trial. All patients received a single shot of ZA prior to HSCT conditioning.
All patients also received vitamin D and calcium supplements, and pre-menopausal women received hormone replacement therapy.
Depending on their risk of bone loss, patients received individualized ZA therapy after transplant. Researchers assessed the patients’ bone density at days 100, 180, 270, and 365 post-HSCT. Patients at high risk of bone loss received additional doses of ZA.
Risks for bone loss included bone mineral density (BMD) reduction of 5% or greater compared to baseline, prednisolone exposure of 1 mg/kg/d or greater for 2 weeks, or prednisolone exposure of 10 mg/d or more for 6 weeks
The primary endpoint of the study was the change in FN BMD at days 100 and 365 after HSCT compared to baseline.
The researchers also compared patients’ FN BMD with 35 untreated historical controls assessed at the same time points.
Patient characteristics
Seventy patients were alive and had not relapsed at day 100. Most (87%) were 60 years or younger, 60% were male, 53% had myeloid disease, 43% lymphoid, and 4% other disease.
“The most common indications for transplant were acute myeloid leukemia and acute lymphoblastic leukemia,” Wong said, “which, together, formed over 50% of the entire cohort.”
Seventy percent of patients were ECOG status 0 or 1, and 30% were 2 or greater.
Most (59%) had received myeloablative conditioning, the predominant regimens being busulfan/cyclophosphamide or cyclophosphamide/total-body irradiation. And the most common reduced-intensity conditioning regimen was fludarabine/melphalan.
Fifty-six percent of patients had a sibling donor, and 43% had a matched unrelated donor.
Thirty-eight percent of patients developed acute GVHD—19% grade 1, and 19% grade 2 to 3.
“Of note,” Wong said, “no patient developed grade 4 acute GVHD.”
Patients received a median of 2 ZA doses (range, 1–4), including the pre-transplant dose.
Sixty patients received at least 1 dose of ZA between day 100 and day 365, including 33% who received multiple doses.
At day 100, 33 patients received additional ZA. At day 180, 27 patients received additional ZA, including 8 patients who did not have it at day 100. And at day 270, 18 patients had additional ZA, including 1 patient who had no additional ZA at earlier time points.
Results
At day 100, there was no statistically significant change in FN bone density compared with baseline. The mean change was -2.6% (range, -6.6% to 1.4%).
For patients with acute GVHD, however, the change in bone density was significant (P=0.03). Patients with grade 1-2 GVHD had a mean change of -1.6% ± 3.7%, and patients with grade 3-4 GVHD had a mean change of -8.5% ± 11.2%.
Sixty-five patients were available for the day 365 efficacy analysis.
Bone density did not change significantly between day 100 and 365 for the entire group.
“By day 365,” Wong noted, “there was a net loss of bone density of -2.9%.”
But by day 365, patients with extensive chronic GVHD had significantly more bone loss compared with patients who had no chronic GVHD (P=0.03).
Age, sex, duration of cyclosporine, and mean steroid dose were not associated with a change in bone density at day 100 or 365, although there was a trend for an association between high steroid exposure and increased bone loss (P=0.07).
When the researchers compared the patients to untreated historical controls, patients who received ZA had significantly less bone loss at day 100 (P=0.001) and day 365 (P<0.0001).
The researchers observed no serious adverse events with ZA.
Wong concluded that patients with extensive GVHD are a “high-risk cohort that needs augmented therapies.” ![]()
ORLANDO, FL—A single, 4 mg-dose of the bisphosphonate zoledronic acid (ZA) prior to allogeneic hematopoietic stem cell transplant (HSCT) prevents femoral neck (FN) bone loss at day 100 in patients with lymphoid or myeloid malignancies, according to new research.
The study also suggests that patients who receive risk-adapted ZA therapy after transplant can experience significant reductions in bone loss between days 100 and 365.
However, patients with acute and chronic graft-versus-host disease (GVHD) continue to be at risk of bone loss.
Eric Wong, of the Royal Melbourne Hospital in Parkville, Australia, presented these findings at the 2017 BMT Tandem Meetings (abstract 53) on behalf of the Australasian Leukaemia and Lymphoma Group.
“Previous studies have demonstrated that efforts to prevent bone loss through calcium and vitamin D supplementation as well as hormone-replacement therapy alone have been ineffective in preventing bone loss,” Wong explained.
And monthly pamidronate begun prior to HSCT reduces, but does not prevent, FN bone density loss.
So Wong and his colleagues began a trial of ZA, which is approximately 100-fold more potent than pamidronate.
Study design
The researchers enrolled 82 patients into the phase 2 ALLG BM07 trial. All patients received a single shot of ZA prior to HSCT conditioning.
All patients also received vitamin D and calcium supplements, and pre-menopausal women received hormone replacement therapy.
Depending on their risk of bone loss, patients received individualized ZA therapy after transplant. Researchers assessed the patients’ bone density at days 100, 180, 270, and 365 post-HSCT. Patients at high risk of bone loss received additional doses of ZA.
Risks for bone loss included bone mineral density (BMD) reduction of 5% or greater compared to baseline, prednisolone exposure of 1 mg/kg/d or greater for 2 weeks, or prednisolone exposure of 10 mg/d or more for 6 weeks
The primary endpoint of the study was the change in FN BMD at days 100 and 365 after HSCT compared to baseline.
The researchers also compared patients’ FN BMD with 35 untreated historical controls assessed at the same time points.
Patient characteristics
Seventy patients were alive and had not relapsed at day 100. Most (87%) were 60 years or younger, 60% were male, 53% had myeloid disease, 43% lymphoid, and 4% other disease.
“The most common indications for transplant were acute myeloid leukemia and acute lymphoblastic leukemia,” Wong said, “which, together, formed over 50% of the entire cohort.”
Seventy percent of patients were ECOG status 0 or 1, and 30% were 2 or greater.
Most (59%) had received myeloablative conditioning, the predominant regimens being busulfan/cyclophosphamide or cyclophosphamide/total-body irradiation. And the most common reduced-intensity conditioning regimen was fludarabine/melphalan.
Fifty-six percent of patients had a sibling donor, and 43% had a matched unrelated donor.
Thirty-eight percent of patients developed acute GVHD—19% grade 1, and 19% grade 2 to 3.
“Of note,” Wong said, “no patient developed grade 4 acute GVHD.”
Patients received a median of 2 ZA doses (range, 1–4), including the pre-transplant dose.
Sixty patients received at least 1 dose of ZA between day 100 and day 365, including 33% who received multiple doses.
At day 100, 33 patients received additional ZA. At day 180, 27 patients received additional ZA, including 8 patients who did not have it at day 100. And at day 270, 18 patients had additional ZA, including 1 patient who had no additional ZA at earlier time points.
Results
At day 100, there was no statistically significant change in FN bone density compared with baseline. The mean change was -2.6% (range, -6.6% to 1.4%).
For patients with acute GVHD, however, the change in bone density was significant (P=0.03). Patients with grade 1-2 GVHD had a mean change of -1.6% ± 3.7%, and patients with grade 3-4 GVHD had a mean change of -8.5% ± 11.2%.
Sixty-five patients were available for the day 365 efficacy analysis.
Bone density did not change significantly between day 100 and 365 for the entire group.
“By day 365,” Wong noted, “there was a net loss of bone density of -2.9%.”
But by day 365, patients with extensive chronic GVHD had significantly more bone loss compared with patients who had no chronic GVHD (P=0.03).
Age, sex, duration of cyclosporine, and mean steroid dose were not associated with a change in bone density at day 100 or 365, although there was a trend for an association between high steroid exposure and increased bone loss (P=0.07).
When the researchers compared the patients to untreated historical controls, patients who received ZA had significantly less bone loss at day 100 (P=0.001) and day 365 (P<0.0001).
The researchers observed no serious adverse events with ZA.
Wong concluded that patients with extensive GVHD are a “high-risk cohort that needs augmented therapies.” ![]()
ORLANDO, FL—A single, 4 mg-dose of the bisphosphonate zoledronic acid (ZA) prior to allogeneic hematopoietic stem cell transplant (HSCT) prevents femoral neck (FN) bone loss at day 100 in patients with lymphoid or myeloid malignancies, according to new research.
The study also suggests that patients who receive risk-adapted ZA therapy after transplant can experience significant reductions in bone loss between days 100 and 365.
However, patients with acute and chronic graft-versus-host disease (GVHD) continue to be at risk of bone loss.
Eric Wong, of the Royal Melbourne Hospital in Parkville, Australia, presented these findings at the 2017 BMT Tandem Meetings (abstract 53) on behalf of the Australasian Leukaemia and Lymphoma Group.
“Previous studies have demonstrated that efforts to prevent bone loss through calcium and vitamin D supplementation as well as hormone-replacement therapy alone have been ineffective in preventing bone loss,” Wong explained.
And monthly pamidronate begun prior to HSCT reduces, but does not prevent, FN bone density loss.
So Wong and his colleagues began a trial of ZA, which is approximately 100-fold more potent than pamidronate.
Study design
The researchers enrolled 82 patients into the phase 2 ALLG BM07 trial. All patients received a single shot of ZA prior to HSCT conditioning.
All patients also received vitamin D and calcium supplements, and pre-menopausal women received hormone replacement therapy.
Depending on their risk of bone loss, patients received individualized ZA therapy after transplant. Researchers assessed the patients’ bone density at days 100, 180, 270, and 365 post-HSCT. Patients at high risk of bone loss received additional doses of ZA.
Risks for bone loss included bone mineral density (BMD) reduction of 5% or greater compared to baseline, prednisolone exposure of 1 mg/kg/d or greater for 2 weeks, or prednisolone exposure of 10 mg/d or more for 6 weeks
The primary endpoint of the study was the change in FN BMD at days 100 and 365 after HSCT compared to baseline.
The researchers also compared patients’ FN BMD with 35 untreated historical controls assessed at the same time points.
Patient characteristics
Seventy patients were alive and had not relapsed at day 100. Most (87%) were 60 years or younger, 60% were male, 53% had myeloid disease, 43% lymphoid, and 4% other disease.
“The most common indications for transplant were acute myeloid leukemia and acute lymphoblastic leukemia,” Wong said, “which, together, formed over 50% of the entire cohort.”
Seventy percent of patients were ECOG status 0 or 1, and 30% were 2 or greater.
Most (59%) had received myeloablative conditioning, the predominant regimens being busulfan/cyclophosphamide or cyclophosphamide/total-body irradiation. And the most common reduced-intensity conditioning regimen was fludarabine/melphalan.
Fifty-six percent of patients had a sibling donor, and 43% had a matched unrelated donor.
Thirty-eight percent of patients developed acute GVHD—19% grade 1, and 19% grade 2 to 3.
“Of note,” Wong said, “no patient developed grade 4 acute GVHD.”
Patients received a median of 2 ZA doses (range, 1–4), including the pre-transplant dose.
Sixty patients received at least 1 dose of ZA between day 100 and day 365, including 33% who received multiple doses.
At day 100, 33 patients received additional ZA. At day 180, 27 patients received additional ZA, including 8 patients who did not have it at day 100. And at day 270, 18 patients had additional ZA, including 1 patient who had no additional ZA at earlier time points.
Results
At day 100, there was no statistically significant change in FN bone density compared with baseline. The mean change was -2.6% (range, -6.6% to 1.4%).
For patients with acute GVHD, however, the change in bone density was significant (P=0.03). Patients with grade 1-2 GVHD had a mean change of -1.6% ± 3.7%, and patients with grade 3-4 GVHD had a mean change of -8.5% ± 11.2%.
Sixty-five patients were available for the day 365 efficacy analysis.
Bone density did not change significantly between day 100 and 365 for the entire group.
“By day 365,” Wong noted, “there was a net loss of bone density of -2.9%.”
But by day 365, patients with extensive chronic GVHD had significantly more bone loss compared with patients who had no chronic GVHD (P=0.03).
Age, sex, duration of cyclosporine, and mean steroid dose were not associated with a change in bone density at day 100 or 365, although there was a trend for an association between high steroid exposure and increased bone loss (P=0.07).
When the researchers compared the patients to untreated historical controls, patients who received ZA had significantly less bone loss at day 100 (P=0.001) and day 365 (P<0.0001).
The researchers observed no serious adverse events with ZA.
Wong concluded that patients with extensive GVHD are a “high-risk cohort that needs augmented therapies.” ![]()
Antithrombotic drugs may raise risk of subdural hematoma

Use of antithrombotic drugs is associated with a higher risk of subdural hematoma, according to a case-control study of more than 400,000 individuals in Denmark.
The highest risk of subdural hematoma was observed in patients who were taking both a vitamin K antagonist (VKA) and clopidogrel.
The study also showed that the incidence of subdural hematoma increased from 2000 to 2015, which appeared to be associated with the increased use of antithrombotic drugs over that time period.
David Gaist, MD, PhD, of Odense University Hospital and the University of Southern Denmark, and his colleagues conducted this research and reported the results in JAMA.
The researchers evaluated 10,010 patients, ages 20 to 89, with a first-ever subdural hematoma diagnosis from 2000 to 2015. The patients were matched to 400,380 individuals from the general population.
Among the patients with subdural hematoma (average age, 69), 47% were taking antithrombotic medications.
The researchers used conditional logistic regression models to estimate the association between antithrombotic drugs and subdural hematoma risk, adjusting for a range of factors. With the following data, they adjusted for age, sex, calendar period, comorbidity, education level, and income level.
The team found that current low-dose aspirin use was associated with a low risk of subdural hematoma (adjusted odds ratio [OR]=1.24).
Current clopidogrel (OR=1.80) and direct oral anticoagulant (OR=1.55) use were both associated with a moderate risk.
And current VKA use was associated with the highest risk of all the agents when given alone (OR=3.69).
Concurrent use of more than one antithrombotic drug was also associated with a high subdural hematoma risk:
- Low-dose aspirin and clopidogrel (OR=2.45)
- Low-dose aspirin and direct oral anticoagulant (OR=2.52)
- Low-dose aspirin and VKA (OR=4.00)
- Clopidogrel and VKA (OR=7.93).
There was one exception to this. Low-dose aspirin combined with the antiplatelet drug dipyridamole was associated with a risk similar to that of low-dose aspirin alone (OR=1.17).
The researchers noted that the prevalence of antithrombotic drug use increased in the general population over the time period studied, from 31.0 per 1000 individuals in 2000 to 76.9 per 1000 individuals in 2015 (P<0.001 for trend).
The overall subdural hematoma incidence rate increased as well, from 10.9 per 100,000 person-years in 2000 to 19.0 per 100,000 person-years in 2015 (P<0.001 for trend).
The largest increase in subdural hematoma incidence was among patients older than 75 years of age—from 55.1 per 100,000 person-years to 99.7 per 100,000 person-years (P<0.001 for trend). ![]()
Use of antithrombotic drugs is associated with a higher risk of subdural hematoma, according to a case-control study of more than 400,000 individuals in Denmark.
The highest risk of subdural hematoma was observed in patients who were taking both a vitamin K antagonist (VKA) and clopidogrel.
The study also showed that the incidence of subdural hematoma increased from 2000 to 2015, which appeared to be associated with the increased use of antithrombotic drugs over that time period.
David Gaist, MD, PhD, of Odense University Hospital and the University of Southern Denmark, and his colleagues conducted this research and reported the results in JAMA.
The researchers evaluated 10,010 patients, ages 20 to 89, with a first-ever subdural hematoma diagnosis from 2000 to 2015. The patients were matched to 400,380 individuals from the general population.
Among the patients with subdural hematoma (average age, 69), 47% were taking antithrombotic medications.
The researchers used conditional logistic regression models to estimate the association between antithrombotic drugs and subdural hematoma risk, adjusting for a range of factors. With the following data, they adjusted for age, sex, calendar period, comorbidity, education level, and income level.
The team found that current low-dose aspirin use was associated with a low risk of subdural hematoma (adjusted odds ratio [OR]=1.24).
Current clopidogrel (OR=1.80) and direct oral anticoagulant (OR=1.55) use were both associated with a moderate risk.
And current VKA use was associated with the highest risk of all the agents when given alone (OR=3.69).
Concurrent use of more than one antithrombotic drug was also associated with a high subdural hematoma risk:
- Low-dose aspirin and clopidogrel (OR=2.45)
- Low-dose aspirin and direct oral anticoagulant (OR=2.52)
- Low-dose aspirin and VKA (OR=4.00)
- Clopidogrel and VKA (OR=7.93).
There was one exception to this. Low-dose aspirin combined with the antiplatelet drug dipyridamole was associated with a risk similar to that of low-dose aspirin alone (OR=1.17).
The researchers noted that the prevalence of antithrombotic drug use increased in the general population over the time period studied, from 31.0 per 1000 individuals in 2000 to 76.9 per 1000 individuals in 2015 (P<0.001 for trend).
The overall subdural hematoma incidence rate increased as well, from 10.9 per 100,000 person-years in 2000 to 19.0 per 100,000 person-years in 2015 (P<0.001 for trend).
The largest increase in subdural hematoma incidence was among patients older than 75 years of age—from 55.1 per 100,000 person-years to 99.7 per 100,000 person-years (P<0.001 for trend). ![]()
Use of antithrombotic drugs is associated with a higher risk of subdural hematoma, according to a case-control study of more than 400,000 individuals in Denmark.
The highest risk of subdural hematoma was observed in patients who were taking both a vitamin K antagonist (VKA) and clopidogrel.
The study also showed that the incidence of subdural hematoma increased from 2000 to 2015, which appeared to be associated with the increased use of antithrombotic drugs over that time period.
David Gaist, MD, PhD, of Odense University Hospital and the University of Southern Denmark, and his colleagues conducted this research and reported the results in JAMA.
The researchers evaluated 10,010 patients, ages 20 to 89, with a first-ever subdural hematoma diagnosis from 2000 to 2015. The patients were matched to 400,380 individuals from the general population.
Among the patients with subdural hematoma (average age, 69), 47% were taking antithrombotic medications.
The researchers used conditional logistic regression models to estimate the association between antithrombotic drugs and subdural hematoma risk, adjusting for a range of factors. With the following data, they adjusted for age, sex, calendar period, comorbidity, education level, and income level.
The team found that current low-dose aspirin use was associated with a low risk of subdural hematoma (adjusted odds ratio [OR]=1.24).
Current clopidogrel (OR=1.80) and direct oral anticoagulant (OR=1.55) use were both associated with a moderate risk.
And current VKA use was associated with the highest risk of all the agents when given alone (OR=3.69).
Concurrent use of more than one antithrombotic drug was also associated with a high subdural hematoma risk:
- Low-dose aspirin and clopidogrel (OR=2.45)
- Low-dose aspirin and direct oral anticoagulant (OR=2.52)
- Low-dose aspirin and VKA (OR=4.00)
- Clopidogrel and VKA (OR=7.93).
There was one exception to this. Low-dose aspirin combined with the antiplatelet drug dipyridamole was associated with a risk similar to that of low-dose aspirin alone (OR=1.17).
The researchers noted that the prevalence of antithrombotic drug use increased in the general population over the time period studied, from 31.0 per 1000 individuals in 2000 to 76.9 per 1000 individuals in 2015 (P<0.001 for trend).
The overall subdural hematoma incidence rate increased as well, from 10.9 per 100,000 person-years in 2000 to 19.0 per 100,000 person-years in 2015 (P<0.001 for trend).
The largest increase in subdural hematoma incidence was among patients older than 75 years of age—from 55.1 per 100,000 person-years to 99.7 per 100,000 person-years (P<0.001 for trend). ![]()
Hemophilia repository open to US scientists

A new resource has been made available for US-based scientists studying hemophilia A and B.
The resource—known as the My Life, Our Future Research Repository—is a collection of genetic data and blood samples that are linked to phenotypic data from more than 5000 hemophilia patients in the US.
Academic and industry scientists within the US can now access the repository but must apply to do so.
Scientists outside of the US should have access to the repository in 2018.
My Life, Our Future is a program founded by the American Thrombosis and Hemostasis Network, Bloodworks Northwest, the National Hemophilia Foundation, and Bioverativ.
My Life, Our Future was launched in 2012 to increase genotyping among individuals with hemophilia A and B, as well as potential and confirmed carriers of the disorder. The program will be available at participating hemophilia treatment centers through the end of 2017.
My Life, Our Future provides hemophilia patients (and potential/confirmed carriers) with free genotyping, and patients can then consent to add their de-identified genetic data and blood sample to the My Life, Our Future Research Repository.
Eighty-three percent of genotyped patients have contributed their data and blood samples. This is more than 5000 individuals representing more than 25% of the US hemophilia population.
The information provided by these individuals has led to the discovery of more than 600 new genetic variants that cause hemophilia.
“Hemophilia is a complex disorder, and, prior to today, there was no genetic and phenotypic database of this scale aiding scientific innovation,” said Barbara Konkle, MD, associate chief scientific officer of Bloodworks Northwest and principal investigator for My Life, Our Future.
“With this new resource, researchers now have one source for the genetic data, repository samples, and clinical data needed to investigate the many unanswered questions about hemophilia.”
Accessing the repository
To access the samples and data in the repository, scientists must first submit a letter of intent to the American Thrombosis and Hemostasis Network.
Scientists may then be asked to submit a full research proposal, which will be evaluated by a research review committee—a group of experts including a molecular pathologist, genetic counselor, research scientist, molecular biologist, genetic epidemiologist, molecular epidemiologist, geneticist, patient representative, and hematologists.
Approved scientists will be selected based on the scientific merit of their proposals and level of benefit to those with bleeding disorders.
For details on how to apply, visit ATHN.org/MLOF. ![]()
A new resource has been made available for US-based scientists studying hemophilia A and B.
The resource—known as the My Life, Our Future Research Repository—is a collection of genetic data and blood samples that are linked to phenotypic data from more than 5000 hemophilia patients in the US.
Academic and industry scientists within the US can now access the repository but must apply to do so.
Scientists outside of the US should have access to the repository in 2018.
My Life, Our Future is a program founded by the American Thrombosis and Hemostasis Network, Bloodworks Northwest, the National Hemophilia Foundation, and Bioverativ.
My Life, Our Future was launched in 2012 to increase genotyping among individuals with hemophilia A and B, as well as potential and confirmed carriers of the disorder. The program will be available at participating hemophilia treatment centers through the end of 2017.
My Life, Our Future provides hemophilia patients (and potential/confirmed carriers) with free genotyping, and patients can then consent to add their de-identified genetic data and blood sample to the My Life, Our Future Research Repository.
Eighty-three percent of genotyped patients have contributed their data and blood samples. This is more than 5000 individuals representing more than 25% of the US hemophilia population.
The information provided by these individuals has led to the discovery of more than 600 new genetic variants that cause hemophilia.
“Hemophilia is a complex disorder, and, prior to today, there was no genetic and phenotypic database of this scale aiding scientific innovation,” said Barbara Konkle, MD, associate chief scientific officer of Bloodworks Northwest and principal investigator for My Life, Our Future.
“With this new resource, researchers now have one source for the genetic data, repository samples, and clinical data needed to investigate the many unanswered questions about hemophilia.”
Accessing the repository
To access the samples and data in the repository, scientists must first submit a letter of intent to the American Thrombosis and Hemostasis Network.
Scientists may then be asked to submit a full research proposal, which will be evaluated by a research review committee—a group of experts including a molecular pathologist, genetic counselor, research scientist, molecular biologist, genetic epidemiologist, molecular epidemiologist, geneticist, patient representative, and hematologists.
Approved scientists will be selected based on the scientific merit of their proposals and level of benefit to those with bleeding disorders.
For details on how to apply, visit ATHN.org/MLOF. ![]()
A new resource has been made available for US-based scientists studying hemophilia A and B.
The resource—known as the My Life, Our Future Research Repository—is a collection of genetic data and blood samples that are linked to phenotypic data from more than 5000 hemophilia patients in the US.
Academic and industry scientists within the US can now access the repository but must apply to do so.
Scientists outside of the US should have access to the repository in 2018.
My Life, Our Future is a program founded by the American Thrombosis and Hemostasis Network, Bloodworks Northwest, the National Hemophilia Foundation, and Bioverativ.
My Life, Our Future was launched in 2012 to increase genotyping among individuals with hemophilia A and B, as well as potential and confirmed carriers of the disorder. The program will be available at participating hemophilia treatment centers through the end of 2017.
My Life, Our Future provides hemophilia patients (and potential/confirmed carriers) with free genotyping, and patients can then consent to add their de-identified genetic data and blood sample to the My Life, Our Future Research Repository.
Eighty-three percent of genotyped patients have contributed their data and blood samples. This is more than 5000 individuals representing more than 25% of the US hemophilia population.
The information provided by these individuals has led to the discovery of more than 600 new genetic variants that cause hemophilia.
“Hemophilia is a complex disorder, and, prior to today, there was no genetic and phenotypic database of this scale aiding scientific innovation,” said Barbara Konkle, MD, associate chief scientific officer of Bloodworks Northwest and principal investigator for My Life, Our Future.
“With this new resource, researchers now have one source for the genetic data, repository samples, and clinical data needed to investigate the many unanswered questions about hemophilia.”
Accessing the repository
To access the samples and data in the repository, scientists must first submit a letter of intent to the American Thrombosis and Hemostasis Network.
Scientists may then be asked to submit a full research proposal, which will be evaluated by a research review committee—a group of experts including a molecular pathologist, genetic counselor, research scientist, molecular biologist, genetic epidemiologist, molecular epidemiologist, geneticist, patient representative, and hematologists.
Approved scientists will be selected based on the scientific merit of their proposals and level of benefit to those with bleeding disorders.
For details on how to apply, visit ATHN.org/MLOF. ![]()
CCSs’ subsequent cancer risk decreased from ’70s to ’90s

Childhood cancer survivors (CCSs) who were diagnosed in the 1990s have a lower risk of subsequent malignancies than CCSs diagnosed in the 1970s, according to research published in JAMA.
The data suggest this outcome is associated with a reduction in the overall use and median dose of therapeutic radiation over time.
Past research has shown an association between therapeutic radiation and the development of subsequent neoplasms in CCSs. Studies have also linked specific chemotherapeutic agents to subsequent neoplasms.
This information has been used to modify childhood cancer treatment over time, with the hope of reducing the risk of subsequent neoplasms while maintaining or improving 5-year survival.
To assess the effects of these treatment modifications, Gregory Armstrong, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted a study of CCSs.
The researchers evaluated 23,603 five-year CCSs (mean age at diagnosis, 7.7 years) treated at pediatric hospitals in the US and Canada from 1970 through 1999, with follow-up through December 2015.
The most common initial diagnoses were acute lymphoblastic leukemia (35.1%), Hodgkin lymphoma (11.1%), and astrocytoma (9.6%).
Subsequent neoplasms, malignancies
At a mean follow-up of 20.5 years, 1639 CCSs had experienced 3115 subsequent neoplasms, including 1026 malignancies, 233 benign meningiomas, and 1856 non-melanoma skin cancers. The most common neoplasms were breast and thyroid cancers.
The 15-year cumulative incidence of subsequent neoplasms decreased by decade of diagnosis. The incidence was 2.9% for patients diagnosed in the 1970s, 2.4% for those diagnosed in the ’80s, and 1.5% for those diagnosed in the ’90s. For the 1970s vs 1980s, the P value was 0.02. For the 1970s vs 1990s and for the 1980s vs 1990s, the P value was <0.001.
The 15-year cumulative incidence of subsequent malignancies also decreased by decade of diagnosis—2.1% for the ’70s, 1.7% for the ’80s, and 1.3% for the ’90s. The P value was <0.001 for the ’70s vs the ’90s and the ’80s vs the ’90s.
Risk factors
In multivariable analyses, female CCSs had a higher rate of subsequent neoplasms (including malignancies) than males.
In addition, high doses of alkylating agents and platinum agents were associated with increased rates of subsequent malignancies.
The researchers noted that, although there was a decrease in the median cumulative dose of alkylating agents over time, the proportion of CCSs receiving these agents increased. And both the median cumulative dose of platinum agents and the proportion of CCSs receiving these agents increased from the ’70s to the ’90s.
Finally, therapeutic radiation was associated with increased rates of subsequent malignant neoplasms, meningiomas, and non-melanoma skin cancers.
This corresponded with the researchers’ findings that the proportion of individuals receiving radiation and the median dose of radiation both decreased over time.
The proportion of individuals receiving any radiation therapy was 77.7% in the ’70s, 56.7% in the ’80s, and 36.8% in the ’90s. The median dose of radiation was 30.0 Gy in the ’70s, 24.0 Gy in the ’80s, and 26.0 Gy in the ’90s.
“The most ominous late effect of pediatric cancer treatment is a second malignancy,” Dr Armstrong said. “This study shows efforts to reduce the late effects of treatment are paying off. The risk of second cancers for survivors increases with age, so it is good to see the reduction emerging early in survivorship while survivors are still young.” ![]()
Childhood cancer survivors (CCSs) who were diagnosed in the 1990s have a lower risk of subsequent malignancies than CCSs diagnosed in the 1970s, according to research published in JAMA.
The data suggest this outcome is associated with a reduction in the overall use and median dose of therapeutic radiation over time.
Past research has shown an association between therapeutic radiation and the development of subsequent neoplasms in CCSs. Studies have also linked specific chemotherapeutic agents to subsequent neoplasms.
This information has been used to modify childhood cancer treatment over time, with the hope of reducing the risk of subsequent neoplasms while maintaining or improving 5-year survival.
To assess the effects of these treatment modifications, Gregory Armstrong, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted a study of CCSs.
The researchers evaluated 23,603 five-year CCSs (mean age at diagnosis, 7.7 years) treated at pediatric hospitals in the US and Canada from 1970 through 1999, with follow-up through December 2015.
The most common initial diagnoses were acute lymphoblastic leukemia (35.1%), Hodgkin lymphoma (11.1%), and astrocytoma (9.6%).
Subsequent neoplasms, malignancies
At a mean follow-up of 20.5 years, 1639 CCSs had experienced 3115 subsequent neoplasms, including 1026 malignancies, 233 benign meningiomas, and 1856 non-melanoma skin cancers. The most common neoplasms were breast and thyroid cancers.
The 15-year cumulative incidence of subsequent neoplasms decreased by decade of diagnosis. The incidence was 2.9% for patients diagnosed in the 1970s, 2.4% for those diagnosed in the ’80s, and 1.5% for those diagnosed in the ’90s. For the 1970s vs 1980s, the P value was 0.02. For the 1970s vs 1990s and for the 1980s vs 1990s, the P value was <0.001.
The 15-year cumulative incidence of subsequent malignancies also decreased by decade of diagnosis—2.1% for the ’70s, 1.7% for the ’80s, and 1.3% for the ’90s. The P value was <0.001 for the ’70s vs the ’90s and the ’80s vs the ’90s.
Risk factors
In multivariable analyses, female CCSs had a higher rate of subsequent neoplasms (including malignancies) than males.
In addition, high doses of alkylating agents and platinum agents were associated with increased rates of subsequent malignancies.
The researchers noted that, although there was a decrease in the median cumulative dose of alkylating agents over time, the proportion of CCSs receiving these agents increased. And both the median cumulative dose of platinum agents and the proportion of CCSs receiving these agents increased from the ’70s to the ’90s.
Finally, therapeutic radiation was associated with increased rates of subsequent malignant neoplasms, meningiomas, and non-melanoma skin cancers.
This corresponded with the researchers’ findings that the proportion of individuals receiving radiation and the median dose of radiation both decreased over time.
The proportion of individuals receiving any radiation therapy was 77.7% in the ’70s, 56.7% in the ’80s, and 36.8% in the ’90s. The median dose of radiation was 30.0 Gy in the ’70s, 24.0 Gy in the ’80s, and 26.0 Gy in the ’90s.
“The most ominous late effect of pediatric cancer treatment is a second malignancy,” Dr Armstrong said. “This study shows efforts to reduce the late effects of treatment are paying off. The risk of second cancers for survivors increases with age, so it is good to see the reduction emerging early in survivorship while survivors are still young.” ![]()
Childhood cancer survivors (CCSs) who were diagnosed in the 1990s have a lower risk of subsequent malignancies than CCSs diagnosed in the 1970s, according to research published in JAMA.
The data suggest this outcome is associated with a reduction in the overall use and median dose of therapeutic radiation over time.
Past research has shown an association between therapeutic radiation and the development of subsequent neoplasms in CCSs. Studies have also linked specific chemotherapeutic agents to subsequent neoplasms.
This information has been used to modify childhood cancer treatment over time, with the hope of reducing the risk of subsequent neoplasms while maintaining or improving 5-year survival.
To assess the effects of these treatment modifications, Gregory Armstrong, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted a study of CCSs.
The researchers evaluated 23,603 five-year CCSs (mean age at diagnosis, 7.7 years) treated at pediatric hospitals in the US and Canada from 1970 through 1999, with follow-up through December 2015.
The most common initial diagnoses were acute lymphoblastic leukemia (35.1%), Hodgkin lymphoma (11.1%), and astrocytoma (9.6%).
Subsequent neoplasms, malignancies
At a mean follow-up of 20.5 years, 1639 CCSs had experienced 3115 subsequent neoplasms, including 1026 malignancies, 233 benign meningiomas, and 1856 non-melanoma skin cancers. The most common neoplasms were breast and thyroid cancers.
The 15-year cumulative incidence of subsequent neoplasms decreased by decade of diagnosis. The incidence was 2.9% for patients diagnosed in the 1970s, 2.4% for those diagnosed in the ’80s, and 1.5% for those diagnosed in the ’90s. For the 1970s vs 1980s, the P value was 0.02. For the 1970s vs 1990s and for the 1980s vs 1990s, the P value was <0.001.
The 15-year cumulative incidence of subsequent malignancies also decreased by decade of diagnosis—2.1% for the ’70s, 1.7% for the ’80s, and 1.3% for the ’90s. The P value was <0.001 for the ’70s vs the ’90s and the ’80s vs the ’90s.
Risk factors
In multivariable analyses, female CCSs had a higher rate of subsequent neoplasms (including malignancies) than males.
In addition, high doses of alkylating agents and platinum agents were associated with increased rates of subsequent malignancies.
The researchers noted that, although there was a decrease in the median cumulative dose of alkylating agents over time, the proportion of CCSs receiving these agents increased. And both the median cumulative dose of platinum agents and the proportion of CCSs receiving these agents increased from the ’70s to the ’90s.
Finally, therapeutic radiation was associated with increased rates of subsequent malignant neoplasms, meningiomas, and non-melanoma skin cancers.
This corresponded with the researchers’ findings that the proportion of individuals receiving radiation and the median dose of radiation both decreased over time.
The proportion of individuals receiving any radiation therapy was 77.7% in the ’70s, 56.7% in the ’80s, and 36.8% in the ’90s. The median dose of radiation was 30.0 Gy in the ’70s, 24.0 Gy in the ’80s, and 26.0 Gy in the ’90s.
“The most ominous late effect of pediatric cancer treatment is a second malignancy,” Dr Armstrong said. “This study shows efforts to reduce the late effects of treatment are paying off. The risk of second cancers for survivors increases with age, so it is good to see the reduction emerging early in survivorship while survivors are still young.” ![]()
March 2017 Digital Edition
Click here to access the March 2017 Digital Edition.
Table of Contents
- Celebrating Federal Social Work!
- FDA Updates
- Screening for Symptomatic Mefloquine Exposure Among Veterans With Chronic Psychiatric Symptoms
- Medication Adherence and OR Efficiency
- Applying a Time-Out and Standardized Report Form in Anesthesia Handoffs
- Oxybutynin Treatment for Hyperhidrosis in Spinal Cord Injury Patients
- Odontogenic Sinusitis
- Generalized Vaccinia After Smallpox Vaccination With Epstein Barr Infection
- Colonic Diaphragm Disease: An Important NSAID Complication to Know
- The Impact of Obesity on the Efficacy of Simvastatin for Lowering LDL-C
- Improving Caregiver Knowledge of Support Resources
- Walter Reed: A Legacy of Leadership and Service

Click here to access the March 2017 Digital Edition.
Table of Contents
- Celebrating Federal Social Work!
- FDA Updates
- Screening for Symptomatic Mefloquine Exposure Among Veterans With Chronic Psychiatric Symptoms
- Medication Adherence and OR Efficiency
- Applying a Time-Out and Standardized Report Form in Anesthesia Handoffs
- Oxybutynin Treatment for Hyperhidrosis in Spinal Cord Injury Patients
- Odontogenic Sinusitis
- Generalized Vaccinia After Smallpox Vaccination With Epstein Barr Infection
- Colonic Diaphragm Disease: An Important NSAID Complication to Know
- The Impact of Obesity on the Efficacy of Simvastatin for Lowering LDL-C
- Improving Caregiver Knowledge of Support Resources
- Walter Reed: A Legacy of Leadership and Service
Click here to access the March 2017 Digital Edition.
Table of Contents
- Celebrating Federal Social Work!
- FDA Updates
- Screening for Symptomatic Mefloquine Exposure Among Veterans With Chronic Psychiatric Symptoms
- Medication Adherence and OR Efficiency
- Applying a Time-Out and Standardized Report Form in Anesthesia Handoffs
- Oxybutynin Treatment for Hyperhidrosis in Spinal Cord Injury Patients
- Odontogenic Sinusitis
- Generalized Vaccinia After Smallpox Vaccination With Epstein Barr Infection
- Colonic Diaphragm Disease: An Important NSAID Complication to Know
- The Impact of Obesity on the Efficacy of Simvastatin for Lowering LDL-C
- Improving Caregiver Knowledge of Support Resources
- Walter Reed: A Legacy of Leadership and Service

A Faster Way to Diagnose Cushing Syndrome
Diagnosing Cushing syndrome (CS) can take 24 hours of complicated and repeated analyses of blood and urine, brain imaging, and tissue samples from sinuses. But that may soon be in the past. The NIH researchers have found that measuring cortisol levels in hair samples can do the same job, faster.
Related: National ALS Biorepository Opens
Patients with CS have a high level of cortisol, perhaps from a tumor of the pituitary or adrenal glands or as an adverse effect from medications. In a study, 36 participants—30 with CS, 6 without—provided hair samples divided into 3 equal segments. The researchers found that the segments closest to the scalp had the most cortisol (96.6 ± 267.7 pg/mg for CS patients, vs 14.1 ± 9.2 pg/mg in control patients). Those segments’ cortisol content correlated closest with the majority of the initial biochemical tests, including in blood taken at night (when cortisol levels normally drop).
Related: Testosterone Replacement Therapy: Playing Catch-up With Patients
The study was small, CS is rare, and it’s hard to recruit large numbers of patients. Still, the researchers believe it is the largest of its kind to compare hair cortisol levels with diagnostic tests in patients with CS. “Our results are encouraging,” said Mihail Zilbermint, MD, the study’s senior author, and an endocrinologist at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development. “We are hopeful that hair analysis may ultimately prove useful as a less-invasive screening test for Cushing syndrome or in helping to confirm the diagnosis.” The researchers suggest that the test also is a convenient alternative with the “unique ability” for retrospective evaluation of hypercortisolemia over months.
Diagnosing Cushing syndrome (CS) can take 24 hours of complicated and repeated analyses of blood and urine, brain imaging, and tissue samples from sinuses. But that may soon be in the past. The NIH researchers have found that measuring cortisol levels in hair samples can do the same job, faster.
Related: National ALS Biorepository Opens
Patients with CS have a high level of cortisol, perhaps from a tumor of the pituitary or adrenal glands or as an adverse effect from medications. In a study, 36 participants—30 with CS, 6 without—provided hair samples divided into 3 equal segments. The researchers found that the segments closest to the scalp had the most cortisol (96.6 ± 267.7 pg/mg for CS patients, vs 14.1 ± 9.2 pg/mg in control patients). Those segments’ cortisol content correlated closest with the majority of the initial biochemical tests, including in blood taken at night (when cortisol levels normally drop).
Related: Testosterone Replacement Therapy: Playing Catch-up With Patients
The study was small, CS is rare, and it’s hard to recruit large numbers of patients. Still, the researchers believe it is the largest of its kind to compare hair cortisol levels with diagnostic tests in patients with CS. “Our results are encouraging,” said Mihail Zilbermint, MD, the study’s senior author, and an endocrinologist at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development. “We are hopeful that hair analysis may ultimately prove useful as a less-invasive screening test for Cushing syndrome or in helping to confirm the diagnosis.” The researchers suggest that the test also is a convenient alternative with the “unique ability” for retrospective evaluation of hypercortisolemia over months.
Diagnosing Cushing syndrome (CS) can take 24 hours of complicated and repeated analyses of blood and urine, brain imaging, and tissue samples from sinuses. But that may soon be in the past. The NIH researchers have found that measuring cortisol levels in hair samples can do the same job, faster.
Related: National ALS Biorepository Opens
Patients with CS have a high level of cortisol, perhaps from a tumor of the pituitary or adrenal glands or as an adverse effect from medications. In a study, 36 participants—30 with CS, 6 without—provided hair samples divided into 3 equal segments. The researchers found that the segments closest to the scalp had the most cortisol (96.6 ± 267.7 pg/mg for CS patients, vs 14.1 ± 9.2 pg/mg in control patients). Those segments’ cortisol content correlated closest with the majority of the initial biochemical tests, including in blood taken at night (when cortisol levels normally drop).
Related: Testosterone Replacement Therapy: Playing Catch-up With Patients
The study was small, CS is rare, and it’s hard to recruit large numbers of patients. Still, the researchers believe it is the largest of its kind to compare hair cortisol levels with diagnostic tests in patients with CS. “Our results are encouraging,” said Mihail Zilbermint, MD, the study’s senior author, and an endocrinologist at NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development. “We are hopeful that hair analysis may ultimately prove useful as a less-invasive screening test for Cushing syndrome or in helping to confirm the diagnosis.” The researchers suggest that the test also is a convenient alternative with the “unique ability” for retrospective evaluation of hypercortisolemia over months.
IHS Gives Pharmacy Students Hands-On Experience
The IHS has partnered with 3 top American universities to give pharmacy students an opportunity to get real-life work experience and potentially careers at IHS facilities.
Related: Dangerous Staff Shortages in the IHS
In the IHS Advanced Pharmacy Practice Experience Program, PharmD candidates at Howard University, Purdue University, and the University of Southern California will join students from more than 80 universities in 39 states to complete rotations at IHS direct service facilities. “Many return to start their career in providing quality health care to the American Indian and Alaska Native community,” said Mary Smith, IHS principal deputy director.
“My experience with IHS as a student inspired me to apply to work here when I graduated,” said Fengyee Zhou, now a pharmacist at the IHS Whiteriver Indian Hospital in Arizona. “The level of teamwork among all health care disciplines and the extent to which pharmacists engage in patient care activities brought me back to Whiteriver.”
Related: What s the VA? The Largest Educator of Health Care Professionals in the U.S.
The IHS also offers internships, externships, rotations, and residencies to pharmacy, behavioral health, dentistry, optometry, nursing, and medical students.
The IHS has partnered with 3 top American universities to give pharmacy students an opportunity to get real-life work experience and potentially careers at IHS facilities.
Related: Dangerous Staff Shortages in the IHS
In the IHS Advanced Pharmacy Practice Experience Program, PharmD candidates at Howard University, Purdue University, and the University of Southern California will join students from more than 80 universities in 39 states to complete rotations at IHS direct service facilities. “Many return to start their career in providing quality health care to the American Indian and Alaska Native community,” said Mary Smith, IHS principal deputy director.
“My experience with IHS as a student inspired me to apply to work here when I graduated,” said Fengyee Zhou, now a pharmacist at the IHS Whiteriver Indian Hospital in Arizona. “The level of teamwork among all health care disciplines and the extent to which pharmacists engage in patient care activities brought me back to Whiteriver.”
Related: What s the VA? The Largest Educator of Health Care Professionals in the U.S.
The IHS also offers internships, externships, rotations, and residencies to pharmacy, behavioral health, dentistry, optometry, nursing, and medical students.
The IHS has partnered with 3 top American universities to give pharmacy students an opportunity to get real-life work experience and potentially careers at IHS facilities.
Related: Dangerous Staff Shortages in the IHS
In the IHS Advanced Pharmacy Practice Experience Program, PharmD candidates at Howard University, Purdue University, and the University of Southern California will join students from more than 80 universities in 39 states to complete rotations at IHS direct service facilities. “Many return to start their career in providing quality health care to the American Indian and Alaska Native community,” said Mary Smith, IHS principal deputy director.
“My experience with IHS as a student inspired me to apply to work here when I graduated,” said Fengyee Zhou, now a pharmacist at the IHS Whiteriver Indian Hospital in Arizona. “The level of teamwork among all health care disciplines and the extent to which pharmacists engage in patient care activities brought me back to Whiteriver.”
Related: What s the VA? The Largest Educator of Health Care Professionals in the U.S.
The IHS also offers internships, externships, rotations, and residencies to pharmacy, behavioral health, dentistry, optometry, nursing, and medical students.
Understanding the brexpiprazole therapeutic window: Why more isn’t always better
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
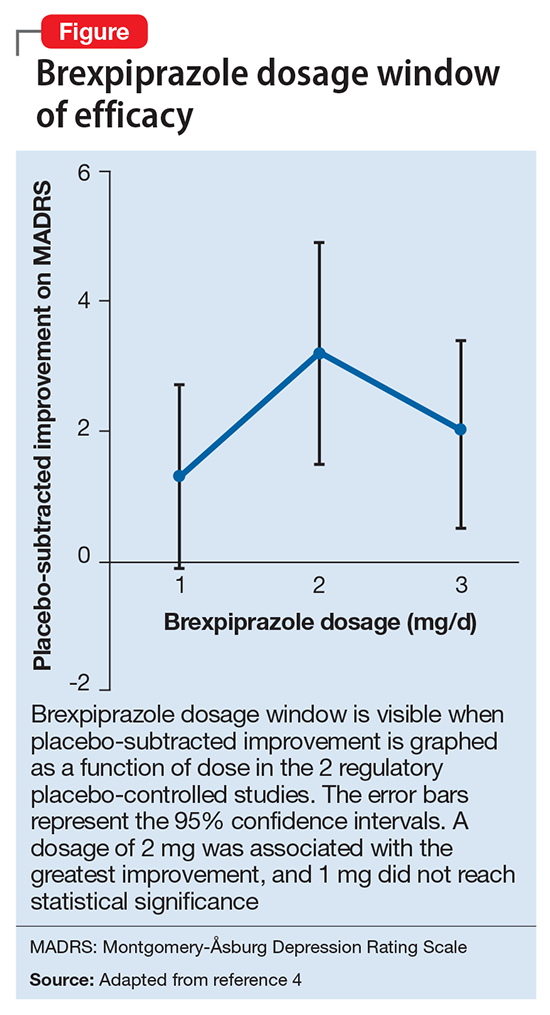
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
Dosage windows could be difficult to understand pharmacologically, but for a partial agonist the presumed mechanism could be more evident. Clinicians should be aware that more is not always better, meaning that with partial agonist drugs a higher dosage might not lead to greater patient response. With brexpiprazole, a dopamine D2 partial agonist FDA-approved for schizophrenia and an adjunct for major depressive disorder (MDD),1 moderation is best because of
Recommended dosage
Two placebo-controlled studies2,3 examined brexpiprazole dosages of 1, 2, and 3 mg/d. The recommended dosage of 2 mg/d for MDD was determined by changes in Montgomery-Åsburg Depression Rating Scale scores (Figure).4 Lower dosages of 1 mg/d did not reach statistical significance, and 3 mg/d were less effective than the intermediate dosage of 2 mg/d. This result suggests a window of efficacy for brexpiprazole for MDD. This therapeutic window likely applies to most patients; however, patient-specific variables could alter the optimum dosage.
Dosage window
Brexpiprazole has high affinity for dopamine D2, D3, serotonin 5-HT1A, 5-HT2A, norepinephrine α1B, and α2 Creceptors. At relatively low drug concentrations, brexpiprazole achieves high receptor occupancy. At receptors for which brexpiprazole is a partial agonist (5-HT1A, D2, D3) the drug blocks the receptor and stimulates it at a fraction of the endogenous neurotransmitter. With a very high affinity agent, the endogenous neurotransmitter could be completely excluded from interacting with these receptors if brexpiprazole occupancy is high. At lower dosages, the drug occupies only a fraction of the receptors, allowing the endogenous neurotransmitters to continue interacting with their receptors, thereby magnifying the signal of that receptor above baseline.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
1. FDA approves Rexulti (brexpiprazole) as adjunctive treatment for adults with major depressive disorder and as a treatment for adults with schizophrenia [news release]. Valby, Denmark; Tokyo, Japan: H. Lundbeck A/S (Lundbeck); Otsuka Pharmaceutical Co., Ltd; July 11, 2015. http://investor.lundbeck.com/ releasedetail.cfm?Release ID=921621. Accessed October 3, 2015.
2. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9): 1232-1240.
3. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebocontrolled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
4. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
Hepatitis C among the mentally ill: Review and treatment update
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.

CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
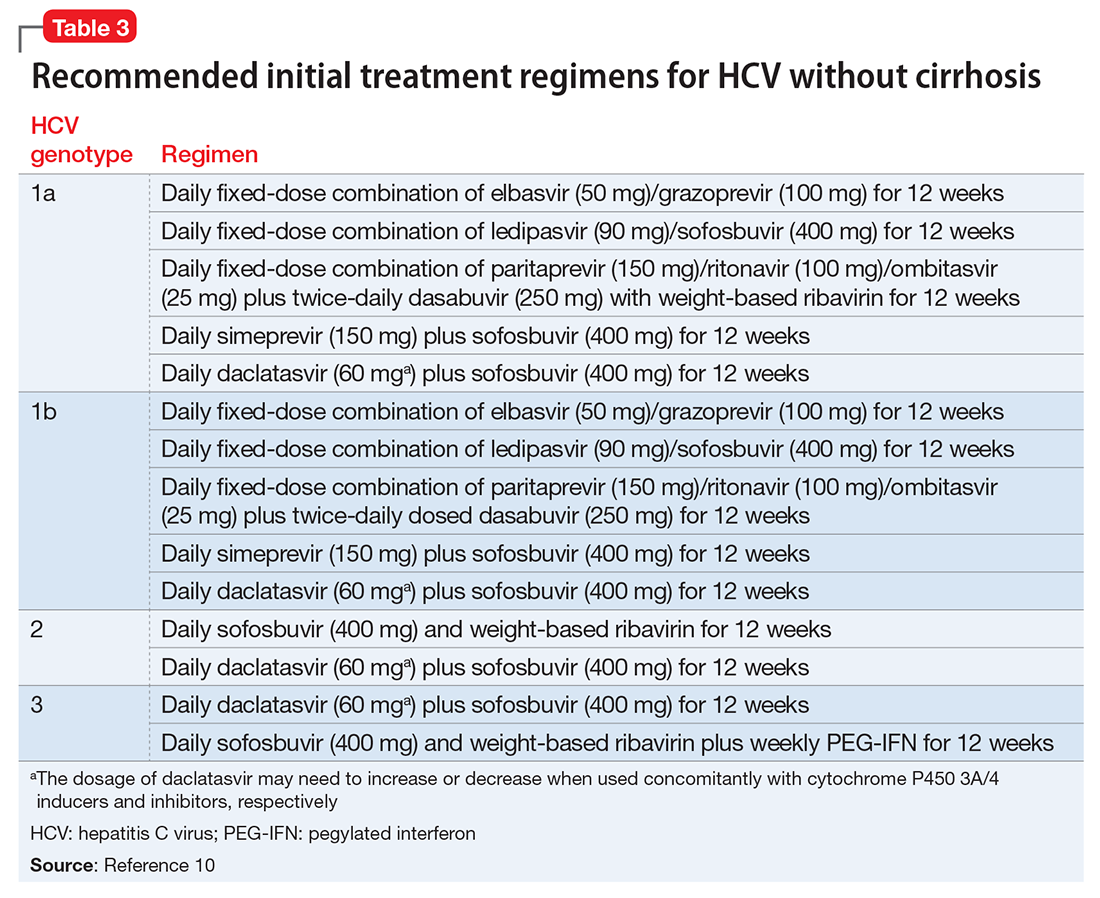
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.
CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
At approximately 3 to 4 million patients, hepatitis C virus (HCV) is the most common viral hepatitis in the United States. Patients with mental illness are disproportionately affected by HCV and the management of their disease poses particular challenges.
HCV is commonly transmitted via IV drug use and blood transfusions; transmission through sexual contact is rare. Most patients with HCV are asymptomatic, although some do develop symptoms of acute hepatitis. Most HCV infections become chronic, with a high incidence of liver failure requiring liver transplantation.
Hepatitis refers to inflammation of the liver, which could have various etiologies, including viral infections, alcohol abuse, or autoimmune disease. Viral hepatitis refers to infection from 5 distinct groups of virus, coined A through E.1 This article will focus on chronic HCV (Table 1).
CASE Bipolar disorder, stress, history of IV drug use
Ms. S, age 48, has bipolar I disorder and has been hospitalized 4 times in the past, including once for a suicide attempt. She has 3 children and works as a cashier. Her psychiatric symptoms have been stable on lurasidone, 80 mg/d, and escitalopram, 10 mg/d. Recently, Ms. S has been under more stress at her job. Sometimes she misses doses of her medication, and then becomes more irritable and impulsive. Her husband, noting that she has used IV heroin in the past, comes with her today and is concerned that she is “not acting right.” What is Ms. S’s risk for HCV?
HCV in mental illness
Compared with the general population, HCV is more prevalent among chronically mentally ill persons. In one study, HCV occurred twice as often in men vs women with chronic mental illness.2 Up to 50% of patients with HCV have a history of mental illness and nearly 90% have a history of substance use disorders.3 Among 668 chronically mentally ill patients at 4 public sector clinics, risk factors for HCV were common and included use of injection drugs (>20%), sharing needles (14%), and crack cocaine use (>20%).4 Higher rates of HCV were reported in hospitalized patients with schizophrenia and comorbid psychoactive substance abuse in Japan.5 Because of the high prevalence in this population, it is essential to assess for substance use disorders. Employing a non-judgmental approach with motivational interviewing techniques can be effective.6
Individuals with mental illness should be screened for HCV risk factors, such as unprotected intercourse with high-risk partners and sharing needles used for illicit drug use. Patients frequently underreport these activities. At-risk individuals should undergo laboratory testing for the HIV-1 antibody, hepatitis C antibodies, and hepatitis B antibodies. Mental health providers should counsel patients about risk reduction (eg, avoiding unprotected sexual intercourse and sharing of drug paraphernalia). Educating patients about complications of viral hepatitis, such as liver failure, could be motivation to change risky behaviors.
CASE continued
During your interview with Ms. S, she becomes irritable and tells you that you are asking too many questions. It is clear that she is not taking her medications consistently, but she agrees to do so because she does not want to lose custody of her children. She denies current use of heroin but her husband says, “I don’t know what she is doing.” In addition to advising her on reducing risk factors, you order appropriate screening tests, including hepatitis and HIV antibody tests.
Screening guidelines
The U.S. Preventive Services Task Force and the CDC both recommend a 1-time screening for HCV in asymptomatic or low-risk patients born between 1945 and 1965.1,7 Furthermore, both organizations recommend screening for HCV in persons at high risk, including:
- those with a history of injection drug use
- persons with recognizable exposure, such as needlesticks
- persons who received blood transfusions before 1992
- medical conditions, such as long-term dialysis.
There is no vaccine for HCV; however, patients with HCV should receive vaccination against hepatitis B.
Diagnosis
Acute symptoms include fever, fatigue, headache, cough, nausea, and vomiting. Jaundice could develop, often accompanied by pain in the right upper quadrant. If there is suspicion of viral hepatitis, psychiatrists can initiate the laboratory evaluation. Chronic hepatitis, on the other hand, often is asymptomatic, although stigmata of chronic liver disease (eg, jaundice, ascites, peripheral edema) might be detected on physical exam.8 Elevated serum transaminases are seen with acute viral hepatitis, although levels could vary in chronic cases. Serologic detection of anti-HCV antibodies establishes a HCV diagnosis.
Treatment recommendations
All patients who test positive for HCV should be evaluated and treated by a hepatologist. Goals of therapy are to reduce complications from chronic viral hepatitis, including cirrhosis and hepatic failure. Duration and optimal regimen depends on the HCV genotype.8 Treatment outcomes are measured by virological parameters, including serum aminotransferases, HCV RNA levels, and histology. The most important parameter in treating chronic HCV is the sustained virological response (SVR), which is the absence of HCV RNA 12 weeks after completing therapy.9
Treatment is recommended for all persons with chronic HCV infection, according to current treatment guidelines, which are updated regularly by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America.10 Until recently, treatment consisted of IV pegylated interferon (PEG-IFN) in combination with oral ribavirin. Success rates with this regimen are approximately 40% to 50%. The advent of direct-acting antivirals (DAAs) has revolutionized treatment of chronic HCV. These agents include simeprevir, sofosbuvir, ledipasvir, and the combination of ombitasvir-paritaprevir-ritonavir plus dasabuvir (brand name, Viekira Pak). Advantages of these agents are oral administration, high treatment success rates (>90%), shorter treatment duration (12 weeks vs up to 48 weeks with older regimens), and few serious adverse effects9-11; drawbacks include the pricing of these regimens, which could cost upward of ≥$100,000 for a 12-week course, and a lack of coverage under some health insurance plans.12 The manufacturers of 2 agents, telaprevir and boceprevir, removed them from the market because of decreased demand related to their unfavorable side-effect profile and the availability of better tolerated agents.
Treatment considerations for interferon in psychiatric patients
Various neuropsychiatric symptoms have been reported with the use of PEG-IFN. The range of reported symptoms include:
- depressed mood
- anxiety
- hostility
- slowness
- fatigue
- sleep disturbance
- lethargy
- irritability
- emotional lability
- social withdrawal
- poor concentration.13,14
Depressive symptoms can present as early as 1 month after starting treatment, but typically occur at 8 to 12 weeks. A systematic review and meta-analysis of 26 observational studies found a cumulative 25% risk of interferon (IFN)-induced depression in the general HCV population.15 Risk factors for IFN-induced depression include:
- female sex
- history of major depression or other psychiatric disorder
- low educational level
- the presence of baseline subthreshold depressive symptoms.
Because of the risk of inducing depression, there was initial hesitation with providing IFN treatment to patients with psychiatric disorders. However, there is evidence that individuals with chronic psychiatric illness can be treated safely with IFN-based regimens and achieve results similar to non-psychiatric populations.16,17 For example, patients with schizophrenia in a small Veterans Affairs database who received IFN for HCV did not experience higher rates of symptoms of schizophrenia, depression, or mania over 8 years of follow-up.18 Furthermore, those with schizophrenia were just as likely to reach SVR as patients without psychiatric illness.19 Other encouraging results have been reported in depressed patients. One study found similar rates of treatment completion and SVR in patients with a history of major depressive disorder compared with those without depression.20 No difference in frequency of neuropsychiatric side effects was found between the groups.
Presence of a psychiatric disorder is no longer an absolute contraindication to IFN treatment for HCV. Optimal control of psychiatric symptoms should be attained in all patients before starting HCV treatment, and close clinical monitoring is warranted. A review of 9 studies showed benefit of antidepressants for HCV patients with elevated baseline depression or a history of IFN-induced depression.21 The largest body of evidence supports the safety and efficacy of selective serotonin reuptake inhibitors for treating IFN-induced depression. Although no antidepressants are FDA-approved for this indication, the best-studied agents include citalopram, escitalopram, sertraline, and paroxetine.
A review of 6 studies on using antidepressants to prevent IFN-induced depression concluded there was inadequate evidence to support this approach in all patients.22 Pretreatment primarily is indicated for those with elevated depressive symptoms at baseline or those with a history of IFN-induced depression. The prevailing approach to IFN-induced depression assessment, prevention, and treatment is summarized in Table 2.
CASE continued
Ms. S tests positive for the HCV antibody but negative for HIV and hepatitis B. She immediately receives the hepatitis B vaccine series. Her sister discourages her from receiving treatment for HCV, warning her, “it will make you crazy depressed.” As a result, Ms. S avoids following up with the hepatologist. Her psychiatrist, aware that she now was taking her psychotropic medication and seeing that her mood is stable, educates her about new treatment options for HCV that do not cause depression. Ms. S finally agrees to see a hepatologist to discuss her treatment options.
IFN-free regimens
With the arrival of the DAAs, the potential now exists to use IFN-free treatment regimens,10 which could eliminate concerns about IFN-induced depression.
Clinical trials of the DAAs and real-world use so far do not indicate an elevated risk for neuropsychiatric symptoms, including depression.11 As a result, more patients with severe psychiatric illness likely will be eligible to receive treatment for HCV. However, as clinical experience builds with these new agents, it is important to monitor the experience of patients with psychiatric comorbidity. Current treatment guidelines for HCV genotype 1, which is most common in the United States, do not include IFN-based regimens.10 Treatment of genotype 3, which affects 6% of the U.S. population, still includes IFN. Therefore, the risk of IFN-induced depression still exists for some patients with HCV. Table 310 describes current treatment regimens in use for HCV without cirrhosis (see Related Resources for treating HCV with cirrhosis).
Evolving role of the psychiatrist
The availability of shorter, better-tolerated regimens means that the psychiatric contraindications to HCV treatment will be eased. With the emergence of non-IFN treatment regimens, the role of mental health providers could shift toward assisting with treatment adherence, monitoring drug–drug interactions, and managing comorbid substance use disorders.10
The psychiatrist’s role might shift away from the psychosocial assessment of factors affecting treatment eligibility, such as IFN-associated depressive symptoms. Clinical focus will likely shift to supporting adherence to HCV treatment regimens.23 Because depression and substance use disorders are risk factors for non-adherence, mental health providers may be called upon to optimize treatment of these conditions before beginning DAA regimens. A multi-dose regimen might be complicated for those with severe mental illness, and increased psychiatric and community support could be needed in these patients.23 Furthermore, models of care that integrate an HCV specialist with psychiatric care have demonstrated benefits.6,23 Long-term follow-up with a mental health provider will be key to provide ongoing psychiatric support, especially for those who do not achieve SVR.
Psychotropic drug–drug interactions with DAAs
Both sofosbuvir and ledipasvir are substrates of P-glycoprotein and not metabolized by cytochrome P450 (CYP) enzymes.24 Therefore, there are no known contraindications with psychotropic medications. However, co-administration of P-glycoprotein inducers, such as St. John’s wort, could reduce sofosbuvir and ledipasvir levels leading to reduced therapeutic efficacy.
Because it has been used for many years as an HIV treatment, drug interactions with ritonavir have been well-described. This agent is a “pan-inhibitor” and inhibits the CYP3A4, 2D6, 2C9, and 2C19 enzymes and could increase levels of any psychotropic metabolized by these enzymes.25 After several weeks of treatment, it also could induce CYP3A4, which could lead to reduced efficacy of oral contraceptives because ethinylestradiol is metabolized by CYP3A4. Ritonavir is primarily metabolized by CYP3A4 (and CYP2D6 to a smaller degree). Carbamazepine induces CYP3A4, which may lead to decreased levels of ritonavir.23 This, in turn, could reduce the likelihood of attaining SVR and successful treatment of HCV.
Boceprevir, telaprevir, and simeprevir inhibit CYP3A4 to varying degrees and therefore could affect psychotropic medications metabolized by this enzyme.23,26,27 These DAAs are metabolized by CYP3A4; therefore CYP3A4 inducers, such as carbamazepine, could lower DAA blood levels, increasing risk of HCV treatment failure and viral resistance.
Daclatasvir is a substrate of CYP3A4 and an inhibitor of P-glycoprotein.28 Concomitant buprenorphine or buprenorphine/naloxone levels may be increased, although the manufacturer does not recommend dosage adjustment. Elbasvir and grazoprevir are metabolized by CYP3A4.29 Drug–drug interactions therefore may result when administered with either CYP3A4 inducers or inhibitors.
CASE Conclusion
Ms. S sees her new hepatologist, Dr. Smith. She decides to try a 12-week course of ledipasvir/sofosbuvir. Dr. Smith collaborates frequently with Ms. S’s psychiatrist to discuss her case and to help monitor her psychiatric symptoms. She follows up closely with her psychiatrist for symptom monitoring and to help ensure treatment compliance. Ms. S does well with the IFN-free treatment regimen and experiences no worsening of her psychiatric symptoms during treatment.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
1. Centers for Disease Control and Prevention. Viral hepatitis. http://www.cdc.gov/hepatitis. Updated December 9, 2016. Accessed February 9, 2017.
2. Butterfield MI, Bosworth HB, Meador KG, et al. Five-Site Health and Risk Study Research Committee. Gender differences in hepatitis C infection and risks among persons with severe mental illness. Psychiatr Serv. 2003;54(6):848-853.
3. Rifai MA, Gleason OC, Sabouni D. Psychiatric care of the patient with hepatitis C: a review of the literature. Prim Care Companion J Clin Psychiatry. 2010;12(6):PCC.09r00877. doi: 10.4088/PCC.09r00877whi.
4. Dinwiddie SH, Shicker L, Newman T. Prevalence of hepatitis C among psychiatric patients in the public sector. Am J Psychiatry. 2003;160(1):172-174.
5. Nakamura Y, Koh M, Miyoshi E, et al. High prevalence of the hepatitis C virus infection among the inpatients of schizophrenia and psychoactive substance abuse in Japan. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(3):591-597.
6. Sockalingam S, Blank D, Banga CA, et al. A novel program for treating patients with trimorbidity: hepatitis C, serious mental illness, and substance abuse. Eur J Gastroenterol Hepatol. 2013;25(12):1377-1384.
7. U.S. Preventive Services Task Force. Screening for hepatitis C virus infection: recommendation summary. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/hepatitis-c-screening. Published June 2013. Accessed February 9, 2017.
8. Longo DL, Fauci AS, Kasper DL. Harrison’s principles of internal medicine. 18th ed. New York, NY: McGraw-Hill; 2012.
9. Belousova V, Abd-Rabou AA, Mousa SA. Recent advances and future directions in the management of hepatitis C infections. Pharmacol Ther. 2015;145:92-102.
10. American Association for the Study of Liver Diseases (AASLD); The Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. http://www.hcvguidelines.org. Accessed February 9, 2017.
11. Rowan PJ, Bhulani N. Psychosocial assessment and monitoring in the new era of non-interferon-alpha hepatitis C treatments. World J Hepatol. 2015;7(19):2209-2213.
12. Good Rx, Inc. http://www.goodrx.com. Accessed October 9, 2015.
13. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
14. Lotrich FE, Rabinovitz M, Gironda P, et al. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63(2):131-135.
15. Udina M, Castellví P, Moreno-España J, et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J Clin Psychiatry. 2012;73(8):1128-1138.
16. Mustafa MZ, Schofield J, Mills PR, et al. The efficacy and safety of treating hepatitis C in patients with a diagnosis of schizophrenia. J Viral Hepat. 2014;21(7):e48-e51.
17. Huckans M, Mitchell A, Pavawalla S, et al. The influence of antiviral therapy on psychiatric symptoms among patients with hepatitis C and schizophrenia. Antivir Ther. 2010;15(1):111-119.
18. Huckans MS, Blackwell AD, Harms TA, et al. Management of hepatitis C disease among VA patients with schizophrenia and substance use disorders. Psychiatr Serv. 2006;57(3):403-406.
19. Huckans M, Mitchell A, Ruimy S, et al. Antiviral therapy completion and response rates among hepatitis C patients with and without schizophrenia. Schizophr Bull. 2010;36(1):165-172.
20. Hauser P, Morasco BJ, Linke A, et al. Antiviral completion rates and sustained viral response in hepatitis C patient with and without preexisting major depressive disorder. Psychosomatics. 2009;50(5):500-505.
21. Sockalingam S, Abbey SE. Managing depression during hepatitis C treatment. Can J Psychiatry. 2009;54(9):614-625.
22. Galvão-de Almeida A, Guindalini C, Batista-Neves S, et al. Can antidepressants prevent interferon-alpha-induced depression? A review of the literature. Gen Hosp Psychiatry. 2010;32(4):401-405.
23. Sockalingam S, Sheehan K, Feld JJ, et al. Psychiatric care during hepatitis c treatment: the changing role of psychiatrists in the era of direct-acting antivirals. Am J Psychiatry. 2015;172(6):512-516.
24. Harvoni [package insert]. Foster City, CA: Gilead Sciences, Inc.; 2016.
25. Wynn GH, Oesterheld, JR, Cozza KL, et al. Clinical manual of drug interactions principles for medical practice. Arlington, VA: American Psychiatric Publishing; 2009.
26. Olysio [package insert]. Titusville, NJ: Janssen Therapeutics; 2016.
27. Victrelis [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
28. Daklinza [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2016.
29. Zepatier [package insert]. Whitehouse Station, NJ: Merck & Co.; 2017.
Social withdrawal and confusion in an inmate with schizoaffective disorder
CASE Withdrawn and confused
Mr. J, age 54, is brought to the emergency department from a correctional treatment facility where he is reported to be displaying new, unusual behavior. He has a history of schizoaffective disorder, which has been stable with haloperidol, 10 mg/d, for more than a year.
Although previously Mr. J openly discussed his long-standing delusions about the FBI coming to release him from prison, he no longer mentions this or any other delusional beliefs, and has become less communicative with staff and peers. Mr. J no longer accompanies the other patients to the cafeteria for meals and eats in his room alone and appears to be losing weight. He says, “I do not feel good,” but otherwise does not communicate spontaneously. Intermittently, he is irritable, without known triggers. The staff notices that Mr. J often lays on his bed, sometimes in a fetal position. Over time, he becomes confused and is seen attempting to open his room door with a toothbrush. His personal hygiene is poor, and he often urinates through his clothes, on the floor, and in his bed. Recently, Mr. J’s eczema has worsened. His gait has become unsteady, and he has orthostasis.
What could be causing these new symptoms?
a) worsening schizoaffective disorder
b) illicit drug use in the prison
c) atypical dementia
d) cardiac etiology
The author’s observations
The differential diagnosis for Mr. J appeared to be wide and without specific etiology. Because of the complex types of symptoms that Mr. J was experiencing, the emergency department managed his care and specialty clinic referrals were ordered.
It was reported that Mr. J started complaining of lightheadedness a few months ago, which worsened (unsteady gait, near falls). In the context of Mr. J’s history of lightheadedness and orthostasis, the cardiology clinic ordered a tilt table test, which was within normal limits:
- 70º head-up tilt: blood pressure, 91/67 to 102/62 mm Hg, and pulse, 70 to 79 beats per minute (bpm)
- with isoproterenol, 1 μg/minute: blood pressure, 90/66 to 110/70 mm Hg, and pulse, 77 to 124 bpm
- with isoproterenol, 2 μg/minute: blood pressure, 98/58 to 111/66 mm Hg, and pulse, 121 to 134 bpm.
The neurologist’s diagnostic impression was atypical dementia; however, Mr. J showed no memory deficits. Parkinsonism also was considered, but Mr. J had no unilateral tremor, masked facies, or micrographia. Mr. J showed some restriction in his movement, but he was not bradykinetic. The team suspected haloperidol could be causing his stiff movement.
Although it was possible that Mr. J’s schizoaffective disorder was worsening and led to the new symptoms, Mr. J appeared to be less delusional because he was no longer talking to the staff about his delusions. There seemed to be no outward evidence of progression of psychotic symptoms.
Mr. J had a history of substance abuse, including alcohol, cocaine, and Cannabis. Although prison inmates have been known to manufacture and drink “hooch,” the new symptoms Mr. J was experiencing were severe enough that his social interactions with other inmates diminished substantially. Because Mr. J had not been communicating with the other inmates and had no recent visitors, the team felt that it was unlikely that drugs were causing these symptoms. Also, a urine drug screen for cocaine, amphetamines, benzodiazepines, Cannabis, and opioids was negative.
HISTORY Substance use, violence
Mr. J was diagnosed with bipolar disorder at age 18. After later hospitalizations, his diagnosis was changed to schizoaffective disorder as a matter of diagnostic clarification. He has a long history of non-compliance with treatment, homelessness, and drug abuse.
Mr. J is serving a 20-year sentence for first-degree reckless homicide. A year after he was incarcerated, Mr. J was sent to a specialized mental health facility for inmates whose illness cannot be managed in a typical correctional setting. While at the treatment facility, Mr. J was non-compliant with medications and because of concerns about dangerousness and psychosis, the court found probable cause for involuntary commitment.
His medication regimen is trihexyphenidyl, 2 mg/d, for extrapyramidal symptoms; haloperidol, 10 mg/d, for psychosis; trazodone, 150 mg/d, for insomnia; vitamin D3, 2,000 IU/d; vitamin E, 400 IU/d, for symptoms of tardive dyskinesia; IM ziprasidone, 20 mg, because he refused oral haliperidol; and hydrocortisone cream 1% for eczema.
EVALUATION Additional tests
Mr. J’s blood pressure is 124/72 mm Hg, and pulse, 104 bpm, laying down; blood pressure, 110/84 mm Hg, and pulse, 112 bpm, sitting; and blood pressure, 108/82 mm Hg, and pulse, 129 bpm, standing. With repeated readings: blood pressure, 128/84 mm Hg, and pulse, 98 bpm, laying down; blood pressure, 125/86 mm Hg, and pulse, 113 bpm, sitting; and blood pressure, 105/76 mm Hg, and pulse, 130 bpm, standing.
Laboratory tests, including complete blood count, chemistry panel, thyroid-stimulating hormone, are within normal limits. The team feels that the investigation for an etiology for Mr. J’s symptoms needs to be more exhaustive and additional tests are ordered, including vitamin levels (C, B1, B12, B6), rapid plasma reagin for syphilis, and arbovirus testing (eastern equine encephalitis virus, western equine encephalitis, West Nile virus, La Crosse encephalitis, St. Louis encephalitis), which are negative.
What’s the next best step in managing Mr. J’s care?
a) adjust his medication
b) eliminate a mediation
c) order further testing
The author’s observations
To determine if Mr. J’s new-onset symptoms might be related to the progression of his psychiatric illness, the haloperidol dosage was increased to 20 mg/d; however, we saw no positive response to this change. His tardive dyskinesia symptoms (bruxism and other oral buccal movements) worsened. Haloperidol was reduced to 10 mg/d.
Trihexyphenidyl then was suspected to contribute to Mr. J’s confusion. Unfortunately, lowering the dosage of trihexyphenidyl to 1 mg/d, did not affect Mr. J’s current symptoms and exacerbated extrapyramidal symptoms.
The treatment team then questioned if porphyria—known as the “little imitator”—might be considered because of the variety of symptoms without an etiology, despite extensive testing. A 24-hour urine collection was ordered.
What is the correct method of collecting a urine sample for porphyrins?
a) collect a small sample and expose it to light before testing
b) collect a 24-hour sample with the sample kept in ambient temperature and light
c) collect a 24-hour sample with the sample kept on ice in a light-blocking container and frozen when sent to the laboratory
EVALUATION Diagnosis revealed
The 24-hour urine collection is obtained. However, it needed to be collected twice, because the first sample was not a full sample. Interestingly, the first sample, which is exposed to light and not kept on ice, turned dark in color. The second sample is obtained properly and sent to the laboratory. When the laboratory results are returned (Figure 1), Mr. J is diagnosed with hereditary coproporphyria (HCP).

The author’s observations
There are several types of porphyria, each associated with a different step in the chain of enzymes associated with synthesis of a heme molecule in the mitochondria. A defect in any single enzyme step will create a build up of porphyrins—a precursor to heme molecules—in erythrocytes or hepatic cells.
It is important to differentiate hepatic from erythropoietic porphyrias. The acute porphyrias (acute intermittent porphyria [AIP], HCP, and variegate porphyria generally are hepatic in origin with neuropsychiatric and neurovisceral symptoms. Cutaneous porphyrias originate in bone marrow and therefore are erythropoietic. However, there are exceptions such as porphyria cutanea tarda (PCT), which is hepatic in origin but the manifestations mainly are cutaneous1 (Figure 2).2

Although acute porphyria originates in the liver, it is a neuropsychiatric illness. In these cases, excess porphyrins cannot cross the blood–brain barrier and are neurotoxic. Clinicians can look for abnormalities in the liver via liver function tests, but liver parenchyma is not damaged by these enzyme precursors. During an acute porphyic attack, patients could experience symptoms such as:
- muscle spasms (commonly abdominal, but can be any muscle group)
- confusion
- disorientation
- autonomic instability
- lightheadness
- disorientation
- diarrhea
- light sensitivity
- dermatologic conditions
- weakness (particularly peripheral weakness)
- hypesthesia
- allodynia
- severe nausea and vomiting
- emotional lability
- psychosis as well as general malaise.
The attack could result in death.
Mr. J had many differing symptoms and was evaluated by several specialty providers. He had a chronic dermatologic condition; he was confused, disoriented, and complained of nausea, weakness, orthostasis, and loose stools. With the variety of possible symptoms that patients such as Mr. J could experience, one can see why it would lead to many different providers being involved in the diagnosis. It is not uncommon for psychiatrists to be the last providers to care for such patients who could have been evaluated by hematology, cardiology, gastroenterology, dermatology, and/or neurology.
Hereditary coproporphyria
The team considered hepatic porphyias because of new-onset symptoms of mood lability, confusion, orthostasis, unsteady gait, weakness, dermatologic conditions on hands not responsive to treatment, and general malaise. Mr. J was diagnosed with HCP, a type of porphyria caused by a defect in coproporphyrinogen oxidase that leads to an accumulation of coproporphyrinogen III. This precursor, as are many porphyrin precursors, is neurotoxic, leading to neurovisceral or neuropsychiatric effects. Although in Mr. J’s case the coproporphyrinogen III value from the 24-hour drug screen was only modestly elevated, it has been noted that levels of excreted prophyrins do not necessarily correlate with symptom severity.3
In the past, porphyria testing was performed using the Watson-Schwartz test, which used Ehrlich’s reagent to precipitate porphyrins in a urine sample,4 and was used as a “bedside” test. Interestingly, porphyrins—not the iron found in the heme molecule—are precipitated in this test and cause the reddish-purple coloration of the urine sample. When quantitative testing was developed, a 24-hour sample of urine—kept on ice and away from ambient light, later to be frozen when sent to the laboratory—became the standard tool for testing for porphyrins. Now DNA testing can be used to diagnose HCP.
OUTCOME Symptoms resolve
Mr. J is started on loxapine, 20 mg at bedtime, and his symptoms resolve within 2 weeks. He maintains some baseline delusional ideation consistent with his history of schizoaffective disorder, but he is more social, his personal hygiene improves, he attends groups, eats in the cafeteria with his peers, and is no longer confused.
The author’s observations
In the 1950s, chlorpromazine was used to treat AIP.5 Mr. J received loxapine, a mid-potency first-generation antipsychotic, although it has been this author’s observation that high-potency first-generation antipsychotics are not effective for treating porphyria.
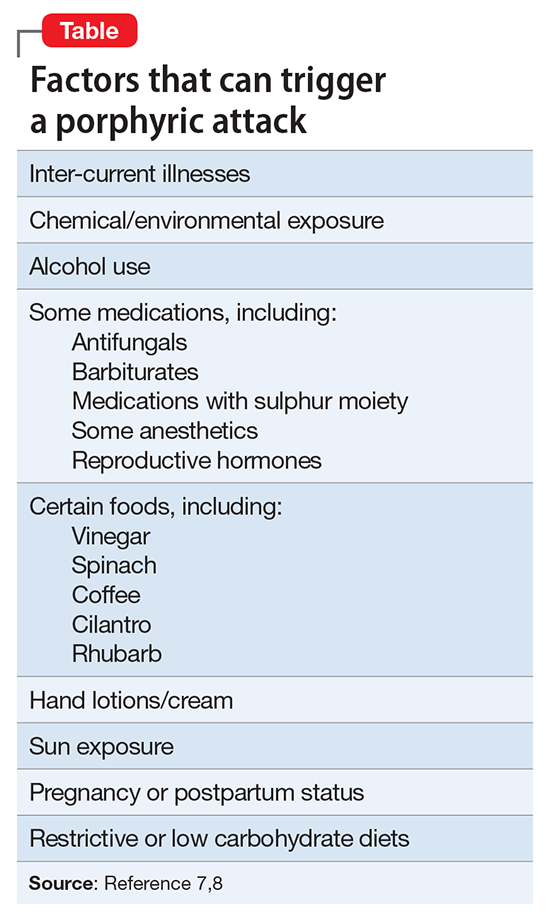
1. NIH: National Human Genome Research Institute. Learning about porphyria. https://www.genome.gov/19016728/learning-about-porphyria/learning-about-porphyria. Accessed February 23, 2017.
2. Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723-736.
3. Peters HA, Gocmen A, Cripps DJ, et al. Epidemiology of hexachlorobenzene-induced porphyria in Turkey: clinical and laboratory follow-up after 25 years. Arch Neurol. 1982;39(12):744-749.
4. The Watsonschwartz test. JAMA. 1966;195(6):481.
5. Brunton L, Chabner BA, Knollman B. Goodman & Gilman’s the pharmacological basis of therapeutics. 12th ed. New York, NY: McGraw-Hill Professional; 2010.
6. Broomfield B. Acute Intermittent porphyria treated with chlorpromazine. Proc R Soc Med. 1962;55(9):799-800.
7. Hunter JA, Khan SA, Hope E, et al. Hereditary coproporphyria. Photosensitivity, jaundice and neuropsychiatric manifestations associated with pregnancy. Br J Dermatol. 1971;84(4):301-310.
8. Bonkovsky HL, Maddukuri V. Merck Manual. http://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/porphyrias/overview-of-porphyrias. Accessed February 2, 2017.
9. Alexopoulos GS, Streim J, Carpenter D, et al; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry. 2004;65(suppl 2):5-99; discussion 100-102; quiz 103-104.
10. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
11. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
12. Rector NA, Beck AT. Cognitive behavioral therapy for schizophrenia: an empirical review. J Nerv Ment Dis. 2012;200(10):832-839.
13. Stobbe J, Mulder NC, Roosenschoon BJ, et al. Assertive community treatment for elderly people with severe mental illness. BMC Psychiatry. 2010;10:84.
14. Hennekens CH, Hennekens AR, Hollar D, et al. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150(6):1115-1121.
15. Bushe CJ, Taylor M, Haukka J. Mortality in schizophrenia: a measurable clinical point. J Psychopharmacol. 2010;24 (suppl 4):17-25.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia, and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
17. Nasrallah HA, Targum SD, Tandon R, et al. Defining and measuring clinical effectiveness in the treatment of schizophrenia. Psychiatr Serv. 2005;56(3):273-282.
18. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale (BPRS): recent developments in ascertainment and scaling. Psychopharmacol Bull. 1988;24:97-99.
19. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276.
20. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4): 247-251.
21. Guy W. ECDEU Assessment manual for psychopharmacology revised, 1976. Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; National Institute of Mental Health Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976.
22. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672-676.
23. Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand. 1970;45(212):11-19.
24. Dott SG, Weiden P, Hopwood P, et al. An innovative approach to clinical communication in schizophrenia: the Approaches to Schizophrenia Communication checklists. CNS Spectr. 2001;6(4):333-338.
25. Dott SG, Knesevich J, Miller A, et al. Using the ASC program: a training guide. J Psychiatr Pract. 2001;7(1): 64-68.
26. Barker S, Barron N, McFarland BH, et al. Multnomah Community Ability Scale: user’s manual. Portland, OR: Western Mental Health Research Center, Oregon Health Sciences University; 1994.
27. Lehman AF. A quality of life interview for the chronically mentally ill. Eval Program Plann. 1988;11(1):51-62.
28. Heinrichs DW, Hanlon TE, Carpenter WT Jr. The Quality of Life Scale: an instrument for rating the schizophrenic deficit syndrome. Schizophr Bull. 1984;10(3):388-398.
29. Becker M, Diamond R, Sainfort F. A new patient focused index for measuring quality of life in persons with severe and persistent mental illness. Qual Life Res. 1993;2(4):239-251.
30. Liberman RP, Kopelowicz A, Ventura J, et al. Operational criteria and factors related to recovery from schizophrenia. Int Rev Psychiatry. 2009;14(4):256-272.
CASE Withdrawn and confused
Mr. J, age 54, is brought to the emergency department from a correctional treatment facility where he is reported to be displaying new, unusual behavior. He has a history of schizoaffective disorder, which has been stable with haloperidol, 10 mg/d, for more than a year.
Although previously Mr. J openly discussed his long-standing delusions about the FBI coming to release him from prison, he no longer mentions this or any other delusional beliefs, and has become less communicative with staff and peers. Mr. J no longer accompanies the other patients to the cafeteria for meals and eats in his room alone and appears to be losing weight. He says, “I do not feel good,” but otherwise does not communicate spontaneously. Intermittently, he is irritable, without known triggers. The staff notices that Mr. J often lays on his bed, sometimes in a fetal position. Over time, he becomes confused and is seen attempting to open his room door with a toothbrush. His personal hygiene is poor, and he often urinates through his clothes, on the floor, and in his bed. Recently, Mr. J’s eczema has worsened. His gait has become unsteady, and he has orthostasis.
What could be causing these new symptoms?
a) worsening schizoaffective disorder
b) illicit drug use in the prison
c) atypical dementia
d) cardiac etiology
The author’s observations
The differential diagnosis for Mr. J appeared to be wide and without specific etiology. Because of the complex types of symptoms that Mr. J was experiencing, the emergency department managed his care and specialty clinic referrals were ordered.
It was reported that Mr. J started complaining of lightheadedness a few months ago, which worsened (unsteady gait, near falls). In the context of Mr. J’s history of lightheadedness and orthostasis, the cardiology clinic ordered a tilt table test, which was within normal limits:
- 70º head-up tilt: blood pressure, 91/67 to 102/62 mm Hg, and pulse, 70 to 79 beats per minute (bpm)
- with isoproterenol, 1 μg/minute: blood pressure, 90/66 to 110/70 mm Hg, and pulse, 77 to 124 bpm
- with isoproterenol, 2 μg/minute: blood pressure, 98/58 to 111/66 mm Hg, and pulse, 121 to 134 bpm.
The neurologist’s diagnostic impression was atypical dementia; however, Mr. J showed no memory deficits. Parkinsonism also was considered, but Mr. J had no unilateral tremor, masked facies, or micrographia. Mr. J showed some restriction in his movement, but he was not bradykinetic. The team suspected haloperidol could be causing his stiff movement.
Although it was possible that Mr. J’s schizoaffective disorder was worsening and led to the new symptoms, Mr. J appeared to be less delusional because he was no longer talking to the staff about his delusions. There seemed to be no outward evidence of progression of psychotic symptoms.
Mr. J had a history of substance abuse, including alcohol, cocaine, and Cannabis. Although prison inmates have been known to manufacture and drink “hooch,” the new symptoms Mr. J was experiencing were severe enough that his social interactions with other inmates diminished substantially. Because Mr. J had not been communicating with the other inmates and had no recent visitors, the team felt that it was unlikely that drugs were causing these symptoms. Also, a urine drug screen for cocaine, amphetamines, benzodiazepines, Cannabis, and opioids was negative.
HISTORY Substance use, violence
Mr. J was diagnosed with bipolar disorder at age 18. After later hospitalizations, his diagnosis was changed to schizoaffective disorder as a matter of diagnostic clarification. He has a long history of non-compliance with treatment, homelessness, and drug abuse.
Mr. J is serving a 20-year sentence for first-degree reckless homicide. A year after he was incarcerated, Mr. J was sent to a specialized mental health facility for inmates whose illness cannot be managed in a typical correctional setting. While at the treatment facility, Mr. J was non-compliant with medications and because of concerns about dangerousness and psychosis, the court found probable cause for involuntary commitment.
His medication regimen is trihexyphenidyl, 2 mg/d, for extrapyramidal symptoms; haloperidol, 10 mg/d, for psychosis; trazodone, 150 mg/d, for insomnia; vitamin D3, 2,000 IU/d; vitamin E, 400 IU/d, for symptoms of tardive dyskinesia; IM ziprasidone, 20 mg, because he refused oral haliperidol; and hydrocortisone cream 1% for eczema.
EVALUATION Additional tests
Mr. J’s blood pressure is 124/72 mm Hg, and pulse, 104 bpm, laying down; blood pressure, 110/84 mm Hg, and pulse, 112 bpm, sitting; and blood pressure, 108/82 mm Hg, and pulse, 129 bpm, standing. With repeated readings: blood pressure, 128/84 mm Hg, and pulse, 98 bpm, laying down; blood pressure, 125/86 mm Hg, and pulse, 113 bpm, sitting; and blood pressure, 105/76 mm Hg, and pulse, 130 bpm, standing.
Laboratory tests, including complete blood count, chemistry panel, thyroid-stimulating hormone, are within normal limits. The team feels that the investigation for an etiology for Mr. J’s symptoms needs to be more exhaustive and additional tests are ordered, including vitamin levels (C, B1, B12, B6), rapid plasma reagin for syphilis, and arbovirus testing (eastern equine encephalitis virus, western equine encephalitis, West Nile virus, La Crosse encephalitis, St. Louis encephalitis), which are negative.
What’s the next best step in managing Mr. J’s care?
a) adjust his medication
b) eliminate a mediation
c) order further testing
The author’s observations
To determine if Mr. J’s new-onset symptoms might be related to the progression of his psychiatric illness, the haloperidol dosage was increased to 20 mg/d; however, we saw no positive response to this change. His tardive dyskinesia symptoms (bruxism and other oral buccal movements) worsened. Haloperidol was reduced to 10 mg/d.
Trihexyphenidyl then was suspected to contribute to Mr. J’s confusion. Unfortunately, lowering the dosage of trihexyphenidyl to 1 mg/d, did not affect Mr. J’s current symptoms and exacerbated extrapyramidal symptoms.
The treatment team then questioned if porphyria—known as the “little imitator”—might be considered because of the variety of symptoms without an etiology, despite extensive testing. A 24-hour urine collection was ordered.
What is the correct method of collecting a urine sample for porphyrins?
a) collect a small sample and expose it to light before testing
b) collect a 24-hour sample with the sample kept in ambient temperature and light
c) collect a 24-hour sample with the sample kept on ice in a light-blocking container and frozen when sent to the laboratory
EVALUATION Diagnosis revealed
The 24-hour urine collection is obtained. However, it needed to be collected twice, because the first sample was not a full sample. Interestingly, the first sample, which is exposed to light and not kept on ice, turned dark in color. The second sample is obtained properly and sent to the laboratory. When the laboratory results are returned (Figure 1), Mr. J is diagnosed with hereditary coproporphyria (HCP).
The author’s observations
There are several types of porphyria, each associated with a different step in the chain of enzymes associated with synthesis of a heme molecule in the mitochondria. A defect in any single enzyme step will create a build up of porphyrins—a precursor to heme molecules—in erythrocytes or hepatic cells.
It is important to differentiate hepatic from erythropoietic porphyrias. The acute porphyrias (acute intermittent porphyria [AIP], HCP, and variegate porphyria generally are hepatic in origin with neuropsychiatric and neurovisceral symptoms. Cutaneous porphyrias originate in bone marrow and therefore are erythropoietic. However, there are exceptions such as porphyria cutanea tarda (PCT), which is hepatic in origin but the manifestations mainly are cutaneous1 (Figure 2).2
Although acute porphyria originates in the liver, it is a neuropsychiatric illness. In these cases, excess porphyrins cannot cross the blood–brain barrier and are neurotoxic. Clinicians can look for abnormalities in the liver via liver function tests, but liver parenchyma is not damaged by these enzyme precursors. During an acute porphyic attack, patients could experience symptoms such as:
- muscle spasms (commonly abdominal, but can be any muscle group)
- confusion
- disorientation
- autonomic instability
- lightheadness
- disorientation
- diarrhea
- light sensitivity
- dermatologic conditions
- weakness (particularly peripheral weakness)
- hypesthesia
- allodynia
- severe nausea and vomiting
- emotional lability
- psychosis as well as general malaise.
The attack could result in death.
Mr. J had many differing symptoms and was evaluated by several specialty providers. He had a chronic dermatologic condition; he was confused, disoriented, and complained of nausea, weakness, orthostasis, and loose stools. With the variety of possible symptoms that patients such as Mr. J could experience, one can see why it would lead to many different providers being involved in the diagnosis. It is not uncommon for psychiatrists to be the last providers to care for such patients who could have been evaluated by hematology, cardiology, gastroenterology, dermatology, and/or neurology.
Hereditary coproporphyria
The team considered hepatic porphyias because of new-onset symptoms of mood lability, confusion, orthostasis, unsteady gait, weakness, dermatologic conditions on hands not responsive to treatment, and general malaise. Mr. J was diagnosed with HCP, a type of porphyria caused by a defect in coproporphyrinogen oxidase that leads to an accumulation of coproporphyrinogen III. This precursor, as are many porphyrin precursors, is neurotoxic, leading to neurovisceral or neuropsychiatric effects. Although in Mr. J’s case the coproporphyrinogen III value from the 24-hour drug screen was only modestly elevated, it has been noted that levels of excreted prophyrins do not necessarily correlate with symptom severity.3
In the past, porphyria testing was performed using the Watson-Schwartz test, which used Ehrlich’s reagent to precipitate porphyrins in a urine sample,4 and was used as a “bedside” test. Interestingly, porphyrins—not the iron found in the heme molecule—are precipitated in this test and cause the reddish-purple coloration of the urine sample. When quantitative testing was developed, a 24-hour sample of urine—kept on ice and away from ambient light, later to be frozen when sent to the laboratory—became the standard tool for testing for porphyrins. Now DNA testing can be used to diagnose HCP.
OUTCOME Symptoms resolve
Mr. J is started on loxapine, 20 mg at bedtime, and his symptoms resolve within 2 weeks. He maintains some baseline delusional ideation consistent with his history of schizoaffective disorder, but he is more social, his personal hygiene improves, he attends groups, eats in the cafeteria with his peers, and is no longer confused.
The author’s observations
In the 1950s, chlorpromazine was used to treat AIP.5 Mr. J received loxapine, a mid-potency first-generation antipsychotic, although it has been this author’s observation that high-potency first-generation antipsychotics are not effective for treating porphyria.
CASE Withdrawn and confused
Mr. J, age 54, is brought to the emergency department from a correctional treatment facility where he is reported to be displaying new, unusual behavior. He has a history of schizoaffective disorder, which has been stable with haloperidol, 10 mg/d, for more than a year.
Although previously Mr. J openly discussed his long-standing delusions about the FBI coming to release him from prison, he no longer mentions this or any other delusional beliefs, and has become less communicative with staff and peers. Mr. J no longer accompanies the other patients to the cafeteria for meals and eats in his room alone and appears to be losing weight. He says, “I do not feel good,” but otherwise does not communicate spontaneously. Intermittently, he is irritable, without known triggers. The staff notices that Mr. J often lays on his bed, sometimes in a fetal position. Over time, he becomes confused and is seen attempting to open his room door with a toothbrush. His personal hygiene is poor, and he often urinates through his clothes, on the floor, and in his bed. Recently, Mr. J’s eczema has worsened. His gait has become unsteady, and he has orthostasis.
What could be causing these new symptoms?
a) worsening schizoaffective disorder
b) illicit drug use in the prison
c) atypical dementia
d) cardiac etiology
The author’s observations
The differential diagnosis for Mr. J appeared to be wide and without specific etiology. Because of the complex types of symptoms that Mr. J was experiencing, the emergency department managed his care and specialty clinic referrals were ordered.
It was reported that Mr. J started complaining of lightheadedness a few months ago, which worsened (unsteady gait, near falls). In the context of Mr. J’s history of lightheadedness and orthostasis, the cardiology clinic ordered a tilt table test, which was within normal limits:
- 70º head-up tilt: blood pressure, 91/67 to 102/62 mm Hg, and pulse, 70 to 79 beats per minute (bpm)
- with isoproterenol, 1 μg/minute: blood pressure, 90/66 to 110/70 mm Hg, and pulse, 77 to 124 bpm
- with isoproterenol, 2 μg/minute: blood pressure, 98/58 to 111/66 mm Hg, and pulse, 121 to 134 bpm.
The neurologist’s diagnostic impression was atypical dementia; however, Mr. J showed no memory deficits. Parkinsonism also was considered, but Mr. J had no unilateral tremor, masked facies, or micrographia. Mr. J showed some restriction in his movement, but he was not bradykinetic. The team suspected haloperidol could be causing his stiff movement.
Although it was possible that Mr. J’s schizoaffective disorder was worsening and led to the new symptoms, Mr. J appeared to be less delusional because he was no longer talking to the staff about his delusions. There seemed to be no outward evidence of progression of psychotic symptoms.
Mr. J had a history of substance abuse, including alcohol, cocaine, and Cannabis. Although prison inmates have been known to manufacture and drink “hooch,” the new symptoms Mr. J was experiencing were severe enough that his social interactions with other inmates diminished substantially. Because Mr. J had not been communicating with the other inmates and had no recent visitors, the team felt that it was unlikely that drugs were causing these symptoms. Also, a urine drug screen for cocaine, amphetamines, benzodiazepines, Cannabis, and opioids was negative.
HISTORY Substance use, violence
Mr. J was diagnosed with bipolar disorder at age 18. After later hospitalizations, his diagnosis was changed to schizoaffective disorder as a matter of diagnostic clarification. He has a long history of non-compliance with treatment, homelessness, and drug abuse.
Mr. J is serving a 20-year sentence for first-degree reckless homicide. A year after he was incarcerated, Mr. J was sent to a specialized mental health facility for inmates whose illness cannot be managed in a typical correctional setting. While at the treatment facility, Mr. J was non-compliant with medications and because of concerns about dangerousness and psychosis, the court found probable cause for involuntary commitment.
His medication regimen is trihexyphenidyl, 2 mg/d, for extrapyramidal symptoms; haloperidol, 10 mg/d, for psychosis; trazodone, 150 mg/d, for insomnia; vitamin D3, 2,000 IU/d; vitamin E, 400 IU/d, for symptoms of tardive dyskinesia; IM ziprasidone, 20 mg, because he refused oral haliperidol; and hydrocortisone cream 1% for eczema.
EVALUATION Additional tests
Mr. J’s blood pressure is 124/72 mm Hg, and pulse, 104 bpm, laying down; blood pressure, 110/84 mm Hg, and pulse, 112 bpm, sitting; and blood pressure, 108/82 mm Hg, and pulse, 129 bpm, standing. With repeated readings: blood pressure, 128/84 mm Hg, and pulse, 98 bpm, laying down; blood pressure, 125/86 mm Hg, and pulse, 113 bpm, sitting; and blood pressure, 105/76 mm Hg, and pulse, 130 bpm, standing.
Laboratory tests, including complete blood count, chemistry panel, thyroid-stimulating hormone, are within normal limits. The team feels that the investigation for an etiology for Mr. J’s symptoms needs to be more exhaustive and additional tests are ordered, including vitamin levels (C, B1, B12, B6), rapid plasma reagin for syphilis, and arbovirus testing (eastern equine encephalitis virus, western equine encephalitis, West Nile virus, La Crosse encephalitis, St. Louis encephalitis), which are negative.
What’s the next best step in managing Mr. J’s care?
a) adjust his medication
b) eliminate a mediation
c) order further testing
The author’s observations
To determine if Mr. J’s new-onset symptoms might be related to the progression of his psychiatric illness, the haloperidol dosage was increased to 20 mg/d; however, we saw no positive response to this change. His tardive dyskinesia symptoms (bruxism and other oral buccal movements) worsened. Haloperidol was reduced to 10 mg/d.
Trihexyphenidyl then was suspected to contribute to Mr. J’s confusion. Unfortunately, lowering the dosage of trihexyphenidyl to 1 mg/d, did not affect Mr. J’s current symptoms and exacerbated extrapyramidal symptoms.
The treatment team then questioned if porphyria—known as the “little imitator”—might be considered because of the variety of symptoms without an etiology, despite extensive testing. A 24-hour urine collection was ordered.
What is the correct method of collecting a urine sample for porphyrins?
a) collect a small sample and expose it to light before testing
b) collect a 24-hour sample with the sample kept in ambient temperature and light
c) collect a 24-hour sample with the sample kept on ice in a light-blocking container and frozen when sent to the laboratory
EVALUATION Diagnosis revealed
The 24-hour urine collection is obtained. However, it needed to be collected twice, because the first sample was not a full sample. Interestingly, the first sample, which is exposed to light and not kept on ice, turned dark in color. The second sample is obtained properly and sent to the laboratory. When the laboratory results are returned (Figure 1), Mr. J is diagnosed with hereditary coproporphyria (HCP).
The author’s observations
There are several types of porphyria, each associated with a different step in the chain of enzymes associated with synthesis of a heme molecule in the mitochondria. A defect in any single enzyme step will create a build up of porphyrins—a precursor to heme molecules—in erythrocytes or hepatic cells.
It is important to differentiate hepatic from erythropoietic porphyrias. The acute porphyrias (acute intermittent porphyria [AIP], HCP, and variegate porphyria generally are hepatic in origin with neuropsychiatric and neurovisceral symptoms. Cutaneous porphyrias originate in bone marrow and therefore are erythropoietic. However, there are exceptions such as porphyria cutanea tarda (PCT), which is hepatic in origin but the manifestations mainly are cutaneous1 (Figure 2).2
Although acute porphyria originates in the liver, it is a neuropsychiatric illness. In these cases, excess porphyrins cannot cross the blood–brain barrier and are neurotoxic. Clinicians can look for abnormalities in the liver via liver function tests, but liver parenchyma is not damaged by these enzyme precursors. During an acute porphyic attack, patients could experience symptoms such as:
- muscle spasms (commonly abdominal, but can be any muscle group)
- confusion
- disorientation
- autonomic instability
- lightheadness
- disorientation
- diarrhea
- light sensitivity
- dermatologic conditions
- weakness (particularly peripheral weakness)
- hypesthesia
- allodynia
- severe nausea and vomiting
- emotional lability
- psychosis as well as general malaise.
The attack could result in death.
Mr. J had many differing symptoms and was evaluated by several specialty providers. He had a chronic dermatologic condition; he was confused, disoriented, and complained of nausea, weakness, orthostasis, and loose stools. With the variety of possible symptoms that patients such as Mr. J could experience, one can see why it would lead to many different providers being involved in the diagnosis. It is not uncommon for psychiatrists to be the last providers to care for such patients who could have been evaluated by hematology, cardiology, gastroenterology, dermatology, and/or neurology.
Hereditary coproporphyria
The team considered hepatic porphyias because of new-onset symptoms of mood lability, confusion, orthostasis, unsteady gait, weakness, dermatologic conditions on hands not responsive to treatment, and general malaise. Mr. J was diagnosed with HCP, a type of porphyria caused by a defect in coproporphyrinogen oxidase that leads to an accumulation of coproporphyrinogen III. This precursor, as are many porphyrin precursors, is neurotoxic, leading to neurovisceral or neuropsychiatric effects. Although in Mr. J’s case the coproporphyrinogen III value from the 24-hour drug screen was only modestly elevated, it has been noted that levels of excreted prophyrins do not necessarily correlate with symptom severity.3
In the past, porphyria testing was performed using the Watson-Schwartz test, which used Ehrlich’s reagent to precipitate porphyrins in a urine sample,4 and was used as a “bedside” test. Interestingly, porphyrins—not the iron found in the heme molecule—are precipitated in this test and cause the reddish-purple coloration of the urine sample. When quantitative testing was developed, a 24-hour sample of urine—kept on ice and away from ambient light, later to be frozen when sent to the laboratory—became the standard tool for testing for porphyrins. Now DNA testing can be used to diagnose HCP.
OUTCOME Symptoms resolve
Mr. J is started on loxapine, 20 mg at bedtime, and his symptoms resolve within 2 weeks. He maintains some baseline delusional ideation consistent with his history of schizoaffective disorder, but he is more social, his personal hygiene improves, he attends groups, eats in the cafeteria with his peers, and is no longer confused.
The author’s observations
In the 1950s, chlorpromazine was used to treat AIP.5 Mr. J received loxapine, a mid-potency first-generation antipsychotic, although it has been this author’s observation that high-potency first-generation antipsychotics are not effective for treating porphyria.
1. NIH: National Human Genome Research Institute. Learning about porphyria. https://www.genome.gov/19016728/learning-about-porphyria/learning-about-porphyria. Accessed February 23, 2017.
2. Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723-736.
3. Peters HA, Gocmen A, Cripps DJ, et al. Epidemiology of hexachlorobenzene-induced porphyria in Turkey: clinical and laboratory follow-up after 25 years. Arch Neurol. 1982;39(12):744-749.
4. The Watsonschwartz test. JAMA. 1966;195(6):481.
5. Brunton L, Chabner BA, Knollman B. Goodman & Gilman’s the pharmacological basis of therapeutics. 12th ed. New York, NY: McGraw-Hill Professional; 2010.
6. Broomfield B. Acute Intermittent porphyria treated with chlorpromazine. Proc R Soc Med. 1962;55(9):799-800.
7. Hunter JA, Khan SA, Hope E, et al. Hereditary coproporphyria. Photosensitivity, jaundice and neuropsychiatric manifestations associated with pregnancy. Br J Dermatol. 1971;84(4):301-310.
8. Bonkovsky HL, Maddukuri V. Merck Manual. http://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/porphyrias/overview-of-porphyrias. Accessed February 2, 2017.
9. Alexopoulos GS, Streim J, Carpenter D, et al; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry. 2004;65(suppl 2):5-99; discussion 100-102; quiz 103-104.
10. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
11. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
12. Rector NA, Beck AT. Cognitive behavioral therapy for schizophrenia: an empirical review. J Nerv Ment Dis. 2012;200(10):832-839.
13. Stobbe J, Mulder NC, Roosenschoon BJ, et al. Assertive community treatment for elderly people with severe mental illness. BMC Psychiatry. 2010;10:84.
14. Hennekens CH, Hennekens AR, Hollar D, et al. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150(6):1115-1121.
15. Bushe CJ, Taylor M, Haukka J. Mortality in schizophrenia: a measurable clinical point. J Psychopharmacol. 2010;24 (suppl 4):17-25.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia, and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
17. Nasrallah HA, Targum SD, Tandon R, et al. Defining and measuring clinical effectiveness in the treatment of schizophrenia. Psychiatr Serv. 2005;56(3):273-282.
18. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale (BPRS): recent developments in ascertainment and scaling. Psychopharmacol Bull. 1988;24:97-99.
19. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276.
20. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4): 247-251.
21. Guy W. ECDEU Assessment manual for psychopharmacology revised, 1976. Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; National Institute of Mental Health Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976.
22. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672-676.
23. Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand. 1970;45(212):11-19.
24. Dott SG, Weiden P, Hopwood P, et al. An innovative approach to clinical communication in schizophrenia: the Approaches to Schizophrenia Communication checklists. CNS Spectr. 2001;6(4):333-338.
25. Dott SG, Knesevich J, Miller A, et al. Using the ASC program: a training guide. J Psychiatr Pract. 2001;7(1): 64-68.
26. Barker S, Barron N, McFarland BH, et al. Multnomah Community Ability Scale: user’s manual. Portland, OR: Western Mental Health Research Center, Oregon Health Sciences University; 1994.
27. Lehman AF. A quality of life interview for the chronically mentally ill. Eval Program Plann. 1988;11(1):51-62.
28. Heinrichs DW, Hanlon TE, Carpenter WT Jr. The Quality of Life Scale: an instrument for rating the schizophrenic deficit syndrome. Schizophr Bull. 1984;10(3):388-398.
29. Becker M, Diamond R, Sainfort F. A new patient focused index for measuring quality of life in persons with severe and persistent mental illness. Qual Life Res. 1993;2(4):239-251.
30. Liberman RP, Kopelowicz A, Ventura J, et al. Operational criteria and factors related to recovery from schizophrenia. Int Rev Psychiatry. 2009;14(4):256-272.
1. NIH: National Human Genome Research Institute. Learning about porphyria. https://www.genome.gov/19016728/learning-about-porphyria/learning-about-porphyria. Accessed February 23, 2017.
2. Ajioka RS, Phillips JD, Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta. 2006;1763(7):723-736.
3. Peters HA, Gocmen A, Cripps DJ, et al. Epidemiology of hexachlorobenzene-induced porphyria in Turkey: clinical and laboratory follow-up after 25 years. Arch Neurol. 1982;39(12):744-749.
4. The Watsonschwartz test. JAMA. 1966;195(6):481.
5. Brunton L, Chabner BA, Knollman B. Goodman & Gilman’s the pharmacological basis of therapeutics. 12th ed. New York, NY: McGraw-Hill Professional; 2010.
6. Broomfield B. Acute Intermittent porphyria treated with chlorpromazine. Proc R Soc Med. 1962;55(9):799-800.
7. Hunter JA, Khan SA, Hope E, et al. Hereditary coproporphyria. Photosensitivity, jaundice and neuropsychiatric manifestations associated with pregnancy. Br J Dermatol. 1971;84(4):301-310.
8. Bonkovsky HL, Maddukuri V. Merck Manual. http://www.merckmanuals.com/professional/endocrine-and-metabolic-disorders/porphyrias/overview-of-porphyrias. Accessed February 2, 2017.
9. Alexopoulos GS, Streim J, Carpenter D, et al; Expert Consensus Panel for Using Antipsychotic Drugs in Older Patients. Using antipsychotic agents in older patients. J Clin Psychiatry. 2004;65(suppl 2):5-99; discussion 100-102; quiz 103-104.
10. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry. 1999;56(3):241-247.
11. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
12. Rector NA, Beck AT. Cognitive behavioral therapy for schizophrenia: an empirical review. J Nerv Ment Dis. 2012;200(10):832-839.
13. Stobbe J, Mulder NC, Roosenschoon BJ, et al. Assertive community treatment for elderly people with severe mental illness. BMC Psychiatry. 2010;10:84.
14. Hennekens CH, Hennekens AR, Hollar D, et al. Schizophrenia and increased risks of cardiovascular disease. Am Heart J. 2005;150(6):1115-1121.
15. Bushe CJ, Taylor M, Haukka J. Mortality in schizophrenia: a measurable clinical point. J Psychopharmacol. 2010;24 (suppl 4):17-25.
16. Nasrallah HA, Meyer JM, Goff DC, et al. Low rates of treatment for hypertension, dyslipidemia, and diabetes in schizophrenia: data from the CATIE schizophrenia trial sample at baseline. Schizophr Res. 2006;86(1-3):15-22.
17. Nasrallah HA, Targum SD, Tandon R, et al. Defining and measuring clinical effectiveness in the treatment of schizophrenia. Psychiatr Serv. 2005;56(3):273-282.
18. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale (BPRS): recent developments in ascertainment and scaling. Psychopharmacol Bull. 1988;24:97-99.
19. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261-276.
20. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4): 247-251.
21. Guy W. ECDEU Assessment manual for psychopharmacology revised, 1976. Rockville, MD: US Department of Health, Education, and Welfare; Public Health Service; Alcohol, Drug Abuse, and Mental Health Administration; National Institute of Mental Health Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976.
22. Barnes TR. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154:672-676.
23. Simpson GM, Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand. 1970;45(212):11-19.
24. Dott SG, Weiden P, Hopwood P, et al. An innovative approach to clinical communication in schizophrenia: the Approaches to Schizophrenia Communication checklists. CNS Spectr. 2001;6(4):333-338.
25. Dott SG, Knesevich J, Miller A, et al. Using the ASC program: a training guide. J Psychiatr Pract. 2001;7(1): 64-68.
26. Barker S, Barron N, McFarland BH, et al. Multnomah Community Ability Scale: user’s manual. Portland, OR: Western Mental Health Research Center, Oregon Health Sciences University; 1994.
27. Lehman AF. A quality of life interview for the chronically mentally ill. Eval Program Plann. 1988;11(1):51-62.
28. Heinrichs DW, Hanlon TE, Carpenter WT Jr. The Quality of Life Scale: an instrument for rating the schizophrenic deficit syndrome. Schizophr Bull. 1984;10(3):388-398.
29. Becker M, Diamond R, Sainfort F. A new patient focused index for measuring quality of life in persons with severe and persistent mental illness. Qual Life Res. 1993;2(4):239-251.
30. Liberman RP, Kopelowicz A, Ventura J, et al. Operational criteria and factors related to recovery from schizophrenia. Int Rev Psychiatry. 2009;14(4):256-272.