User login
Transplantation of donor bone marrow or umbilical cord blood partially corrected the collagen deficiency in five of seven children with recessive dystrophic epidermolysis bullosa who underwent the experimental therapy, greatly ameliorating their severe symptoms by improving their skin and mucosal integrity, according to a recent report in the Aug. 12 issue of the New England Journal of Medicine.
In the phase II clinical trial, six of the patients were alive at 130-799 days after the procedure; their rates of recovery and ultimate outcomes varied. Two showed rapid and dramatic clinical improvement in wound healing and mucocutaneous blistering, two showed marked improvement, and one showed slow, modest improvement. Another patient had a recurrence of blistering after 2 months of substantial improvement, said Dr. John E. Wagner of the University of Minnesota Health Center, Minneapolis, and his associates.
One of the patients who responded to the transplantation died from opportunistic infections on day 183. And one of the patients died before bone marrow infusion could be performed, from complications of the conditioning immunomyeloablative chemotherapy.
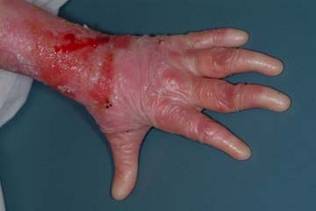
Epidermolysis bullosa (EB) refers to a group of inherited skin diseases characterized by painful erosions and blisters on skin and mucosal membranes, induced by mild trauma. Recessive dystrophic EB is one of the most severe forms of the disease, present at birth and often eventually resulting in esophageal strictures, mutilating scars, local and systemic infections, joint contractures, fusion of fingers and toes, and aggressive squamous-cell carcinomas. Median survival is only 30 years.
EB is caused by loss-of-function mutations in the gene that encodes for collagen type VII (C7). The mutations cause a severe decrease in the expression of C7, "a collagen localized at the dermal-epidermal junction" that contributes to the formation of "anchoring fibrils that tether the epidermal basement membrane to the dermal matrix," noted the investigators. When C7 is not expressed properly, these fibrils fail to form properly, "and epidermal-dermal adherence is lost beneath the lamina densa of the basement membrane," Dr. Wagner and his colleagues explained.
"To date, the care of patients with recessive dystrophic EB has been palliative and restricted to the treatment of individual wounds," they added.
The investigators explored whether bone marrow or cord blood transplantation might correct the collagen mutations systemically in a murine model. When that proved successful, they undertook the clinical trial in the seven pediatric patients (aged 15 months to 14.5 years). All had extensive cutaneous disease and four had severe mucosal disease requiring esophageal dilation and placement of a gastrostomy tube for nutritional support. Five patients had severe mitten deformities, four required wheelchairs, two had renal impairment, and four had severe iron-deficiency anemia.
One patient died before transplantation from hemorrhagic cardiomyopathy "that was probably due to cyclophosphamide cardiotoxicity," and a second had to delay transplantation until different complications from the conditioning chemotherapy resolved. At transplantation, five patients received unfiltered marrow stem cells from a human leukocyte antigen – identical but healthy sibling, and one of these five also received umbilical cord blood from the same donor. A sixth patient received umbilical cord blood from an unrelated donor.
All six patients showed improved wound healing and decreased mucocutaneous blistering within the first 100 days, with three showing marked improvement within 30 days. The percentage of affected body surface area was reduced significantly in three patients, as assessed by clinician and parent reports and by documented reductions in the need for bandages.
Four patients were able to discontinue all immunosuppressive therapy and the fifth has tolerated tapering of cyclosporine, according to the investigators.
Skin biopsy specimens showed increases in C7 immunoreactivity at the dermal-epidermal junction after transplantation in all six patients. Testing with an anti-C7 antibody showed an increase in C7 expression over time in five of the six.
At baseline, electron micrographs had shown "a complete absence of mature anchoring fibrils." After transplantation, five of the six patients showed "scanty, wispy structures under the lamina densa," which could represent rudimentary anchoring fibrils or fragmented elastic fibrils. More study is needed to further characterize these structures. None of the micrographs showed the morphologic hallmarks of normal anchoring fibrils, the investigators reported (N. Engl. J. Med. 2010;363:629-39).
"Unexpectedly, we detected substantial proportions of donor cells in the skin and mucosa after treatment; these proportions varied over time and with the location of the biopsy site," they wrote.
Although the precise identity and function of these donor cells has yet to be determined, "we favor the possibility that these healthy donor cells residing in the skin secrete C7 and that the secreted C7 is subsequently incorporated into the lamina densa at the dermal-epidermal junction," Dr. Wagner and his colleagues noted,
"Substantial efforts are under way to understand the physiology of the apparent clinical response after bone marrow transplantation and to identify the stem-cell population responsible for this effect," they wrote.
Until this trial was performed, it was not known whether patients with preexisting mucocutaneous disease could tolerate the conditioning regimens used to prepare for allogeneic bone marrow transplantation. In particular, mucositis was feared because it is a common side effect even in patients without mucocutaneous disease. "The unique skin and mucosal membrane defects of this disease pose a particular challenge to any bone marrow transplantation program," the investigators added.
Yet only one of the six patients developed severe cutaneous toxicity. All patients developed mucositis, but the condition responded to therapy. "Notably, no patient had uncontrolled cellulitis, despite pretransplantation bacterial or fungal skin colonization," they noted.
Other adverse events included transient hyperbilirubinemia (four patients) and renal insufficiency requiring 3 days of hemodialysis (two patients). No patient developed acute or chronic graft-vs.-host disease.
"Clearly, much remains to be learned regarding the mechanism of the apparent functional correction as well as the long-term risks and benefits of this therapeutic approach, including the risk of squamous-cell carcinoma, which may occur after chemotherapy or as a result of incomplete correction of the underlying disease," Dr. Wagner and his colleagues noted.
"Already, we and others are considering modifications to enhance safety, such as coinfusion of mesenchymal stromal cells or the use of reduced-dose conditioning before bone marrow transplantation," they added.
This study was supported by the University of Minnesota Academic Health Center; the National institutes of Health; the Ministry of Health, Labor, and Welfare of Japan; the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Epidermolysis Bullosa (Liao Family) Research Fund; the Sarah Rose Mooreland EB Fund; and the Children’s Cancer Research Fund. One of Dr. Wagner’s associates reported previous ties to Johnson & Johnson, Procter & Gamble, Novartis, Astellas, and Allergan.
Despite the still unresolved clinical and scientific issues with this experimental therapy, the systemic approach to this genetic skin disease "represents a leap forward," wrote Dr. Leena Bruckner-Tuderman in an accompanying editorial (N. Engl. J. Med. 2010;363:680-2).
The study by Dr. Wagner and his colleagues "gives cautious hope that effective therapy of recessive dystrophic EB and other genetic skin diseases may one day be available," noted Dr. Bruckner-Tuderman, is in the department of dermatology at the University of Freiburg (Germany) Medical Center.
Future research should focus on the extent and duration of the therapeutic effects, particularly on whether higher C7 levels in the skin and improvements in mucocutaneous integrity can be sustained over the long term. In addition, more objective methods to assess treatment response are needed, as parental and clinical observation can be "quite subjective."
Dr. Wagner and his colleagues demonstrated that some patients with mucocutaneous fragility can tolerate the conditioning regimen and other procedures and medications needed for bone marrow transplantation. But they also showed that some cannot, and that the life-threatening adverse effects must be weighed carefully against potential benefits.
In their report, the investigators did not specifically address the issue of subject age in this clinical trial. Although some may consider it questionable to test an experimental treatment in children, it was important in this instance to conduct the test among patients in optimal clinical condition.
In recessive dystrophic EB, the severe secondary symptoms accrue with time, so it is reasonable to perform the transplantation as early as possible, "with the aim of preventing severe scarring, deformities, and also, ultimately squamous-cell carcinomas."
She reported no financial disclosures.
Transplantation of donor bone marrow or umbilical cord blood partially corrected the collagen deficiency in five of seven children with recessive dystrophic epidermolysis bullosa who underwent the experimental therapy, greatly ameliorating their severe symptoms by improving their skin and mucosal integrity, according to a recent report in the Aug. 12 issue of the New England Journal of Medicine.
In the phase II clinical trial, six of the patients were alive at 130-799 days after the procedure; their rates of recovery and ultimate outcomes varied. Two showed rapid and dramatic clinical improvement in wound healing and mucocutaneous blistering, two showed marked improvement, and one showed slow, modest improvement. Another patient had a recurrence of blistering after 2 months of substantial improvement, said Dr. John E. Wagner of the University of Minnesota Health Center, Minneapolis, and his associates.
One of the patients who responded to the transplantation died from opportunistic infections on day 183. And one of the patients died before bone marrow infusion could be performed, from complications of the conditioning immunomyeloablative chemotherapy.
Epidermolysis bullosa (EB) refers to a group of inherited skin diseases characterized by painful erosions and blisters on skin and mucosal membranes, induced by mild trauma. Recessive dystrophic EB is one of the most severe forms of the disease, present at birth and often eventually resulting in esophageal strictures, mutilating scars, local and systemic infections, joint contractures, fusion of fingers and toes, and aggressive squamous-cell carcinomas. Median survival is only 30 years.
EB is caused by loss-of-function mutations in the gene that encodes for collagen type VII (C7). The mutations cause a severe decrease in the expression of C7, "a collagen localized at the dermal-epidermal junction" that contributes to the formation of "anchoring fibrils that tether the epidermal basement membrane to the dermal matrix," noted the investigators. When C7 is not expressed properly, these fibrils fail to form properly, "and epidermal-dermal adherence is lost beneath the lamina densa of the basement membrane," Dr. Wagner and his colleagues explained.
"To date, the care of patients with recessive dystrophic EB has been palliative and restricted to the treatment of individual wounds," they added.
The investigators explored whether bone marrow or cord blood transplantation might correct the collagen mutations systemically in a murine model. When that proved successful, they undertook the clinical trial in the seven pediatric patients (aged 15 months to 14.5 years). All had extensive cutaneous disease and four had severe mucosal disease requiring esophageal dilation and placement of a gastrostomy tube for nutritional support. Five patients had severe mitten deformities, four required wheelchairs, two had renal impairment, and four had severe iron-deficiency anemia.
One patient died before transplantation from hemorrhagic cardiomyopathy "that was probably due to cyclophosphamide cardiotoxicity," and a second had to delay transplantation until different complications from the conditioning chemotherapy resolved. At transplantation, five patients received unfiltered marrow stem cells from a human leukocyte antigen – identical but healthy sibling, and one of these five also received umbilical cord blood from the same donor. A sixth patient received umbilical cord blood from an unrelated donor.
All six patients showed improved wound healing and decreased mucocutaneous blistering within the first 100 days, with three showing marked improvement within 30 days. The percentage of affected body surface area was reduced significantly in three patients, as assessed by clinician and parent reports and by documented reductions in the need for bandages.
Four patients were able to discontinue all immunosuppressive therapy and the fifth has tolerated tapering of cyclosporine, according to the investigators.
Skin biopsy specimens showed increases in C7 immunoreactivity at the dermal-epidermal junction after transplantation in all six patients. Testing with an anti-C7 antibody showed an increase in C7 expression over time in five of the six.
At baseline, electron micrographs had shown "a complete absence of mature anchoring fibrils." After transplantation, five of the six patients showed "scanty, wispy structures under the lamina densa," which could represent rudimentary anchoring fibrils or fragmented elastic fibrils. More study is needed to further characterize these structures. None of the micrographs showed the morphologic hallmarks of normal anchoring fibrils, the investigators reported (N. Engl. J. Med. 2010;363:629-39).
"Unexpectedly, we detected substantial proportions of donor cells in the skin and mucosa after treatment; these proportions varied over time and with the location of the biopsy site," they wrote.
Although the precise identity and function of these donor cells has yet to be determined, "we favor the possibility that these healthy donor cells residing in the skin secrete C7 and that the secreted C7 is subsequently incorporated into the lamina densa at the dermal-epidermal junction," Dr. Wagner and his colleagues noted,
"Substantial efforts are under way to understand the physiology of the apparent clinical response after bone marrow transplantation and to identify the stem-cell population responsible for this effect," they wrote.
Until this trial was performed, it was not known whether patients with preexisting mucocutaneous disease could tolerate the conditioning regimens used to prepare for allogeneic bone marrow transplantation. In particular, mucositis was feared because it is a common side effect even in patients without mucocutaneous disease. "The unique skin and mucosal membrane defects of this disease pose a particular challenge to any bone marrow transplantation program," the investigators added.
Yet only one of the six patients developed severe cutaneous toxicity. All patients developed mucositis, but the condition responded to therapy. "Notably, no patient had uncontrolled cellulitis, despite pretransplantation bacterial or fungal skin colonization," they noted.
Other adverse events included transient hyperbilirubinemia (four patients) and renal insufficiency requiring 3 days of hemodialysis (two patients). No patient developed acute or chronic graft-vs.-host disease.
"Clearly, much remains to be learned regarding the mechanism of the apparent functional correction as well as the long-term risks and benefits of this therapeutic approach, including the risk of squamous-cell carcinoma, which may occur after chemotherapy or as a result of incomplete correction of the underlying disease," Dr. Wagner and his colleagues noted.
"Already, we and others are considering modifications to enhance safety, such as coinfusion of mesenchymal stromal cells or the use of reduced-dose conditioning before bone marrow transplantation," they added.
This study was supported by the University of Minnesota Academic Health Center; the National institutes of Health; the Ministry of Health, Labor, and Welfare of Japan; the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Epidermolysis Bullosa (Liao Family) Research Fund; the Sarah Rose Mooreland EB Fund; and the Children’s Cancer Research Fund. One of Dr. Wagner’s associates reported previous ties to Johnson & Johnson, Procter & Gamble, Novartis, Astellas, and Allergan.
Despite the still unresolved clinical and scientific issues with this experimental therapy, the systemic approach to this genetic skin disease "represents a leap forward," wrote Dr. Leena Bruckner-Tuderman in an accompanying editorial (N. Engl. J. Med. 2010;363:680-2).
The study by Dr. Wagner and his colleagues "gives cautious hope that effective therapy of recessive dystrophic EB and other genetic skin diseases may one day be available," noted Dr. Bruckner-Tuderman, is in the department of dermatology at the University of Freiburg (Germany) Medical Center.
Future research should focus on the extent and duration of the therapeutic effects, particularly on whether higher C7 levels in the skin and improvements in mucocutaneous integrity can be sustained over the long term. In addition, more objective methods to assess treatment response are needed, as parental and clinical observation can be "quite subjective."
Dr. Wagner and his colleagues demonstrated that some patients with mucocutaneous fragility can tolerate the conditioning regimen and other procedures and medications needed for bone marrow transplantation. But they also showed that some cannot, and that the life-threatening adverse effects must be weighed carefully against potential benefits.
In their report, the investigators did not specifically address the issue of subject age in this clinical trial. Although some may consider it questionable to test an experimental treatment in children, it was important in this instance to conduct the test among patients in optimal clinical condition.
In recessive dystrophic EB, the severe secondary symptoms accrue with time, so it is reasonable to perform the transplantation as early as possible, "with the aim of preventing severe scarring, deformities, and also, ultimately squamous-cell carcinomas."
She reported no financial disclosures.
Transplantation of donor bone marrow or umbilical cord blood partially corrected the collagen deficiency in five of seven children with recessive dystrophic epidermolysis bullosa who underwent the experimental therapy, greatly ameliorating their severe symptoms by improving their skin and mucosal integrity, according to a recent report in the Aug. 12 issue of the New England Journal of Medicine.
In the phase II clinical trial, six of the patients were alive at 130-799 days after the procedure; their rates of recovery and ultimate outcomes varied. Two showed rapid and dramatic clinical improvement in wound healing and mucocutaneous blistering, two showed marked improvement, and one showed slow, modest improvement. Another patient had a recurrence of blistering after 2 months of substantial improvement, said Dr. John E. Wagner of the University of Minnesota Health Center, Minneapolis, and his associates.
One of the patients who responded to the transplantation died from opportunistic infections on day 183. And one of the patients died before bone marrow infusion could be performed, from complications of the conditioning immunomyeloablative chemotherapy.
Epidermolysis bullosa (EB) refers to a group of inherited skin diseases characterized by painful erosions and blisters on skin and mucosal membranes, induced by mild trauma. Recessive dystrophic EB is one of the most severe forms of the disease, present at birth and often eventually resulting in esophageal strictures, mutilating scars, local and systemic infections, joint contractures, fusion of fingers and toes, and aggressive squamous-cell carcinomas. Median survival is only 30 years.
EB is caused by loss-of-function mutations in the gene that encodes for collagen type VII (C7). The mutations cause a severe decrease in the expression of C7, "a collagen localized at the dermal-epidermal junction" that contributes to the formation of "anchoring fibrils that tether the epidermal basement membrane to the dermal matrix," noted the investigators. When C7 is not expressed properly, these fibrils fail to form properly, "and epidermal-dermal adherence is lost beneath the lamina densa of the basement membrane," Dr. Wagner and his colleagues explained.
"To date, the care of patients with recessive dystrophic EB has been palliative and restricted to the treatment of individual wounds," they added.
The investigators explored whether bone marrow or cord blood transplantation might correct the collagen mutations systemically in a murine model. When that proved successful, they undertook the clinical trial in the seven pediatric patients (aged 15 months to 14.5 years). All had extensive cutaneous disease and four had severe mucosal disease requiring esophageal dilation and placement of a gastrostomy tube for nutritional support. Five patients had severe mitten deformities, four required wheelchairs, two had renal impairment, and four had severe iron-deficiency anemia.
One patient died before transplantation from hemorrhagic cardiomyopathy "that was probably due to cyclophosphamide cardiotoxicity," and a second had to delay transplantation until different complications from the conditioning chemotherapy resolved. At transplantation, five patients received unfiltered marrow stem cells from a human leukocyte antigen – identical but healthy sibling, and one of these five also received umbilical cord blood from the same donor. A sixth patient received umbilical cord blood from an unrelated donor.
All six patients showed improved wound healing and decreased mucocutaneous blistering within the first 100 days, with three showing marked improvement within 30 days. The percentage of affected body surface area was reduced significantly in three patients, as assessed by clinician and parent reports and by documented reductions in the need for bandages.
Four patients were able to discontinue all immunosuppressive therapy and the fifth has tolerated tapering of cyclosporine, according to the investigators.
Skin biopsy specimens showed increases in C7 immunoreactivity at the dermal-epidermal junction after transplantation in all six patients. Testing with an anti-C7 antibody showed an increase in C7 expression over time in five of the six.
At baseline, electron micrographs had shown "a complete absence of mature anchoring fibrils." After transplantation, five of the six patients showed "scanty, wispy structures under the lamina densa," which could represent rudimentary anchoring fibrils or fragmented elastic fibrils. More study is needed to further characterize these structures. None of the micrographs showed the morphologic hallmarks of normal anchoring fibrils, the investigators reported (N. Engl. J. Med. 2010;363:629-39).
"Unexpectedly, we detected substantial proportions of donor cells in the skin and mucosa after treatment; these proportions varied over time and with the location of the biopsy site," they wrote.
Although the precise identity and function of these donor cells has yet to be determined, "we favor the possibility that these healthy donor cells residing in the skin secrete C7 and that the secreted C7 is subsequently incorporated into the lamina densa at the dermal-epidermal junction," Dr. Wagner and his colleagues noted,
"Substantial efforts are under way to understand the physiology of the apparent clinical response after bone marrow transplantation and to identify the stem-cell population responsible for this effect," they wrote.
Until this trial was performed, it was not known whether patients with preexisting mucocutaneous disease could tolerate the conditioning regimens used to prepare for allogeneic bone marrow transplantation. In particular, mucositis was feared because it is a common side effect even in patients without mucocutaneous disease. "The unique skin and mucosal membrane defects of this disease pose a particular challenge to any bone marrow transplantation program," the investigators added.
Yet only one of the six patients developed severe cutaneous toxicity. All patients developed mucositis, but the condition responded to therapy. "Notably, no patient had uncontrolled cellulitis, despite pretransplantation bacterial or fungal skin colonization," they noted.
Other adverse events included transient hyperbilirubinemia (four patients) and renal insufficiency requiring 3 days of hemodialysis (two patients). No patient developed acute or chronic graft-vs.-host disease.
"Clearly, much remains to be learned regarding the mechanism of the apparent functional correction as well as the long-term risks and benefits of this therapeutic approach, including the risk of squamous-cell carcinoma, which may occur after chemotherapy or as a result of incomplete correction of the underlying disease," Dr. Wagner and his colleagues noted.
"Already, we and others are considering modifications to enhance safety, such as coinfusion of mesenchymal stromal cells or the use of reduced-dose conditioning before bone marrow transplantation," they added.
This study was supported by the University of Minnesota Academic Health Center; the National institutes of Health; the Ministry of Health, Labor, and Welfare of Japan; the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Epidermolysis Bullosa (Liao Family) Research Fund; the Sarah Rose Mooreland EB Fund; and the Children’s Cancer Research Fund. One of Dr. Wagner’s associates reported previous ties to Johnson & Johnson, Procter & Gamble, Novartis, Astellas, and Allergan.
Despite the still unresolved clinical and scientific issues with this experimental therapy, the systemic approach to this genetic skin disease "represents a leap forward," wrote Dr. Leena Bruckner-Tuderman in an accompanying editorial (N. Engl. J. Med. 2010;363:680-2).
The study by Dr. Wagner and his colleagues "gives cautious hope that effective therapy of recessive dystrophic EB and other genetic skin diseases may one day be available," noted Dr. Bruckner-Tuderman, is in the department of dermatology at the University of Freiburg (Germany) Medical Center.
Future research should focus on the extent and duration of the therapeutic effects, particularly on whether higher C7 levels in the skin and improvements in mucocutaneous integrity can be sustained over the long term. In addition, more objective methods to assess treatment response are needed, as parental and clinical observation can be "quite subjective."
Dr. Wagner and his colleagues demonstrated that some patients with mucocutaneous fragility can tolerate the conditioning regimen and other procedures and medications needed for bone marrow transplantation. But they also showed that some cannot, and that the life-threatening adverse effects must be weighed carefully against potential benefits.
In their report, the investigators did not specifically address the issue of subject age in this clinical trial. Although some may consider it questionable to test an experimental treatment in children, it was important in this instance to conduct the test among patients in optimal clinical condition.
In recessive dystrophic EB, the severe secondary symptoms accrue with time, so it is reasonable to perform the transplantation as early as possible, "with the aim of preventing severe scarring, deformities, and also, ultimately squamous-cell carcinomas."
She reported no financial disclosures.