User login
Hematology drugs on the fast track

Credit: Bill Branson
The US Food and Drug Administration (FDA) has granted fast track designation to two hematology drugs: the monoclonal antibody MOR208 to treat relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and the antifibrotic agent PRM-151 to treat myelofibrosis (MF).
The FDA’s fast track program aims to expedite the development and review of drugs that have the potential to fill an unmet medical need in serious or life-threatening conditions.
MOR208
MOR208 is a humanized monoclonal antibody targeting CD19. It is under development by MorphoSys AG to treat B-cell malignancies. The program is in phase 2 clinical development in chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), and non-Hodgkin lymphoma.
Preclinical research with MOR208 revealed that it can trigger natural killer cell-mediated lysis of ALL cells. The drug had lytic activity against ALL cells from both adult and pediatric patients.
In a phase 1 study, MOR208 exhibited preliminary efficacy in patients with high-risk, heavily pretreated CLL, prompting responses in 67% of patients. Researchers said toxicity was acceptable, but infusion reactions were common.
“First results of our ongoing phase 2 trial, which we will present at this year’s ASH conference in December, have helped to identify diffuse large B-cell lymphoma as a valuable development opportunity for MOR208,” said Arndt Schottelius, chief development officer of MorphoSys AG.
“We are therefore delighted to have received the fast track designation for further development of MOR208 in DLBCL. The more frequent interactions with the FDA that this enables will help us to expedite the development of MOR208 in this particular subset of non-Hodgkin’s lymphoma patients.”
PRM-151
PRM-151 is a recombinant form of an endogenous human protein, pentraxin-2, that is specifically active at the site of tissue damage. PRM-151 is an agonist that acts as a monocyte/macrophage differentiation factor to prevent and potentially reverse fibrosis.
The drug has shown broad anti-fibrotic activity in preclinical models of fibrotic disease, including pulmonary fibrosis, acute and chronic nephropathy, liver fibrosis, and age-related macular degeneration.
PRM-151 has orphan designation in the US for MF and in both the US and European Union for the treatment of idiopathic pulmonary fibrosis.
The FDA’s fast track designation for PRM-151 covers primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
“This designation validates our perspective that there is a clear and compelling need for a novel mechanism for the treatment of myelofibrosis that specifically targets the underlying fibrotic processes of the disease,” said Beth Tréhu, MD, FACP, chief medical officer of Promedior Inc., the company developing PRM-151.
“We will continue to work expeditiously to advance this program through the clinic and look forward to presenting the full data set from the first stage of our phase 2 study later this year.”
Preliminary data from the phase 2 study of PRM-151 demonstrated benefits across all clinically relevant measures of MF, including decreases in bone marrow fibrosis, symptom responses, improvements in hemoglobin and platelets, and reductions in spleen size.
The drug also appeared to be well-tolerated and did not prompt myelosuppression. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has granted fast track designation to two hematology drugs: the monoclonal antibody MOR208 to treat relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and the antifibrotic agent PRM-151 to treat myelofibrosis (MF).
The FDA’s fast track program aims to expedite the development and review of drugs that have the potential to fill an unmet medical need in serious or life-threatening conditions.
MOR208
MOR208 is a humanized monoclonal antibody targeting CD19. It is under development by MorphoSys AG to treat B-cell malignancies. The program is in phase 2 clinical development in chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), and non-Hodgkin lymphoma.
Preclinical research with MOR208 revealed that it can trigger natural killer cell-mediated lysis of ALL cells. The drug had lytic activity against ALL cells from both adult and pediatric patients.
In a phase 1 study, MOR208 exhibited preliminary efficacy in patients with high-risk, heavily pretreated CLL, prompting responses in 67% of patients. Researchers said toxicity was acceptable, but infusion reactions were common.
“First results of our ongoing phase 2 trial, which we will present at this year’s ASH conference in December, have helped to identify diffuse large B-cell lymphoma as a valuable development opportunity for MOR208,” said Arndt Schottelius, chief development officer of MorphoSys AG.
“We are therefore delighted to have received the fast track designation for further development of MOR208 in DLBCL. The more frequent interactions with the FDA that this enables will help us to expedite the development of MOR208 in this particular subset of non-Hodgkin’s lymphoma patients.”
PRM-151
PRM-151 is a recombinant form of an endogenous human protein, pentraxin-2, that is specifically active at the site of tissue damage. PRM-151 is an agonist that acts as a monocyte/macrophage differentiation factor to prevent and potentially reverse fibrosis.
The drug has shown broad anti-fibrotic activity in preclinical models of fibrotic disease, including pulmonary fibrosis, acute and chronic nephropathy, liver fibrosis, and age-related macular degeneration.
PRM-151 has orphan designation in the US for MF and in both the US and European Union for the treatment of idiopathic pulmonary fibrosis.
The FDA’s fast track designation for PRM-151 covers primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
“This designation validates our perspective that there is a clear and compelling need for a novel mechanism for the treatment of myelofibrosis that specifically targets the underlying fibrotic processes of the disease,” said Beth Tréhu, MD, FACP, chief medical officer of Promedior Inc., the company developing PRM-151.
“We will continue to work expeditiously to advance this program through the clinic and look forward to presenting the full data set from the first stage of our phase 2 study later this year.”
Preliminary data from the phase 2 study of PRM-151 demonstrated benefits across all clinically relevant measures of MF, including decreases in bone marrow fibrosis, symptom responses, improvements in hemoglobin and platelets, and reductions in spleen size.
The drug also appeared to be well-tolerated and did not prompt myelosuppression. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has granted fast track designation to two hematology drugs: the monoclonal antibody MOR208 to treat relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and the antifibrotic agent PRM-151 to treat myelofibrosis (MF).
The FDA’s fast track program aims to expedite the development and review of drugs that have the potential to fill an unmet medical need in serious or life-threatening conditions.
MOR208
MOR208 is a humanized monoclonal antibody targeting CD19. It is under development by MorphoSys AG to treat B-cell malignancies. The program is in phase 2 clinical development in chronic lymphocytic leukemia (CLL), acute lymphoblastic leukemia (ALL), and non-Hodgkin lymphoma.
Preclinical research with MOR208 revealed that it can trigger natural killer cell-mediated lysis of ALL cells. The drug had lytic activity against ALL cells from both adult and pediatric patients.
In a phase 1 study, MOR208 exhibited preliminary efficacy in patients with high-risk, heavily pretreated CLL, prompting responses in 67% of patients. Researchers said toxicity was acceptable, but infusion reactions were common.
“First results of our ongoing phase 2 trial, which we will present at this year’s ASH conference in December, have helped to identify diffuse large B-cell lymphoma as a valuable development opportunity for MOR208,” said Arndt Schottelius, chief development officer of MorphoSys AG.
“We are therefore delighted to have received the fast track designation for further development of MOR208 in DLBCL. The more frequent interactions with the FDA that this enables will help us to expedite the development of MOR208 in this particular subset of non-Hodgkin’s lymphoma patients.”
PRM-151
PRM-151 is a recombinant form of an endogenous human protein, pentraxin-2, that is specifically active at the site of tissue damage. PRM-151 is an agonist that acts as a monocyte/macrophage differentiation factor to prevent and potentially reverse fibrosis.
The drug has shown broad anti-fibrotic activity in preclinical models of fibrotic disease, including pulmonary fibrosis, acute and chronic nephropathy, liver fibrosis, and age-related macular degeneration.
PRM-151 has orphan designation in the US for MF and in both the US and European Union for the treatment of idiopathic pulmonary fibrosis.
The FDA’s fast track designation for PRM-151 covers primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
“This designation validates our perspective that there is a clear and compelling need for a novel mechanism for the treatment of myelofibrosis that specifically targets the underlying fibrotic processes of the disease,” said Beth Tréhu, MD, FACP, chief medical officer of Promedior Inc., the company developing PRM-151.
“We will continue to work expeditiously to advance this program through the clinic and look forward to presenting the full data set from the first stage of our phase 2 study later this year.”
Preliminary data from the phase 2 study of PRM-151 demonstrated benefits across all clinically relevant measures of MF, including decreases in bone marrow fibrosis, symptom responses, improvements in hemoglobin and platelets, and reductions in spleen size.
The drug also appeared to be well-tolerated and did not prompt myelosuppression. ![]()
Artificial platelets halt bleeding faster

Credit: Andre E.X. Brown
Artificial platelet mimics can halt bleeding in mouse models 65% faster than nature can on its own, a new study has shown.
These platelet-like nanoparticles (PLNs) mimic the shape, size, flexibility, and surface chemistry of real blood platelets.
Researchers believe these design factors together are important for inducing clots to form faster at vascular injury sites while preventing harmful clots from forming indiscriminately elsewhere in the body.
The new technology is reported in ACS Nano.
Study investigator Anirban Sen Gupta, PhD, of Case Western Reserve in Cleveland, Ohio, previously designed peptide-based surface chemistries that mimic the clot-relevant activities of real platelets.
Building on this work, Dr Sen Gupta has now focused on incorporating morphological and mechanical cues that are naturally present in platelets to further refine the design.
“Morphological and mechanical factors influence the margination of natural platelets to the blood vessel wall, and only when they are near the wall can the critical clot-promoting chemical interactions take place,” he said.
These natural cues motivated Dr Sen Gupta to team up with Samir Mitragotri, PhD, of the University of California Santa Barbara, whose lab recently developed albumin-based technologies to make particles that mimic the geometry and mechanical properties of red blood cells and platelets.
Together, the researchers developed PLNs that combine morphological, mechanical, and surface chemical properties of natural platelets.
The team believes this refined design will be able to simulate natural platelet’s ability to collide effectively with larger and softer red blood cells in systemic blood flow. The collisions cause margination—pushing the platelets out of the main flow and closer to the blood vessel wall—increasing the probability of interacting with an injury site.
The surface coatings enable the PLNs to anchor to injury-site-specific proteins, von Willebrand factor and collagen, while inducing the natural and artificial platelets to aggregate faster at the injury site.
When the researchers injected the PLNs in mice, the artificial platelets formed clots at the site of injury 3 times faster than natural platelets alone in control mice.
The ability to interact selectively with injury site proteins, as well as the ability to remain mechanically flexible like natural platelets, enables these PLNs to safely ride through the smallest of blood vessels without causing unwanted clots. PLNs that don’t aggregate in a clot and continue to circulate are metabolized in 1 to 2 days.
The researchers believe their new artificial platelet design may be even more effective in larger-volume blood flows, where margination to the blood vessel wall is more prominent. They expect to soon begin testing those capabilities.
If the PLNs prove effective in humans, they could be used to stem bleeding in patients with clotting disorders, those suffering from traumatic injury, and patients undergoing surgeries.
The technology might also be used to deliver drugs to target sites in patients suffering from atherosclerosis, thrombosis, or other platelet-involved pathologic conditions. Dr Sen Gupta believes the PLNs could even be used to deliver cancer drugs to metastatic tumors that have high platelet interactions. ![]()
Credit: Andre E.X. Brown
Artificial platelet mimics can halt bleeding in mouse models 65% faster than nature can on its own, a new study has shown.
These platelet-like nanoparticles (PLNs) mimic the shape, size, flexibility, and surface chemistry of real blood platelets.
Researchers believe these design factors together are important for inducing clots to form faster at vascular injury sites while preventing harmful clots from forming indiscriminately elsewhere in the body.
The new technology is reported in ACS Nano.
Study investigator Anirban Sen Gupta, PhD, of Case Western Reserve in Cleveland, Ohio, previously designed peptide-based surface chemistries that mimic the clot-relevant activities of real platelets.
Building on this work, Dr Sen Gupta has now focused on incorporating morphological and mechanical cues that are naturally present in platelets to further refine the design.
“Morphological and mechanical factors influence the margination of natural platelets to the blood vessel wall, and only when they are near the wall can the critical clot-promoting chemical interactions take place,” he said.
These natural cues motivated Dr Sen Gupta to team up with Samir Mitragotri, PhD, of the University of California Santa Barbara, whose lab recently developed albumin-based technologies to make particles that mimic the geometry and mechanical properties of red blood cells and platelets.
Together, the researchers developed PLNs that combine morphological, mechanical, and surface chemical properties of natural platelets.
The team believes this refined design will be able to simulate natural platelet’s ability to collide effectively with larger and softer red blood cells in systemic blood flow. The collisions cause margination—pushing the platelets out of the main flow and closer to the blood vessel wall—increasing the probability of interacting with an injury site.
The surface coatings enable the PLNs to anchor to injury-site-specific proteins, von Willebrand factor and collagen, while inducing the natural and artificial platelets to aggregate faster at the injury site.
When the researchers injected the PLNs in mice, the artificial platelets formed clots at the site of injury 3 times faster than natural platelets alone in control mice.
The ability to interact selectively with injury site proteins, as well as the ability to remain mechanically flexible like natural platelets, enables these PLNs to safely ride through the smallest of blood vessels without causing unwanted clots. PLNs that don’t aggregate in a clot and continue to circulate are metabolized in 1 to 2 days.
The researchers believe their new artificial platelet design may be even more effective in larger-volume blood flows, where margination to the blood vessel wall is more prominent. They expect to soon begin testing those capabilities.
If the PLNs prove effective in humans, they could be used to stem bleeding in patients with clotting disorders, those suffering from traumatic injury, and patients undergoing surgeries.
The technology might also be used to deliver drugs to target sites in patients suffering from atherosclerosis, thrombosis, or other platelet-involved pathologic conditions. Dr Sen Gupta believes the PLNs could even be used to deliver cancer drugs to metastatic tumors that have high platelet interactions. ![]()
Credit: Andre E.X. Brown
Artificial platelet mimics can halt bleeding in mouse models 65% faster than nature can on its own, a new study has shown.
These platelet-like nanoparticles (PLNs) mimic the shape, size, flexibility, and surface chemistry of real blood platelets.
Researchers believe these design factors together are important for inducing clots to form faster at vascular injury sites while preventing harmful clots from forming indiscriminately elsewhere in the body.
The new technology is reported in ACS Nano.
Study investigator Anirban Sen Gupta, PhD, of Case Western Reserve in Cleveland, Ohio, previously designed peptide-based surface chemistries that mimic the clot-relevant activities of real platelets.
Building on this work, Dr Sen Gupta has now focused on incorporating morphological and mechanical cues that are naturally present in platelets to further refine the design.
“Morphological and mechanical factors influence the margination of natural platelets to the blood vessel wall, and only when they are near the wall can the critical clot-promoting chemical interactions take place,” he said.
These natural cues motivated Dr Sen Gupta to team up with Samir Mitragotri, PhD, of the University of California Santa Barbara, whose lab recently developed albumin-based technologies to make particles that mimic the geometry and mechanical properties of red blood cells and platelets.
Together, the researchers developed PLNs that combine morphological, mechanical, and surface chemical properties of natural platelets.
The team believes this refined design will be able to simulate natural platelet’s ability to collide effectively with larger and softer red blood cells in systemic blood flow. The collisions cause margination—pushing the platelets out of the main flow and closer to the blood vessel wall—increasing the probability of interacting with an injury site.
The surface coatings enable the PLNs to anchor to injury-site-specific proteins, von Willebrand factor and collagen, while inducing the natural and artificial platelets to aggregate faster at the injury site.
When the researchers injected the PLNs in mice, the artificial platelets formed clots at the site of injury 3 times faster than natural platelets alone in control mice.
The ability to interact selectively with injury site proteins, as well as the ability to remain mechanically flexible like natural platelets, enables these PLNs to safely ride through the smallest of blood vessels without causing unwanted clots. PLNs that don’t aggregate in a clot and continue to circulate are metabolized in 1 to 2 days.
The researchers believe their new artificial platelet design may be even more effective in larger-volume blood flows, where margination to the blood vessel wall is more prominent. They expect to soon begin testing those capabilities.
If the PLNs prove effective in humans, they could be used to stem bleeding in patients with clotting disorders, those suffering from traumatic injury, and patients undergoing surgeries.
The technology might also be used to deliver drugs to target sites in patients suffering from atherosclerosis, thrombosis, or other platelet-involved pathologic conditions. Dr Sen Gupta believes the PLNs could even be used to deliver cancer drugs to metastatic tumors that have high platelet interactions. ![]()
Team discovers key aspects of HSC development

New research suggests proinflammatory signaling is crucial to the creation of hematopoietic stem cells (HSCs) during embryonic development, a finding that could help scientists reproduce HSCs for therapeutic use.
Researchers discovered that TNFR2, via TNFα, activates the Notch and NF-kB signaling pathways to establish HSC fate, which suggests inflammatory signaling is required for HSC generation.
The group also found that primitive neutrophils are the major source of TNFa. So it seems these cells are crucial to HSC development as well.
The researchers described these findings in Cell.
“The development of some mature cell lineages from iPSCs [induced pluripotent stem cells], such as cardiac and neural, has been reasonably straightforward, but not with HSCs,” said principal investigator David Traver, PhD, of the University of California, San Diego School of Medicine.
“This is likely due, at least in part, to not fully understanding all of the factors used by the embryo to generate HSCs. We believe the discovery that proinflammatory cues are important in vivo will help us recapitulate instruction of HSC fate in vitro from iPSCs.”
For this study, Dr Traver and his colleagues decided to examine the role of TNFα in HSC development, extending previous research by Victoriano Mulero, PhD, of the University of Murcia in Spain.
Dr Mulero reported that TNFα is important in the function of the embryonic vascular system. And, in animal models where TNF function was absent, blood defects resulted.
Raquel Espin-Palazon, PhD, a researcher in Dr Traver’s lab and a former colleague of Dr Mulero’s, determined that TNFα is required for the emergence of HSCs during embryogenesis in zebrafish.
Dr Traver said the finding was completely unexpected because HSCs emerge relatively early in embryonic formation, when the developing organism is considered to be largely sterile and devoid of infection.
“Thus, there was no expectation that proinflammatory signaling would be active at this time or in the blood-forming regions,” Dr Traver said. “Equally surprising, we found that a population of embryonic myeloid cells, which are transient cells produced before HSCs arise, are the producers of the TNFα needed to establish HSC fate.”
“So it turns out that a small subset of myeloid cells that persist for only a few days in development are necessary to help generate the lineal precursors of the entire adult blood-forming system.” ![]()
New research suggests proinflammatory signaling is crucial to the creation of hematopoietic stem cells (HSCs) during embryonic development, a finding that could help scientists reproduce HSCs for therapeutic use.
Researchers discovered that TNFR2, via TNFα, activates the Notch and NF-kB signaling pathways to establish HSC fate, which suggests inflammatory signaling is required for HSC generation.
The group also found that primitive neutrophils are the major source of TNFa. So it seems these cells are crucial to HSC development as well.
The researchers described these findings in Cell.
“The development of some mature cell lineages from iPSCs [induced pluripotent stem cells], such as cardiac and neural, has been reasonably straightforward, but not with HSCs,” said principal investigator David Traver, PhD, of the University of California, San Diego School of Medicine.
“This is likely due, at least in part, to not fully understanding all of the factors used by the embryo to generate HSCs. We believe the discovery that proinflammatory cues are important in vivo will help us recapitulate instruction of HSC fate in vitro from iPSCs.”
For this study, Dr Traver and his colleagues decided to examine the role of TNFα in HSC development, extending previous research by Victoriano Mulero, PhD, of the University of Murcia in Spain.
Dr Mulero reported that TNFα is important in the function of the embryonic vascular system. And, in animal models where TNF function was absent, blood defects resulted.
Raquel Espin-Palazon, PhD, a researcher in Dr Traver’s lab and a former colleague of Dr Mulero’s, determined that TNFα is required for the emergence of HSCs during embryogenesis in zebrafish.
Dr Traver said the finding was completely unexpected because HSCs emerge relatively early in embryonic formation, when the developing organism is considered to be largely sterile and devoid of infection.
“Thus, there was no expectation that proinflammatory signaling would be active at this time or in the blood-forming regions,” Dr Traver said. “Equally surprising, we found that a population of embryonic myeloid cells, which are transient cells produced before HSCs arise, are the producers of the TNFα needed to establish HSC fate.”
“So it turns out that a small subset of myeloid cells that persist for only a few days in development are necessary to help generate the lineal precursors of the entire adult blood-forming system.” ![]()
New research suggests proinflammatory signaling is crucial to the creation of hematopoietic stem cells (HSCs) during embryonic development, a finding that could help scientists reproduce HSCs for therapeutic use.
Researchers discovered that TNFR2, via TNFα, activates the Notch and NF-kB signaling pathways to establish HSC fate, which suggests inflammatory signaling is required for HSC generation.
The group also found that primitive neutrophils are the major source of TNFa. So it seems these cells are crucial to HSC development as well.
The researchers described these findings in Cell.
“The development of some mature cell lineages from iPSCs [induced pluripotent stem cells], such as cardiac and neural, has been reasonably straightforward, but not with HSCs,” said principal investigator David Traver, PhD, of the University of California, San Diego School of Medicine.
“This is likely due, at least in part, to not fully understanding all of the factors used by the embryo to generate HSCs. We believe the discovery that proinflammatory cues are important in vivo will help us recapitulate instruction of HSC fate in vitro from iPSCs.”
For this study, Dr Traver and his colleagues decided to examine the role of TNFα in HSC development, extending previous research by Victoriano Mulero, PhD, of the University of Murcia in Spain.
Dr Mulero reported that TNFα is important in the function of the embryonic vascular system. And, in animal models where TNF function was absent, blood defects resulted.
Raquel Espin-Palazon, PhD, a researcher in Dr Traver’s lab and a former colleague of Dr Mulero’s, determined that TNFα is required for the emergence of HSCs during embryogenesis in zebrafish.
Dr Traver said the finding was completely unexpected because HSCs emerge relatively early in embryonic formation, when the developing organism is considered to be largely sterile and devoid of infection.
“Thus, there was no expectation that proinflammatory signaling would be active at this time or in the blood-forming regions,” Dr Traver said. “Equally surprising, we found that a population of embryonic myeloid cells, which are transient cells produced before HSCs arise, are the producers of the TNFα needed to establish HSC fate.”
“So it turns out that a small subset of myeloid cells that persist for only a few days in development are necessary to help generate the lineal precursors of the entire adult blood-forming system.” ![]()
Agent reverses effects of edoxaban

Credit: Kevin MacKenzie
An investigational anticoagulant reversal agent known as PER977 can restore hemostasis following edoxaban administration, according to a phase 1/2 trial.
The study included 80 healthy subjects who received PER977 or placebo 3 hours after they received a dose of edoxaban.
PER977 decreased anticoagulation to within 10% of baseline levels within 10 to 30 minutes of administration, an effect that took 12 to 15 hours in placebo-treated subjects.
Jack Ansell, MD, of Hofstra North Shore–LIJ School of Medicine in Hempstead, New York, and his colleagues reported these results in a letter to NEJM. The research was funded by Perosphere, the company developing PER977.
PER977 is a small molecule that can bind to unfractionated heparin, low-molecular-weight heparin, edoxaban, rivaroxaban, apixaban, and dabigatran. In preclinical studies, PER977 reversed anticoagulation with each of the new oral anticoagulants.
Dr Ansell and his colleagues wanted to determine the safety and efficacy of PER977 when given to healthy subjects after edoxaban. So the team enrolled 80 subjects and gave them placebo or single, escalating doses of PER977 (5 mg to 300 mg) 3 hours after a 60 mg dose of edoxaban.
After edoxaban administration, the mean whole-blood clotting time (WBCT) increased 37% from baseline. In placebo-treated subjects, it took 12 to 15 hours for the mean WBCT to return to within 10% above the baseline level.
In subjects who received PER977 at 100 mg to 300 mg, it took 10 minutes or less to reach the same level. And the WBCT remained within 10% above or below the baseline value for 24 hours, with no rebound and no infusions needed.
The researchers also assessed clots while measuring WBCT to determine the mean fibrin-fiber diameter. They found that edoxaban significantly reduced the mean fibrin-fiber diameter relative to baseline, from about 250 nm to about 125 nm (P<0.001).
But the mean fibrin-fiber diameter was restored to normal 30 minutes after subjects received 100 mg to 300 mg of PER977.
In addition, the researchers saw no evidence of procoagulant activity after PER977 administration, as assessed by levels of D-dimer, prothrombin fragment 1.2, and tissue factor pathway inhibitor.
The team noted a few adverse events that may have been related to PER977, including transient, mild perioral and facial flushing, dysgeusia, and moderate headache.
One subject experienced a moderate muscle cramp and elevation in creatinine phosphokinase levels, but the researchers believe this was not related to PER977.
“[T]he fact that PER977 was shown to be safe, well tolerated, and, most importantly, effective in reversing edoxaban, one of the new factor Xa oral anticoagulants, is a major step forward in developing a readily available and simple-to-use reversal agent for the new oral anticoagulants,” Dr Ansell said. ![]()
Credit: Kevin MacKenzie
An investigational anticoagulant reversal agent known as PER977 can restore hemostasis following edoxaban administration, according to a phase 1/2 trial.
The study included 80 healthy subjects who received PER977 or placebo 3 hours after they received a dose of edoxaban.
PER977 decreased anticoagulation to within 10% of baseline levels within 10 to 30 minutes of administration, an effect that took 12 to 15 hours in placebo-treated subjects.
Jack Ansell, MD, of Hofstra North Shore–LIJ School of Medicine in Hempstead, New York, and his colleagues reported these results in a letter to NEJM. The research was funded by Perosphere, the company developing PER977.
PER977 is a small molecule that can bind to unfractionated heparin, low-molecular-weight heparin, edoxaban, rivaroxaban, apixaban, and dabigatran. In preclinical studies, PER977 reversed anticoagulation with each of the new oral anticoagulants.
Dr Ansell and his colleagues wanted to determine the safety and efficacy of PER977 when given to healthy subjects after edoxaban. So the team enrolled 80 subjects and gave them placebo or single, escalating doses of PER977 (5 mg to 300 mg) 3 hours after a 60 mg dose of edoxaban.
After edoxaban administration, the mean whole-blood clotting time (WBCT) increased 37% from baseline. In placebo-treated subjects, it took 12 to 15 hours for the mean WBCT to return to within 10% above the baseline level.
In subjects who received PER977 at 100 mg to 300 mg, it took 10 minutes or less to reach the same level. And the WBCT remained within 10% above or below the baseline value for 24 hours, with no rebound and no infusions needed.
The researchers also assessed clots while measuring WBCT to determine the mean fibrin-fiber diameter. They found that edoxaban significantly reduced the mean fibrin-fiber diameter relative to baseline, from about 250 nm to about 125 nm (P<0.001).
But the mean fibrin-fiber diameter was restored to normal 30 minutes after subjects received 100 mg to 300 mg of PER977.
In addition, the researchers saw no evidence of procoagulant activity after PER977 administration, as assessed by levels of D-dimer, prothrombin fragment 1.2, and tissue factor pathway inhibitor.
The team noted a few adverse events that may have been related to PER977, including transient, mild perioral and facial flushing, dysgeusia, and moderate headache.
One subject experienced a moderate muscle cramp and elevation in creatinine phosphokinase levels, but the researchers believe this was not related to PER977.
“[T]he fact that PER977 was shown to be safe, well tolerated, and, most importantly, effective in reversing edoxaban, one of the new factor Xa oral anticoagulants, is a major step forward in developing a readily available and simple-to-use reversal agent for the new oral anticoagulants,” Dr Ansell said. ![]()
Credit: Kevin MacKenzie
An investigational anticoagulant reversal agent known as PER977 can restore hemostasis following edoxaban administration, according to a phase 1/2 trial.
The study included 80 healthy subjects who received PER977 or placebo 3 hours after they received a dose of edoxaban.
PER977 decreased anticoagulation to within 10% of baseline levels within 10 to 30 minutes of administration, an effect that took 12 to 15 hours in placebo-treated subjects.
Jack Ansell, MD, of Hofstra North Shore–LIJ School of Medicine in Hempstead, New York, and his colleagues reported these results in a letter to NEJM. The research was funded by Perosphere, the company developing PER977.
PER977 is a small molecule that can bind to unfractionated heparin, low-molecular-weight heparin, edoxaban, rivaroxaban, apixaban, and dabigatran. In preclinical studies, PER977 reversed anticoagulation with each of the new oral anticoagulants.
Dr Ansell and his colleagues wanted to determine the safety and efficacy of PER977 when given to healthy subjects after edoxaban. So the team enrolled 80 subjects and gave them placebo or single, escalating doses of PER977 (5 mg to 300 mg) 3 hours after a 60 mg dose of edoxaban.
After edoxaban administration, the mean whole-blood clotting time (WBCT) increased 37% from baseline. In placebo-treated subjects, it took 12 to 15 hours for the mean WBCT to return to within 10% above the baseline level.
In subjects who received PER977 at 100 mg to 300 mg, it took 10 minutes or less to reach the same level. And the WBCT remained within 10% above or below the baseline value for 24 hours, with no rebound and no infusions needed.
The researchers also assessed clots while measuring WBCT to determine the mean fibrin-fiber diameter. They found that edoxaban significantly reduced the mean fibrin-fiber diameter relative to baseline, from about 250 nm to about 125 nm (P<0.001).
But the mean fibrin-fiber diameter was restored to normal 30 minutes after subjects received 100 mg to 300 mg of PER977.
In addition, the researchers saw no evidence of procoagulant activity after PER977 administration, as assessed by levels of D-dimer, prothrombin fragment 1.2, and tissue factor pathway inhibitor.
The team noted a few adverse events that may have been related to PER977, including transient, mild perioral and facial flushing, dysgeusia, and moderate headache.
One subject experienced a moderate muscle cramp and elevation in creatinine phosphokinase levels, but the researchers believe this was not related to PER977.
“[T]he fact that PER977 was shown to be safe, well tolerated, and, most importantly, effective in reversing edoxaban, one of the new factor Xa oral anticoagulants, is a major step forward in developing a readily available and simple-to-use reversal agent for the new oral anticoagulants,” Dr Ansell said. ![]()
Aspartame, sweetened drinks don’t increase risk of NHL

beverages at the supermarket
Consuming aspartame and drinking sweetened beverages do not increase a person’s risk of developing non-Hodgkin lymphoma (NHL), according to research published in the Journal of Nutrition.
Investigators analyzed information from more than 100,000 men and women in the US.
The results suggested that neither aspartame intake nor the consumption of sugar-sweetened or artificially sweetened beverages were associated with an increased risk of NHL.
Marjorie L. McCullough, SCD, RD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this research, analyzing data from the nutrition cohort of the Cancer Prevention Study II, an assessment of cancer incidence
and mortality in the US.
Study subjects first completed a questionnaire in 1992, noting information related to diet and other lifestyle factors. They completed follow-up questionnaires in 1999 and 2003, which included questions related to the consumption of sugar-sweetened and artificially sweetened beverages, as well as tabletop sweeteners containing aspartame.
Among the 100,442 adult men and women who provided information on diet and lifestyle factors in 1999, there were 1196 NHL cases verified during a 10-year follow-up period.
The investigators assessed the risk of NHL associated with sweetened beverage and aspartame consumption, adjusted for the subjects’ smoking status, body mass index, and history of diabetes.
The analysis revealed that, in women and men combined, there was no association between NHL risk and the consumption of 1 or more servings (355 mL) of artificially sweetened beverages. Compared to nondrinkers, subjects who drank artificially sweetened beverages had a risk ratio (RR) of 0.92 (P=0.14).
Similarly, there was no association between NHL risk and sugar-sweetened beverages. Compared to nondrinkers, the RR for sugar-sweetened beverage drinkers was 1.10 (P=0.62).
Furthermore, subjects’ overall aspartame intake, which was estimated from artificially sweetened carbonated beverage consumption and the use of aspartame packets, was not associated with NHL risk.
The RR was 1.02 (P=0.69) for the top quintile (which had a median aspartame intake of 145 mg per day) vs the bottom quintile (which had a median aspartame intake of 0 mg per day).
The investigators also found that associations between disease and sweetened beverage consumption or aspartame intake were generally null for specific NHL subtypes, including multiple myeloma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, follicular lymphoma, and other B-cell lymphomas.
“The study supports the decades of research that have continued to find that aspartame is safe for use in foods and beverages,” said Haley Stevens, PhD, President of the Calorie Control Council. “It also supports the conclusions of the National Cancer Institute, who have determined that aspartame does not increase a person’s risk of developing cancer.” ![]()
beverages at the supermarket
Consuming aspartame and drinking sweetened beverages do not increase a person’s risk of developing non-Hodgkin lymphoma (NHL), according to research published in the Journal of Nutrition.
Investigators analyzed information from more than 100,000 men and women in the US.
The results suggested that neither aspartame intake nor the consumption of sugar-sweetened or artificially sweetened beverages were associated with an increased risk of NHL.
Marjorie L. McCullough, SCD, RD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this research, analyzing data from the nutrition cohort of the Cancer Prevention Study II, an assessment of cancer incidence
and mortality in the US.
Study subjects first completed a questionnaire in 1992, noting information related to diet and other lifestyle factors. They completed follow-up questionnaires in 1999 and 2003, which included questions related to the consumption of sugar-sweetened and artificially sweetened beverages, as well as tabletop sweeteners containing aspartame.
Among the 100,442 adult men and women who provided information on diet and lifestyle factors in 1999, there were 1196 NHL cases verified during a 10-year follow-up period.
The investigators assessed the risk of NHL associated with sweetened beverage and aspartame consumption, adjusted for the subjects’ smoking status, body mass index, and history of diabetes.
The analysis revealed that, in women and men combined, there was no association between NHL risk and the consumption of 1 or more servings (355 mL) of artificially sweetened beverages. Compared to nondrinkers, subjects who drank artificially sweetened beverages had a risk ratio (RR) of 0.92 (P=0.14).
Similarly, there was no association between NHL risk and sugar-sweetened beverages. Compared to nondrinkers, the RR for sugar-sweetened beverage drinkers was 1.10 (P=0.62).
Furthermore, subjects’ overall aspartame intake, which was estimated from artificially sweetened carbonated beverage consumption and the use of aspartame packets, was not associated with NHL risk.
The RR was 1.02 (P=0.69) for the top quintile (which had a median aspartame intake of 145 mg per day) vs the bottom quintile (which had a median aspartame intake of 0 mg per day).
The investigators also found that associations between disease and sweetened beverage consumption or aspartame intake were generally null for specific NHL subtypes, including multiple myeloma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, follicular lymphoma, and other B-cell lymphomas.
“The study supports the decades of research that have continued to find that aspartame is safe for use in foods and beverages,” said Haley Stevens, PhD, President of the Calorie Control Council. “It also supports the conclusions of the National Cancer Institute, who have determined that aspartame does not increase a person’s risk of developing cancer.” ![]()
beverages at the supermarket
Consuming aspartame and drinking sweetened beverages do not increase a person’s risk of developing non-Hodgkin lymphoma (NHL), according to research published in the Journal of Nutrition.
Investigators analyzed information from more than 100,000 men and women in the US.
The results suggested that neither aspartame intake nor the consumption of sugar-sweetened or artificially sweetened beverages were associated with an increased risk of NHL.
Marjorie L. McCullough, SCD, RD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this research, analyzing data from the nutrition cohort of the Cancer Prevention Study II, an assessment of cancer incidence
and mortality in the US.
Study subjects first completed a questionnaire in 1992, noting information related to diet and other lifestyle factors. They completed follow-up questionnaires in 1999 and 2003, which included questions related to the consumption of sugar-sweetened and artificially sweetened beverages, as well as tabletop sweeteners containing aspartame.
Among the 100,442 adult men and women who provided information on diet and lifestyle factors in 1999, there were 1196 NHL cases verified during a 10-year follow-up period.
The investigators assessed the risk of NHL associated with sweetened beverage and aspartame consumption, adjusted for the subjects’ smoking status, body mass index, and history of diabetes.
The analysis revealed that, in women and men combined, there was no association between NHL risk and the consumption of 1 or more servings (355 mL) of artificially sweetened beverages. Compared to nondrinkers, subjects who drank artificially sweetened beverages had a risk ratio (RR) of 0.92 (P=0.14).
Similarly, there was no association between NHL risk and sugar-sweetened beverages. Compared to nondrinkers, the RR for sugar-sweetened beverage drinkers was 1.10 (P=0.62).
Furthermore, subjects’ overall aspartame intake, which was estimated from artificially sweetened carbonated beverage consumption and the use of aspartame packets, was not associated with NHL risk.
The RR was 1.02 (P=0.69) for the top quintile (which had a median aspartame intake of 145 mg per day) vs the bottom quintile (which had a median aspartame intake of 0 mg per day).
The investigators also found that associations between disease and sweetened beverage consumption or aspartame intake were generally null for specific NHL subtypes, including multiple myeloma, diffuse large B-cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, follicular lymphoma, and other B-cell lymphomas.
“The study supports the decades of research that have continued to find that aspartame is safe for use in foods and beverages,” said Haley Stevens, PhD, President of the Calorie Control Council. “It also supports the conclusions of the National Cancer Institute, who have determined that aspartame does not increase a person’s risk of developing cancer.” ![]()
RNAi therapy may eliminate resistance in B-ALL
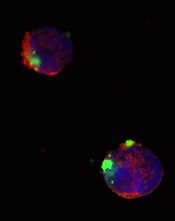
CD22ΔE12-siRNA nanoparticle
Credit: Fatih Uckun
Researchers have identified a molecular target for RNA interference (RNAi) therapy, and they believe that, by targeting this molecular lesion, they may be able to eliminate drug resistance in B-lineage acute lymphoblastic leukemia (ALL).
“We knew that we could kill chemotherapy-resistant leukemia cells if we only knew what made them so resistant,” said study author Fatih Uckun, MD, PhD, of the Children’s Hospital Los Angeles in California.
“Once we determined the mechanism, the next step was obvious—to rationally design a drug that would take out that specific target.”
The target is a defective gene that results in the production of abnormal CD22—CD22ΔE12. The researchers found that pediatric and adult B-lineage lymphoid malignancies are characterized by a high incidence of CD22ΔE12.
The group’s experiments revealed a causal link between CD22ΔE12 and the aggressive biology of B-ALL cells. When the researchers knocked down CD22ΔE12 in primary B-ALL cells using small interfering RNA (siRNA), they observed inhibition of clonogenicity.
The team therefore theorized that the siRNA-mediated depletion of CD22ΔE12 might be useful for treating B-ALL.
So they designed a nanoparticle formulation of CD22ΔE12-siRNA in which a polypeptide functions as the delivery system. The particle has a diameter of 100 nanometers.
Experiments showed that the nanoparticle could deliver its siRNA cargo into the cytosol of ALL-1 cells, thereby depleting CD22ΔE12 and inhibiting leukemic cell growth in vitro.
The researchers said that further development of this nanoparticle or other nanoformulation platforms for CD22ΔE12-siRNA could facilitate an effective therapeutic RNAi strategy to treat aggressive or chemotherapy-resistant B-lineage lymphoid malignancies.
“The goal is to translate our recent research discoveries in nanotechnology and biotherapy into effective patient-tailored treatment programs for the most common form of childhood cancer,” Dr Uckun said.
He and his colleagues described this research in EBioMedicine. ![]()
CD22ΔE12-siRNA nanoparticle
Credit: Fatih Uckun
Researchers have identified a molecular target for RNA interference (RNAi) therapy, and they believe that, by targeting this molecular lesion, they may be able to eliminate drug resistance in B-lineage acute lymphoblastic leukemia (ALL).
“We knew that we could kill chemotherapy-resistant leukemia cells if we only knew what made them so resistant,” said study author Fatih Uckun, MD, PhD, of the Children’s Hospital Los Angeles in California.
“Once we determined the mechanism, the next step was obvious—to rationally design a drug that would take out that specific target.”
The target is a defective gene that results in the production of abnormal CD22—CD22ΔE12. The researchers found that pediatric and adult B-lineage lymphoid malignancies are characterized by a high incidence of CD22ΔE12.
The group’s experiments revealed a causal link between CD22ΔE12 and the aggressive biology of B-ALL cells. When the researchers knocked down CD22ΔE12 in primary B-ALL cells using small interfering RNA (siRNA), they observed inhibition of clonogenicity.
The team therefore theorized that the siRNA-mediated depletion of CD22ΔE12 might be useful for treating B-ALL.
So they designed a nanoparticle formulation of CD22ΔE12-siRNA in which a polypeptide functions as the delivery system. The particle has a diameter of 100 nanometers.
Experiments showed that the nanoparticle could deliver its siRNA cargo into the cytosol of ALL-1 cells, thereby depleting CD22ΔE12 and inhibiting leukemic cell growth in vitro.
The researchers said that further development of this nanoparticle or other nanoformulation platforms for CD22ΔE12-siRNA could facilitate an effective therapeutic RNAi strategy to treat aggressive or chemotherapy-resistant B-lineage lymphoid malignancies.
“The goal is to translate our recent research discoveries in nanotechnology and biotherapy into effective patient-tailored treatment programs for the most common form of childhood cancer,” Dr Uckun said.
He and his colleagues described this research in EBioMedicine. ![]()
CD22ΔE12-siRNA nanoparticle
Credit: Fatih Uckun
Researchers have identified a molecular target for RNA interference (RNAi) therapy, and they believe that, by targeting this molecular lesion, they may be able to eliminate drug resistance in B-lineage acute lymphoblastic leukemia (ALL).
“We knew that we could kill chemotherapy-resistant leukemia cells if we only knew what made them so resistant,” said study author Fatih Uckun, MD, PhD, of the Children’s Hospital Los Angeles in California.
“Once we determined the mechanism, the next step was obvious—to rationally design a drug that would take out that specific target.”
The target is a defective gene that results in the production of abnormal CD22—CD22ΔE12. The researchers found that pediatric and adult B-lineage lymphoid malignancies are characterized by a high incidence of CD22ΔE12.
The group’s experiments revealed a causal link between CD22ΔE12 and the aggressive biology of B-ALL cells. When the researchers knocked down CD22ΔE12 in primary B-ALL cells using small interfering RNA (siRNA), they observed inhibition of clonogenicity.
The team therefore theorized that the siRNA-mediated depletion of CD22ΔE12 might be useful for treating B-ALL.
So they designed a nanoparticle formulation of CD22ΔE12-siRNA in which a polypeptide functions as the delivery system. The particle has a diameter of 100 nanometers.
Experiments showed that the nanoparticle could deliver its siRNA cargo into the cytosol of ALL-1 cells, thereby depleting CD22ΔE12 and inhibiting leukemic cell growth in vitro.
The researchers said that further development of this nanoparticle or other nanoformulation platforms for CD22ΔE12-siRNA could facilitate an effective therapeutic RNAi strategy to treat aggressive or chemotherapy-resistant B-lineage lymphoid malignancies.
“The goal is to translate our recent research discoveries in nanotechnology and biotherapy into effective patient-tailored treatment programs for the most common form of childhood cancer,” Dr Uckun said.
He and his colleagues described this research in EBioMedicine. ![]()
Dabigatran increases major, GI bleeding in AF

Dabigatran poses a greater risk of major bleeding and gastrointestinal (GI) bleeding compared to warfarin, according to a study of patients with atrial fibrillation.
The incidence of GI and major bleeding with dabigatran was particularly high in African Americans, patients with chronic kidney disease (CKD), patients age 75
and older, and those with 7 or more comorbidities.
On the other hand, the risk of intracranial hemorrhage was higher in most warfarin-treated patients. African Americans were the exception to this rule.
Researchers reported these results in JAMA Internal Medicine alongside a related editorial.
“Dabigatran was introduced in 2010 and, at the time of approval, it was the only available alternative to warfarin,” noted study author Yuting Zhang, PhD, of the University of Pittsburgh in Pennsylvania.
“Warfarin dosing can be tricky, and regular monitoring with blood tests is required, so doctors and patients were glad to have a drug that was easier to manage. But some recent studies suggest that dabigatran is associated with a higher risk of bleeding.”
To investigate that possibility, Dr Zhang and her colleagues reviewed pharmacy and medical claims data from 2010 and 2011 of a random national sample of Medicare beneficiaries. The team tracked 1302 dabigatran users and 8102 warfarin users to see whether they experienced bleeding episodes.
The researchers classified bleeding events as major, such as intracranial bleeding or GI bleeding requiring a hospital or emergency room stay, or minor, such as GI bleeding that was treated on an outpatient basis or nose bleeds.
The incidence of any bleeding was significantly higher in dabigatran-treated patients than in warfarin-treated patients (32.7% vs 26.5%, P<0.001). The same was true of major bleeding (9% vs 5.9%, P<0.001) and GI bleeding (17.4% vs 10%, P<0.001).
However, the rate of intracranial bleeding was higher with warfarin than with dabigatran (1.8% vs 0.6%, P<0.001), as was the rate of epistaxis (3.1% vs 2%, P=0.002) and not-otherwise-specified hemorrhage (5.9% vs 4.4%, P=0.003).
All other types of bleeding were more frequent in the dabigatran group than the warfarin group. This included hematuria (12% vs 8.8%, P<0.001), vaginal bleeding (0.7% vs 0.3%, P=0.003), hemarthrosis (0.5% vs 0.2%, P=0.007), and hemoptysis (2% vs 1.4%, P=0.03).
The researchers also looked at bleeding episodes in 4 high-risk subgroups: patients who were 75 years of age and older, African Americans, patients with CKD, and those with 7 or more co-existing medical problems.
The risk of major bleeding was higher among dabigatran-treated patients belonging to these subgroups. The hazard ratios (HRs) were 1.60 for patients 75 and older, 2.12 for African Americans, 2.07 for patients with CKD, and 1.88 for patients with 7 or more comorbidities.
Likewise, the risk of GI bleeding was higher among dabigatran-treated patients in the subgroups. The HRs were 1.85 for patients 75 and older, 2.38 for African Americans, 1.81 for patients with CKD, and 1.81 for patients with 7 or more comorbidities.
“These findings indicate that physicians should be cautious when prescribing dabigatran, particularly to African Americans and patients with kidney impairments,” said study author Inmaculada Hernandez, PharmD, of the University of Pittsburgh.
“Also, the incidence of gastrointestinal bleeding was high in all the subgroups, so we recommend doctors explain to patients how to detect it so that it can be treated promptly.”
The researchers also noted that the risk of intracranial bleeding varied among the subgroups. The HRs for dabigatran vs warfarin were 0.10 for patients 75 and older, 2.29 for African Americans, 0.71 for patients with CKD, and 0.59 for patients with 7 or more comorbidities.
“We plan to examine 2012 data to monitor the risk of stroke for patients on dabigatran, which is the primary indication for taking the blood thinner,” Dr Zhang said. “It’s possible that, for some patients, a greater reduction in the risk of stroke will outweigh the higher risk of bleeding with dabigatran compared to warfarin.” ![]()
Dabigatran poses a greater risk of major bleeding and gastrointestinal (GI) bleeding compared to warfarin, according to a study of patients with atrial fibrillation.
The incidence of GI and major bleeding with dabigatran was particularly high in African Americans, patients with chronic kidney disease (CKD), patients age 75
and older, and those with 7 or more comorbidities.
On the other hand, the risk of intracranial hemorrhage was higher in most warfarin-treated patients. African Americans were the exception to this rule.
Researchers reported these results in JAMA Internal Medicine alongside a related editorial.
“Dabigatran was introduced in 2010 and, at the time of approval, it was the only available alternative to warfarin,” noted study author Yuting Zhang, PhD, of the University of Pittsburgh in Pennsylvania.
“Warfarin dosing can be tricky, and regular monitoring with blood tests is required, so doctors and patients were glad to have a drug that was easier to manage. But some recent studies suggest that dabigatran is associated with a higher risk of bleeding.”
To investigate that possibility, Dr Zhang and her colleagues reviewed pharmacy and medical claims data from 2010 and 2011 of a random national sample of Medicare beneficiaries. The team tracked 1302 dabigatran users and 8102 warfarin users to see whether they experienced bleeding episodes.
The researchers classified bleeding events as major, such as intracranial bleeding or GI bleeding requiring a hospital or emergency room stay, or minor, such as GI bleeding that was treated on an outpatient basis or nose bleeds.
The incidence of any bleeding was significantly higher in dabigatran-treated patients than in warfarin-treated patients (32.7% vs 26.5%, P<0.001). The same was true of major bleeding (9% vs 5.9%, P<0.001) and GI bleeding (17.4% vs 10%, P<0.001).
However, the rate of intracranial bleeding was higher with warfarin than with dabigatran (1.8% vs 0.6%, P<0.001), as was the rate of epistaxis (3.1% vs 2%, P=0.002) and not-otherwise-specified hemorrhage (5.9% vs 4.4%, P=0.003).
All other types of bleeding were more frequent in the dabigatran group than the warfarin group. This included hematuria (12% vs 8.8%, P<0.001), vaginal bleeding (0.7% vs 0.3%, P=0.003), hemarthrosis (0.5% vs 0.2%, P=0.007), and hemoptysis (2% vs 1.4%, P=0.03).
The researchers also looked at bleeding episodes in 4 high-risk subgroups: patients who were 75 years of age and older, African Americans, patients with CKD, and those with 7 or more co-existing medical problems.
The risk of major bleeding was higher among dabigatran-treated patients belonging to these subgroups. The hazard ratios (HRs) were 1.60 for patients 75 and older, 2.12 for African Americans, 2.07 for patients with CKD, and 1.88 for patients with 7 or more comorbidities.
Likewise, the risk of GI bleeding was higher among dabigatran-treated patients in the subgroups. The HRs were 1.85 for patients 75 and older, 2.38 for African Americans, 1.81 for patients with CKD, and 1.81 for patients with 7 or more comorbidities.
“These findings indicate that physicians should be cautious when prescribing dabigatran, particularly to African Americans and patients with kidney impairments,” said study author Inmaculada Hernandez, PharmD, of the University of Pittsburgh.
“Also, the incidence of gastrointestinal bleeding was high in all the subgroups, so we recommend doctors explain to patients how to detect it so that it can be treated promptly.”
The researchers also noted that the risk of intracranial bleeding varied among the subgroups. The HRs for dabigatran vs warfarin were 0.10 for patients 75 and older, 2.29 for African Americans, 0.71 for patients with CKD, and 0.59 for patients with 7 or more comorbidities.
“We plan to examine 2012 data to monitor the risk of stroke for patients on dabigatran, which is the primary indication for taking the blood thinner,” Dr Zhang said. “It’s possible that, for some patients, a greater reduction in the risk of stroke will outweigh the higher risk of bleeding with dabigatran compared to warfarin.” ![]()
Dabigatran poses a greater risk of major bleeding and gastrointestinal (GI) bleeding compared to warfarin, according to a study of patients with atrial fibrillation.
The incidence of GI and major bleeding with dabigatran was particularly high in African Americans, patients with chronic kidney disease (CKD), patients age 75
and older, and those with 7 or more comorbidities.
On the other hand, the risk of intracranial hemorrhage was higher in most warfarin-treated patients. African Americans were the exception to this rule.
Researchers reported these results in JAMA Internal Medicine alongside a related editorial.
“Dabigatran was introduced in 2010 and, at the time of approval, it was the only available alternative to warfarin,” noted study author Yuting Zhang, PhD, of the University of Pittsburgh in Pennsylvania.
“Warfarin dosing can be tricky, and regular monitoring with blood tests is required, so doctors and patients were glad to have a drug that was easier to manage. But some recent studies suggest that dabigatran is associated with a higher risk of bleeding.”
To investigate that possibility, Dr Zhang and her colleagues reviewed pharmacy and medical claims data from 2010 and 2011 of a random national sample of Medicare beneficiaries. The team tracked 1302 dabigatran users and 8102 warfarin users to see whether they experienced bleeding episodes.
The researchers classified bleeding events as major, such as intracranial bleeding or GI bleeding requiring a hospital or emergency room stay, or minor, such as GI bleeding that was treated on an outpatient basis or nose bleeds.
The incidence of any bleeding was significantly higher in dabigatran-treated patients than in warfarin-treated patients (32.7% vs 26.5%, P<0.001). The same was true of major bleeding (9% vs 5.9%, P<0.001) and GI bleeding (17.4% vs 10%, P<0.001).
However, the rate of intracranial bleeding was higher with warfarin than with dabigatran (1.8% vs 0.6%, P<0.001), as was the rate of epistaxis (3.1% vs 2%, P=0.002) and not-otherwise-specified hemorrhage (5.9% vs 4.4%, P=0.003).
All other types of bleeding were more frequent in the dabigatran group than the warfarin group. This included hematuria (12% vs 8.8%, P<0.001), vaginal bleeding (0.7% vs 0.3%, P=0.003), hemarthrosis (0.5% vs 0.2%, P=0.007), and hemoptysis (2% vs 1.4%, P=0.03).
The researchers also looked at bleeding episodes in 4 high-risk subgroups: patients who were 75 years of age and older, African Americans, patients with CKD, and those with 7 or more co-existing medical problems.
The risk of major bleeding was higher among dabigatran-treated patients belonging to these subgroups. The hazard ratios (HRs) were 1.60 for patients 75 and older, 2.12 for African Americans, 2.07 for patients with CKD, and 1.88 for patients with 7 or more comorbidities.
Likewise, the risk of GI bleeding was higher among dabigatran-treated patients in the subgroups. The HRs were 1.85 for patients 75 and older, 2.38 for African Americans, 1.81 for patients with CKD, and 1.81 for patients with 7 or more comorbidities.
“These findings indicate that physicians should be cautious when prescribing dabigatran, particularly to African Americans and patients with kidney impairments,” said study author Inmaculada Hernandez, PharmD, of the University of Pittsburgh.
“Also, the incidence of gastrointestinal bleeding was high in all the subgroups, so we recommend doctors explain to patients how to detect it so that it can be treated promptly.”
The researchers also noted that the risk of intracranial bleeding varied among the subgroups. The HRs for dabigatran vs warfarin were 0.10 for patients 75 and older, 2.29 for African Americans, 0.71 for patients with CKD, and 0.59 for patients with 7 or more comorbidities.
“We plan to examine 2012 data to monitor the risk of stroke for patients on dabigatran, which is the primary indication for taking the blood thinner,” Dr Zhang said. “It’s possible that, for some patients, a greater reduction in the risk of stroke will outweigh the higher risk of bleeding with dabigatran compared to warfarin.”
NICE recommends ofatumumab in CLL

Credit: Linda Bartlett
The UK’s National Institute for Health and Care Excellence (NICE) has issued a preliminary draft guidance recommending ofatumumab (Arzerra) for use in patients with chronic lymphocytic leukemia (CLL).
The agency is recommending the anti-CD20 monoclonal antibody in combination with chlorambucil for patients with untreated CLL who are ineligible for treatment with fludarabine combination therapy and for whom bendamustine is unsuitable.
NICE believes ofatumumab is a cost-effective use of National Health Service (NHS) resources for this patient population, as GlaxoSmithKline, the company developing ofatumumab, has agreed to provide the drug at a reduced price.
The company has agreed with the Department of Health that the size of the discount be confidential.
Clinical effectiveness
“The information provided by GlaxoSmithKline, who market the drug, showed that ofatumumab with chlorambucil is a clinically effective treatment option for those people unable to take fludarabine combination therapy or bendamustine,” said Sir Andrew Dillon, NICE chief executive.
In the phase 3 COMPLEMENT 1 trial, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
A NICE advisory committee also considered the use of ofatumumab in combination with bendamustine. But the committee said that, due to limited clinical evidence and the absence of cost-effectiveness estimates, it could not make a recommendation on this combination.
In a phase 2 trial known as OMB115991, ofatumumab plus bendamustine elicited an overall response rate of 95% and a complete response rate of 43%.
Cost-effectiveness
Ofatumumab’s list price is £182 for a 100 mg vial and £1820 for a 1000 mg vial. Assuming 6 cycles and no drug wastage, the mean cost of a treatment course for ofatumumab at its list price is £11,466 for 6300 mg.
GlaxoSmithKline has agreed to a patient access scheme with the Department of Health that makes ofatumumab available with a discount on the list price, though the exact amount is confidential.
The NICE advisory committee said the most plausible cost-effectiveness estimate for ofatumumab plus chlorambucil compared with chlorambucil alone using the
ofatumumab patient access scheme price was £26,000 per quality-adjusted life year gained.
Consultees, including GlaxoSmithKline, healthcare professionals, and members of the public, can now comment on NICE’s preliminary draft guidance. It will be available for public consultation until November 25, 2014.
Until the final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance on a technology, it replaces local recommendations.
Credit: Linda Bartlett
The UK’s National Institute for Health and Care Excellence (NICE) has issued a preliminary draft guidance recommending ofatumumab (Arzerra) for use in patients with chronic lymphocytic leukemia (CLL).
The agency is recommending the anti-CD20 monoclonal antibody in combination with chlorambucil for patients with untreated CLL who are ineligible for treatment with fludarabine combination therapy and for whom bendamustine is unsuitable.
NICE believes ofatumumab is a cost-effective use of National Health Service (NHS) resources for this patient population, as GlaxoSmithKline, the company developing ofatumumab, has agreed to provide the drug at a reduced price.
The company has agreed with the Department of Health that the size of the discount be confidential.
Clinical effectiveness
“The information provided by GlaxoSmithKline, who market the drug, showed that ofatumumab with chlorambucil is a clinically effective treatment option for those people unable to take fludarabine combination therapy or bendamustine,” said Sir Andrew Dillon, NICE chief executive.
In the phase 3 COMPLEMENT 1 trial, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
A NICE advisory committee also considered the use of ofatumumab in combination with bendamustine. But the committee said that, due to limited clinical evidence and the absence of cost-effectiveness estimates, it could not make a recommendation on this combination.
In a phase 2 trial known as OMB115991, ofatumumab plus bendamustine elicited an overall response rate of 95% and a complete response rate of 43%.
Cost-effectiveness
Ofatumumab’s list price is £182 for a 100 mg vial and £1820 for a 1000 mg vial. Assuming 6 cycles and no drug wastage, the mean cost of a treatment course for ofatumumab at its list price is £11,466 for 6300 mg.
GlaxoSmithKline has agreed to a patient access scheme with the Department of Health that makes ofatumumab available with a discount on the list price, though the exact amount is confidential.
The NICE advisory committee said the most plausible cost-effectiveness estimate for ofatumumab plus chlorambucil compared with chlorambucil alone using the
ofatumumab patient access scheme price was £26,000 per quality-adjusted life year gained.
Consultees, including GlaxoSmithKline, healthcare professionals, and members of the public, can now comment on NICE’s preliminary draft guidance. It will be available for public consultation until November 25, 2014.
Until the final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance on a technology, it replaces local recommendations.
Credit: Linda Bartlett
The UK’s National Institute for Health and Care Excellence (NICE) has issued a preliminary draft guidance recommending ofatumumab (Arzerra) for use in patients with chronic lymphocytic leukemia (CLL).
The agency is recommending the anti-CD20 monoclonal antibody in combination with chlorambucil for patients with untreated CLL who are ineligible for treatment with fludarabine combination therapy and for whom bendamustine is unsuitable.
NICE believes ofatumumab is a cost-effective use of National Health Service (NHS) resources for this patient population, as GlaxoSmithKline, the company developing ofatumumab, has agreed to provide the drug at a reduced price.
The company has agreed with the Department of Health that the size of the discount be confidential.
Clinical effectiveness
“The information provided by GlaxoSmithKline, who market the drug, showed that ofatumumab with chlorambucil is a clinically effective treatment option for those people unable to take fludarabine combination therapy or bendamustine,” said Sir Andrew Dillon, NICE chief executive.
In the phase 3 COMPLEMENT 1 trial, ofatumumab plus chlorambucil improved progression-free survival compared to chlorambucil alone. The median times were 22.4 months and 13.1 months, respectively, and the hazard ratio was 0.57 (P<0.001).
A NICE advisory committee also considered the use of ofatumumab in combination with bendamustine. But the committee said that, due to limited clinical evidence and the absence of cost-effectiveness estimates, it could not make a recommendation on this combination.
In a phase 2 trial known as OMB115991, ofatumumab plus bendamustine elicited an overall response rate of 95% and a complete response rate of 43%.
Cost-effectiveness
Ofatumumab’s list price is £182 for a 100 mg vial and £1820 for a 1000 mg vial. Assuming 6 cycles and no drug wastage, the mean cost of a treatment course for ofatumumab at its list price is £11,466 for 6300 mg.
GlaxoSmithKline has agreed to a patient access scheme with the Department of Health that makes ofatumumab available with a discount on the list price, though the exact amount is confidential.
The NICE advisory committee said the most plausible cost-effectiveness estimate for ofatumumab plus chlorambucil compared with chlorambucil alone using the
ofatumumab patient access scheme price was £26,000 per quality-adjusted life year gained.
Consultees, including GlaxoSmithKline, healthcare professionals, and members of the public, can now comment on NICE’s preliminary draft guidance. It will be available for public consultation until November 25, 2014.
Until the final guidance is issued, NHS bodies should make decisions locally on the funding of specific treatments. Once NICE issues its final guidance on a technology, it replaces local recommendations.
FDA lifts clinical hold on imetelstat
The US Food and Drug Administration (FDA) has removed the full clinical hold placed on the investigational new drug application for the telomerase inhibitor imetelstat.
The hold, which was placed in March, suspended a phase 2 study of imetelstat in patients with essential thrombocythemia (ET) or polycythemia vera (PV), as well as a phase 2 study of the drug in patients with multiple myeloma (MM).
The hold also delayed a planned phase 2 trial in patients with myelofibrosis (MF).
And it temporarily suspended an investigator-sponsored trial of imetelstat in MF. The FDA lifted the hold on the investigator-sponsored trial in June.
The FDA halted these trials due to reports of persistent, low-grade liver function test (LFT) abnormalities observed in the phase 2 study of ET/PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The FDA expressed concern about whether these LFT abnormalities are reversible.
Now, data provided by the Geron Corporation, the company developing imetelstat, has convinced the FDA to lift the hold on all trials.
The FDA said the proposed clinical development plan for imetelstat, which is focused on high-risk myeloid disorders such as MF, is acceptable. Geron Corporation has said it does not intend to conduct further studies with, or develop imetelstat for, patients with ET or PV.
To address the clinical hold, the FDA required Geron to provide follow-up information from imetelstat-treated patients who experienced LFT abnormalities until such abnormalities resolved to normal or baseline.
Geron obtained follow-up information from patients in the previously ongoing company-sponsored phase 2 trials in ET/PV and MM. These data were submitted to the FDA as part of the company’s complete response.
The company’s analysis of these data showed that, in the ET/PV trial, LFT abnormalities resolved to normal or baseline in 14 of 18 follow-up patients. For the remaining 4 patients, at the time of the data cut-off, 3 patients showed improvement in LFT abnormalities, and 1 patient had unresolved LFT abnormalities. Two of the remaining 4 patients continue in follow-up.
In the MM trial, LFT abnormalities resolved to normal or baseline in all 9 follow-up patients. In addition, no emergent hepatic adverse events were reported during follow-up for either study.
The FDA also requested information regarding the reversibility of liver toxicity after chronic imetelstat administration in animals. Geron submitted data from its non-clinical toxicology studies, which included a 6-month study in mice and a 9-month study in cynomolgus monkeys.
In these studies, no clinical pathology changes indicative of hepatocellular injury were observed, and no clear signal of LFT abnormalities were identified.
With the clinical hold lifted, a multicenter phase 2 trial in MF is projected to begin in the first half of 2015.
The US Food and Drug Administration (FDA) has removed the full clinical hold placed on the investigational new drug application for the telomerase inhibitor imetelstat.
The hold, which was placed in March, suspended a phase 2 study of imetelstat in patients with essential thrombocythemia (ET) or polycythemia vera (PV), as well as a phase 2 study of the drug in patients with multiple myeloma (MM).
The hold also delayed a planned phase 2 trial in patients with myelofibrosis (MF).
And it temporarily suspended an investigator-sponsored trial of imetelstat in MF. The FDA lifted the hold on the investigator-sponsored trial in June.
The FDA halted these trials due to reports of persistent, low-grade liver function test (LFT) abnormalities observed in the phase 2 study of ET/PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The FDA expressed concern about whether these LFT abnormalities are reversible.
Now, data provided by the Geron Corporation, the company developing imetelstat, has convinced the FDA to lift the hold on all trials.
The FDA said the proposed clinical development plan for imetelstat, which is focused on high-risk myeloid disorders such as MF, is acceptable. Geron Corporation has said it does not intend to conduct further studies with, or develop imetelstat for, patients with ET or PV.
To address the clinical hold, the FDA required Geron to provide follow-up information from imetelstat-treated patients who experienced LFT abnormalities until such abnormalities resolved to normal or baseline.
Geron obtained follow-up information from patients in the previously ongoing company-sponsored phase 2 trials in ET/PV and MM. These data were submitted to the FDA as part of the company’s complete response.
The company’s analysis of these data showed that, in the ET/PV trial, LFT abnormalities resolved to normal or baseline in 14 of 18 follow-up patients. For the remaining 4 patients, at the time of the data cut-off, 3 patients showed improvement in LFT abnormalities, and 1 patient had unresolved LFT abnormalities. Two of the remaining 4 patients continue in follow-up.
In the MM trial, LFT abnormalities resolved to normal or baseline in all 9 follow-up patients. In addition, no emergent hepatic adverse events were reported during follow-up for either study.
The FDA also requested information regarding the reversibility of liver toxicity after chronic imetelstat administration in animals. Geron submitted data from its non-clinical toxicology studies, which included a 6-month study in mice and a 9-month study in cynomolgus monkeys.
In these studies, no clinical pathology changes indicative of hepatocellular injury were observed, and no clear signal of LFT abnormalities were identified.
With the clinical hold lifted, a multicenter phase 2 trial in MF is projected to begin in the first half of 2015.
The US Food and Drug Administration (FDA) has removed the full clinical hold placed on the investigational new drug application for the telomerase inhibitor imetelstat.
The hold, which was placed in March, suspended a phase 2 study of imetelstat in patients with essential thrombocythemia (ET) or polycythemia vera (PV), as well as a phase 2 study of the drug in patients with multiple myeloma (MM).
The hold also delayed a planned phase 2 trial in patients with myelofibrosis (MF).
And it temporarily suspended an investigator-sponsored trial of imetelstat in MF. The FDA lifted the hold on the investigator-sponsored trial in June.
The FDA halted these trials due to reports of persistent, low-grade liver function test (LFT) abnormalities observed in the phase 2 study of ET/PV patients and the potential risk of chronic liver injury following long-term exposure to imetelstat. The FDA expressed concern about whether these LFT abnormalities are reversible.
Now, data provided by the Geron Corporation, the company developing imetelstat, has convinced the FDA to lift the hold on all trials.
The FDA said the proposed clinical development plan for imetelstat, which is focused on high-risk myeloid disorders such as MF, is acceptable. Geron Corporation has said it does not intend to conduct further studies with, or develop imetelstat for, patients with ET or PV.
To address the clinical hold, the FDA required Geron to provide follow-up information from imetelstat-treated patients who experienced LFT abnormalities until such abnormalities resolved to normal or baseline.
Geron obtained follow-up information from patients in the previously ongoing company-sponsored phase 2 trials in ET/PV and MM. These data were submitted to the FDA as part of the company’s complete response.
The company’s analysis of these data showed that, in the ET/PV trial, LFT abnormalities resolved to normal or baseline in 14 of 18 follow-up patients. For the remaining 4 patients, at the time of the data cut-off, 3 patients showed improvement in LFT abnormalities, and 1 patient had unresolved LFT abnormalities. Two of the remaining 4 patients continue in follow-up.
In the MM trial, LFT abnormalities resolved to normal or baseline in all 9 follow-up patients. In addition, no emergent hepatic adverse events were reported during follow-up for either study.
The FDA also requested information regarding the reversibility of liver toxicity after chronic imetelstat administration in animals. Geron submitted data from its non-clinical toxicology studies, which included a 6-month study in mice and a 9-month study in cynomolgus monkeys.
In these studies, no clinical pathology changes indicative of hepatocellular injury were observed, and no clear signal of LFT abnormalities were identified.
With the clinical hold lifted, a multicenter phase 2 trial in MF is projected to begin in the first half of 2015.
Obesity affects toxicity of immunotherapy
Immunotherapy that can be effective against tumors in young, thin mice can be lethal to obese ones, according to research published in The Journal of Experimental Medicine.
Investigators conducted experiments in mouse models to determine if there is a subset of cancer patients for whom certain immunotherapies might be especially toxic.
The group found a potential link between body fat and the risk of toxicity from some types of immunotherapy.
“Cancer is primarily considered a disease of the aged, and yet preclinical studies generally use young, lean animal models that may not be reflective of the ‘typical’ cancer patient,” said study author Annie Mirsoian, a PhD candidate at the University of California, Davis in Sacramento.
“Aging is a dynamic process that is characterized by increases in inflammatory factors, as well as a shift in body composition where there is a gradual loss of lean muscle mass and an increase in fat accumulation, which effect how the immune system functions.”
Mirsoian and her colleagues sought to determine if, by adjusting the mouse model to more closely reflect the cancer patient phenotype (advanced age and overweight), they could better understand the discrepancies between animal study outcomes and those in patients in the clinic.
So the team examined aged mice on standard diets and compared those to aged mice that were calorie-restricted throughout their lives.
The investigators found that calorie restriction plays a protective role against toxicity. When aged mice ate their standard diet freely throughout life, they became obese and ultimately experienced lethal adverse reactions after receiving a systemic immunotherapy regimen.
“We know that people who are obese in general are at higher risk for complications from surgery, radiation, and chemotherapy,” said study author Arta Monjazeb, MD, PhD, of the University of California, Davis.
“We know that obese people have higher levels of inflammatory markers in their blood, but there is a lack of data examining the effects of obesity on cancer treatment outcomes.”
In follow-up experiments, the investigators found that young mice that are obese also endure similar toxic consequences, demonstrating that fat is a critical factor in toxic responses to stimulatory anticancer immunotherapy regimens.
“It is important to note, however, that the aged mice on standard diets succumbed to lethality at a quicker rate than young obese mice,” Mirsoian said. “Although our data demonstrate that obesity plays a central role in the development of adverse effects, future studies will focus on examining the aged immune system and cellular characteristics that may have enhanced the sensitivity of these mice to inflammation.”
This study follows an earlier paper, which demonstrated that while young, lean mice tolerate immunotherapy regimens without toxicity, the same regimen for an aged cohort resulted in lethal consequences. The new paper takes a deeper look into how fat deposition throughout aging can be critical in determining treatment tolerance and efficacy.
“Obesity has become an epidemic in our society and is now also affecting younger populations,” Mirsoian said. “Therefore, it’s likely that what the ‘typical’ cancer patient looks like will change. Our findings demonstrate the importance of having preclinical animal models that reflect the clinical scenario.”
“Changing the characteristics of our mouse models allowed for a more accurate determination of possible adverse reactions to therapy and more closely modeled what has been reported in the clinic with stimulatory immunotherapies.”
The investigators said factors like age, fat content, and the types of infections experienced throughout life together shape how the immune system reacts, and they will continue to work on improving their modeling system to reflect these changes.
They project that improvements in mouse modeling may help produce data that can better modulate treatment choices for patients, as well as identify early which patients would benefit from the inclusion of drugs to prevent adverse reactions while maintaining anticancer efficacy.
Immunotherapy that can be effective against tumors in young, thin mice can be lethal to obese ones, according to research published in The Journal of Experimental Medicine.
Investigators conducted experiments in mouse models to determine if there is a subset of cancer patients for whom certain immunotherapies might be especially toxic.
The group found a potential link between body fat and the risk of toxicity from some types of immunotherapy.
“Cancer is primarily considered a disease of the aged, and yet preclinical studies generally use young, lean animal models that may not be reflective of the ‘typical’ cancer patient,” said study author Annie Mirsoian, a PhD candidate at the University of California, Davis in Sacramento.
“Aging is a dynamic process that is characterized by increases in inflammatory factors, as well as a shift in body composition where there is a gradual loss of lean muscle mass and an increase in fat accumulation, which effect how the immune system functions.”
Mirsoian and her colleagues sought to determine if, by adjusting the mouse model to more closely reflect the cancer patient phenotype (advanced age and overweight), they could better understand the discrepancies between animal study outcomes and those in patients in the clinic.
So the team examined aged mice on standard diets and compared those to aged mice that were calorie-restricted throughout their lives.
The investigators found that calorie restriction plays a protective role against toxicity. When aged mice ate their standard diet freely throughout life, they became obese and ultimately experienced lethal adverse reactions after receiving a systemic immunotherapy regimen.
“We know that people who are obese in general are at higher risk for complications from surgery, radiation, and chemotherapy,” said study author Arta Monjazeb, MD, PhD, of the University of California, Davis.
“We know that obese people have higher levels of inflammatory markers in their blood, but there is a lack of data examining the effects of obesity on cancer treatment outcomes.”
In follow-up experiments, the investigators found that young mice that are obese also endure similar toxic consequences, demonstrating that fat is a critical factor in toxic responses to stimulatory anticancer immunotherapy regimens.
“It is important to note, however, that the aged mice on standard diets succumbed to lethality at a quicker rate than young obese mice,” Mirsoian said. “Although our data demonstrate that obesity plays a central role in the development of adverse effects, future studies will focus on examining the aged immune system and cellular characteristics that may have enhanced the sensitivity of these mice to inflammation.”
This study follows an earlier paper, which demonstrated that while young, lean mice tolerate immunotherapy regimens without toxicity, the same regimen for an aged cohort resulted in lethal consequences. The new paper takes a deeper look into how fat deposition throughout aging can be critical in determining treatment tolerance and efficacy.
“Obesity has become an epidemic in our society and is now also affecting younger populations,” Mirsoian said. “Therefore, it’s likely that what the ‘typical’ cancer patient looks like will change. Our findings demonstrate the importance of having preclinical animal models that reflect the clinical scenario.”
“Changing the characteristics of our mouse models allowed for a more accurate determination of possible adverse reactions to therapy and more closely modeled what has been reported in the clinic with stimulatory immunotherapies.”
The investigators said factors like age, fat content, and the types of infections experienced throughout life together shape how the immune system reacts, and they will continue to work on improving their modeling system to reflect these changes.
They project that improvements in mouse modeling may help produce data that can better modulate treatment choices for patients, as well as identify early which patients would benefit from the inclusion of drugs to prevent adverse reactions while maintaining anticancer efficacy.
Immunotherapy that can be effective against tumors in young, thin mice can be lethal to obese ones, according to research published in The Journal of Experimental Medicine.
Investigators conducted experiments in mouse models to determine if there is a subset of cancer patients for whom certain immunotherapies might be especially toxic.
The group found a potential link between body fat and the risk of toxicity from some types of immunotherapy.
“Cancer is primarily considered a disease of the aged, and yet preclinical studies generally use young, lean animal models that may not be reflective of the ‘typical’ cancer patient,” said study author Annie Mirsoian, a PhD candidate at the University of California, Davis in Sacramento.
“Aging is a dynamic process that is characterized by increases in inflammatory factors, as well as a shift in body composition where there is a gradual loss of lean muscle mass and an increase in fat accumulation, which effect how the immune system functions.”
Mirsoian and her colleagues sought to determine if, by adjusting the mouse model to more closely reflect the cancer patient phenotype (advanced age and overweight), they could better understand the discrepancies between animal study outcomes and those in patients in the clinic.
So the team examined aged mice on standard diets and compared those to aged mice that were calorie-restricted throughout their lives.
The investigators found that calorie restriction plays a protective role against toxicity. When aged mice ate their standard diet freely throughout life, they became obese and ultimately experienced lethal adverse reactions after receiving a systemic immunotherapy regimen.
“We know that people who are obese in general are at higher risk for complications from surgery, radiation, and chemotherapy,” said study author Arta Monjazeb, MD, PhD, of the University of California, Davis.
“We know that obese people have higher levels of inflammatory markers in their blood, but there is a lack of data examining the effects of obesity on cancer treatment outcomes.”
In follow-up experiments, the investigators found that young mice that are obese also endure similar toxic consequences, demonstrating that fat is a critical factor in toxic responses to stimulatory anticancer immunotherapy regimens.
“It is important to note, however, that the aged mice on standard diets succumbed to lethality at a quicker rate than young obese mice,” Mirsoian said. “Although our data demonstrate that obesity plays a central role in the development of adverse effects, future studies will focus on examining the aged immune system and cellular characteristics that may have enhanced the sensitivity of these mice to inflammation.”
This study follows an earlier paper, which demonstrated that while young, lean mice tolerate immunotherapy regimens without toxicity, the same regimen for an aged cohort resulted in lethal consequences. The new paper takes a deeper look into how fat deposition throughout aging can be critical in determining treatment tolerance and efficacy.
“Obesity has become an epidemic in our society and is now also affecting younger populations,” Mirsoian said. “Therefore, it’s likely that what the ‘typical’ cancer patient looks like will change. Our findings demonstrate the importance of having preclinical animal models that reflect the clinical scenario.”
“Changing the characteristics of our mouse models allowed for a more accurate determination of possible adverse reactions to therapy and more closely modeled what has been reported in the clinic with stimulatory immunotherapies.”
The investigators said factors like age, fat content, and the types of infections experienced throughout life together shape how the immune system reacts, and they will continue to work on improving their modeling system to reflect these changes.
They project that improvements in mouse modeling may help produce data that can better modulate treatment choices for patients, as well as identify early which patients would benefit from the inclusion of drugs to prevent adverse reactions while maintaining anticancer efficacy.