User login
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
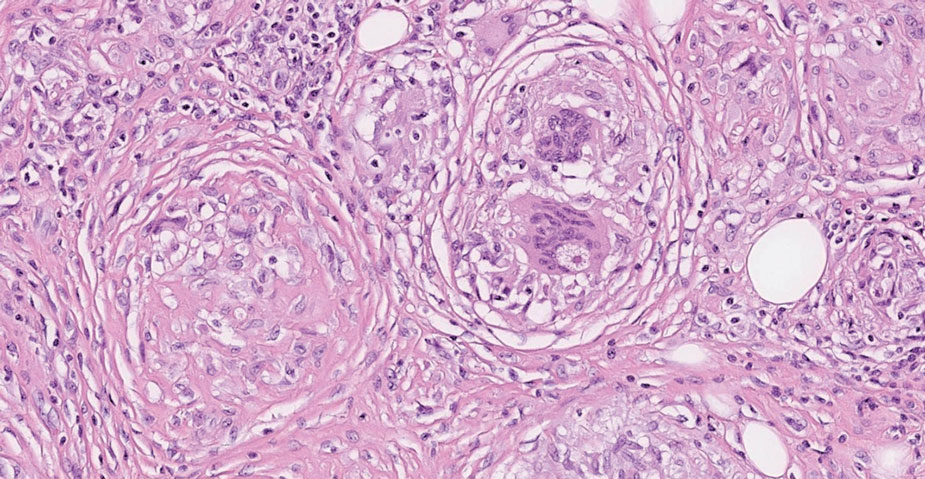
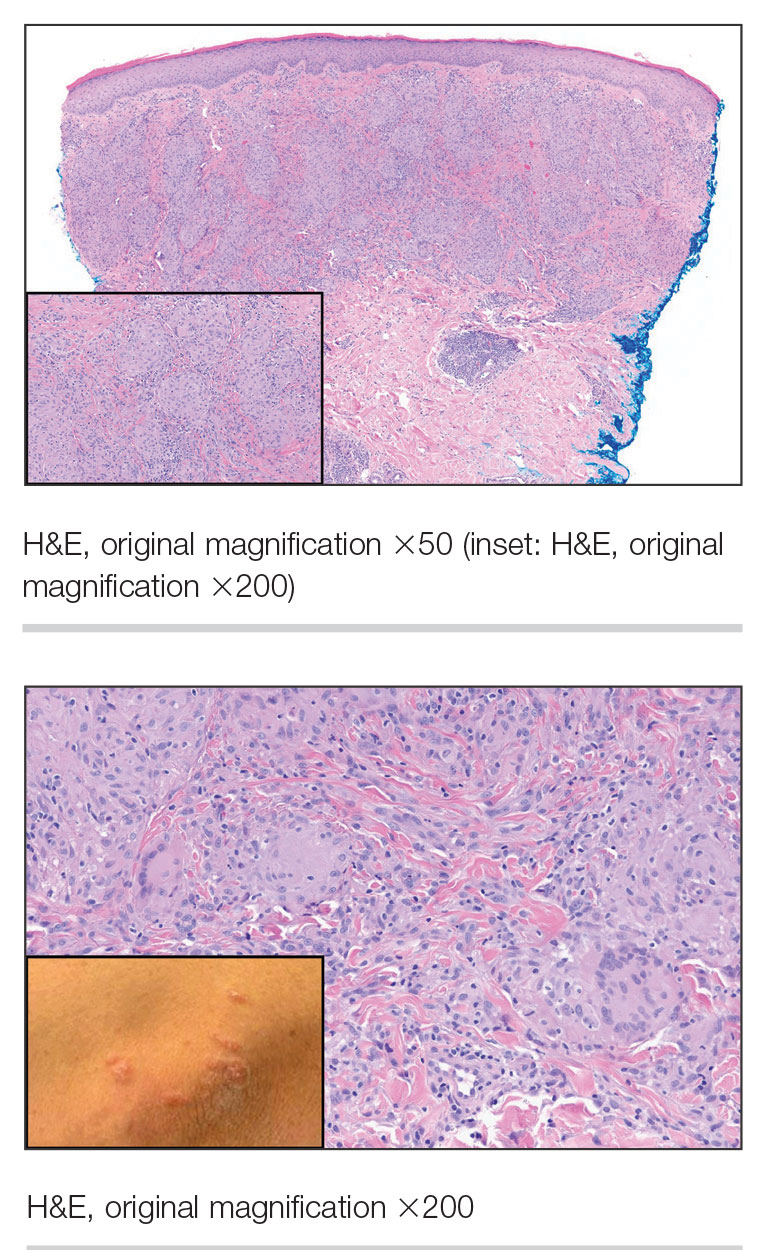
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
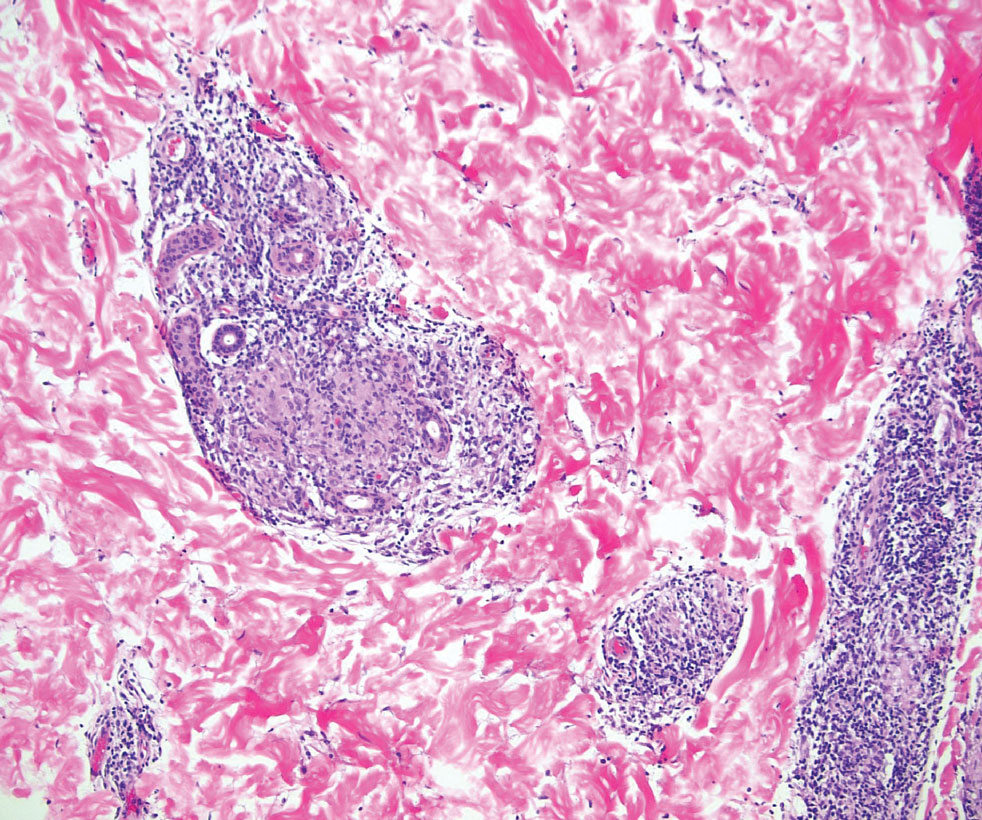
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
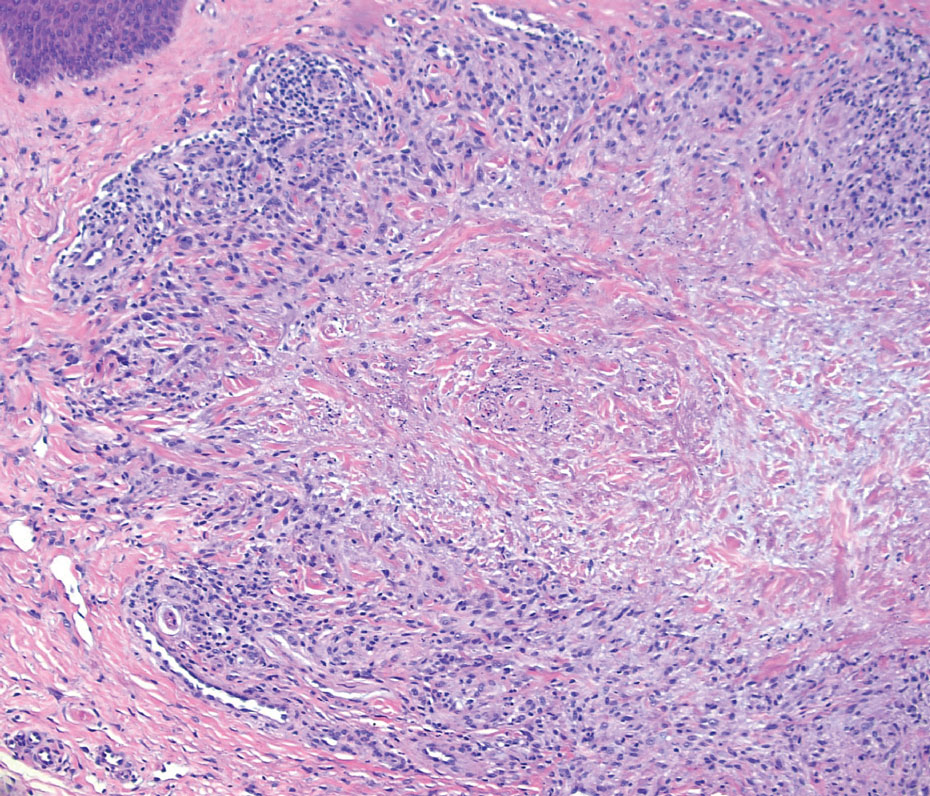
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
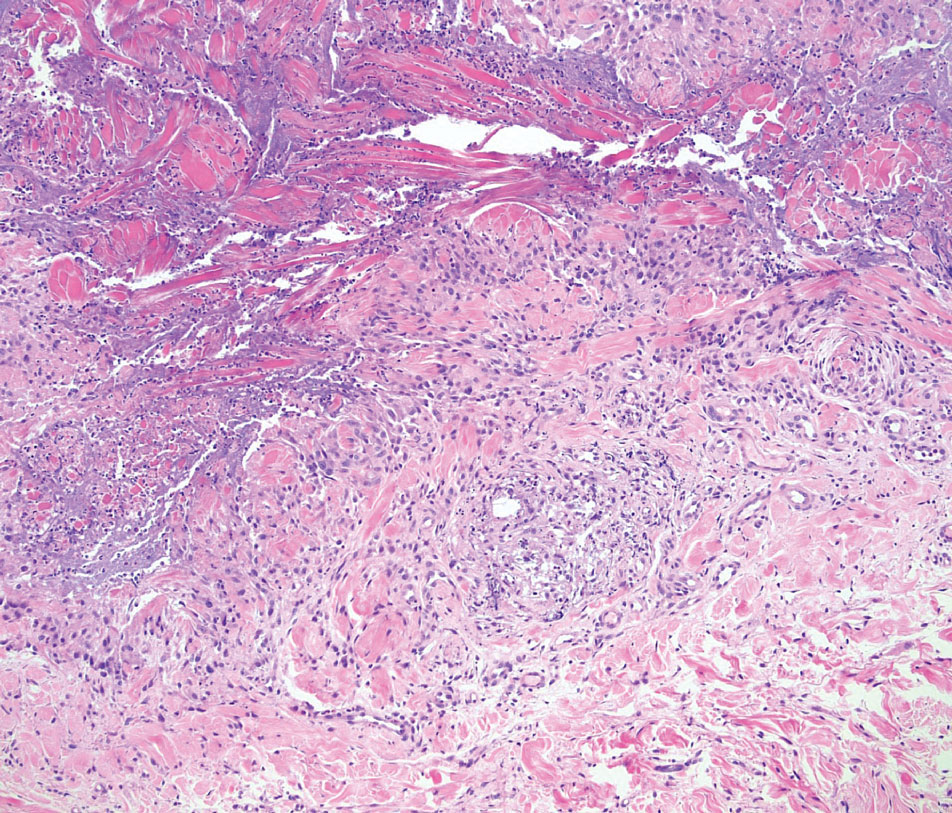
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
THE DIAGNOSIS: Sarcoidlike Reaction
Sarcoidlike reaction (SLR) is a rare cutaneous immune-related adverse event characterized by a multisystem granulomatous reaction indistinguishable from sarcoidosis but temporally associated with a trigger.1 Drug-induced SLR typically involves the mediastinal or hilar lymph nodes, with frequent involvement of the lungs and skin; cutaneous manifestations typically encompass erythematous papulonodular eruptions on the trunk and extremities.1-3 Sarcoidosis predominantly affects middle-aged women of African American or Scandinavian descent; genetic predisposition likely is a contributing factor.4 Unlike sarcoidosis, SLR is linked to various triggers such as medication or malignancy.
Immune checkpoint inhibitors (ICIs), particularly anti–PD-1 agents, have been linked to SLR through overexpression of proinflammatory cytokines, resulting in excessive T-helper 1 cell and macrophage activation and granulomatous eruption; notably, cutaneous immune-related adverse events often are correlated with greater treatment efficacy.5,6 Overall, anticancer therapy–induced SLR is most commonly reported in patients receiving ICIs for melanoma but it also has been described with ICI therapy for other cancers and with chemotherapy for melanoma. 1,3 Although most cases demonstrate both cutaneous and extracutaneous involvement, approximately 13 reported cases have been exclusively cutaneous.1 Recognition of SLR is important because misdiagnosis as true sarcoidosis may prompt unnecessary testing or therapy; furthermore, distinction from tumor progression is critical.3 The lesions can mimic other granulomatous or inflammatory dermatoses, posing a diagnostic challenge.
On histopathology, SLR typically demonstrates well-formed, noncaseating dermal granulomas composed of epithelioid histiocytes and Langhans or foreign-body giant cells, a sparse lymphocytic rim, and few plasma cells.2,4 Immunohistochemistry shows CD68-positive histiocytes predominating within the granulomas. Asteroid and Schaumann bodies occasionally are present.7 Special stains will be negative for microorganisms. Sarcoidosis manifests essentially identically from both a clinical and histopathologic perspective (Figure 1). Temporal association with an offending agent and symptomatic resolution following drug cessation remain the most reliable features for distinguishing SLR from sarcoidosis.7
Tuberculoid leprosy is a chronic infectious disease caused by Mycobacterium leprae (found most commonly in tropical regions) and manifesting as localized hypopigmented macules or papules with raised erythematous margins.8 Histopathologically, lesions show well-formed granulomas composed of epithelioid histiocytes and Langhans giant cells without necrosis, surrounded by a prominent lymphocytic rim (Figure 2).9 Rarely, focal caseous necrosis occurs, particularly in involved nerves.10 Hallmark features include enlarged cutaneous nerves surrounded by dermal granulomas and absence of bacilli on special stains; eccrine glands are infrequently involved.9 Standard treatment is 6 months of combination therapy with dapsone and rifampin.
Generalized granuloma annulare is an inflammatory dermatosis manifesting as diffuse erythematous annular papules, classically on the trunk and extremities.11 It predominantly affects individuals in their fifth and sixth decades of life and may be drug induced.2 Histopathology may reveal palisaded granulomas with central necrobiotic collagen, intercalating histiocytes, and interstitial mucin (Figure 3).2 Pathology also may show interstitial histiocytes and lymphocytes intercalating between collagen bundles with increased mucin but absent palisading or necrobiosis or a mixed pattern.2,12 Alcian blue or colloidal iron stains highlight mucin to help distinguish from other granulomatous processes. Multinucleated giant cells are rare. The nonnecrobiotic histologic pattern can mimic sarcoidosis, necessitating clinical correlation for correct diagnosis.13 Certain cases show genetic predisposition, such as HLA-B35, with a relapsing course often requiring combined systemic immunosuppression and phototherapy.14
Granulomatosis with polyangiitis is a systemic vasculitis that classically manifests as palpable purpura on the lower extremities, often with ulceration. Localized erythematous papules on the extensor surfaces may occur less commonly.15 Pathogenesis involves antineutrophil cytoplasmic antibodies inducing neutrophil degranulation, release of reactive oxygen species and proinflammatory cytokines, and subsequent endothelial damage.15 Histopathology shows necrotizing granulomatous inflammation and necrotizing vasculitis of small and medium vessels with nuclear debris.15 Poorly formed granulomas containing abundant neutrophils and mixed perivascular inflammatory infiltrates may be seen with or without vasculitis (Figure 4). Systemic features commonly include chronic rhinosinusitis, pauci-immune glomerulonephritis, and pulmonary nodules.15 Pharmacotherapy includes glucocorticoids combined with a glucocorticoid-sparing agent.
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
- Mazumder A, Mehrmal S, Chaudhry S. Immunotherapy-induced exclusively cutaneous sarcoid-like reaction. BMJ Case Rep. 2023;16:E252766. doi:10.1136/bcr-2022-252766
- Shah N, Shah M, Drucker AM, et al. Granulomatous cutaneous drug eruptions: a systematic review. Am J Clin Dermatol. 2021;22:39-53. doi:10.1007/s40257-020-00566-4
- Nykaza I, Murciano-Goroff YR, Desilets A, et al. Sarcoid-like reactions in patients treated with checkpoint inhibitors for advanced solid tumors. Oncologist. 2025;30:oyaf017. doi:10.1093/oncolo /oyaf017
- Tana C, Donatiello I, Caputo A, et al. Clinical features, histopathology and differential diagnosis of sarcoidosis. Cells. 2021;11:59. doi:10.3390/cells11010059
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361. doi:10.1007/s40257-017-0336-3
- Diaz-Perez JA, Beveridge MG, Victor TA, et al. Granulomatous and lichenoid dermatitis after IgG4 anti-PD-1 monoclonal antibody therapy for advanced cancer. J Cutan Pathol. 2018;45:434-438. doi:10.1111/cup.13133
- Chopra A, Nautiyal A, Kalkanis A, et al. Drug-induced sarcoidosis-like reactions. Chest. 2018;154:664-677. doi:10.1016 /j.chest.2018.03.056
- Froes LAR Jr, Sotto MN, Trindade MAB. Leprosy: clinical and immunopathological characteristics. An Bras Dermatol. 2022;97:338-347. doi:10.1016/j.abd.2021.08.006
- Magaña M, Vargas Bornacini MF, Landeta-Sa AP, et al. Lucio phenomenon: a review. Am J Dermatopathol. 2025;47:1-8. doi:10.1097 /DAD.0000000000002833
- Jayalakshmy PS, Prasad PH, Kamala VV, et al. Segmental necrotizing granulomatous neuritis: a rare manifestation of Hansen disease-report of 2 cases. Case Rep Dermatol Med. 2012;2012:758093. doi:10.1155/2012/758093
- Lee JH, Cho S. Resolution of refractory generalized granuloma annulare after treatment with alitretinoin. JAAD Case Rep. 2022;24:38-41. doi:10.1016/j.jdcr.2022.04.006
- Yun JH, Lee JY, Kim MK, et al. Clinical and pathological features of generalized granuloma annulare with their correlation: a retrospective multicenter study in Korea. Ann Dermatol. 2009; 21:113-119. doi:10.5021/ad.2009.21.2.113
- Cohen PR, Carlos CA. Granuloma annulare mimicking sarcoidosis: report of patient with localized granuloma annulare whose skin lesions show 3 clinical morphologies and 2 histology patterns. Am J Dermatopathol. 2015;37:547-550. doi:10.1097/DAD.0000000000000125
- Rankin BD, Haber RM. Familial granuloma annulare: first report of occurrence in a father and daughter and updated review of the literature. JAAD Case Rep. 2021;17:61-64. doi:10.1016 /j.jdcr.2021.09.023
- Rout P, Garlapati P, Qurie A. Granulomatosis with polyangiitis. StatPearls (Internet). Updated August 31, 2024. Accessed May 4, 2026. https://www.ncbi.nlm.nih.gov/books/NBK557827/
Pink Papulonodular Eruption on the Trunk and Arms
Pink Papulonodular Eruption on the Trunk and Arms
A 47-year-old man with a history of chronic kidney disease and bilateral clear cell renal cell carcinoma who was undergoing treatment with adjuvant pembrolizumab presented to the dermatology department with a scattered papulonodular eruption of several weeks’ duration. Physical examination revealed pink papules and nodules with coalescing erythema over the trunk and upper extremities, most pronounced on the right elbow (bottom [inset]). A 4-mm punch biopsy demonstrated dermal granulomatous inflammation. Special stains were negative for microorganisms. Computed tomography of the chest revealed a new subpleural nodule and new hilar lymphadenopathy.

Eosinophilic Pustular Folliculitis in the Setting of Untreated Chronic Lymphocytic Leukemia
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.

A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.

Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.
A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.
Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
To the Editor:
Eosinophilic pustular folliculitis (EPF) is a noninfectious dermatosis that typically manifests as recurrent follicular papulopustules that generally affect the face and occasionally the trunk and arms. There are several subtypes of EPF: classic EPF (Ofuji disease), infancy-associated EPF, and immunosuppression-associated EPF.1,2 We report a rare case of EPF in the setting of untreated chronic lymphocytic leukemia (CLL), a subtype of immunosuppression-associated EPF that has been associated with hematologic malignancy EPF (HM-EPF).3-5
A 69-year-old woman presented with diffusely scattered, pruritic, erythematous, erosive lesions on the back, arms, legs, and forehead (Figure 1) of 4 months’ duration, as well as an ulcerative lesion on the left third toe due to a suspected insect bite. She had a history of untreated CLL that was diagnosed 2 years prior. The patient was empirically started on clindamycin for presumed infection of the toe. A punch biopsy of the left wrist revealed superficial and deep dermal perivascular and interstitial inflammatory infiltrates composed of lymphocytes, histiocytes, and numerous eosinophils in association with edema and necrosis. Histopathology was overall most consistent with an exuberant arthropod reaction; however, at 2-week follow-up, the patient reported that the pustular lesions improved upon starting antibiotics, which raised concerns for a bacterial process. The patient initially was continued on clindamycin given subjective improvement but was later switched to daptomycin, as she developed clindamycin-resistant methicillin-resistant Staphylococcus aureus osteomyelitis from the necrotic toe.
A month later, the patient returned with new papules and pustules on the arms and trunk. A repeat biopsy showed notable dermal collections comprised predominantly of neutrophils and eosinophils as well as involvement of follicular structures by dense inflammation (Figure 2). Immunohistochemistry demonstrated a predominant population of small CD3+ T cells, which raised concern for cutaneous T-cell lymphoma. However, retention of CD5 expression made this less likely. Few scattered CD20+ B cells with limited CD23 reactivity and without CD5 co-expression were detected, which ruled out cutaneous involvement of the patient’s CLL. Bacterial culture and Grocott methenamine-silver, Gram, acid-fast bacilli, and periodic acid-Schiff stains were negative. Polymerase chain reaction testing for varicella-zoster virus and herpes simplex virus also were negative. Thus, a diagnosis of EPF secondary to CLL was favored, as an infectious process also was unlikely. The patient was started on triamcinolone cream 0.1% with gradual improvement.
Cases of HM-EPF predominantly have been reported in patients who have undergone chemotherapy, bone marrow transplantation, or hematopoietic stem cell transplantation. Furthermore, a vast majority of these cases have been reported in older males.3-16 In a retrospective study of more than 750 patients with established CLL, Agnew et al7 identified 125 different skin complications in 40 patients. Of this subset, only a small number (2/40) were associated with eosinophilic folliculitis, with 1 case noted in a middle-aged woman with a history of CLL treatment.7 Moreover, Motaparthi et al4 reported 3 additional cases of HM-EPF, with all patients identified as middle-aged men who were treated with chemotherapy for underlying CLL. Our patient represents a case of EPF in the context of untreated CLL in a woman.
Although topical corticosteroids remain the first-line treatment for EPF, a survey study conducted across 67 hospitals in Japan indicated that antibiotics were moderately or highly effective in 79% of EPF patients (n=143).17 This association may explain the subjective improvement reported by our patient upon starting clindamycin. Furthermore, in HIV-associated EPF, high-dose cetirizine, itraconazole, and metronidazole have been successful when topical therapies have failed.18 Although the precise pathogenesis of EPF is unknown, histopathologic features, clinical appearance, and identification of the accurate EPF subtype can still prove valuable in informing empiric treatment strategies. Consequently, the initial histopathologic diagnosis of an arthropod bite reaction in our patient highlights the importance of clinical correlation and additional ancillary studies in the determination of EPF vs other inflammatory dermatoses that manifest microscopically with lymphocytic infiltrates, prominent eosinophils, and follicular involvement.4 The histopathologic features of EPF demonstrate considerable overlap with eosinophilic dermatosis of hematologic malignancy (also known as eosinophilic dermatosis of myeloproliferative disease). It is suspected that eosinophilic dermatosis of hematologic malignancy and EPF may exist on a spectrum, and additional cases may improve categorization of these entities.19
In conclusion, this report adds to the medical practitioner’s awareness of EPF manifestations in patients with underlying CLL, an infrequently reported subtype of HM-EPF.
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
- Fujiyama T, Tokura Y. Clinical and histopathological differential diagnosis of eosinophilic pustular folliculitis. J Dermatol. 2013;40:419-423. doi:10.1111/1346-8138.12125
- Katoh M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. doi:10.1111/1346-8138.12008
- Takamura S, Teraki Y. Eosinophilic pustular folliculitis associated with hematological disorders: a report of two cases and review of Japanese literature. J Dermatol. 2016;43:432-435. doi: 10.1111/1346-8138.13088
- Motaparthi K, Kapil J, Hsu S. Eosinophilic folliculitis in association with chronic lymphocytic leukemia: a clinicopathologic series. JAAD Case Rep. 2017;3:263-268. doi:10.1016/j.jdcr.2017.03.007
- Lambert J, Berneman Z, Dockx P, et al. Eosinophilic pustular folliculitis and B-cell chronic lymphatic leukaemia. Dermatology. 1994;189(suppl 2):58-59. doi:10.1159/000246994
- Patrizi A, Chieregato C, Visani G, et al. Leukaemia-associated eosinophilic folliculitis (Ofuji’s disease). J Eur Acad Dermatol Venereol. 2004;18:596-598. doi:10.1111/j.1468-3083.2004.00982.x
- Agnew KL, Ruchlemer R, Catovsky D, et al. Cutaneous findings in chronic lymphocytic leukaemia. Br J Dermatol. 2004;150:1129-1135. doi:10.1111/j.1365-2133.2004.05982.x
- Zitelli K, Fernandes N, Adams BB. Eosinophilic folliculitis occurring after stem cell transplant for acute lymphoblastic leukemia: a case report and review. Int J Dermatol. 2015;54:785-789. doi:10.1111/j.1365-2133.2004.05982.x
- Goiriz R, Guhl-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36. doi:10.1111/j.1600-0560.2006.00725.x
- Bhandare PC, Ghodge RR, Bhobe MR, et al. Eosinophilic pustular folliculitis post chemotherapy in a patient of non-Hodgkins lymphoma: a case report. Indian J Dermatol. 2015;60:521. doi:10.4103/0019-5154.164432
- Sugaya M, Suga H, Miyagaki T, et al. Eosinophilic pustular folliculitis associated with Sézary syndrome. Clin Exp Dermatol. 2014;39:536-538. doi:10.1111/ced.12315
- Keida T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplantation. J Dermatol. 2004;31:21-26. doi:10.1111/j.1346-8138.2004.tb00499.x
- Ota M, Shimizu T, Hashino S, et al. Eosinophilic folliculitis in a patient after allogeneic bone marrow transplantation: case report and review of the literature. Am J Hematol. 2004;76:295-296. doi:10.1002/ajh.20080
- Vassallo C, Ciocca O, Arcaini L, et al. Eosinophilic folliculitis occurring in a patient affected by Hodgkin lymphoma. Int J Dermatol. 2002;41:298-300. doi:10.1046/j.1365-4362.2002.01356_6.x
- Evans TR, Mansi JL, Bull R, et al. Eosinophilic folliculitis occurring after bone marrow autograft in a patient with non-Hodgkin’s lymphoma. Cancer. 1994;73:2512-2514. doi:10.1002/1097-0142(19940515)73:10<2512::aid-cncr2820731010>3.0.co;2-s
- Patrizi A, Di Lernia V, Neri I, et al. Eosinophilic pustular folliculitis (Ofuji’s disease) and non-Hodgkin lymphoma. Acta Derm Venereol. 1992;72:146-147.
- Ono S, Yamamoto Y, Otsuka A, et al. Evaluation of the effectiveness of antibiotics against eosinophilic pustular folliculitis. Case Rep Dermatol. 2013;5:144-147. doi:10.1159/000351330
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis. Am J Clin Dermatol. 2004;5:189-197. doi:10.2165/00128071-200405030-00007
- Bailey CAR, Laurain DA, Sheinbein DM, et al. Eosinophilic folliculitis, eosinophilic dermatosis of hematologic malignancy and acneiform follicular mucinosis: two case reports and a review of the literature highlighting the spectrum of histopathology. J Cutan Pathol. 2021;48:439-450. doi:10.1111/cup.13932
Practice Points
- Eosinophilic pustular folliculitis (EPF) is associated with an immunosuppressed state, as in patients with underlying hematologic malignancy.
- Topical corticosteroids remain the first-line treatment for EPF; however, antimicrobial agents have been used with moderate success when topical therapies have failed.
