User login
Official Newspaper of the American College of Surgeons
UnitedHealth warns of marketplace exit: Start of a trend or push for White House action?
UnitedHealthGroup laid out a litany of reasons on Nov. 19 as to why it might stop selling individual health insurance through federal and state markets in 2017 – a move some see as an effort to compel the Obama administration to ease regulations and make good on promised payments.
Those problems, including low participation by healthy people, have led to financial losses, according to UnitedHealth. If not addressed, similar issues could affect other insurers, causing more to exit the market in the coming years, some Wall Street analysts and policy experts said.

Many said they anticipate the federal government will act to forestall widespread departures, particularly because continued withdrawals could be politically explosive during an election year.
A key piece of President Obama’s signature health care law, the online marketplaces, also called exchanges, opened in 2014 for people who buy their own insurance because they don’t get it through their jobs. Enrollment, while growing, has fallen short of capturing the share of the eligible uninsured that was anticipated. This year, the marketplaces saw enrollment of more than 9 million customers, although the law’s expansion of Medicaid enrollment in many states also has played a large role in reducing the overall number of uninsured.
Only a month ago, United sounded more optimistic about business on the exchanges. But on Nov. 19, the insurer said it would cut its earnings forecast and projected hundreds of millions in losses stemming from the policies it sells through the health law’s marketplaces.
The turnaround led some analysts to ask the insurer what had changed.
Stephen Hemsley, UnitedHealth chief executive officer, said too many healthy people dropped coverage and noted slower than expected enrollment. A major factor, he added, was far higher costs for those who signed up for 2015 coverage under special exemptions after the general open-enrollment period ended. Those exemptions included, for example, people who lost their insurance, moved, or suffered a hardship, such as an eviction or had their utilities turned off. United said it did not see a similar increase in costs for people who bought policies from private brokers or websites instead of the government marketplaces after open enrollment, suggesting the reason was partly that the company’s eligibility assessments were more thorough.
The firm did not say it would halt sales in 2017 but warned that it would strongly consider doing so based on what happens in the next few months.
“We cannot sustain these losses,” Mr. Hemsley said on a phone call with Wall Street analysts. “We can’t subsidize a marketplace that doesn’t appear at the moment to be sustaining itself.”
Although it’s the nation’s largest insurer, United captured only a small percentage of consumers who currently have coverage through the Affordable Care Act marketplace, in part because it sat out the first year of enrollment and really ramped up only for this year’s coverage.
While seen as a serious challenge to the health care law, United’s decision alone doesn’t mark the death knell for the exchanges. In remarks to analysts and press reports on Nov. 19, Aetna and Kaiser Permanente re-affirmed their commitment to selling through the marketplaces.
Ben Wakana, a spokesman for the Health and Human Services department, defended the government marketplaces, noting that 9 of 10 of policyholders re-enrolling have a choice of three or more insurers for next year. “The reality is we continue to see more people signing up for health insurance and more issuers entering the marketplaces, and at the end of January, we believe we’ll be looking at another successful open enrollment – just like the last two,” he said. “[Thursday’s] statement by one issuer is not indicative of the marketplace’s strength and viability.”
But insurers, including Humana, Aetna, and some of the large Blue Cross Blue Shield plans, were losing money or barely breaking even on their marketplace business, according to earnings reports.
“If there are no changes, all the large publicly traded companies will end up leaving,” said Ana Gupte, analyst with Leerink Partners. “But I would be very surprised if [the Department of Health & Human Services] doesn’t do something to accommodate their issues.”
Those options would be limited to what the agency could do without congressional action, many analysts said. Still, that could include relaxing some regulations or reconsidering some of the exemptions that allow people to sign up after the open-enrollment period.
Former insurance executive and consultant Robert Laszewski said the administration needs to relax the rules to give insurers more flexibility to design plans that would attract healthier people. He said the costs – including deductibles and premiums – were too high for many people, particularly those with few medical needs.
“Disproportionately, the sick are signing up and the healthy are dropping out,” said Mr. Laszewski, adding that alternative plans with fewer benefits but lower costs should be made available.
Economist Len Nichols cautioned, however, that most of the law’s benefit requirements – taken individually – add little to the cost of a plan. Removing the bigger-ticket requirements, such as coverage for maternity care, would leave consumers without adequate coverage, said Nichols, who directs the George Mason University Center for Health Research and Ethics in Fairfax, Va.
Mr. Nichols, Ms. Gupte, and other analysts agree with the industry’s trade lobby, which says one thing the administration could do is make good on a promise to pay insurers under a temporary program designed to redistribute profits from some insurers that did especially well to offset losses others experienced in the marketplace plans. That program, however, has paid only about 13 cents on the dollar of what was promised, mainly because fewer insurers than expected made money.
Earlier this month, HHS Secretary Sylvia Burwell said the administration is exploring ways it might be able to help make those payments, although such a move comes too late to save many of the dozen insurance cooperatives that have announced they will pull out of the market in January. The less-than-anticipated payments are often cited as a main factor in the co-ops demise.
UnitedHealthGroup laid out a litany of reasons on Nov. 19 as to why it might stop selling individual health insurance through federal and state markets in 2017 – a move some see as an effort to compel the Obama administration to ease regulations and make good on promised payments.
Those problems, including low participation by healthy people, have led to financial losses, according to UnitedHealth. If not addressed, similar issues could affect other insurers, causing more to exit the market in the coming years, some Wall Street analysts and policy experts said.
Many said they anticipate the federal government will act to forestall widespread departures, particularly because continued withdrawals could be politically explosive during an election year.
A key piece of President Obama’s signature health care law, the online marketplaces, also called exchanges, opened in 2014 for people who buy their own insurance because they don’t get it through their jobs. Enrollment, while growing, has fallen short of capturing the share of the eligible uninsured that was anticipated. This year, the marketplaces saw enrollment of more than 9 million customers, although the law’s expansion of Medicaid enrollment in many states also has played a large role in reducing the overall number of uninsured.
Only a month ago, United sounded more optimistic about business on the exchanges. But on Nov. 19, the insurer said it would cut its earnings forecast and projected hundreds of millions in losses stemming from the policies it sells through the health law’s marketplaces.
The turnaround led some analysts to ask the insurer what had changed.
Stephen Hemsley, UnitedHealth chief executive officer, said too many healthy people dropped coverage and noted slower than expected enrollment. A major factor, he added, was far higher costs for those who signed up for 2015 coverage under special exemptions after the general open-enrollment period ended. Those exemptions included, for example, people who lost their insurance, moved, or suffered a hardship, such as an eviction or had their utilities turned off. United said it did not see a similar increase in costs for people who bought policies from private brokers or websites instead of the government marketplaces after open enrollment, suggesting the reason was partly that the company’s eligibility assessments were more thorough.
The firm did not say it would halt sales in 2017 but warned that it would strongly consider doing so based on what happens in the next few months.
“We cannot sustain these losses,” Mr. Hemsley said on a phone call with Wall Street analysts. “We can’t subsidize a marketplace that doesn’t appear at the moment to be sustaining itself.”
Although it’s the nation’s largest insurer, United captured only a small percentage of consumers who currently have coverage through the Affordable Care Act marketplace, in part because it sat out the first year of enrollment and really ramped up only for this year’s coverage.
While seen as a serious challenge to the health care law, United’s decision alone doesn’t mark the death knell for the exchanges. In remarks to analysts and press reports on Nov. 19, Aetna and Kaiser Permanente re-affirmed their commitment to selling through the marketplaces.
Ben Wakana, a spokesman for the Health and Human Services department, defended the government marketplaces, noting that 9 of 10 of policyholders re-enrolling have a choice of three or more insurers for next year. “The reality is we continue to see more people signing up for health insurance and more issuers entering the marketplaces, and at the end of January, we believe we’ll be looking at another successful open enrollment – just like the last two,” he said. “[Thursday’s] statement by one issuer is not indicative of the marketplace’s strength and viability.”
But insurers, including Humana, Aetna, and some of the large Blue Cross Blue Shield plans, were losing money or barely breaking even on their marketplace business, according to earnings reports.
“If there are no changes, all the large publicly traded companies will end up leaving,” said Ana Gupte, analyst with Leerink Partners. “But I would be very surprised if [the Department of Health & Human Services] doesn’t do something to accommodate their issues.”
Those options would be limited to what the agency could do without congressional action, many analysts said. Still, that could include relaxing some regulations or reconsidering some of the exemptions that allow people to sign up after the open-enrollment period.
Former insurance executive and consultant Robert Laszewski said the administration needs to relax the rules to give insurers more flexibility to design plans that would attract healthier people. He said the costs – including deductibles and premiums – were too high for many people, particularly those with few medical needs.
“Disproportionately, the sick are signing up and the healthy are dropping out,” said Mr. Laszewski, adding that alternative plans with fewer benefits but lower costs should be made available.
Economist Len Nichols cautioned, however, that most of the law’s benefit requirements – taken individually – add little to the cost of a plan. Removing the bigger-ticket requirements, such as coverage for maternity care, would leave consumers without adequate coverage, said Nichols, who directs the George Mason University Center for Health Research and Ethics in Fairfax, Va.
Mr. Nichols, Ms. Gupte, and other analysts agree with the industry’s trade lobby, which says one thing the administration could do is make good on a promise to pay insurers under a temporary program designed to redistribute profits from some insurers that did especially well to offset losses others experienced in the marketplace plans. That program, however, has paid only about 13 cents on the dollar of what was promised, mainly because fewer insurers than expected made money.
Earlier this month, HHS Secretary Sylvia Burwell said the administration is exploring ways it might be able to help make those payments, although such a move comes too late to save many of the dozen insurance cooperatives that have announced they will pull out of the market in January. The less-than-anticipated payments are often cited as a main factor in the co-ops demise.
UnitedHealthGroup laid out a litany of reasons on Nov. 19 as to why it might stop selling individual health insurance through federal and state markets in 2017 – a move some see as an effort to compel the Obama administration to ease regulations and make good on promised payments.
Those problems, including low participation by healthy people, have led to financial losses, according to UnitedHealth. If not addressed, similar issues could affect other insurers, causing more to exit the market in the coming years, some Wall Street analysts and policy experts said.
Many said they anticipate the federal government will act to forestall widespread departures, particularly because continued withdrawals could be politically explosive during an election year.
A key piece of President Obama’s signature health care law, the online marketplaces, also called exchanges, opened in 2014 for people who buy their own insurance because they don’t get it through their jobs. Enrollment, while growing, has fallen short of capturing the share of the eligible uninsured that was anticipated. This year, the marketplaces saw enrollment of more than 9 million customers, although the law’s expansion of Medicaid enrollment in many states also has played a large role in reducing the overall number of uninsured.
Only a month ago, United sounded more optimistic about business on the exchanges. But on Nov. 19, the insurer said it would cut its earnings forecast and projected hundreds of millions in losses stemming from the policies it sells through the health law’s marketplaces.
The turnaround led some analysts to ask the insurer what had changed.
Stephen Hemsley, UnitedHealth chief executive officer, said too many healthy people dropped coverage and noted slower than expected enrollment. A major factor, he added, was far higher costs for those who signed up for 2015 coverage under special exemptions after the general open-enrollment period ended. Those exemptions included, for example, people who lost their insurance, moved, or suffered a hardship, such as an eviction or had their utilities turned off. United said it did not see a similar increase in costs for people who bought policies from private brokers or websites instead of the government marketplaces after open enrollment, suggesting the reason was partly that the company’s eligibility assessments were more thorough.
The firm did not say it would halt sales in 2017 but warned that it would strongly consider doing so based on what happens in the next few months.
“We cannot sustain these losses,” Mr. Hemsley said on a phone call with Wall Street analysts. “We can’t subsidize a marketplace that doesn’t appear at the moment to be sustaining itself.”
Although it’s the nation’s largest insurer, United captured only a small percentage of consumers who currently have coverage through the Affordable Care Act marketplace, in part because it sat out the first year of enrollment and really ramped up only for this year’s coverage.
While seen as a serious challenge to the health care law, United’s decision alone doesn’t mark the death knell for the exchanges. In remarks to analysts and press reports on Nov. 19, Aetna and Kaiser Permanente re-affirmed their commitment to selling through the marketplaces.
Ben Wakana, a spokesman for the Health and Human Services department, defended the government marketplaces, noting that 9 of 10 of policyholders re-enrolling have a choice of three or more insurers for next year. “The reality is we continue to see more people signing up for health insurance and more issuers entering the marketplaces, and at the end of January, we believe we’ll be looking at another successful open enrollment – just like the last two,” he said. “[Thursday’s] statement by one issuer is not indicative of the marketplace’s strength and viability.”
But insurers, including Humana, Aetna, and some of the large Blue Cross Blue Shield plans, were losing money or barely breaking even on their marketplace business, according to earnings reports.
“If there are no changes, all the large publicly traded companies will end up leaving,” said Ana Gupte, analyst with Leerink Partners. “But I would be very surprised if [the Department of Health & Human Services] doesn’t do something to accommodate their issues.”
Those options would be limited to what the agency could do without congressional action, many analysts said. Still, that could include relaxing some regulations or reconsidering some of the exemptions that allow people to sign up after the open-enrollment period.
Former insurance executive and consultant Robert Laszewski said the administration needs to relax the rules to give insurers more flexibility to design plans that would attract healthier people. He said the costs – including deductibles and premiums – were too high for many people, particularly those with few medical needs.
“Disproportionately, the sick are signing up and the healthy are dropping out,” said Mr. Laszewski, adding that alternative plans with fewer benefits but lower costs should be made available.
Economist Len Nichols cautioned, however, that most of the law’s benefit requirements – taken individually – add little to the cost of a plan. Removing the bigger-ticket requirements, such as coverage for maternity care, would leave consumers without adequate coverage, said Nichols, who directs the George Mason University Center for Health Research and Ethics in Fairfax, Va.
Mr. Nichols, Ms. Gupte, and other analysts agree with the industry’s trade lobby, which says one thing the administration could do is make good on a promise to pay insurers under a temporary program designed to redistribute profits from some insurers that did especially well to offset losses others experienced in the marketplace plans. That program, however, has paid only about 13 cents on the dollar of what was promised, mainly because fewer insurers than expected made money.
Earlier this month, HHS Secretary Sylvia Burwell said the administration is exploring ways it might be able to help make those payments, although such a move comes too late to save many of the dozen insurance cooperatives that have announced they will pull out of the market in January. The less-than-anticipated payments are often cited as a main factor in the co-ops demise.

Artificial pancreas improved glycemia after islet cell transplant
A closed-loop insulin pump with continuous glucose monitor produced significantly better blood glucose control in patients who have received islet cell transplants after pancreatectomy, compared with regular insulin injections, a pilot study has found.
Fourteen adults who received auto-islet transplants after pancreatectomy for chronic pancreatitis were randomized either to receive a closed-loop insulin pump system or the usual treatment of multiple insulin injections for 72 hours after transition from intravenous to subcutaneous insulin following surgery.
Researchers observed a significantly lower mean serum glucose in the insulin pump group, compared with the control group, with the highest average serum glucose level in individual patients in the pump group still being lower than the lowest average in the control group.
These improvements in glycemia were not associated with an increased risk of hypoglycemia in the closed-loop pump group and patients in the closed-loop pump group also required a significantly lower total daily insulin dose than did the control group, according to a paper published Nov. 20 in the American Journal of Transplantation.
“Success of islet engraftment is heavily dependent on maintenance of narrow-range euglycemia in the post-transplant period,” wrote Dr. Gregory P. Forlenza of the University of Minnesota Medical Center, Minneapolis, and his coauthors (Am J Transplant. 2015 Nov 20. doi: 10.1111/ajt.13539).
“This technology was shown in this study to provide some statistically and clinically significant improvements in glycemic parameters in adults after TP [total pancreatectomy] with IAT [intraportal islet autotransplantation] without producing associated increased episodes of hypoglycemia or adverse events.”
The study was funded by the Vikings Children’s Research Fund and the University of Minnesota. Medtronic Diabetes provided supplies as part of an investigator-initiated grant. No conflicts of interest were declared.
A closed-loop insulin pump with continuous glucose monitor produced significantly better blood glucose control in patients who have received islet cell transplants after pancreatectomy, compared with regular insulin injections, a pilot study has found.
Fourteen adults who received auto-islet transplants after pancreatectomy for chronic pancreatitis were randomized either to receive a closed-loop insulin pump system or the usual treatment of multiple insulin injections for 72 hours after transition from intravenous to subcutaneous insulin following surgery.
Researchers observed a significantly lower mean serum glucose in the insulin pump group, compared with the control group, with the highest average serum glucose level in individual patients in the pump group still being lower than the lowest average in the control group.
These improvements in glycemia were not associated with an increased risk of hypoglycemia in the closed-loop pump group and patients in the closed-loop pump group also required a significantly lower total daily insulin dose than did the control group, according to a paper published Nov. 20 in the American Journal of Transplantation.
“Success of islet engraftment is heavily dependent on maintenance of narrow-range euglycemia in the post-transplant period,” wrote Dr. Gregory P. Forlenza of the University of Minnesota Medical Center, Minneapolis, and his coauthors (Am J Transplant. 2015 Nov 20. doi: 10.1111/ajt.13539).
“This technology was shown in this study to provide some statistically and clinically significant improvements in glycemic parameters in adults after TP [total pancreatectomy] with IAT [intraportal islet autotransplantation] without producing associated increased episodes of hypoglycemia or adverse events.”
The study was funded by the Vikings Children’s Research Fund and the University of Minnesota. Medtronic Diabetes provided supplies as part of an investigator-initiated grant. No conflicts of interest were declared.
A closed-loop insulin pump with continuous glucose monitor produced significantly better blood glucose control in patients who have received islet cell transplants after pancreatectomy, compared with regular insulin injections, a pilot study has found.
Fourteen adults who received auto-islet transplants after pancreatectomy for chronic pancreatitis were randomized either to receive a closed-loop insulin pump system or the usual treatment of multiple insulin injections for 72 hours after transition from intravenous to subcutaneous insulin following surgery.
Researchers observed a significantly lower mean serum glucose in the insulin pump group, compared with the control group, with the highest average serum glucose level in individual patients in the pump group still being lower than the lowest average in the control group.
These improvements in glycemia were not associated with an increased risk of hypoglycemia in the closed-loop pump group and patients in the closed-loop pump group also required a significantly lower total daily insulin dose than did the control group, according to a paper published Nov. 20 in the American Journal of Transplantation.
“Success of islet engraftment is heavily dependent on maintenance of narrow-range euglycemia in the post-transplant period,” wrote Dr. Gregory P. Forlenza of the University of Minnesota Medical Center, Minneapolis, and his coauthors (Am J Transplant. 2015 Nov 20. doi: 10.1111/ajt.13539).
“This technology was shown in this study to provide some statistically and clinically significant improvements in glycemic parameters in adults after TP [total pancreatectomy] with IAT [intraportal islet autotransplantation] without producing associated increased episodes of hypoglycemia or adverse events.”
The study was funded by the Vikings Children’s Research Fund and the University of Minnesota. Medtronic Diabetes provided supplies as part of an investigator-initiated grant. No conflicts of interest were declared.
FROM THE AMERICAN JOURNAL OF TRANSPLANTATION
Key clinical point: A closed-loop insulin pump with a continuous glucose monitor offers significantly better blood glucose control in patients who have received islet cell transplants after pancreatectomy.
Major finding: Closed-loop insulin pumps were associated with a significantly lower mean serum glucose, compared with multiple daily insulin injections.
Data source: Randomized, controlled pilot study in 14 patients receiving auto-islet transplants after pancreatectomy.
Disclosures: The study was funded by the Vikings Children’s Research Fund and the University of Minnesota. Medtronic Diabetes provided supplies as part of an investigator-initiated grant. No conflicts of interest were declared.
Reoperation risk doubled in Roux-en-Y over sleeve gastrectomy
CHICAGO – Patients undergoing Roux-en-Y gastric bypass are twice as likely to need a reoperation as those having sleeve gastrectomy, according to ACS NSQIP data.
Reoperation among Roux-en-Y patients was associated with a 10-fold increase in mortality over sleeve gastrectomy (1.2% vs. 0.1%; P less than .01) and a 3-fold increase in length of stay (6 days vs. 2 days; P less than .01), Dr. Matthew Whealon reported at the American College of Surgeons Clinical Congress
The results are consistent with prior contemporary analyses using ACS National Surgical Quality Improvement Program (NSQIP) data reporting reoperation rates of 2.5%-5.1% for Roux-en-Y gastric bypass (RYGB) and 1.6%-3% for sleeve gastrectomy. Those analyses, however, did not include the reasons for reoperation, as these data were not available until the 2012 database release, he said.

With these data now in hand, lead author Dr. Mark Hanna and his fellow investigators at the University of California, Irvine, identified 36,757 adults in the 2012-2013 database who underwent RYGB (n = 19,597) or sleeve gastrectomy (n = 17,160) for morbid obesity and performed multivariate regression analyses to identify risk factors associated with reoperation.
In all, 518 RYGB patients and 231 sleeve gastrectomy patients required an unplanned return to the operating room (2.6% vs. 1.3%), Dr. Whealon said. The mean time from the index procedure to reoperation was 7.6 days and 7.1 days, respectively.
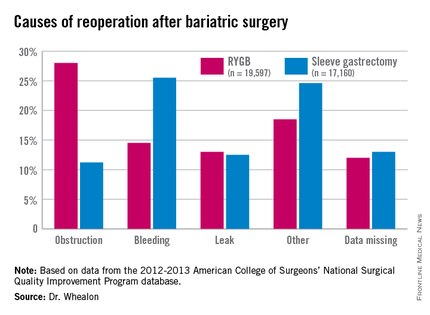
Obstruction was the biggest driver of reoperation following RYGB, accounting for 28% of reoperations. Other causes were bleeding (14.5%), leak (13%), and other unspecified reasons (18.5%), with data missing in 12%.
Bleeding was the most common indication for reoperation after sleeve gastrectomy (25.5%), followed by other unspecified reasons(24.6%), missing data (13%), leak (12.55%), and obstruction (11.2%), he said.
In adjusted multivariate analyses, factors that significantly increased the risk for reoperation were heart failure (adjusted odds ratio, 2.3), dependent functional status (aOR, 2.1), RYGB (aOR, 1.94), chronic obstructive pulmonary disease (aOR, 1.7), open operation (aOR, 1.6), and male sex (aOR, 1.1). The P values were less than .05 for all comparisons.
Factors not significant for reoperation included body mass index, age, smoking status, bleeding disorder, steroid use, dialysis, hypertension, diabetes, preoperative sepsis, emergent admission, elective operation, and preoperative weight loss.
While bariatric surgery remains a safe operation with low mortality and reoperation rates, additional studies are needed, because of the increased mortality associated with reoperation, to identify ways to mitigate these complications, Dr. Whealon said.
Limitations of the study were that ICD-9 codes for postoperative hemorrhage could not differentiate between intra-abdominal and gastrointestinal bleeding, the database is subject to coding errors, and missing data may have introduced bias into the study, he noted.
Discussant Dr. Matthew Goldblatt of the Medical College of Wisconsin in Milwaukee commented that use of the ACS MBSAQIP (Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program) database would have avoided some of the coding errors for reoperation.
He also questioned whether the average 7-day return to surgery interval reflects the use of endoscopy, as few surgeons would wait that long if, as the analysis suggests, a primary reason for reoperation was postoperative bleeding.
Endoscopy was included in the reoperations, Dr. Whealon said, but he could not speak to the exact percentage it comprised.
Finally, Dr. Goldblatt said, “the patients that you identified as being the highest risk for complication, as is often the case in these reviews, are really the ones most likely to gain the most from the procedure. … So how can people avoid operating on these patients when they are the ones that can get the most out of it?”
Dr. Whealon agreed that high-risk patients have the most to gain and suggested that “optimizing their comorbid conditions before operation will help reduce their risk.”
The authors reported having no conflicts of interest.
CHICAGO – Patients undergoing Roux-en-Y gastric bypass are twice as likely to need a reoperation as those having sleeve gastrectomy, according to ACS NSQIP data.
Reoperation among Roux-en-Y patients was associated with a 10-fold increase in mortality over sleeve gastrectomy (1.2% vs. 0.1%; P less than .01) and a 3-fold increase in length of stay (6 days vs. 2 days; P less than .01), Dr. Matthew Whealon reported at the American College of Surgeons Clinical Congress
The results are consistent with prior contemporary analyses using ACS National Surgical Quality Improvement Program (NSQIP) data reporting reoperation rates of 2.5%-5.1% for Roux-en-Y gastric bypass (RYGB) and 1.6%-3% for sleeve gastrectomy. Those analyses, however, did not include the reasons for reoperation, as these data were not available until the 2012 database release, he said.
With these data now in hand, lead author Dr. Mark Hanna and his fellow investigators at the University of California, Irvine, identified 36,757 adults in the 2012-2013 database who underwent RYGB (n = 19,597) or sleeve gastrectomy (n = 17,160) for morbid obesity and performed multivariate regression analyses to identify risk factors associated with reoperation.
In all, 518 RYGB patients and 231 sleeve gastrectomy patients required an unplanned return to the operating room (2.6% vs. 1.3%), Dr. Whealon said. The mean time from the index procedure to reoperation was 7.6 days and 7.1 days, respectively.
Obstruction was the biggest driver of reoperation following RYGB, accounting for 28% of reoperations. Other causes were bleeding (14.5%), leak (13%), and other unspecified reasons (18.5%), with data missing in 12%.
Bleeding was the most common indication for reoperation after sleeve gastrectomy (25.5%), followed by other unspecified reasons(24.6%), missing data (13%), leak (12.55%), and obstruction (11.2%), he said.
In adjusted multivariate analyses, factors that significantly increased the risk for reoperation were heart failure (adjusted odds ratio, 2.3), dependent functional status (aOR, 2.1), RYGB (aOR, 1.94), chronic obstructive pulmonary disease (aOR, 1.7), open operation (aOR, 1.6), and male sex (aOR, 1.1). The P values were less than .05 for all comparisons.
Factors not significant for reoperation included body mass index, age, smoking status, bleeding disorder, steroid use, dialysis, hypertension, diabetes, preoperative sepsis, emergent admission, elective operation, and preoperative weight loss.
While bariatric surgery remains a safe operation with low mortality and reoperation rates, additional studies are needed, because of the increased mortality associated with reoperation, to identify ways to mitigate these complications, Dr. Whealon said.
Limitations of the study were that ICD-9 codes for postoperative hemorrhage could not differentiate between intra-abdominal and gastrointestinal bleeding, the database is subject to coding errors, and missing data may have introduced bias into the study, he noted.
Discussant Dr. Matthew Goldblatt of the Medical College of Wisconsin in Milwaukee commented that use of the ACS MBSAQIP (Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program) database would have avoided some of the coding errors for reoperation.
He also questioned whether the average 7-day return to surgery interval reflects the use of endoscopy, as few surgeons would wait that long if, as the analysis suggests, a primary reason for reoperation was postoperative bleeding.
Endoscopy was included in the reoperations, Dr. Whealon said, but he could not speak to the exact percentage it comprised.
Finally, Dr. Goldblatt said, “the patients that you identified as being the highest risk for complication, as is often the case in these reviews, are really the ones most likely to gain the most from the procedure. … So how can people avoid operating on these patients when they are the ones that can get the most out of it?”
Dr. Whealon agreed that high-risk patients have the most to gain and suggested that “optimizing their comorbid conditions before operation will help reduce their risk.”
The authors reported having no conflicts of interest.
CHICAGO – Patients undergoing Roux-en-Y gastric bypass are twice as likely to need a reoperation as those having sleeve gastrectomy, according to ACS NSQIP data.
Reoperation among Roux-en-Y patients was associated with a 10-fold increase in mortality over sleeve gastrectomy (1.2% vs. 0.1%; P less than .01) and a 3-fold increase in length of stay (6 days vs. 2 days; P less than .01), Dr. Matthew Whealon reported at the American College of Surgeons Clinical Congress
The results are consistent with prior contemporary analyses using ACS National Surgical Quality Improvement Program (NSQIP) data reporting reoperation rates of 2.5%-5.1% for Roux-en-Y gastric bypass (RYGB) and 1.6%-3% for sleeve gastrectomy. Those analyses, however, did not include the reasons for reoperation, as these data were not available until the 2012 database release, he said.
With these data now in hand, lead author Dr. Mark Hanna and his fellow investigators at the University of California, Irvine, identified 36,757 adults in the 2012-2013 database who underwent RYGB (n = 19,597) or sleeve gastrectomy (n = 17,160) for morbid obesity and performed multivariate regression analyses to identify risk factors associated with reoperation.
In all, 518 RYGB patients and 231 sleeve gastrectomy patients required an unplanned return to the operating room (2.6% vs. 1.3%), Dr. Whealon said. The mean time from the index procedure to reoperation was 7.6 days and 7.1 days, respectively.
Obstruction was the biggest driver of reoperation following RYGB, accounting for 28% of reoperations. Other causes were bleeding (14.5%), leak (13%), and other unspecified reasons (18.5%), with data missing in 12%.
Bleeding was the most common indication for reoperation after sleeve gastrectomy (25.5%), followed by other unspecified reasons(24.6%), missing data (13%), leak (12.55%), and obstruction (11.2%), he said.
In adjusted multivariate analyses, factors that significantly increased the risk for reoperation were heart failure (adjusted odds ratio, 2.3), dependent functional status (aOR, 2.1), RYGB (aOR, 1.94), chronic obstructive pulmonary disease (aOR, 1.7), open operation (aOR, 1.6), and male sex (aOR, 1.1). The P values were less than .05 for all comparisons.
Factors not significant for reoperation included body mass index, age, smoking status, bleeding disorder, steroid use, dialysis, hypertension, diabetes, preoperative sepsis, emergent admission, elective operation, and preoperative weight loss.
While bariatric surgery remains a safe operation with low mortality and reoperation rates, additional studies are needed, because of the increased mortality associated with reoperation, to identify ways to mitigate these complications, Dr. Whealon said.
Limitations of the study were that ICD-9 codes for postoperative hemorrhage could not differentiate between intra-abdominal and gastrointestinal bleeding, the database is subject to coding errors, and missing data may have introduced bias into the study, he noted.
Discussant Dr. Matthew Goldblatt of the Medical College of Wisconsin in Milwaukee commented that use of the ACS MBSAQIP (Metabolic and Bariatric Surgery Accreditation and Quality Improvement Program) database would have avoided some of the coding errors for reoperation.
He also questioned whether the average 7-day return to surgery interval reflects the use of endoscopy, as few surgeons would wait that long if, as the analysis suggests, a primary reason for reoperation was postoperative bleeding.
Endoscopy was included in the reoperations, Dr. Whealon said, but he could not speak to the exact percentage it comprised.
Finally, Dr. Goldblatt said, “the patients that you identified as being the highest risk for complication, as is often the case in these reviews, are really the ones most likely to gain the most from the procedure. … So how can people avoid operating on these patients when they are the ones that can get the most out of it?”
Dr. Whealon agreed that high-risk patients have the most to gain and suggested that “optimizing their comorbid conditions before operation will help reduce their risk.”
The authors reported having no conflicts of interest.
AT THE ACS CLINICAL CONGRESS
Key clinical point: Patients undergoing Roux-en-Y gastric bypass were twice as likely to need a reoperation as with sleeve gastrectomy, and reoperation increased morbidity 10-fold.
Major finding: The reoperation rate for Roux-en-Y gastric bypass was 2.6% vs. 1.3% for sleeve gastrectomy.
Data source: An ACS NSQIP database analysis of 36,757 patients undergoing bariatric surgery.
Disclosures: The authors reported having no conflicts of interest.

Myth of the Month: Does Colace work?
Myth: Docusate is a stool softener and helps with constipation.
A 60-year-old man is injured in a fall and breaks four ribs. He is in severe pain and is prescribed oxycodone and naproxen for pain. What treatment would you prescribe to help decrease problems with constipation?
A. Docusate.
B. Docusate and polyethylene glycol.
C. Psyllium.
D. Polyethylene glycol.
Constipation is extremely common, occurring in up to 20%-25% of the elderly population and 90% of patients treated with opioids. The formal definition of constipation is fewer than three bowel movements per week. Patients are concerned with other symptoms as well, including hard stool consistency and the feeling of incomplete evacuation.
An extremely commonly prescribed medication for patients with symptoms of constipation/hard stool passage is docusate (Colace). This medication is often a part of bowel programs for institutionalized/hospitalized patients, as well as being frequently prescribed when patients are treated with opiates.
Does it work?

Docusate is frequently prescribed as a “stool softener,” but does it increase water content in stool? In a randomized, controlled trial of docusate vs. psyllium, 170 adult patients with chronic constipation received either 5.1 g twice a day of psyllium or 100 mg twice a day of docusate (Aliment Pharmacol Ther. 1998 May;12[5]:491-7).
Psyllium was superior in its effect on stool frequency, stool water content, total stool output, and the combination of several objective measures of constipation. Compared with baseline, psyllium increased stool water content by 2.33%, vs .01% for docusate (P =. 007), and stool weight was increased in the group treated with psyllium, compared with docusate-treated patients (359.9 g/week vs. 271.9 g/week, respectively; P = .005). Docusate does not appear to have any effect on stool water content or amount of stool.
In a study of constipation treatment in patients receiving opioids, Dr. Yoko Tarumi and her colleagues studied 74 patients admitted to hospice units (J Pain Symptom Manage. 2013 Jan;45[1]:2-13). A total of 74 patients were randomized to receive docusate 100 mg twice a day plus senna, or placebo plus senna. Once the study was started, inclusion criteria were broadened to include hospice patients with nonmalignant disease and patients who were not on opioids.
Almost all patients in the study did receive opioids (94% of the docusate patients and 100% of placebo-treated patients). There were no significant between the groups in stool volume, frequency, consistency, or in perceived completeness of evacuation.
In a randomized, controlled study of elderly patients on a medicine ward, 34 patients were randomized to docusate or control (no laxatives)(J Chronic Dis. 1976 Jan;29[1]:59-63). There was no difference in frequency or quality of stools between groups.
A systematic review of the usefulness of docusate in chronically ill patients concluded that the widespread use of docusate for the treatment of constipation in palliative-care patients is based on inadequate experimental evidence (J Pain Symptom Manage. 2000 Feb;19[2]:130-6).
The Canadian Agency for Drugs and Technologies in Health concluded “the available evidence suggests that docusate is no more effective than placebo in the prevention or management of constipation” (Dioctyl sulfosuccinate or docusate [calcium or sodium] for the prevention or management of constipation: a review of the clinical effectiveness. Canadian Agency for Drugs and Technologies in Health; 2014 Jun 26).
Dr. Davendra Ramkumar and his colleagues published a systematic review of drug trials for the treatment of constipation in 2005 (Am J Gastroenterol. 2005 Apr;100[4]:936-71). Only polyethylene glycol and tegaserod received grade A evidence for published trials. Psyllium and lactulose received grade B evidence. Docusate received a level 3, grade C for evidence (poor quality evidence, poor evidence to support a recommendation for or against the use of the modality).
I have been surprised at how docusate has been the most commonly prescribed laxative agent. Polyethylene glycol or psyllium are better evidence-based options. Docusate is often prescribed as a stool softener, and it has even less evidence that it softens stool than its poor evidence as a laxative.
Acknowledgments
My thanks to the late Dr. David Saunders for teaching me 30 years ago that docusate was not a helpful option for the management of constipation, and to Sarah Steinkruger for doing much of the research that was used in this column.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.
Myth: Docusate is a stool softener and helps with constipation.
A 60-year-old man is injured in a fall and breaks four ribs. He is in severe pain and is prescribed oxycodone and naproxen for pain. What treatment would you prescribe to help decrease problems with constipation?
A. Docusate.
B. Docusate and polyethylene glycol.
C. Psyllium.
D. Polyethylene glycol.
Constipation is extremely common, occurring in up to 20%-25% of the elderly population and 90% of patients treated with opioids. The formal definition of constipation is fewer than three bowel movements per week. Patients are concerned with other symptoms as well, including hard stool consistency and the feeling of incomplete evacuation.
An extremely commonly prescribed medication for patients with symptoms of constipation/hard stool passage is docusate (Colace). This medication is often a part of bowel programs for institutionalized/hospitalized patients, as well as being frequently prescribed when patients are treated with opiates.
Does it work?
Docusate is frequently prescribed as a “stool softener,” but does it increase water content in stool? In a randomized, controlled trial of docusate vs. psyllium, 170 adult patients with chronic constipation received either 5.1 g twice a day of psyllium or 100 mg twice a day of docusate (Aliment Pharmacol Ther. 1998 May;12[5]:491-7).
Psyllium was superior in its effect on stool frequency, stool water content, total stool output, and the combination of several objective measures of constipation. Compared with baseline, psyllium increased stool water content by 2.33%, vs .01% for docusate (P =. 007), and stool weight was increased in the group treated with psyllium, compared with docusate-treated patients (359.9 g/week vs. 271.9 g/week, respectively; P = .005). Docusate does not appear to have any effect on stool water content or amount of stool.
In a study of constipation treatment in patients receiving opioids, Dr. Yoko Tarumi and her colleagues studied 74 patients admitted to hospice units (J Pain Symptom Manage. 2013 Jan;45[1]:2-13). A total of 74 patients were randomized to receive docusate 100 mg twice a day plus senna, or placebo plus senna. Once the study was started, inclusion criteria were broadened to include hospice patients with nonmalignant disease and patients who were not on opioids.
Almost all patients in the study did receive opioids (94% of the docusate patients and 100% of placebo-treated patients). There were no significant between the groups in stool volume, frequency, consistency, or in perceived completeness of evacuation.
In a randomized, controlled study of elderly patients on a medicine ward, 34 patients were randomized to docusate or control (no laxatives)(J Chronic Dis. 1976 Jan;29[1]:59-63). There was no difference in frequency or quality of stools between groups.
A systematic review of the usefulness of docusate in chronically ill patients concluded that the widespread use of docusate for the treatment of constipation in palliative-care patients is based on inadequate experimental evidence (J Pain Symptom Manage. 2000 Feb;19[2]:130-6).
The Canadian Agency for Drugs and Technologies in Health concluded “the available evidence suggests that docusate is no more effective than placebo in the prevention or management of constipation” (Dioctyl sulfosuccinate or docusate [calcium or sodium] for the prevention or management of constipation: a review of the clinical effectiveness. Canadian Agency for Drugs and Technologies in Health; 2014 Jun 26).
Dr. Davendra Ramkumar and his colleagues published a systematic review of drug trials for the treatment of constipation in 2005 (Am J Gastroenterol. 2005 Apr;100[4]:936-71). Only polyethylene glycol and tegaserod received grade A evidence for published trials. Psyllium and lactulose received grade B evidence. Docusate received a level 3, grade C for evidence (poor quality evidence, poor evidence to support a recommendation for or against the use of the modality).
I have been surprised at how docusate has been the most commonly prescribed laxative agent. Polyethylene glycol or psyllium are better evidence-based options. Docusate is often prescribed as a stool softener, and it has even less evidence that it softens stool than its poor evidence as a laxative.
Acknowledgments
My thanks to the late Dr. David Saunders for teaching me 30 years ago that docusate was not a helpful option for the management of constipation, and to Sarah Steinkruger for doing much of the research that was used in this column.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.
Myth: Docusate is a stool softener and helps with constipation.
A 60-year-old man is injured in a fall and breaks four ribs. He is in severe pain and is prescribed oxycodone and naproxen for pain. What treatment would you prescribe to help decrease problems with constipation?
A. Docusate.
B. Docusate and polyethylene glycol.
C. Psyllium.
D. Polyethylene glycol.
Constipation is extremely common, occurring in up to 20%-25% of the elderly population and 90% of patients treated with opioids. The formal definition of constipation is fewer than three bowel movements per week. Patients are concerned with other symptoms as well, including hard stool consistency and the feeling of incomplete evacuation.
An extremely commonly prescribed medication for patients with symptoms of constipation/hard stool passage is docusate (Colace). This medication is often a part of bowel programs for institutionalized/hospitalized patients, as well as being frequently prescribed when patients are treated with opiates.
Does it work?
Docusate is frequently prescribed as a “stool softener,” but does it increase water content in stool? In a randomized, controlled trial of docusate vs. psyllium, 170 adult patients with chronic constipation received either 5.1 g twice a day of psyllium or 100 mg twice a day of docusate (Aliment Pharmacol Ther. 1998 May;12[5]:491-7).
Psyllium was superior in its effect on stool frequency, stool water content, total stool output, and the combination of several objective measures of constipation. Compared with baseline, psyllium increased stool water content by 2.33%, vs .01% for docusate (P =. 007), and stool weight was increased in the group treated with psyllium, compared with docusate-treated patients (359.9 g/week vs. 271.9 g/week, respectively; P = .005). Docusate does not appear to have any effect on stool water content or amount of stool.
In a study of constipation treatment in patients receiving opioids, Dr. Yoko Tarumi and her colleagues studied 74 patients admitted to hospice units (J Pain Symptom Manage. 2013 Jan;45[1]:2-13). A total of 74 patients were randomized to receive docusate 100 mg twice a day plus senna, or placebo plus senna. Once the study was started, inclusion criteria were broadened to include hospice patients with nonmalignant disease and patients who were not on opioids.
Almost all patients in the study did receive opioids (94% of the docusate patients and 100% of placebo-treated patients). There were no significant between the groups in stool volume, frequency, consistency, or in perceived completeness of evacuation.
In a randomized, controlled study of elderly patients on a medicine ward, 34 patients were randomized to docusate or control (no laxatives)(J Chronic Dis. 1976 Jan;29[1]:59-63). There was no difference in frequency or quality of stools between groups.
A systematic review of the usefulness of docusate in chronically ill patients concluded that the widespread use of docusate for the treatment of constipation in palliative-care patients is based on inadequate experimental evidence (J Pain Symptom Manage. 2000 Feb;19[2]:130-6).
The Canadian Agency for Drugs and Technologies in Health concluded “the available evidence suggests that docusate is no more effective than placebo in the prevention or management of constipation” (Dioctyl sulfosuccinate or docusate [calcium or sodium] for the prevention or management of constipation: a review of the clinical effectiveness. Canadian Agency for Drugs and Technologies in Health; 2014 Jun 26).
Dr. Davendra Ramkumar and his colleagues published a systematic review of drug trials for the treatment of constipation in 2005 (Am J Gastroenterol. 2005 Apr;100[4]:936-71). Only polyethylene glycol and tegaserod received grade A evidence for published trials. Psyllium and lactulose received grade B evidence. Docusate received a level 3, grade C for evidence (poor quality evidence, poor evidence to support a recommendation for or against the use of the modality).
I have been surprised at how docusate has been the most commonly prescribed laxative agent. Polyethylene glycol or psyllium are better evidence-based options. Docusate is often prescribed as a stool softener, and it has even less evidence that it softens stool than its poor evidence as a laxative.
Acknowledgments
My thanks to the late Dr. David Saunders for teaching me 30 years ago that docusate was not a helpful option for the management of constipation, and to Sarah Steinkruger for doing much of the research that was used in this column.
Dr. Paauw is professor of medicine in the division of general internal medicine at the University of Washington, Seattle, and he serves as third-year medical student clerkship director at the University of Washington. Contact Dr. Paauw at dpaauw@uw.edu.

The Calling of Rural Surgery
Recruitment of surgeons to rural hospitals is a challenge for many communities. As the current workforce of rural surgeons reaches retirement age, this challenge will only become more acute.
The American College of Surgeons (ACS) and Corrado Studios have collaborated to create “The Calling of Rural Surgery,” a 6-minute recruitment video to inspire interest and appreciation of rural surgery as a career choice. This film is a peek into the world of surgeons practicing in small towns and rural areas. It premiered at the Rural Surgeons Open Forum and Oweida Scholarship Presentation at the ACS Clinical Congress 2015 in Chicago.

The video producer, Meredith Corrado, said, “Our goals are not just to recruit surgeons to practice in the rural and underserved areas in America [but to] put our message in front of fledgling surgeons deciding on a career path, [and] those experienced surgeons looking for change.”
Ms. Corrado interviewed ACS fellows, members of the college’s Advisory Council for Rural Surgery, past ACS governors, and an ACS past vice president. Her father, Dr. Joseph Corrado, a surgeon practicing in Mexico, Mo., was a source of insight into the work life of rural surgeons
Dr. Corrado said, “The problem of succession will be critical, especially for hospitals like my own with only one or two surgeons. One thing that makes it particularly hard is that we want the best. My friends and neighbors are counting on my judgment, so I’m picky.”
He wants potential recruits to know that rural practice is “extremely rewarding. You are an integral part of a community in all aspects: economically, socially and medically. You have the ability to choose from a wide variety in how broad you want your practice to be.” In addition, he is confident that exposure to the rural surgical practice will draw the right recruits. “Once a surgeon is able to see all the surgeries we get to do and how rewarding our lives are, the right person will not be able to walk away.”
View the video at www.ruralsurgeonsfilm.com.
Recruitment of surgeons to rural hospitals is a challenge for many communities. As the current workforce of rural surgeons reaches retirement age, this challenge will only become more acute.
The American College of Surgeons (ACS) and Corrado Studios have collaborated to create “The Calling of Rural Surgery,” a 6-minute recruitment video to inspire interest and appreciation of rural surgery as a career choice. This film is a peek into the world of surgeons practicing in small towns and rural areas. It premiered at the Rural Surgeons Open Forum and Oweida Scholarship Presentation at the ACS Clinical Congress 2015 in Chicago.
The video producer, Meredith Corrado, said, “Our goals are not just to recruit surgeons to practice in the rural and underserved areas in America [but to] put our message in front of fledgling surgeons deciding on a career path, [and] those experienced surgeons looking for change.”
Ms. Corrado interviewed ACS fellows, members of the college’s Advisory Council for Rural Surgery, past ACS governors, and an ACS past vice president. Her father, Dr. Joseph Corrado, a surgeon practicing in Mexico, Mo., was a source of insight into the work life of rural surgeons
Dr. Corrado said, “The problem of succession will be critical, especially for hospitals like my own with only one or two surgeons. One thing that makes it particularly hard is that we want the best. My friends and neighbors are counting on my judgment, so I’m picky.”
He wants potential recruits to know that rural practice is “extremely rewarding. You are an integral part of a community in all aspects: economically, socially and medically. You have the ability to choose from a wide variety in how broad you want your practice to be.” In addition, he is confident that exposure to the rural surgical practice will draw the right recruits. “Once a surgeon is able to see all the surgeries we get to do and how rewarding our lives are, the right person will not be able to walk away.”
View the video at www.ruralsurgeonsfilm.com.
Recruitment of surgeons to rural hospitals is a challenge for many communities. As the current workforce of rural surgeons reaches retirement age, this challenge will only become more acute.
The American College of Surgeons (ACS) and Corrado Studios have collaborated to create “The Calling of Rural Surgery,” a 6-minute recruitment video to inspire interest and appreciation of rural surgery as a career choice. This film is a peek into the world of surgeons practicing in small towns and rural areas. It premiered at the Rural Surgeons Open Forum and Oweida Scholarship Presentation at the ACS Clinical Congress 2015 in Chicago.
The video producer, Meredith Corrado, said, “Our goals are not just to recruit surgeons to practice in the rural and underserved areas in America [but to] put our message in front of fledgling surgeons deciding on a career path, [and] those experienced surgeons looking for change.”
Ms. Corrado interviewed ACS fellows, members of the college’s Advisory Council for Rural Surgery, past ACS governors, and an ACS past vice president. Her father, Dr. Joseph Corrado, a surgeon practicing in Mexico, Mo., was a source of insight into the work life of rural surgeons
Dr. Corrado said, “The problem of succession will be critical, especially for hospitals like my own with only one or two surgeons. One thing that makes it particularly hard is that we want the best. My friends and neighbors are counting on my judgment, so I’m picky.”
He wants potential recruits to know that rural practice is “extremely rewarding. You are an integral part of a community in all aspects: economically, socially and medically. You have the ability to choose from a wide variety in how broad you want your practice to be.” In addition, he is confident that exposure to the rural surgical practice will draw the right recruits. “Once a surgeon is able to see all the surgeries we get to do and how rewarding our lives are, the right person will not be able to walk away.”
View the video at www.ruralsurgeonsfilm.com.

Law & Medicine: To whom do doctors owe a duty?
Question: A doctor may owe a duty of care in the setting of:
A. A cyber relationship.
B. A special relationship.
C. Both A and B.
D. Neither A nor B.
Answer: C. Ascertaining whether a defendant owes a duty to a claimant is the first inquiry in the tort of negligence. To say there is no duty owed is to deny liability altogether, however obvious the breach or horrendous the foreseeable injuries.
Thus, duty is used as a filter mechanism to reduce frivolous suits or otherwise control the tide of litigation, to prevent “liability in an indeterminate amount for an indeterminate time to an indeterminate class.”
Duty in the context of medical negligence is not usually in dispute, as it is plainly owed by a doctor to his or her patient. It arises out of the doctor-patient relationship. Whether a relationship has been formed in the first place is a threshold inquiry. Where a doctor accepts a patient who is seeking his or her services, the relationship is readily evident. Duty is also established when the doctor begins the evaluation process in a typical encounter.
However, a phone inquiry by a potential patient, without more, may be insufficient to create this relationship, although this may depend on the nature of the phone conversation and the doctor’s response.
Likewise, a “curbside” consultation sought by a colleague does not normally translate into a duty for the doctor offering the opinion. Presumably casual advice given freely and understood as such at social gatherings does not add up to a doctor-patient relationship, and courts will look to reasonableness as the touchstone in deciding whether such a relationship was ever formed.
Still, there are some medical situations where a legitimate question of duty can be raised. With the growth of electronic medical records and communication, medical encounters in cyberspace will emerge as an increasing source of litigation.
Internet liability can be far reaching. In addition to risks governing negligence, informed consent, and privacy/confidentiality, there are additional issues of product liability, cross-border jurisdictional conflicts, and others.
The threshold question when assessing cyberspace liability arising, for example, from the use of doctor-operated medical websites concerns duty, because its existence or denial will determine whether the case can go forward in the first place. Although not the typical office or hospital patient, a plaintiff may argue successfully that a doctor-patient relationship had nonetheless been formed in cyberspace.
It is possible that such a relationship will be found in some circumstances, relevant factors being knowledge of names of subscribers, frequency of interactions, specificity of queries, and so on. In particular, a subscription fee is likely to be construed as evidence of soliciting and accepting a more committed interaction, so it places the operator of the website at greater legal risk. A specific disclaimer is a standard precaution but may not be enough to definitively protect against a lawsuit.
Courts have ruled in favor of plaintiffs despite the absence of face-to-face interaction with a physician. In one case, a doctor speaking to a patient from the emergency department was deemed to have formed a doctor-patient relationship (O’Neill v. Montefiore Hospital, 11 A.D.2d 132 (N.Y.A.D. 1 Dept. 1960). In another, an on-call neurologist’s telephone advice to the treating doctor likewise raised the issue of legal duty (Lection v. Dyll, 65 S.W.3d 696 (Tex. App. Dallas 2001).
The state of Hawaii now permits telehealth services to be reimbursable, notwithstanding the absence of face-to-face contact (HI Rev Stat § 431:10A-116.3[a]). With this law, an online encounter will likely translate into a professional relationship – with corresponding legal duty of due care.
In the case of a Good Samaritan physician – i.e., one who offers gratuitous aid to a stranger in need of medical assistance – courts are unlikely to find a professional relationship, because there is no common law duty to help a stranger.
However, once treatment has begun, there is a duty not to make matters worse. So, all 50 states have enacted Good Samaritan statutes, which protect against liability arising out of negligent rescue. Note that statutory protection is generally excluded for Good Samaritan acts performed within a hospital setting, under the theory that doctors have an ongoing relationship with the hospital and are already obligated to provide emergency care within its walls.
Another category of legal duty concerns nonpatient third parties. The complaint may relate to a failure to warn family members of a patient’s contagious disease, or the transmissible condition may have been missed and an innocent third party was injured as a result.
Another situation where duty to a third party might arise is the learning of a credible threat of harm directed at a named individual. This is famously known as the Tarasoff doctrine, after a California case in which the court imposed a duty on a college psychologist to directly warn an intended victim of harm by his patient – even though that meant breaching confidentiality of a professional relationship, and the victim was a nonpatient third party (Tarasoff v. Regents of University of California, 551 P.2d 334 [Cal. 1976]).
A doctor may also incur liability for automobile injuries sustained by one other than his or her own patient. In a Hawaii case, a car suddenly veered across five lanes of traffic, striking an 11-year-old bystander. The driver alleged that the prescription medication prazosin caused him to lose control of the car.
In ruling that the health care provider was liable to the injured bystander, the Hawaii Supreme Court held that physicians have a duty to warn their patients of potential adverse medication effects, and this responsibility should extend to third parties (McKenzie v. Hawaii Permanente Medical Group, 47 P.3d 1209 [Haw. 2002]).
A foreseeable and unreasonable risk of harm is an important factor, but not the only decisive factor, in construing the existence of a legal duty. Under some circumstances, the term “special relationship” has been employed based on a consideration of “existing social values, customs, and considerations of policy.”
In a Massachusetts case, a family practitioner had failed to warn his patient of the risk of diabetic drugs when operating a vehicle. Just 45 minutes after the patient’s discharge from the hospital, he developed hypoglycemia, losing consciousness and injuring a motorcyclist who then sued the doctor. The court used the “special relationship” rationale in ruling that the doctor owed a duty to the motorcyclist (Arsenault v. McConarty, 21 Mass. L. Rptr. 500 [2006]).
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at siang@hawaii.edu.
Question: A doctor may owe a duty of care in the setting of:
A. A cyber relationship.
B. A special relationship.
C. Both A and B.
D. Neither A nor B.
Answer: C. Ascertaining whether a defendant owes a duty to a claimant is the first inquiry in the tort of negligence. To say there is no duty owed is to deny liability altogether, however obvious the breach or horrendous the foreseeable injuries.
Thus, duty is used as a filter mechanism to reduce frivolous suits or otherwise control the tide of litigation, to prevent “liability in an indeterminate amount for an indeterminate time to an indeterminate class.”
Duty in the context of medical negligence is not usually in dispute, as it is plainly owed by a doctor to his or her patient. It arises out of the doctor-patient relationship. Whether a relationship has been formed in the first place is a threshold inquiry. Where a doctor accepts a patient who is seeking his or her services, the relationship is readily evident. Duty is also established when the doctor begins the evaluation process in a typical encounter.
However, a phone inquiry by a potential patient, without more, may be insufficient to create this relationship, although this may depend on the nature of the phone conversation and the doctor’s response.
Likewise, a “curbside” consultation sought by a colleague does not normally translate into a duty for the doctor offering the opinion. Presumably casual advice given freely and understood as such at social gatherings does not add up to a doctor-patient relationship, and courts will look to reasonableness as the touchstone in deciding whether such a relationship was ever formed.
Still, there are some medical situations where a legitimate question of duty can be raised. With the growth of electronic medical records and communication, medical encounters in cyberspace will emerge as an increasing source of litigation.
Internet liability can be far reaching. In addition to risks governing negligence, informed consent, and privacy/confidentiality, there are additional issues of product liability, cross-border jurisdictional conflicts, and others.
The threshold question when assessing cyberspace liability arising, for example, from the use of doctor-operated medical websites concerns duty, because its existence or denial will determine whether the case can go forward in the first place. Although not the typical office or hospital patient, a plaintiff may argue successfully that a doctor-patient relationship had nonetheless been formed in cyberspace.
It is possible that such a relationship will be found in some circumstances, relevant factors being knowledge of names of subscribers, frequency of interactions, specificity of queries, and so on. In particular, a subscription fee is likely to be construed as evidence of soliciting and accepting a more committed interaction, so it places the operator of the website at greater legal risk. A specific disclaimer is a standard precaution but may not be enough to definitively protect against a lawsuit.
Courts have ruled in favor of plaintiffs despite the absence of face-to-face interaction with a physician. In one case, a doctor speaking to a patient from the emergency department was deemed to have formed a doctor-patient relationship (O’Neill v. Montefiore Hospital, 11 A.D.2d 132 (N.Y.A.D. 1 Dept. 1960). In another, an on-call neurologist’s telephone advice to the treating doctor likewise raised the issue of legal duty (Lection v. Dyll, 65 S.W.3d 696 (Tex. App. Dallas 2001).
The state of Hawaii now permits telehealth services to be reimbursable, notwithstanding the absence of face-to-face contact (HI Rev Stat § 431:10A-116.3[a]). With this law, an online encounter will likely translate into a professional relationship – with corresponding legal duty of due care.
In the case of a Good Samaritan physician – i.e., one who offers gratuitous aid to a stranger in need of medical assistance – courts are unlikely to find a professional relationship, because there is no common law duty to help a stranger.
However, once treatment has begun, there is a duty not to make matters worse. So, all 50 states have enacted Good Samaritan statutes, which protect against liability arising out of negligent rescue. Note that statutory protection is generally excluded for Good Samaritan acts performed within a hospital setting, under the theory that doctors have an ongoing relationship with the hospital and are already obligated to provide emergency care within its walls.
Another category of legal duty concerns nonpatient third parties. The complaint may relate to a failure to warn family members of a patient’s contagious disease, or the transmissible condition may have been missed and an innocent third party was injured as a result.
Another situation where duty to a third party might arise is the learning of a credible threat of harm directed at a named individual. This is famously known as the Tarasoff doctrine, after a California case in which the court imposed a duty on a college psychologist to directly warn an intended victim of harm by his patient – even though that meant breaching confidentiality of a professional relationship, and the victim was a nonpatient third party (Tarasoff v. Regents of University of California, 551 P.2d 334 [Cal. 1976]).
A doctor may also incur liability for automobile injuries sustained by one other than his or her own patient. In a Hawaii case, a car suddenly veered across five lanes of traffic, striking an 11-year-old bystander. The driver alleged that the prescription medication prazosin caused him to lose control of the car.
In ruling that the health care provider was liable to the injured bystander, the Hawaii Supreme Court held that physicians have a duty to warn their patients of potential adverse medication effects, and this responsibility should extend to third parties (McKenzie v. Hawaii Permanente Medical Group, 47 P.3d 1209 [Haw. 2002]).
A foreseeable and unreasonable risk of harm is an important factor, but not the only decisive factor, in construing the existence of a legal duty. Under some circumstances, the term “special relationship” has been employed based on a consideration of “existing social values, customs, and considerations of policy.”
In a Massachusetts case, a family practitioner had failed to warn his patient of the risk of diabetic drugs when operating a vehicle. Just 45 minutes after the patient’s discharge from the hospital, he developed hypoglycemia, losing consciousness and injuring a motorcyclist who then sued the doctor. The court used the “special relationship” rationale in ruling that the doctor owed a duty to the motorcyclist (Arsenault v. McConarty, 21 Mass. L. Rptr. 500 [2006]).
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at siang@hawaii.edu.
Question: A doctor may owe a duty of care in the setting of:
A. A cyber relationship.
B. A special relationship.
C. Both A and B.
D. Neither A nor B.
Answer: C. Ascertaining whether a defendant owes a duty to a claimant is the first inquiry in the tort of negligence. To say there is no duty owed is to deny liability altogether, however obvious the breach or horrendous the foreseeable injuries.
Thus, duty is used as a filter mechanism to reduce frivolous suits or otherwise control the tide of litigation, to prevent “liability in an indeterminate amount for an indeterminate time to an indeterminate class.”
Duty in the context of medical negligence is not usually in dispute, as it is plainly owed by a doctor to his or her patient. It arises out of the doctor-patient relationship. Whether a relationship has been formed in the first place is a threshold inquiry. Where a doctor accepts a patient who is seeking his or her services, the relationship is readily evident. Duty is also established when the doctor begins the evaluation process in a typical encounter.
However, a phone inquiry by a potential patient, without more, may be insufficient to create this relationship, although this may depend on the nature of the phone conversation and the doctor’s response.
Likewise, a “curbside” consultation sought by a colleague does not normally translate into a duty for the doctor offering the opinion. Presumably casual advice given freely and understood as such at social gatherings does not add up to a doctor-patient relationship, and courts will look to reasonableness as the touchstone in deciding whether such a relationship was ever formed.
Still, there are some medical situations where a legitimate question of duty can be raised. With the growth of electronic medical records and communication, medical encounters in cyberspace will emerge as an increasing source of litigation.
Internet liability can be far reaching. In addition to risks governing negligence, informed consent, and privacy/confidentiality, there are additional issues of product liability, cross-border jurisdictional conflicts, and others.
The threshold question when assessing cyberspace liability arising, for example, from the use of doctor-operated medical websites concerns duty, because its existence or denial will determine whether the case can go forward in the first place. Although not the typical office or hospital patient, a plaintiff may argue successfully that a doctor-patient relationship had nonetheless been formed in cyberspace.
It is possible that such a relationship will be found in some circumstances, relevant factors being knowledge of names of subscribers, frequency of interactions, specificity of queries, and so on. In particular, a subscription fee is likely to be construed as evidence of soliciting and accepting a more committed interaction, so it places the operator of the website at greater legal risk. A specific disclaimer is a standard precaution but may not be enough to definitively protect against a lawsuit.
Courts have ruled in favor of plaintiffs despite the absence of face-to-face interaction with a physician. In one case, a doctor speaking to a patient from the emergency department was deemed to have formed a doctor-patient relationship (O’Neill v. Montefiore Hospital, 11 A.D.2d 132 (N.Y.A.D. 1 Dept. 1960). In another, an on-call neurologist’s telephone advice to the treating doctor likewise raised the issue of legal duty (Lection v. Dyll, 65 S.W.3d 696 (Tex. App. Dallas 2001).
The state of Hawaii now permits telehealth services to be reimbursable, notwithstanding the absence of face-to-face contact (HI Rev Stat § 431:10A-116.3[a]). With this law, an online encounter will likely translate into a professional relationship – with corresponding legal duty of due care.
In the case of a Good Samaritan physician – i.e., one who offers gratuitous aid to a stranger in need of medical assistance – courts are unlikely to find a professional relationship, because there is no common law duty to help a stranger.
However, once treatment has begun, there is a duty not to make matters worse. So, all 50 states have enacted Good Samaritan statutes, which protect against liability arising out of negligent rescue. Note that statutory protection is generally excluded for Good Samaritan acts performed within a hospital setting, under the theory that doctors have an ongoing relationship with the hospital and are already obligated to provide emergency care within its walls.
Another category of legal duty concerns nonpatient third parties. The complaint may relate to a failure to warn family members of a patient’s contagious disease, or the transmissible condition may have been missed and an innocent third party was injured as a result.
Another situation where duty to a third party might arise is the learning of a credible threat of harm directed at a named individual. This is famously known as the Tarasoff doctrine, after a California case in which the court imposed a duty on a college psychologist to directly warn an intended victim of harm by his patient – even though that meant breaching confidentiality of a professional relationship, and the victim was a nonpatient third party (Tarasoff v. Regents of University of California, 551 P.2d 334 [Cal. 1976]).
A doctor may also incur liability for automobile injuries sustained by one other than his or her own patient. In a Hawaii case, a car suddenly veered across five lanes of traffic, striking an 11-year-old bystander. The driver alleged that the prescription medication prazosin caused him to lose control of the car.
In ruling that the health care provider was liable to the injured bystander, the Hawaii Supreme Court held that physicians have a duty to warn their patients of potential adverse medication effects, and this responsibility should extend to third parties (McKenzie v. Hawaii Permanente Medical Group, 47 P.3d 1209 [Haw. 2002]).
A foreseeable and unreasonable risk of harm is an important factor, but not the only decisive factor, in construing the existence of a legal duty. Under some circumstances, the term “special relationship” has been employed based on a consideration of “existing social values, customs, and considerations of policy.”
In a Massachusetts case, a family practitioner had failed to warn his patient of the risk of diabetic drugs when operating a vehicle. Just 45 minutes after the patient’s discharge from the hospital, he developed hypoglycemia, losing consciousness and injuring a motorcyclist who then sued the doctor. The court used the “special relationship” rationale in ruling that the doctor owed a duty to the motorcyclist (Arsenault v. McConarty, 21 Mass. L. Rptr. 500 [2006]).
Dr. Tan is emeritus professor of medicine and former adjunct professor of law at the University of Hawaii, and currently directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at siang@hawaii.edu.

Transplant waiting-list registrations dropped after direct acting–antiviral approval
SAN FRANCISCO – The number of new waiting-list registrations for liver transplantation among patients with hepatitis C virus declined significantly after the introduction of second-generation direct-acting antiviral agents, according to a review of United Network for Organ Sharing data.
New waiting-list registrations (NWRs) through March 31, 2015 – based on the most recent data available as of September 2015 – ranged from 740 to 976 per month between August 2012 and March 2015, and a review of the data show that HCV-specific NWRs declined by 23% overall in the 15 months after the Food and Drug Administration approved simeprevir (Nov. 22, 2013) and sofosbuvir (Dec. 6, 2013) vs. the 15 months prior (from 34.8% to 26.8%), Dr. Ryan B. Perumpail reported at the annual meeting of the American Association for the Study of Liver Diseases.
Further, the proportion of HCV patients without hepatocellular carcinoma among new waiting-list registrations declined by 33% (from 23.0% to 15.4%) and there was a significant decline in NWRs for liver transplants in nonhepatocellular-carcinoma HCV patients from January 2014 to March 2015 (153 per month) vs. August 2012 to October 2013 (mean 188 per month), for a mean difference of 35.0, said Dr. Perumpail of Stanford University Medical Center, Palo Alto, Calif.
Among the HCV-related NWRs for liver transplantation, the proportion without hepatocellular carcinoma declined 12.5% (from 66.3% to 58.0%).
Direct-acting antiviral therapy has been prioritized in patients with HCV-related cirrhosis in an effort to slow clinical disease progress and to induce regression of hepatic histologic damage, Dr. Perumpail explained.
Thought limited by the retrospective design of the study, the findings suggest that the use of direct-acting antivirals contributed to a downward trend in NWRs for liver transplantation among HCV patients without hepatocellular carcinoma.
Dr. Perumpail reported having no disclosures.
SAN FRANCISCO – The number of new waiting-list registrations for liver transplantation among patients with hepatitis C virus declined significantly after the introduction of second-generation direct-acting antiviral agents, according to a review of United Network for Organ Sharing data.
New waiting-list registrations (NWRs) through March 31, 2015 – based on the most recent data available as of September 2015 – ranged from 740 to 976 per month between August 2012 and March 2015, and a review of the data show that HCV-specific NWRs declined by 23% overall in the 15 months after the Food and Drug Administration approved simeprevir (Nov. 22, 2013) and sofosbuvir (Dec. 6, 2013) vs. the 15 months prior (from 34.8% to 26.8%), Dr. Ryan B. Perumpail reported at the annual meeting of the American Association for the Study of Liver Diseases.
Further, the proportion of HCV patients without hepatocellular carcinoma among new waiting-list registrations declined by 33% (from 23.0% to 15.4%) and there was a significant decline in NWRs for liver transplants in nonhepatocellular-carcinoma HCV patients from January 2014 to March 2015 (153 per month) vs. August 2012 to October 2013 (mean 188 per month), for a mean difference of 35.0, said Dr. Perumpail of Stanford University Medical Center, Palo Alto, Calif.
Among the HCV-related NWRs for liver transplantation, the proportion without hepatocellular carcinoma declined 12.5% (from 66.3% to 58.0%).
Direct-acting antiviral therapy has been prioritized in patients with HCV-related cirrhosis in an effort to slow clinical disease progress and to induce regression of hepatic histologic damage, Dr. Perumpail explained.
Thought limited by the retrospective design of the study, the findings suggest that the use of direct-acting antivirals contributed to a downward trend in NWRs for liver transplantation among HCV patients without hepatocellular carcinoma.
Dr. Perumpail reported having no disclosures.
SAN FRANCISCO – The number of new waiting-list registrations for liver transplantation among patients with hepatitis C virus declined significantly after the introduction of second-generation direct-acting antiviral agents, according to a review of United Network for Organ Sharing data.
New waiting-list registrations (NWRs) through March 31, 2015 – based on the most recent data available as of September 2015 – ranged from 740 to 976 per month between August 2012 and March 2015, and a review of the data show that HCV-specific NWRs declined by 23% overall in the 15 months after the Food and Drug Administration approved simeprevir (Nov. 22, 2013) and sofosbuvir (Dec. 6, 2013) vs. the 15 months prior (from 34.8% to 26.8%), Dr. Ryan B. Perumpail reported at the annual meeting of the American Association for the Study of Liver Diseases.
Further, the proportion of HCV patients without hepatocellular carcinoma among new waiting-list registrations declined by 33% (from 23.0% to 15.4%) and there was a significant decline in NWRs for liver transplants in nonhepatocellular-carcinoma HCV patients from January 2014 to March 2015 (153 per month) vs. August 2012 to October 2013 (mean 188 per month), for a mean difference of 35.0, said Dr. Perumpail of Stanford University Medical Center, Palo Alto, Calif.
Among the HCV-related NWRs for liver transplantation, the proportion without hepatocellular carcinoma declined 12.5% (from 66.3% to 58.0%).
Direct-acting antiviral therapy has been prioritized in patients with HCV-related cirrhosis in an effort to slow clinical disease progress and to induce regression of hepatic histologic damage, Dr. Perumpail explained.
Thought limited by the retrospective design of the study, the findings suggest that the use of direct-acting antivirals contributed to a downward trend in NWRs for liver transplantation among HCV patients without hepatocellular carcinoma.
Dr. Perumpail reported having no disclosures.
AT THE LIVER MEETING 2015
Key clinical point: New waiting-list registrations for liver transplantation in patients with hepatitis C virus declined significantly after introduction of second-generation direct-acting antiviral agents.
Major finding: HCV-specific waiting-list registrations declined by 23% in the 15 months after vs. before FDA approval of simeprevir and sofosbuvir (34.8% vs. 26.8%).
Data source: A review of United Network for Organ Sharing data.
Disclosures: Dr. Perumpail reported having no disclosures.
Senate panel targets drug prices during FDA commissioner nomination hearing
WASHINGTON – Drug pricing was front and center as a Senate panel convened to review the nomination of Dr. Robert M. Califf as commissioner of the Food and Drug Administration.
During a Nov. 17 hearing of the Senate Health, Education, Labor, and Pensions (HELP) Committee, many members keyed in on the role of the FDA in trying to bring down drug prices.

When asked about the price of prescription drugs, Dr. Califf was clear in stating that the agency’s role was not to set prices, but suggested that the FDA could have an impact on pricing by making the approval process for both brand and generic products more efficient, including revamping the clinical trials needed for approval to make them cheaper and more inclusive, while improving the quality of the data produced.
“We think that we can do trials that are actually bigger and include more patients that are more representative for a much lower cost,” Dr. Califf said, responding to Sen. Orrin Hatch’s (R-Utah) concerns about length of time of data exclusivity and fears that calls for shortening it might lead to higher drug prices as companies try to recoup development costs.
He also pointed to the use of electronic health records as a way to broaden clinical trials at a lower cost.
“The key here is using electronic health records that we already have,” Dr. Califf said, adding that once interoperability issues are addressed, EHRs will enable manufacturers to develop therapies “at a much lower cost with better information about safety and efficacy.”
In looking at rising generic prices, Committee Chairman Lamar Alexander (R-Tenn) focused on generic approval times, noting that generic manufacturers estimated that it took on average 48 months for a generic to get approval in 2014, up from 30 months in 2011.
A more rapid approval, “if safe and effective, might have some effect on lowering drug prices,” Sen. Alexander said.
Dr. Califf responded that FDA is already ahead of the generic user fee goals that have been set and the agency has been clearing the more simple applications from its backlog.
“But we have this backlog of applications that are requiring a back-and-forth because we want generic drugs to be just as safe and effective as the innovative drugs and when the applications [are not complete and there are questions] those get held up,” he responded, adding that he was “confident” that once the backlog is cleared over the next several years, generic applications will be processed quickly.
However, his answers were not convincing to at least one member. Presidential hopeful Sen. Bernie Sanders (I-Vt.) was not convinced Dr. Califf will do enough in this area.
“At a time when millions of Americans cannot afford to purchase the prescription drugs they need, we need a new leader at the FDA who is prepared to stand up to the pharmaceutical companies and work to substantially lower drug prices. Unfortunately, I have come to the conclusion that Dr. Califf is not that person,” Sanders said in an Oct. 9 statement.
Sen. Sanders reaffirmed his objection, saying that nothing he heard during the hearing has changed his opinions.
And while he openly objected to the nomination, both the chairman and ranking member of the committee both openly endorsed the White House nominee.
In his prepared remarks, Mr. Alexander noted that his staff “has spent 2 months carefully reviewing everything you submitted and has not found anything that would call into doubt your ability to lead the FDA fairly and impartially.”
Likewise, Ranking Member Patty Murray (D-Wash.) said during the hearing that after “careful consideration and review, I am confident that Dr. Califf would contribute leadership and expertise as we work to find new ways to advance medical innovation for patients and families, and improve the health and well-being across this country. He is a strong nominee for the role of FDA commissioner.”
In his opening statement, Dr. Califf outlined his priorities if confirmed as commissioner, including strengthening and better supporting FDA’s workforce, finishing work in an number of areas – including improving the nation’s food supply, tobacco regulation, better combating antibiotic resistant bacteria, medical countermeasure development, and the White House’s Precision Medicine Initiative – and “further develop the science base that informs FDA’s decision making.”
During his testimony, he also said there is “a need to work on postmarket surveillance of devices,” suggesting that a similar program, the FDA’s Sentinel Initiative drug surveillance program, be developed. Dr. Califf also said a new pathway is needed for approving drug/device combination products.
In the area of medical apps, Dr. Califf said there needs to be more clarity as to where the line for regulation should be. As an example, he said that an app that monitors heart rate does not need much oversight, but if that app were connected to an implantable defibrillator, that would require more regulatory oversight.
Sen. Sanders cited Dr. Califf’s ties to the pharmaceutical industry as the prime reason for opposing the nomination.
Dr. Califf noted that for any of the research he had conducted, even when primarily funded from industry, he was clear that manufacturers, while being able to make suggestions for analysis and publication of results, did not have final say, and he said that numerous studies were rejected because companies would not agree to full access to databases or independence with the final publication of results.
One area that was not raised during the hearing, but was raised by the Project on Government Oversight regarding the ROCKET AF trial of rivaroxaban (Xarelto), for which he was co-primary investigator (N Engl J Med. 2011 Sep 8;365:883-91).
“Dr. Califf’s handling of the Xarelto trial raises concerns about his judgment in overseeing the pharmaceutical industry.” POGO Executive Director Danielle Brian said in a statement. “As senators consider Dr. Califf’s confirmation to run the FDA, they should ask tough questions about the Xarelto clinical trial.”
FDA primary reviewers recommended against approval based on a number of issues with the trial, but FDA ultimately decided in favor of bringing the drug to market.
A cardiologist by training, Dr. Califf has received support from the American College of Cardiology.
In an Oct. 21, 2015, letter to the Senate HELP Committee, ACC called Dr. Califf “the right person to lead the FDA as commissioner based on his impressive medical knowledge, clinical research experience, and visionary leadership abilities.”
Dr. Califf joined the FDA in February as deputy commissioner of medical products and currently provides executive leadership to FDA’s Center for Drug Evaluation and Research, the Center for Biologics Evaluation and Research, the Center for Devices and Radiological Health, and the Center for Tobacco Products.
He also oversees FDA’s Office of Special Medical Programs and plays a critical role in providing high-level advice and policy direction on the agency’s medical product and tobacco priorities.
Prior to coming to FDA, he was vice chancellor of clinical and translational research and professor of medicine in the division of cardiology at Duke University in Durham, N.C. He has held multiple positions at the university, including director of the Duke Translational Medicine Institute and founding director of the Duke Clinical Research Institute.
He also served as a member of FDA’s Cardiovascular and Renal Drugs Advisory Committee and the agency’s Science Board Subcommittee on Science and Technology.
“We believe that with Dr. Califf’s diverse background, and his exemplary knowledge of clinical and translational medicine, he will continue to improve the FDA’s drug approval process while ensuring that patients are receiving the safest and most effective treatments as quickly as possible. We urge his immediate confirmation,” a coalitions of 50 medical and advocacy groups, including the American Academy of Pediatrics, American Society of Clinical Oncology, American Association of Cancer Research, National Organization for Rare Disorders, and the Personalized Medicine Coalition, said in an Oct. 29, 2015, letter to the Senate HELP Committee.
WASHINGTON – Drug pricing was front and center as a Senate panel convened to review the nomination of Dr. Robert M. Califf as commissioner of the Food and Drug Administration.
During a Nov. 17 hearing of the Senate Health, Education, Labor, and Pensions (HELP) Committee, many members keyed in on the role of the FDA in trying to bring down drug prices.
When asked about the price of prescription drugs, Dr. Califf was clear in stating that the agency’s role was not to set prices, but suggested that the FDA could have an impact on pricing by making the approval process for both brand and generic products more efficient, including revamping the clinical trials needed for approval to make them cheaper and more inclusive, while improving the quality of the data produced.
“We think that we can do trials that are actually bigger and include more patients that are more representative for a much lower cost,” Dr. Califf said, responding to Sen. Orrin Hatch’s (R-Utah) concerns about length of time of data exclusivity and fears that calls for shortening it might lead to higher drug prices as companies try to recoup development costs.
He also pointed to the use of electronic health records as a way to broaden clinical trials at a lower cost.
“The key here is using electronic health records that we already have,” Dr. Califf said, adding that once interoperability issues are addressed, EHRs will enable manufacturers to develop therapies “at a much lower cost with better information about safety and efficacy.”
In looking at rising generic prices, Committee Chairman Lamar Alexander (R-Tenn) focused on generic approval times, noting that generic manufacturers estimated that it took on average 48 months for a generic to get approval in 2014, up from 30 months in 2011.
A more rapid approval, “if safe and effective, might have some effect on lowering drug prices,” Sen. Alexander said.
Dr. Califf responded that FDA is already ahead of the generic user fee goals that have been set and the agency has been clearing the more simple applications from its backlog.
“But we have this backlog of applications that are requiring a back-and-forth because we want generic drugs to be just as safe and effective as the innovative drugs and when the applications [are not complete and there are questions] those get held up,” he responded, adding that he was “confident” that once the backlog is cleared over the next several years, generic applications will be processed quickly.
However, his answers were not convincing to at least one member. Presidential hopeful Sen. Bernie Sanders (I-Vt.) was not convinced Dr. Califf will do enough in this area.
“At a time when millions of Americans cannot afford to purchase the prescription drugs they need, we need a new leader at the FDA who is prepared to stand up to the pharmaceutical companies and work to substantially lower drug prices. Unfortunately, I have come to the conclusion that Dr. Califf is not that person,” Sanders said in an Oct. 9 statement.
Sen. Sanders reaffirmed his objection, saying that nothing he heard during the hearing has changed his opinions.
And while he openly objected to the nomination, both the chairman and ranking member of the committee both openly endorsed the White House nominee.
In his prepared remarks, Mr. Alexander noted that his staff “has spent 2 months carefully reviewing everything you submitted and has not found anything that would call into doubt your ability to lead the FDA fairly and impartially.”
Likewise, Ranking Member Patty Murray (D-Wash.) said during the hearing that after “careful consideration and review, I am confident that Dr. Califf would contribute leadership and expertise as we work to find new ways to advance medical innovation for patients and families, and improve the health and well-being across this country. He is a strong nominee for the role of FDA commissioner.”
In his opening statement, Dr. Califf outlined his priorities if confirmed as commissioner, including strengthening and better supporting FDA’s workforce, finishing work in an number of areas – including improving the nation’s food supply, tobacco regulation, better combating antibiotic resistant bacteria, medical countermeasure development, and the White House’s Precision Medicine Initiative – and “further develop the science base that informs FDA’s decision making.”
During his testimony, he also said there is “a need to work on postmarket surveillance of devices,” suggesting that a similar program, the FDA’s Sentinel Initiative drug surveillance program, be developed. Dr. Califf also said a new pathway is needed for approving drug/device combination products.
In the area of medical apps, Dr. Califf said there needs to be more clarity as to where the line for regulation should be. As an example, he said that an app that monitors heart rate does not need much oversight, but if that app were connected to an implantable defibrillator, that would require more regulatory oversight.
Sen. Sanders cited Dr. Califf’s ties to the pharmaceutical industry as the prime reason for opposing the nomination.
Dr. Califf noted that for any of the research he had conducted, even when primarily funded from industry, he was clear that manufacturers, while being able to make suggestions for analysis and publication of results, did not have final say, and he said that numerous studies were rejected because companies would not agree to full access to databases or independence with the final publication of results.
One area that was not raised during the hearing, but was raised by the Project on Government Oversight regarding the ROCKET AF trial of rivaroxaban (Xarelto), for which he was co-primary investigator (N Engl J Med. 2011 Sep 8;365:883-91).
“Dr. Califf’s handling of the Xarelto trial raises concerns about his judgment in overseeing the pharmaceutical industry.” POGO Executive Director Danielle Brian said in a statement. “As senators consider Dr. Califf’s confirmation to run the FDA, they should ask tough questions about the Xarelto clinical trial.”
FDA primary reviewers recommended against approval based on a number of issues with the trial, but FDA ultimately decided in favor of bringing the drug to market.
A cardiologist by training, Dr. Califf has received support from the American College of Cardiology.
In an Oct. 21, 2015, letter to the Senate HELP Committee, ACC called Dr. Califf “the right person to lead the FDA as commissioner based on his impressive medical knowledge, clinical research experience, and visionary leadership abilities.”
Dr. Califf joined the FDA in February as deputy commissioner of medical products and currently provides executive leadership to FDA’s Center for Drug Evaluation and Research, the Center for Biologics Evaluation and Research, the Center for Devices and Radiological Health, and the Center for Tobacco Products.
He also oversees FDA’s Office of Special Medical Programs and plays a critical role in providing high-level advice and policy direction on the agency’s medical product and tobacco priorities.
Prior to coming to FDA, he was vice chancellor of clinical and translational research and professor of medicine in the division of cardiology at Duke University in Durham, N.C. He has held multiple positions at the university, including director of the Duke Translational Medicine Institute and founding director of the Duke Clinical Research Institute.
He also served as a member of FDA’s Cardiovascular and Renal Drugs Advisory Committee and the agency’s Science Board Subcommittee on Science and Technology.
“We believe that with Dr. Califf’s diverse background, and his exemplary knowledge of clinical and translational medicine, he will continue to improve the FDA’s drug approval process while ensuring that patients are receiving the safest and most effective treatments as quickly as possible. We urge his immediate confirmation,” a coalitions of 50 medical and advocacy groups, including the American Academy of Pediatrics, American Society of Clinical Oncology, American Association of Cancer Research, National Organization for Rare Disorders, and the Personalized Medicine Coalition, said in an Oct. 29, 2015, letter to the Senate HELP Committee.
WASHINGTON – Drug pricing was front and center as a Senate panel convened to review the nomination of Dr. Robert M. Califf as commissioner of the Food and Drug Administration.
During a Nov. 17 hearing of the Senate Health, Education, Labor, and Pensions (HELP) Committee, many members keyed in on the role of the FDA in trying to bring down drug prices.
When asked about the price of prescription drugs, Dr. Califf was clear in stating that the agency’s role was not to set prices, but suggested that the FDA could have an impact on pricing by making the approval process for both brand and generic products more efficient, including revamping the clinical trials needed for approval to make them cheaper and more inclusive, while improving the quality of the data produced.
“We think that we can do trials that are actually bigger and include more patients that are more representative for a much lower cost,” Dr. Califf said, responding to Sen. Orrin Hatch’s (R-Utah) concerns about length of time of data exclusivity and fears that calls for shortening it might lead to higher drug prices as companies try to recoup development costs.
He also pointed to the use of electronic health records as a way to broaden clinical trials at a lower cost.
“The key here is using electronic health records that we already have,” Dr. Califf said, adding that once interoperability issues are addressed, EHRs will enable manufacturers to develop therapies “at a much lower cost with better information about safety and efficacy.”
In looking at rising generic prices, Committee Chairman Lamar Alexander (R-Tenn) focused on generic approval times, noting that generic manufacturers estimated that it took on average 48 months for a generic to get approval in 2014, up from 30 months in 2011.
A more rapid approval, “if safe and effective, might have some effect on lowering drug prices,” Sen. Alexander said.
Dr. Califf responded that FDA is already ahead of the generic user fee goals that have been set and the agency has been clearing the more simple applications from its backlog.
“But we have this backlog of applications that are requiring a back-and-forth because we want generic drugs to be just as safe and effective as the innovative drugs and when the applications [are not complete and there are questions] those get held up,” he responded, adding that he was “confident” that once the backlog is cleared over the next several years, generic applications will be processed quickly.
However, his answers were not convincing to at least one member. Presidential hopeful Sen. Bernie Sanders (I-Vt.) was not convinced Dr. Califf will do enough in this area.
“At a time when millions of Americans cannot afford to purchase the prescription drugs they need, we need a new leader at the FDA who is prepared to stand up to the pharmaceutical companies and work to substantially lower drug prices. Unfortunately, I have come to the conclusion that Dr. Califf is not that person,” Sanders said in an Oct. 9 statement.
Sen. Sanders reaffirmed his objection, saying that nothing he heard during the hearing has changed his opinions.
And while he openly objected to the nomination, both the chairman and ranking member of the committee both openly endorsed the White House nominee.
In his prepared remarks, Mr. Alexander noted that his staff “has spent 2 months carefully reviewing everything you submitted and has not found anything that would call into doubt your ability to lead the FDA fairly and impartially.”
Likewise, Ranking Member Patty Murray (D-Wash.) said during the hearing that after “careful consideration and review, I am confident that Dr. Califf would contribute leadership and expertise as we work to find new ways to advance medical innovation for patients and families, and improve the health and well-being across this country. He is a strong nominee for the role of FDA commissioner.”
In his opening statement, Dr. Califf outlined his priorities if confirmed as commissioner, including strengthening and better supporting FDA’s workforce, finishing work in an number of areas – including improving the nation’s food supply, tobacco regulation, better combating antibiotic resistant bacteria, medical countermeasure development, and the White House’s Precision Medicine Initiative – and “further develop the science base that informs FDA’s decision making.”
During his testimony, he also said there is “a need to work on postmarket surveillance of devices,” suggesting that a similar program, the FDA’s Sentinel Initiative drug surveillance program, be developed. Dr. Califf also said a new pathway is needed for approving drug/device combination products.
In the area of medical apps, Dr. Califf said there needs to be more clarity as to where the line for regulation should be. As an example, he said that an app that monitors heart rate does not need much oversight, but if that app were connected to an implantable defibrillator, that would require more regulatory oversight.
Sen. Sanders cited Dr. Califf’s ties to the pharmaceutical industry as the prime reason for opposing the nomination.
Dr. Califf noted that for any of the research he had conducted, even when primarily funded from industry, he was clear that manufacturers, while being able to make suggestions for analysis and publication of results, did not have final say, and he said that numerous studies were rejected because companies would not agree to full access to databases or independence with the final publication of results.
One area that was not raised during the hearing, but was raised by the Project on Government Oversight regarding the ROCKET AF trial of rivaroxaban (Xarelto), for which he was co-primary investigator (N Engl J Med. 2011 Sep 8;365:883-91).
“Dr. Califf’s handling of the Xarelto trial raises concerns about his judgment in overseeing the pharmaceutical industry.” POGO Executive Director Danielle Brian said in a statement. “As senators consider Dr. Califf’s confirmation to run the FDA, they should ask tough questions about the Xarelto clinical trial.”
FDA primary reviewers recommended against approval based on a number of issues with the trial, but FDA ultimately decided in favor of bringing the drug to market.
A cardiologist by training, Dr. Califf has received support from the American College of Cardiology.
In an Oct. 21, 2015, letter to the Senate HELP Committee, ACC called Dr. Califf “the right person to lead the FDA as commissioner based on his impressive medical knowledge, clinical research experience, and visionary leadership abilities.”
Dr. Califf joined the FDA in February as deputy commissioner of medical products and currently provides executive leadership to FDA’s Center for Drug Evaluation and Research, the Center for Biologics Evaluation and Research, the Center for Devices and Radiological Health, and the Center for Tobacco Products.
He also oversees FDA’s Office of Special Medical Programs and plays a critical role in providing high-level advice and policy direction on the agency’s medical product and tobacco priorities.
Prior to coming to FDA, he was vice chancellor of clinical and translational research and professor of medicine in the division of cardiology at Duke University in Durham, N.C. He has held multiple positions at the university, including director of the Duke Translational Medicine Institute and founding director of the Duke Clinical Research Institute.
He also served as a member of FDA’s Cardiovascular and Renal Drugs Advisory Committee and the agency’s Science Board Subcommittee on Science and Technology.
“We believe that with Dr. Califf’s diverse background, and his exemplary knowledge of clinical and translational medicine, he will continue to improve the FDA’s drug approval process while ensuring that patients are receiving the safest and most effective treatments as quickly as possible. We urge his immediate confirmation,” a coalitions of 50 medical and advocacy groups, including the American Academy of Pediatrics, American Society of Clinical Oncology, American Association of Cancer Research, National Organization for Rare Disorders, and the Personalized Medicine Coalition, said in an Oct. 29, 2015, letter to the Senate HELP Committee.
AT THE SENATE HELP COMMITTEE HEARING

VIDEO: Survival benefits, relapse risks in liver transplant for alcoholic hepatitis
SAN FRANCISCO – Are policies requiring 6 months of abstinence before liver transplantation in severe alcoholic hepatitis justified, given the potential survival advantage that earlier transplantation offers?
Alcoholic hepatitis has an “exceptionally high mortality rate,” noted Dr. Brian Lee of Johns Hopkins University, Baltimore. But a policy requiring 6 months of abstinence before liver transplantation in patients with severe alcoholic hepatitis “is possibly even causing a precondition that’s death for these patients.”
In an interview at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Lee discussed a study in which early liver transplantation in 40 patients with severe alcoholic hepatitis achieved a 100% survival rate at 1 year, with a 22% alcohol relapse rate.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Are policies requiring 6 months of abstinence before liver transplantation in severe alcoholic hepatitis justified, given the potential survival advantage that earlier transplantation offers?
Alcoholic hepatitis has an “exceptionally high mortality rate,” noted Dr. Brian Lee of Johns Hopkins University, Baltimore. But a policy requiring 6 months of abstinence before liver transplantation in patients with severe alcoholic hepatitis “is possibly even causing a precondition that’s death for these patients.”
In an interview at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Lee discussed a study in which early liver transplantation in 40 patients with severe alcoholic hepatitis achieved a 100% survival rate at 1 year, with a 22% alcohol relapse rate.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Are policies requiring 6 months of abstinence before liver transplantation in severe alcoholic hepatitis justified, given the potential survival advantage that earlier transplantation offers?
Alcoholic hepatitis has an “exceptionally high mortality rate,” noted Dr. Brian Lee of Johns Hopkins University, Baltimore. But a policy requiring 6 months of abstinence before liver transplantation in patients with severe alcoholic hepatitis “is possibly even causing a precondition that’s death for these patients.”
In an interview at the annual meeting of the American Association for the Study of Liver Diseases, Dr. Lee discussed a study in which early liver transplantation in 40 patients with severe alcoholic hepatitis achieved a 100% survival rate at 1 year, with a 22% alcohol relapse rate.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE LIVER MEETING 2015

Complications climb with revisional surgery after adjustable gastric banding
LOS ANGELES – Revisional surgery after failed adjustable gastric banding (AGB) is associated with an increased risk of adverse events and resource utilization, according to Dr. Steven Poplawski, medical director for Barix Clinics in Ypsilanti, Mich.
His conclusion is based on safety outcomes at 30 days for 55,237 patients who underwent primary bariatric surgery and 1,417 patients who underwent AGB revision between June 2006 and July 2015 in the Michigan Bariatric Surgery Collaborative. Patients were excluded from the retrospective evaluation if they had urgent/emergent procedures or more than one previous bariatric operation.

The primary bariatric surgery was Roux-en-Y gastric bypass (RYGB) in 43%, sleeve gastrectomy in 37%, AGB in 19%, and biliopancreatic diversion with duodenal switch (BPD/DS) in 1%.
The patients turned to AGB revision for the same reasons that have prompted its dramatic decline in utilization: weight loss failure (38%), band complications (33%), or both (29%). AGB revisional procedures were sleeve gastrectomy in 54%, RYGB in 35%, AGB in 9%, and BPD/DS in 2%.
Patients undergoing band-to-RYGB conversions had significantly more serious complications, compared with primary RYGB procedures (10.2% vs. 4.4%; P less than .0001), reoperations (5.1% vs. 2.3%; P = .0001), and hospital readmissions (10% vs. 7.3%; P = .0064), and a nonsignificant trend toward more leaks (1.3% vs. 0.7%; P = .13), Dr. Poplawski reported.
Patients undergoing band-to-sleeve conversions had significantly more serious complications when compared with primary sleeve gastrectomy (5% vs. 1.8%; P less than .0001), reoperations (3.3% vs. 1%; P less than .0001), and leaks (1.5% vs. 0.5%; P = .0001), and a nonsignificant trend toward more readmissions (7.7% vs. 4.6%; P = .0685).
Outcomes were not reported for the smaller number of patients undergoing AGB-to-AGB or AGB-to-BPD/DS conversions.
A secondary analysis was performed examining a one-stage versus a two-stage procedure in 525 patients undergoing revisional surgery for weight loss failure only. The only safety outcome to show a significant difference at 30 days was hospital readmissions in the RYGB-conversion group, favoring the one-stage over the two-stage procedure (7.1% vs. 11.5%; P = .0164).
“Clearly, the benefit of a one-stage procedure versus a two-stage procedure is unclear in the way it was studied here,” Dr. Poplawski said at Obesity Week 2015, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
“You can read in some reports that [surgeons] do all [of these procedures as] two-stage because they think it’s safer, but I don’t know that there’s much support for that here. I think it’s reasonable to get most of them done in one stage because there’s also two hospitalizations, two periods of convalescence, and when we talk about the complications of the two-stage operation we aren’t even including the costs to remove the initial band, which are not insignificant,” he noted.
Dr. Raul Rosenthal of the Cleveland Clinic in Weston, Fla., who comoderated the session, said the takeaway message is that “reoperative surgery pays a price. No matter how you look at it, one stage, two stages, with bands or sleeve, you’re going to get in trouble.”
LOS ANGELES – Revisional surgery after failed adjustable gastric banding (AGB) is associated with an increased risk of adverse events and resource utilization, according to Dr. Steven Poplawski, medical director for Barix Clinics in Ypsilanti, Mich.
His conclusion is based on safety outcomes at 30 days for 55,237 patients who underwent primary bariatric surgery and 1,417 patients who underwent AGB revision between June 2006 and July 2015 in the Michigan Bariatric Surgery Collaborative. Patients were excluded from the retrospective evaluation if they had urgent/emergent procedures or more than one previous bariatric operation.
The primary bariatric surgery was Roux-en-Y gastric bypass (RYGB) in 43%, sleeve gastrectomy in 37%, AGB in 19%, and biliopancreatic diversion with duodenal switch (BPD/DS) in 1%.
The patients turned to AGB revision for the same reasons that have prompted its dramatic decline in utilization: weight loss failure (38%), band complications (33%), or both (29%). AGB revisional procedures were sleeve gastrectomy in 54%, RYGB in 35%, AGB in 9%, and BPD/DS in 2%.
Patients undergoing band-to-RYGB conversions had significantly more serious complications, compared with primary RYGB procedures (10.2% vs. 4.4%; P less than .0001), reoperations (5.1% vs. 2.3%; P = .0001), and hospital readmissions (10% vs. 7.3%; P = .0064), and a nonsignificant trend toward more leaks (1.3% vs. 0.7%; P = .13), Dr. Poplawski reported.
Patients undergoing band-to-sleeve conversions had significantly more serious complications when compared with primary sleeve gastrectomy (5% vs. 1.8%; P less than .0001), reoperations (3.3% vs. 1%; P less than .0001), and leaks (1.5% vs. 0.5%; P = .0001), and a nonsignificant trend toward more readmissions (7.7% vs. 4.6%; P = .0685).
Outcomes were not reported for the smaller number of patients undergoing AGB-to-AGB or AGB-to-BPD/DS conversions.
A secondary analysis was performed examining a one-stage versus a two-stage procedure in 525 patients undergoing revisional surgery for weight loss failure only. The only safety outcome to show a significant difference at 30 days was hospital readmissions in the RYGB-conversion group, favoring the one-stage over the two-stage procedure (7.1% vs. 11.5%; P = .0164).
“Clearly, the benefit of a one-stage procedure versus a two-stage procedure is unclear in the way it was studied here,” Dr. Poplawski said at Obesity Week 2015, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
“You can read in some reports that [surgeons] do all [of these procedures as] two-stage because they think it’s safer, but I don’t know that there’s much support for that here. I think it’s reasonable to get most of them done in one stage because there’s also two hospitalizations, two periods of convalescence, and when we talk about the complications of the two-stage operation we aren’t even including the costs to remove the initial band, which are not insignificant,” he noted.
Dr. Raul Rosenthal of the Cleveland Clinic in Weston, Fla., who comoderated the session, said the takeaway message is that “reoperative surgery pays a price. No matter how you look at it, one stage, two stages, with bands or sleeve, you’re going to get in trouble.”
LOS ANGELES – Revisional surgery after failed adjustable gastric banding (AGB) is associated with an increased risk of adverse events and resource utilization, according to Dr. Steven Poplawski, medical director for Barix Clinics in Ypsilanti, Mich.
His conclusion is based on safety outcomes at 30 days for 55,237 patients who underwent primary bariatric surgery and 1,417 patients who underwent AGB revision between June 2006 and July 2015 in the Michigan Bariatric Surgery Collaborative. Patients were excluded from the retrospective evaluation if they had urgent/emergent procedures or more than one previous bariatric operation.
The primary bariatric surgery was Roux-en-Y gastric bypass (RYGB) in 43%, sleeve gastrectomy in 37%, AGB in 19%, and biliopancreatic diversion with duodenal switch (BPD/DS) in 1%.
The patients turned to AGB revision for the same reasons that have prompted its dramatic decline in utilization: weight loss failure (38%), band complications (33%), or both (29%). AGB revisional procedures were sleeve gastrectomy in 54%, RYGB in 35%, AGB in 9%, and BPD/DS in 2%.
Patients undergoing band-to-RYGB conversions had significantly more serious complications, compared with primary RYGB procedures (10.2% vs. 4.4%; P less than .0001), reoperations (5.1% vs. 2.3%; P = .0001), and hospital readmissions (10% vs. 7.3%; P = .0064), and a nonsignificant trend toward more leaks (1.3% vs. 0.7%; P = .13), Dr. Poplawski reported.
Patients undergoing band-to-sleeve conversions had significantly more serious complications when compared with primary sleeve gastrectomy (5% vs. 1.8%; P less than .0001), reoperations (3.3% vs. 1%; P less than .0001), and leaks (1.5% vs. 0.5%; P = .0001), and a nonsignificant trend toward more readmissions (7.7% vs. 4.6%; P = .0685).
Outcomes were not reported for the smaller number of patients undergoing AGB-to-AGB or AGB-to-BPD/DS conversions.
A secondary analysis was performed examining a one-stage versus a two-stage procedure in 525 patients undergoing revisional surgery for weight loss failure only. The only safety outcome to show a significant difference at 30 days was hospital readmissions in the RYGB-conversion group, favoring the one-stage over the two-stage procedure (7.1% vs. 11.5%; P = .0164).
“Clearly, the benefit of a one-stage procedure versus a two-stage procedure is unclear in the way it was studied here,” Dr. Poplawski said at Obesity Week 2015, presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
“You can read in some reports that [surgeons] do all [of these procedures as] two-stage because they think it’s safer, but I don’t know that there’s much support for that here. I think it’s reasonable to get most of them done in one stage because there’s also two hospitalizations, two periods of convalescence, and when we talk about the complications of the two-stage operation we aren’t even including the costs to remove the initial band, which are not insignificant,” he noted.
Dr. Raul Rosenthal of the Cleveland Clinic in Weston, Fla., who comoderated the session, said the takeaway message is that “reoperative surgery pays a price. No matter how you look at it, one stage, two stages, with bands or sleeve, you’re going to get in trouble.”
AT OBESITY WEEK 2015
Key clinical point: Conversions of adjustable gastric bands are associated with more 30-day adverse events.
Major finding: Serious complications were higher in AGB–to–Roux-en-Y gastric bypass conversions than in primary RYGB (10.2% vs. 4.4%; P less than .0001) and in AGB-to-sleeve gastrectomy conversions, compared with primary sleeves (5% vs. 1.8%; P less than .0001).
Data source: Retrospective study of 55,237 patients who underwent primary bariatric surgery and 1,417 patients who underwent adjustable gastric band revision.
Disclosures: Dr. Poplawski reported having no disclosures.
