User login
Chemists discover true structure of anticancer agent

Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Credit: NIH
Chemists say they have determined the correct structure of a compound that has shown activity against lymphoma and a range of other cancers.
Their research, published in Angewandte Chemie, focused on a compound called TIC10.
The team showed that TIC10’s structure differs subtly from a version described by another group last year, and the previous structure associated with TIC10 actually describes a molecule that lacks TIC10’s anticancer activity.
The newly identified structure describes a molecule with potent anticancer effects in animals, representing a new family of biologically active structures that can now be explored for possible therapeutic uses.
“This new structure should generate much interest in the cancer research community,” said study author Kim D. Janda, PhD, of The Scripps Research Institute in La Jolla, California.
Antitumor potential
TIC10 was first described in Science Translational Medicine in early 2013. The authors identified the compound, within a library of thousands of molecules maintained by the National Cancer Institute (NCI), for its ability to boost cells’ production of the natural antitumor protein TRAIL. (TIC10 stands for TRAIL-inducing compound #10.)
As a small molecule, TIC10 would be easier to deliver in a therapy than the TRAIL protein itself. The paper’s authors reported that TIC10 was orally active and dramatically shrank a variety of tumors in mice.
Tumors can develop resistance to TRAIL, but Dr Janda had been studying compounds that defeat this resistance. The news about TIC10 therefore got his attention.
“I thought, ‘They have this molecule for upregulating TRAIL, and we have these molecules that can overcome tumor-cell TRAIL resistance—the combination could be important,’” he said.
The original publication on TIC10 included a figure showing its predicted structure. So Dr Janda asked one of his postdoctoral researchers, Jonathan Lockner, to make TIC10 using that information.
Although the original TIC10 research team had seemingly confirmed the predicted structure with mass spectrometry, no one had published a thorough characterization of the TIC10 molecule.
“There were no nuclear magnetic resonance data or X-ray crystallography data, and there was definitely no procedure for the synthesis,” Dr Lockner said. “My background was chemistry, though, so I was able to find a way to synthesize it starting from simple compounds.”
Surprising inactivity
There was just one problem with Dr Lockner’s newly synthesized “TIC10.” When tested, it failed to induce TRAIL expression in cells, even at high doses.
“Of course, I was nervous,” Dr Lockner said. “As a chemist, you never want to make a mistake and give biologists the wrong material.”
To try and verify they had the right material, Dr Janda’s team obtained a sample of TIC10 directly from the NCI.
“When we got that sample and tested it, we saw that it had the expected TRAIL-upregulating effect,” said Nicholas Jacob, a graduate student in the Janda Lab and coauthor of the new paper.
“That prompted us to look more closely at the structures of these 2 compounds.”
The researchers spent months characterizing their own synthesized material and the NCI material, using an array of sophisticated structural analysis tools. They also tested the 2 compounds’ biological effects.
The team eventually concluded that the TIC10 compound from the NCI library does boost TRAIL production in cells and remains promising as the basis for anticancer therapies, but it does not have the structure that was originally published.
The right structure
The originally published structure has a core made of 3 carbon-nitrogen rings in a straight line and does not induce TRAIL activity. The correct, TRAIL-inducing structure differs subtly, with an end ring that sticks out at an angle.
In chemists’ parlance, the 2 compounds are constitutional isomers: a linear imidazolinopyrimidinone and an angular imidazolinopyrimidinone.
And Dr Lockner found that the angular, TRAIL-inducing structure was easier to synthesize than the one originally described.
Now, with the correct molecule in hand and a solid understanding of its structure and synthesis, Dr Janda and his team are moving forward with their original plan to study TIC10 in combination with TRAIL-resistance-thwarting molecules as an anticancer therapy.
The therapeutic implications of TIC10 may even go beyond cancer, according to the researchers. The angular core of the TRAIL-inducing molecule Dr Janda’s team discovered is a novel type of a biologically active structure, or pharmacophore, from which chemists may now be able to build a new class of candidate drugs, possibly for a variety of ailments. ![]()
Drug gains orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.” ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.” ![]()
The US Food and Drug Administration (FDA) has granted orphan designation to selinexor (KPT-330) for the treatment of diffuse large B-cell lymphoma (DLBCL).
The drug elicited responses in patients with non-Hodgkin lymphoma (NHL), including DLBCL, in an ongoing phase 1 study.
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1). This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation for DLBCL qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
The FDA has also granted selinexor orphan status for the treatment of acute myeloid leukemia.
Phase 1 study
Researchers evaluated selinexor in an ongoing phase 1 study of patients with NHL or chronic lymphocytic leukemia (CLL) and presented results at the 2013 ASH Annual Meeting (abstract 90).
At that time, the study included 18 patients with NHL or CLL. They had a median age of 66.5 years and had received a median of 4.5 prior treatment regimens.
Patients received selinexor at 6 different dose levels. There were no clinically significant cumulative toxicities or cases of major organ dysfunction, and the maximum-tolerated dose was not reached. Researchers continued dosing at 35 mg/m2 twice weekly.
Ten patients experienced drug-related grade 3/4 adverse events, including thrombocytopenia without bleeding (n=6), neutropenia (n=5), dehydration (n=1), syncope (n=1), hypotension (n=1), and fatigue (n=1).
The most common grade 1/2 events were anorexia (n=10), fatigue (n=9), diarrhea (n=6), vomiting (n=6), neutropenia (n=5), malaise (n=3), anemia (n=3), and weight loss (n=3).
Response was evaluable in 15 patients. Eighty percent of patients, all of whom had progressive disease on study entry, experienced tumor shrinkage or disease stabilization on selinexor. The other 20% of patients progressed.
Of 3 patients with DLBCL, 1 progressed, 1 had stable disease, and 1 achieved 93% tumor shrinkage.
“We are encouraged by the response data in patients with DLBCL who have received selinexor in our ongoing phase 1 clinical trial in advanced hematological malignancies,” said Michael G. Kauffman, MD, PhD, Karyopharm’s Chief Executive Officer.
“We plan to present updated clinical data for selinexor across multiple indications, including DLBCL, at ASCO 2014.” ![]()
Cancer trial publications often omit minority accrual rates

Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology. ![]()
Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology. ![]()
Credit: Rhoda Baer
A review of clinical trial data from 2012 suggests Hispanic patients are underrepresented in US cancer studies, and many trial publications fail to provide information on patients’ racial/ethnic backgrounds.
Researchers analyzed 159 reports of phase 2 and 3 trials and found that roughly 21% included information on the number of minority patients enrolled.
About 8% of the publications included data on the number of Hispanic patients enrolled.
And from this data, the investigators found the accrual rate for Hispanic patients was about 4%.
According to the researchers, this lack of information and low representation inhibits physicians’ ability to provide optimal treatment to Hispanic cancer patients and patients belonging to other minority groups.
“We have a major responsibility to ensure adequate representation,” said study author Ian M. Thompson Jr, MD, of The University of Texas Health Science Center at San Antonio.
“How else will we know how best to treat our patients, and how else are we going to reduce the health disparities in [the Hispanic] population?”
Dr Thompson and his colleagues have a particular interest in the Hispanic population because 58% of San Antonio residents are Hispanic, as are 68% of residents in South Texas as a whole.
So the investigators wanted to determine Hispanic accrual rates in randomized trials of cancer patients. The team evaluated data from phase 2 and 3 cancer trials published in 2012. They focused on studies that were considered likely to change the standard of care and were published in “high-impact” journals.
The researchers identified 159 trials—68 phase 2 studies and 91 phase 3 studies. They discovered that 33 of the trial publications—about 21%—disclosed data on minority accrual. And 13 publications—about 8%—included data on the accrual of Hispanic cancer patients.
Of the 4154 patients enrolled on those 13 trials, 162 were Hispanic, which translates to an overall accrual rate of 3.9%. The enrollment of Hispanic patients ranged from 1 patient (0.5%) in a phase 2 trial of lung cancer to 17 patients (26%) in a phase 2 study of acute lymphoblastic leukemia.
“Fundamentally, in the most recent published cancer clinical trials, either the number and proportion of Hispanics are not reported or are far below their actual representation in the national population,” Dr Thompson summarized.
“For institutions like ours that serve a ‘minority-majority’ population, it’s a major responsibility for us to ensure adequate representation so that we can tell our patients how they can best be treated and how we can reduce the disparities of this rapidly growing population.”
Dr Thompson and his colleagues described this research in the Journal of Clinical Oncology. ![]()
mAb gets breakthrough designation for MM

Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression. ![]()
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression. ![]()
Credit: Linda Bartlett
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for elotuzumab, a humanized monoclonal antibody
(mAb).
The designation is for elotuzumab used in combination with lenalidomide and dexamethasone to treat patients with multiple myeloma (MM) who have received 1 or more prior therapies.
The FDA’s decision is based on findings from a phase 2 trial in which MM patients received that treatment combination.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions. For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
About elotuzumab
Elotuzumab is a humanized IgG1 mAb targeting signaling lymphocyte activation molecule (SLAMF7, also known as CS1), a glycoprotein expressed on myeloma and natural killer cells but not detectable in normal tissue.
Researchers are investigating whether, through both direct activation and engagement of natural killer cells, elotuzumab may selectively target and kill SLAMF7-expressing myeloma cells.
Elotuzumab is under investigation as a monotherapy in smoldering myeloma and in combination with other therapies in first-line and relapsed or refractory MM.
A clinical development program for the mAb is underway, including phase 3 trials in first-line MM (ELOQUENT-1) and relapsed or refractory MM (ELOQUENT-2). The agent is also under investigation in a randomized, phase 2 study of bortezomib and dexamethasone in patients with relapsed or refractory MM.
Elotuzumab is under development by AbbVie and Bristol-Myers Squibb.
Phase 2 trial results
The breakthrough therapy designation for elotuzumab is based on results of a randomized, phase 2 trial presented at the EHA 2013 Annual Congress (abstract 14). Study investigators tested 2 doses of the mAb in combination with lenalidomide and low-dose dexamethasone in patients with previously treated MM.
Patients were randomized 1:1 to receive elotuzumab at 10 mg/kg or 20 mg/kg (intravenous infusion on days 1, 8, 15, and 22 of a 28-day cycle in the first 2 cycles and then days 1 and 15 of subsequent cycles) in combination with oral lenalidomide at 25 mg/day on days 1 to 21 and oral dexamethasone at 40 mg/week. Patients were treated until their disease progressed or they developed unacceptable toxicity.
In the 10 mg/kg arm (n=36), which is the dose used in the ongoing phase 3 trials, the median progression-free survival was 33 months, after a median follow-up of 20.8 months. And the objective response rate was 92%.
In the 20 mg/kg arm (n=37), the median progression-free survival was 18 months, after a median follow-up of 17.1 months. And the objective response rate was 76%.
The safety data were consistent with previously reported results for elotuzumab from this trial. In patients receiving elotuzumab at 10 mg/kg or 20 mg/kg, most treatment-emergent adverse events occurred within 18 months of starting therapy.
The most common grade 3/4 adverse events for the 10 mg/kg and 20 mg/kg arms, respectively, were lymphopenia (26% and 9%), neutropenia (21% and 22%), thrombocytopenia (21% and 17%), anemia (13% and 12%), leukopenia (8% and 7%), hyperglycemia (5% and 12%), pneumonia (8% and 5%), diarrhea (10% and 5%), fatigue (8% and 9%), and hypokalemia (8% and 5%).
Two deaths occurred on study. One patient died of pneumonia, multiple organ failure, and sepsis. The other died of disease progression. ![]()
Team reports way to reduce sickling, SCD progression

Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.” ![]()
Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.” ![]()
Credit: Graham Colm
Through a series of preclinical experiments, scientists discovered they could reduce the sickling of red blood cells (RBCs) and slow the progression of sickle cell disease (SCD).
In a mouse model of SCD and blood samples from SCD patients, the researchers reduced sickling by manipulating sphingosine-1-phosphate (S1P) and sphingosine kinase 1 (SPHK1).
Yang Xia, MD, PhD, of The University of Texas Health Science Center at Houston, and colleagues described this work in The Journal of Clinical Investigation.
The scientists first discovered that S1P, a lipid enriched and stored in RBCs, is elevated in mice with SCD. Further investigation revealed that elevated SPHK1 activity underlies the increased levels of S1P and contributes to RBC sickling.
To confirm SPHK1’s role in SCD, the researchers tested 2 SPHK1 inhibitors, SK1-I and PF-543, in cells from mice with SCD. Both agents successfully inhibited SPHK1 activity and reduced S1P levels in a dose-dependent manner, but PF-543 demonstrated greater potency.
In subsequent experiments, PF-543 decreased intravascular hemolysis and reduced inflammation in mice with SCD. The treatment also decreased tissue injury and splenomegaly and increased survival in the mice.
When the scientists knocked down SPHK1 in hematopoietic stem cells (HSCs), they observed decreased sickling and reticulocytes in SCD chimeras, resulting from a reduction in S1P levels.
Knocking down SPHK1 in HSCs also decreased intravascular hemolysis, prolonged RBC life span, reduced inflammation, decreased splenomegaly and tissue injury, and increased survival in the mice.
Experiments in cells from patients with SCD showed that SPHK1 activity and S1P levels were elevated and directly contributed to sickling. So the researchers decided to evaluate how PF-543 would affect these cells.
Treating cells with PF-543 significantly inhibited hypoxia-induced SPHK1 activity and prevented the elevation of S1P in a dose-dependent manner. The treatment also significantly reduced the percentage of sickled cells in a dose-dependent manner.
Finally, the scientists found that S1P-induced sickling was independent of S1P receptor activation. S1P receptor antagonists did not inhibit hypoxia-induced sickling in cells from SCD patients. And treatment with S1P did not enhance sickling under hypoxic conditions.
“This work could lead to novel treatments for sickle cell disease,” said study author Harinder Juneja, MD, of The University of Texas Health Science Center. “The study provides a better understanding of the pathogenesis of the disease and reveals a new therapeutic target.”
Coauthor Rod Kellems, PhD, also of The University of Texas Health Science Center, added, “This research provides insight into how red blood cells work, revealing that SPHK1-mediated elevation of S1P contributes to sickling and promotes disease progression and highlights potential therapeutic opportunities for sickle cell disease.” ![]()
Inhibitor gets breakthrough designation for HL
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the investigational PD-1 immune checkpoint inhibitor nivolumab to treat Hodgkin lymphoma (HL) in patients who have failed autologous stem cell transplant and treatment with brentuximab vedotin.
The FDA’s decision is based on data from a cohort of HL patients in an ongoing phase 1b study of patients with relapsed or refractory hematologic malignancies.
According to the FDA, breakthrough designation is intended to expedite the development and review of drugs for serious or life-threatening conditions.
For a treatment to receive this designation, there must be preliminary clinical evidence that demonstrates the drug may offer substantial improvement over currently available therapy on at least 1 clinically significant endpoint.
Nivolumab is an investigational agent that binds to the checkpoint receptor PD-1 expressed on activated T cells. Researchers are investigating whether, by blocking this pathway, nivolumab would enable the immune system to resume its ability to recognize, attack, and destroy cancer cells.
Nivolumab is under investigation in multiple tumor types as monotherapy or in combination with other therapies. There are 35 trials of the agent underway, in which more than 7000 patients have been enrolled.
The breakthrough designation for nivolumab in HL is based on results of a 2-part phase 1 study, which have not been made public.
The researchers planned to enroll 100 patients with relapsed or refractory hematologic malignancies on this study. For the dose-escalation portion, the team planned to treat successive cohorts of patients using a 6+3 escalation design.
Patients would receive 1 mg/kg or 3 mg/kg of intravenous nivolumab every 2 weeks (although the first dose would be followed by a 3-week evaluation period) for 2 years, with the potential for an additional year of therapy for patients who progress.
Then, the researchers would enroll 5 cohorts of 16 patients representing the following tumor sites: HL/primary mediastinal B-cell lymphoma, multiple myeloma, B-cell lymphoma, T-cell lymphoma, and chronic myelogenous leukemia.
These patients would receive nivolumab at the maximum-tolerated dose identified in the first part of the study.
A poster describing the study plan was presented at the 2013 ASCO Annual Meeting (abstract TPS3113). The study is funded by Bristol-Myers Squibb, the company developing nivolumab. ![]()
Method can help predict utility of tPA
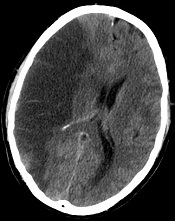
Credit: Lucien Monfils
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke. ![]()
Credit: Lucien Monfils
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke. ![]()
Credit: Lucien Monfils
Researchers say they’ve developed a technique that can predict—with 95% accuracy—which stroke patients will benefit from tissue-type plasminogen activator (tPA) and which will suffer from intracranial hemorrhage if treated.
The team devised a method that uses standard MRI scans to measure damage to the blood-brain barrier.
If further tests confirm the method’s accuracy, the researchers say it could allow for more precise use of intravenous tPA.
“If we are able to replicate our findings in more patients, it will indicate we are able to identify which people are likely to have bad outcomes, improving the drug’s safety and also potentially allowing us to give the drug to patients who currently go untreated,” said study author Richard Leigh, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
Dr Leigh and his colleagues described their technique in Stroke.
The group’s method is a computer program that lets physicians see how much gadolinium, the contrast material injected into a patient’s vein during an MRI scan, has leaked into the brain tissue from surrounding blood vessels.
By quantifying this damage in 75 stroke patients, the researchers identified a threshold for determining how much leakage was dangerous.
Then, they applied this threshold to those 75 records to determine how well it would predict who had suffered a brain hemorrhage and who had not. The test correctly predicted the outcome with 95% accuracy.
The researchers noted that, until now, physicians haven’t been able to predict with any precision which patients are likely to suffer a drug-related bleed and which are not. In these situations, if physicians knew the extent of the damage to the blood-brain barrier, they would be able to more safely administer treatment.
Typically, physicians do a CT scan of a stroke victim to see if he or she has visible bleeding before administering tPA. Dr Leigh said his computer program, which works with an MRI scan instead, can detect subtle changes to the blood-brain barrier that are otherwise impossible to see.
If his findings hold up, Dr Leigh said, “We should probably be doing MRI scans in every stroke patient before we give tPA.”
He conceded that an MRI scan does take longer to conduct in most institutions than a CT scan. But if the benefits of getting tPA into the right patients outweigh the harms of waiting a little longer to get MRI results, physicians should consider changing their practice, according to Dr Leigh.
“If we could eliminate all intracranial hemorrhages, it would be worth it,” he said.
Dr Leigh is now analyzing data from patients who received other treatments for stroke outside the typical time window, in some cases many hours after the FDA-approved cutoff for tPA. It’s possible, he said, that some patients who come to the hospital many hours after a stroke can still benefit from tPA, the only FDA-approved treatment for ischemic stroke.
Drug granted orphan designation for AML

The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan designation for the treatment of acute myeloid leukemia (AML).
Selinexor is a selective inhibitor of nuclear transport that functions by binding to the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which is thought to cause apoptosis in cancer cells while largely sparing normal cells.
The FDA grants orphan designation to promote the development of drugs that target conditions affecting 200,000 or fewer US patients annually and are expected to provide significant therapeutic advantage over existing treatments.
Selinexor’s orphan designation qualifies the drug’s developer, Karyopharm Therapeutics, Inc., for benefits that apply across all stages of development, including an accelerated approval process, 7 years of market exclusivity following marketing approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
Promising early results
Selinexor has shown promising results in a phase 1 trial of older patients with relapsed or refractory AML. The study, which was sponsored by Karyopharm, was published in Blood.
Researchers enrolled 16 patients with relapsed or refractory AML. The median age was 71 years, and the median number of prior therapeutic regimens was 2.
Patients received 8 to 10 doses of selinexor on a 4-week cycle across 2 dose levels, 16.8 mg/m2 to 23 mg/m2 (with additional cohorts ongoing).
The researchers reported no dose-limiting toxicity, no clinically significant cumulative toxicities, and no major organ dysfunction.
However, 4 patients experienced drug-related grade 3/4 adverse events, including hypotension (n=1), increased AST (n=1), hypokalemia (n=1), nausea (n=1), headache (n=1), and fatigue (n=1).
The most common grade 1/2 toxicities were nausea (9/17; 53%), anorexia (8/17; 47%), vomiting (6/17; 35%), fatigue (5/17; 29%), weight loss (5/17; 29%), and diarrhea (3/17; 18%). But these events were manageable.
Fourteen of the patients were evaluable for response. Two (14%) achieved a complete response with full hematologic recovery, and 2 (14%) achieved a complete response without hematologic recovery. Four patients (29%) had stable disease for more than 30 days, and 6 (43%) experienced progression.
Other trials of selinexor in AML
Karyopharm’s development plans for selinexor in AML include a number of additional studies.
In a phase 2 trial, researchers will evaluate selinexor monotherapy in older patients with AML. The study will enroll patients 60 years of age or older with relapsed or refractory AML who are ineligible for intensive chemotherapy and/or transplant.
In another study, researchers will evaluate selinexor in combination with decitabine for patients with relapsed, refractory, or newly diagnosed AML. The study will enroll up to 42 patients aged 60 or older who are ineligible for intensive chemotherapy.
Lastly, researchers are planning a study of selinexor in pediatric leukemia patients. The goal of this study is to determine the oral dosing, toxicity, and preliminary clinical activity of selinexor in pediatric patients. It will enroll up to 28 children with relapsed or refractory AML or acute lymphoblastic leukemia.
C difficile vaccine generates immune response
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013.
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013.
on a blood agar plate
Credit: CDC
BOSTON—Results of a phase 2 study suggest an investigational vaccine may be able to prevent Clostridium difficile infection (CDI).
The vaccine generated an immune response against C difficile toxins A and B. And adverse reactions were generally mild and of short duration.
Researchers presented these results at the 114th General Meeting of the American Society for Microbiology. The study was sponsored by Sanofi, the company developing the vaccine.
“C diff infection threatens the many people who frequently use antibiotics, as well as older hospitalized patients and residents in long-term healthcare facilities,” said study investigator Jamshid Saleh, MD of the Northern California Clinical Research Center in Redding, California.
“It would be great if we could offer patients a way to help prevent this contagious and debilitating disease versus just treating it after it happens.”
To find out if Sanofi’s vaccine would do the job, Dr Saleh and his colleagues conducted a randomized, multicenter trial split into 2 stages.
The first stage, conducted with 455 volunteers, was placebo-controlled, double-blind, and designed for dose and formulation selection. The second stage, which included 206 additional volunteers, was designed to compare the dose and formulation chosen in the first stage against 2 alternate dosing schedules.
Volunteers ranged in age from 40 to 75 years and were at risk of CDI due to impending hospitalization or residence in a long-term healthcare facility.
In stage 1, volunteers were randomized into 1 of 5 study groups: high-dose or low-dose vaccine, either with or without adjuvant, or placebo. Each formulation was administered on days 0, 7, and 30.
Researchers measured immune responses using both enzyme linked immunosorbent assay (ELISA), which assesses antitoxin A and B immunoglobulin G (IgG) concentrations, and toxin neutralization activity (TNA), which measures antitoxin A and B neutralizing activity.
Composite ELISA ranking analysis determined that the high-dose plus adjuvant vaccine formulation (group 3) generated the greatest immune response over a 60-day period. ELISA results also showed a 4-fold increase in the development of detectable antibodies for both toxins A and B.
So the researchers selected the high-dose plus adjuvant vaccine formulation for further study in stage 2 of the trial. They compared its use across 3 schedules: days 0, 7, and 30 (group 3, n=101); days 0, 7, and 180 (group 6, n=103); and days 0, 30, and 180 (group 7, n=103). The team analyzed subjects on days 0, 7, 14, 30, 60, 180, and 210.
There were increased immune responses in all vaccine groups and with each dose, according to ELISA and TNA. Overall, group 3 demonstrated the most favorable immune profile over the 30-, 60- and 180-day periods, particularly in volunteers aged 65-75 years.
The safety profile of all vaccine doses was deemed acceptable throughout the study. Reactions were monitored until day 210 and were generally grade 1, of short duration, did not lead to study discontinuation, and were not considered clinically significant.
“Sanofi Pasteur’s investigational vaccine stimulates a person’s immune system to fight C diff toxins upon exposure and, ultimately, may help prevent a future CDI from occurring,” Dr Saleh said.
“Like other toxoid vaccines—such as tetanus, diphtheria, and whooping cough—this investigational vaccine targets the symptom-causing toxins generated by C diff bacteria and could be an important public health measure to help protect individuals from CDI.”
Based on the phase 2 results, researchers started a phase 3 trial in August 2013.
Olive oil may protect against adverse vascular effects
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.”
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.”
SAN DIEGO—Taking olive oil supplements may counteract some of the adverse cardiovascular effects of air pollution, according to a new study.
“Exposure to airborne particulate matter can lead to endothelial dysfunction, a condition in which the endothelium of blood vessels does not function normally, which is a risk factor for clinical cardiovascular events and progression of atherosclerosis,” said Haiyan Tong,
MD, PhD, of the US Environmental Protection Agency.
“As olive oil and fish oil are known to have beneficial effects on endothelial dysfunction, we examined whether use of these supplements would counteract the adverse cardiovascular effects of exposure to concentrated ambient particulate matter in a controlled setting.”
Dr Tong and his colleagues presented their findings at the American Thoracic Society’s 2014 International Conference (abstract 55100).
Their study involved 42 healthy adults who were randomized to receive 3 g/day of olive oil, fish oil, or no supplements for 4 weeks. Subjects then underwent controlled, 2-hour exposures to filtered air, followed on the next day by exposure to fine/ultrafine concentrated ambient particulate matter (CAP, mean mass concentration 253±16 µg/m3) in a controlled-exposure chamber.
The researchers assessed endothelial function by sonographic measurement of flow-mediated dilation of the brachial artery before, immediately after, and 20 hours after exposure to air and CAP. They also measured blood markers of vasoconstriction and fibrinolysis.
Immediately after exposure to CAP, there were significant particulate matter mass-dependent reductions in flow-mediated dilation in the control group (-19.4±8.4% per 100 µg/m3 increase in CAP concentration relative to pre-filtered air levels) and the fish oil group (-13.7±5.3%), but the decrease in the olive oil group was not significant (-7.6±6.8%).
Tissue plasminogen activator increased immediately after CAP exposure in the olive oil group (11.6±5%), and this effect persisted for up to 20 hours.
Olive oil supplementation also ameliorated changes in blood markers associated with vasoconstriction and fibrinolysis, while fish oil supplementation had no effect on endothelial function or fibrinolysis after CAP exposure.
“Our study suggests that use of olive oil supplements may protect against the adverse vascular effects of exposure to air pollution particles,” Dr Tong said. “If these results are replicated in further studies, use of these supplements might offer a safe, low-cost, and effective means of counteracting some of the health consequences of exposure to air pollution.”