User login
Study reveals potential strategy for treating CML
Image courtesy of UCSD
New discoveries concerning a well-known tumor suppressor protein could help advance the diagnosis and treatment of chronic myeloid leukemia (CML), according to researchers.
They found that levels of the protein, BRCA1, are significantly decreased in advanced phases of CML, the expression of BCR-ABL1 correlates with decreased levels of BRCA1, and this downregulation of BRCA1 is caused by the inhibition of BRCA1 messenger RNA (mRNA) translation.
These discoveries explain the mechanism that supports CML development and uncover its weakness, the investigators said. They reported their findings in Cell Cycle.
“Our data demonstrated that BRCA1 synthesis is diminished in [the] advanced stage[s] of CML,” said study author Paulina Podszywałow-Bartnicka, PhD, of the Nencki Institute in Warsaw, Poland.
“The gene coding for BRCA1 protein is not mutated. However, BRCA1 mRNA, which is necessary for the protein production, is aggregated and stored in protein complexes [and], thus, not available for the protein synthesis.”
To gain more insight into this phenomenon, the investigators looked at 2 mRNA-binding proteins, HuR and TIAR. They found that BCR-ABL1 promoted cytosolic localization of TIAR and HuR, the proteins’ binding to BRCA1 mRNA, and formation of the TIAR-HuR complex.
The researchers also found that HuR positively regulated BRCA1 mRNA stability and translation, while TIAR negatively regulated BRCA1 translation.
TIAR-dependent downregulation of BRCA1 was a result of endoplasmic reticulum stress, which is activated in BCR-ABL1 expressing cells. And experiments showed that silencing TIAR in CML cells elevated BRCA1 levels.
This suggests that TIAR-mediated repression of BRCA1 mRNA translation is responsible for the downregulation of BRCA1 observed in BCR-ABL1-positive leukemia cells.
The investigators said this research indicates that BRCA1 deficiency, which supports CML, can be also used as a weapon against the disease.
“When a cell has damaged one signaling pathway or one gene, it may function properly due to alternative pathways . . . ,” explained study author Tomasz Skorski, MD, PhD, of Temple University School of Medicine in Philadelphia, Pennsylvania.
“Only when this alternative pathway is inhibited [do cells] lose the ability to survive. As we know that one of the [DNA double-strand break] repair pathways which depend on BRCA1 is blocked in leukemia cells, we can try to find the alternative, parallel pathway and inhibit it as well.”
This will induce apoptosis via synthetic lethality, but only in leukemia cells, because healthy cells still have functional BRCA1-dependent signaling. Dr Skorski noted that therapies based on BRCA1 deficiency are currently under investigation in clinical trials. ![]()
Image courtesy of UCSD
New discoveries concerning a well-known tumor suppressor protein could help advance the diagnosis and treatment of chronic myeloid leukemia (CML), according to researchers.
They found that levels of the protein, BRCA1, are significantly decreased in advanced phases of CML, the expression of BCR-ABL1 correlates with decreased levels of BRCA1, and this downregulation of BRCA1 is caused by the inhibition of BRCA1 messenger RNA (mRNA) translation.
These discoveries explain the mechanism that supports CML development and uncover its weakness, the investigators said. They reported their findings in Cell Cycle.
“Our data demonstrated that BRCA1 synthesis is diminished in [the] advanced stage[s] of CML,” said study author Paulina Podszywałow-Bartnicka, PhD, of the Nencki Institute in Warsaw, Poland.
“The gene coding for BRCA1 protein is not mutated. However, BRCA1 mRNA, which is necessary for the protein production, is aggregated and stored in protein complexes [and], thus, not available for the protein synthesis.”
To gain more insight into this phenomenon, the investigators looked at 2 mRNA-binding proteins, HuR and TIAR. They found that BCR-ABL1 promoted cytosolic localization of TIAR and HuR, the proteins’ binding to BRCA1 mRNA, and formation of the TIAR-HuR complex.
The researchers also found that HuR positively regulated BRCA1 mRNA stability and translation, while TIAR negatively regulated BRCA1 translation.
TIAR-dependent downregulation of BRCA1 was a result of endoplasmic reticulum stress, which is activated in BCR-ABL1 expressing cells. And experiments showed that silencing TIAR in CML cells elevated BRCA1 levels.
This suggests that TIAR-mediated repression of BRCA1 mRNA translation is responsible for the downregulation of BRCA1 observed in BCR-ABL1-positive leukemia cells.
The investigators said this research indicates that BRCA1 deficiency, which supports CML, can be also used as a weapon against the disease.
“When a cell has damaged one signaling pathway or one gene, it may function properly due to alternative pathways . . . ,” explained study author Tomasz Skorski, MD, PhD, of Temple University School of Medicine in Philadelphia, Pennsylvania.
“Only when this alternative pathway is inhibited [do cells] lose the ability to survive. As we know that one of the [DNA double-strand break] repair pathways which depend on BRCA1 is blocked in leukemia cells, we can try to find the alternative, parallel pathway and inhibit it as well.”
This will induce apoptosis via synthetic lethality, but only in leukemia cells, because healthy cells still have functional BRCA1-dependent signaling. Dr Skorski noted that therapies based on BRCA1 deficiency are currently under investigation in clinical trials. ![]()
Image courtesy of UCSD
New discoveries concerning a well-known tumor suppressor protein could help advance the diagnosis and treatment of chronic myeloid leukemia (CML), according to researchers.
They found that levels of the protein, BRCA1, are significantly decreased in advanced phases of CML, the expression of BCR-ABL1 correlates with decreased levels of BRCA1, and this downregulation of BRCA1 is caused by the inhibition of BRCA1 messenger RNA (mRNA) translation.
These discoveries explain the mechanism that supports CML development and uncover its weakness, the investigators said. They reported their findings in Cell Cycle.
“Our data demonstrated that BRCA1 synthesis is diminished in [the] advanced stage[s] of CML,” said study author Paulina Podszywałow-Bartnicka, PhD, of the Nencki Institute in Warsaw, Poland.
“The gene coding for BRCA1 protein is not mutated. However, BRCA1 mRNA, which is necessary for the protein production, is aggregated and stored in protein complexes [and], thus, not available for the protein synthesis.”
To gain more insight into this phenomenon, the investigators looked at 2 mRNA-binding proteins, HuR and TIAR. They found that BCR-ABL1 promoted cytosolic localization of TIAR and HuR, the proteins’ binding to BRCA1 mRNA, and formation of the TIAR-HuR complex.
The researchers also found that HuR positively regulated BRCA1 mRNA stability and translation, while TIAR negatively regulated BRCA1 translation.
TIAR-dependent downregulation of BRCA1 was a result of endoplasmic reticulum stress, which is activated in BCR-ABL1 expressing cells. And experiments showed that silencing TIAR in CML cells elevated BRCA1 levels.
This suggests that TIAR-mediated repression of BRCA1 mRNA translation is responsible for the downregulation of BRCA1 observed in BCR-ABL1-positive leukemia cells.
The investigators said this research indicates that BRCA1 deficiency, which supports CML, can be also used as a weapon against the disease.
“When a cell has damaged one signaling pathway or one gene, it may function properly due to alternative pathways . . . ,” explained study author Tomasz Skorski, MD, PhD, of Temple University School of Medicine in Philadelphia, Pennsylvania.
“Only when this alternative pathway is inhibited [do cells] lose the ability to survive. As we know that one of the [DNA double-strand break] repair pathways which depend on BRCA1 is blocked in leukemia cells, we can try to find the alternative, parallel pathway and inhibit it as well.”
This will induce apoptosis via synthetic lethality, but only in leukemia cells, because healthy cells still have functional BRCA1-dependent signaling. Dr Skorski noted that therapies based on BRCA1 deficiency are currently under investigation in clinical trials. ![]()
FDA grants drug orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal toll-like receptors (TLRs) 7, 8, and 9, for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions that affect fewer than 200,000 people in the US.
The designation will provide Idera Pharmaceuticals, the company developing IMO-8400, with certain incentives. These include eligibility for federal grants, research and development tax credits, and a 7-year period of marketing exclusivity if the product is approved.
Relevant research
Preclinical research published in Leukemia showed that the MYD88 L265P oncogenic mutation is an independent prognostic factor in DLBCL. In DLBCL patients with this mutation, TLR signaling is over-activated, which enables tumor cell survival and proliferation.
Data presented at the 2014 AACR Annual Meeting showed that IMO-8400 inhibited the survival and proliferation of DLBCL cells and Waldenstrom’s macroglobulinemia (WM) cells harboring the MYD88 L265P mutation.
A phase 1 trial of IMO-8400 presented at the 2013 FOCIS Annual Meeting showed the drug was active and well-tolerated in 42 healthy subjects. IMO-8400 was given at single and multiple escalating doses up to 0.6 mg/kg for 4 weeks. The drug inhibited immune responses mediated by TLRs 7, 8, and 9.
Idera Pharmaceuticals is currently conducting a phase 1/2 trial of IMO-8400 in patients with relapsed or refractory DLBCL who harbor the MYD88 L265P mutation. The protocol includes 3 dose-escalation cohorts in which IMO-8400 is administered subcutaneously.
Idera is also pursuing clinical development of IMO-8400 in WM. The FDA recently granted the drug orphan designation to treat WM. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal toll-like receptors (TLRs) 7, 8, and 9, for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions that affect fewer than 200,000 people in the US.
The designation will provide Idera Pharmaceuticals, the company developing IMO-8400, with certain incentives. These include eligibility for federal grants, research and development tax credits, and a 7-year period of marketing exclusivity if the product is approved.
Relevant research
Preclinical research published in Leukemia showed that the MYD88 L265P oncogenic mutation is an independent prognostic factor in DLBCL. In DLBCL patients with this mutation, TLR signaling is over-activated, which enables tumor cell survival and proliferation.
Data presented at the 2014 AACR Annual Meeting showed that IMO-8400 inhibited the survival and proliferation of DLBCL cells and Waldenstrom’s macroglobulinemia (WM) cells harboring the MYD88 L265P mutation.
A phase 1 trial of IMO-8400 presented at the 2013 FOCIS Annual Meeting showed the drug was active and well-tolerated in 42 healthy subjects. IMO-8400 was given at single and multiple escalating doses up to 0.6 mg/kg for 4 weeks. The drug inhibited immune responses mediated by TLRs 7, 8, and 9.
Idera Pharmaceuticals is currently conducting a phase 1/2 trial of IMO-8400 in patients with relapsed or refractory DLBCL who harbor the MYD88 L265P mutation. The protocol includes 3 dose-escalation cohorts in which IMO-8400 is administered subcutaneously.
Idera is also pursuing clinical development of IMO-8400 in WM. The FDA recently granted the drug orphan designation to treat WM. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for IMO-8400, an antagonist of the endosomal toll-like receptors (TLRs) 7, 8, and 9, for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs intended to treat conditions that affect fewer than 200,000 people in the US.
The designation will provide Idera Pharmaceuticals, the company developing IMO-8400, with certain incentives. These include eligibility for federal grants, research and development tax credits, and a 7-year period of marketing exclusivity if the product is approved.
Relevant research
Preclinical research published in Leukemia showed that the MYD88 L265P oncogenic mutation is an independent prognostic factor in DLBCL. In DLBCL patients with this mutation, TLR signaling is over-activated, which enables tumor cell survival and proliferation.
Data presented at the 2014 AACR Annual Meeting showed that IMO-8400 inhibited the survival and proliferation of DLBCL cells and Waldenstrom’s macroglobulinemia (WM) cells harboring the MYD88 L265P mutation.
A phase 1 trial of IMO-8400 presented at the 2013 FOCIS Annual Meeting showed the drug was active and well-tolerated in 42 healthy subjects. IMO-8400 was given at single and multiple escalating doses up to 0.6 mg/kg for 4 weeks. The drug inhibited immune responses mediated by TLRs 7, 8, and 9.
Idera Pharmaceuticals is currently conducting a phase 1/2 trial of IMO-8400 in patients with relapsed or refractory DLBCL who harbor the MYD88 L265P mutation. The protocol includes 3 dose-escalation cohorts in which IMO-8400 is administered subcutaneously.
Idera is also pursuing clinical development of IMO-8400 in WM. The FDA recently granted the drug orphan designation to treat WM. ![]()
Most childhood cancer survivors have morbidities
Photo by Logan Tuttle
New research suggests the prevalence of childhood cancer survivors in the US has increased, and the majority of pediatric patients who have survived 5
or more years beyond cancer diagnosis may have at least one chronic health condition.
About 70% of the childhood cancer survivors studied were estimated to have a mild or moderate chronic condition, and nearly a third were estimated to have a severe, disabling, or life-threatening chronic condition.
“Our study findings highlight that a singular focus on curing cancer yields an incomplete picture of childhood cancer survivorship,” said study author Siobhan M. Phillips, PhD, of Northwestern University Feinberg School of Medicine in Chicago, Illinois.
“The burden of chronic conditions in this population is profound, both in occurrence and severity. Efforts to understand how to effectively decrease morbidity burden and incorporate effective care coordination and rehabilitation models to optimize longevity and well-being in this population should be a priority.”
Dr Phillips and her colleagues reported their findings in Cancer Epidemiology, Biomarkers & Prevention.
The researchers analyzed data on cancer incidence and survival for children who were diagnosed with cancer between 0 and 19 years of age. The data had been recorded between 1975 and 2011 in 9 different US Surveillance, Epidemiology, and End Results (SEER) registries.
The team also used data from the Childhood Cancer Survivor Study (CCSS) cohort, which had information on a range of potential adverse and late effects of cancer treatment from more than 14,000 long-term survivors of childhood cancers who were treated at 26 cancer centers across the US and Canada.
The investigators first obtained estimates of the probability of each measure of morbidity from CCSS and then multiplied these estimates by the relevant number of survivors in the US estimated from the SEER data.
The researchers estimated the number of childhood cancer survivors in the US to be 388,501, which is an increase of 59,849 from the previous estimate made in 2005 by a team from the National Cancer Institute.
Of these patients, about 84% had survived 5 or more years post-diagnosis, and about 45% had survived for 20 years or more.
About 70% of the survivors were estimated to have a mild or moderate chronic condition (grade 1-2), and about 32% were estimated to have a severe, disabling, or life-threatening chronic condition (grade 3-4).
An estimated 35% of the survivors, ages 20 to 49, had neurocognitive dysfunction. About 13% to 17% of survivors in this age group had self-reported functional impairment, activity limitations, impaired mental health, pain, or anxiety/fear.
“We know that many of these morbidities are at least somewhat modifiable in the general population,” Dr Phillips noted. “However, we don’t know if typical population guidelines for preventive behaviors apply to [childhood cancer survivors].”
“We need to develop a better understanding of the multilevel factors—including, but not limited to, physical activity, diet, and treatment characteristics—which influence childhood cancer survivors’ susceptibility to these morbidities in order to effectively prevent and delay their onset.” ![]()
Photo by Logan Tuttle
New research suggests the prevalence of childhood cancer survivors in the US has increased, and the majority of pediatric patients who have survived 5
or more years beyond cancer diagnosis may have at least one chronic health condition.
About 70% of the childhood cancer survivors studied were estimated to have a mild or moderate chronic condition, and nearly a third were estimated to have a severe, disabling, or life-threatening chronic condition.
“Our study findings highlight that a singular focus on curing cancer yields an incomplete picture of childhood cancer survivorship,” said study author Siobhan M. Phillips, PhD, of Northwestern University Feinberg School of Medicine in Chicago, Illinois.
“The burden of chronic conditions in this population is profound, both in occurrence and severity. Efforts to understand how to effectively decrease morbidity burden and incorporate effective care coordination and rehabilitation models to optimize longevity and well-being in this population should be a priority.”
Dr Phillips and her colleagues reported their findings in Cancer Epidemiology, Biomarkers & Prevention.
The researchers analyzed data on cancer incidence and survival for children who were diagnosed with cancer between 0 and 19 years of age. The data had been recorded between 1975 and 2011 in 9 different US Surveillance, Epidemiology, and End Results (SEER) registries.
The team also used data from the Childhood Cancer Survivor Study (CCSS) cohort, which had information on a range of potential adverse and late effects of cancer treatment from more than 14,000 long-term survivors of childhood cancers who were treated at 26 cancer centers across the US and Canada.
The investigators first obtained estimates of the probability of each measure of morbidity from CCSS and then multiplied these estimates by the relevant number of survivors in the US estimated from the SEER data.
The researchers estimated the number of childhood cancer survivors in the US to be 388,501, which is an increase of 59,849 from the previous estimate made in 2005 by a team from the National Cancer Institute.
Of these patients, about 84% had survived 5 or more years post-diagnosis, and about 45% had survived for 20 years or more.
About 70% of the survivors were estimated to have a mild or moderate chronic condition (grade 1-2), and about 32% were estimated to have a severe, disabling, or life-threatening chronic condition (grade 3-4).
An estimated 35% of the survivors, ages 20 to 49, had neurocognitive dysfunction. About 13% to 17% of survivors in this age group had self-reported functional impairment, activity limitations, impaired mental health, pain, or anxiety/fear.
“We know that many of these morbidities are at least somewhat modifiable in the general population,” Dr Phillips noted. “However, we don’t know if typical population guidelines for preventive behaviors apply to [childhood cancer survivors].”
“We need to develop a better understanding of the multilevel factors—including, but not limited to, physical activity, diet, and treatment characteristics—which influence childhood cancer survivors’ susceptibility to these morbidities in order to effectively prevent and delay their onset.” ![]()
Photo by Logan Tuttle
New research suggests the prevalence of childhood cancer survivors in the US has increased, and the majority of pediatric patients who have survived 5
or more years beyond cancer diagnosis may have at least one chronic health condition.
About 70% of the childhood cancer survivors studied were estimated to have a mild or moderate chronic condition, and nearly a third were estimated to have a severe, disabling, or life-threatening chronic condition.
“Our study findings highlight that a singular focus on curing cancer yields an incomplete picture of childhood cancer survivorship,” said study author Siobhan M. Phillips, PhD, of Northwestern University Feinberg School of Medicine in Chicago, Illinois.
“The burden of chronic conditions in this population is profound, both in occurrence and severity. Efforts to understand how to effectively decrease morbidity burden and incorporate effective care coordination and rehabilitation models to optimize longevity and well-being in this population should be a priority.”
Dr Phillips and her colleagues reported their findings in Cancer Epidemiology, Biomarkers & Prevention.
The researchers analyzed data on cancer incidence and survival for children who were diagnosed with cancer between 0 and 19 years of age. The data had been recorded between 1975 and 2011 in 9 different US Surveillance, Epidemiology, and End Results (SEER) registries.
The team also used data from the Childhood Cancer Survivor Study (CCSS) cohort, which had information on a range of potential adverse and late effects of cancer treatment from more than 14,000 long-term survivors of childhood cancers who were treated at 26 cancer centers across the US and Canada.
The investigators first obtained estimates of the probability of each measure of morbidity from CCSS and then multiplied these estimates by the relevant number of survivors in the US estimated from the SEER data.
The researchers estimated the number of childhood cancer survivors in the US to be 388,501, which is an increase of 59,849 from the previous estimate made in 2005 by a team from the National Cancer Institute.
Of these patients, about 84% had survived 5 or more years post-diagnosis, and about 45% had survived for 20 years or more.
About 70% of the survivors were estimated to have a mild or moderate chronic condition (grade 1-2), and about 32% were estimated to have a severe, disabling, or life-threatening chronic condition (grade 3-4).
An estimated 35% of the survivors, ages 20 to 49, had neurocognitive dysfunction. About 13% to 17% of survivors in this age group had self-reported functional impairment, activity limitations, impaired mental health, pain, or anxiety/fear.
“We know that many of these morbidities are at least somewhat modifiable in the general population,” Dr Phillips noted. “However, we don’t know if typical population guidelines for preventive behaviors apply to [childhood cancer survivors].”
“We need to develop a better understanding of the multilevel factors—including, but not limited to, physical activity, diet, and treatment characteristics—which influence childhood cancer survivors’ susceptibility to these morbidities in order to effectively prevent and delay their onset.” ![]()
Vitamin D levels linked to outcomes in FL

Vitamin D deficiency may negatively impact outcomes in patients with follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
The study showed that FL patients with vitamin D deficiency had inferior progression-free survival (PFS) and overall survival (OS) compared to patients with higher vitamin D levels.
According to researchers, this suggests serum vitamin D might be the first potentially modifiable factor to be associated with survival in FL.
However, additional research is needed to determine the effects of vitamin D supplementation in these patients.
Jonathan W. Friedberg, MD, of the Wilmot Cancer Institute at the University of Rochester in New York, and his colleagues conducted this research, analyzing data from 2 cohorts of FL patients.
One cohort consisted of patients derived from 3 SWOG trials (S9800, S9911, and S0016), and the other consisted of patients from a Lymphoma Study Association (LYSA) trial known as PRIMA.
SWOG patients had received CHOP chemotherapy plus an anti-CD20 antibody (rituximab or iodine-131 tositumomab), and LYSA patients had received rituximab plus chemotherapy (and were randomized to rituximab maintenance or observation).
SWOG cohort
After a median follow-up of 5.4 years, patients with vitamin D deficiency (defined as <20 ng/mL) had significantly inferior PFS (hazard ratio [HR]=2.00; P=0.011) and OS (HR=3.57; P=0.003) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (IPI), body mass index, and latitude (≥ vs <35°N). For PFS, the adjusted HR was 1.97 (P=0.023). And for OS, the adjusted HR was 4.16 (P=0.002).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but neither result was significant.
LYSA cohort
After a median follow-up of 6.6 years, patients with vitamin D deficiency (defined as <10 ng/mL) had significantly inferior PFS (HR=1.66; P=0.013) but not OS (HR=1.84; P=0.14) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (FLIPI), body mass index, latitude (Europe vs Australia), and hemoglobin. For PFS, the adjusted HR was 1.50 (P=0.095). For OS, the adjusted HR was 1.92 (P=0.192).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but only the association with OS reached statistical significance (HR=5.32; P=0.037).
Dr Friedberg said that, taken together, these results suggest vitamin D levels may be a modifiable factor associated with prognosis in patients with FL.
“Our data, replicated internationally, supports other published observations linking vitamin D deficiency with inferior cancer outcomes,” he said. “However, the mechanisms of this link are likely complex and require further study.” ![]()
Vitamin D deficiency may negatively impact outcomes in patients with follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
The study showed that FL patients with vitamin D deficiency had inferior progression-free survival (PFS) and overall survival (OS) compared to patients with higher vitamin D levels.
According to researchers, this suggests serum vitamin D might be the first potentially modifiable factor to be associated with survival in FL.
However, additional research is needed to determine the effects of vitamin D supplementation in these patients.
Jonathan W. Friedberg, MD, of the Wilmot Cancer Institute at the University of Rochester in New York, and his colleagues conducted this research, analyzing data from 2 cohorts of FL patients.
One cohort consisted of patients derived from 3 SWOG trials (S9800, S9911, and S0016), and the other consisted of patients from a Lymphoma Study Association (LYSA) trial known as PRIMA.
SWOG patients had received CHOP chemotherapy plus an anti-CD20 antibody (rituximab or iodine-131 tositumomab), and LYSA patients had received rituximab plus chemotherapy (and were randomized to rituximab maintenance or observation).
SWOG cohort
After a median follow-up of 5.4 years, patients with vitamin D deficiency (defined as <20 ng/mL) had significantly inferior PFS (hazard ratio [HR]=2.00; P=0.011) and OS (HR=3.57; P=0.003) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (IPI), body mass index, and latitude (≥ vs <35°N). For PFS, the adjusted HR was 1.97 (P=0.023). And for OS, the adjusted HR was 4.16 (P=0.002).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but neither result was significant.
LYSA cohort
After a median follow-up of 6.6 years, patients with vitamin D deficiency (defined as <10 ng/mL) had significantly inferior PFS (HR=1.66; P=0.013) but not OS (HR=1.84; P=0.14) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (FLIPI), body mass index, latitude (Europe vs Australia), and hemoglobin. For PFS, the adjusted HR was 1.50 (P=0.095). For OS, the adjusted HR was 1.92 (P=0.192).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but only the association with OS reached statistical significance (HR=5.32; P=0.037).
Dr Friedberg said that, taken together, these results suggest vitamin D levels may be a modifiable factor associated with prognosis in patients with FL.
“Our data, replicated internationally, supports other published observations linking vitamin D deficiency with inferior cancer outcomes,” he said. “However, the mechanisms of this link are likely complex and require further study.” ![]()
Vitamin D deficiency may negatively impact outcomes in patients with follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
The study showed that FL patients with vitamin D deficiency had inferior progression-free survival (PFS) and overall survival (OS) compared to patients with higher vitamin D levels.
According to researchers, this suggests serum vitamin D might be the first potentially modifiable factor to be associated with survival in FL.
However, additional research is needed to determine the effects of vitamin D supplementation in these patients.
Jonathan W. Friedberg, MD, of the Wilmot Cancer Institute at the University of Rochester in New York, and his colleagues conducted this research, analyzing data from 2 cohorts of FL patients.
One cohort consisted of patients derived from 3 SWOG trials (S9800, S9911, and S0016), and the other consisted of patients from a Lymphoma Study Association (LYSA) trial known as PRIMA.
SWOG patients had received CHOP chemotherapy plus an anti-CD20 antibody (rituximab or iodine-131 tositumomab), and LYSA patients had received rituximab plus chemotherapy (and were randomized to rituximab maintenance or observation).
SWOG cohort
After a median follow-up of 5.4 years, patients with vitamin D deficiency (defined as <20 ng/mL) had significantly inferior PFS (hazard ratio [HR]=2.00; P=0.011) and OS (HR=3.57; P=0.003) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (IPI), body mass index, and latitude (≥ vs <35°N). For PFS, the adjusted HR was 1.97 (P=0.023). And for OS, the adjusted HR was 4.16 (P=0.002).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but neither result was significant.
LYSA cohort
After a median follow-up of 6.6 years, patients with vitamin D deficiency (defined as <10 ng/mL) had significantly inferior PFS (HR=1.66; P=0.013) but not OS (HR=1.84; P=0.14) compared to patients with higher vitamin D levels.
Results were similar when the researchers adjusted for other variables, such as prognostic index (FLIPI), body mass index, latitude (Europe vs Australia), and hemoglobin. For PFS, the adjusted HR was 1.50 (P=0.095). For OS, the adjusted HR was 1.92 (P=0.192).
Multivariable analysis of vitamin D by tertile confirmed that the lowest tertile of vitamin D was associated with a greater increase in the risk of either progression or death, but only the association with OS reached statistical significance (HR=5.32; P=0.037).
Dr Friedberg said that, taken together, these results suggest vitamin D levels may be a modifiable factor associated with prognosis in patients with FL.
“Our data, replicated internationally, supports other published observations linking vitamin D deficiency with inferior cancer outcomes,” he said. “However, the mechanisms of this link are likely complex and require further study.” ![]()
FDA approves new formulation of iron overload drug
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com. ![]()
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com. ![]()
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com. ![]()
Predicting treatment response in CMML

Photo courtesy of
University of Michigan
Newly identified molecular signatures may allow us to predict which patients with chronic myelomonocytic leukemia (CMML) will respond to treatment, according to a study published in The Journal of Clinical Investigation.
Finding effective biomarkers is particularly crucial for CMML, the investigators said, because current treatment is slow-acting. Patients must often undergo as much as 6 months of treatment before there are signs of response.
“The slow kinetics is what gets us,” said study author Maria E. Figueroa, MD, of the University of Michigan Medical School in Ann Arbor.
“It’s not just one week or one dose to see signs of response. A good biomarker test could potentially prevent patients who are unlikely to respond from receiving prolonged, unwarranted treatments.”
With this in mind, Dr Figueroa and her colleagues used next-generation sequencing techniques to analyze 40 CMML samples from patients treated with the DNA methyltransferase inhibitor decitabine.
The researchers found 167 differentially methylated regions of DNA at baseline that distinguished patients who responded to decitabine from those who did not. There was a methylation difference of 25% or more between responders and nonresponders.
The investigators speculated that the baseline differences in DNA methylation could be used to predict treatment response at diagnosis.
So they used the percentage of cytosine methylation at each genomic location among the 40 patients as potential predictors and applied a machine-learning approach to build an epigenetic classifier.
The investigators tested the classifier in 28 additional CMML samples and found it was 87% accurate in predicting a patient’s response to decitabine.
The researchers also investigated why nonresponders were resistant to decitabine and found that 2 proteins, CXCL4 and CXCL7, were overexpressed in nonresponders. In cells exposed to high levels of these chemokines, the effects of decitabine were blocked.
“We are pursuing this to understand why these proteins block the effect of the drug and whether we can develop a new compound that could be used along with decitabine to turn nonresponders into responders,” Dr Figueroa said.
The researchers are also working to refine and translate their findings into a viable biomarker test that could be used in the clinic. ![]()
Photo courtesy of
University of Michigan
Newly identified molecular signatures may allow us to predict which patients with chronic myelomonocytic leukemia (CMML) will respond to treatment, according to a study published in The Journal of Clinical Investigation.
Finding effective biomarkers is particularly crucial for CMML, the investigators said, because current treatment is slow-acting. Patients must often undergo as much as 6 months of treatment before there are signs of response.
“The slow kinetics is what gets us,” said study author Maria E. Figueroa, MD, of the University of Michigan Medical School in Ann Arbor.
“It’s not just one week or one dose to see signs of response. A good biomarker test could potentially prevent patients who are unlikely to respond from receiving prolonged, unwarranted treatments.”
With this in mind, Dr Figueroa and her colleagues used next-generation sequencing techniques to analyze 40 CMML samples from patients treated with the DNA methyltransferase inhibitor decitabine.
The researchers found 167 differentially methylated regions of DNA at baseline that distinguished patients who responded to decitabine from those who did not. There was a methylation difference of 25% or more between responders and nonresponders.
The investigators speculated that the baseline differences in DNA methylation could be used to predict treatment response at diagnosis.
So they used the percentage of cytosine methylation at each genomic location among the 40 patients as potential predictors and applied a machine-learning approach to build an epigenetic classifier.
The investigators tested the classifier in 28 additional CMML samples and found it was 87% accurate in predicting a patient’s response to decitabine.
The researchers also investigated why nonresponders were resistant to decitabine and found that 2 proteins, CXCL4 and CXCL7, were overexpressed in nonresponders. In cells exposed to high levels of these chemokines, the effects of decitabine were blocked.
“We are pursuing this to understand why these proteins block the effect of the drug and whether we can develop a new compound that could be used along with decitabine to turn nonresponders into responders,” Dr Figueroa said.
The researchers are also working to refine and translate their findings into a viable biomarker test that could be used in the clinic. ![]()
Photo courtesy of
University of Michigan
Newly identified molecular signatures may allow us to predict which patients with chronic myelomonocytic leukemia (CMML) will respond to treatment, according to a study published in The Journal of Clinical Investigation.
Finding effective biomarkers is particularly crucial for CMML, the investigators said, because current treatment is slow-acting. Patients must often undergo as much as 6 months of treatment before there are signs of response.
“The slow kinetics is what gets us,” said study author Maria E. Figueroa, MD, of the University of Michigan Medical School in Ann Arbor.
“It’s not just one week or one dose to see signs of response. A good biomarker test could potentially prevent patients who are unlikely to respond from receiving prolonged, unwarranted treatments.”
With this in mind, Dr Figueroa and her colleagues used next-generation sequencing techniques to analyze 40 CMML samples from patients treated with the DNA methyltransferase inhibitor decitabine.
The researchers found 167 differentially methylated regions of DNA at baseline that distinguished patients who responded to decitabine from those who did not. There was a methylation difference of 25% or more between responders and nonresponders.
The investigators speculated that the baseline differences in DNA methylation could be used to predict treatment response at diagnosis.
So they used the percentage of cytosine methylation at each genomic location among the 40 patients as potential predictors and applied a machine-learning approach to build an epigenetic classifier.
The investigators tested the classifier in 28 additional CMML samples and found it was 87% accurate in predicting a patient’s response to decitabine.
The researchers also investigated why nonresponders were resistant to decitabine and found that 2 proteins, CXCL4 and CXCL7, were overexpressed in nonresponders. In cells exposed to high levels of these chemokines, the effects of decitabine were blocked.
“We are pursuing this to understand why these proteins block the effect of the drug and whether we can develop a new compound that could be used along with decitabine to turn nonresponders into responders,” Dr Figueroa said.
The researchers are also working to refine and translate their findings into a viable biomarker test that could be used in the clinic. ![]()
Immunotherapy gets orphan designation

Photo by Graham Colm
The US Food and Drug Administration (FDA) has granted orphan designation to an immunotherapy known as CMD-003, which is under development to treat Epstein-Barr-virus (EBV)-positive non-Hodgkin lymphomas.
CMD-003 consists of T cells derived from blood samples that are activated and expanded through a proprietary process developed for commercial-scale use.
Researchers have treated more than 250 patients with prototypes of CMD-003. And the prototypes have produced promising results in a range of malignancies.
CMD-003 is under development by Cell Medica and the Center for Cell and Gene Therapy (CAGT) at Baylor College of Medicine, Texas Children’s Hospital, and Houston Methodist Hospital.
Orphan designation from the FDA will provide CMD-003’s developers with several benefits, including accessibility to grants to support clinical development, 7 years of market exclusivity if the treatment is approved in the US, and tax credits on US clinical trials.
CMD-003 prototype
Researchers have not published any trials of CMD-003, but they have studied other EBV-specific T-cell products related to CMD-003.
In their most recent study, published in the Journal of Clinical Oncology, the researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission from their disease at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of them died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery. ![]()
Photo by Graham Colm
The US Food and Drug Administration (FDA) has granted orphan designation to an immunotherapy known as CMD-003, which is under development to treat Epstein-Barr-virus (EBV)-positive non-Hodgkin lymphomas.
CMD-003 consists of T cells derived from blood samples that are activated and expanded through a proprietary process developed for commercial-scale use.
Researchers have treated more than 250 patients with prototypes of CMD-003. And the prototypes have produced promising results in a range of malignancies.
CMD-003 is under development by Cell Medica and the Center for Cell and Gene Therapy (CAGT) at Baylor College of Medicine, Texas Children’s Hospital, and Houston Methodist Hospital.
Orphan designation from the FDA will provide CMD-003’s developers with several benefits, including accessibility to grants to support clinical development, 7 years of market exclusivity if the treatment is approved in the US, and tax credits on US clinical trials.
CMD-003 prototype
Researchers have not published any trials of CMD-003, but they have studied other EBV-specific T-cell products related to CMD-003.
In their most recent study, published in the Journal of Clinical Oncology, the researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission from their disease at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of them died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery. ![]()
Photo by Graham Colm
The US Food and Drug Administration (FDA) has granted orphan designation to an immunotherapy known as CMD-003, which is under development to treat Epstein-Barr-virus (EBV)-positive non-Hodgkin lymphomas.
CMD-003 consists of T cells derived from blood samples that are activated and expanded through a proprietary process developed for commercial-scale use.
Researchers have treated more than 250 patients with prototypes of CMD-003. And the prototypes have produced promising results in a range of malignancies.
CMD-003 is under development by Cell Medica and the Center for Cell and Gene Therapy (CAGT) at Baylor College of Medicine, Texas Children’s Hospital, and Houston Methodist Hospital.
Orphan designation from the FDA will provide CMD-003’s developers with several benefits, including accessibility to grants to support clinical development, 7 years of market exclusivity if the treatment is approved in the US, and tax credits on US clinical trials.
CMD-003 prototype
Researchers have not published any trials of CMD-003, but they have studied other EBV-specific T-cell products related to CMD-003.
In their most recent study, published in the Journal of Clinical Oncology, the researchers administered cytotoxic T lymphocytes (CTLs) in 50 patients with EBV-associated Hodgkin or non-Hodgkin lymphoma.
Twenty-nine of the patients were in remission when they received CTL infusions, but they were at a high risk of relapse. The remaining 21 patients had relapsed or refractory disease at the time of CTL infusion.
Twenty-seven of the patients who received CTLs as an adjuvant treatment remained in remission from their disease at 3.1 years after treatment.
Their 2-year event-free survival rate was 82%. None of them died of lymphoma, but 9 died from complications associated with the chemotherapy and radiation they had received.
Of the 21 patients with relapsed or refractory disease, 13 responded to CTL infusions, and 11 patients achieved a complete response. In this group, the 2-year event-free survival rate was about 50%.
The researchers said there were no toxicities that were definitively related to CTL infusion.
One patient had central nervous system deterioration 2 weeks after infusion. This was attributed to disease progression but could possibly have been treatment-related.
Another patient developed respiratory complications about 4 weeks after a second CTL infusion that may have been treatment-related. However, the researchers attributed it to an intercurrent infection, and the patient made a complete recovery.
Compounds could treat MLL leukemia

and Jolanta Grembecka, PhD
Photo courtesy of the
University of Michigan
Two small-molecule inhibitors can fight aggressive, acute leukemias by targeting a protein-protein interaction, according to preclinical research published in Cancer Cell.
The compounds, MI-463 and MI-503, work by inhibiting the interaction between menin and mixed-lineage leukemia (MLL) fusion proteins.
Menin binds to the N-terminal fragment of MLL retained in all MLL fusion proteins, and the fusion proteins require menin for leukemogenic activity.
That’s why Jolanta Grembecka, PhD, and Tomasz Cierpicki, PhD, both of the University of Michigan in Ann Arbor, have been working for several years to identify small-molecule inhibitors that would block the MLL-menin interaction.
“The MLL-menin interaction is a good drug target because it’s the primary driver in [MLL] leukemia,” Dr Grembecka said. “By blocking this interaction, it’s very likely to stop the cancer.”
With that in mind, Dr Grembecka and her colleagues tested 2 compounds they developed, MI-463 and MI-503, in cell lines and mice with MLL leukemia. The compounds blocked the MLL-menin interaction without affecting normal hematopoiesis.
The team also noted that both compounds demonstrated metabolic stability and favorable pharmacokinetic profiles.
“Against all odds, we decided to explore finding a way to block the MLL-menin interaction with small molecules,” Dr Cierpicki said. “From nothing, we have been able to identify and greatly improve a compound and show that it’s got valuable potential in blocking MLL fusion leukemia in animal models.”
In a separate study published in Nature Medicine, the researchers discovered that menin and MLL play a role in androgen receptor signaling, a key driver of prostate cancer.
The team found that MI-503 and MI-136, another inhibitor of the menin-MLL interaction, were both active against castration-resistant prostate cancer in vitro and in vivo.
The researchers said they will continue to investigate the role of MLL in castration-resistant prostate cancer. And they plan to further refine their inhibitors and put the compounds through more advanced preclinical testing in MLL leukemia.
and Jolanta Grembecka, PhD
Photo courtesy of the
University of Michigan
Two small-molecule inhibitors can fight aggressive, acute leukemias by targeting a protein-protein interaction, according to preclinical research published in Cancer Cell.
The compounds, MI-463 and MI-503, work by inhibiting the interaction between menin and mixed-lineage leukemia (MLL) fusion proteins.
Menin binds to the N-terminal fragment of MLL retained in all MLL fusion proteins, and the fusion proteins require menin for leukemogenic activity.
That’s why Jolanta Grembecka, PhD, and Tomasz Cierpicki, PhD, both of the University of Michigan in Ann Arbor, have been working for several years to identify small-molecule inhibitors that would block the MLL-menin interaction.
“The MLL-menin interaction is a good drug target because it’s the primary driver in [MLL] leukemia,” Dr Grembecka said. “By blocking this interaction, it’s very likely to stop the cancer.”
With that in mind, Dr Grembecka and her colleagues tested 2 compounds they developed, MI-463 and MI-503, in cell lines and mice with MLL leukemia. The compounds blocked the MLL-menin interaction without affecting normal hematopoiesis.
The team also noted that both compounds demonstrated metabolic stability and favorable pharmacokinetic profiles.
“Against all odds, we decided to explore finding a way to block the MLL-menin interaction with small molecules,” Dr Cierpicki said. “From nothing, we have been able to identify and greatly improve a compound and show that it’s got valuable potential in blocking MLL fusion leukemia in animal models.”
In a separate study published in Nature Medicine, the researchers discovered that menin and MLL play a role in androgen receptor signaling, a key driver of prostate cancer.
The team found that MI-503 and MI-136, another inhibitor of the menin-MLL interaction, were both active against castration-resistant prostate cancer in vitro and in vivo.
The researchers said they will continue to investigate the role of MLL in castration-resistant prostate cancer. And they plan to further refine their inhibitors and put the compounds through more advanced preclinical testing in MLL leukemia.
and Jolanta Grembecka, PhD
Photo courtesy of the
University of Michigan
Two small-molecule inhibitors can fight aggressive, acute leukemias by targeting a protein-protein interaction, according to preclinical research published in Cancer Cell.
The compounds, MI-463 and MI-503, work by inhibiting the interaction between menin and mixed-lineage leukemia (MLL) fusion proteins.
Menin binds to the N-terminal fragment of MLL retained in all MLL fusion proteins, and the fusion proteins require menin for leukemogenic activity.
That’s why Jolanta Grembecka, PhD, and Tomasz Cierpicki, PhD, both of the University of Michigan in Ann Arbor, have been working for several years to identify small-molecule inhibitors that would block the MLL-menin interaction.
“The MLL-menin interaction is a good drug target because it’s the primary driver in [MLL] leukemia,” Dr Grembecka said. “By blocking this interaction, it’s very likely to stop the cancer.”
With that in mind, Dr Grembecka and her colleagues tested 2 compounds they developed, MI-463 and MI-503, in cell lines and mice with MLL leukemia. The compounds blocked the MLL-menin interaction without affecting normal hematopoiesis.
The team also noted that both compounds demonstrated metabolic stability and favorable pharmacokinetic profiles.
“Against all odds, we decided to explore finding a way to block the MLL-menin interaction with small molecules,” Dr Cierpicki said. “From nothing, we have been able to identify and greatly improve a compound and show that it’s got valuable potential in blocking MLL fusion leukemia in animal models.”
In a separate study published in Nature Medicine, the researchers discovered that menin and MLL play a role in androgen receptor signaling, a key driver of prostate cancer.
The team found that MI-503 and MI-136, another inhibitor of the menin-MLL interaction, were both active against castration-resistant prostate cancer in vitro and in vivo.
The researchers said they will continue to investigate the role of MLL in castration-resistant prostate cancer. And they plan to further refine their inhibitors and put the compounds through more advanced preclinical testing in MLL leukemia.
Findings may aid development of antithrombotic drugs
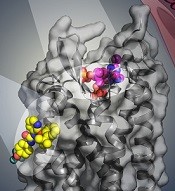
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects.
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects.
BPTU (yellow) bind to P2Y1R
Image courtesy of
Yekaterina Kadyshevskaya
Researchers say they have identified 2 disparate ligand-binding sites in the human P2Y1 receptor (P2Y1R), which plays a critical role in thrombosis.
Their research has provided a detailed molecular map of P2Y1R, a G protein-coupled receptor (GPCR), in complex with a nucleotide antagonist MRS2500 and a non-nucleotide antagonist BPTU.
The researchers believe their findings, published in Nature, could aid the development of new antithrombotic drugs.
Beili Wu, PhD, of the Shanghai Institute of Materia Medica in China, and her colleagues noted that the human purinergic receptors P2Y1R and P2Y12R play a major physiological role in adenosine 5′-diphosphate (ADP)-mediated platelet aggregation, an important component of thrombosis.
Although most of the available antithrombotic drugs act on P2Y12R, research has suggested that P2Y1R may be a promising antithrombotic drug target. In addition, P2Y1R inhibitors may offer a safety advantage over P2Y12R inhibitors by reducing the risk of bleeding. However, efforts to develop new drugs have been impeded by poor understanding of receptor-ligand interaction.
“The P2Y1R structures [we mapped] help us understand how this receptor and different types of experimental drugs interact at the molecular level and could enable further exploration to design new and safer antithrombotic drugs with reduced adverse effects,” Dr Wu said.
She and her colleagues found that the nucleotide ligand MRS2500 recognizes a binding site within the transmembrane bundle of P2Y1R. And it is different in shape and location from the nucleotide-binding site in P2Y12R.
“It is amazing to observe that 2 GPCRs recognize the same ligand in such different ways,” Dr Wu said. “The finding highlights the diversity of signal recognition mechanisms in GPCRs, and this is of great value to drug design for each receptor with high selectivity.”
The researchers also found that, instead of interacting within the transmembrane bundle, the non-nucleotide ligand BPTU binds to a pocket on the outer interface of P2Y1R embedded in the cell membrane.
This is the first structurally characterized, selective, and high-affinity GPCR ligand located entirely outside of the helical bundle, and it represents a new paradigm in ligand binding to alter signaling in GPCRs, according to the researchers.
The team believes this new understanding of the P2Y1R structure provides opportunities to broaden the scope of future GPCR drug discovery to target novel sites outside of the conventional GPCR ligand-binding pocket, which may improve drug selectivity and reduce side effects.
FDA strengthens warnings for anemia drug

Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program.
Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program.
Photo by Bill Branson
The US Food and Drug Administration (FDA) has strengthened an existing warning that serious, potentially fatal, allergic reactions can occur with the anemia drug Feraheme (ferumoxytol).
The FDA changed the drug’s prescribing information and approved a boxed warning detailing this risk.
The agency also added a new contraindication, which advises against the use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product.
The FDA said it is continuing to monitor and evaluate the risk of serious allergic reactions with all IV iron products, and the agency will update the public as new information becomes available.
About Feraheme
Feraheme is an IV iron replacement product used to treat iron-deficiency anemia in patients with chronic kidney disease. Like other IV iron products, Feraheme may only be given where emergency personnel and equipment are immediately available to treat the potentially life-threatening allergic reactions that can occur with treatment.
All IV iron products carry a risk of potentially life-threatening allergic reactions. At the time of Feraheme’s approval in 2009, this risk was described in the “Warnings and Precautions” section of the drug label.
Since then, serious reactions, including deaths, have occurred, despite the proper use of therapies to treat these reactions and emergency resuscitation measures.
Serious reactions reported
In the initial clinical trials of Feraheme, conducted predominantly in patients with chronic kidney disease, serious hypersensitivity reactions were reported in 0.2% (3/1726) of patients receiving Feraheme.
Other adverse reactions potentially associated with hypersensitivity (eg, pruritus, rash, urticaria, or wheezing) were reported in 3.7% (63/1726) of these patients.
In other trials that did not include patients with chronic kidney disease, moderate to severe hypersensitivity reactions, including anaphylaxis, were reported in 2.6% (26/1014) of patients treated with Feraheme.
Since the approval of Feraheme on June 30, 2009, cases of serious hypersensitivity reactions, including death, have occurred.
A search of the FDA Adverse Event Reporting System database revealed 79 cases of anaphylactic reactions associated with Feraheme administration, reported from the time of approval to June 30, 2014. Of the 79 cases, 18 were fatal, despite immediate medical intervention and emergency resuscitation attempts.
The 79 patients ranged in age from 19 to 96 years. In nearly half of all cases, the anaphylactic reactions occurred with the first dose of Feraheme. For approximately 75% (60/79) of the cases, the reaction began during the infusion or within 5 minutes after administration was complete.
Frequently reported symptoms included cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing. Of the 79 patients, 43% (34/79) had a medical history of drug allergy, and 24% had a history of multiple drug allergies.
Recommendations for administering Feraheme
Initial symptoms of fatal and serious hypersensitivity reactions associated with Feraheme may include hypotension, syncope, unresponsiveness, and cardiac/cardiorespiratory arrest, with or without signs of rash.
All IV iron products carry a risk of anaphylaxis, so these products should be administered only in patients who require IV iron therapy.
Feraheme is only approved for use in adults with iron-deficiency anemia in the setting of chronic kidney disease. The drug is contraindicated in patients with a history of hypersensitivity to Feraheme or any other IV iron product.
Only administer Feraheme and other IV iron products when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Feraheme should only be administered as an IV infusion in 50-200 mL of 0.9% sodium chloride or 5% dextrose over a minimum period of 15 minutes following dilution. Do not administer Feraheme by undiluted IV injection.
Closely monitor patients for signs and symptoms of hypersensitivity reactions, including monitoring blood pressure and pulse during administration and for at least 30 minutes following each infusion of Feraheme.
Elderly patients 65 years of age and older with multiple or serious comorbidities who experience hypersensitivity reactions, hypotension, or both following administration of Feraheme may have more severe outcomes.
Advise patients to immediately report any signs and symptoms of hypersensitivity that may develop during and following Feraheme administration, such as respiratory distress, hypotension, dizziness or lightheadedness, edema, rash, or itching. Advise patients to seek immediate medical attention if these signs and symptoms occur.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions, hypotension, or both, such as chemotherapeutic agents or monoclonal antibodies.
Report adverse events involving Feraheme to the FDA’s MedWatch Program.