User login
Inhibitor gets breakthrough designation for CLL
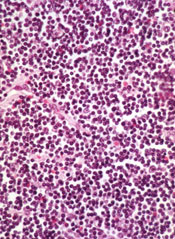
Though a safety issue previously slowed development of the BCL-2 inhibitor venetoclax (ABT-199), the drug is now moving through the pipeline.
The US Food and Drug Administration (FDA) has granted venetoclax breakthrough therapy designation to treat patients with relapsed or refractory chronic lymphocytic leukemia (CLL), including those with 17p deletion.
The drug has proven active against CLL and other hematologic malignancies.
However, it is also known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Now, the FDA has granted the drug breakthrough designation, which is intended to expedite the development and review of drugs indicated for serious or life-threatening conditions.
The criteria for breakthrough designation include preliminary clinical evidence suggesting the drug may offer substantial improvement on at least one clinically significant endpoint compared to available therapy.
Venetoclax in CLL/SLL
Results presented at the 2014 EHA Congress suggested that venetoclax can be effective in patients with CLL/small lymphocytic lymphoma (SLL), and certain measures can reduce the risk of TLS.
Researchers reported that modifying the dosing schedule of venetoclax, administering TLS prophylaxis, and monitoring patients can decrease or eliminate the risk of TLS. And venetoclax can produce responses in patients with high-risk disease.
In a phase 1 trial, the researchers tested venetoclax monotherapy in 105 patients with high-risk CLL/SLL. Seventy-eight patients were evaluable for treatment response as of April 2014. Nineteen of these patients had del (17p), 41 were fludarabine-refractory, and 24 had unmutated IGHV.
The response rate was 77% overall, 79% among patients with del (17p), 76% in patients who were fludarabine-refractory, and 75% in those with unmutated IGHV. The complete response rates were 23%, 26%, 22%, and 29%, respectively.
The median progression-free survival was about 18 months overall, but the median progression-free survival had not been reached for patients treated at or above 400 mg.
Seven patients developed TLS. One of these patients died, and 1 required dialysis. At the time of analysis, there were no cases of TLS among the 49 patients who received TLS prophylaxis and were given venetoclax according to the modified dosing schedule.
Common treatment-emergent adverse events included diarrhea (40%), neutropenia (36%), and nausea (35%). Grade 3/4 neutropenia occurred in 33% of patients, and febrile neutropenia occurred in 4%.
Thirty-seven patients discontinued treatment—22 due to progressive disease, 12 due to adverse events, and 3 for other reasons (1 required warfarin, and 2 proceeded to transplant).
Venetoclax is now being tested in phase 2 and 3 trials of CLL, as well as trials in other hematologic malignancies. Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Though a safety issue previously slowed development of the BCL-2 inhibitor venetoclax (ABT-199), the drug is now moving through the pipeline.
The US Food and Drug Administration (FDA) has granted venetoclax breakthrough therapy designation to treat patients with relapsed or refractory chronic lymphocytic leukemia (CLL), including those with 17p deletion.
The drug has proven active against CLL and other hematologic malignancies.
However, it is also known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Now, the FDA has granted the drug breakthrough designation, which is intended to expedite the development and review of drugs indicated for serious or life-threatening conditions.
The criteria for breakthrough designation include preliminary clinical evidence suggesting the drug may offer substantial improvement on at least one clinically significant endpoint compared to available therapy.
Venetoclax in CLL/SLL
Results presented at the 2014 EHA Congress suggested that venetoclax can be effective in patients with CLL/small lymphocytic lymphoma (SLL), and certain measures can reduce the risk of TLS.
Researchers reported that modifying the dosing schedule of venetoclax, administering TLS prophylaxis, and monitoring patients can decrease or eliminate the risk of TLS. And venetoclax can produce responses in patients with high-risk disease.
In a phase 1 trial, the researchers tested venetoclax monotherapy in 105 patients with high-risk CLL/SLL. Seventy-eight patients were evaluable for treatment response as of April 2014. Nineteen of these patients had del (17p), 41 were fludarabine-refractory, and 24 had unmutated IGHV.
The response rate was 77% overall, 79% among patients with del (17p), 76% in patients who were fludarabine-refractory, and 75% in those with unmutated IGHV. The complete response rates were 23%, 26%, 22%, and 29%, respectively.
The median progression-free survival was about 18 months overall, but the median progression-free survival had not been reached for patients treated at or above 400 mg.
Seven patients developed TLS. One of these patients died, and 1 required dialysis. At the time of analysis, there were no cases of TLS among the 49 patients who received TLS prophylaxis and were given venetoclax according to the modified dosing schedule.
Common treatment-emergent adverse events included diarrhea (40%), neutropenia (36%), and nausea (35%). Grade 3/4 neutropenia occurred in 33% of patients, and febrile neutropenia occurred in 4%.
Thirty-seven patients discontinued treatment—22 due to progressive disease, 12 due to adverse events, and 3 for other reasons (1 required warfarin, and 2 proceeded to transplant).
Venetoclax is now being tested in phase 2 and 3 trials of CLL, as well as trials in other hematologic malignancies. Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Though a safety issue previously slowed development of the BCL-2 inhibitor venetoclax (ABT-199), the drug is now moving through the pipeline.
The US Food and Drug Administration (FDA) has granted venetoclax breakthrough therapy designation to treat patients with relapsed or refractory chronic lymphocytic leukemia (CLL), including those with 17p deletion.
The drug has proven active against CLL and other hematologic malignancies.
However, it is also known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Now, the FDA has granted the drug breakthrough designation, which is intended to expedite the development and review of drugs indicated for serious or life-threatening conditions.
The criteria for breakthrough designation include preliminary clinical evidence suggesting the drug may offer substantial improvement on at least one clinically significant endpoint compared to available therapy.
Venetoclax in CLL/SLL
Results presented at the 2014 EHA Congress suggested that venetoclax can be effective in patients with CLL/small lymphocytic lymphoma (SLL), and certain measures can reduce the risk of TLS.
Researchers reported that modifying the dosing schedule of venetoclax, administering TLS prophylaxis, and monitoring patients can decrease or eliminate the risk of TLS. And venetoclax can produce responses in patients with high-risk disease.
In a phase 1 trial, the researchers tested venetoclax monotherapy in 105 patients with high-risk CLL/SLL. Seventy-eight patients were evaluable for treatment response as of April 2014. Nineteen of these patients had del (17p), 41 were fludarabine-refractory, and 24 had unmutated IGHV.
The response rate was 77% overall, 79% among patients with del (17p), 76% in patients who were fludarabine-refractory, and 75% in those with unmutated IGHV. The complete response rates were 23%, 26%, 22%, and 29%, respectively.
The median progression-free survival was about 18 months overall, but the median progression-free survival had not been reached for patients treated at or above 400 mg.
Seven patients developed TLS. One of these patients died, and 1 required dialysis. At the time of analysis, there were no cases of TLS among the 49 patients who received TLS prophylaxis and were given venetoclax according to the modified dosing schedule.
Common treatment-emergent adverse events included diarrhea (40%), neutropenia (36%), and nausea (35%). Grade 3/4 neutropenia occurred in 33% of patients, and febrile neutropenia occurred in 4%.
Thirty-seven patients discontinued treatment—22 due to progressive disease, 12 due to adverse events, and 3 for other reasons (1 required warfarin, and 2 proceeded to transplant).
Venetoclax is now being tested in phase 2 and 3 trials of CLL, as well as trials in other hematologic malignancies. Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Drug can alleviate transfusion dependence in non-del-5q MDS

Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Reversing glucocorticoid resistance in ALL

Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
Defining the role of TAMs in DLBCL
pseudopodia to engulf particles
PHILADELPHIA—New research suggests the prognostic value of tumor-associated macrophages (TAMs) is disease-specific as well as treatment-specific.
Investigators set out to determine if TAMs have a negative prognostic impact in diffuse large B-cell lymphoma (DLBCL), as previous studies produced conflicting results.
The team found that higher TAM levels are associated with worse survival in DLBCL, but only in patients who do not receive rituximab. The drug can overcome the poor prognosis TAMs confer in DLBCL.
Eri Matsuki, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, New York, and her colleagues presented these findings in a poster at the AACR Annual Meeting 2015 (abstract 2371*).
To ascertain the role of TAMs in DLBCL, the investigators analyzed specimens from 103 DLBCL patients, 61 of whom received rituximab, 33 who did not, and 9 whose rituximab status was unknown. The team first looked at the expression of CD163 as a marker of TAMs.
“CD163 is more specific to M2-type macrophages, which have pro-tumor effects, compared to a more pan-macrophage marker which is widely used—CD68,” Dr Matsuki noted.
She and her colleagues found that a high level of CD163-positive cells (more than 150) was significantly associated with advanced-stage disease (P=0.016), non-GCB DLBCL (P=0.0071), and higher expression of c-Myc (P=0.0022).
“Our interpretation of that at this point is that, the more aggressive the tumor, the more likely that it would induce macrophages into the tissue,” Dr Matsuki said.
The investigators then assessed the impact of rituximab use. Among patients who didn’t receive rituximab, having a high level of CD163-positive cells (more than 150) was associated with inferior overall survival (OS, P=0.022). However, if patients did receive rituximab, there was no significant difference in OS.
Dr Matsuki said this supports previous studies showing that the prognostic effect of TAMs diminishes with rituximab use, as well as the in vitro finding that M2 macrophages exhibit increased phagocytosis of rituximab-opsonized tumor cells.
“So overall, what we’re seeing is that . . . the negative influence of TAMs can be overcome with rituximab use,” she summarized.
Dr Matsuki and her colleagues also looked at the patients’ lymphocyte-to-monocyte-ratio (LMR) because TAMs partly arise from peripheral blood monocytes.
The team found that having an LMR higher than 2.77 was significantly associated with superior OS (P=0.03) in patients who did not receive rituximab. And there was a trend toward improved OS with a higher LMR in patients who did receive the drug (P=0.07).
The investigators believe the differences they observed in the prognostic value of CD163 and LMR could be explained by the fact that TAMs are derived from both circulating monocytes and resident macrophages.
Dr Matsuki said this research has improved her group’s understanding of DLBCL, but they are still working to identify additional biomarkers associated with prognosis in this disease. ![]()
*Information in the abstract differs from that presented at the meeting.
pseudopodia to engulf particles
PHILADELPHIA—New research suggests the prognostic value of tumor-associated macrophages (TAMs) is disease-specific as well as treatment-specific.
Investigators set out to determine if TAMs have a negative prognostic impact in diffuse large B-cell lymphoma (DLBCL), as previous studies produced conflicting results.
The team found that higher TAM levels are associated with worse survival in DLBCL, but only in patients who do not receive rituximab. The drug can overcome the poor prognosis TAMs confer in DLBCL.
Eri Matsuki, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, New York, and her colleagues presented these findings in a poster at the AACR Annual Meeting 2015 (abstract 2371*).
To ascertain the role of TAMs in DLBCL, the investigators analyzed specimens from 103 DLBCL patients, 61 of whom received rituximab, 33 who did not, and 9 whose rituximab status was unknown. The team first looked at the expression of CD163 as a marker of TAMs.
“CD163 is more specific to M2-type macrophages, which have pro-tumor effects, compared to a more pan-macrophage marker which is widely used—CD68,” Dr Matsuki noted.
She and her colleagues found that a high level of CD163-positive cells (more than 150) was significantly associated with advanced-stage disease (P=0.016), non-GCB DLBCL (P=0.0071), and higher expression of c-Myc (P=0.0022).
“Our interpretation of that at this point is that, the more aggressive the tumor, the more likely that it would induce macrophages into the tissue,” Dr Matsuki said.
The investigators then assessed the impact of rituximab use. Among patients who didn’t receive rituximab, having a high level of CD163-positive cells (more than 150) was associated with inferior overall survival (OS, P=0.022). However, if patients did receive rituximab, there was no significant difference in OS.
Dr Matsuki said this supports previous studies showing that the prognostic effect of TAMs diminishes with rituximab use, as well as the in vitro finding that M2 macrophages exhibit increased phagocytosis of rituximab-opsonized tumor cells.
“So overall, what we’re seeing is that . . . the negative influence of TAMs can be overcome with rituximab use,” she summarized.
Dr Matsuki and her colleagues also looked at the patients’ lymphocyte-to-monocyte-ratio (LMR) because TAMs partly arise from peripheral blood monocytes.
The team found that having an LMR higher than 2.77 was significantly associated with superior OS (P=0.03) in patients who did not receive rituximab. And there was a trend toward improved OS with a higher LMR in patients who did receive the drug (P=0.07).
The investigators believe the differences they observed in the prognostic value of CD163 and LMR could be explained by the fact that TAMs are derived from both circulating monocytes and resident macrophages.
Dr Matsuki said this research has improved her group’s understanding of DLBCL, but they are still working to identify additional biomarkers associated with prognosis in this disease. ![]()
*Information in the abstract differs from that presented at the meeting.
pseudopodia to engulf particles
PHILADELPHIA—New research suggests the prognostic value of tumor-associated macrophages (TAMs) is disease-specific as well as treatment-specific.
Investigators set out to determine if TAMs have a negative prognostic impact in diffuse large B-cell lymphoma (DLBCL), as previous studies produced conflicting results.
The team found that higher TAM levels are associated with worse survival in DLBCL, but only in patients who do not receive rituximab. The drug can overcome the poor prognosis TAMs confer in DLBCL.
Eri Matsuki, MD, PhD, of Memorial Sloan-Kettering Cancer Center in New York, New York, and her colleagues presented these findings in a poster at the AACR Annual Meeting 2015 (abstract 2371*).
To ascertain the role of TAMs in DLBCL, the investigators analyzed specimens from 103 DLBCL patients, 61 of whom received rituximab, 33 who did not, and 9 whose rituximab status was unknown. The team first looked at the expression of CD163 as a marker of TAMs.
“CD163 is more specific to M2-type macrophages, which have pro-tumor effects, compared to a more pan-macrophage marker which is widely used—CD68,” Dr Matsuki noted.
She and her colleagues found that a high level of CD163-positive cells (more than 150) was significantly associated with advanced-stage disease (P=0.016), non-GCB DLBCL (P=0.0071), and higher expression of c-Myc (P=0.0022).
“Our interpretation of that at this point is that, the more aggressive the tumor, the more likely that it would induce macrophages into the tissue,” Dr Matsuki said.
The investigators then assessed the impact of rituximab use. Among patients who didn’t receive rituximab, having a high level of CD163-positive cells (more than 150) was associated with inferior overall survival (OS, P=0.022). However, if patients did receive rituximab, there was no significant difference in OS.
Dr Matsuki said this supports previous studies showing that the prognostic effect of TAMs diminishes with rituximab use, as well as the in vitro finding that M2 macrophages exhibit increased phagocytosis of rituximab-opsonized tumor cells.
“So overall, what we’re seeing is that . . . the negative influence of TAMs can be overcome with rituximab use,” she summarized.
Dr Matsuki and her colleagues also looked at the patients’ lymphocyte-to-monocyte-ratio (LMR) because TAMs partly arise from peripheral blood monocytes.
The team found that having an LMR higher than 2.77 was significantly associated with superior OS (P=0.03) in patients who did not receive rituximab. And there was a trend toward improved OS with a higher LMR in patients who did receive the drug (P=0.07).
The investigators believe the differences they observed in the prognostic value of CD163 and LMR could be explained by the fact that TAMs are derived from both circulating monocytes and resident macrophages.
Dr Matsuki said this research has improved her group’s understanding of DLBCL, but they are still working to identify additional biomarkers associated with prognosis in this disease. ![]()
*Information in the abstract differs from that presented at the meeting.
New model simulates human MM

Photo by Ben Skála
By growing human tumors on chicken embryos, scientists have created a new model system for screening drugs to treat multiple myeloma (MM).
Several MM drug candidates have shown promising activity in this system, according to the researchers.
Gerold Untergasser, PhD, of Innsbruck Medical University in Austria, and his colleagues described how they developed the system in the Journal of Visualized Experiments.
First, the researchers prepared cultures of the MM cell lines OPM2 and RPMI 8226, as well as human mesenchymal stem cells. They then transfected MM cells with enhanced green fluorescent protein, making the cells easy to observe through a fluorescence microscope.
Next, the team cultured MM cells with mesenchymal cells and collagen to create 3-dimensional cell spheres, thereby simulating the natural microenvironment of the tumor.
The researchers then transferred their cell spheres to the outer membrane of chicken embryos. This choriallantoid membrane provides a suitable base for growing miniature human tumors in culture.
The team has introduced anti-myeloma drugs, such as bortezomib, into this system and assessed the drugs’ ability to target MM cells and prevent tumor growth and angiogenesis. They were also able to evaluate toxicity.
“The chicken egg is much easier to handle and cheaper than mice, and it reduces the number of animal experiments,” Dr Untergasser said. “In our new video publication, we give detailed information on how our system works.”
“We provide an easy-to-repeat protocol for broad use within the research community. Our vision for the future is that a separate test is performed for each patient to determine which drug is best suitable.” ![]()
Photo by Ben Skála
By growing human tumors on chicken embryos, scientists have created a new model system for screening drugs to treat multiple myeloma (MM).
Several MM drug candidates have shown promising activity in this system, according to the researchers.
Gerold Untergasser, PhD, of Innsbruck Medical University in Austria, and his colleagues described how they developed the system in the Journal of Visualized Experiments.
First, the researchers prepared cultures of the MM cell lines OPM2 and RPMI 8226, as well as human mesenchymal stem cells. They then transfected MM cells with enhanced green fluorescent protein, making the cells easy to observe through a fluorescence microscope.
Next, the team cultured MM cells with mesenchymal cells and collagen to create 3-dimensional cell spheres, thereby simulating the natural microenvironment of the tumor.
The researchers then transferred their cell spheres to the outer membrane of chicken embryos. This choriallantoid membrane provides a suitable base for growing miniature human tumors in culture.
The team has introduced anti-myeloma drugs, such as bortezomib, into this system and assessed the drugs’ ability to target MM cells and prevent tumor growth and angiogenesis. They were also able to evaluate toxicity.
“The chicken egg is much easier to handle and cheaper than mice, and it reduces the number of animal experiments,” Dr Untergasser said. “In our new video publication, we give detailed information on how our system works.”
“We provide an easy-to-repeat protocol for broad use within the research community. Our vision for the future is that a separate test is performed for each patient to determine which drug is best suitable.” ![]()
Photo by Ben Skála
By growing human tumors on chicken embryos, scientists have created a new model system for screening drugs to treat multiple myeloma (MM).
Several MM drug candidates have shown promising activity in this system, according to the researchers.
Gerold Untergasser, PhD, of Innsbruck Medical University in Austria, and his colleagues described how they developed the system in the Journal of Visualized Experiments.
First, the researchers prepared cultures of the MM cell lines OPM2 and RPMI 8226, as well as human mesenchymal stem cells. They then transfected MM cells with enhanced green fluorescent protein, making the cells easy to observe through a fluorescence microscope.
Next, the team cultured MM cells with mesenchymal cells and collagen to create 3-dimensional cell spheres, thereby simulating the natural microenvironment of the tumor.
The researchers then transferred their cell spheres to the outer membrane of chicken embryos. This choriallantoid membrane provides a suitable base for growing miniature human tumors in culture.
The team has introduced anti-myeloma drugs, such as bortezomib, into this system and assessed the drugs’ ability to target MM cells and prevent tumor growth and angiogenesis. They were also able to evaluate toxicity.
“The chicken egg is much easier to handle and cheaper than mice, and it reduces the number of animal experiments,” Dr Untergasser said. “In our new video publication, we give detailed information on how our system works.”
“We provide an easy-to-repeat protocol for broad use within the research community. Our vision for the future is that a separate test is performed for each patient to determine which drug is best suitable.” ![]()
Inhibitor improves OS in poor-prognosis MDS

WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
Drug shows promise for lower-risk MDS

WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS.
Whole blood better for cardiac surgery in young children

Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery.
Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery.
Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery.
Study shows importance of VTE screening

Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge.
Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge.
Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge.
FDA approves new drug for hemophilia B

The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com.
The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com.
The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com.