User login
Explaining obesity in cancer survivors

Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Antimalarial drug unavailable, CDC says

Photo courtesy of the FDA
The antimalarial drug chloroquine is not currently available from US suppliers, according to the Centers for Disease Control and Prevention (CDC).
The agency said it will provide updates as more information becomes available from the Food and Drug Administration.
Chloroquine is used as malaria treatment and prophylaxis, but hydroxychloroquine sulfate can be prescribed in place of chloroquine when indicated.
Healthcare providers who need assistance diagnosing or managing suspected or confirmed cases of malaria can call the CDC Malaria Hotline at 1-855-856-4713 (Monday through Friday, 9 am to 5pm, Eastern time).
For emergency consultation after hours, providers can call 1-770-488-7100 and ask to speak with a CDC Malaria Branch clinician. ![]()
Photo courtesy of the FDA
The antimalarial drug chloroquine is not currently available from US suppliers, according to the Centers for Disease Control and Prevention (CDC).
The agency said it will provide updates as more information becomes available from the Food and Drug Administration.
Chloroquine is used as malaria treatment and prophylaxis, but hydroxychloroquine sulfate can be prescribed in place of chloroquine when indicated.
Healthcare providers who need assistance diagnosing or managing suspected or confirmed cases of malaria can call the CDC Malaria Hotline at 1-855-856-4713 (Monday through Friday, 9 am to 5pm, Eastern time).
For emergency consultation after hours, providers can call 1-770-488-7100 and ask to speak with a CDC Malaria Branch clinician. ![]()
Photo courtesy of the FDA
The antimalarial drug chloroquine is not currently available from US suppliers, according to the Centers for Disease Control and Prevention (CDC).
The agency said it will provide updates as more information becomes available from the Food and Drug Administration.
Chloroquine is used as malaria treatment and prophylaxis, but hydroxychloroquine sulfate can be prescribed in place of chloroquine when indicated.
Healthcare providers who need assistance diagnosing or managing suspected or confirmed cases of malaria can call the CDC Malaria Hotline at 1-855-856-4713 (Monday through Friday, 9 am to 5pm, Eastern time).
For emergency consultation after hours, providers can call 1-770-488-7100 and ask to speak with a CDC Malaria Branch clinician. ![]()
Impotence drug could prevent malaria transmission
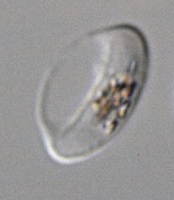
blood cell that has stiffened
after treatment
© 2015 Ramdani et al.
The erectile dysfunction drug sildenafil (Viagra) could prevent transmission of the malaria parasite, according to research published in PLOS Pathogens.
Investigators found that sildenafil increases the stiffness of erythrocytes infected by the parasite Plasmodium falciparum.
This allows the cells to be eliminated from the bloodstream and may therefore reduce transmission of the malaria parasite from humans to mosquitoes.
The investigators noted that P falciparum has a complex developmental cycle that is completed partly in humans and partly in mosquitoes. Treatments for malaria target the asexual forms of this parasite that cause symptoms, but not the sexual forms transmitted from a human to a mosquito.
Malaria eradication therefore necessitates new types of treatments against sexual forms of the parasite in order to block transmission and prevent dissemination of the disease.
The sexual forms of P falciparum develop in human erythrocytes sequestered in the bone marrow before they are released into the blood. They are then accessible to mosquitoes, which can ingest them when they bite.
Circulating erythrocytes are deformable, thus preventing their clearance via the spleen. And gametocyte-infected erythrocytes can easily pass through the spleen and persist for several days in the blood circulation.
With this in mind, Ghania Ramdani, of Université Paris Descartes in France, and colleagues sought to stiffen the infected erythrocytes so they would be removed from circulation.
The team found that the deformability of gametocyte-infected erythrocytes is regulated by a signaling pathway that involves cAMP. When cAMP molecules accumulate, an erythrocyte becomes stiffer. And cAMP is degraded by the enzyme phosphodiesterase, which promotes erythrocyte deformability.
Using an in vitro model reproducing filtration by the spleen, the investigators were able to identify several pharmacological agents that inhibit phosophodiesterases and can therefore increase the stiffness of infected erythrocytes.
One of these agents is sildenafil. The team showed that a standard dose of the drug had the potential to increase the stiffness of sexual forms of the parasite and therefore favor the elimination of infected erythrocytes from the circulation.
This discovery could lead to new ways to stop the spread of malaria, the investigators said. They believe that modifying the active substance in sildenafil to block its erectile effect, or testing similar agents devoid of this effect, could indeed result in a treatment to prevent transmission of the parasite from humans to mosquitoes. ![]()
blood cell that has stiffened
after treatment
© 2015 Ramdani et al.
The erectile dysfunction drug sildenafil (Viagra) could prevent transmission of the malaria parasite, according to research published in PLOS Pathogens.
Investigators found that sildenafil increases the stiffness of erythrocytes infected by the parasite Plasmodium falciparum.
This allows the cells to be eliminated from the bloodstream and may therefore reduce transmission of the malaria parasite from humans to mosquitoes.
The investigators noted that P falciparum has a complex developmental cycle that is completed partly in humans and partly in mosquitoes. Treatments for malaria target the asexual forms of this parasite that cause symptoms, but not the sexual forms transmitted from a human to a mosquito.
Malaria eradication therefore necessitates new types of treatments against sexual forms of the parasite in order to block transmission and prevent dissemination of the disease.
The sexual forms of P falciparum develop in human erythrocytes sequestered in the bone marrow before they are released into the blood. They are then accessible to mosquitoes, which can ingest them when they bite.
Circulating erythrocytes are deformable, thus preventing their clearance via the spleen. And gametocyte-infected erythrocytes can easily pass through the spleen and persist for several days in the blood circulation.
With this in mind, Ghania Ramdani, of Université Paris Descartes in France, and colleagues sought to stiffen the infected erythrocytes so they would be removed from circulation.
The team found that the deformability of gametocyte-infected erythrocytes is regulated by a signaling pathway that involves cAMP. When cAMP molecules accumulate, an erythrocyte becomes stiffer. And cAMP is degraded by the enzyme phosphodiesterase, which promotes erythrocyte deformability.
Using an in vitro model reproducing filtration by the spleen, the investigators were able to identify several pharmacological agents that inhibit phosophodiesterases and can therefore increase the stiffness of infected erythrocytes.
One of these agents is sildenafil. The team showed that a standard dose of the drug had the potential to increase the stiffness of sexual forms of the parasite and therefore favor the elimination of infected erythrocytes from the circulation.
This discovery could lead to new ways to stop the spread of malaria, the investigators said. They believe that modifying the active substance in sildenafil to block its erectile effect, or testing similar agents devoid of this effect, could indeed result in a treatment to prevent transmission of the parasite from humans to mosquitoes. ![]()
blood cell that has stiffened
after treatment
© 2015 Ramdani et al.
The erectile dysfunction drug sildenafil (Viagra) could prevent transmission of the malaria parasite, according to research published in PLOS Pathogens.
Investigators found that sildenafil increases the stiffness of erythrocytes infected by the parasite Plasmodium falciparum.
This allows the cells to be eliminated from the bloodstream and may therefore reduce transmission of the malaria parasite from humans to mosquitoes.
The investigators noted that P falciparum has a complex developmental cycle that is completed partly in humans and partly in mosquitoes. Treatments for malaria target the asexual forms of this parasite that cause symptoms, but not the sexual forms transmitted from a human to a mosquito.
Malaria eradication therefore necessitates new types of treatments against sexual forms of the parasite in order to block transmission and prevent dissemination of the disease.
The sexual forms of P falciparum develop in human erythrocytes sequestered in the bone marrow before they are released into the blood. They are then accessible to mosquitoes, which can ingest them when they bite.
Circulating erythrocytes are deformable, thus preventing their clearance via the spleen. And gametocyte-infected erythrocytes can easily pass through the spleen and persist for several days in the blood circulation.
With this in mind, Ghania Ramdani, of Université Paris Descartes in France, and colleagues sought to stiffen the infected erythrocytes so they would be removed from circulation.
The team found that the deformability of gametocyte-infected erythrocytes is regulated by a signaling pathway that involves cAMP. When cAMP molecules accumulate, an erythrocyte becomes stiffer. And cAMP is degraded by the enzyme phosphodiesterase, which promotes erythrocyte deformability.
Using an in vitro model reproducing filtration by the spleen, the investigators were able to identify several pharmacological agents that inhibit phosophodiesterases and can therefore increase the stiffness of infected erythrocytes.
One of these agents is sildenafil. The team showed that a standard dose of the drug had the potential to increase the stiffness of sexual forms of the parasite and therefore favor the elimination of infected erythrocytes from the circulation.
This discovery could lead to new ways to stop the spread of malaria, the investigators said. They believe that modifying the active substance in sildenafil to block its erectile effect, or testing similar agents devoid of this effect, could indeed result in a treatment to prevent transmission of the parasite from humans to mosquitoes. ![]()
Inhibitor may benefit certain ALL patients
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
Malaria vaccine proves partially protective
Photo by James Gathany
A new malaria vaccine candidate proved partially effective in adult males living in an area of low malaria transmission.
The T-cell vaccine consists of the recombinant viral vectors chimpanzee adenovirus 63 (ChAd63) and modified vaccinia Ankara (MVA), both encoding the multiple epitope string and thrombospondin-related adhesion protein (ME-TRAP), a fusion of protein fragments found on the surface of the malaria parasite Plasmodium falciparum.
In what’s known as a prime-boost strategy, an initial dose of the vaccine “primes” the immune system by exposing it to the malaria antigen and is followed by a vaccine booster, which re-stimulates the immune system to further solidify immunity.
Caroline Ogwang, of the Kenya Medical Research Institute–Wellcome Trust Research Programme in Kilifi, Kenya, and her colleagues described their trial of the vaccine in Science Translational Medicine.
The researchers tested the vaccine in an area of low malaria transmission in Kenya. They enrolled 121 healthy adult men and randomized them to receive the malaria vaccine or a control rabies vaccine.
The subjects were also treated with antimalarial drugs to clear any previous infection and were closely monitored for 8 weeks for P falciparum infection.
The malaria vaccine appeared to be safe, prompting no serious adverse events. The most common local adverse event associated with both ChAd63 ME-TRAP and MVA ME-TRAP was mild to moderate pain that lasted from a few hours to 3 days.
Subjects also experienced various systemic adverse events of mild to moderate intensity that lasted from a few hours to 5 days. Other events were reported within 30 days of vaccination as well, but these were not considered vaccine-related.
As for efficacy, blood tests revealed that the vaccine activated a strong immune response from T cells. For at least 2 weeks after vaccination, the vaccine showed partial efficacy in protecting against malaria infection.
The vaccine reduced subjects’ risk of infection by 67% (P=0.002) during the 8-week monitoring period. And the researchers said T-cell responses to TRAP peptides 21 to 30 were significantly associated with protection (hazard ratio=0.24, P=0.016).
The team is continuing clinical testing of the vaccine, including possible combinations with other vaccination strategies to increase efficacy. ![]()
Photo by James Gathany
A new malaria vaccine candidate proved partially effective in adult males living in an area of low malaria transmission.
The T-cell vaccine consists of the recombinant viral vectors chimpanzee adenovirus 63 (ChAd63) and modified vaccinia Ankara (MVA), both encoding the multiple epitope string and thrombospondin-related adhesion protein (ME-TRAP), a fusion of protein fragments found on the surface of the malaria parasite Plasmodium falciparum.
In what’s known as a prime-boost strategy, an initial dose of the vaccine “primes” the immune system by exposing it to the malaria antigen and is followed by a vaccine booster, which re-stimulates the immune system to further solidify immunity.
Caroline Ogwang, of the Kenya Medical Research Institute–Wellcome Trust Research Programme in Kilifi, Kenya, and her colleagues described their trial of the vaccine in Science Translational Medicine.
The researchers tested the vaccine in an area of low malaria transmission in Kenya. They enrolled 121 healthy adult men and randomized them to receive the malaria vaccine or a control rabies vaccine.
The subjects were also treated with antimalarial drugs to clear any previous infection and were closely monitored for 8 weeks for P falciparum infection.
The malaria vaccine appeared to be safe, prompting no serious adverse events. The most common local adverse event associated with both ChAd63 ME-TRAP and MVA ME-TRAP was mild to moderate pain that lasted from a few hours to 3 days.
Subjects also experienced various systemic adverse events of mild to moderate intensity that lasted from a few hours to 5 days. Other events were reported within 30 days of vaccination as well, but these were not considered vaccine-related.
As for efficacy, blood tests revealed that the vaccine activated a strong immune response from T cells. For at least 2 weeks after vaccination, the vaccine showed partial efficacy in protecting against malaria infection.
The vaccine reduced subjects’ risk of infection by 67% (P=0.002) during the 8-week monitoring period. And the researchers said T-cell responses to TRAP peptides 21 to 30 were significantly associated with protection (hazard ratio=0.24, P=0.016).
The team is continuing clinical testing of the vaccine, including possible combinations with other vaccination strategies to increase efficacy. ![]()
Photo by James Gathany
A new malaria vaccine candidate proved partially effective in adult males living in an area of low malaria transmission.
The T-cell vaccine consists of the recombinant viral vectors chimpanzee adenovirus 63 (ChAd63) and modified vaccinia Ankara (MVA), both encoding the multiple epitope string and thrombospondin-related adhesion protein (ME-TRAP), a fusion of protein fragments found on the surface of the malaria parasite Plasmodium falciparum.
In what’s known as a prime-boost strategy, an initial dose of the vaccine “primes” the immune system by exposing it to the malaria antigen and is followed by a vaccine booster, which re-stimulates the immune system to further solidify immunity.
Caroline Ogwang, of the Kenya Medical Research Institute–Wellcome Trust Research Programme in Kilifi, Kenya, and her colleagues described their trial of the vaccine in Science Translational Medicine.
The researchers tested the vaccine in an area of low malaria transmission in Kenya. They enrolled 121 healthy adult men and randomized them to receive the malaria vaccine or a control rabies vaccine.
The subjects were also treated with antimalarial drugs to clear any previous infection and were closely monitored for 8 weeks for P falciparum infection.
The malaria vaccine appeared to be safe, prompting no serious adverse events. The most common local adverse event associated with both ChAd63 ME-TRAP and MVA ME-TRAP was mild to moderate pain that lasted from a few hours to 3 days.
Subjects also experienced various systemic adverse events of mild to moderate intensity that lasted from a few hours to 5 days. Other events were reported within 30 days of vaccination as well, but these were not considered vaccine-related.
As for efficacy, blood tests revealed that the vaccine activated a strong immune response from T cells. For at least 2 weeks after vaccination, the vaccine showed partial efficacy in protecting against malaria infection.
The vaccine reduced subjects’ risk of infection by 67% (P=0.002) during the 8-week monitoring period. And the researchers said T-cell responses to TRAP peptides 21 to 30 were significantly associated with protection (hazard ratio=0.24, P=0.016).
The team is continuing clinical testing of the vaccine, including possible combinations with other vaccination strategies to increase efficacy. ![]()
Protein linked to leukemia, breast cancer

Photo courtesy of
The Scripps Research Institute
Overexpression of the cyclin E protein may cause leukemia and breast cancer, according to research published in Current Biology.
The study suggested that overexpression of cyclin E slows down DNA replication and introduces potentially harmful oncogenic mutations when cells divide.
“Overexpression of cyclin E is one route to cancer,” said study author Steven Reed, PhD, of The Scripps Research Institute in La Jolla, California.
Dr Reed and his colleagues originally discovered cyclin E, and their previous studies showed that abnormally high levels of cyclin E are associated with chromosome instability, increasing the chances that a chromosome will acquire more mutations as it divides.
The researchers found that cyclin E is frequently overexpressed in cancer cells, and that overexpression is linked to a decreased survival rate for breast cancer patients.
However, until they conducted the current study, the team didn’t know exactly how cyclin E introduces chromosome instability and errors into DNA.
DNA ‘tug-of-war’
The researchers investigated the role of cyclin E by comparing normal human mammary cells with mammary cells forced to overexpress cyclin E at the same levels seen in some breast cancer cells.
They found that DNA replication took significantly longer in the cyclin E-deregulated cells. In fact, the cells seemed to enter the next stage of cell division before the DNA was even done replicating. A small number (n=16) of very specific regions on the chromosomes frequently failed to complete replication.
The researchers then screened the cyclin E-deregulated cells for errors later in the cell division process. And they found that chromosomes of the deregulated cells’ daughter cells stuck together in the spots where replication had not finished.
“You could see a tug-of-war going on,” Dr Reed said. “That would cause either the chromosome to tear or both chromosomes to go to one side.”
The researchers spotted abnormal DNA “bridges” tying daughter cells together, as well as cells in which chunks of chromosomes ripped away and floated nearby. After these abnormal divisions took place, a third of the cyclin E-deregulated cells showed DNA deletions at the previously identified regions where replication failed.
The link to cancers
Next, the researchers investigated how the genetic instability from DNA deletions in cyclin E-deregulated cells could contribute to cancer. Many of the sites with DNA deletions were areas in which DNA was already known to be fragile or difficult to replicate.
Using a database of tumor DNA sequences, the team found that 6 of the 16 DNA regions they had identified in their cell-based studies showed damage in breast tumors that could be directly linked to cyclin E overexpression.
In addition, an area commonly damaged in cyclin E-deregulated cells matched up with an area commonly rearranged in mixed-lineage leukemia, where cyclin E had already been shown to be a contributing factor.
One of the unanswered questions posed by this work is how cells are allowed to divide before all the chromosomes are completely replicated. It was believed that “checkpoints” exist to prevent this from happening.
Dr Reed thinks these unreplicated regions are small enough to bypass the cellular checkpoints and keep cells dividing and accumulating potentially harmful mutations.
His team’s next step is to sequence the entire genomes of cells that undergo damage from cyclin E overexpression to understand exactly how the deletions contribute to cancer. ![]()
Photo courtesy of
The Scripps Research Institute
Overexpression of the cyclin E protein may cause leukemia and breast cancer, according to research published in Current Biology.
The study suggested that overexpression of cyclin E slows down DNA replication and introduces potentially harmful oncogenic mutations when cells divide.
“Overexpression of cyclin E is one route to cancer,” said study author Steven Reed, PhD, of The Scripps Research Institute in La Jolla, California.
Dr Reed and his colleagues originally discovered cyclin E, and their previous studies showed that abnormally high levels of cyclin E are associated with chromosome instability, increasing the chances that a chromosome will acquire more mutations as it divides.
The researchers found that cyclin E is frequently overexpressed in cancer cells, and that overexpression is linked to a decreased survival rate for breast cancer patients.
However, until they conducted the current study, the team didn’t know exactly how cyclin E introduces chromosome instability and errors into DNA.
DNA ‘tug-of-war’
The researchers investigated the role of cyclin E by comparing normal human mammary cells with mammary cells forced to overexpress cyclin E at the same levels seen in some breast cancer cells.
They found that DNA replication took significantly longer in the cyclin E-deregulated cells. In fact, the cells seemed to enter the next stage of cell division before the DNA was even done replicating. A small number (n=16) of very specific regions on the chromosomes frequently failed to complete replication.
The researchers then screened the cyclin E-deregulated cells for errors later in the cell division process. And they found that chromosomes of the deregulated cells’ daughter cells stuck together in the spots where replication had not finished.
“You could see a tug-of-war going on,” Dr Reed said. “That would cause either the chromosome to tear or both chromosomes to go to one side.”
The researchers spotted abnormal DNA “bridges” tying daughter cells together, as well as cells in which chunks of chromosomes ripped away and floated nearby. After these abnormal divisions took place, a third of the cyclin E-deregulated cells showed DNA deletions at the previously identified regions where replication failed.
The link to cancers
Next, the researchers investigated how the genetic instability from DNA deletions in cyclin E-deregulated cells could contribute to cancer. Many of the sites with DNA deletions were areas in which DNA was already known to be fragile or difficult to replicate.
Using a database of tumor DNA sequences, the team found that 6 of the 16 DNA regions they had identified in their cell-based studies showed damage in breast tumors that could be directly linked to cyclin E overexpression.
In addition, an area commonly damaged in cyclin E-deregulated cells matched up with an area commonly rearranged in mixed-lineage leukemia, where cyclin E had already been shown to be a contributing factor.
One of the unanswered questions posed by this work is how cells are allowed to divide before all the chromosomes are completely replicated. It was believed that “checkpoints” exist to prevent this from happening.
Dr Reed thinks these unreplicated regions are small enough to bypass the cellular checkpoints and keep cells dividing and accumulating potentially harmful mutations.
His team’s next step is to sequence the entire genomes of cells that undergo damage from cyclin E overexpression to understand exactly how the deletions contribute to cancer. ![]()
Photo courtesy of
The Scripps Research Institute
Overexpression of the cyclin E protein may cause leukemia and breast cancer, according to research published in Current Biology.
The study suggested that overexpression of cyclin E slows down DNA replication and introduces potentially harmful oncogenic mutations when cells divide.
“Overexpression of cyclin E is one route to cancer,” said study author Steven Reed, PhD, of The Scripps Research Institute in La Jolla, California.
Dr Reed and his colleagues originally discovered cyclin E, and their previous studies showed that abnormally high levels of cyclin E are associated with chromosome instability, increasing the chances that a chromosome will acquire more mutations as it divides.
The researchers found that cyclin E is frequently overexpressed in cancer cells, and that overexpression is linked to a decreased survival rate for breast cancer patients.
However, until they conducted the current study, the team didn’t know exactly how cyclin E introduces chromosome instability and errors into DNA.
DNA ‘tug-of-war’
The researchers investigated the role of cyclin E by comparing normal human mammary cells with mammary cells forced to overexpress cyclin E at the same levels seen in some breast cancer cells.
They found that DNA replication took significantly longer in the cyclin E-deregulated cells. In fact, the cells seemed to enter the next stage of cell division before the DNA was even done replicating. A small number (n=16) of very specific regions on the chromosomes frequently failed to complete replication.
The researchers then screened the cyclin E-deregulated cells for errors later in the cell division process. And they found that chromosomes of the deregulated cells’ daughter cells stuck together in the spots where replication had not finished.
“You could see a tug-of-war going on,” Dr Reed said. “That would cause either the chromosome to tear or both chromosomes to go to one side.”
The researchers spotted abnormal DNA “bridges” tying daughter cells together, as well as cells in which chunks of chromosomes ripped away and floated nearby. After these abnormal divisions took place, a third of the cyclin E-deregulated cells showed DNA deletions at the previously identified regions where replication failed.
The link to cancers
Next, the researchers investigated how the genetic instability from DNA deletions in cyclin E-deregulated cells could contribute to cancer. Many of the sites with DNA deletions were areas in which DNA was already known to be fragile or difficult to replicate.
Using a database of tumor DNA sequences, the team found that 6 of the 16 DNA regions they had identified in their cell-based studies showed damage in breast tumors that could be directly linked to cyclin E overexpression.
In addition, an area commonly damaged in cyclin E-deregulated cells matched up with an area commonly rearranged in mixed-lineage leukemia, where cyclin E had already been shown to be a contributing factor.
One of the unanswered questions posed by this work is how cells are allowed to divide before all the chromosomes are completely replicated. It was believed that “checkpoints” exist to prevent this from happening.
Dr Reed thinks these unreplicated regions are small enough to bypass the cellular checkpoints and keep cells dividing and accumulating potentially harmful mutations.
His team’s next step is to sequence the entire genomes of cells that undergo damage from cyclin E overexpression to understand exactly how the deletions contribute to cancer. ![]()
GIST patients have higher risk of NHL, other cancers

Photo courtesy of CDC
Patients with gastrointestinal stromal tumors (GIST) have an increased risk of developing non-Hodgkin lymphoma (NHL) and other cancers, a new study suggests.
About 1 in 6 of the patients studied were diagnosed with an additional malignancy.
The patients had an increased risk of other sarcomas, NHL, carcinoid tumors, melanoma, and colorectal, esophageal, pancreatic, hepatobiliary, non-small cell lung, prostate, and renal cell cancers.
“Only 5% of patients with gastrointestinal stromal tumors have a hereditary disorder that predisposes them to develop multiple benign and malignant tumors,” said study author Jason K. Sicklick, MD, of the University of California San Diego Moores Cancer Center.
“The research indicates that these patients may develop cancers outside of these syndromes, but the exact mechanisms are not yet known.”
Dr Sicklick and his colleagues described their research in Cancer.
The team analyzed 6112 GIST patients and found that 1047 of them (17.1%) had additional cancers.
When compared to the general US population, patients had a 44% increased prevalence of cancers occurring before a GIST diagnosis and a 66% higher risk of developing cancers after GIST diagnosis.
That corresponds to a standardized prevalence ratio (SPR) of 1.44 (risk before GIST diagnosis) and a standardized incidence ratio (SIR) of 1.66 (risk after GIST diagnosis).
Both before and after GIST diagnosis, patients had a significantly increased risk of NHL (SPR=1.69, SIR=1.76), other sarcomas (SPR=5.24, SIR=4.02), neuroendocrine-carcinoid tumors (SPR=3.56, SIR=4.79), and colorectal adenocarcinoma (SPR=1.51, SIR=2.16).
Before GIST diagnosis, patients had an increased risk of esophageal adenocarcinoma (SPR=12.0), bladder adenocarcinoma (SPR=7.51), melanoma (SPR=1.46), and prostate adenocarcinoma (SPR=1.20).
And after GIST diagnosis, they had an increased risk of ovarian carcinoma (SIR=8.72), small intestine adenocarcinoma (SIR=5.89), papillary thyroid cancer (SIR=5.16), renal cell carcinoma (SIR=4.46), hepatobiliary adenocarcinoma (SIR=3.10), gastric adenocarcinoma (SIR=2.70), pancreatic adenocarcinoma (SIR=2.03), uterine adenocarcinoma (SIR=1.96), non-small cell lung cancer (SIR=1.74), and transitional cell carcinoma of the bladder (SIR=1.65).
The researchers said further studies are needed to understand the connection between GIST and other cancers, but these findings may have clinical implications.
“Patients diagnosed with gastrointestinal stromal tumors may warrant consideration for additional screenings based on the other cancers that they are most susceptible to contract,” said James D. Murphy, MD, also of the University of California San Diego Moores Cancer Center. ![]()
Photo courtesy of CDC
Patients with gastrointestinal stromal tumors (GIST) have an increased risk of developing non-Hodgkin lymphoma (NHL) and other cancers, a new study suggests.
About 1 in 6 of the patients studied were diagnosed with an additional malignancy.
The patients had an increased risk of other sarcomas, NHL, carcinoid tumors, melanoma, and colorectal, esophageal, pancreatic, hepatobiliary, non-small cell lung, prostate, and renal cell cancers.
“Only 5% of patients with gastrointestinal stromal tumors have a hereditary disorder that predisposes them to develop multiple benign and malignant tumors,” said study author Jason K. Sicklick, MD, of the University of California San Diego Moores Cancer Center.
“The research indicates that these patients may develop cancers outside of these syndromes, but the exact mechanisms are not yet known.”
Dr Sicklick and his colleagues described their research in Cancer.
The team analyzed 6112 GIST patients and found that 1047 of them (17.1%) had additional cancers.
When compared to the general US population, patients had a 44% increased prevalence of cancers occurring before a GIST diagnosis and a 66% higher risk of developing cancers after GIST diagnosis.
That corresponds to a standardized prevalence ratio (SPR) of 1.44 (risk before GIST diagnosis) and a standardized incidence ratio (SIR) of 1.66 (risk after GIST diagnosis).
Both before and after GIST diagnosis, patients had a significantly increased risk of NHL (SPR=1.69, SIR=1.76), other sarcomas (SPR=5.24, SIR=4.02), neuroendocrine-carcinoid tumors (SPR=3.56, SIR=4.79), and colorectal adenocarcinoma (SPR=1.51, SIR=2.16).
Before GIST diagnosis, patients had an increased risk of esophageal adenocarcinoma (SPR=12.0), bladder adenocarcinoma (SPR=7.51), melanoma (SPR=1.46), and prostate adenocarcinoma (SPR=1.20).
And after GIST diagnosis, they had an increased risk of ovarian carcinoma (SIR=8.72), small intestine adenocarcinoma (SIR=5.89), papillary thyroid cancer (SIR=5.16), renal cell carcinoma (SIR=4.46), hepatobiliary adenocarcinoma (SIR=3.10), gastric adenocarcinoma (SIR=2.70), pancreatic adenocarcinoma (SIR=2.03), uterine adenocarcinoma (SIR=1.96), non-small cell lung cancer (SIR=1.74), and transitional cell carcinoma of the bladder (SIR=1.65).
The researchers said further studies are needed to understand the connection between GIST and other cancers, but these findings may have clinical implications.
“Patients diagnosed with gastrointestinal stromal tumors may warrant consideration for additional screenings based on the other cancers that they are most susceptible to contract,” said James D. Murphy, MD, also of the University of California San Diego Moores Cancer Center. ![]()
Photo courtesy of CDC
Patients with gastrointestinal stromal tumors (GIST) have an increased risk of developing non-Hodgkin lymphoma (NHL) and other cancers, a new study suggests.
About 1 in 6 of the patients studied were diagnosed with an additional malignancy.
The patients had an increased risk of other sarcomas, NHL, carcinoid tumors, melanoma, and colorectal, esophageal, pancreatic, hepatobiliary, non-small cell lung, prostate, and renal cell cancers.
“Only 5% of patients with gastrointestinal stromal tumors have a hereditary disorder that predisposes them to develop multiple benign and malignant tumors,” said study author Jason K. Sicklick, MD, of the University of California San Diego Moores Cancer Center.
“The research indicates that these patients may develop cancers outside of these syndromes, but the exact mechanisms are not yet known.”
Dr Sicklick and his colleagues described their research in Cancer.
The team analyzed 6112 GIST patients and found that 1047 of them (17.1%) had additional cancers.
When compared to the general US population, patients had a 44% increased prevalence of cancers occurring before a GIST diagnosis and a 66% higher risk of developing cancers after GIST diagnosis.
That corresponds to a standardized prevalence ratio (SPR) of 1.44 (risk before GIST diagnosis) and a standardized incidence ratio (SIR) of 1.66 (risk after GIST diagnosis).
Both before and after GIST diagnosis, patients had a significantly increased risk of NHL (SPR=1.69, SIR=1.76), other sarcomas (SPR=5.24, SIR=4.02), neuroendocrine-carcinoid tumors (SPR=3.56, SIR=4.79), and colorectal adenocarcinoma (SPR=1.51, SIR=2.16).
Before GIST diagnosis, patients had an increased risk of esophageal adenocarcinoma (SPR=12.0), bladder adenocarcinoma (SPR=7.51), melanoma (SPR=1.46), and prostate adenocarcinoma (SPR=1.20).
And after GIST diagnosis, they had an increased risk of ovarian carcinoma (SIR=8.72), small intestine adenocarcinoma (SIR=5.89), papillary thyroid cancer (SIR=5.16), renal cell carcinoma (SIR=4.46), hepatobiliary adenocarcinoma (SIR=3.10), gastric adenocarcinoma (SIR=2.70), pancreatic adenocarcinoma (SIR=2.03), uterine adenocarcinoma (SIR=1.96), non-small cell lung cancer (SIR=1.74), and transitional cell carcinoma of the bladder (SIR=1.65).
The researchers said further studies are needed to understand the connection between GIST and other cancers, but these findings may have clinical implications.
“Patients diagnosed with gastrointestinal stromal tumors may warrant consideration for additional screenings based on the other cancers that they are most susceptible to contract,” said James D. Murphy, MD, also of the University of California San Diego Moores Cancer Center.
Study reveals ‘doorway’ into RBCs
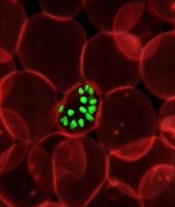
infecting an RBC
Photo courtesy of St. Jude
Children’s Research Hospital
A protein on the surface of red blood cells (RBCs) serves as an essential entry point for malaria parasite invasion, according to researchers.
They found the presence of this protein, CD55, was critical to the Plasmodium falciparum parasite’s ability to attach itself to the RBC surface.
The team believes this discovery, published in Science, opens up a promising new avenue for developing therapies to treat and prevent malaria.
“Plasmodium falciparum malaria parasites have evolved several key-like molecules to enter into human red blood cells through different door-like host receptors,” said study author Manoj Duraisingh, PhD, of the Harvard T. H. Chan School of Public Health in Boston, Massachusetts.
“Hence, if one red blood cell door is blocked, the parasite finds another way to enter. We have now identified an essential host factor which, when removed, prevents all parasite strains from entering red blood cells.”
The researchers accomplished this by developing a new technique to tap into a relatively unexplored area: identifying characteristics of a host RBC that make it susceptible to parasites. RBCs are difficult targets for such efforts as they lack a nucleus, which makes genetic manipulation impossible.
So the team transformed stem cells into RBCs, which allowed them to conduct a genetic screen for host determinants of P falciparum infection. They found that malaria parasites failed to attach properly to the surface of RBCs that lacked CD55.
The protein was required for invasion in all tested strains of the parasite, including those developed in a lab and those isolated from patients. This makes CD55 a primary candidate for intervention, the researchers said.
“The discovery of CD55 as an essential host factor for P falciparum raises the intriguing possibility of host-directed therapeutics for malaria, as is used in HIV,” said study author Elizabeth Egan, MD, PhD, also of the Harvard T. H. Chan School of Public Health.
“CD55 also gives us a hook with which to search for new parasite proteins important for invasion, which could serve as vaccine targets.”
infecting an RBC
Photo courtesy of St. Jude
Children’s Research Hospital
A protein on the surface of red blood cells (RBCs) serves as an essential entry point for malaria parasite invasion, according to researchers.
They found the presence of this protein, CD55, was critical to the Plasmodium falciparum parasite’s ability to attach itself to the RBC surface.
The team believes this discovery, published in Science, opens up a promising new avenue for developing therapies to treat and prevent malaria.
“Plasmodium falciparum malaria parasites have evolved several key-like molecules to enter into human red blood cells through different door-like host receptors,” said study author Manoj Duraisingh, PhD, of the Harvard T. H. Chan School of Public Health in Boston, Massachusetts.
“Hence, if one red blood cell door is blocked, the parasite finds another way to enter. We have now identified an essential host factor which, when removed, prevents all parasite strains from entering red blood cells.”
The researchers accomplished this by developing a new technique to tap into a relatively unexplored area: identifying characteristics of a host RBC that make it susceptible to parasites. RBCs are difficult targets for such efforts as they lack a nucleus, which makes genetic manipulation impossible.
So the team transformed stem cells into RBCs, which allowed them to conduct a genetic screen for host determinants of P falciparum infection. They found that malaria parasites failed to attach properly to the surface of RBCs that lacked CD55.
The protein was required for invasion in all tested strains of the parasite, including those developed in a lab and those isolated from patients. This makes CD55 a primary candidate for intervention, the researchers said.
“The discovery of CD55 as an essential host factor for P falciparum raises the intriguing possibility of host-directed therapeutics for malaria, as is used in HIV,” said study author Elizabeth Egan, MD, PhD, also of the Harvard T. H. Chan School of Public Health.
“CD55 also gives us a hook with which to search for new parasite proteins important for invasion, which could serve as vaccine targets.”
infecting an RBC
Photo courtesy of St. Jude
Children’s Research Hospital
A protein on the surface of red blood cells (RBCs) serves as an essential entry point for malaria parasite invasion, according to researchers.
They found the presence of this protein, CD55, was critical to the Plasmodium falciparum parasite’s ability to attach itself to the RBC surface.
The team believes this discovery, published in Science, opens up a promising new avenue for developing therapies to treat and prevent malaria.
“Plasmodium falciparum malaria parasites have evolved several key-like molecules to enter into human red blood cells through different door-like host receptors,” said study author Manoj Duraisingh, PhD, of the Harvard T. H. Chan School of Public Health in Boston, Massachusetts.
“Hence, if one red blood cell door is blocked, the parasite finds another way to enter. We have now identified an essential host factor which, when removed, prevents all parasite strains from entering red blood cells.”
The researchers accomplished this by developing a new technique to tap into a relatively unexplored area: identifying characteristics of a host RBC that make it susceptible to parasites. RBCs are difficult targets for such efforts as they lack a nucleus, which makes genetic manipulation impossible.
So the team transformed stem cells into RBCs, which allowed them to conduct a genetic screen for host determinants of P falciparum infection. They found that malaria parasites failed to attach properly to the surface of RBCs that lacked CD55.
The protein was required for invasion in all tested strains of the parasite, including those developed in a lab and those isolated from patients. This makes CD55 a primary candidate for intervention, the researchers said.
“The discovery of CD55 as an essential host factor for P falciparum raises the intriguing possibility of host-directed therapeutics for malaria, as is used in HIV,” said study author Elizabeth Egan, MD, PhD, also of the Harvard T. H. Chan School of Public Health.
“CD55 also gives us a hook with which to search for new parasite proteins important for invasion, which could serve as vaccine targets.”
Tool identifies CNAs other algorithms miss

Image courtesy of NIGMS
A new tool can detect genetic alterations that have proven difficult to identify, according to research published in Nature Methods.
The tool is an algorithm called CONSERTING (Copy Number Segmentation by Regression Tree in Next-Generation Sequencing).
Researchers created CONSERTING to improve their ability to detect copy number alterations (CNAs) in the information generated by whole-genome sequencing techniques.
The group showed that CONSERTING could detect CNAs with better accuracy and sensitivity than other techniques, including 4 published algorithms used to recognize CNAs in whole-genome sequencing data.
The data the team analyzed encompassed the normal and tumor genomes from 43 children and adults with leukemia, brain tumors, melanoma, and retinoblastoma.
“CONSERTING helped us identify alterations that other algorithms missed, including previously undetected chromosomal rearrangements and copy number alterations present in a small percentage of tumor cells,” said study author Xiang Chen, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“[C]ONSERTING identified copy number alterations in children with 100 times greater precision and 10 times greater precision in adults,” added Jinghui Zhang, PhD, also of St. Jude.
Using CONSERTING, the researchers discovered genetic alterations driving pediatric leukemia, low-grade glioma, glioblastoma, and retinoblastoma.
The algorithm also helped the team identify genetic changes that are present in a small percentage of a tumor’s cells. The alterations may be the key to understanding why tumors sometimes return after treatment, they said.
Dr Zhang said CONSERTING should make it easier to track the evolution of tumors with complex genetic rearrangements.
St. Jude has made CONSERTING available to researchers free of charge. The software, user manual, and related data can be downloaded from http://www.stjuderesearch.org/site/lab/zhang.
St. Jude researchers have also developed a cloud version of CONSERTING and related tools that can be accessed through Amazon Web Services. Instead of downloading CONSERTING, scientists can upload data for analysis.
Creating CONSERTING
Work on CONSERTING began in 2010, shortly after the St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project was launched. The Pediatric Cancer Genome Project used next-generation, whole-genome sequencing to study some of the most aggressive and least understood childhood cancers.
Early on in the project, researchers realized that existing analytic methods often missed duplications or deletions of DNA segments, particularly small changes that involve a handful of genes and provide insight into the origins of a patient’s cancer.
CONSERTING has now been used to analyze data for the Pediatric Cancer Genome Project. The project includes the normal and cancer genomes of 700 pediatric cancer patients with 21 different cancer subtypes.
CONSERTING combines a method of data analysis called regression tree, which is a machine-learning algorithm, with next-generation, whole-genome sequencing. Machine learning capitalizes on advances in computing to design algorithms that repeatedly and rapidly analyze large, complex sets of data and unearth unexpected insights.
“This combination has provided us with a powerful tool for recognizing copy number alterations, even those present in relatively few cells or in tumor samples that include normal cells along with tumor cells,” Dr Zhang said.
Next-generation, whole-genome sequencing involves breaking the human genome into about 1 billion pieces that are copied and reassembled using the normal genome as a template.
CONSERTING software compensates for gaps and variations in sequencing data. The sequencing data is integrated with information about the chromosomal rearrangements to find CNAs and identify their origins in the genome.
Image courtesy of NIGMS
A new tool can detect genetic alterations that have proven difficult to identify, according to research published in Nature Methods.
The tool is an algorithm called CONSERTING (Copy Number Segmentation by Regression Tree in Next-Generation Sequencing).
Researchers created CONSERTING to improve their ability to detect copy number alterations (CNAs) in the information generated by whole-genome sequencing techniques.
The group showed that CONSERTING could detect CNAs with better accuracy and sensitivity than other techniques, including 4 published algorithms used to recognize CNAs in whole-genome sequencing data.
The data the team analyzed encompassed the normal and tumor genomes from 43 children and adults with leukemia, brain tumors, melanoma, and retinoblastoma.
“CONSERTING helped us identify alterations that other algorithms missed, including previously undetected chromosomal rearrangements and copy number alterations present in a small percentage of tumor cells,” said study author Xiang Chen, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“[C]ONSERTING identified copy number alterations in children with 100 times greater precision and 10 times greater precision in adults,” added Jinghui Zhang, PhD, also of St. Jude.
Using CONSERTING, the researchers discovered genetic alterations driving pediatric leukemia, low-grade glioma, glioblastoma, and retinoblastoma.
The algorithm also helped the team identify genetic changes that are present in a small percentage of a tumor’s cells. The alterations may be the key to understanding why tumors sometimes return after treatment, they said.
Dr Zhang said CONSERTING should make it easier to track the evolution of tumors with complex genetic rearrangements.
St. Jude has made CONSERTING available to researchers free of charge. The software, user manual, and related data can be downloaded from http://www.stjuderesearch.org/site/lab/zhang.
St. Jude researchers have also developed a cloud version of CONSERTING and related tools that can be accessed through Amazon Web Services. Instead of downloading CONSERTING, scientists can upload data for analysis.
Creating CONSERTING
Work on CONSERTING began in 2010, shortly after the St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project was launched. The Pediatric Cancer Genome Project used next-generation, whole-genome sequencing to study some of the most aggressive and least understood childhood cancers.
Early on in the project, researchers realized that existing analytic methods often missed duplications or deletions of DNA segments, particularly small changes that involve a handful of genes and provide insight into the origins of a patient’s cancer.
CONSERTING has now been used to analyze data for the Pediatric Cancer Genome Project. The project includes the normal and cancer genomes of 700 pediatric cancer patients with 21 different cancer subtypes.
CONSERTING combines a method of data analysis called regression tree, which is a machine-learning algorithm, with next-generation, whole-genome sequencing. Machine learning capitalizes on advances in computing to design algorithms that repeatedly and rapidly analyze large, complex sets of data and unearth unexpected insights.
“This combination has provided us with a powerful tool for recognizing copy number alterations, even those present in relatively few cells or in tumor samples that include normal cells along with tumor cells,” Dr Zhang said.
Next-generation, whole-genome sequencing involves breaking the human genome into about 1 billion pieces that are copied and reassembled using the normal genome as a template.
CONSERTING software compensates for gaps and variations in sequencing data. The sequencing data is integrated with information about the chromosomal rearrangements to find CNAs and identify their origins in the genome.
Image courtesy of NIGMS
A new tool can detect genetic alterations that have proven difficult to identify, according to research published in Nature Methods.
The tool is an algorithm called CONSERTING (Copy Number Segmentation by Regression Tree in Next-Generation Sequencing).
Researchers created CONSERTING to improve their ability to detect copy number alterations (CNAs) in the information generated by whole-genome sequencing techniques.
The group showed that CONSERTING could detect CNAs with better accuracy and sensitivity than other techniques, including 4 published algorithms used to recognize CNAs in whole-genome sequencing data.
The data the team analyzed encompassed the normal and tumor genomes from 43 children and adults with leukemia, brain tumors, melanoma, and retinoblastoma.
“CONSERTING helped us identify alterations that other algorithms missed, including previously undetected chromosomal rearrangements and copy number alterations present in a small percentage of tumor cells,” said study author Xiang Chen, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“[C]ONSERTING identified copy number alterations in children with 100 times greater precision and 10 times greater precision in adults,” added Jinghui Zhang, PhD, also of St. Jude.
Using CONSERTING, the researchers discovered genetic alterations driving pediatric leukemia, low-grade glioma, glioblastoma, and retinoblastoma.
The algorithm also helped the team identify genetic changes that are present in a small percentage of a tumor’s cells. The alterations may be the key to understanding why tumors sometimes return after treatment, they said.
Dr Zhang said CONSERTING should make it easier to track the evolution of tumors with complex genetic rearrangements.
St. Jude has made CONSERTING available to researchers free of charge. The software, user manual, and related data can be downloaded from http://www.stjuderesearch.org/site/lab/zhang.
St. Jude researchers have also developed a cloud version of CONSERTING and related tools that can be accessed through Amazon Web Services. Instead of downloading CONSERTING, scientists can upload data for analysis.
Creating CONSERTING
Work on CONSERTING began in 2010, shortly after the St. Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project was launched. The Pediatric Cancer Genome Project used next-generation, whole-genome sequencing to study some of the most aggressive and least understood childhood cancers.
Early on in the project, researchers realized that existing analytic methods often missed duplications or deletions of DNA segments, particularly small changes that involve a handful of genes and provide insight into the origins of a patient’s cancer.
CONSERTING has now been used to analyze data for the Pediatric Cancer Genome Project. The project includes the normal and cancer genomes of 700 pediatric cancer patients with 21 different cancer subtypes.
CONSERTING combines a method of data analysis called regression tree, which is a machine-learning algorithm, with next-generation, whole-genome sequencing. Machine learning capitalizes on advances in computing to design algorithms that repeatedly and rapidly analyze large, complex sets of data and unearth unexpected insights.
“This combination has provided us with a powerful tool for recognizing copy number alterations, even those present in relatively few cells or in tumor samples that include normal cells along with tumor cells,” Dr Zhang said.
Next-generation, whole-genome sequencing involves breaking the human genome into about 1 billion pieces that are copied and reassembled using the normal genome as a template.
CONSERTING software compensates for gaps and variations in sequencing data. The sequencing data is integrated with information about the chromosomal rearrangements to find CNAs and identify their origins in the genome.
Agent preferentially targets FLT3-ITD AML

PHILADELPHIA—Preclinical research suggests a novel agent has preferential activity in acute myeloid leukemia (AML) with FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations.
The agent, VNLG-152, proved more cytotoxic in AML cell lines and patient samples with FLT3-ITD than in samples and cell lines with wild-type FLT3.
Exactly how and why this occurs remains somewhat of a mystery, however.
Sheetal Karne, MD, of the University of Maryland School of Medicine in Baltimore, and her colleagues detailed this mystery in a poster presentation at the AACR Annual Meeting 2015 (abstract 5408*).
Dr Karne noted that VNLG-152 targets translation by promoting the degradation of MAPK-interacting kinases (Mnks).
“[VNLG-152] has been previously published as functioning in Mnk degradation, which has been shown in triple-negative breast cancer and prostate cancer—in vivo and in vitro,” she said. “Our hypothesis was that, since [the drug] worked via decreasing translation, it would function in leukemia cells and, specifically, in leukemic cells with ITD mutations.”
So the investigators tested VNLG-152 in samples from AML patients, as well as both murine and human cell lines. They found that VNLG-152 was more cytotoxic in the presence of FLT3-ITD mutations, as evidenced by low micromolar IC50 concentrations.
The IC50 concentration was 3.4 μM in Ba/F3-ITD cells and 5.8 μM in Ba/F3-WT cells, which are murine cells transfected with human FLT3-ITD and wild-type FLT3, respectively. Similarly, the IC50 concentration was 1.8 μM in 32D-ITD cells and 18.2 μM in 32D-WT cells.
In the human FLT3-ITD AML cell lines MV4-11 and MOLM-14, IC50 concentrations were 2.3 μM and 4.2 μM, respectively. But concentrations were greater than 10 µM in the wild-type FLT3 human cell lines HL60 and U937.
In patient samples, the IC50 concentration was 1.0 μM in FLT3-ITD AML and 7.5 μM in AML with wild-type FLT3.
In additional tests with murine cell lines, the investigators found that VNLG-152 inhibits the growth of Ba/F3-ITD and Ba/F3-WT cells. But the drug induces apoptosis in these cell lines only when given in high concentrations.
Looking into the mechanism of VNLG-152, the investigators found that the drug decreased Mnk-1 expression in Ba/F3-ITD and Ba/F3-WT cell lines.
“We saw that VNLG-152 worked via degradation of Mnk, but it was the same in both wild-type and ITD, so we’re still looking for an explanation as to what caused this difference,” Dr Karne said.
She and her colleagues believe the Mnk degradation inhibits the phosphorylation of eukaryotic translation initiation factor 4E (eIF4E), a downstream target of FLT3-ITD. But they are still investigating that possibility.
The team is also hoping to test VNLG-152 in combination with other drugs, such as FLT3 inhibitors or chemotherapeutic agents.
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Preclinical research suggests a novel agent has preferential activity in acute myeloid leukemia (AML) with FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations.
The agent, VNLG-152, proved more cytotoxic in AML cell lines and patient samples with FLT3-ITD than in samples and cell lines with wild-type FLT3.
Exactly how and why this occurs remains somewhat of a mystery, however.
Sheetal Karne, MD, of the University of Maryland School of Medicine in Baltimore, and her colleagues detailed this mystery in a poster presentation at the AACR Annual Meeting 2015 (abstract 5408*).
Dr Karne noted that VNLG-152 targets translation by promoting the degradation of MAPK-interacting kinases (Mnks).
“[VNLG-152] has been previously published as functioning in Mnk degradation, which has been shown in triple-negative breast cancer and prostate cancer—in vivo and in vitro,” she said. “Our hypothesis was that, since [the drug] worked via decreasing translation, it would function in leukemia cells and, specifically, in leukemic cells with ITD mutations.”
So the investigators tested VNLG-152 in samples from AML patients, as well as both murine and human cell lines. They found that VNLG-152 was more cytotoxic in the presence of FLT3-ITD mutations, as evidenced by low micromolar IC50 concentrations.
The IC50 concentration was 3.4 μM in Ba/F3-ITD cells and 5.8 μM in Ba/F3-WT cells, which are murine cells transfected with human FLT3-ITD and wild-type FLT3, respectively. Similarly, the IC50 concentration was 1.8 μM in 32D-ITD cells and 18.2 μM in 32D-WT cells.
In the human FLT3-ITD AML cell lines MV4-11 and MOLM-14, IC50 concentrations were 2.3 μM and 4.2 μM, respectively. But concentrations were greater than 10 µM in the wild-type FLT3 human cell lines HL60 and U937.
In patient samples, the IC50 concentration was 1.0 μM in FLT3-ITD AML and 7.5 μM in AML with wild-type FLT3.
In additional tests with murine cell lines, the investigators found that VNLG-152 inhibits the growth of Ba/F3-ITD and Ba/F3-WT cells. But the drug induces apoptosis in these cell lines only when given in high concentrations.
Looking into the mechanism of VNLG-152, the investigators found that the drug decreased Mnk-1 expression in Ba/F3-ITD and Ba/F3-WT cell lines.
“We saw that VNLG-152 worked via degradation of Mnk, but it was the same in both wild-type and ITD, so we’re still looking for an explanation as to what caused this difference,” Dr Karne said.
She and her colleagues believe the Mnk degradation inhibits the phosphorylation of eukaryotic translation initiation factor 4E (eIF4E), a downstream target of FLT3-ITD. But they are still investigating that possibility.
The team is also hoping to test VNLG-152 in combination with other drugs, such as FLT3 inhibitors or chemotherapeutic agents.
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Preclinical research suggests a novel agent has preferential activity in acute myeloid leukemia (AML) with FMS-like tyrosine kinase 3 internal tandem duplication (FLT3-ITD) mutations.
The agent, VNLG-152, proved more cytotoxic in AML cell lines and patient samples with FLT3-ITD than in samples and cell lines with wild-type FLT3.
Exactly how and why this occurs remains somewhat of a mystery, however.
Sheetal Karne, MD, of the University of Maryland School of Medicine in Baltimore, and her colleagues detailed this mystery in a poster presentation at the AACR Annual Meeting 2015 (abstract 5408*).
Dr Karne noted that VNLG-152 targets translation by promoting the degradation of MAPK-interacting kinases (Mnks).
“[VNLG-152] has been previously published as functioning in Mnk degradation, which has been shown in triple-negative breast cancer and prostate cancer—in vivo and in vitro,” she said. “Our hypothesis was that, since [the drug] worked via decreasing translation, it would function in leukemia cells and, specifically, in leukemic cells with ITD mutations.”
So the investigators tested VNLG-152 in samples from AML patients, as well as both murine and human cell lines. They found that VNLG-152 was more cytotoxic in the presence of FLT3-ITD mutations, as evidenced by low micromolar IC50 concentrations.
The IC50 concentration was 3.4 μM in Ba/F3-ITD cells and 5.8 μM in Ba/F3-WT cells, which are murine cells transfected with human FLT3-ITD and wild-type FLT3, respectively. Similarly, the IC50 concentration was 1.8 μM in 32D-ITD cells and 18.2 μM in 32D-WT cells.
In the human FLT3-ITD AML cell lines MV4-11 and MOLM-14, IC50 concentrations were 2.3 μM and 4.2 μM, respectively. But concentrations were greater than 10 µM in the wild-type FLT3 human cell lines HL60 and U937.
In patient samples, the IC50 concentration was 1.0 μM in FLT3-ITD AML and 7.5 μM in AML with wild-type FLT3.
In additional tests with murine cell lines, the investigators found that VNLG-152 inhibits the growth of Ba/F3-ITD and Ba/F3-WT cells. But the drug induces apoptosis in these cell lines only when given in high concentrations.
Looking into the mechanism of VNLG-152, the investigators found that the drug decreased Mnk-1 expression in Ba/F3-ITD and Ba/F3-WT cell lines.
“We saw that VNLG-152 worked via degradation of Mnk, but it was the same in both wild-type and ITD, so we’re still looking for an explanation as to what caused this difference,” Dr Karne said.
She and her colleagues believe the Mnk degradation inhibits the phosphorylation of eukaryotic translation initiation factor 4E (eIF4E), a downstream target of FLT3-ITD. But they are still investigating that possibility.
The team is also hoping to test VNLG-152 in combination with other drugs, such as FLT3 inhibitors or chemotherapeutic agents.
*Information in the abstract differs from that presented at the meeting.