User login
Accuracy of blood test results varies

Photo by Graham Colm
A comparison of commercially available blood tests has revealed more variability than expected, according to researchers.
The group compared basic blood tests run by commercial laboratories and found the testing service, type of test, and time of collection all influenced the accuracy of results.
Given that these tests can be used for disease diagnosis or to determine whether a patient’s medication is working, the researchers said this study highlights the importance of knowing the accuracy and variability of blood test results.
“While most of the variability we found was within clinically accepted ranges, there were several cases where inaccurate results would have led to incorrect medical decisions,” said Joel Dudley, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“We hope this study will inspire the biomedical community to take a critical look at all testing variables to ensure that lab results are as robust and reproducible as possible.”
Dr Dudley and his colleagues described this study in the Journal of Clinical Investigation.
The researchers collected peripheral blood samples from 60 healthy adults at 4 separate time points within a 6.5-hour window. The samples were collected in Phoenix, Arizona, at an ambulatory clinic and at retail outlets with point-of-care services.
The team collected 14 samples per subject and used those samples to compare 22 common clinical lab tests conducted at 3 commercial labs. One lab, Theranos, offered blood tests obtained from a finger prick, and the other 2, Quest and LabCorp, required standard venipuncture draws.
More than half of the test results showed significant differences between test providers. Of the 22 tests, 15 (68%) showed significant variability between labs (P<0.002).
Triglyceride levels and red blood cell counts were among the most consistent results, while white blood cell counts and overall cholesterol levels were among the most variable.
Test results from Theranos were flagged by Theranos as abnormal 1.6 times more often than tests from LabCorp or Quest (P<0.0001). The percentages for measurements outside their normal range were 8.3% for LabCorp, 7.5% for Quest, and 12.2% for Theranos.
In addition, the researchers noted that, although they controlled subjects’ eating and physical activity, data from blood samples collected earlier in the day were sometimes significantly different from samples taken from the same subjects later in the day.
There were significant difference between measurements collected at time points 1 and 2 vs time points 3 and 4 for 13 of the 22 tests (P<0.002).
“These testing disparities occurred despite rigorous laboratory certification and proficiency standards designed to ensure consistency,” said study author Eric Schadt, PhD, of Mount Sinai.
“Our results suggest the need for greater transparency in lab technologies and procedures, as well as a much more thorough investigation of biological mechanisms that may contribute to more dynamic levels than we currently understand.” ![]()
Photo by Graham Colm
A comparison of commercially available blood tests has revealed more variability than expected, according to researchers.
The group compared basic blood tests run by commercial laboratories and found the testing service, type of test, and time of collection all influenced the accuracy of results.
Given that these tests can be used for disease diagnosis or to determine whether a patient’s medication is working, the researchers said this study highlights the importance of knowing the accuracy and variability of blood test results.
“While most of the variability we found was within clinically accepted ranges, there were several cases where inaccurate results would have led to incorrect medical decisions,” said Joel Dudley, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“We hope this study will inspire the biomedical community to take a critical look at all testing variables to ensure that lab results are as robust and reproducible as possible.”
Dr Dudley and his colleagues described this study in the Journal of Clinical Investigation.
The researchers collected peripheral blood samples from 60 healthy adults at 4 separate time points within a 6.5-hour window. The samples were collected in Phoenix, Arizona, at an ambulatory clinic and at retail outlets with point-of-care services.
The team collected 14 samples per subject and used those samples to compare 22 common clinical lab tests conducted at 3 commercial labs. One lab, Theranos, offered blood tests obtained from a finger prick, and the other 2, Quest and LabCorp, required standard venipuncture draws.
More than half of the test results showed significant differences between test providers. Of the 22 tests, 15 (68%) showed significant variability between labs (P<0.002).
Triglyceride levels and red blood cell counts were among the most consistent results, while white blood cell counts and overall cholesterol levels were among the most variable.
Test results from Theranos were flagged by Theranos as abnormal 1.6 times more often than tests from LabCorp or Quest (P<0.0001). The percentages for measurements outside their normal range were 8.3% for LabCorp, 7.5% for Quest, and 12.2% for Theranos.
In addition, the researchers noted that, although they controlled subjects’ eating and physical activity, data from blood samples collected earlier in the day were sometimes significantly different from samples taken from the same subjects later in the day.
There were significant difference between measurements collected at time points 1 and 2 vs time points 3 and 4 for 13 of the 22 tests (P<0.002).
“These testing disparities occurred despite rigorous laboratory certification and proficiency standards designed to ensure consistency,” said study author Eric Schadt, PhD, of Mount Sinai.
“Our results suggest the need for greater transparency in lab technologies and procedures, as well as a much more thorough investigation of biological mechanisms that may contribute to more dynamic levels than we currently understand.” ![]()
Photo by Graham Colm
A comparison of commercially available blood tests has revealed more variability than expected, according to researchers.
The group compared basic blood tests run by commercial laboratories and found the testing service, type of test, and time of collection all influenced the accuracy of results.
Given that these tests can be used for disease diagnosis or to determine whether a patient’s medication is working, the researchers said this study highlights the importance of knowing the accuracy and variability of blood test results.
“While most of the variability we found was within clinically accepted ranges, there were several cases where inaccurate results would have led to incorrect medical decisions,” said Joel Dudley, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York.
“We hope this study will inspire the biomedical community to take a critical look at all testing variables to ensure that lab results are as robust and reproducible as possible.”
Dr Dudley and his colleagues described this study in the Journal of Clinical Investigation.
The researchers collected peripheral blood samples from 60 healthy adults at 4 separate time points within a 6.5-hour window. The samples were collected in Phoenix, Arizona, at an ambulatory clinic and at retail outlets with point-of-care services.
The team collected 14 samples per subject and used those samples to compare 22 common clinical lab tests conducted at 3 commercial labs. One lab, Theranos, offered blood tests obtained from a finger prick, and the other 2, Quest and LabCorp, required standard venipuncture draws.
More than half of the test results showed significant differences between test providers. Of the 22 tests, 15 (68%) showed significant variability between labs (P<0.002).
Triglyceride levels and red blood cell counts were among the most consistent results, while white blood cell counts and overall cholesterol levels were among the most variable.
Test results from Theranos were flagged by Theranos as abnormal 1.6 times more often than tests from LabCorp or Quest (P<0.0001). The percentages for measurements outside their normal range were 8.3% for LabCorp, 7.5% for Quest, and 12.2% for Theranos.
In addition, the researchers noted that, although they controlled subjects’ eating and physical activity, data from blood samples collected earlier in the day were sometimes significantly different from samples taken from the same subjects later in the day.
There were significant difference between measurements collected at time points 1 and 2 vs time points 3 and 4 for 13 of the 22 tests (P<0.002).
“These testing disparities occurred despite rigorous laboratory certification and proficiency standards designed to ensure consistency,” said study author Eric Schadt, PhD, of Mount Sinai.
“Our results suggest the need for greater transparency in lab technologies and procedures, as well as a much more thorough investigation of biological mechanisms that may contribute to more dynamic levels than we currently understand.” ![]()
High costs limit CML patients’ access to TKIs

Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said. ![]()
Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said. ![]()
Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said. ![]()
iPSCs can differentiate into functional lymphocytes

Image from the Salk Institute
Researchers say they have generated induced pluripotent stem cells (iPSCs) that can differentiate into multiple lineages of functional lymphocytes.
The team noted that lymphohematopoietic stem cells (L-HSCs) generated from self-somatic cell-derived iPSCs could potentially be used to treat hematologic disorders, but no one has generated “truly functional” L-HSCs from iPSCs.
So the researchers set out to determine whether iPSCs have the inherent potential to generate multiple lineages of functional, terminally differentiated lymphocytes.
They described this work in Stem Cells and Development.
The researchers said they used tetraploid embryo complementation to provide a normal environment for the differentiation of L-HSCs from iPSCs and embryonic stem cells (ESCs). The team then compared lymphocytes derived from iPSCs, ESCs, and naïve isogenic C57BL/6 mice.
The researchers found that iPSC-derived lymphocytes expressed normal levels of major histocompatibility complex-I. Levels were comparable in iPSC-derived lymphocytes, ESC-derived lymphocytes, and lymphocytes from the control mice.
In addition, iPSC-derived lymphocytes were able to differentiate into multiple cell types—CD4+ T cells, CD8+ T cells, regulatory T cells, B cells, and natural killer cells.
Lymphocytes generated from iPSCs and lymphocytes generated from ESCs had the same capacity as lymphocytes from the control mice to proliferate and secrete chemical signals, such as cytokines.
All 3 types of lymphocytes proliferated under allogeneic stimulation but not under syngeneic stimulation. And the researchers found similar levels of IL-2, IL-4, IL-6, IL-10, IL-17, TNF, and IFN-γ in iPSC, ESC, and C57BL/6 lymphocyte culture supernatants.
The team also found that lymphocytes generated by iPSC-derived bone marrow cells could repopulate the hematopoietic systems of lethally irradiated recipient mice.
The iPSC bone marrow cells proved as effective as ESC-derived bone marrow cells and wild-type bone marrow cells. All 3 types of cells negated lymphocyte storage exhaustion in the spleen and peripheral blood.
In addition, there were no major phenotypic or behavioral abnormalities in any of the mice more than 1 month after cell transplantation.
The researchers said this work shows that truly functional lymphocytes can be generated from iPSCs, and it supports the clinical application of iPSC technology to develop treatments for hematologic disorders. ![]()
Image from the Salk Institute
Researchers say they have generated induced pluripotent stem cells (iPSCs) that can differentiate into multiple lineages of functional lymphocytes.
The team noted that lymphohematopoietic stem cells (L-HSCs) generated from self-somatic cell-derived iPSCs could potentially be used to treat hematologic disorders, but no one has generated “truly functional” L-HSCs from iPSCs.
So the researchers set out to determine whether iPSCs have the inherent potential to generate multiple lineages of functional, terminally differentiated lymphocytes.
They described this work in Stem Cells and Development.
The researchers said they used tetraploid embryo complementation to provide a normal environment for the differentiation of L-HSCs from iPSCs and embryonic stem cells (ESCs). The team then compared lymphocytes derived from iPSCs, ESCs, and naïve isogenic C57BL/6 mice.
The researchers found that iPSC-derived lymphocytes expressed normal levels of major histocompatibility complex-I. Levels were comparable in iPSC-derived lymphocytes, ESC-derived lymphocytes, and lymphocytes from the control mice.
In addition, iPSC-derived lymphocytes were able to differentiate into multiple cell types—CD4+ T cells, CD8+ T cells, regulatory T cells, B cells, and natural killer cells.
Lymphocytes generated from iPSCs and lymphocytes generated from ESCs had the same capacity as lymphocytes from the control mice to proliferate and secrete chemical signals, such as cytokines.
All 3 types of lymphocytes proliferated under allogeneic stimulation but not under syngeneic stimulation. And the researchers found similar levels of IL-2, IL-4, IL-6, IL-10, IL-17, TNF, and IFN-γ in iPSC, ESC, and C57BL/6 lymphocyte culture supernatants.
The team also found that lymphocytes generated by iPSC-derived bone marrow cells could repopulate the hematopoietic systems of lethally irradiated recipient mice.
The iPSC bone marrow cells proved as effective as ESC-derived bone marrow cells and wild-type bone marrow cells. All 3 types of cells negated lymphocyte storage exhaustion in the spleen and peripheral blood.
In addition, there were no major phenotypic or behavioral abnormalities in any of the mice more than 1 month after cell transplantation.
The researchers said this work shows that truly functional lymphocytes can be generated from iPSCs, and it supports the clinical application of iPSC technology to develop treatments for hematologic disorders. ![]()
Image from the Salk Institute
Researchers say they have generated induced pluripotent stem cells (iPSCs) that can differentiate into multiple lineages of functional lymphocytes.
The team noted that lymphohematopoietic stem cells (L-HSCs) generated from self-somatic cell-derived iPSCs could potentially be used to treat hematologic disorders, but no one has generated “truly functional” L-HSCs from iPSCs.
So the researchers set out to determine whether iPSCs have the inherent potential to generate multiple lineages of functional, terminally differentiated lymphocytes.
They described this work in Stem Cells and Development.
The researchers said they used tetraploid embryo complementation to provide a normal environment for the differentiation of L-HSCs from iPSCs and embryonic stem cells (ESCs). The team then compared lymphocytes derived from iPSCs, ESCs, and naïve isogenic C57BL/6 mice.
The researchers found that iPSC-derived lymphocytes expressed normal levels of major histocompatibility complex-I. Levels were comparable in iPSC-derived lymphocytes, ESC-derived lymphocytes, and lymphocytes from the control mice.
In addition, iPSC-derived lymphocytes were able to differentiate into multiple cell types—CD4+ T cells, CD8+ T cells, regulatory T cells, B cells, and natural killer cells.
Lymphocytes generated from iPSCs and lymphocytes generated from ESCs had the same capacity as lymphocytes from the control mice to proliferate and secrete chemical signals, such as cytokines.
All 3 types of lymphocytes proliferated under allogeneic stimulation but not under syngeneic stimulation. And the researchers found similar levels of IL-2, IL-4, IL-6, IL-10, IL-17, TNF, and IFN-γ in iPSC, ESC, and C57BL/6 lymphocyte culture supernatants.
The team also found that lymphocytes generated by iPSC-derived bone marrow cells could repopulate the hematopoietic systems of lethally irradiated recipient mice.
The iPSC bone marrow cells proved as effective as ESC-derived bone marrow cells and wild-type bone marrow cells. All 3 types of cells negated lymphocyte storage exhaustion in the spleen and peripheral blood.
In addition, there were no major phenotypic or behavioral abnormalities in any of the mice more than 1 month after cell transplantation.
The researchers said this work shows that truly functional lymphocytes can be generated from iPSCs, and it supports the clinical application of iPSC technology to develop treatments for hematologic disorders. ![]()
A better method for detecting amyloidosis?
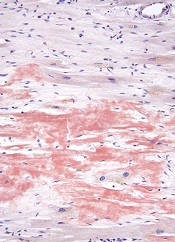
A novel molecular probe can detect amyloidosis at least as well as—and perhaps even better than—traditional methods, according to research published in Amyloid: The Journal of Protein Folding Disorders.
Investigators found that a luminescent conjugated oligothiophene, h-FTAA, allowed them to correctly identify amyloidosis in every sample tested.
But results also suggested h-FTAA may be more sensitive than traditional methods used to diagnose amyloidosis, as h-FTAA detected small amyloid deposits in samples that were previously determined to be amyloid-free.
The investigators said this suggests h-FTAA could be used to detect amyloidosis before symptoms present, leading to faster treatment.
“Given the sensitivity of the probe, we think this would make an excellent complement to traditional methods and could eventually be a replacement,” said study author Per Hammarström, PhD, of Linköping University in Sweden.
Dr Hammarström and his colleagues screened amyloid-containing tissues from 107 patients who had their amyloidosis verified by Congo red staining and/or immunohistochemistry, as well as tissues from 32 negative control cases.
The results showed that h-FTAA could detect amyloidosis with 100% sensitivity, identifying amyloid deposits in all 107 patients.
However, h-FTAA also detected microdeposits of amyloid-like protein aggregates in 5 of the control samples that were negative according to Congo red.
The investigators said they don’t know the clinical significance of these “false-positive” lesions. However, because h-FTAA fluorescence is 1 magnitude brighter than Congo red and because the staining is performed 4 magnitudes lower than the concentration of dye, the team believes these 5 cases may have been beyond detection by Congo red and h-FTAA may be a more sensitive technique.
They therefore concluded that h-FTAA could potentially be used as a complementary technique for accurate detection of amyloid in routine surgical pathology settings, for the detection of prodromal amyloidosis, and for the discovery of new amyloid-like protein aggregates. ![]()
A novel molecular probe can detect amyloidosis at least as well as—and perhaps even better than—traditional methods, according to research published in Amyloid: The Journal of Protein Folding Disorders.
Investigators found that a luminescent conjugated oligothiophene, h-FTAA, allowed them to correctly identify amyloidosis in every sample tested.
But results also suggested h-FTAA may be more sensitive than traditional methods used to diagnose amyloidosis, as h-FTAA detected small amyloid deposits in samples that were previously determined to be amyloid-free.
The investigators said this suggests h-FTAA could be used to detect amyloidosis before symptoms present, leading to faster treatment.
“Given the sensitivity of the probe, we think this would make an excellent complement to traditional methods and could eventually be a replacement,” said study author Per Hammarström, PhD, of Linköping University in Sweden.
Dr Hammarström and his colleagues screened amyloid-containing tissues from 107 patients who had their amyloidosis verified by Congo red staining and/or immunohistochemistry, as well as tissues from 32 negative control cases.
The results showed that h-FTAA could detect amyloidosis with 100% sensitivity, identifying amyloid deposits in all 107 patients.
However, h-FTAA also detected microdeposits of amyloid-like protein aggregates in 5 of the control samples that were negative according to Congo red.
The investigators said they don’t know the clinical significance of these “false-positive” lesions. However, because h-FTAA fluorescence is 1 magnitude brighter than Congo red and because the staining is performed 4 magnitudes lower than the concentration of dye, the team believes these 5 cases may have been beyond detection by Congo red and h-FTAA may be a more sensitive technique.
They therefore concluded that h-FTAA could potentially be used as a complementary technique for accurate detection of amyloid in routine surgical pathology settings, for the detection of prodromal amyloidosis, and for the discovery of new amyloid-like protein aggregates. ![]()
A novel molecular probe can detect amyloidosis at least as well as—and perhaps even better than—traditional methods, according to research published in Amyloid: The Journal of Protein Folding Disorders.
Investigators found that a luminescent conjugated oligothiophene, h-FTAA, allowed them to correctly identify amyloidosis in every sample tested.
But results also suggested h-FTAA may be more sensitive than traditional methods used to diagnose amyloidosis, as h-FTAA detected small amyloid deposits in samples that were previously determined to be amyloid-free.
The investigators said this suggests h-FTAA could be used to detect amyloidosis before symptoms present, leading to faster treatment.
“Given the sensitivity of the probe, we think this would make an excellent complement to traditional methods and could eventually be a replacement,” said study author Per Hammarström, PhD, of Linköping University in Sweden.
Dr Hammarström and his colleagues screened amyloid-containing tissues from 107 patients who had their amyloidosis verified by Congo red staining and/or immunohistochemistry, as well as tissues from 32 negative control cases.
The results showed that h-FTAA could detect amyloidosis with 100% sensitivity, identifying amyloid deposits in all 107 patients.
However, h-FTAA also detected microdeposits of amyloid-like protein aggregates in 5 of the control samples that were negative according to Congo red.
The investigators said they don’t know the clinical significance of these “false-positive” lesions. However, because h-FTAA fluorescence is 1 magnitude brighter than Congo red and because the staining is performed 4 magnitudes lower than the concentration of dye, the team believes these 5 cases may have been beyond detection by Congo red and h-FTAA may be a more sensitive technique.
They therefore concluded that h-FTAA could potentially be used as a complementary technique for accurate detection of amyloid in routine surgical pathology settings, for the detection of prodromal amyloidosis, and for the discovery of new amyloid-like protein aggregates. ![]()
Drug ‘not powerful enough’ to treat CTCL

mycosis fungoides
Results of a phase 2 trial suggest the drug APO866 is not suitable for the treatment of relapsed or refractory cutaneous T-cell lymphoma (CTCL).
Researchers said APO866 had “a reasonable toxic effect,” but it was “not powerful enough,” so the trial was stopped early.
Two of the 14 patients studied achieved a partial response during the trial, but there were no complete responses, and most patients withdrew from the study early.
The researchers described these results in a letter to JAMA Dermatology. The study was initially sponsored by Apoxis SA and later by TopoTarget A/S, which is now known as Onxeo after merging with BioAlliance Pharma.
According to Onxeo, APO866 is an injectable molecule that induces apoptosis by inhibiting the biosynthesis of NAD+ from niacinamide, which is essential for the cellular metabolism, protein modification, and calcium-dependent messenger synthesis.
In the phase 2 trial, researchers tested APO866 in 14 patients with relapsed or refractory CTCL. The patients were 19 to 83 years of age, and half were female.
Eight patients had mycosis fungoides, 3 had Sézary syndrome, 1 had CD30+ anaplastic large-cell lymphoma, 1 had poikilodermic mycosis fungoides, and 1 had CD30- nonepidermotropic CTCL. One patient had stage IB disease, 2 had stage IIA, 3 had stage IIB, and 8 had stage IVA.
The patients received a continuous intravenous infusion, via pump, of APO866 at 0.126 mg/m2/h over the course of 96 hours. Patients received this treatment every 28 days for a total of 3 cycles.
Five patients completed all 3 treatment cycles and had no major protocol violations. Nine patients discontinued treatment early due to consent withdrawal (n=2), early disease progression (n=5), or adverse events (AEs, n=2).
At week 8, 1 patient had achieved a partial response to treatment, 4 patients had stable disease, 5 had progressed, and 4 patients were not evaluable because they had withdrawn from the study.
At week 16, 1 patient had a partial response (not the same patient as at week 8), 4 patients had stable disease, and 9 patients were not evaluable due to withdrawal.
There were a total of 141 AEs, and 77 of these were considered related to APO866. Most patients (n=12) had mild to moderate AEs, but there were 7 serious AEs thought to be treatment-related. These included pyrexia, lymphopenia (n=2), spondylitis, Staphylococcal sepsis, rhabdomyolysis, and thrombocytopenia.
There were 4 deaths, but they were not considered drug-related.
The researchers said these results suggest APO866 should not be pursued as a treatment for CTCL. However, as the drug induces immunosuppression and has insulin-mimicking effects, it might prove useful for treating other conditions. ![]()
mycosis fungoides
Results of a phase 2 trial suggest the drug APO866 is not suitable for the treatment of relapsed or refractory cutaneous T-cell lymphoma (CTCL).
Researchers said APO866 had “a reasonable toxic effect,” but it was “not powerful enough,” so the trial was stopped early.
Two of the 14 patients studied achieved a partial response during the trial, but there were no complete responses, and most patients withdrew from the study early.
The researchers described these results in a letter to JAMA Dermatology. The study was initially sponsored by Apoxis SA and later by TopoTarget A/S, which is now known as Onxeo after merging with BioAlliance Pharma.
According to Onxeo, APO866 is an injectable molecule that induces apoptosis by inhibiting the biosynthesis of NAD+ from niacinamide, which is essential for the cellular metabolism, protein modification, and calcium-dependent messenger synthesis.
In the phase 2 trial, researchers tested APO866 in 14 patients with relapsed or refractory CTCL. The patients were 19 to 83 years of age, and half were female.
Eight patients had mycosis fungoides, 3 had Sézary syndrome, 1 had CD30+ anaplastic large-cell lymphoma, 1 had poikilodermic mycosis fungoides, and 1 had CD30- nonepidermotropic CTCL. One patient had stage IB disease, 2 had stage IIA, 3 had stage IIB, and 8 had stage IVA.
The patients received a continuous intravenous infusion, via pump, of APO866 at 0.126 mg/m2/h over the course of 96 hours. Patients received this treatment every 28 days for a total of 3 cycles.
Five patients completed all 3 treatment cycles and had no major protocol violations. Nine patients discontinued treatment early due to consent withdrawal (n=2), early disease progression (n=5), or adverse events (AEs, n=2).
At week 8, 1 patient had achieved a partial response to treatment, 4 patients had stable disease, 5 had progressed, and 4 patients were not evaluable because they had withdrawn from the study.
At week 16, 1 patient had a partial response (not the same patient as at week 8), 4 patients had stable disease, and 9 patients were not evaluable due to withdrawal.
There were a total of 141 AEs, and 77 of these were considered related to APO866. Most patients (n=12) had mild to moderate AEs, but there were 7 serious AEs thought to be treatment-related. These included pyrexia, lymphopenia (n=2), spondylitis, Staphylococcal sepsis, rhabdomyolysis, and thrombocytopenia.
There were 4 deaths, but they were not considered drug-related.
The researchers said these results suggest APO866 should not be pursued as a treatment for CTCL. However, as the drug induces immunosuppression and has insulin-mimicking effects, it might prove useful for treating other conditions. ![]()
mycosis fungoides
Results of a phase 2 trial suggest the drug APO866 is not suitable for the treatment of relapsed or refractory cutaneous T-cell lymphoma (CTCL).
Researchers said APO866 had “a reasonable toxic effect,” but it was “not powerful enough,” so the trial was stopped early.
Two of the 14 patients studied achieved a partial response during the trial, but there were no complete responses, and most patients withdrew from the study early.
The researchers described these results in a letter to JAMA Dermatology. The study was initially sponsored by Apoxis SA and later by TopoTarget A/S, which is now known as Onxeo after merging with BioAlliance Pharma.
According to Onxeo, APO866 is an injectable molecule that induces apoptosis by inhibiting the biosynthesis of NAD+ from niacinamide, which is essential for the cellular metabolism, protein modification, and calcium-dependent messenger synthesis.
In the phase 2 trial, researchers tested APO866 in 14 patients with relapsed or refractory CTCL. The patients were 19 to 83 years of age, and half were female.
Eight patients had mycosis fungoides, 3 had Sézary syndrome, 1 had CD30+ anaplastic large-cell lymphoma, 1 had poikilodermic mycosis fungoides, and 1 had CD30- nonepidermotropic CTCL. One patient had stage IB disease, 2 had stage IIA, 3 had stage IIB, and 8 had stage IVA.
The patients received a continuous intravenous infusion, via pump, of APO866 at 0.126 mg/m2/h over the course of 96 hours. Patients received this treatment every 28 days for a total of 3 cycles.
Five patients completed all 3 treatment cycles and had no major protocol violations. Nine patients discontinued treatment early due to consent withdrawal (n=2), early disease progression (n=5), or adverse events (AEs, n=2).
At week 8, 1 patient had achieved a partial response to treatment, 4 patients had stable disease, 5 had progressed, and 4 patients were not evaluable because they had withdrawn from the study.
At week 16, 1 patient had a partial response (not the same patient as at week 8), 4 patients had stable disease, and 9 patients were not evaluable due to withdrawal.
There were a total of 141 AEs, and 77 of these were considered related to APO866. Most patients (n=12) had mild to moderate AEs, but there were 7 serious AEs thought to be treatment-related. These included pyrexia, lymphopenia (n=2), spondylitis, Staphylococcal sepsis, rhabdomyolysis, and thrombocytopenia.
There were 4 deaths, but they were not considered drug-related.
The researchers said these results suggest APO866 should not be pursued as a treatment for CTCL. However, as the drug induces immunosuppression and has insulin-mimicking effects, it might prove useful for treating other conditions. ![]()
Research helps explain how malaria evolved

Photo by Holly Lutz
A study published in Molecular Phylogenetics and Evolution has revealed a new hypothesis on the evolution of malaria.
Researchers tested malarial DNA found in birds, bats, and other small mammals from 5 East African countries and found evidence to suggest that malaria has its roots in bird hosts.
It then spread to bats and on to other mammals.
“We can’t begin to understand how malaria spread to humans until we understand its evolutionary history,” said Holly Lutz, a doctoral candidate at Cornell University in Ithaca, New York.
“In learning about its past, we may be better able to understand the effects it has on us.”
Lutz and her colleagues took blood samples from hundreds of East African birds, bats, and other small mammals and screened the blood for malaria parasites.
When they found malaria, the team took samples of the parasites’ DNA and sequenced it to identify mutations in the genetic code. From there, the researchers performed phylogenetic analyses to determine how different malaria species are related.
In analyzing the genetic codes of the parasites, the team was able to find places where the DNA differed from one species to the next. Then, the researchers used computing software to determine how the different species evolved and how they’re related to each other.
“[B]y looking at patterns of mutations in the DNA of the different malaria species, we’re able to see when it branched off from one host group into another,” Lutz explained. “It started out as a parasite in birds, and then it evolved to colonize bats, and from there, it’s evolved to affect other mammals.”
In addition to shedding light on the way malaria was able to evolve and spread, the study provides information about the manner in which animals and their parasites are connected.
“Each of these individual vertebrates is an ecosystem in and of itself,” Lutz said. “In learning more about how parasites live within their hosts, who is infecting who, and how these organisms coexist in these living, breathing ecosystems, we can learn more about how they are connected to and affected by the natural environments that we share with animals and plants.”
The researchers noted that this study doesn’t have direct implications for malaria treatment in humans. However, the team believes that having a better understanding of malaria’s evolutionary history could help scientists anticipate how it will change and evolve in the future. ![]()
Photo by Holly Lutz
A study published in Molecular Phylogenetics and Evolution has revealed a new hypothesis on the evolution of malaria.
Researchers tested malarial DNA found in birds, bats, and other small mammals from 5 East African countries and found evidence to suggest that malaria has its roots in bird hosts.
It then spread to bats and on to other mammals.
“We can’t begin to understand how malaria spread to humans until we understand its evolutionary history,” said Holly Lutz, a doctoral candidate at Cornell University in Ithaca, New York.
“In learning about its past, we may be better able to understand the effects it has on us.”
Lutz and her colleagues took blood samples from hundreds of East African birds, bats, and other small mammals and screened the blood for malaria parasites.
When they found malaria, the team took samples of the parasites’ DNA and sequenced it to identify mutations in the genetic code. From there, the researchers performed phylogenetic analyses to determine how different malaria species are related.
In analyzing the genetic codes of the parasites, the team was able to find places where the DNA differed from one species to the next. Then, the researchers used computing software to determine how the different species evolved and how they’re related to each other.
“[B]y looking at patterns of mutations in the DNA of the different malaria species, we’re able to see when it branched off from one host group into another,” Lutz explained. “It started out as a parasite in birds, and then it evolved to colonize bats, and from there, it’s evolved to affect other mammals.”
In addition to shedding light on the way malaria was able to evolve and spread, the study provides information about the manner in which animals and their parasites are connected.
“Each of these individual vertebrates is an ecosystem in and of itself,” Lutz said. “In learning more about how parasites live within their hosts, who is infecting who, and how these organisms coexist in these living, breathing ecosystems, we can learn more about how they are connected to and affected by the natural environments that we share with animals and plants.”
The researchers noted that this study doesn’t have direct implications for malaria treatment in humans. However, the team believes that having a better understanding of malaria’s evolutionary history could help scientists anticipate how it will change and evolve in the future. ![]()
Photo by Holly Lutz
A study published in Molecular Phylogenetics and Evolution has revealed a new hypothesis on the evolution of malaria.
Researchers tested malarial DNA found in birds, bats, and other small mammals from 5 East African countries and found evidence to suggest that malaria has its roots in bird hosts.
It then spread to bats and on to other mammals.
“We can’t begin to understand how malaria spread to humans until we understand its evolutionary history,” said Holly Lutz, a doctoral candidate at Cornell University in Ithaca, New York.
“In learning about its past, we may be better able to understand the effects it has on us.”
Lutz and her colleagues took blood samples from hundreds of East African birds, bats, and other small mammals and screened the blood for malaria parasites.
When they found malaria, the team took samples of the parasites’ DNA and sequenced it to identify mutations in the genetic code. From there, the researchers performed phylogenetic analyses to determine how different malaria species are related.
In analyzing the genetic codes of the parasites, the team was able to find places where the DNA differed from one species to the next. Then, the researchers used computing software to determine how the different species evolved and how they’re related to each other.
“[B]y looking at patterns of mutations in the DNA of the different malaria species, we’re able to see when it branched off from one host group into another,” Lutz explained. “It started out as a parasite in birds, and then it evolved to colonize bats, and from there, it’s evolved to affect other mammals.”
In addition to shedding light on the way malaria was able to evolve and spread, the study provides information about the manner in which animals and their parasites are connected.
“Each of these individual vertebrates is an ecosystem in and of itself,” Lutz said. “In learning more about how parasites live within their hosts, who is infecting who, and how these organisms coexist in these living, breathing ecosystems, we can learn more about how they are connected to and affected by the natural environments that we share with animals and plants.”
The researchers noted that this study doesn’t have direct implications for malaria treatment in humans. However, the team believes that having a better understanding of malaria’s evolutionary history could help scientists anticipate how it will change and evolve in the future. ![]()
Study: Dying at home doesn’t mean dying sooner
patient’s hand
Choosing to die at home does not hasten death for patients with terminal cancer, according to a study published in Cancer.
The research showed that cancer patients who died at home lived at least as long as patients who spent their last days in hospitals.
Investigators say these results suggest oncologists should not hesitate to refer patients for home-based palliative care simply because less medical treatment may be provided.
“The cancer patient and family tend to be concerned that the quality of medical treatment provided at home will be inferior to that given in a hospital and that survival might be shortened,” said study author Jun Hamano, MD, of the University of Tsukuba in Japan.
“However, our finding—that home death does not actually have a negative influence on the survival of cancer patients at all and, rather, may have a positive influence—could suggest that the patient and family can choose the place of death in terms of their preference and values.”
Dr Hamano and his colleagues conducted this research by prospectively studying 2069 patients—1582 receiving hospital-based palliative care and 487 receiving home-based palliative care.
In all, 1607 patients died in the hospital, and 462 died at home.
Among patients thought to have only days to live, the survival of those who died at home was significantly longer than the survival of those who died in a hospital. The estimated median survival times were 13 days and 9 days, respectively (P<0.006).
Similarly, survival was significantly longer in the home group than the hospital group among patients thought to have weeks to live. The estimated median survival times were 36 days and 29 days, respectively (P<0.007).
There was no significant difference between the home and hospital groups among patients thought to have months to live. The estimated median survival times were 59 days and 62 days, respectively (P=0.925).
Finally, analyses suggested the place of death had a significant influence on the survival time in both unadjusted and adjusted models. The hazard ratios were 0.86 (P<0.01) and 0.87 (P=0.01), respectively.
Based on these findings, Dr Hamano concluded that, “Patients, families, and clinicians should be reassured that good home hospice care does not shorten patient life and even may achieve longer survival.” ![]()
patient’s hand
Choosing to die at home does not hasten death for patients with terminal cancer, according to a study published in Cancer.
The research showed that cancer patients who died at home lived at least as long as patients who spent their last days in hospitals.
Investigators say these results suggest oncologists should not hesitate to refer patients for home-based palliative care simply because less medical treatment may be provided.
“The cancer patient and family tend to be concerned that the quality of medical treatment provided at home will be inferior to that given in a hospital and that survival might be shortened,” said study author Jun Hamano, MD, of the University of Tsukuba in Japan.
“However, our finding—that home death does not actually have a negative influence on the survival of cancer patients at all and, rather, may have a positive influence—could suggest that the patient and family can choose the place of death in terms of their preference and values.”
Dr Hamano and his colleagues conducted this research by prospectively studying 2069 patients—1582 receiving hospital-based palliative care and 487 receiving home-based palliative care.
In all, 1607 patients died in the hospital, and 462 died at home.
Among patients thought to have only days to live, the survival of those who died at home was significantly longer than the survival of those who died in a hospital. The estimated median survival times were 13 days and 9 days, respectively (P<0.006).
Similarly, survival was significantly longer in the home group than the hospital group among patients thought to have weeks to live. The estimated median survival times were 36 days and 29 days, respectively (P<0.007).
There was no significant difference between the home and hospital groups among patients thought to have months to live. The estimated median survival times were 59 days and 62 days, respectively (P=0.925).
Finally, analyses suggested the place of death had a significant influence on the survival time in both unadjusted and adjusted models. The hazard ratios were 0.86 (P<0.01) and 0.87 (P=0.01), respectively.
Based on these findings, Dr Hamano concluded that, “Patients, families, and clinicians should be reassured that good home hospice care does not shorten patient life and even may achieve longer survival.” ![]()
patient’s hand
Choosing to die at home does not hasten death for patients with terminal cancer, according to a study published in Cancer.
The research showed that cancer patients who died at home lived at least as long as patients who spent their last days in hospitals.
Investigators say these results suggest oncologists should not hesitate to refer patients for home-based palliative care simply because less medical treatment may be provided.
“The cancer patient and family tend to be concerned that the quality of medical treatment provided at home will be inferior to that given in a hospital and that survival might be shortened,” said study author Jun Hamano, MD, of the University of Tsukuba in Japan.
“However, our finding—that home death does not actually have a negative influence on the survival of cancer patients at all and, rather, may have a positive influence—could suggest that the patient and family can choose the place of death in terms of their preference and values.”
Dr Hamano and his colleagues conducted this research by prospectively studying 2069 patients—1582 receiving hospital-based palliative care and 487 receiving home-based palliative care.
In all, 1607 patients died in the hospital, and 462 died at home.
Among patients thought to have only days to live, the survival of those who died at home was significantly longer than the survival of those who died in a hospital. The estimated median survival times were 13 days and 9 days, respectively (P<0.006).
Similarly, survival was significantly longer in the home group than the hospital group among patients thought to have weeks to live. The estimated median survival times were 36 days and 29 days, respectively (P<0.007).
There was no significant difference between the home and hospital groups among patients thought to have months to live. The estimated median survival times were 59 days and 62 days, respectively (P=0.925).
Finally, analyses suggested the place of death had a significant influence on the survival time in both unadjusted and adjusted models. The hazard ratios were 0.86 (P<0.01) and 0.87 (P=0.01), respectively.
Based on these findings, Dr Hamano concluded that, “Patients, families, and clinicians should be reassured that good home hospice care does not shorten patient life and even may achieve longer survival.”
EC grants gene therapy orphan designation for hemophilia A

The European Commission (EC) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union.
The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April.
BMN 270 also has orphan designation in the US.
The European Commission (EC) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union.
The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April.
BMN 270 also has orphan designation in the US.
The European Commission (EC) has granted orphan designation to BMN 270, an investigational gene therapy, for the treatment of patients with hemophilia A.
BMN 270 is an adeno-associated virus-factor VIII (FVIII) vector designed to restore FVIII plasma concentrations in these patients.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union.
The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
BMN 270 is under development by BioMarin Pharmaceutical Inc.
BioMarin is conducting a phase 1/2 study to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
Researchers are assessing the safety of a single infusion of BMN 270 and the change in FVIII expression level from baseline to 16 weeks after infusion.
The group is also assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses.
Patients will be monitored for safety and durability of effect for 5 years. BioMarin plans to provide an update on this trial in April.
BMN 270 also has orphan designation in the US.
Drug exhibits preclinical activity against MDS

Researchers have found the fusion protein APG101 can rescue erythropoiesis in bone marrow samples from patients with lower-risk myelodysplastic syndromes (MDS).
Previous research suggested that CD95—a receptor that can induce apoptosis when triggered by the CD95 ligand—is overexpressed in two-thirds of patients with lower-risk MDS, and overexpression of CD95 is predictive of a lower response to erythropoiesis-stimulating agents (ESAs).
APG101, which consists of the extracellular domain of the CD95 receptor and the Fc domain of an IgG antibody, is designed to block the CD95 ligand.
The new study showed that APG101 can inhibit apoptosis in erythrocyte precursor cells and improve their overall proliferation rate. The drug increased the number of burst-forming unit-erythroid (BFU-E) progenitors in samples from MDS patients with low BFU-E numbers at baseline.
“APG101 added to cellular assays efficiently rescued the growth of erythroid progenitors in MDS patients harboring a profound defect of erythropoiesis, independent of the expression level of CD95 or CD95 ligand,” said Michaela Fontenay, MD, PhD, of the Institut Cochin in Paris, France.
Dr Fontenay and her colleagues described these results in Oncotarget. The research was funded by Apogenix, the company developing APG101.
By comparing bone marrow samples from MDS patients and healthy control subjects, the researchers found that CD95, but not CD95 ligand, was overexpressed in patients with lower-risk MDS.
Further analysis revealed that a patient’s CD95 expression level at diagnosis could predict response to an ESA. Specifically, CD95 overexpression was predictive of a lower response rate to ESAs in patients with low- or intermediate-1-risk MDS.
Next, the researchers tested bone marrow erythroid progenitors from 3 control subjects and 5 patients with MDS and found that CD95 expression increased during MDS erythroid progenitor amplification but remained lower in control cultures.
On day 5 of culture, the mean number of BFU-Es was significantly lower in MDS patient samples than in controls. And treatment with APG101 prompted a dose-dependent increase in BFU-E growth in MDS samples but not in controls.
When the researchers added APG101 (at 10 μg/mL) to the cultures over 7 days, they observed an improvement in the proliferation of erythroblasts but no significant effect on the kinetics of differentiation.
They also found that APG101 reduced apoptosis in immature precursors by 30% but had no effect on apoptosis in mature precursors.
Baseline BFU-E number affects response
The researchers then assessed the effects of APG101 in samples from 5 control subjects and 20 MDS patients with varying responses to ESAs and varying baseline levels of BFU-Es.
Fifteen of the MDS patients had a significantly lower number of baseline BFU-Es than controls (P=0.005), but 5 MDS patients had a mean number of BFU-Es that was comparable to controls (P=0.429). There was no significant difference in WHO classification or CD95 expression between the 2 groups of MDS patients (P=0.612).
However, 11 of the 15 patients with low erythropoiesis had received an ESA, and all of them were resistant to this treatment. In all, 15 of the MDS patients had received an ESA, and 14 were resistant to it (6 primary and 8 secondary).
The researchers found that APG101 induced a dose-dependent increase of BFU-Es in samples from the 15 MDS patients with low erythropoiesis but not in samples from the 5 patients with normal erythropoiesis or in the control samples (P<0.001).
The team said that a low BFU-E number at baseline was significantly associated with in vitro response to APG101 among the 20 MDS patients (P=0.031) and the 14 ESA-resistant patients (P=0.027).
Further investigation confirmed that a low clonogenic progenitor number at baseline, but not the level of CD95 or CD95 ligand expression, was predictive of the response to APG101.
“This study provides a rationale for further clinical investigation of this potential new therapeutic option in patients with severely impaired erythropoiesis who are resistant to erythropoiesis-stimulating agents,” Dr Fontenay said.
Apogenix has conducted a phase 1 trial of APG101 in transfusion-dependent patients with low- to intermediate-1-risk MDS. The company expects the results of this trial will be available in the coming months.
Researchers have found the fusion protein APG101 can rescue erythropoiesis in bone marrow samples from patients with lower-risk myelodysplastic syndromes (MDS).
Previous research suggested that CD95—a receptor that can induce apoptosis when triggered by the CD95 ligand—is overexpressed in two-thirds of patients with lower-risk MDS, and overexpression of CD95 is predictive of a lower response to erythropoiesis-stimulating agents (ESAs).
APG101, which consists of the extracellular domain of the CD95 receptor and the Fc domain of an IgG antibody, is designed to block the CD95 ligand.
The new study showed that APG101 can inhibit apoptosis in erythrocyte precursor cells and improve their overall proliferation rate. The drug increased the number of burst-forming unit-erythroid (BFU-E) progenitors in samples from MDS patients with low BFU-E numbers at baseline.
“APG101 added to cellular assays efficiently rescued the growth of erythroid progenitors in MDS patients harboring a profound defect of erythropoiesis, independent of the expression level of CD95 or CD95 ligand,” said Michaela Fontenay, MD, PhD, of the Institut Cochin in Paris, France.
Dr Fontenay and her colleagues described these results in Oncotarget. The research was funded by Apogenix, the company developing APG101.
By comparing bone marrow samples from MDS patients and healthy control subjects, the researchers found that CD95, but not CD95 ligand, was overexpressed in patients with lower-risk MDS.
Further analysis revealed that a patient’s CD95 expression level at diagnosis could predict response to an ESA. Specifically, CD95 overexpression was predictive of a lower response rate to ESAs in patients with low- or intermediate-1-risk MDS.
Next, the researchers tested bone marrow erythroid progenitors from 3 control subjects and 5 patients with MDS and found that CD95 expression increased during MDS erythroid progenitor amplification but remained lower in control cultures.
On day 5 of culture, the mean number of BFU-Es was significantly lower in MDS patient samples than in controls. And treatment with APG101 prompted a dose-dependent increase in BFU-E growth in MDS samples but not in controls.
When the researchers added APG101 (at 10 μg/mL) to the cultures over 7 days, they observed an improvement in the proliferation of erythroblasts but no significant effect on the kinetics of differentiation.
They also found that APG101 reduced apoptosis in immature precursors by 30% but had no effect on apoptosis in mature precursors.
Baseline BFU-E number affects response
The researchers then assessed the effects of APG101 in samples from 5 control subjects and 20 MDS patients with varying responses to ESAs and varying baseline levels of BFU-Es.
Fifteen of the MDS patients had a significantly lower number of baseline BFU-Es than controls (P=0.005), but 5 MDS patients had a mean number of BFU-Es that was comparable to controls (P=0.429). There was no significant difference in WHO classification or CD95 expression between the 2 groups of MDS patients (P=0.612).
However, 11 of the 15 patients with low erythropoiesis had received an ESA, and all of them were resistant to this treatment. In all, 15 of the MDS patients had received an ESA, and 14 were resistant to it (6 primary and 8 secondary).
The researchers found that APG101 induced a dose-dependent increase of BFU-Es in samples from the 15 MDS patients with low erythropoiesis but not in samples from the 5 patients with normal erythropoiesis or in the control samples (P<0.001).
The team said that a low BFU-E number at baseline was significantly associated with in vitro response to APG101 among the 20 MDS patients (P=0.031) and the 14 ESA-resistant patients (P=0.027).
Further investigation confirmed that a low clonogenic progenitor number at baseline, but not the level of CD95 or CD95 ligand expression, was predictive of the response to APG101.
“This study provides a rationale for further clinical investigation of this potential new therapeutic option in patients with severely impaired erythropoiesis who are resistant to erythropoiesis-stimulating agents,” Dr Fontenay said.
Apogenix has conducted a phase 1 trial of APG101 in transfusion-dependent patients with low- to intermediate-1-risk MDS. The company expects the results of this trial will be available in the coming months.
Researchers have found the fusion protein APG101 can rescue erythropoiesis in bone marrow samples from patients with lower-risk myelodysplastic syndromes (MDS).
Previous research suggested that CD95—a receptor that can induce apoptosis when triggered by the CD95 ligand—is overexpressed in two-thirds of patients with lower-risk MDS, and overexpression of CD95 is predictive of a lower response to erythropoiesis-stimulating agents (ESAs).
APG101, which consists of the extracellular domain of the CD95 receptor and the Fc domain of an IgG antibody, is designed to block the CD95 ligand.
The new study showed that APG101 can inhibit apoptosis in erythrocyte precursor cells and improve their overall proliferation rate. The drug increased the number of burst-forming unit-erythroid (BFU-E) progenitors in samples from MDS patients with low BFU-E numbers at baseline.
“APG101 added to cellular assays efficiently rescued the growth of erythroid progenitors in MDS patients harboring a profound defect of erythropoiesis, independent of the expression level of CD95 or CD95 ligand,” said Michaela Fontenay, MD, PhD, of the Institut Cochin in Paris, France.
Dr Fontenay and her colleagues described these results in Oncotarget. The research was funded by Apogenix, the company developing APG101.
By comparing bone marrow samples from MDS patients and healthy control subjects, the researchers found that CD95, but not CD95 ligand, was overexpressed in patients with lower-risk MDS.
Further analysis revealed that a patient’s CD95 expression level at diagnosis could predict response to an ESA. Specifically, CD95 overexpression was predictive of a lower response rate to ESAs in patients with low- or intermediate-1-risk MDS.
Next, the researchers tested bone marrow erythroid progenitors from 3 control subjects and 5 patients with MDS and found that CD95 expression increased during MDS erythroid progenitor amplification but remained lower in control cultures.
On day 5 of culture, the mean number of BFU-Es was significantly lower in MDS patient samples than in controls. And treatment with APG101 prompted a dose-dependent increase in BFU-E growth in MDS samples but not in controls.
When the researchers added APG101 (at 10 μg/mL) to the cultures over 7 days, they observed an improvement in the proliferation of erythroblasts but no significant effect on the kinetics of differentiation.
They also found that APG101 reduced apoptosis in immature precursors by 30% but had no effect on apoptosis in mature precursors.
Baseline BFU-E number affects response
The researchers then assessed the effects of APG101 in samples from 5 control subjects and 20 MDS patients with varying responses to ESAs and varying baseline levels of BFU-Es.
Fifteen of the MDS patients had a significantly lower number of baseline BFU-Es than controls (P=0.005), but 5 MDS patients had a mean number of BFU-Es that was comparable to controls (P=0.429). There was no significant difference in WHO classification or CD95 expression between the 2 groups of MDS patients (P=0.612).
However, 11 of the 15 patients with low erythropoiesis had received an ESA, and all of them were resistant to this treatment. In all, 15 of the MDS patients had received an ESA, and 14 were resistant to it (6 primary and 8 secondary).
The researchers found that APG101 induced a dose-dependent increase of BFU-Es in samples from the 15 MDS patients with low erythropoiesis but not in samples from the 5 patients with normal erythropoiesis or in the control samples (P<0.001).
The team said that a low BFU-E number at baseline was significantly associated with in vitro response to APG101 among the 20 MDS patients (P=0.031) and the 14 ESA-resistant patients (P=0.027).
Further investigation confirmed that a low clonogenic progenitor number at baseline, but not the level of CD95 or CD95 ligand expression, was predictive of the response to APG101.
“This study provides a rationale for further clinical investigation of this potential new therapeutic option in patients with severely impaired erythropoiesis who are resistant to erythropoiesis-stimulating agents,” Dr Fontenay said.
Apogenix has conducted a phase 1 trial of APG101 in transfusion-dependent patients with low- to intermediate-1-risk MDS. The company expects the results of this trial will be available in the coming months.
Lenalidomide prolongs PFS, not OS, in rel/ref MCL

Photo by Esther Dyson
Results of the phase 2 SPRINT trial suggest lenalidomide compares favorably to other single-agent therapies for patients with relapsed/refractory mantle cell lymphoma (MCL).
Patients who received lenalidomide had a significantly higher overall response rate and significantly longer progression-free survival (PFS) compared to patients who received single-agent therapy chosen by investigators.
However, there was no significant difference between the treatment arms with regard to overall survival (OS).
In addition, there were more treatment-related adverse events (AEs) in the lenalidomide arm than the investigator’s choice arm.
These results were published in The Lancet Oncology. The study was funded by Celgene Corporation, the company developing lenalidomide.
“The SPRINT study provided the first head-to-head, randomized, controlled study of single-agent lenalidomide compared with a range of single-agent comparators in previously treated MCL,” said study author Marek Trneny, MD, of Charles University Hospital in Prague, Czech Republic.
“The study demonstrated a statistically significant reduction in the risk of disease progression or death for lenalidomide over investigator’s choice in patients with relapsed/refractory MCL.”
The study enrolled 254 patients with relapsed/refractory MCL who were ineligible for intensive chemotherapy or hematopoietic stem cell transplant. The patients’ median age was 68.5 (range, 44-88), and they had received a median of 2 previous treatment regimens (range, 1-≥4).
Treatment
The patients were randomized (2:1) to receive lenalidomide orally at 25 mg on days 1-21 every 28 days until progressive disease or intolerability (n=170) or single-agent investigator’s choice (n=84), which included rituximab, gemcitabine, fludarabine, chlorambucil, and cytarabine. Patients who progressed on investigator’s choice could cross over to the lenalidomide arm.
In all, 167 patients in the lenalidomide arm and 83 patients in the investigator’s choice arm received at least 1 dose of treatment. In the investigator’s choice arm, 33% of patients received rituximab, 24% got gemcitabine, 22% fludarabine, 13% chlorambucil, and 8% cytarabine.
After disease progression on investigator’s choice, 46% of patients crossed over to the lenalidomide arm.
Before cycle 6, half of the patients in the lenalidomide arm and two-thirds of those in the investigator’s choice arm had discontinued treatment. After discontinuation, 46% of patients in the lenalidomide arm and 50% in the investigator’s choice arm received 1 or more new antilymphoma therapies (excluding crossover patients).
The investigators said the proportion of patients who responded to subsequent therapies or had progressive disease did not substantially differ whether they had previously been randomized to lenalidomide or investigator’s choice.
Efficacy
As of the data cutoff point (March 7, 2014), the median follow-up was 15.9 months (range, 7.6 to 31.7).
The overall response rate was 40% in the lenalidomide arm and 11% in the investigator’s choice arm (P<0.001). The complete response rates were 5% and 0%, respectively (P=0.04). The median duration of response was 16 months and 10.4 months, respectively.
Lenalidomide significantly prolonged PFS. The median PFS was 8.7 months in the lenalidomide arm and 5.2 months in the investigator’s choice arm. The hazard ratio was 0.61 (P=0.004).
There was no significant difference in OS between the treatment arms. The median OS was 27.8 months in the lenalidomide arm and 21.2 months in the investigator’s choice arm. The hazard ratio was 0.89 (P=0.45)
A total of 128 patients (50%) died. Most deaths occurred during follow-up, and the most frequent cause of death was underlying lymphoma.
Safety
The incidence of treatment-related AEs was 84% in the lenalidomide arm and 60% in the investigator’s choice arm.
The most common grade 3 or higher AEs—in the lenalidomide and investigator’s choice arms, respectively—were neutropenia (44% vs 34%) without increased risk of infection, thrombocytopenia (18% vs 28%), leukopenia (8% vs 11%), and anemia (8% vs 7%).
The incidence of any-grade nasopharyngitis was 15% in the lenalidomide arm and 6% in the investigator’s choice arm. The incidence of upper respiratory tract infection was 12% and 6%, respectively. Febrile neutropenia was reported in 6% and 2% of patients, respectively.
Growth factors were used more often in the lenalidomide arm than the investigator’s choice arm—30% and 23%, respectively. But platelet transfusions were used less often in the lenalidomide arm—4% and 11%, respectively.
Photo by Esther Dyson
Results of the phase 2 SPRINT trial suggest lenalidomide compares favorably to other single-agent therapies for patients with relapsed/refractory mantle cell lymphoma (MCL).
Patients who received lenalidomide had a significantly higher overall response rate and significantly longer progression-free survival (PFS) compared to patients who received single-agent therapy chosen by investigators.
However, there was no significant difference between the treatment arms with regard to overall survival (OS).
In addition, there were more treatment-related adverse events (AEs) in the lenalidomide arm than the investigator’s choice arm.
These results were published in The Lancet Oncology. The study was funded by Celgene Corporation, the company developing lenalidomide.
“The SPRINT study provided the first head-to-head, randomized, controlled study of single-agent lenalidomide compared with a range of single-agent comparators in previously treated MCL,” said study author Marek Trneny, MD, of Charles University Hospital in Prague, Czech Republic.
“The study demonstrated a statistically significant reduction in the risk of disease progression or death for lenalidomide over investigator’s choice in patients with relapsed/refractory MCL.”
The study enrolled 254 patients with relapsed/refractory MCL who were ineligible for intensive chemotherapy or hematopoietic stem cell transplant. The patients’ median age was 68.5 (range, 44-88), and they had received a median of 2 previous treatment regimens (range, 1-≥4).
Treatment
The patients were randomized (2:1) to receive lenalidomide orally at 25 mg on days 1-21 every 28 days until progressive disease or intolerability (n=170) or single-agent investigator’s choice (n=84), which included rituximab, gemcitabine, fludarabine, chlorambucil, and cytarabine. Patients who progressed on investigator’s choice could cross over to the lenalidomide arm.
In all, 167 patients in the lenalidomide arm and 83 patients in the investigator’s choice arm received at least 1 dose of treatment. In the investigator’s choice arm, 33% of patients received rituximab, 24% got gemcitabine, 22% fludarabine, 13% chlorambucil, and 8% cytarabine.
After disease progression on investigator’s choice, 46% of patients crossed over to the lenalidomide arm.
Before cycle 6, half of the patients in the lenalidomide arm and two-thirds of those in the investigator’s choice arm had discontinued treatment. After discontinuation, 46% of patients in the lenalidomide arm and 50% in the investigator’s choice arm received 1 or more new antilymphoma therapies (excluding crossover patients).
The investigators said the proportion of patients who responded to subsequent therapies or had progressive disease did not substantially differ whether they had previously been randomized to lenalidomide or investigator’s choice.
Efficacy
As of the data cutoff point (March 7, 2014), the median follow-up was 15.9 months (range, 7.6 to 31.7).
The overall response rate was 40% in the lenalidomide arm and 11% in the investigator’s choice arm (P<0.001). The complete response rates were 5% and 0%, respectively (P=0.04). The median duration of response was 16 months and 10.4 months, respectively.
Lenalidomide significantly prolonged PFS. The median PFS was 8.7 months in the lenalidomide arm and 5.2 months in the investigator’s choice arm. The hazard ratio was 0.61 (P=0.004).
There was no significant difference in OS between the treatment arms. The median OS was 27.8 months in the lenalidomide arm and 21.2 months in the investigator’s choice arm. The hazard ratio was 0.89 (P=0.45)
A total of 128 patients (50%) died. Most deaths occurred during follow-up, and the most frequent cause of death was underlying lymphoma.
Safety
The incidence of treatment-related AEs was 84% in the lenalidomide arm and 60% in the investigator’s choice arm.
The most common grade 3 or higher AEs—in the lenalidomide and investigator’s choice arms, respectively—were neutropenia (44% vs 34%) without increased risk of infection, thrombocytopenia (18% vs 28%), leukopenia (8% vs 11%), and anemia (8% vs 7%).
The incidence of any-grade nasopharyngitis was 15% in the lenalidomide arm and 6% in the investigator’s choice arm. The incidence of upper respiratory tract infection was 12% and 6%, respectively. Febrile neutropenia was reported in 6% and 2% of patients, respectively.
Growth factors were used more often in the lenalidomide arm than the investigator’s choice arm—30% and 23%, respectively. But platelet transfusions were used less often in the lenalidomide arm—4% and 11%, respectively.
Photo by Esther Dyson
Results of the phase 2 SPRINT trial suggest lenalidomide compares favorably to other single-agent therapies for patients with relapsed/refractory mantle cell lymphoma (MCL).
Patients who received lenalidomide had a significantly higher overall response rate and significantly longer progression-free survival (PFS) compared to patients who received single-agent therapy chosen by investigators.
However, there was no significant difference between the treatment arms with regard to overall survival (OS).
In addition, there were more treatment-related adverse events (AEs) in the lenalidomide arm than the investigator’s choice arm.
These results were published in The Lancet Oncology. The study was funded by Celgene Corporation, the company developing lenalidomide.
“The SPRINT study provided the first head-to-head, randomized, controlled study of single-agent lenalidomide compared with a range of single-agent comparators in previously treated MCL,” said study author Marek Trneny, MD, of Charles University Hospital in Prague, Czech Republic.
“The study demonstrated a statistically significant reduction in the risk of disease progression or death for lenalidomide over investigator’s choice in patients with relapsed/refractory MCL.”
The study enrolled 254 patients with relapsed/refractory MCL who were ineligible for intensive chemotherapy or hematopoietic stem cell transplant. The patients’ median age was 68.5 (range, 44-88), and they had received a median of 2 previous treatment regimens (range, 1-≥4).
Treatment
The patients were randomized (2:1) to receive lenalidomide orally at 25 mg on days 1-21 every 28 days until progressive disease or intolerability (n=170) or single-agent investigator’s choice (n=84), which included rituximab, gemcitabine, fludarabine, chlorambucil, and cytarabine. Patients who progressed on investigator’s choice could cross over to the lenalidomide arm.
In all, 167 patients in the lenalidomide arm and 83 patients in the investigator’s choice arm received at least 1 dose of treatment. In the investigator’s choice arm, 33% of patients received rituximab, 24% got gemcitabine, 22% fludarabine, 13% chlorambucil, and 8% cytarabine.
After disease progression on investigator’s choice, 46% of patients crossed over to the lenalidomide arm.
Before cycle 6, half of the patients in the lenalidomide arm and two-thirds of those in the investigator’s choice arm had discontinued treatment. After discontinuation, 46% of patients in the lenalidomide arm and 50% in the investigator’s choice arm received 1 or more new antilymphoma therapies (excluding crossover patients).
The investigators said the proportion of patients who responded to subsequent therapies or had progressive disease did not substantially differ whether they had previously been randomized to lenalidomide or investigator’s choice.
Efficacy
As of the data cutoff point (March 7, 2014), the median follow-up was 15.9 months (range, 7.6 to 31.7).
The overall response rate was 40% in the lenalidomide arm and 11% in the investigator’s choice arm (P<0.001). The complete response rates were 5% and 0%, respectively (P=0.04). The median duration of response was 16 months and 10.4 months, respectively.
Lenalidomide significantly prolonged PFS. The median PFS was 8.7 months in the lenalidomide arm and 5.2 months in the investigator’s choice arm. The hazard ratio was 0.61 (P=0.004).
There was no significant difference in OS between the treatment arms. The median OS was 27.8 months in the lenalidomide arm and 21.2 months in the investigator’s choice arm. The hazard ratio was 0.89 (P=0.45)
A total of 128 patients (50%) died. Most deaths occurred during follow-up, and the most frequent cause of death was underlying lymphoma.
Safety
The incidence of treatment-related AEs was 84% in the lenalidomide arm and 60% in the investigator’s choice arm.
The most common grade 3 or higher AEs—in the lenalidomide and investigator’s choice arms, respectively—were neutropenia (44% vs 34%) without increased risk of infection, thrombocytopenia (18% vs 28%), leukopenia (8% vs 11%), and anemia (8% vs 7%).
The incidence of any-grade nasopharyngitis was 15% in the lenalidomide arm and 6% in the investigator’s choice arm. The incidence of upper respiratory tract infection was 12% and 6%, respectively. Febrile neutropenia was reported in 6% and 2% of patients, respectively.
Growth factors were used more often in the lenalidomide arm than the investigator’s choice arm—30% and 23%, respectively. But platelet transfusions were used less often in the lenalidomide arm—4% and 11%, respectively.