User login
Young HCT survivors have increased risk for frailty
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()
for transplant
Photo credit: Chad McNeeley
Frailty among young adult hematopoietic cell transplant (HCT) survivors is high and approaches that of a community-based elderly population, according to results of the Bone Marrow Transplant Survivor Study.
Of the 998 HCT participants, frailty exceeded 8%, and they were 8.4 times more likely to be frail than their siblings.
Investigators defined frailty as exhibiting 3 or more of the following traits: clinically underweight, exhaustion, low energy expenditure, slow walking speed, and muscle weakness.
Because HCT recipients are exposed to high-intensity chemotherapy, radiation, and immunosuppression at points before, during, and after transplant, the investigators set out to determine whether non-elderly HCT recipients who have survived 2 years or more after transplant were at a higher risk of frailty compared with a sibling comparator group.
Smita Bhatia, MD, of the University of Alabama at Birmingham, and colleagues conducted the study of HCT survivors between the ages of 18 and 64 and compared the results to a sibling control group. The authors also looked at the subsequent mortality of HCT survivors.
They reported their findings in JAMA Oncology.
The 998 HCT survivors who participated in the study received their transplants at City of Hope in Duarte, California, or at the University of Minnesota in Minneapolis, between 1974 and 1998. The survivors and the 297 siblings completed questionnaires between February 1999 and June 2005.
Demographics
The HCT survivors were a mean age of 42.5 years, and 911 (93%) had health insurance coverage.
This was comparable to the sibling controls, who were a mean age of 43.8 years (P=0.09) and 279 (95%) had health insurance coverage.
However, more siblings were female (64%), non-Hispanic white (88%), college graduates (56%), and 92% had annual household incomes of $20,000 or more.
In the HCT survivor group, 46% were female (P<0.001), 81% non-Hispanic white (P=0.004), 49% college graduates (P<0.001), and 80% had annual incomes of $20,000 or more (P<0.001).
HCT survivors were a mean age of 33.8 years when they had their transplants and the interval between HCT and participation in the study was 8.7 years.
Hematologic malignancies were the major diagnoses leading to HCT. Twenty-three percent had a primary diagnosis of chronic myeloid leukemia, 24% had acute myeloid leukemia, 19% had non-Hodgkin lymphoma, 10% had acute lymphoblastic leukemia, and 9% had Hodgkin lymphoma.
Seventy-seven percent of the HCT survivors had total body irradiation, and 300 of the 562 who received allogeneic transplants had chronic GVHD, with 24% of them reporting active GVHD at the time they participated in the study.
Frailty
Only 2 siblings (0.7%) considered themselves frail compared to 84 (8.4%) HCT survivors.
More survivors were underweight and reported low energy expenditure compared to the sibling group, but the differences were not statistically significant, P=0.26 and P=0.14, respectively.
However, significantly more survivors reported exhaustion (P<0.001), slowness (P<0.001), and weakness (P<0.001) compared to the sibling group,
The investigators then adjusted the data for age at study participation, sex, race/ethnicity, education, household income, health insurance, presence of grades 3 or 4 chronic health conditions, and transplant institution. They then found the HCT survivors to be 8.35 times more likely to be frail than their siblings (P=0.003).
HCT survivors with low annual incomes (P=0.03), less than a college education (P=0.002), with grades 3 or 4 chronic health conditions (P=0.02), with multiple myeloma (P=0.05), or with resolved chronic (P=0.04) or active chronic GVHD (P<0.001) were more likely to be frail compared to the other HCT survivors.
Mortality
The investigators followed the patients for a median of 10.3 years from the time participants completed the survey. At that time, 182 (18%) patients had died.
The 10-year cumulative all-cause mortality was 39.3% for patients with frailty and 14.7% for patients without frailty (P<0.001).
The 10-year cumulative relapse-related mortality was 15.5% among frail HCT patients and 4.5% for non-frail HCT patients.
And the 10-year cumulative non-relapse mortality was also higher among frail HCT recipients, 23.9% compared to 10.2% of the non-frail HCT recipients (P<0.001).
Multivariate analysis revealed that frailty was associated with a 2.76-fold increase in death. The variables included age at study participation, sex, presence of grades 3 to 4 chronic health conditions, primary diagnosis, annual household income, and risk of relapse at transplant.
The investigators concluded that the therapies transplant patients undergo and post-transplant complications constitute a substantial stressor, placing HCT survivors at risk for frailty and premature aging.
“These findings demonstrate the need for interventions,” they added, “including personalized assessments and multidisciplinary efforts targeting both pre-frail and frail individuals to improve outcomes.” ![]()
Two compounds show promise against Zika virus

Photo courtesy of
Muhammad Mahdi Karim
Two compounds have shown activity against the Zika virus, according to Biotech Biotron, a company that develops compounds to fight viral diseases such as HIV and hepatitis C. The two compounds from its library killed the Zika virus in vitro, as determined by an independent USA laboratory facility.
“These early results are encouraging,” Michelle Miller, PhD, of Biotron, said. “Identification of these active compounds in our library is a starting point for designing potent drugs against Zika.”
At present, there is no approved vaccine or treatment for Zika virus, whose common symptoms include fever, rash, joint pain, and conjunctivitis.
While the symptoms are generally mild, Zika infection during pregnancy has been associated with microcephaly and other severe brain defects in the newborn.
In addition, Zika infection may be associated with an increased risk of Guillain-Barré syndrome, which is being investigated by the Centers for Disease Control and Prevention.
Biotron is planning to carry out more tests on the Zika virus to determine whether the compounds are likely to be safe and effective in humans.
The Zika virus is primarily spread by infected mosquitoes. But exposure to an infected person’s blood or other body fluids may also result in transmission. ![]()
Photo courtesy of
Muhammad Mahdi Karim
Two compounds have shown activity against the Zika virus, according to Biotech Biotron, a company that develops compounds to fight viral diseases such as HIV and hepatitis C. The two compounds from its library killed the Zika virus in vitro, as determined by an independent USA laboratory facility.
“These early results are encouraging,” Michelle Miller, PhD, of Biotron, said. “Identification of these active compounds in our library is a starting point for designing potent drugs against Zika.”
At present, there is no approved vaccine or treatment for Zika virus, whose common symptoms include fever, rash, joint pain, and conjunctivitis.
While the symptoms are generally mild, Zika infection during pregnancy has been associated with microcephaly and other severe brain defects in the newborn.
In addition, Zika infection may be associated with an increased risk of Guillain-Barré syndrome, which is being investigated by the Centers for Disease Control and Prevention.
Biotron is planning to carry out more tests on the Zika virus to determine whether the compounds are likely to be safe and effective in humans.
The Zika virus is primarily spread by infected mosquitoes. But exposure to an infected person’s blood or other body fluids may also result in transmission. ![]()
Photo courtesy of
Muhammad Mahdi Karim
Two compounds have shown activity against the Zika virus, according to Biotech Biotron, a company that develops compounds to fight viral diseases such as HIV and hepatitis C. The two compounds from its library killed the Zika virus in vitro, as determined by an independent USA laboratory facility.
“These early results are encouraging,” Michelle Miller, PhD, of Biotron, said. “Identification of these active compounds in our library is a starting point for designing potent drugs against Zika.”
At present, there is no approved vaccine or treatment for Zika virus, whose common symptoms include fever, rash, joint pain, and conjunctivitis.
While the symptoms are generally mild, Zika infection during pregnancy has been associated with microcephaly and other severe brain defects in the newborn.
In addition, Zika infection may be associated with an increased risk of Guillain-Barré syndrome, which is being investigated by the Centers for Disease Control and Prevention.
Biotron is planning to carry out more tests on the Zika virus to determine whether the compounds are likely to be safe and effective in humans.
The Zika virus is primarily spread by infected mosquitoes. But exposure to an infected person’s blood or other body fluids may also result in transmission. ![]()
Platelet transfusions do not reduce IVH risk in VLBW infants
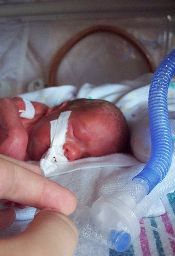
A retrospective, multicenter study of 972 very-low-birth-weight (VLBW) infants treated in 6 US neonatal intensive care units (NICUs) has shown that platelet transfusions do not significantly affect the incidence of intraventricular hemorrhage (IVH).
Thrombocytopenia is a risk factor for IVH, but investigators found no correlation between its severity and risk for IVH. Nor did they find platelet transfusions to have a significant effect on the incidence of IVH.
To describe platelet transfusion practices in US NICUs, senior author Martha Sola-Visner, MD, of Boston Children’s Hospital in Massachusetts, and colleagues studied NICU admissions from January 1, 2006, to December 31, 2007. They collected the last data on December 4, 2008.
Of the 972 VLBW infants, 231 (23.8%) received at least 1 platelet transfusion. And more males received transfusions (61%) than females.
Infants who received transfusions were more premature at 26.3 weeks’ gestation age compared with 28.8 weeks for those who did not receive transfusions, P<0.001.
Transfused infants were also smaller, with a mean birth weight of 805 g compared with 1113 g in the group that did not receive a transfusion, P<0.001.
Platelet transfusions
The 231 transfused infants received a total of 1002 platelet transfusions, with a mean of 4.3 per infant (range 1 to 63 transfusions).
Forty-one percent of infants had transfusions during the first 7 days of life only, amounting to 281 transfusions; 32.9% had transfusions after the first 7 days only, and 26.4% had transfusions during both periods. Seven hundred twenty-one transfusions were administered after day 7.
Almost two thirds of the transfusions, 65.4% or 653 of 998 transfusions, were given to infants who had a pre-transfusion platelet count of at least 50,000/μL.
The investigators poined out that this finding “was in contrast to UK NICUs,” where transfusions are administered at a median platelet count of 27,000/μL.
Illness severity
The investigators found significant differences among NICU sites in terms of clinical markers for transfusions.
Overall, 189 VLBW infants had platelet counts less than 100,000/μL in the first 7 days of life for a total of 402 days. And at least 1 platelet transfusion was given on 212 of those days. Of these, 198 transfusions (93.4%) had a marker of severe illness or bleeding.
On the other hand, of the 190 patient days without a transfusion, 113 (59.5%) had at least 1 of these markers (P<0.001).
Thrombocytopenia and IVH risk
The investigators evaluated the risk for IVH based on the lowest platelet count before the diagnosis of IVH was made.
They found that infants with thrombocytopenia were at higher risk for IVH, with a hazard ratio of 2.17 for any platelet count less than 150,000/μL (P<0.001).
Nevertheless, for the 314 infants with at least 1 platelet count less than 150,000/μL during the first 7 days of life, they found no association between severity of thrombocytopenia and the risk for subsequent IVH (P=0.70).
Transfusion and IVH risk
To determine whether platelet transfusions protected VLBW infants from IVH during their first 7 days of life, the investigators performed a Cox regression analysis in 756 infants.
They found that 134 infants (17.7%) had an IVH, including 62 (8.2%) with grade III or IV. So in the unadjusted model, they found a significant association between platelet transfusion and subsequent IVH, P=0.004.
However, when they adjusted the model for clinical covariates, only infants with grade III or IV IVH had a significantly greater risk with platelet transfusion, P=0.01.
Clinical covariates included sex, gestational age less than 28 weeks, 5-minute Apgar score less than 7, antenatal corticosteroid treatment, and pregnancy-induced hypertension as an indication for delivery.
The investigators also adjusted the model for clinical covariates and nadir platelet count of less than 15,000/μL. In this model, platelet transfusion became nonsignificant, even for IVH of grade III or IV.
The investigators noted that the degree to which their results are generalizable to infants with more severe thrombocytopenia is unclear, since infants in this analysis often had transfusions at platelet levels between 50,000/μL and 150,000/μL. They also collected the data approximately 8 years ago, and transfusion practices may have changed since then.
The 6 NICU study sites included Boston Children’s Hospital, Boston, Massachusetts; University of Iowa Children’s Hospital, Iowa City, Iowa; and 4 NICUs affiliated with Intermountain Health Care in Utah.
The investigators published their findings in JAMA Pediatrics. ![]()
A retrospective, multicenter study of 972 very-low-birth-weight (VLBW) infants treated in 6 US neonatal intensive care units (NICUs) has shown that platelet transfusions do not significantly affect the incidence of intraventricular hemorrhage (IVH).
Thrombocytopenia is a risk factor for IVH, but investigators found no correlation between its severity and risk for IVH. Nor did they find platelet transfusions to have a significant effect on the incidence of IVH.
To describe platelet transfusion practices in US NICUs, senior author Martha Sola-Visner, MD, of Boston Children’s Hospital in Massachusetts, and colleagues studied NICU admissions from January 1, 2006, to December 31, 2007. They collected the last data on December 4, 2008.
Of the 972 VLBW infants, 231 (23.8%) received at least 1 platelet transfusion. And more males received transfusions (61%) than females.
Infants who received transfusions were more premature at 26.3 weeks’ gestation age compared with 28.8 weeks for those who did not receive transfusions, P<0.001.
Transfused infants were also smaller, with a mean birth weight of 805 g compared with 1113 g in the group that did not receive a transfusion, P<0.001.
Platelet transfusions
The 231 transfused infants received a total of 1002 platelet transfusions, with a mean of 4.3 per infant (range 1 to 63 transfusions).
Forty-one percent of infants had transfusions during the first 7 days of life only, amounting to 281 transfusions; 32.9% had transfusions after the first 7 days only, and 26.4% had transfusions during both periods. Seven hundred twenty-one transfusions were administered after day 7.
Almost two thirds of the transfusions, 65.4% or 653 of 998 transfusions, were given to infants who had a pre-transfusion platelet count of at least 50,000/μL.
The investigators poined out that this finding “was in contrast to UK NICUs,” where transfusions are administered at a median platelet count of 27,000/μL.
Illness severity
The investigators found significant differences among NICU sites in terms of clinical markers for transfusions.
Overall, 189 VLBW infants had platelet counts less than 100,000/μL in the first 7 days of life for a total of 402 days. And at least 1 platelet transfusion was given on 212 of those days. Of these, 198 transfusions (93.4%) had a marker of severe illness or bleeding.
On the other hand, of the 190 patient days without a transfusion, 113 (59.5%) had at least 1 of these markers (P<0.001).
Thrombocytopenia and IVH risk
The investigators evaluated the risk for IVH based on the lowest platelet count before the diagnosis of IVH was made.
They found that infants with thrombocytopenia were at higher risk for IVH, with a hazard ratio of 2.17 for any platelet count less than 150,000/μL (P<0.001).
Nevertheless, for the 314 infants with at least 1 platelet count less than 150,000/μL during the first 7 days of life, they found no association between severity of thrombocytopenia and the risk for subsequent IVH (P=0.70).
Transfusion and IVH risk
To determine whether platelet transfusions protected VLBW infants from IVH during their first 7 days of life, the investigators performed a Cox regression analysis in 756 infants.
They found that 134 infants (17.7%) had an IVH, including 62 (8.2%) with grade III or IV. So in the unadjusted model, they found a significant association between platelet transfusion and subsequent IVH, P=0.004.
However, when they adjusted the model for clinical covariates, only infants with grade III or IV IVH had a significantly greater risk with platelet transfusion, P=0.01.
Clinical covariates included sex, gestational age less than 28 weeks, 5-minute Apgar score less than 7, antenatal corticosteroid treatment, and pregnancy-induced hypertension as an indication for delivery.
The investigators also adjusted the model for clinical covariates and nadir platelet count of less than 15,000/μL. In this model, platelet transfusion became nonsignificant, even for IVH of grade III or IV.
The investigators noted that the degree to which their results are generalizable to infants with more severe thrombocytopenia is unclear, since infants in this analysis often had transfusions at platelet levels between 50,000/μL and 150,000/μL. They also collected the data approximately 8 years ago, and transfusion practices may have changed since then.
The 6 NICU study sites included Boston Children’s Hospital, Boston, Massachusetts; University of Iowa Children’s Hospital, Iowa City, Iowa; and 4 NICUs affiliated with Intermountain Health Care in Utah.
The investigators published their findings in JAMA Pediatrics. ![]()
A retrospective, multicenter study of 972 very-low-birth-weight (VLBW) infants treated in 6 US neonatal intensive care units (NICUs) has shown that platelet transfusions do not significantly affect the incidence of intraventricular hemorrhage (IVH).
Thrombocytopenia is a risk factor for IVH, but investigators found no correlation between its severity and risk for IVH. Nor did they find platelet transfusions to have a significant effect on the incidence of IVH.
To describe platelet transfusion practices in US NICUs, senior author Martha Sola-Visner, MD, of Boston Children’s Hospital in Massachusetts, and colleagues studied NICU admissions from January 1, 2006, to December 31, 2007. They collected the last data on December 4, 2008.
Of the 972 VLBW infants, 231 (23.8%) received at least 1 platelet transfusion. And more males received transfusions (61%) than females.
Infants who received transfusions were more premature at 26.3 weeks’ gestation age compared with 28.8 weeks for those who did not receive transfusions, P<0.001.
Transfused infants were also smaller, with a mean birth weight of 805 g compared with 1113 g in the group that did not receive a transfusion, P<0.001.
Platelet transfusions
The 231 transfused infants received a total of 1002 platelet transfusions, with a mean of 4.3 per infant (range 1 to 63 transfusions).
Forty-one percent of infants had transfusions during the first 7 days of life only, amounting to 281 transfusions; 32.9% had transfusions after the first 7 days only, and 26.4% had transfusions during both periods. Seven hundred twenty-one transfusions were administered after day 7.
Almost two thirds of the transfusions, 65.4% or 653 of 998 transfusions, were given to infants who had a pre-transfusion platelet count of at least 50,000/μL.
The investigators poined out that this finding “was in contrast to UK NICUs,” where transfusions are administered at a median platelet count of 27,000/μL.
Illness severity
The investigators found significant differences among NICU sites in terms of clinical markers for transfusions.
Overall, 189 VLBW infants had platelet counts less than 100,000/μL in the first 7 days of life for a total of 402 days. And at least 1 platelet transfusion was given on 212 of those days. Of these, 198 transfusions (93.4%) had a marker of severe illness or bleeding.
On the other hand, of the 190 patient days without a transfusion, 113 (59.5%) had at least 1 of these markers (P<0.001).
Thrombocytopenia and IVH risk
The investigators evaluated the risk for IVH based on the lowest platelet count before the diagnosis of IVH was made.
They found that infants with thrombocytopenia were at higher risk for IVH, with a hazard ratio of 2.17 for any platelet count less than 150,000/μL (P<0.001).
Nevertheless, for the 314 infants with at least 1 platelet count less than 150,000/μL during the first 7 days of life, they found no association between severity of thrombocytopenia and the risk for subsequent IVH (P=0.70).
Transfusion and IVH risk
To determine whether platelet transfusions protected VLBW infants from IVH during their first 7 days of life, the investigators performed a Cox regression analysis in 756 infants.
They found that 134 infants (17.7%) had an IVH, including 62 (8.2%) with grade III or IV. So in the unadjusted model, they found a significant association between platelet transfusion and subsequent IVH, P=0.004.
However, when they adjusted the model for clinical covariates, only infants with grade III or IV IVH had a significantly greater risk with platelet transfusion, P=0.01.
Clinical covariates included sex, gestational age less than 28 weeks, 5-minute Apgar score less than 7, antenatal corticosteroid treatment, and pregnancy-induced hypertension as an indication for delivery.
The investigators also adjusted the model for clinical covariates and nadir platelet count of less than 15,000/μL. In this model, platelet transfusion became nonsignificant, even for IVH of grade III or IV.
The investigators noted that the degree to which their results are generalizable to infants with more severe thrombocytopenia is unclear, since infants in this analysis often had transfusions at platelet levels between 50,000/μL and 150,000/μL. They also collected the data approximately 8 years ago, and transfusion practices may have changed since then.
The 6 NICU study sites included Boston Children’s Hospital, Boston, Massachusetts; University of Iowa Children’s Hospital, Iowa City, Iowa; and 4 NICUs affiliated with Intermountain Health Care in Utah.
The investigators published their findings in JAMA Pediatrics. ![]()
Negative RT-PCR result doesn’t exclude Zika infection
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Photo by Jeremy L. Grisham
The Centers for Disease Control and Prevention (CDC) has issued an interim guidance on how to interpret results of the Zika virus antibody test.
Over the past few weeks, the FDA has authorized the use of new Zika tests, including RealStar® Zika Virus RT-PCR Kit U.S., Zika Virus RNA Qualitative Real-Time RT-PCR test, and the Trioplex Real-time RT-PCR Assay.
The CDC is now updating its guidance, since a negative real time reverse transcription-polymerase chain reaction (rRT-PCR) test does not necessarily rule out Zika infection.
In these cases, the CDC recommends immunoglobulin (Ig) M and neutralizing antibody testing, which can identify additional recent Zika virus infections.
The Zika antibody test, however, is difficult to interpret because of cross-reactivity with other flaviviruses. Zika is a mosquito-borne flavivirus that is closely related to dengue, West Nile, Japanese encephalitis, and yellow fever viruses.
The cross-reactivity can preclude identification of the specific infecting virus, particularly if a person was previously infected with or vaccinated against a related flavivirus. And appropriate clinical management is dependent upon proper identification of the virus.
If IgM test results are positive, equivocal, or inconclusive, the CDC recommends performing a plaque reduction neutralization test (PRNT) to confirm the diagnosis.
However, in people who have been previously infected with or vaccinated against a related flavivirus, even a 4-fold higher titer by PRNT may not be able to discriminate between anti-Zika virus antibodies and cross-reacting antibodies.
Therefore, the CDC now recommends an even more conservative approach to interpreting PRNT results to reduce the possibility of missing the diagnosis of either Zika or dengue virus infection.
The US Food and Drug Administration issued in February an Emergency Use Authorization for the CDC Zika IgM Antibody Capture Enzyme-Linked Immunosorbent Assay (Zika MAC-ELISA) for antibody testing.
It is used to detect Zika virus IgM antibodies in serum or cerebrospinal fluid from people with suspect Zika virus disease.
Presumptive positive results should be confirmed with PRNT against Zika, dengue, and other flaviviruses.
Equivocal and inconclusive results that are not resolved by re-testing should also have PRNT performed to rule out false-positive results.
For more information on interpretation of the Zika virus antibody test results, see the CDC’s Interim Guidance published as part of the Morbidity and Mortality Weekly Report for 31 May 2016. ![]()
Blood test may aid diagnosis of HELLP

Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
Photo by Nina Matthews
A blood test developed to diagnose a rare genetic blood cell disorder, atypical hemolytic uremic syndrome (aHUS), may also aid in the diagnosis of HELLP syndrome, a life-threatening high blood pressure condition that affects 1% of all pregnant women.
The study, based on blood samples from a small number of women, suggests that aHUS has similar underlying biochemistry to HELLP, which affects hemolysis, elevates liver enzymes, and causes a low platelet count. Both conditions have over activation of the alternative pathway of complement.
At present, no diagnostic blood or biomarker test exists to diagnose HELLP, which is thought to be a severe form of preeclampsia. The condition is diagnosed only by its symptoms.
Senior study author Robert Brodsky, MD, of Johns Hopkins, and his team developed the modified Ham test to diagnose aHUS, a genetic disorder in which abnormal blood clots form in small blood vessels in the kidneys. They published that work last year in Blood.
The two conditions share a number of traits, such as hemolysis, elevated liver enzymes, a low platelet count, kidney dysfunction, high blood pressure, and seizures. This led the investigators to believe that the modified Ham test could also help identify women with HELLP syndrome.
"The clinical implications from an obstetric point of view are potentially huge," said lead study author Arthur Vaught, MD, also of Johns Hopkins. "If this works, we can reduce pre-term deliveries, stays in the neonatal intensive care unit, and other complications for mothers and their babies."
The team analyzed serum samples from 14 women with classic or atypical HELLP syndrome, 7 with severe preeclampsia, 11 women with normal pregnancies, and 8 healthy nonpregnant women. All pregnant women were at least 23 weeks’ gestation, the point at which HELLP symptoms start to arise.
The team evaluated patient sera using terminal product of complement activation (C5b-9). They observed that women with classic or atypical HELLP had increased complement activation compared to nonpregnant controls.
Women with classic HELLP had an average cell killing of 34.3% compared with 26% in women with atypical HELLP, 5% in women with normal pregnancies, and3.3% in women who were not pregnant.
The investigators then added eculizumab to HELLP sera to see whether the agent could inhibit complement activation. They found that mixing HELLP serum with eculizumab-containing serum significantly decreased cell killing compared with HELLP serum alone.
The kill rate in women with classic or atypical HELLP decreased from 34% to 5% with eculizumab.
Eculizumab (Soliris), manufactured by Alexion Pharmaceuticals, is a monoclonal antibody approved by the US Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) and aHUS.
Further investigation is required to confirm these findings, but thus far, the investigators believe the modified Ham assay may assist in diagnosing the HELLP syndrome and corroborate its relationship to aHUS.
The current study by Vaught et al is published in Experimental Hematology. ![]()
AYAs still fare worse than kids with leukemia, lymphoma

patient and her father
Photo by Rhoda Baer
Adolescents and young adults (AYAs) are less likely than children to survive 8 relatively common types of cancer, according to a long-running study of cancer survival across Europe.
The study showed that AYAs had significantly worse survival rates than children if they were diagnosed with acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), Hodgkin or non-Hodgkin lymphoma (NHL), and 4 types of solid tumor malignancies.
The study’s authors say that variations in survival between age groups are due to a number of factors, including delays in diagnosis and treatment, a lack of treatment guidelines and clinical trials specifically for AYAs, and differences in the biology of some cancers.
“The good news is that the number of children, adolescents, and young adults surviving for at least 5 years after diagnosis has risen steadily over time in Europe,” said author Annalisa Trama, PhD, of The National Institute of Cancer (Istituto Nazionale dei Tumori: Fondazione IRCCS) in Milan, Italy.
“Across all cancers, the level of improvement is similar in these age groups. This contrasts with earlier results that adolescents and young adults diagnosed up to the 1990s were lagging behind children in terms of survival.”
“However, we found that adolescents and young adults still tend to die earlier than children for several cancers common to these age groups, particularly blood cancers like leukemias and non-Hodgkin’s lymphoma.”
Dr Trama and her colleagues reported these findings in The Lancet Oncology.
The researchers compared survival between AYAs (ages 15 to 39), children (ages 0 to 14), and adults (ages 40 to 69) who were diagnosed from 2000 to 2007 and followed up to at least 2008.
The team analyzed data from population-based cancer registries covering all or part of 27 European countries* and estimated 5-year survival for 56,505 cancer cases in children; 312,483 in AYAs; and 3,567,383 in adults. The researchers also analyzed changes in survival over time from 1999 to 2007.
For AYAs, survival at 5 years from diagnosis for all cancers combined was 82% for 2005-2007, which is up from 79% for 1999-2001 (P<0.0001). In children, survival improved from 76% to 79% over the same time period (P<0.0001).
Survival improved significantly in children and AYAs for ALL (P<0.0001) and NHL (P<0.0001 in AYAs and P=0.023 in children). On the other hand, between 1999 and 2007, survival rates remained unchanged for AYAs with AML (around 50%).
Overall, AYAs had slightly better 5-year survival than children because they were diagnosed more often with cancers with fairly good prognoses—Hodgkin lymphoma, NHL, germ cell tumors, melanoma, thyroid cancer, and breast cancer.
However, the overall survival rates conceal differences between specific cancers. Survival was significantly worse for AYAs than for children when it came to 8 relatively common cancers affecting both age groups:
- ALL—55.6% for AYAs and 85.8% for children (P<0.0001)
- AML—49.8% and 60.5%, respectively (P<0.0001)
- Hodgkin lymphoma—92.9% and 95.1%, respectively (P<0.0001)
- NHL—77.4% and 83.0%, respectively (P<0.0001)
- Astrocytomas—46.4% and 61.9%, respectively (P<0.0001)
- Ewing’s sarcoma of bone—49.3% and 66.6%, respectively (P<0.0001)
- Rhabdomyosarcoma—37.8% and 66.6%, respectively (P<0.0001)
- Osteosarcoma—61.5% and 66.8%, respectively (P=0.011).
AYAs had a survival advantage over adults for almost all major cancers affecting both age groups, supporting the idea that younger patients with few other illnesses are likely to fare better than older patients.
There are only 2 types of cancer for which AYAs were at a survival disadvantage—breast (83.5% vs 87.0%) and prostate (79.9% vs 89.8%).
Dr Trama and her colleagues pointed out that this analysis pre-dates recent initiatives to improve outcomes for AYAs that have been implemented in several European countries.
“The European Network for Teenagers and Young Adults with Cancer is advocating collaboration between pediatric and adult oncologists, greater access to clinical trials and research to improve treatments for this specific age group, as well as developing adolescent and young adult-specific practice guidelines, encouraging healthier lifestyles and the greater involvement of patients and patients support groups,” Dr Trama said.
“This study will provide an important starting point from which to evaluate whether these initiatives will reduce the gulf in survival between European adolescents and young adults and children with cancer.” ![]()
*Finland, Iceland, Norway, Sweden, England, Ireland, Northern Ireland, Scotland, Wales, Austria, Belgium, France, Germany, Netherlands, Switzerland, Croatia, Italy, Malta, Portugal, Slovenia, Spain, Bulgaria, Estonia, Latvia, Lithuania, Poland, and Slovakia
patient and her father
Photo by Rhoda Baer
Adolescents and young adults (AYAs) are less likely than children to survive 8 relatively common types of cancer, according to a long-running study of cancer survival across Europe.
The study showed that AYAs had significantly worse survival rates than children if they were diagnosed with acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), Hodgkin or non-Hodgkin lymphoma (NHL), and 4 types of solid tumor malignancies.
The study’s authors say that variations in survival between age groups are due to a number of factors, including delays in diagnosis and treatment, a lack of treatment guidelines and clinical trials specifically for AYAs, and differences in the biology of some cancers.
“The good news is that the number of children, adolescents, and young adults surviving for at least 5 years after diagnosis has risen steadily over time in Europe,” said author Annalisa Trama, PhD, of The National Institute of Cancer (Istituto Nazionale dei Tumori: Fondazione IRCCS) in Milan, Italy.
“Across all cancers, the level of improvement is similar in these age groups. This contrasts with earlier results that adolescents and young adults diagnosed up to the 1990s were lagging behind children in terms of survival.”
“However, we found that adolescents and young adults still tend to die earlier than children for several cancers common to these age groups, particularly blood cancers like leukemias and non-Hodgkin’s lymphoma.”
Dr Trama and her colleagues reported these findings in The Lancet Oncology.
The researchers compared survival between AYAs (ages 15 to 39), children (ages 0 to 14), and adults (ages 40 to 69) who were diagnosed from 2000 to 2007 and followed up to at least 2008.
The team analyzed data from population-based cancer registries covering all or part of 27 European countries* and estimated 5-year survival for 56,505 cancer cases in children; 312,483 in AYAs; and 3,567,383 in adults. The researchers also analyzed changes in survival over time from 1999 to 2007.
For AYAs, survival at 5 years from diagnosis for all cancers combined was 82% for 2005-2007, which is up from 79% for 1999-2001 (P<0.0001). In children, survival improved from 76% to 79% over the same time period (P<0.0001).
Survival improved significantly in children and AYAs for ALL (P<0.0001) and NHL (P<0.0001 in AYAs and P=0.023 in children). On the other hand, between 1999 and 2007, survival rates remained unchanged for AYAs with AML (around 50%).
Overall, AYAs had slightly better 5-year survival than children because they were diagnosed more often with cancers with fairly good prognoses—Hodgkin lymphoma, NHL, germ cell tumors, melanoma, thyroid cancer, and breast cancer.
However, the overall survival rates conceal differences between specific cancers. Survival was significantly worse for AYAs than for children when it came to 8 relatively common cancers affecting both age groups:
- ALL—55.6% for AYAs and 85.8% for children (P<0.0001)
- AML—49.8% and 60.5%, respectively (P<0.0001)
- Hodgkin lymphoma—92.9% and 95.1%, respectively (P<0.0001)
- NHL—77.4% and 83.0%, respectively (P<0.0001)
- Astrocytomas—46.4% and 61.9%, respectively (P<0.0001)
- Ewing’s sarcoma of bone—49.3% and 66.6%, respectively (P<0.0001)
- Rhabdomyosarcoma—37.8% and 66.6%, respectively (P<0.0001)
- Osteosarcoma—61.5% and 66.8%, respectively (P=0.011).
AYAs had a survival advantage over adults for almost all major cancers affecting both age groups, supporting the idea that younger patients with few other illnesses are likely to fare better than older patients.
There are only 2 types of cancer for which AYAs were at a survival disadvantage—breast (83.5% vs 87.0%) and prostate (79.9% vs 89.8%).
Dr Trama and her colleagues pointed out that this analysis pre-dates recent initiatives to improve outcomes for AYAs that have been implemented in several European countries.
“The European Network for Teenagers and Young Adults with Cancer is advocating collaboration between pediatric and adult oncologists, greater access to clinical trials and research to improve treatments for this specific age group, as well as developing adolescent and young adult-specific practice guidelines, encouraging healthier lifestyles and the greater involvement of patients and patients support groups,” Dr Trama said.
“This study will provide an important starting point from which to evaluate whether these initiatives will reduce the gulf in survival between European adolescents and young adults and children with cancer.” ![]()
*Finland, Iceland, Norway, Sweden, England, Ireland, Northern Ireland, Scotland, Wales, Austria, Belgium, France, Germany, Netherlands, Switzerland, Croatia, Italy, Malta, Portugal, Slovenia, Spain, Bulgaria, Estonia, Latvia, Lithuania, Poland, and Slovakia
patient and her father
Photo by Rhoda Baer
Adolescents and young adults (AYAs) are less likely than children to survive 8 relatively common types of cancer, according to a long-running study of cancer survival across Europe.
The study showed that AYAs had significantly worse survival rates than children if they were diagnosed with acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), Hodgkin or non-Hodgkin lymphoma (NHL), and 4 types of solid tumor malignancies.
The study’s authors say that variations in survival between age groups are due to a number of factors, including delays in diagnosis and treatment, a lack of treatment guidelines and clinical trials specifically for AYAs, and differences in the biology of some cancers.
“The good news is that the number of children, adolescents, and young adults surviving for at least 5 years after diagnosis has risen steadily over time in Europe,” said author Annalisa Trama, PhD, of The National Institute of Cancer (Istituto Nazionale dei Tumori: Fondazione IRCCS) in Milan, Italy.
“Across all cancers, the level of improvement is similar in these age groups. This contrasts with earlier results that adolescents and young adults diagnosed up to the 1990s were lagging behind children in terms of survival.”
“However, we found that adolescents and young adults still tend to die earlier than children for several cancers common to these age groups, particularly blood cancers like leukemias and non-Hodgkin’s lymphoma.”
Dr Trama and her colleagues reported these findings in The Lancet Oncology.
The researchers compared survival between AYAs (ages 15 to 39), children (ages 0 to 14), and adults (ages 40 to 69) who were diagnosed from 2000 to 2007 and followed up to at least 2008.
The team analyzed data from population-based cancer registries covering all or part of 27 European countries* and estimated 5-year survival for 56,505 cancer cases in children; 312,483 in AYAs; and 3,567,383 in adults. The researchers also analyzed changes in survival over time from 1999 to 2007.
For AYAs, survival at 5 years from diagnosis for all cancers combined was 82% for 2005-2007, which is up from 79% for 1999-2001 (P<0.0001). In children, survival improved from 76% to 79% over the same time period (P<0.0001).
Survival improved significantly in children and AYAs for ALL (P<0.0001) and NHL (P<0.0001 in AYAs and P=0.023 in children). On the other hand, between 1999 and 2007, survival rates remained unchanged for AYAs with AML (around 50%).
Overall, AYAs had slightly better 5-year survival than children because they were diagnosed more often with cancers with fairly good prognoses—Hodgkin lymphoma, NHL, germ cell tumors, melanoma, thyroid cancer, and breast cancer.
However, the overall survival rates conceal differences between specific cancers. Survival was significantly worse for AYAs than for children when it came to 8 relatively common cancers affecting both age groups:
- ALL—55.6% for AYAs and 85.8% for children (P<0.0001)
- AML—49.8% and 60.5%, respectively (P<0.0001)
- Hodgkin lymphoma—92.9% and 95.1%, respectively (P<0.0001)
- NHL—77.4% and 83.0%, respectively (P<0.0001)
- Astrocytomas—46.4% and 61.9%, respectively (P<0.0001)
- Ewing’s sarcoma of bone—49.3% and 66.6%, respectively (P<0.0001)
- Rhabdomyosarcoma—37.8% and 66.6%, respectively (P<0.0001)
- Osteosarcoma—61.5% and 66.8%, respectively (P=0.011).
AYAs had a survival advantage over adults for almost all major cancers affecting both age groups, supporting the idea that younger patients with few other illnesses are likely to fare better than older patients.
There are only 2 types of cancer for which AYAs were at a survival disadvantage—breast (83.5% vs 87.0%) and prostate (79.9% vs 89.8%).
Dr Trama and her colleagues pointed out that this analysis pre-dates recent initiatives to improve outcomes for AYAs that have been implemented in several European countries.
“The European Network for Teenagers and Young Adults with Cancer is advocating collaboration between pediatric and adult oncologists, greater access to clinical trials and research to improve treatments for this specific age group, as well as developing adolescent and young adult-specific practice guidelines, encouraging healthier lifestyles and the greater involvement of patients and patients support groups,” Dr Trama said.
“This study will provide an important starting point from which to evaluate whether these initiatives will reduce the gulf in survival between European adolescents and young adults and children with cancer.” ![]()
*Finland, Iceland, Norway, Sweden, England, Ireland, Northern Ireland, Scotland, Wales, Austria, Belgium, France, Germany, Netherlands, Switzerland, Croatia, Italy, Malta, Portugal, Slovenia, Spain, Bulgaria, Estonia, Latvia, Lithuania, Poland, and Slovakia
CHMP advises against approving MM drug

multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors. ![]()
multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors. ![]()
multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors.
Drug may provide benefit for VTE prevention
Results of the phase 3 APEX study suggest extended treatment with betrixaban may provide a benefit over standard-duration enoxaparin as thromboprophylaxis for patients with acute medical illnesses.
The study’s primary efficacy outcome was a composite of asymptomatic, proximal deep-vein thrombosis (DVT) and symptomatic venous thromboembolism (VTE).
Among patients with an elevated D-dimer level, there was no significant difference between the treatment arms with regard to the primary efficacy outcome.
However, there was a significant difference between the treatment arms in the entire study cohort and among a cohort of patients who either had an elevated D-dimer level or were 75 and older.
“In a pre-specified subgroup of medically ill patients who were D-dimer positive, extended-duration betrixaban demonstrated a reduction in VTE events approaching statistical significance,” explained study author C. Michael Gibson, MD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“In the pre-specified exploratory analyses of central lab D-dimer values and in progressively larger cohorts, including all study patients, the data demonstrated a consistent and significant reduction in VTE with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms.”
These results were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting in Montpellier, France, and published simultaneously in NEJM.
This study was supported by Portola Pharmaceuticals, the company developing betrixaban.
The trial enrolled 7513 patients who were hospitalized for acute medical illnesses. The patients were randomized to receive:
- Subcutaneous enoxaparin (40 mg once daily) for 10 ± 4 days plus oral placebo for 35 to 42 days (n=3754)
- Or subcutaneous placebo for 10 ± 4 days plus oral betrixaban (80 mg once daily) for 35 to 42 days (n=3759).
Results
The researchers performed sequential analyses in 3 prespecified cohorts:
- Cohort 1—Patients with an elevated D-dimer level
- Cohort 2—Patients with an elevated D-dimer level or an age of at least 75 years
- All enrolled patients.
The statistical analysis plan specified that if the between-group difference in any of the 3 analyses was not significant, the other analyses would be considered exploratory.
In cohort 1, the primary efficacy outcome (a composite of asymptomatic proximal DVT and symptomatic VTE) occurred in 6.9% of patients receiving betrixaban and 8.5% receiving enoxaparin. The relative risk (RR) was 0.81 (95% CI, 0.65 to 1.00; P=0.054).
In cohort 2, the primary efficacy outcome occurred in 5.6% of patients receiving betrixaban and 7.1% receiving enoxaparin. The RR was 0.80 (95% CI, 0.66 to 0.98; P=0.03).
In all patients, the primary efficacy outcome occurred in 5.3% of patients receiving betrixaban and 7.0% receiving enoxaparin. The RR was 0.76 (95% CI, 0.63 to 0.92; P=0.006).
Though the differences between the treatment arms were statistically significant in the overall patient population and in cohort 2, these analyses were considered exploratory because the differences did not reach statistical significance in cohort 1.
The primary safety outcome was major bleeding. In the overall population, major bleeding occurred in 0.7% of patients receiving betrixaban and 0.6% of patients receiving enoxaparin. The RR was 1.19 (95% CI, 0.67 to 2.12; P=0.55).
The secondary safety outcome was major or clinically relevant nonmajor bleeding. It occurred in 3.1% of patients receiving betrixaban and 1.6% of patients receiving enoxaparin. The RR was 1.97 (95% CI, 1.44 to 2.68; P<0.001).
“The APEX study results show consistent evidence that VTE events can be reduced with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms,” said study author Alexander T. Cohen, MBBS, MD, of Guy’s and St. Thomas’ Hospitals in London, UK.
“This is particularly true for the most clinically relevant symptomatic disease, where we observed a 30% to 45% reduction in events over the duration of the study. Such meaningful results in an area where there is currently no available recommended therapy offer important potential benefits for public health worldwide.”
Results of the phase 3 APEX study suggest extended treatment with betrixaban may provide a benefit over standard-duration enoxaparin as thromboprophylaxis for patients with acute medical illnesses.
The study’s primary efficacy outcome was a composite of asymptomatic, proximal deep-vein thrombosis (DVT) and symptomatic venous thromboembolism (VTE).
Among patients with an elevated D-dimer level, there was no significant difference between the treatment arms with regard to the primary efficacy outcome.
However, there was a significant difference between the treatment arms in the entire study cohort and among a cohort of patients who either had an elevated D-dimer level or were 75 and older.
“In a pre-specified subgroup of medically ill patients who were D-dimer positive, extended-duration betrixaban demonstrated a reduction in VTE events approaching statistical significance,” explained study author C. Michael Gibson, MD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“In the pre-specified exploratory analyses of central lab D-dimer values and in progressively larger cohorts, including all study patients, the data demonstrated a consistent and significant reduction in VTE with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms.”
These results were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting in Montpellier, France, and published simultaneously in NEJM.
This study was supported by Portola Pharmaceuticals, the company developing betrixaban.
The trial enrolled 7513 patients who were hospitalized for acute medical illnesses. The patients were randomized to receive:
- Subcutaneous enoxaparin (40 mg once daily) for 10 ± 4 days plus oral placebo for 35 to 42 days (n=3754)
- Or subcutaneous placebo for 10 ± 4 days plus oral betrixaban (80 mg once daily) for 35 to 42 days (n=3759).
Results
The researchers performed sequential analyses in 3 prespecified cohorts:
- Cohort 1—Patients with an elevated D-dimer level
- Cohort 2—Patients with an elevated D-dimer level or an age of at least 75 years
- All enrolled patients.
The statistical analysis plan specified that if the between-group difference in any of the 3 analyses was not significant, the other analyses would be considered exploratory.
In cohort 1, the primary efficacy outcome (a composite of asymptomatic proximal DVT and symptomatic VTE) occurred in 6.9% of patients receiving betrixaban and 8.5% receiving enoxaparin. The relative risk (RR) was 0.81 (95% CI, 0.65 to 1.00; P=0.054).
In cohort 2, the primary efficacy outcome occurred in 5.6% of patients receiving betrixaban and 7.1% receiving enoxaparin. The RR was 0.80 (95% CI, 0.66 to 0.98; P=0.03).
In all patients, the primary efficacy outcome occurred in 5.3% of patients receiving betrixaban and 7.0% receiving enoxaparin. The RR was 0.76 (95% CI, 0.63 to 0.92; P=0.006).
Though the differences between the treatment arms were statistically significant in the overall patient population and in cohort 2, these analyses were considered exploratory because the differences did not reach statistical significance in cohort 1.
The primary safety outcome was major bleeding. In the overall population, major bleeding occurred in 0.7% of patients receiving betrixaban and 0.6% of patients receiving enoxaparin. The RR was 1.19 (95% CI, 0.67 to 2.12; P=0.55).
The secondary safety outcome was major or clinically relevant nonmajor bleeding. It occurred in 3.1% of patients receiving betrixaban and 1.6% of patients receiving enoxaparin. The RR was 1.97 (95% CI, 1.44 to 2.68; P<0.001).
“The APEX study results show consistent evidence that VTE events can be reduced with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms,” said study author Alexander T. Cohen, MBBS, MD, of Guy’s and St. Thomas’ Hospitals in London, UK.
“This is particularly true for the most clinically relevant symptomatic disease, where we observed a 30% to 45% reduction in events over the duration of the study. Such meaningful results in an area where there is currently no available recommended therapy offer important potential benefits for public health worldwide.”
Results of the phase 3 APEX study suggest extended treatment with betrixaban may provide a benefit over standard-duration enoxaparin as thromboprophylaxis for patients with acute medical illnesses.
The study’s primary efficacy outcome was a composite of asymptomatic, proximal deep-vein thrombosis (DVT) and symptomatic venous thromboembolism (VTE).
Among patients with an elevated D-dimer level, there was no significant difference between the treatment arms with regard to the primary efficacy outcome.
However, there was a significant difference between the treatment arms in the entire study cohort and among a cohort of patients who either had an elevated D-dimer level or were 75 and older.
“In a pre-specified subgroup of medically ill patients who were D-dimer positive, extended-duration betrixaban demonstrated a reduction in VTE events approaching statistical significance,” explained study author C. Michael Gibson, MD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“In the pre-specified exploratory analyses of central lab D-dimer values and in progressively larger cohorts, including all study patients, the data demonstrated a consistent and significant reduction in VTE with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms.”
These results were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting in Montpellier, France, and published simultaneously in NEJM.
This study was supported by Portola Pharmaceuticals, the company developing betrixaban.
The trial enrolled 7513 patients who were hospitalized for acute medical illnesses. The patients were randomized to receive:
- Subcutaneous enoxaparin (40 mg once daily) for 10 ± 4 days plus oral placebo for 35 to 42 days (n=3754)
- Or subcutaneous placebo for 10 ± 4 days plus oral betrixaban (80 mg once daily) for 35 to 42 days (n=3759).
Results
The researchers performed sequential analyses in 3 prespecified cohorts:
- Cohort 1—Patients with an elevated D-dimer level
- Cohort 2—Patients with an elevated D-dimer level or an age of at least 75 years
- All enrolled patients.
The statistical analysis plan specified that if the between-group difference in any of the 3 analyses was not significant, the other analyses would be considered exploratory.
In cohort 1, the primary efficacy outcome (a composite of asymptomatic proximal DVT and symptomatic VTE) occurred in 6.9% of patients receiving betrixaban and 8.5% receiving enoxaparin. The relative risk (RR) was 0.81 (95% CI, 0.65 to 1.00; P=0.054).
In cohort 2, the primary efficacy outcome occurred in 5.6% of patients receiving betrixaban and 7.1% receiving enoxaparin. The RR was 0.80 (95% CI, 0.66 to 0.98; P=0.03).
In all patients, the primary efficacy outcome occurred in 5.3% of patients receiving betrixaban and 7.0% receiving enoxaparin. The RR was 0.76 (95% CI, 0.63 to 0.92; P=0.006).
Though the differences between the treatment arms were statistically significant in the overall patient population and in cohort 2, these analyses were considered exploratory because the differences did not reach statistical significance in cohort 1.
The primary safety outcome was major bleeding. In the overall population, major bleeding occurred in 0.7% of patients receiving betrixaban and 0.6% of patients receiving enoxaparin. The RR was 1.19 (95% CI, 0.67 to 2.12; P=0.55).
The secondary safety outcome was major or clinically relevant nonmajor bleeding. It occurred in 3.1% of patients receiving betrixaban and 1.6% of patients receiving enoxaparin. The RR was 1.97 (95% CI, 1.44 to 2.68; P<0.001).
“The APEX study results show consistent evidence that VTE events can be reduced with betrixaban, with no statistical difference in major bleeding between the betrixaban and enoxaparin arms,” said study author Alexander T. Cohen, MBBS, MD, of Guy’s and St. Thomas’ Hospitals in London, UK.
“This is particularly true for the most clinically relevant symptomatic disease, where we observed a 30% to 45% reduction in events over the duration of the study. Such meaningful results in an area where there is currently no available recommended therapy offer important potential benefits for public health worldwide.”
CHMP recommends extending brentuximab approval

Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).
Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).
Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138).
Dose of transplanted HSCs affects their behavior

in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.”
in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.”
in the bone marrow
The dose of hematopoietic stem cells (HSCs) given in a transplant affects how those cells behave in the body, according to research published in Cell Reports.
To track the behavior of transplanted HSCs, researchers “barcoded” individual mouse HSCs with a genetic marker and observed their contributions to hematopoiesis.
The team found that only 20% to 30% of HSCs differentiated into all types of blood cells.
This relatively small group of HSCs produced a disproportionately large amount of blood.
The remaining 70% to 80% of HSCs were more strategic, and their behavior was dependent on the dose of HSCs transplanted.
At higher HSC doses, the “strategic majority” of HSCs opted to differentiate early, producing a balanced array of T cells and B cells. But at low HSC doses, the strategic HSCs prioritized T-cell production.
“The dose of transplanted bone marrow has strong and lasting effects on how HSCs specialize and coordinate their behavior,” said study author Rong Lu, PhD, of the University of Southern California in Los Angeles.
“This suggests that altering transplantation dose could be a tool for improving outcomes for patients—promoting bone marrow engraftment, reducing the risk of infection, and, ultimately, saving lives.”