User login
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.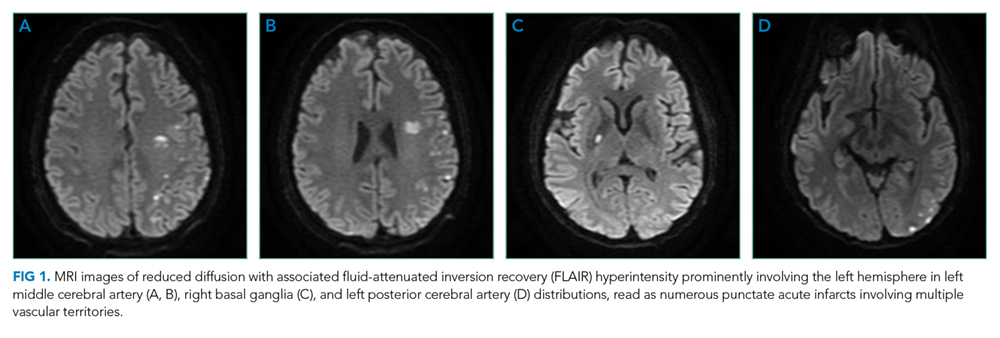
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.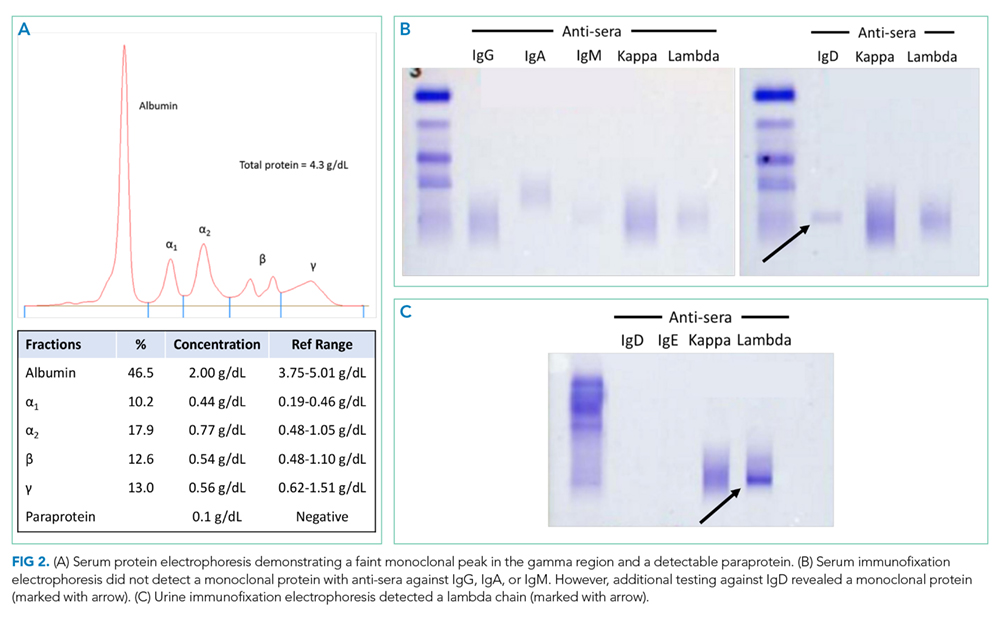
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.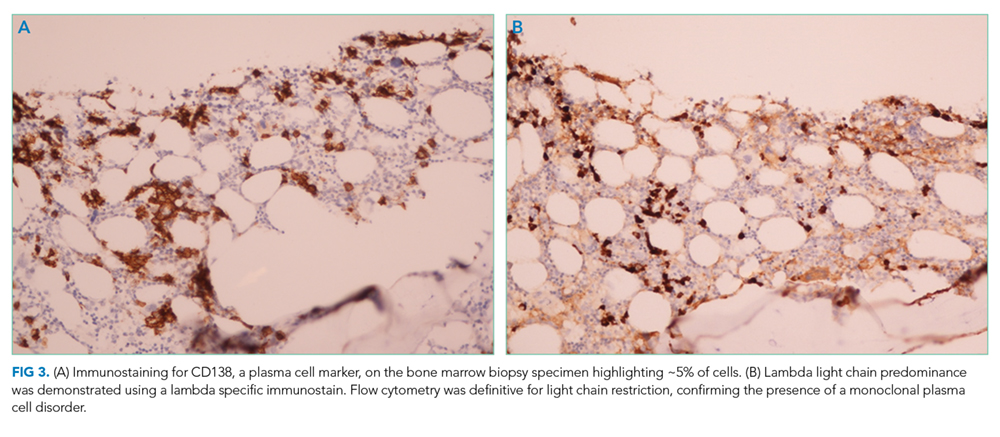
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
A 39-year-old man presented to a neurologist with three weeks of progressive leg weakness associated with numbness in his feet and fingertips. His medical history included hypertriglyceridemia, hypogonadism, and gout. He was taking fenofibrate and colchicine as needed. There was no family history of neurologic issues. He did not smoke or drink alcohol.
The patient appeared well with a heart rate of 76 beats per minute, blood pressure 133/72 mm Hg, temperature 36.6°C, respiratory rate 16 breaths per minute, and oxygen saturation 100% on room air. His cardiopulmonary and abdominal examinations were normal. His skin was warm and dry without rashes. On neurologic examination, upper extremity strength and sensation was normal. Bilateral hip flexion, knee flexion, and knee extension strength was 4/5; bilateral ankle dorsiflexion and plantar flexion strength was 3/5. Reflexes were trace in the arms and absent at the patellae and ankles. He had symmetric, length-dependent reduction in vibration, pinprick, and light touch sensation in his legs.
Peripheral neuropathy presenting with ascending symmetric motor and sensory deficits progressing over three weeks raises the suspicion of an acquired inflammatory demyelinating polyneuropathy (AIDP), a variant of Guillain-Barre Syndrome. Alternative causes of acute polyneuropathy include thiamine (B1) deficiency, vasculitis, sarcoidosis, or malignancy, particularly lymphoma and multiple myeloma. Further evaluation should include electromyography, nerve conduction studies, lumbar puncture with cerebrospinal fluid (CSF) protein, glucose, and cell count differential. Follow-up laboratory testing based on results of the above may include serum protein electrophoresis (SPEP), serum free light chains (sFLC), vitamin B12, human immunodeficiency virus (HIV), hepatitis B and C testing, antinuclear antibody, and erythrocyte sedimentation rate.
Electromyography and nerve conduction studies revealed a sensorimotor mixed axonal/demyelinating polyneuropathy in all extremities. CSF analysis found one white cell per mm3, glucose of 93 mg/dL, and protein of 313 mg/dL. Magnetic resonance imaging (MRI) of the spine without contrast showed normal cord parenchyma. The vitamin B12 level was 441 pg/mL (normal >200 pg/mL). Antibodies to HIV-1, HIV-2, hepatitis C virus, and Borrelia burgdorferi were negative. Serum protein electrophoresis (SPEP) and immunofixation were normal.
The patient received two courses of intravenous immunoglobulin (IVIG) for suspected AIDP. His weakness progressed over the next several weeks to the point that he required a wheelchair.
Progression of symptoms beyond three weeks and lack of response to IVIG are atypical for AIDP. Alternate diagnoses for a sensorimotor polyneuropathy should be considered. Causes of subacute or chronic demyelinating polyneuropathy include inflammatory conditions (chronic inflammatory demyelinating polyneuropathy [CIDP], connective-tissue disorders), paraprotein disorders (myeloma, amyloidosis, lymphoplasmacytic lymphoma), paraneoplastic syndromes, infectious diseases (HIV, Lyme disease), infiltrative disorders (sarcoidosis), medications or toxins, and hereditary disorders. Of these etiologies, the first three seem the most likely given the history and clinical course, the negative HIV and Lyme testing, and the absence of exposures and family history. Normal SPEP and immunofixation make paraprotein disorders less likely, but sFLC testing should be sent to evaluate for a light chain-only paraprotein. A paraneoplastic antibody panel and a CT of the chest, abdomen, and pelvis should be ordered to evaluate for sarcoidosis, lymphoma, or other malignancies. Although a peripheral nerve biopsy would further classify the polyneuropathy, it is of low diagnostic yield in patients with subacute and chronic distal symmetric polyneuropathies and is associated with significant morbidity. In the absence of history or physical exam findings to narrow the differential diagnosis for polyneuropathy, testing for paraneoplastic antibodies and imaging is appropriate.
The patient tested negative for antiganglioside GM1 and antimyelin-associated glycoprotein antibodies. Urine arsenic, lead, and mercury levels were normal. Tests for serum antinuclear antibody, rapid plasmin reagin, and a paraneoplastic neuropathy panel including amphiphysin antibody, CV2 antibody, and Hu auto-antibody were negative. Repeat electrodiagnostic testing was consistent with CIDP. The patient received prednisone 60 mg daily for six weeks and was then tapered to 30 mg daily over six weeks. Concurrently, he underwent twelve cycles of plasma exchange. His strength improved, and he could walk with a cane; however, weakness recurred when steroids were further tapered.
He was maintained on prednisone 50 mg daily. Over the next year, the patient’s lower extremities became flaccid and severely atrophied. He developed hyperpigmented patches on his trunk, severe gastroesophageal reflux disease (GERD), dysphonia, and gynecomastia. He had lost 60 pounds since symptom onset. He was prescribed levothyroxine for subclinical hypothyroidism (thyroid stimulating hormone 12.63 µIU/mL [normal 0.10-5.50 µIU/mL], free thyroxine 0.8 ng/dL [0.8-1.7 ng/dL]).
At this point, the diagnosis of CIDP should be questioned, and additional investigation is warranted. Although improvement was initially observed with plasma exchange and steroids, subsequent progression of symptoms despite prednisone suggests a nonimmune-mediated etiology, such as a neoplastic or infiltrative process. Conversely, negative serologic testing for paraneoplastic antibodies may be due to an antibody that has not been well characterized.
While prednisone could explain GERD and gynecomastia, the weight loss, dysphonia, and subclinical hypothyroidism may offer clues to the diagnosis underlying the neurological symptoms. Weight loss raises suspicion of a hypercatabolic process such as cancer, cachexia, systemic inflammation, heart failure, or chronic obstructive pulmonary disease. Causes of dysphonia relevant to this presentation include neurologic dysfunction related to malignant invasion of the vagus nerve or demyelinating disease. Subclinical hypothyroidism due to chronic autoimmune thyroiditis seems most likely in the absence of a medication effect or thyroid injury, yet infiltrative disorders of the thyroid (eg, amyloidosis, sarcoidosis, lymphoma) should also be considered. A diagnosis that unifies the neurologic and nonneurologic findings would be desirable; lymphoma with paraneoplastic peripheral neuropathy manifesting as CIDP seems most likely. As of yet, CT of the chest, abdomen, and pelvis or an 18-Fluoro-deoxyglucose positron emission tomography (FDG-PET) scan have not been obtained and would be helpful to evaluate for underlying malignancy. Further evaluation for a paraprotein disorder that includes sFLC is also still indicated to rule out a paraneoplastic disorder that may be associated with polyneuropathy.
Repeat SPEP and serum immunofixation were normal. sFLC assay showed elevated levels of both kappa and lambda light chains with a ratio of 0.61 (reference range: 0.26-1.25). Urine protein electrophoresis (UPEP) from a 24-hour specimen showed a homogenous band in the gamma region, but urine immunofixation demonstrated polyclonal light chains. The plasma vascular endothelial growth factor (VEGF) level was 612 pg/mL (reference range, 31-86 pg/mL).
CT imaging of the chest, abdomen, and pelvis with contrast demonstrated an enlarged liver and spleen and possible splenic infarcts. A skeletal survey and whole-body FGD-PET scan were normal. The patient declined bone marrow biopsy.
Polyneuropathy secondary to a monoclonal protein was previously considered, and an SPEP was normal. Full evaluation for a monoclonal protein additionally requires sFLC testing. If clinical suspicion remains high after a negative result, 24-hour UPEP and urine immunofixation should be obtained. Normal results in this case argue against the presence of a monoclonal protein.
The presence of a monoclonal protein and polyneuropathy are mandatory diagnostic criteria for POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes), a plasma cell proliferative disorder. Major diagnostic criteria include osteosclerotic bone lesions, Castleman’s disease, and markedly elevated VEGF levels. Castleman’s disease is a lymphoproliferative disorder characterized by angiofollicular lymphoid hyperplasia that results in lymphadenopathy in one or multiple lymph node regions. Imaging studies reveal organomegaly, one of many minor criteria, but not bone lesions or lymphadenopathy. A diagnosis of POEMS syndrome requires the presence of both mandatory, one major, and one minor criteria. Since only one of two of the mandatory criteria are met at this point, a diagnosis of POEMS syndrome cannot be made.
Eighteen months after symptom onset, the patient presented to the emergency department with dyspnea, orthopnea, and lower extremity edema. B-type natriuretic peptide was 1564 pg/mL. Transthoracic echocardiography showed a severely dilated and hypertrophied left ventricle. Left ventricular ejection fraction was 20%. A furosemide infusion was initiated. Angiography of the coronary vessels was not performed. Congo red stain of an abdominal adipose biopsy was negative for amyloid.
On hospital day five, he developed gangrenous changes in his right first toe. CT angiography of the abdomen and lower extremities demonstrated patent three vessel runoff to the foot with an infrarenal aortic thrombus. Heparin infusion was started. On hospital day 10, the patient developed expressive aphasia and somnolence, prompting intubation for airway protection. MRI and MR angiography (MRA) of the brain and cerebral vessels revealed multiple bilateral acute ischemic strokes (Figure 1) without flow limiting stenosis in cerebral vessels.
These clinical developments lead to an important opportunity to rethink this patient’s working diagnosis. The new diagnosis of heart failure in this young patient with polyneuropathy raises suspicion for an infiltrative cardiomyopathy such as amyloidosis, sarcoidosis, or Fabry disease. Of these, Fabry disease is the least likely because it is typically characterized by a painful burning sensation in response to specific triggers. Although polyneuropathy and heart failure may be concurrently observed with both sarcoidosis and amyloidosis, the absence of an apparent arrhythmia make amyloidosis the more likely of these two diagnoses. The development of an arterial thrombus and multiple strokes may represent emboli from a cardiac thrombus.
Cardiac imaging and tissue biopsy of the heart or other affected organs would distinguish between these diagnostic possibilities. An abdominal adipose biopsy negative for amyloid does not rule out amyloidosis, as the test is approximately 80% sensitive when cardiac amyloidosis is present and varies depending on the etiology of the amyloid protein (ie, light chain vs transthyretin). Evaluation of cardiac amyloid in the setting of peripheral neuropathy should include echocardiography (as was performed here) and repeat testing for a monoclonal protein.
If clinical suspicion of a paraprotein-associated disorder remains high and both SPEP and sFLC are normal, it is important to obtain a 24-hour UPEP and immunofixation. A monoclonal protein can be overlooked by SPEP and serum immunofixation if the monoclonal protein is composed only of a light chain or if the monoclonal protein is IgD or IgE. In these rare circumstances, sFLC analysis or 24-hour UPEP and immunofixation should mitigate the potential for a falsely negative SPEP/IFE. These studies are normal in this case, which argues against the presence of a monoclonal protein.
Transesophageal echocardiography showed grade IV atheromatous plaque within the descending thoracic aorta with mobile elements suggesting a superimposed thrombus; there was no intracardiac shunt or thrombus. MRA of the neck and great vessels was normal.
Testing for heparin-induced thrombocytopenia (HIT) was sent due to thrombocytopenia and the presence of thrombosis. An immunoassay for antiheparin-platelet factor 4 (anti-PF4) antibodies was substantially positive (optical density 2.178); however, functional testing with a washed platelet heparin-induced platelet activation assay was negative. Anticoagulation was changed to argatroban due to concern for HIT. Dry gangrenous changes developed in all distal toes on the right foot and three toes on the left foot. A right radial artery thrombus formed at the site of a prior arterial line.
Thrombocytopenia that develops between the fifth and tenth day following heparin exposure in a patient with new thromboses is consistent with HIT. However, the patient’s infrarenal aortic thrombus preceded the initiation of heparin, and negative functional testing undermines the diagnosis of HIT in this case. Therefore, the arterial thromboses may be related to an underlying unifying diagnosis.
A third SPEP showed a 0.1 g/dL M-spike in the gamma region, but standard immunofixation did not reveal a monoclonal protein (Figure 2). However, a specific request for immunofixation testing using IgD antisera detected an IgD heavy chain. A lambda chain comprising 3% of urine protein was detected on 24-hour urine immunofixation but was not detectable by serum immunofixation. Bone marrow biopsy demonstrated plasma cells comprising 5% of bone marrow cellularity (Figure 3); flow cytometry of the aspirate demonstrated an abnormal lambda-restricted plasma cell population.
When a monoclonal protein is identified but does not react with standard antisera to detect IgG, IgM, and IgA, immunofixation with IgD and IgE antisera are necessary to rule out a monoclonal IgD or IgE protein. The underlying IgD isotype coupled with its low abundance made detection of this monoclonal protein especially challenging. With the discovery of a monoclonal protein in the context of polyneuropathy, the mandatory criteria of POEMS syndrome are met. The elevated VEGF level and hypothyroidism meet major and minor criteria, respectively. Arterial thromboses and heart failure are other features that may be observed in cases of POEMS syndrome.
POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) was diagnosed. Prednisone was continued, and weekly cyclophosphamide was initiated. After six weeks, the VEGF level remained elevated, and a neurologic examination showed minimal improvement. Due to poor respiratory muscle strength and difficulty managing secretions, he underwent percutaneous tracheostomy and gastrostomy tube placement. Unfortunately, his condition further deteriorated and he subsequently died of sepsis from pneumonia.
An autopsy revealed acute bronchopneumonia and multiple acute and subacute cerebral infarctions. There was extensive peripheral mixed axonal/demyelinating neuropathy, hepatosplenomegaly, atrophy of the thyroid and adrenal glands, hyperpigmented patches and thickened integument, and severe aortic and coronary atherosclerotic disease with a healed myocardial infarction.
DISCUSSION
POEMS syndrome1 is a rare constellation of clinical and laboratory findings resulting from an underlying plasma cell proliferative disorder. This paraneoplastic syndrome is characterized by the chronic overproduction of proinflammatory and proangiogenic cytokines, including VEGF, which are postulated to drive its manifestations,2 though the exact pathogenesis is not understood. Some of the disease’s most common features are summarized by its name: polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, and skin changes.3
The International Myeloma Working Group (IMWG) diagnostic criteria1 (Table) require the presence of both mandatory criteria (polyneuropathy and monoclonal plasma cell proliferation), plus at least one major and one minor criterion. Delayed diagnosis or misdiagnosis of this protean disorder is often driven by its rarity and clinical overlap with other paraprotein-associated polyneuropathies. These include amyloidosis, cryoglobulinemia, and monoclonal gammopathy of undetermined significance (MGUS), which can all produce antibodies directed against neural antigens. In addition, polyneuropathy is often the first and most striking manifestation of POEMS syndrome, fostering confusion with CIDP as both disorders are subacute, symmetric, motor-dominant, mixed axonal/demyelinating polyneuropathies.4
IgD and IgE monoclonal gammopathies are extremely rare. IgD myeloma, for instance, accounts for 2% of multiple myeloma cases, and IgE myeloma has been reported fewer than 50 times.5 IgD is secreted only in very small amounts, ordinarily representing 0.25% of the immunoglobulins in serum, while the majority is found in the plasma membranes of mature B-cells.6 These monoclonal gammopathies often escape detection for two reasons: (1) the very low paraprotein concentration produces undetectable or small M-protein levels on electrophoresis,5 and (2) immunofixation is routinely performed without antisera against IgD and IgE heavy chains.7
While this case depicts a rare manifestation of a rare disease, the principles underlying its elusive diagnosis are routinely encountered. Recognition of the specific limitations of the SPEP, UPEP, sFLC, and immunofixation tests, outlined below, can assist the hospitalist when suspicion for paraproteinemia is high.
First, low levels of monoclonal proteins may be associated with a normal SPEP. Accordingly, suspicion of a plasma cell dyscrasia should prompt serum immunofixation, even when the electrophoretic pattern appears normal.8
Second, laboratories routinely perform immunofixation with antisera against IgG, IgA, and IgM heavy chains and kappa and lambda light chains, whereas testing with IgD or IgE antisera must be specifically requested. Thus, clinicians should screen for the presence of IgD and IgE in patients with an apparently free monoclonal immunoglobulin light chain in the serum or with a monoclonal serum protein and negative immunofixation. In this case, the paraprotein was not detected on the first two serum electrophoreses, likely due to a low serum concentration, then missed on immunofixation due to a lack of IgD antiserum. On admission to the hospital, this patient had a very low paraprotein concentration (0.1 g/dL) on SPEP, and the lab initially reported negative immunofixation. When asked to test specifically for IgD and IgE, the lab ran a more comprehensive immunofixation revealing IgD heavy chain paraprotein.
Third, this case illustrates the limitations of the sFLC assay. IMWG guidelines specify that sFLC assay in combination with SPEP and serum immunofixation is sufficient to screen for monoclonal plasma cell proliferative disorders other than light chain amyloidosis (which requires all the serum tests as well as 24-hour urine immunofixation).9 Though the sFLC assay has been demonstrated to be more sensitive than urine analysis for detecting monoclonal free light chains,10 it is still subject to false negatives. Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal sFLC,11 the latter of which likely explains why the sFLC ratio was repeatedly normal in this case. In these circumstances, monoclonal free light chains can be identified by urine studies.11 In this case, 24-hour urine immunofixation detected the excess light chain that was not evident on the sFLC assay. Even with these pitfalls in mind, there is still no evident explanation as to why the 24-hour urine studies done prior to the patient’s hospital admission did not reveal a monoclonal light chain.
This case also highlights the thrombotic diathesis in POEMS syndrome. Although the patient was treated with argatroban for suspected HIT, it is likely that the HIT antibody result was a false positive, and his thrombi were better explained by POEMS syndrome in and of itself. Coronary, limb, and cerebral artery thromboses have been linked to POEMS syndrome,12,13 all of which were present in this case. Laboratory testing for HIT involves an immunoassay to detect circulating HIT antibody and a functional assay to measure platelet activity in the presence of patient serum and heparin. The immunoassay binds anti-PF4/heparin complex irrespective of its ability to activate platelets. The presence of nonspecific antibodies may lead to cross-reactions with the immunoassay test components, which has been demonstrated in cases of MGUS.14 In this case, elevated production of monoclonal antibodies by plasma cells may have led to false-positive results. With moderate to high clinical suspicion of HIT, the combination of a positive immunoassay and negative functional assay (as in this case) make the diagnosis of HIT indeterminate.15
TEACHING POINTS
- If a monoclonal protein is suggested by SPEP but cannot be identified by standard immunofixation, request immunofixation for IgD or IgE. Screen patients for IgD and IgE paraproteins before making a diagnosis of light chain multiple myeloma.
- Polyclonal gammopathy or reduced renal clearance with accumulation of free light chains in the serum may mask the presence of low levels of monoclonal FLC and result in a normal sFLC ratio.
- Thrombosis is a less-recognized but documented feature of POEMS syndrome which may be mediated by the overproduction of proinflammatory and proangiogenic cytokines, though the precise pathogenesis is unknown.
Acknowledgment
The authors thank Dr. Theodore Kurtz and Dr. Anne Deucher from the department of laboratory medicine at the University of California, San Francisco for providing their respective expertise in clinical chemistry and hematopathology.
Disclosures
The authors have no conflicts of interests to disclose.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5.
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
1. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-ee548. doi: 10.1016/S1470-2045(14)70442-5. PubMed
2. Watanabe O, Arimura K, Kitajima I, Osame M, Maruyama I. Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome. Lancet. 1996;347(9002):702. doi: 10.1016/S0140-6736(96)91261-1. PubMed
3. Dispenzieri A. How I treat POEMS syndrome. Blood. 2012;119(24):5650-5658. doi: 10.1182/blood-2012-03-378992. PubMed
4. Nasu S, Misawa S, Sekiguchi Y, et al. Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83(5):476-479. doi: 10.1136/jnnp-2011-301706. PubMed
5. Pandey S, Kyle RA. Unusual myelomas: a review of IgD and IgE variants. Oncology. 2013;27(8):798-803. PubMed
6. Vladutiu AO. Immunoglobulin D: properties, measurement, and clinical relevance. Clin Diagn Lab Immunol. 2000;7(2):131-140. doi: 10.1128/CDLI.7.2.131-140.2000. PubMed
7. Sinclair D, Cranfield T. IgD myeloma: A potential missed diagnosis. Ann Clin Biochem. 2001;38(5):564-565. doi: 10.1177/000456320103800517. PubMed
8. Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701-4705. doi: 10.1182/blood-2010-10-299529. PubMed
9. Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215-224. doi: 10.1038/leu.2008.307. PubMed
10. Dejoie T, Attal M, Moreau P, Harousseau JL, Avet-Loiseau H. Comparison of serum free light chain and urine electrophoresis for the detection of the light chain component of monoclonal immunoglobulins in light chain and intact immunoglobulin multiple myeloma. Haematologica. 2016;101(3):356-362. doi: 10.3324/haematol.2015.126797. PubMed
11. Levinson SS. Polyclonal free light chain of Ig may interfere with interpretation of monoclonal free light chain κ/λ ratio. Ann Clin Lab Sci. 2010;40(4):348-353. PubMed
12. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-2506. doi: 10.1182/blood-2002-07-2299. PubMed
13. Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology. 2009;73(16):1308-1312. doi: 10.1212/WNL.0b013e3181bd136b. PubMed
14. Markovic I, Debeljak Z, Bosnjak B, Marijanovic M. False positive immunoassay for heparin-induced thrombocytopenia in the presence of monoclonal gammopathy: a case report. Biochemia Medica. 2017;27(3):030801. doi: 10.11613/BM.2017.030801. PubMed
15. Cuker A, Cines DB. How I treat heparin-induced thrombocytopenia. Blood. 2012;119(10):2209-2218. doi: 10.1182/blood-2011-11-376293. PubMed
© 2019 Society of Hospital Medicine