User login
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”

At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
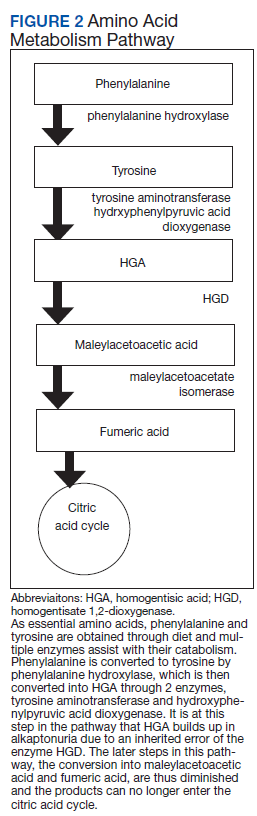
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
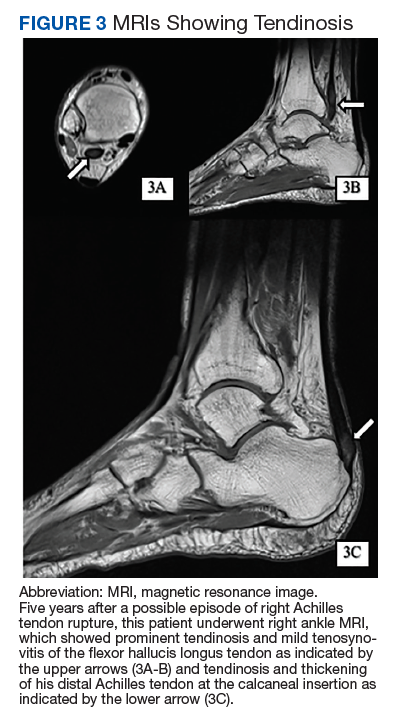
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
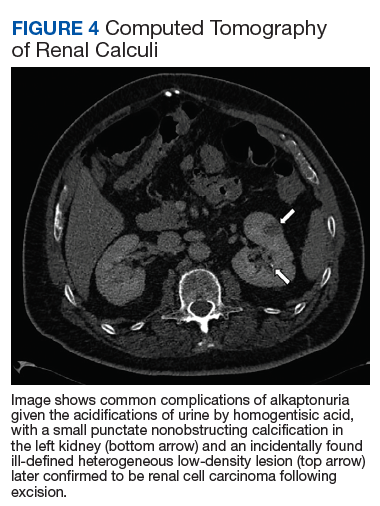
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.