User login
When ‘blue babies’ grow up: What you need to know about tetralogy of Fallot
Children born with tetralogy of Fallot and other congenital heart defects are living longer—long enough for new problems to arise, and, eventually, to present to your clinic. In primary care, the presentation of tetralogy of Fallot is still rare, but it is becoming more common.
Congenital heart disease was once solely a pediatric specialty, but adults who have been treated for these conditions now outnumber children with congenital heart conditions.1–4 More than 85% of infants with congenital heart disease are now expected to reach adulthood.5,6 For those with tetralogy of Fallot, the most common form of cyanotic congenital heart disease, the 40-year survival rate is now at least 90%.5
But these former “blue babies” eventually have serious problems. Most develop pulmonary valve insufficiency (regurgitation), which, over time, can result in right ventricular volume overload, enlargement, and dysfunction. 7–10 These problems lead to arrhythmias, the most significant cause of illness and death in these patients.11–13 Ventricular and atrial arrhythmias occur in up to 35% of patients with tetralogy of Fallot, and over a follow-up period of up to 30 years the incidence of sudden cardiac death is 6%.14
Furthermore, because many patients have no symptoms in early adulthood, they are often lost to follow-up, potentially missing the opportunity to have complications treated before they become irreversible. Recent data suggest that most patients who present with symptoms had stopped seeing a cardiologist about 10 years before.15
The challenge for primary care clinicians is to identify these patients in their practice, to recognize the early signs and symptoms of a worsening condition, and to refer and treat before cardiac damage becomes irreversible.
ONE IN 3,600 LIVE BIRTHS
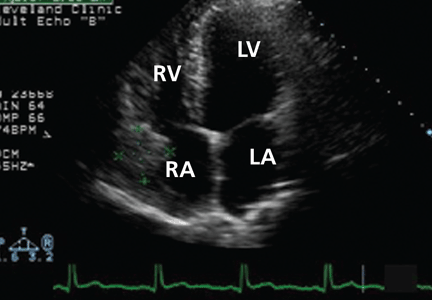
Tetralogy of Fallot occurs in approximately 1 in 3,600 live births or 3.5% of infants born with congenital heart disease.6 It is the most common type of cyanotic congenital heart disease, accounting for 10% of all cases.16 Patients in whom it has been repaired are the biggest group of adults with complex congenital heart disease. At Cleveland Clinic, it is the reason for 23% of new referrals to our adult congenital cardiology clinic, second only to atrial septal defects (33%).
FOUR DISTINCT FEATURES
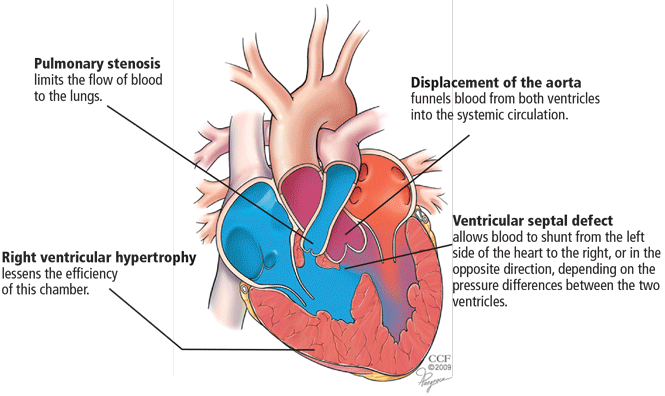
- Pulmonary stenosis (subvalvar, valvar, or both subvalvar and valvar)
- Ventricular septal defect
- Hypertrophy of the right ventricle
- Rightward deviation of the aortic valve, so that it overrides the ventricular septum; this can range from minimal overriding of the aorta and trivial pulmonary stenosis to up to 90% override and frank pulmonary atresia.
The aorta, receiving blood from both ventricles, is usually dilated. It arises from a right-sided arch in about 25% of patients and may override the septum so much that more than 50% of the blood flow comes from the right ventricle.17 In such cases, whether the patient has true tetralogy of Fallot or a double-outlet right ventricle with pulmonic stenosis may be ambiguous. Though controversial, the latter condition is generally distinguished by a ventricular septal defect that is integral to the left ventricular outflow tract and by lack of fibrous continuity between the aortic and mitral valves.18
SURGERY HAS EVOLVED
Surgical repair has been performed since the 1950s, and the perioperative death rate has fallen to less than 1% at most experienced centers.19
In the past, surgeons often placed a shunt between a systemic artery and the pulmonary artery as a palliative measure to improve oxygenation in infants with tetralogy of Fallot, waiting until the child was older to remove the shunt and repair the defects definitively.
Now, however, they generally favor repairing the heart in the initial procedure. This involves patching the ventricular septal defect, widening the infundibulum, and repairing the pulmonary valve or patching the annulus. Transannular patching opens the entire right ventricular outflow tract, but it crosses the pulmonary valve, and this is what eventually results in severe pulmonary insufficiency and its complications.20 For this reason, surgeons at most institutions now favor valve-sparing procedures rather than transannular patching, whenever possible.
WHAT HAPPENS YEARS AFTER SURGICAL REPAIR?
Surgery used to be considered the definitive cure for tetralogy of Fallot. However, problems that arise years later include chronic pulmonary valve insufficiency, obstruction of the right ventricular outflow tract, depressed right ventricular function, residual ventricular septal defect leaks, and arrhythmias.17,21,22 For these reasons, many experts have abandoned the notion that surgical repair is definitive. 23,24
Pulmonary valve insufficiency leads to right ventricular systolic dysfunction
In the past, pulmonary insufficiency was considered relatively benign because most patients tolerate it well for a long time. As these patients age, however, it becomes the core of their problems.1 If severe, it may result in right ventricular volume overload and dilatation, fibrosis, arrhythmia, and myocardial damage, all of which are cumulatively detrimental.25 Right ventricular function and exercise capacity deteriorate, and the tendency toward ventricular arrhythmias develops.26
If the problem is chronic, right ventricular systolic function may remain normal for years, during which most patients remain relatively free of symptoms. In time, however, the compensatory mechanisms of the right ventricular myocardium fail, the right ventricular wall stress (afterload) increases, while the right ventricular ejection fraction decreases. Patients begin to experience symptoms, and if the volume load is not reduced, the dysfunction may become irreversible.27
PULMONARY INSUFFICIENCY PREDISPOSES TO ARRHYTHMIAS, SUDDEN CARDIAC DEATH
Pulmonary insufficiency predisposes to atrial and ventricular arrhythmias, presumably due to progressive enlargement and stretching of the right atrium and ventricle.
Clinically significant atrial arrhythmias, predominantly intra-atrial reentrant tachycardia but also atrial tachycardia and atrial fibrillation, occur in 12% to 35% of patients with repaired tetralogy of Fallot.11,28–31
Ventricular arrhythmias and sudden cardiac death also occur. In one study,1 100% of patients who died suddenly had moderate or severe pulmonary insufficiency, and 94% with ventricular tachycardia had significant pulmonary insufficiency. In contrast, only 49% of patients who were arrhythmia-free had significant pulmonary insufficiency. None of the patients with late sudden death or ventricular tachycardia had undergone late pulmonary valve replacement. This is further supported by a multicenter analysis of patients with repaired tetralogy, which demonstrated that moderate or severe pulmonary insufficiency was the main hemodynamic abnormality in patients with ventricular tachycardia and sudden death.11
In general, the risk of late sudden death is 25 to 100 times higher in patients who survive surgery for congenital heart disease than in age-matched controls, and the risk is even higher for those with cyanotic conditions such as tetralogy of Fallot. In fact, one-third to one-half of deaths in adults with tetralogy of Fallot are sudden.25,32
FINDINGS ON ASSESSMENT
Most patients with tetralogy of Fallot remain free of symptoms for many years. While individual responses to pulmonary insufficiency vary, symptoms generally get worse as the pulmonary insufficiency gets worse. Patients present with a spectrum of complaints, from palpitations to a general decline in function. Late symptoms include exertional dyspnea, palpitations, right heart failure, and syncope.17
Signs of right ventricular failure can include elevated jugular venous pressure, peripheral edema, hepatomegaly, ascites,33 and jugular venous distention with a large a wave.
Heart murmurs
Pulmonary insufficiency causes a low-pitched, brief diastolic murmur. Although often present, it may be short or difficult to hear, even if the regurgitation is severe, because this is “low-pressure” pulmonary insufficiency as opposed to the regurgitation that can occur in patients with pulmonary hypertension. Therefore, this murmur is often missed on physical examination.
There may be an ejection click due to a dilated aorta. An aortic insufficiency murmur may also be present.
A right ventricular outflow murmur is generally audible, along with a pansystolic murmur if a residual ventricular septal defect is also present.
A right-sided aortic arch, present in about 25% of patients with tetralogy of Fallot, may cause a lift below the right sternoclavicular junction.17
Electrocardiographic findings
Electrocardiography commonly shows right ventricular hypertrophy with a right bundle branch block. The longer the QRS duration, the greater the right ventricular volume and mass. Furthermore, a QRS duration greater than 180 ms is a significant marker of risk of ventricular arrhythmias and sudden death.22,34–37
Another feature strongly associated with ventricular arrhythmias and sudden death is the rate of change in the QRS duration. A relatively rapid increase (> 3.5 ms/year) is associated with a significantly higher risk.1 A rapid rate of change may be meaningful even if the QRS duration is not markedly prolonged.11
Reduced heart rate variability also appears to be a marker of risk of sudden cardiac death in these patients.38,39
Imaging studies
Chest radiography typically shows a prominent right ventricular shadow and cardiomegaly.17
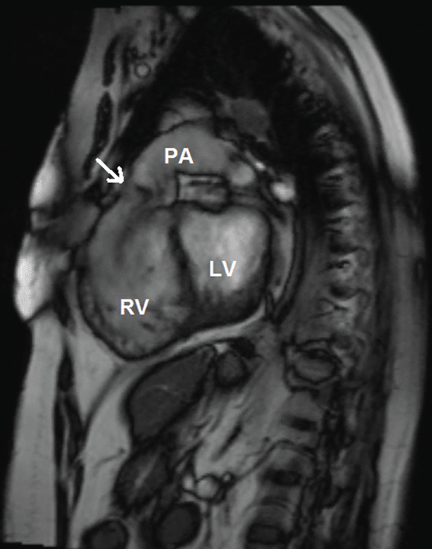
Many centers specializing in congenital heart disease therefore recommend baseline cardiac MRI, even for patients without symptoms.33
PULMONARY VALVE REPLACEMENT IS THE ONLY PROVEN TREATMENT
No study has yet shown that drug therapy alone slows the progression of complications.1 Pulmonary valve replacement is the only treatment proven to reduce right ventricular size and improve right ventricular function in the long term.
The risks of surgery, including the need for repeat operations, must be balanced against the risk of irreversible right ventricular dysfunction and its associated complications. The operative death rate is low, as is the long-term risk of death afterward. Therrien et al12 reported that, in a series of 70 patients who underwent pulmonary valve replacement, the probability of survival was 92% at 5 years and 86% at 10 years.
Surgery appears to reverse or at least arrest the progression of many of the complications associated with pulmonary insufficiency, including tricuspid regurgitation and diastolic dysfunction.17 Its utility in ameliorating ventricular tachycardia, however, remains controversial. One series showed a lower prevalence of tachycardia after pulmonary valve replacement (9% after surgery vs 22% before), but later studies have had more equivocal results.17
When should surgery be done?
There is little controversy about the eventual need for pulmonary valve replacement in most patients. What is controversial is the timing.12,44–47
This issue has been hotly debated. Some believe that pulmonary valve replacement should be done only if evidence of right ventricular dysfunction has developed.17 Others suggest that it be considered earlier and that the onset of symptoms may be a late and suboptimal indication for it.6,8,48,49 Many experts now recommend surgery early, before symptoms of heart failure develop.17 Though surgery has traditionally been recommended if the QRS duration is longer than 180 ms, some believe it should be done before this occurs.11
Arguments for early surgery. In one study, in no patient who had a right ventricular end-diastolic volume greater than 170 mL/m2 (normal ≤ 108) or a right ventricular end-systolic volume greater than 85 mL/m2 (normal ≤ 47) did these numbers return to normal after pulmonary valve replacement.45,50 This suggests a point of irreversible dilatation and a volume threshold beyond which right ventricular function is unlikely to completely improve. Normalization of right ventricular volumes was shown to occur when pulmonary valve replacement was performed before the right ventricular end-diastolic volume reached 160 mL/m2 or the right ventricular end-systolic volume reached 82 mL/m2.47,51
Delaying surgery until symptoms occur may be unfavorable because the long-term outcomes of increased right ventricular volumes and decreased right ventricular ejection fractions after surgery are not known.
Arguments for watchful waiting. There does not seem to be a threshold above which right ventricular volumes do not decrease after surgery—although they may not decrease to the normal range. Pulmonary valve replacement substantially reduced right ventricular dilatation even in patients with very high right ventricular volumes and right ventricular dysfunction, and resulted in an overall improvement in function (measured by New York Heart Association class).47
Late pulmonary valve replacement rapidly improves right ventricular volumes and improves the effective ejection fraction, although its impact on absolute right ventricular function is not as pronounced. The QRS duration shortened after surgery in those in whom it was 180 ms or longer before surgery, although this appeared to be a transient change.52 The prevalence of ventricular tachycardia declined from 22% to 9% and that of atrial fibrillation or flutter declined from 17% to 12%.17,48
A recent study with long-term follow-up has raised questions about the necessity of aggressive early intervention in tetralogy of Fallot. Sixty-seven patients were followed for as long as 27 years after surgery. Forty-five had severe pulmonary insufficiency and severe right ventricular dilatation, and of those, 28 remained free of symptoms and did not undergo pulmonary valve replacement. The authors found that refraining from pulmonary valve replacement in asymptomatic patients with severe pulmonary insufficiency led to no measurable deterioration in 25 of 28 patients.53
The available data do not support pulmonary valve replacement in young patients with mild or moderate right ventricular dilatation, normal right ventricular systolic function, and no additional risk factors.27
Mechanical vs bioprosthetic replacement valves
Once the decision is made to proceed to surgery, the next step is choosing the type of prosthetic valve.
Mechanical valves pose a risk of thrombosis, requiring life-long anticoagulation. To give warfarin (Coumadin) to younger, active people exposes them to the risk of potentially catastrophic bleeding if trauma were to occur. Women who become pregnant are generally at an increased risk of thrombotic complications due to the hypercoagulable state of pregnancy, but the risk of fetal defects is considerable if they receive warfarin.54–56
Bioprosthetic valves generally come in two varieties: preserved and treated human tissue (homografts) and animal tissue (bovine pericardial or porcine, depending on the size required). These can be implanted as isolated valves or as part of a conduit (valve and surrounding tissue).
Bioprosthetic valves eliminate the need for anticoagulation. However, they are not very durable, especially in younger patients, which is worrisome. An estimated 45% of bioprosthetic valves fail by 10 years,57 thus nearly guaranteeing that an otherwise healthy 40-year-old, for example, will need to undergo at least one repeat surgery, and very likely more.
NOVEL THERAPIES
Percutaneous valve replacement
The future of pulmonary valve replacement may lie in percutaneous procedures.
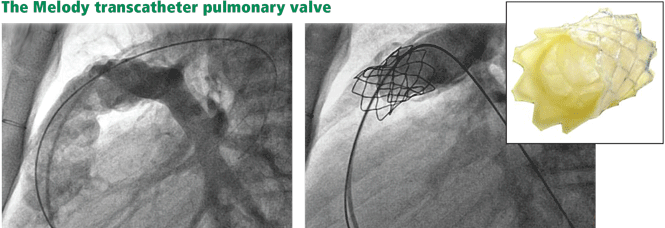
The Melody valve is now approved through a humanitarian device exemption (ie, based on demonstrated safety without proven efficacy) for patients who have a prior pulmonary conduit now complicated by either stenosis or regurgitation.
If percutaneous pulmonary valve replacement proves to have reasonable long-term durability, it has the potential to dramatically shift the balance toward earlier intervention.
Pulmonary vasodilator drugs
Our group is examining whether pharmacologic therapy can alter the clinical outcome in patients with pulmonary insufficiency (due to either tetralogy of Fallot or valvotomy done to treat remote pulmonary stenosis). Specifically, we are using MRI to examine the effects of inhaled nitric oxide, a selective pulmonary vasodilator. Preliminary results suggest that such a strategy may work, and we are designing a trial to examine the longer-term benefit of using an oral drug with similar properties.
- Gregg D, Foster E. Pulmonary insufficiency is the nexus of late complications in tetralogy of Fallot. Curr Cardiol Rep 2007; 9:315–322.
- Khairy P, Hosn JA, Broberg C, et al; Alliance for Adult Research in Congenital Cardiology (AARCC). Multicenter research in adult congenital heart disease. Int J Cardiol 2008; 129:155–159.
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation 2008; 118:e714–e833.
- Perloff JK, Warnes CA. Challenges posed by adults with repaired congenital heart disease. Circulation 2001; 103:2637–2643.
- Hickey EJ, Veldtman G, Bradley TJ, et al. Late risk of outcomes for adults with repaired tetralogy of Fallot from an inception cohort spanning four decades. Eur J Cardiothorac Surg 2009; 35:156–164.
- Apitz C, Webb GD, Redington AN. Tetralogy of Fallot. Lancet 2009; 374:1462–1471.
- Gatzoulis MA, Clark AL, Cullen S, Newman CG, Redington AN. Right ventricular diastolic function 15 to 35 years after repair of tetralogy of Fallot. Restrictive physiology predicts superior exercise performance. Circulation 1995; 91:1775–1781.
- van Straten A, Vliegen HW, Hazekamp MG, et al. Right ventricular function after pulmonary valve replacement in patients with tetralogy of Fallot. Radiology 2004; 233:824–829.
- Bouzas B, Kilner PJ, Gatzoulis MA. Pulmonary regurgitation: not a benign lesion. Eur Heart J 2005; 26:433–439.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet 2000; 356:975–981.
- Therrien J, Siu SC, Harris L, et al. Impact of pulmonary valve replacement on arrhythmia propensity late after repair of tetralogy of Fallot. Circulation 2001; 103:2489–2494.
- Deanfield JE, McKenna WJ, Presbitero P, England D, Graham GR, Hallidie-Smith K. Ventricular arrhythmia in unrepaired and repaired tetralogy of Fallot. Relation to age, timing of repair, and haemodynamic status. Br Heart J 1984; 52:77–81.
- Murphy JG, Gersh BJ, Mair DD, et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 1993; 329:593–599.
- Mackie AS, Ionescu-Ittu R, Therrien J, Pilote L, Abrahamowicz M, Marelli AJ. Children and adults with congenital heart disease lost to follow-up: who and when? Circulation 2009; 120:302–309.
- Pinsky WW, Arciniegas E. Tetralogy of Fallot. Pediatr Clin North Am 1990; 37:179–192.
- Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation 2007; 115:1933–1947.
- Walters HL, Mavroudis C, Tchervenkov CI, Jacobs JP, Lacour-Gayet F, Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: double outlet right ventricle. Ann Thorac Surg 2000; 69(suppl 4):S249–S263.
- Van Arsdell GS, Maharaj GS, Tom J, et al. What is the optimal age for repair of tetralogy of Fallot? Circulation 2000; 102(suppl 3):III123–III129.
- Cheung MM, Konstantinov IE, Redington AN. Late complications of repair of tetralogy of Fallot and indications for pulmonary valve replacement. Semin Thorac Cardiovasc Surg 2005; 17:155–159.
- Babu-Narayan SV, Gatzoulis MA. Management of adults with operated tetralogy of Fallot. Curr Treat Options Cardiovasc Med 2003; 5:389–398.
- Gatzoulis MA, Till JA, Somerville J, Redington AN. Mechanoelectrical interaction in tetralogy of Fallot. QRS prolongation relates to right ventricular size and predicts malignant ventricular arrhythmias and sudden death. Circulation 1995; 92:231–237.
- van Doorn C. The unnatural history of tetralogy of Fallot: surgical repair is not as definitive as previously thought. Heart 2002; 88:447–448.
- Norton KI, Tong C, Glass RB, Nielsen JC. Cardiac MR imaging assessment following tetralogy of Fallot repair. Radiographics 2006; 26:197–211.
- Bhat AH, Sahn DJ. Congenital heart disease never goes away, even when it has been ‘treated’: the adult with congenital heart disease. Curr Opin Pediatr 2004; 16:500–507.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:11–22.
- Khairy P, Dore A, Talajic M, et al. Arrhythmias in adult congenital heart disease. Expert Rev Cardiovasc Ther 2006; 4:83–95.
- Roos-Hesselink J, Perlroth MG, McGhie J, Spitaels S. Atrial arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and echocardiographic findings. Circulation 1995; 91:2214–2219.
- Harrison DA, Siu SC, Hussain F, MacLoghlin CJ, Webb GD, Harris L. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol 2001; 87:584–588.
- Collins KK, Dubin AM. Detecting and diagnosing arrhythmias in adults with congenital heart disease. Curr Cardiol Rep 2003; 5:331–335.
- Silka MJ, Hardy BG, Menashe VD, Morris CD. A population-based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol 1998; 32:245–251.
- Ammash NM, Dearani JA, Burkhart HM, Connolly HM. Pulmonary regurgitation after tetralogy of Fallot repair: clinical features, sequelae, and timing of pulmonary valve replacement. Congenit Heart Dis 2007; 2:386–403.
- Berul CI, Hill SL, Geggel RL, et al. Electrocardiographic markers of late sudden death risk in postoperative tetralogy of Fallot children. J Cardiovasc Electrophysiol 1997; 8:1349–1356.
- Gatzoulis MA, Till JA, Redington AN. Depolarization-repolarization inhomogeneity after repair of tetralogy of Fallot. The substrate for malignant ventricular tachycardia? Circulation 1997; 95:401–404.
- Balaji S, Lau YR, Case CL, Gillette PC. QRS prolongation is associated with inducible ventricular tachycardia after repair of tetralogy of Fallot. Am J Cardiol 1997; 80:160–163.
- Abd El Rahman MY, Abdul-Khaliq H, Vogel M, Alexi-Meskishvili V, Gutberlet M, Lange PE. Relation between right ventricular enlargement, QRS duration, and right ventricular function in patients with tetralogy of Fallot and pulmonary regurgitation after surgical repair. Heart 2000; 84:416–420.
- McLeod KA, Hillis WS, Houston AB, et al. Reduced heart rate variability following repair of tetralogy of Fallot. Heart 1999; 81:656–660.
- Davos CH, Moutafi AC, Alexandridi A, et al. Heart rate turbulence in adults with repaired tetralogy of Fallot. Int J Cardiol 2009; 135:308–314.
- de Roos A, Roest AA. Evaluation of congenital heart disease by magnetic resonance imaging. Eur Radiol 2000; 10:2–6.
- Niezen RA, Helbing WA, van der Wall EE, van der Geest RJ, Rebergen SA, de Roos A. Biventricular systolic function and mass studied with MR imaging in children with pulmonary regurgitation after repair for tetralogy of Fallot. Radiology 1996; 201:135–140.
- Helbing WA, de Roos A. Optimal imaging in assessment of right ventricular function in tetralogy of Fallot with pulmonary regurgitation. Am J Cardiol 1998; 82:1561–1562.
- van der Geest RJ, de Roos A, van der Wall EE, Reiber JH. Quantitative analysis of cardiovascular MR images. Int J Card Imaging 1997; 13:247–258.
- Therrien J, Siu SC, McLaughlin PR, Liu PP, Williams WG, Webb GD. Pulmonary valve replacement in adults late after repair of tetralogy of Fallot: are we operating too late? J Am Coll Cardiol 2000; 36:1670–1675.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Davlouros PA, Karatza AA, Gatzoulis MA, Shore DF. Timing and type of surgery for severe pulmonary regurgitation after repair of tetralogy of Fallot. Int J Cardiol 2004; 97(suppl 1):91–101.
- Oosterhof T, van Straten A, Vliegen HW, et al. Preoperative thresholds for pulmonary valve replacement in patients with corrected tetralogy of Fallot using cardiovascular magnetic resonance. Circulation 2007; 116:545–551.
- Buechel ER, Dave HH, Kellenberger CJ, et al. Remodelling of the right ventricle after early pulmonary valve replacement in children with repaired tetralogy of Fallot: assessment by cardiovascular magnetic resonance. Eur Heart J 2005; 26:2721–2727.
- Henkens IR, van Straten A, Schalij MJ, et al. Predicting outcome of pulmonary valve replacement in adult tetralogy of Fallot patients. Ann Thorac Surg 2007; 83:907–911.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Redington AN. Physiopathology of right ventricular failure. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:3–10.
- Oosterhof T, Vliegen HW, Meijboom FJ, Zwinderman AH, Bouma B, Mulder BJ. Long-term effect of pulmonary valve replacement on QRS duration in patients with corrected tetralogy of Fallot. Heart 2007; 93:506–509.
- Meijboom FJ, Roos-Hesselink JW, McGhie JS, et al. Consequences of a selective approach toward pulmonary valve replacement in adult patients with tetralogy of Fallot and pulmonary regurgitation. J Thorac Cardiovasc Surg 2008; 135:50–55.
- Danik S, Fuster V. The obstetrical patient with a prosthetic heart valve. Obstet Gynecol Clin North Am 2006; 33:481–491.
- Danik S, Fuster V. Anticoagulation in pregnant women with prosthetic heart valves. Mt Sinai J Med 2004; 71:322–329.
- Manso B, Gran F, Pijuán A, et al. Pregnancy and congenital heart disease (article in Spanish). Rev Esp Cardiol 2008; 61:236–243.
- Gallegos RP. Selection of prosthetic heart valves. Curr Treat Options Cardiovasc Med 2006; 8:443–452.
- Bonhoeffer P, Boudjemline Y, Qureshi SA, et al. Percutaneous insertion of the pulmonary valve. J Am Coll Cardiol 2002; 39:1664–1669.
- Khambadkone S, Coats L, Taylor A, et al. Percutaneous pulmonary valve implantation in humans: results in 59 consecutive patients. Circulation 2005; 112:1189–1197.
- Lurz P, Coats L, Khambadkone S, et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation 2008; 117:1964–1972.
Children born with tetralogy of Fallot and other congenital heart defects are living longer—long enough for new problems to arise, and, eventually, to present to your clinic. In primary care, the presentation of tetralogy of Fallot is still rare, but it is becoming more common.
Congenital heart disease was once solely a pediatric specialty, but adults who have been treated for these conditions now outnumber children with congenital heart conditions.1–4 More than 85% of infants with congenital heart disease are now expected to reach adulthood.5,6 For those with tetralogy of Fallot, the most common form of cyanotic congenital heart disease, the 40-year survival rate is now at least 90%.5
But these former “blue babies” eventually have serious problems. Most develop pulmonary valve insufficiency (regurgitation), which, over time, can result in right ventricular volume overload, enlargement, and dysfunction. 7–10 These problems lead to arrhythmias, the most significant cause of illness and death in these patients.11–13 Ventricular and atrial arrhythmias occur in up to 35% of patients with tetralogy of Fallot, and over a follow-up period of up to 30 years the incidence of sudden cardiac death is 6%.14
Furthermore, because many patients have no symptoms in early adulthood, they are often lost to follow-up, potentially missing the opportunity to have complications treated before they become irreversible. Recent data suggest that most patients who present with symptoms had stopped seeing a cardiologist about 10 years before.15
The challenge for primary care clinicians is to identify these patients in their practice, to recognize the early signs and symptoms of a worsening condition, and to refer and treat before cardiac damage becomes irreversible.
ONE IN 3,600 LIVE BIRTHS
Tetralogy of Fallot occurs in approximately 1 in 3,600 live births or 3.5% of infants born with congenital heart disease.6 It is the most common type of cyanotic congenital heart disease, accounting for 10% of all cases.16 Patients in whom it has been repaired are the biggest group of adults with complex congenital heart disease. At Cleveland Clinic, it is the reason for 23% of new referrals to our adult congenital cardiology clinic, second only to atrial septal defects (33%).
FOUR DISTINCT FEATURES
- Pulmonary stenosis (subvalvar, valvar, or both subvalvar and valvar)
- Ventricular septal defect
- Hypertrophy of the right ventricle
- Rightward deviation of the aortic valve, so that it overrides the ventricular septum; this can range from minimal overriding of the aorta and trivial pulmonary stenosis to up to 90% override and frank pulmonary atresia.
The aorta, receiving blood from both ventricles, is usually dilated. It arises from a right-sided arch in about 25% of patients and may override the septum so much that more than 50% of the blood flow comes from the right ventricle.17 In such cases, whether the patient has true tetralogy of Fallot or a double-outlet right ventricle with pulmonic stenosis may be ambiguous. Though controversial, the latter condition is generally distinguished by a ventricular septal defect that is integral to the left ventricular outflow tract and by lack of fibrous continuity between the aortic and mitral valves.18
SURGERY HAS EVOLVED
Surgical repair has been performed since the 1950s, and the perioperative death rate has fallen to less than 1% at most experienced centers.19
In the past, surgeons often placed a shunt between a systemic artery and the pulmonary artery as a palliative measure to improve oxygenation in infants with tetralogy of Fallot, waiting until the child was older to remove the shunt and repair the defects definitively.
Now, however, they generally favor repairing the heart in the initial procedure. This involves patching the ventricular septal defect, widening the infundibulum, and repairing the pulmonary valve or patching the annulus. Transannular patching opens the entire right ventricular outflow tract, but it crosses the pulmonary valve, and this is what eventually results in severe pulmonary insufficiency and its complications.20 For this reason, surgeons at most institutions now favor valve-sparing procedures rather than transannular patching, whenever possible.
WHAT HAPPENS YEARS AFTER SURGICAL REPAIR?
Surgery used to be considered the definitive cure for tetralogy of Fallot. However, problems that arise years later include chronic pulmonary valve insufficiency, obstruction of the right ventricular outflow tract, depressed right ventricular function, residual ventricular septal defect leaks, and arrhythmias.17,21,22 For these reasons, many experts have abandoned the notion that surgical repair is definitive. 23,24
Pulmonary valve insufficiency leads to right ventricular systolic dysfunction
In the past, pulmonary insufficiency was considered relatively benign because most patients tolerate it well for a long time. As these patients age, however, it becomes the core of their problems.1 If severe, it may result in right ventricular volume overload and dilatation, fibrosis, arrhythmia, and myocardial damage, all of which are cumulatively detrimental.25 Right ventricular function and exercise capacity deteriorate, and the tendency toward ventricular arrhythmias develops.26
If the problem is chronic, right ventricular systolic function may remain normal for years, during which most patients remain relatively free of symptoms. In time, however, the compensatory mechanisms of the right ventricular myocardium fail, the right ventricular wall stress (afterload) increases, while the right ventricular ejection fraction decreases. Patients begin to experience symptoms, and if the volume load is not reduced, the dysfunction may become irreversible.27
PULMONARY INSUFFICIENCY PREDISPOSES TO ARRHYTHMIAS, SUDDEN CARDIAC DEATH
Pulmonary insufficiency predisposes to atrial and ventricular arrhythmias, presumably due to progressive enlargement and stretching of the right atrium and ventricle.
Clinically significant atrial arrhythmias, predominantly intra-atrial reentrant tachycardia but also atrial tachycardia and atrial fibrillation, occur in 12% to 35% of patients with repaired tetralogy of Fallot.11,28–31
Ventricular arrhythmias and sudden cardiac death also occur. In one study,1 100% of patients who died suddenly had moderate or severe pulmonary insufficiency, and 94% with ventricular tachycardia had significant pulmonary insufficiency. In contrast, only 49% of patients who were arrhythmia-free had significant pulmonary insufficiency. None of the patients with late sudden death or ventricular tachycardia had undergone late pulmonary valve replacement. This is further supported by a multicenter analysis of patients with repaired tetralogy, which demonstrated that moderate or severe pulmonary insufficiency was the main hemodynamic abnormality in patients with ventricular tachycardia and sudden death.11
In general, the risk of late sudden death is 25 to 100 times higher in patients who survive surgery for congenital heart disease than in age-matched controls, and the risk is even higher for those with cyanotic conditions such as tetralogy of Fallot. In fact, one-third to one-half of deaths in adults with tetralogy of Fallot are sudden.25,32
FINDINGS ON ASSESSMENT
Most patients with tetralogy of Fallot remain free of symptoms for many years. While individual responses to pulmonary insufficiency vary, symptoms generally get worse as the pulmonary insufficiency gets worse. Patients present with a spectrum of complaints, from palpitations to a general decline in function. Late symptoms include exertional dyspnea, palpitations, right heart failure, and syncope.17
Signs of right ventricular failure can include elevated jugular venous pressure, peripheral edema, hepatomegaly, ascites,33 and jugular venous distention with a large a wave.
Heart murmurs
Pulmonary insufficiency causes a low-pitched, brief diastolic murmur. Although often present, it may be short or difficult to hear, even if the regurgitation is severe, because this is “low-pressure” pulmonary insufficiency as opposed to the regurgitation that can occur in patients with pulmonary hypertension. Therefore, this murmur is often missed on physical examination.
There may be an ejection click due to a dilated aorta. An aortic insufficiency murmur may also be present.
A right ventricular outflow murmur is generally audible, along with a pansystolic murmur if a residual ventricular septal defect is also present.
A right-sided aortic arch, present in about 25% of patients with tetralogy of Fallot, may cause a lift below the right sternoclavicular junction.17
Electrocardiographic findings
Electrocardiography commonly shows right ventricular hypertrophy with a right bundle branch block. The longer the QRS duration, the greater the right ventricular volume and mass. Furthermore, a QRS duration greater than 180 ms is a significant marker of risk of ventricular arrhythmias and sudden death.22,34–37
Another feature strongly associated with ventricular arrhythmias and sudden death is the rate of change in the QRS duration. A relatively rapid increase (> 3.5 ms/year) is associated with a significantly higher risk.1 A rapid rate of change may be meaningful even if the QRS duration is not markedly prolonged.11
Reduced heart rate variability also appears to be a marker of risk of sudden cardiac death in these patients.38,39
Imaging studies
Chest radiography typically shows a prominent right ventricular shadow and cardiomegaly.17
Many centers specializing in congenital heart disease therefore recommend baseline cardiac MRI, even for patients without symptoms.33
PULMONARY VALVE REPLACEMENT IS THE ONLY PROVEN TREATMENT
No study has yet shown that drug therapy alone slows the progression of complications.1 Pulmonary valve replacement is the only treatment proven to reduce right ventricular size and improve right ventricular function in the long term.
The risks of surgery, including the need for repeat operations, must be balanced against the risk of irreversible right ventricular dysfunction and its associated complications. The operative death rate is low, as is the long-term risk of death afterward. Therrien et al12 reported that, in a series of 70 patients who underwent pulmonary valve replacement, the probability of survival was 92% at 5 years and 86% at 10 years.
Surgery appears to reverse or at least arrest the progression of many of the complications associated with pulmonary insufficiency, including tricuspid regurgitation and diastolic dysfunction.17 Its utility in ameliorating ventricular tachycardia, however, remains controversial. One series showed a lower prevalence of tachycardia after pulmonary valve replacement (9% after surgery vs 22% before), but later studies have had more equivocal results.17
When should surgery be done?
There is little controversy about the eventual need for pulmonary valve replacement in most patients. What is controversial is the timing.12,44–47
This issue has been hotly debated. Some believe that pulmonary valve replacement should be done only if evidence of right ventricular dysfunction has developed.17 Others suggest that it be considered earlier and that the onset of symptoms may be a late and suboptimal indication for it.6,8,48,49 Many experts now recommend surgery early, before symptoms of heart failure develop.17 Though surgery has traditionally been recommended if the QRS duration is longer than 180 ms, some believe it should be done before this occurs.11
Arguments for early surgery. In one study, in no patient who had a right ventricular end-diastolic volume greater than 170 mL/m2 (normal ≤ 108) or a right ventricular end-systolic volume greater than 85 mL/m2 (normal ≤ 47) did these numbers return to normal after pulmonary valve replacement.45,50 This suggests a point of irreversible dilatation and a volume threshold beyond which right ventricular function is unlikely to completely improve. Normalization of right ventricular volumes was shown to occur when pulmonary valve replacement was performed before the right ventricular end-diastolic volume reached 160 mL/m2 or the right ventricular end-systolic volume reached 82 mL/m2.47,51
Delaying surgery until symptoms occur may be unfavorable because the long-term outcomes of increased right ventricular volumes and decreased right ventricular ejection fractions after surgery are not known.
Arguments for watchful waiting. There does not seem to be a threshold above which right ventricular volumes do not decrease after surgery—although they may not decrease to the normal range. Pulmonary valve replacement substantially reduced right ventricular dilatation even in patients with very high right ventricular volumes and right ventricular dysfunction, and resulted in an overall improvement in function (measured by New York Heart Association class).47
Late pulmonary valve replacement rapidly improves right ventricular volumes and improves the effective ejection fraction, although its impact on absolute right ventricular function is not as pronounced. The QRS duration shortened after surgery in those in whom it was 180 ms or longer before surgery, although this appeared to be a transient change.52 The prevalence of ventricular tachycardia declined from 22% to 9% and that of atrial fibrillation or flutter declined from 17% to 12%.17,48
A recent study with long-term follow-up has raised questions about the necessity of aggressive early intervention in tetralogy of Fallot. Sixty-seven patients were followed for as long as 27 years after surgery. Forty-five had severe pulmonary insufficiency and severe right ventricular dilatation, and of those, 28 remained free of symptoms and did not undergo pulmonary valve replacement. The authors found that refraining from pulmonary valve replacement in asymptomatic patients with severe pulmonary insufficiency led to no measurable deterioration in 25 of 28 patients.53
The available data do not support pulmonary valve replacement in young patients with mild or moderate right ventricular dilatation, normal right ventricular systolic function, and no additional risk factors.27
Mechanical vs bioprosthetic replacement valves
Once the decision is made to proceed to surgery, the next step is choosing the type of prosthetic valve.
Mechanical valves pose a risk of thrombosis, requiring life-long anticoagulation. To give warfarin (Coumadin) to younger, active people exposes them to the risk of potentially catastrophic bleeding if trauma were to occur. Women who become pregnant are generally at an increased risk of thrombotic complications due to the hypercoagulable state of pregnancy, but the risk of fetal defects is considerable if they receive warfarin.54–56
Bioprosthetic valves generally come in two varieties: preserved and treated human tissue (homografts) and animal tissue (bovine pericardial or porcine, depending on the size required). These can be implanted as isolated valves or as part of a conduit (valve and surrounding tissue).
Bioprosthetic valves eliminate the need for anticoagulation. However, they are not very durable, especially in younger patients, which is worrisome. An estimated 45% of bioprosthetic valves fail by 10 years,57 thus nearly guaranteeing that an otherwise healthy 40-year-old, for example, will need to undergo at least one repeat surgery, and very likely more.
NOVEL THERAPIES
Percutaneous valve replacement
The future of pulmonary valve replacement may lie in percutaneous procedures.
The Melody valve is now approved through a humanitarian device exemption (ie, based on demonstrated safety without proven efficacy) for patients who have a prior pulmonary conduit now complicated by either stenosis or regurgitation.
If percutaneous pulmonary valve replacement proves to have reasonable long-term durability, it has the potential to dramatically shift the balance toward earlier intervention.
Pulmonary vasodilator drugs
Our group is examining whether pharmacologic therapy can alter the clinical outcome in patients with pulmonary insufficiency (due to either tetralogy of Fallot or valvotomy done to treat remote pulmonary stenosis). Specifically, we are using MRI to examine the effects of inhaled nitric oxide, a selective pulmonary vasodilator. Preliminary results suggest that such a strategy may work, and we are designing a trial to examine the longer-term benefit of using an oral drug with similar properties.
Children born with tetralogy of Fallot and other congenital heart defects are living longer—long enough for new problems to arise, and, eventually, to present to your clinic. In primary care, the presentation of tetralogy of Fallot is still rare, but it is becoming more common.
Congenital heart disease was once solely a pediatric specialty, but adults who have been treated for these conditions now outnumber children with congenital heart conditions.1–4 More than 85% of infants with congenital heart disease are now expected to reach adulthood.5,6 For those with tetralogy of Fallot, the most common form of cyanotic congenital heart disease, the 40-year survival rate is now at least 90%.5
But these former “blue babies” eventually have serious problems. Most develop pulmonary valve insufficiency (regurgitation), which, over time, can result in right ventricular volume overload, enlargement, and dysfunction. 7–10 These problems lead to arrhythmias, the most significant cause of illness and death in these patients.11–13 Ventricular and atrial arrhythmias occur in up to 35% of patients with tetralogy of Fallot, and over a follow-up period of up to 30 years the incidence of sudden cardiac death is 6%.14
Furthermore, because many patients have no symptoms in early adulthood, they are often lost to follow-up, potentially missing the opportunity to have complications treated before they become irreversible. Recent data suggest that most patients who present with symptoms had stopped seeing a cardiologist about 10 years before.15
The challenge for primary care clinicians is to identify these patients in their practice, to recognize the early signs and symptoms of a worsening condition, and to refer and treat before cardiac damage becomes irreversible.
ONE IN 3,600 LIVE BIRTHS
Tetralogy of Fallot occurs in approximately 1 in 3,600 live births or 3.5% of infants born with congenital heart disease.6 It is the most common type of cyanotic congenital heart disease, accounting for 10% of all cases.16 Patients in whom it has been repaired are the biggest group of adults with complex congenital heart disease. At Cleveland Clinic, it is the reason for 23% of new referrals to our adult congenital cardiology clinic, second only to atrial septal defects (33%).
FOUR DISTINCT FEATURES
- Pulmonary stenosis (subvalvar, valvar, or both subvalvar and valvar)
- Ventricular septal defect
- Hypertrophy of the right ventricle
- Rightward deviation of the aortic valve, so that it overrides the ventricular septum; this can range from minimal overriding of the aorta and trivial pulmonary stenosis to up to 90% override and frank pulmonary atresia.
The aorta, receiving blood from both ventricles, is usually dilated. It arises from a right-sided arch in about 25% of patients and may override the septum so much that more than 50% of the blood flow comes from the right ventricle.17 In such cases, whether the patient has true tetralogy of Fallot or a double-outlet right ventricle with pulmonic stenosis may be ambiguous. Though controversial, the latter condition is generally distinguished by a ventricular septal defect that is integral to the left ventricular outflow tract and by lack of fibrous continuity between the aortic and mitral valves.18
SURGERY HAS EVOLVED
Surgical repair has been performed since the 1950s, and the perioperative death rate has fallen to less than 1% at most experienced centers.19
In the past, surgeons often placed a shunt between a systemic artery and the pulmonary artery as a palliative measure to improve oxygenation in infants with tetralogy of Fallot, waiting until the child was older to remove the shunt and repair the defects definitively.
Now, however, they generally favor repairing the heart in the initial procedure. This involves patching the ventricular septal defect, widening the infundibulum, and repairing the pulmonary valve or patching the annulus. Transannular patching opens the entire right ventricular outflow tract, but it crosses the pulmonary valve, and this is what eventually results in severe pulmonary insufficiency and its complications.20 For this reason, surgeons at most institutions now favor valve-sparing procedures rather than transannular patching, whenever possible.
WHAT HAPPENS YEARS AFTER SURGICAL REPAIR?
Surgery used to be considered the definitive cure for tetralogy of Fallot. However, problems that arise years later include chronic pulmonary valve insufficiency, obstruction of the right ventricular outflow tract, depressed right ventricular function, residual ventricular septal defect leaks, and arrhythmias.17,21,22 For these reasons, many experts have abandoned the notion that surgical repair is definitive. 23,24
Pulmonary valve insufficiency leads to right ventricular systolic dysfunction
In the past, pulmonary insufficiency was considered relatively benign because most patients tolerate it well for a long time. As these patients age, however, it becomes the core of their problems.1 If severe, it may result in right ventricular volume overload and dilatation, fibrosis, arrhythmia, and myocardial damage, all of which are cumulatively detrimental.25 Right ventricular function and exercise capacity deteriorate, and the tendency toward ventricular arrhythmias develops.26
If the problem is chronic, right ventricular systolic function may remain normal for years, during which most patients remain relatively free of symptoms. In time, however, the compensatory mechanisms of the right ventricular myocardium fail, the right ventricular wall stress (afterload) increases, while the right ventricular ejection fraction decreases. Patients begin to experience symptoms, and if the volume load is not reduced, the dysfunction may become irreversible.27
PULMONARY INSUFFICIENCY PREDISPOSES TO ARRHYTHMIAS, SUDDEN CARDIAC DEATH
Pulmonary insufficiency predisposes to atrial and ventricular arrhythmias, presumably due to progressive enlargement and stretching of the right atrium and ventricle.
Clinically significant atrial arrhythmias, predominantly intra-atrial reentrant tachycardia but also atrial tachycardia and atrial fibrillation, occur in 12% to 35% of patients with repaired tetralogy of Fallot.11,28–31
Ventricular arrhythmias and sudden cardiac death also occur. In one study,1 100% of patients who died suddenly had moderate or severe pulmonary insufficiency, and 94% with ventricular tachycardia had significant pulmonary insufficiency. In contrast, only 49% of patients who were arrhythmia-free had significant pulmonary insufficiency. None of the patients with late sudden death or ventricular tachycardia had undergone late pulmonary valve replacement. This is further supported by a multicenter analysis of patients with repaired tetralogy, which demonstrated that moderate or severe pulmonary insufficiency was the main hemodynamic abnormality in patients with ventricular tachycardia and sudden death.11
In general, the risk of late sudden death is 25 to 100 times higher in patients who survive surgery for congenital heart disease than in age-matched controls, and the risk is even higher for those with cyanotic conditions such as tetralogy of Fallot. In fact, one-third to one-half of deaths in adults with tetralogy of Fallot are sudden.25,32
FINDINGS ON ASSESSMENT
Most patients with tetralogy of Fallot remain free of symptoms for many years. While individual responses to pulmonary insufficiency vary, symptoms generally get worse as the pulmonary insufficiency gets worse. Patients present with a spectrum of complaints, from palpitations to a general decline in function. Late symptoms include exertional dyspnea, palpitations, right heart failure, and syncope.17
Signs of right ventricular failure can include elevated jugular venous pressure, peripheral edema, hepatomegaly, ascites,33 and jugular venous distention with a large a wave.
Heart murmurs
Pulmonary insufficiency causes a low-pitched, brief diastolic murmur. Although often present, it may be short or difficult to hear, even if the regurgitation is severe, because this is “low-pressure” pulmonary insufficiency as opposed to the regurgitation that can occur in patients with pulmonary hypertension. Therefore, this murmur is often missed on physical examination.
There may be an ejection click due to a dilated aorta. An aortic insufficiency murmur may also be present.
A right ventricular outflow murmur is generally audible, along with a pansystolic murmur if a residual ventricular septal defect is also present.
A right-sided aortic arch, present in about 25% of patients with tetralogy of Fallot, may cause a lift below the right sternoclavicular junction.17
Electrocardiographic findings
Electrocardiography commonly shows right ventricular hypertrophy with a right bundle branch block. The longer the QRS duration, the greater the right ventricular volume and mass. Furthermore, a QRS duration greater than 180 ms is a significant marker of risk of ventricular arrhythmias and sudden death.22,34–37
Another feature strongly associated with ventricular arrhythmias and sudden death is the rate of change in the QRS duration. A relatively rapid increase (> 3.5 ms/year) is associated with a significantly higher risk.1 A rapid rate of change may be meaningful even if the QRS duration is not markedly prolonged.11
Reduced heart rate variability also appears to be a marker of risk of sudden cardiac death in these patients.38,39
Imaging studies
Chest radiography typically shows a prominent right ventricular shadow and cardiomegaly.17
Many centers specializing in congenital heart disease therefore recommend baseline cardiac MRI, even for patients without symptoms.33
PULMONARY VALVE REPLACEMENT IS THE ONLY PROVEN TREATMENT
No study has yet shown that drug therapy alone slows the progression of complications.1 Pulmonary valve replacement is the only treatment proven to reduce right ventricular size and improve right ventricular function in the long term.
The risks of surgery, including the need for repeat operations, must be balanced against the risk of irreversible right ventricular dysfunction and its associated complications. The operative death rate is low, as is the long-term risk of death afterward. Therrien et al12 reported that, in a series of 70 patients who underwent pulmonary valve replacement, the probability of survival was 92% at 5 years and 86% at 10 years.
Surgery appears to reverse or at least arrest the progression of many of the complications associated with pulmonary insufficiency, including tricuspid regurgitation and diastolic dysfunction.17 Its utility in ameliorating ventricular tachycardia, however, remains controversial. One series showed a lower prevalence of tachycardia after pulmonary valve replacement (9% after surgery vs 22% before), but later studies have had more equivocal results.17
When should surgery be done?
There is little controversy about the eventual need for pulmonary valve replacement in most patients. What is controversial is the timing.12,44–47
This issue has been hotly debated. Some believe that pulmonary valve replacement should be done only if evidence of right ventricular dysfunction has developed.17 Others suggest that it be considered earlier and that the onset of symptoms may be a late and suboptimal indication for it.6,8,48,49 Many experts now recommend surgery early, before symptoms of heart failure develop.17 Though surgery has traditionally been recommended if the QRS duration is longer than 180 ms, some believe it should be done before this occurs.11
Arguments for early surgery. In one study, in no patient who had a right ventricular end-diastolic volume greater than 170 mL/m2 (normal ≤ 108) or a right ventricular end-systolic volume greater than 85 mL/m2 (normal ≤ 47) did these numbers return to normal after pulmonary valve replacement.45,50 This suggests a point of irreversible dilatation and a volume threshold beyond which right ventricular function is unlikely to completely improve. Normalization of right ventricular volumes was shown to occur when pulmonary valve replacement was performed before the right ventricular end-diastolic volume reached 160 mL/m2 or the right ventricular end-systolic volume reached 82 mL/m2.47,51
Delaying surgery until symptoms occur may be unfavorable because the long-term outcomes of increased right ventricular volumes and decreased right ventricular ejection fractions after surgery are not known.
Arguments for watchful waiting. There does not seem to be a threshold above which right ventricular volumes do not decrease after surgery—although they may not decrease to the normal range. Pulmonary valve replacement substantially reduced right ventricular dilatation even in patients with very high right ventricular volumes and right ventricular dysfunction, and resulted in an overall improvement in function (measured by New York Heart Association class).47
Late pulmonary valve replacement rapidly improves right ventricular volumes and improves the effective ejection fraction, although its impact on absolute right ventricular function is not as pronounced. The QRS duration shortened after surgery in those in whom it was 180 ms or longer before surgery, although this appeared to be a transient change.52 The prevalence of ventricular tachycardia declined from 22% to 9% and that of atrial fibrillation or flutter declined from 17% to 12%.17,48
A recent study with long-term follow-up has raised questions about the necessity of aggressive early intervention in tetralogy of Fallot. Sixty-seven patients were followed for as long as 27 years after surgery. Forty-five had severe pulmonary insufficiency and severe right ventricular dilatation, and of those, 28 remained free of symptoms and did not undergo pulmonary valve replacement. The authors found that refraining from pulmonary valve replacement in asymptomatic patients with severe pulmonary insufficiency led to no measurable deterioration in 25 of 28 patients.53
The available data do not support pulmonary valve replacement in young patients with mild or moderate right ventricular dilatation, normal right ventricular systolic function, and no additional risk factors.27
Mechanical vs bioprosthetic replacement valves
Once the decision is made to proceed to surgery, the next step is choosing the type of prosthetic valve.
Mechanical valves pose a risk of thrombosis, requiring life-long anticoagulation. To give warfarin (Coumadin) to younger, active people exposes them to the risk of potentially catastrophic bleeding if trauma were to occur. Women who become pregnant are generally at an increased risk of thrombotic complications due to the hypercoagulable state of pregnancy, but the risk of fetal defects is considerable if they receive warfarin.54–56
Bioprosthetic valves generally come in two varieties: preserved and treated human tissue (homografts) and animal tissue (bovine pericardial or porcine, depending on the size required). These can be implanted as isolated valves or as part of a conduit (valve and surrounding tissue).
Bioprosthetic valves eliminate the need for anticoagulation. However, they are not very durable, especially in younger patients, which is worrisome. An estimated 45% of bioprosthetic valves fail by 10 years,57 thus nearly guaranteeing that an otherwise healthy 40-year-old, for example, will need to undergo at least one repeat surgery, and very likely more.
NOVEL THERAPIES
Percutaneous valve replacement
The future of pulmonary valve replacement may lie in percutaneous procedures.
The Melody valve is now approved through a humanitarian device exemption (ie, based on demonstrated safety without proven efficacy) for patients who have a prior pulmonary conduit now complicated by either stenosis or regurgitation.
If percutaneous pulmonary valve replacement proves to have reasonable long-term durability, it has the potential to dramatically shift the balance toward earlier intervention.
Pulmonary vasodilator drugs
Our group is examining whether pharmacologic therapy can alter the clinical outcome in patients with pulmonary insufficiency (due to either tetralogy of Fallot or valvotomy done to treat remote pulmonary stenosis). Specifically, we are using MRI to examine the effects of inhaled nitric oxide, a selective pulmonary vasodilator. Preliminary results suggest that such a strategy may work, and we are designing a trial to examine the longer-term benefit of using an oral drug with similar properties.
- Gregg D, Foster E. Pulmonary insufficiency is the nexus of late complications in tetralogy of Fallot. Curr Cardiol Rep 2007; 9:315–322.
- Khairy P, Hosn JA, Broberg C, et al; Alliance for Adult Research in Congenital Cardiology (AARCC). Multicenter research in adult congenital heart disease. Int J Cardiol 2008; 129:155–159.
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation 2008; 118:e714–e833.
- Perloff JK, Warnes CA. Challenges posed by adults with repaired congenital heart disease. Circulation 2001; 103:2637–2643.
- Hickey EJ, Veldtman G, Bradley TJ, et al. Late risk of outcomes for adults with repaired tetralogy of Fallot from an inception cohort spanning four decades. Eur J Cardiothorac Surg 2009; 35:156–164.
- Apitz C, Webb GD, Redington AN. Tetralogy of Fallot. Lancet 2009; 374:1462–1471.
- Gatzoulis MA, Clark AL, Cullen S, Newman CG, Redington AN. Right ventricular diastolic function 15 to 35 years after repair of tetralogy of Fallot. Restrictive physiology predicts superior exercise performance. Circulation 1995; 91:1775–1781.
- van Straten A, Vliegen HW, Hazekamp MG, et al. Right ventricular function after pulmonary valve replacement in patients with tetralogy of Fallot. Radiology 2004; 233:824–829.
- Bouzas B, Kilner PJ, Gatzoulis MA. Pulmonary regurgitation: not a benign lesion. Eur Heart J 2005; 26:433–439.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet 2000; 356:975–981.
- Therrien J, Siu SC, Harris L, et al. Impact of pulmonary valve replacement on arrhythmia propensity late after repair of tetralogy of Fallot. Circulation 2001; 103:2489–2494.
- Deanfield JE, McKenna WJ, Presbitero P, England D, Graham GR, Hallidie-Smith K. Ventricular arrhythmia in unrepaired and repaired tetralogy of Fallot. Relation to age, timing of repair, and haemodynamic status. Br Heart J 1984; 52:77–81.
- Murphy JG, Gersh BJ, Mair DD, et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 1993; 329:593–599.
- Mackie AS, Ionescu-Ittu R, Therrien J, Pilote L, Abrahamowicz M, Marelli AJ. Children and adults with congenital heart disease lost to follow-up: who and when? Circulation 2009; 120:302–309.
- Pinsky WW, Arciniegas E. Tetralogy of Fallot. Pediatr Clin North Am 1990; 37:179–192.
- Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation 2007; 115:1933–1947.
- Walters HL, Mavroudis C, Tchervenkov CI, Jacobs JP, Lacour-Gayet F, Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: double outlet right ventricle. Ann Thorac Surg 2000; 69(suppl 4):S249–S263.
- Van Arsdell GS, Maharaj GS, Tom J, et al. What is the optimal age for repair of tetralogy of Fallot? Circulation 2000; 102(suppl 3):III123–III129.
- Cheung MM, Konstantinov IE, Redington AN. Late complications of repair of tetralogy of Fallot and indications for pulmonary valve replacement. Semin Thorac Cardiovasc Surg 2005; 17:155–159.
- Babu-Narayan SV, Gatzoulis MA. Management of adults with operated tetralogy of Fallot. Curr Treat Options Cardiovasc Med 2003; 5:389–398.
- Gatzoulis MA, Till JA, Somerville J, Redington AN. Mechanoelectrical interaction in tetralogy of Fallot. QRS prolongation relates to right ventricular size and predicts malignant ventricular arrhythmias and sudden death. Circulation 1995; 92:231–237.
- van Doorn C. The unnatural history of tetralogy of Fallot: surgical repair is not as definitive as previously thought. Heart 2002; 88:447–448.
- Norton KI, Tong C, Glass RB, Nielsen JC. Cardiac MR imaging assessment following tetralogy of Fallot repair. Radiographics 2006; 26:197–211.
- Bhat AH, Sahn DJ. Congenital heart disease never goes away, even when it has been ‘treated’: the adult with congenital heart disease. Curr Opin Pediatr 2004; 16:500–507.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:11–22.
- Khairy P, Dore A, Talajic M, et al. Arrhythmias in adult congenital heart disease. Expert Rev Cardiovasc Ther 2006; 4:83–95.
- Roos-Hesselink J, Perlroth MG, McGhie J, Spitaels S. Atrial arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and echocardiographic findings. Circulation 1995; 91:2214–2219.
- Harrison DA, Siu SC, Hussain F, MacLoghlin CJ, Webb GD, Harris L. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol 2001; 87:584–588.
- Collins KK, Dubin AM. Detecting and diagnosing arrhythmias in adults with congenital heart disease. Curr Cardiol Rep 2003; 5:331–335.
- Silka MJ, Hardy BG, Menashe VD, Morris CD. A population-based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol 1998; 32:245–251.
- Ammash NM, Dearani JA, Burkhart HM, Connolly HM. Pulmonary regurgitation after tetralogy of Fallot repair: clinical features, sequelae, and timing of pulmonary valve replacement. Congenit Heart Dis 2007; 2:386–403.
- Berul CI, Hill SL, Geggel RL, et al. Electrocardiographic markers of late sudden death risk in postoperative tetralogy of Fallot children. J Cardiovasc Electrophysiol 1997; 8:1349–1356.
- Gatzoulis MA, Till JA, Redington AN. Depolarization-repolarization inhomogeneity after repair of tetralogy of Fallot. The substrate for malignant ventricular tachycardia? Circulation 1997; 95:401–404.
- Balaji S, Lau YR, Case CL, Gillette PC. QRS prolongation is associated with inducible ventricular tachycardia after repair of tetralogy of Fallot. Am J Cardiol 1997; 80:160–163.
- Abd El Rahman MY, Abdul-Khaliq H, Vogel M, Alexi-Meskishvili V, Gutberlet M, Lange PE. Relation between right ventricular enlargement, QRS duration, and right ventricular function in patients with tetralogy of Fallot and pulmonary regurgitation after surgical repair. Heart 2000; 84:416–420.
- McLeod KA, Hillis WS, Houston AB, et al. Reduced heart rate variability following repair of tetralogy of Fallot. Heart 1999; 81:656–660.
- Davos CH, Moutafi AC, Alexandridi A, et al. Heart rate turbulence in adults with repaired tetralogy of Fallot. Int J Cardiol 2009; 135:308–314.
- de Roos A, Roest AA. Evaluation of congenital heart disease by magnetic resonance imaging. Eur Radiol 2000; 10:2–6.
- Niezen RA, Helbing WA, van der Wall EE, van der Geest RJ, Rebergen SA, de Roos A. Biventricular systolic function and mass studied with MR imaging in children with pulmonary regurgitation after repair for tetralogy of Fallot. Radiology 1996; 201:135–140.
- Helbing WA, de Roos A. Optimal imaging in assessment of right ventricular function in tetralogy of Fallot with pulmonary regurgitation. Am J Cardiol 1998; 82:1561–1562.
- van der Geest RJ, de Roos A, van der Wall EE, Reiber JH. Quantitative analysis of cardiovascular MR images. Int J Card Imaging 1997; 13:247–258.
- Therrien J, Siu SC, McLaughlin PR, Liu PP, Williams WG, Webb GD. Pulmonary valve replacement in adults late after repair of tetralogy of Fallot: are we operating too late? J Am Coll Cardiol 2000; 36:1670–1675.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Davlouros PA, Karatza AA, Gatzoulis MA, Shore DF. Timing and type of surgery for severe pulmonary regurgitation after repair of tetralogy of Fallot. Int J Cardiol 2004; 97(suppl 1):91–101.
- Oosterhof T, van Straten A, Vliegen HW, et al. Preoperative thresholds for pulmonary valve replacement in patients with corrected tetralogy of Fallot using cardiovascular magnetic resonance. Circulation 2007; 116:545–551.
- Buechel ER, Dave HH, Kellenberger CJ, et al. Remodelling of the right ventricle after early pulmonary valve replacement in children with repaired tetralogy of Fallot: assessment by cardiovascular magnetic resonance. Eur Heart J 2005; 26:2721–2727.
- Henkens IR, van Straten A, Schalij MJ, et al. Predicting outcome of pulmonary valve replacement in adult tetralogy of Fallot patients. Ann Thorac Surg 2007; 83:907–911.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Redington AN. Physiopathology of right ventricular failure. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:3–10.
- Oosterhof T, Vliegen HW, Meijboom FJ, Zwinderman AH, Bouma B, Mulder BJ. Long-term effect of pulmonary valve replacement on QRS duration in patients with corrected tetralogy of Fallot. Heart 2007; 93:506–509.
- Meijboom FJ, Roos-Hesselink JW, McGhie JS, et al. Consequences of a selective approach toward pulmonary valve replacement in adult patients with tetralogy of Fallot and pulmonary regurgitation. J Thorac Cardiovasc Surg 2008; 135:50–55.
- Danik S, Fuster V. The obstetrical patient with a prosthetic heart valve. Obstet Gynecol Clin North Am 2006; 33:481–491.
- Danik S, Fuster V. Anticoagulation in pregnant women with prosthetic heart valves. Mt Sinai J Med 2004; 71:322–329.
- Manso B, Gran F, Pijuán A, et al. Pregnancy and congenital heart disease (article in Spanish). Rev Esp Cardiol 2008; 61:236–243.
- Gallegos RP. Selection of prosthetic heart valves. Curr Treat Options Cardiovasc Med 2006; 8:443–452.
- Bonhoeffer P, Boudjemline Y, Qureshi SA, et al. Percutaneous insertion of the pulmonary valve. J Am Coll Cardiol 2002; 39:1664–1669.
- Khambadkone S, Coats L, Taylor A, et al. Percutaneous pulmonary valve implantation in humans: results in 59 consecutive patients. Circulation 2005; 112:1189–1197.
- Lurz P, Coats L, Khambadkone S, et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation 2008; 117:1964–1972.
- Gregg D, Foster E. Pulmonary insufficiency is the nexus of late complications in tetralogy of Fallot. Curr Cardiol Rep 2007; 9:315–322.
- Khairy P, Hosn JA, Broberg C, et al; Alliance for Adult Research in Congenital Cardiology (AARCC). Multicenter research in adult congenital heart disease. Int J Cardiol 2008; 129:155–159.
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation 2008; 118:e714–e833.
- Perloff JK, Warnes CA. Challenges posed by adults with repaired congenital heart disease. Circulation 2001; 103:2637–2643.
- Hickey EJ, Veldtman G, Bradley TJ, et al. Late risk of outcomes for adults with repaired tetralogy of Fallot from an inception cohort spanning four decades. Eur J Cardiothorac Surg 2009; 35:156–164.
- Apitz C, Webb GD, Redington AN. Tetralogy of Fallot. Lancet 2009; 374:1462–1471.
- Gatzoulis MA, Clark AL, Cullen S, Newman CG, Redington AN. Right ventricular diastolic function 15 to 35 years after repair of tetralogy of Fallot. Restrictive physiology predicts superior exercise performance. Circulation 1995; 91:1775–1781.
- van Straten A, Vliegen HW, Hazekamp MG, et al. Right ventricular function after pulmonary valve replacement in patients with tetralogy of Fallot. Radiology 2004; 233:824–829.
- Bouzas B, Kilner PJ, Gatzoulis MA. Pulmonary regurgitation: not a benign lesion. Eur Heart J 2005; 26:433–439.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Gatzoulis MA, Balaji S, Webber SA, et al. Risk factors for arrhythmia and sudden cardiac death late after repair of tetralogy of Fallot: a multicentre study. Lancet 2000; 356:975–981.
- Therrien J, Siu SC, Harris L, et al. Impact of pulmonary valve replacement on arrhythmia propensity late after repair of tetralogy of Fallot. Circulation 2001; 103:2489–2494.
- Deanfield JE, McKenna WJ, Presbitero P, England D, Graham GR, Hallidie-Smith K. Ventricular arrhythmia in unrepaired and repaired tetralogy of Fallot. Relation to age, timing of repair, and haemodynamic status. Br Heart J 1984; 52:77–81.
- Murphy JG, Gersh BJ, Mair DD, et al. Long-term outcome in patients undergoing surgical repair of tetralogy of Fallot. N Engl J Med 1993; 329:593–599.
- Mackie AS, Ionescu-Ittu R, Therrien J, Pilote L, Abrahamowicz M, Marelli AJ. Children and adults with congenital heart disease lost to follow-up: who and when? Circulation 2009; 120:302–309.
- Pinsky WW, Arciniegas E. Tetralogy of Fallot. Pediatr Clin North Am 1990; 37:179–192.
- Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation 2007; 115:1933–1947.
- Walters HL, Mavroudis C, Tchervenkov CI, Jacobs JP, Lacour-Gayet F, Jacobs ML. Congenital Heart Surgery Nomenclature and Database Project: double outlet right ventricle. Ann Thorac Surg 2000; 69(suppl 4):S249–S263.
- Van Arsdell GS, Maharaj GS, Tom J, et al. What is the optimal age for repair of tetralogy of Fallot? Circulation 2000; 102(suppl 3):III123–III129.
- Cheung MM, Konstantinov IE, Redington AN. Late complications of repair of tetralogy of Fallot and indications for pulmonary valve replacement. Semin Thorac Cardiovasc Surg 2005; 17:155–159.
- Babu-Narayan SV, Gatzoulis MA. Management of adults with operated tetralogy of Fallot. Curr Treat Options Cardiovasc Med 2003; 5:389–398.
- Gatzoulis MA, Till JA, Somerville J, Redington AN. Mechanoelectrical interaction in tetralogy of Fallot. QRS prolongation relates to right ventricular size and predicts malignant ventricular arrhythmias and sudden death. Circulation 1995; 92:231–237.
- van Doorn C. The unnatural history of tetralogy of Fallot: surgical repair is not as definitive as previously thought. Heart 2002; 88:447–448.
- Norton KI, Tong C, Glass RB, Nielsen JC. Cardiac MR imaging assessment following tetralogy of Fallot repair. Radiographics 2006; 26:197–211.
- Bhat AH, Sahn DJ. Congenital heart disease never goes away, even when it has been ‘treated’: the adult with congenital heart disease. Curr Opin Pediatr 2004; 16:500–507.
- Redington AN. Determinants and assessment of pulmonary regurgitation in tetralogy of Fallot: practice and pitfalls. Cardiol Clin 2006; 24:631–639.
- Geva T. Indications and timing of pulmonary valve replacement after tetralogy of Fallot repair. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:11–22.
- Khairy P, Dore A, Talajic M, et al. Arrhythmias in adult congenital heart disease. Expert Rev Cardiovasc Ther 2006; 4:83–95.
- Roos-Hesselink J, Perlroth MG, McGhie J, Spitaels S. Atrial arrhythmias in adults after repair of tetralogy of Fallot. Correlations with clinical, exercise, and echocardiographic findings. Circulation 1995; 91:2214–2219.
- Harrison DA, Siu SC, Hussain F, MacLoghlin CJ, Webb GD, Harris L. Sustained atrial arrhythmias in adults late after repair of tetralogy of Fallot. Am J Cardiol 2001; 87:584–588.
- Collins KK, Dubin AM. Detecting and diagnosing arrhythmias in adults with congenital heart disease. Curr Cardiol Rep 2003; 5:331–335.
- Silka MJ, Hardy BG, Menashe VD, Morris CD. A population-based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol 1998; 32:245–251.
- Ammash NM, Dearani JA, Burkhart HM, Connolly HM. Pulmonary regurgitation after tetralogy of Fallot repair: clinical features, sequelae, and timing of pulmonary valve replacement. Congenit Heart Dis 2007; 2:386–403.
- Berul CI, Hill SL, Geggel RL, et al. Electrocardiographic markers of late sudden death risk in postoperative tetralogy of Fallot children. J Cardiovasc Electrophysiol 1997; 8:1349–1356.
- Gatzoulis MA, Till JA, Redington AN. Depolarization-repolarization inhomogeneity after repair of tetralogy of Fallot. The substrate for malignant ventricular tachycardia? Circulation 1997; 95:401–404.
- Balaji S, Lau YR, Case CL, Gillette PC. QRS prolongation is associated with inducible ventricular tachycardia after repair of tetralogy of Fallot. Am J Cardiol 1997; 80:160–163.
- Abd El Rahman MY, Abdul-Khaliq H, Vogel M, Alexi-Meskishvili V, Gutberlet M, Lange PE. Relation between right ventricular enlargement, QRS duration, and right ventricular function in patients with tetralogy of Fallot and pulmonary regurgitation after surgical repair. Heart 2000; 84:416–420.
- McLeod KA, Hillis WS, Houston AB, et al. Reduced heart rate variability following repair of tetralogy of Fallot. Heart 1999; 81:656–660.
- Davos CH, Moutafi AC, Alexandridi A, et al. Heart rate turbulence in adults with repaired tetralogy of Fallot. Int J Cardiol 2009; 135:308–314.
- de Roos A, Roest AA. Evaluation of congenital heart disease by magnetic resonance imaging. Eur Radiol 2000; 10:2–6.
- Niezen RA, Helbing WA, van der Wall EE, van der Geest RJ, Rebergen SA, de Roos A. Biventricular systolic function and mass studied with MR imaging in children with pulmonary regurgitation after repair for tetralogy of Fallot. Radiology 1996; 201:135–140.
- Helbing WA, de Roos A. Optimal imaging in assessment of right ventricular function in tetralogy of Fallot with pulmonary regurgitation. Am J Cardiol 1998; 82:1561–1562.
- van der Geest RJ, de Roos A, van der Wall EE, Reiber JH. Quantitative analysis of cardiovascular MR images. Int J Card Imaging 1997; 13:247–258.
- Therrien J, Siu SC, McLaughlin PR, Liu PP, Williams WG, Webb GD. Pulmonary valve replacement in adults late after repair of tetralogy of Fallot: are we operating too late? J Am Coll Cardiol 2000; 36:1670–1675.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Davlouros PA, Karatza AA, Gatzoulis MA, Shore DF. Timing and type of surgery for severe pulmonary regurgitation after repair of tetralogy of Fallot. Int J Cardiol 2004; 97(suppl 1):91–101.
- Oosterhof T, van Straten A, Vliegen HW, et al. Preoperative thresholds for pulmonary valve replacement in patients with corrected tetralogy of Fallot using cardiovascular magnetic resonance. Circulation 2007; 116:545–551.
- Buechel ER, Dave HH, Kellenberger CJ, et al. Remodelling of the right ventricle after early pulmonary valve replacement in children with repaired tetralogy of Fallot: assessment by cardiovascular magnetic resonance. Eur Heart J 2005; 26:2721–2727.
- Henkens IR, van Straten A, Schalij MJ, et al. Predicting outcome of pulmonary valve replacement in adult tetralogy of Fallot patients. Ann Thorac Surg 2007; 83:907–911.
- Therrien J, Provost Y, Merchant N, Williams W, Colman J, Webb G. Optimal timing for pulmonary valve replacement in adults after tetralogy of Fallot repair. Am J Cardiol 2005; 95:779–782.
- Redington AN. Physiopathology of right ventricular failure. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2006:3–10.
- Oosterhof T, Vliegen HW, Meijboom FJ, Zwinderman AH, Bouma B, Mulder BJ. Long-term effect of pulmonary valve replacement on QRS duration in patients with corrected tetralogy of Fallot. Heart 2007; 93:506–509.
- Meijboom FJ, Roos-Hesselink JW, McGhie JS, et al. Consequences of a selective approach toward pulmonary valve replacement in adult patients with tetralogy of Fallot and pulmonary regurgitation. J Thorac Cardiovasc Surg 2008; 135:50–55.
- Danik S, Fuster V. The obstetrical patient with a prosthetic heart valve. Obstet Gynecol Clin North Am 2006; 33:481–491.
- Danik S, Fuster V. Anticoagulation in pregnant women with prosthetic heart valves. Mt Sinai J Med 2004; 71:322–329.
- Manso B, Gran F, Pijuán A, et al. Pregnancy and congenital heart disease (article in Spanish). Rev Esp Cardiol 2008; 61:236–243.
- Gallegos RP. Selection of prosthetic heart valves. Curr Treat Options Cardiovasc Med 2006; 8:443–452.
- Bonhoeffer P, Boudjemline Y, Qureshi SA, et al. Percutaneous insertion of the pulmonary valve. J Am Coll Cardiol 2002; 39:1664–1669.
- Khambadkone S, Coats L, Taylor A, et al. Percutaneous pulmonary valve implantation in humans: results in 59 consecutive patients. Circulation 2005; 112:1189–1197.
- Lurz P, Coats L, Khambadkone S, et al. Percutaneous pulmonary valve implantation: impact of evolving technology and learning curve on clinical outcome. Circulation 2008; 117:1964–1972.
KEY POINTS
- The major long-term complication of tetralogy of Fallot repair is pulmonary valve insufficiency, which leads to right heart failure. Other problems include atrial and ventricular arrhythmias and sudden cardiac death.
- Surgical pulmonary valve replacement is the standard of care, but the optimal time to do this is unclear.
- Novel and experimental therapies include percutaneous pulmonary valve replacement and medical therapy with pulmonary arterial vasodilators.
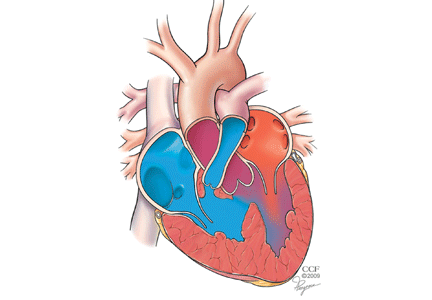
Insulin use does not protect against restenosis in diabetic patients presenting with acute coronary syndrome or symptomatic angina
A 37-year-old man with chest pain, ECG changes, and elevated cardiac enzymes
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
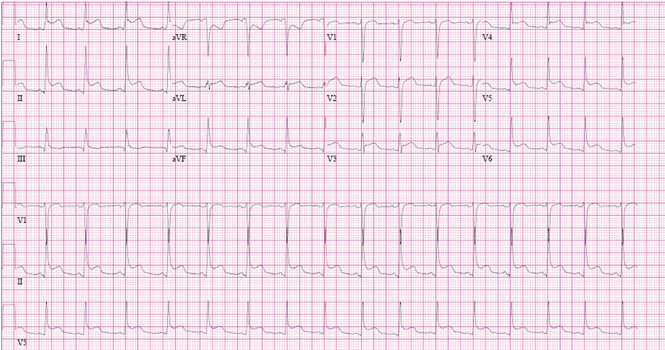
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.

CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
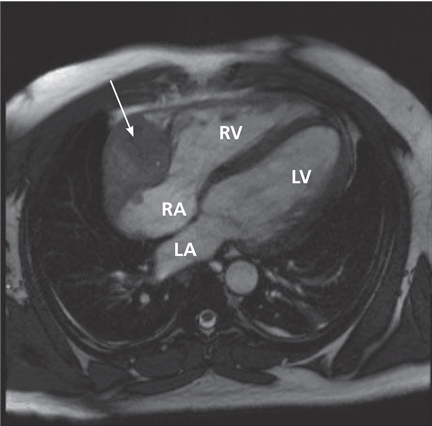
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
A 37-year-old African American man presents to the emergency department with chest pain and dyspnea, which began suddenly 30 minutes ago. The pain is severe, pressure-like, nonradiating, and pleuritic.
His heart rate is 88 beats per minute, blood pressure 135/72 mm Hg, respiratory rate 12 per minute, and oral temperature 38.5°C (101.3°F). His oxygen saturation by pulse oximetry is 99% while breathing room air. He is given sublingual nitroglycerin, but this does not alleviate his pain.
While blood samples are being drawn, we learn more about his history. He has hypertension, for which he takes amlodipine (Norvasc), and gastroesophageal reflux under control with esomeprazole (Nexium). He says he does not have hyperlipidemia, diabetes, or coronary artery disease and his surgical history is unremarkable. He says he does not smoke, rarely drinks, and does not use any drugs. No one in his family has had premature coronary artery disease.
He says he has had similar symptoms in the past few months, which resulted in two emergency room visits. Electrocardiograms at those times were unremarkable, and a stress test was negative for ischemia.
A computed tomographic (CT) scan of the chest was also obtained during one of those visits. The scan was negative for a pulmonary embolus but incidentally showed liver hemangiomas.
The patient’s initial laboratory results are shown in Table 1.
WHAT IS THE CAUSE OF HIS CHEST PAIN?
1. Which is the most likely cause of this patient’s chest pain?
- Acute myocardial infarction
- Acute pericarditis
- Myocarditis
- Pulmonary embolism
- Aortic dissection
- Pneumonia
Acute myocardial infarction. This is a young man with chest pain, ST-segment elevation, and elevated cardiac enzymes. Acute myocardial infarction should always be included in the differential diagnosis of such a patient, as recognizing it early and making an effort to rapidly restore blood flow to the myocardium can greatly improve the clinical outcome. However, particular features in his electrocardiogram and the duration and nature of his chest pain suggest another diagnosis.
Acute pericarditis causes pleuritic chest pain with diffuse ST-segment elevation, and its electrocardiographic changes may be difficult to distinguish from those of ischemia. The features in our patient’s electrocardiogram that point to pericarditis are1:
- ST-segment elevation that is concave upward, occurring in all leads except aVR
- T waves concordant with ST-segment deviation
- PR-segment depression, sparing V1 and aVR
- PR-segment elevation and ST depression in aVR.
Pleuritic chest pain is the most common symptom in acute pericarditis. A prodrome of fever, myalgia, and malaise is also common, especially in younger patients.2 On physical examination, a pericardial friction rub is pathognomonic.
Our patient has most if not all of the classic features of acute pericarditis. Elevated cardiac enzymes, which this patient has, are not a classic feature of pericarditis and are generally considered a marker of cardiac ischemia. However, because the myocardium is adjacent to the pericardium, the acute inflammatory process of acute pericarditis may also result in myocardial injury, resulting in release of creatine kinase-MB.3
An increase in cardiac troponin is also frequently observed in acute pericarditis, reflecting biochemical evidence of inflammatory myocardial cell damage.4 Furthermore, cardiac troponin can be elevated in several other medical conditions,5 such as ischemic heart disease, congestive heart failure, myocarditis, pulmonary embolism, severe pulmonary hypertension, significant left ventricular hypertrophy, renal failure, sepsis, critical illness, and subarachnoid hemorrhage. Therefore, cardiac enzymes are not good markers to distinguish between acute myocardial infarction and acute pericarditis. However, echocardiography is an effective way to help differentiate pericarditis from myocardial ischemia in the setting of elevated troponins and electrocardiographic changes, by determining if wall-motion abnormalities are present or absent.
Hence, the diagnosis of acute pericarditis should take into account the combination of the clinical picture, electrocardiographic findings, and laboratory values. Overreliance on any of these in isolation can lead to misdiagnosis.
Pulmonary embolism is another common cause of acute-onset pleuritic chest pain and dyspnea. Electrocardiographic changes can include ST-segment elevation, and cardiac enzymes can be elevated, although this is uncommon.
Myocarditis is commonly due to infections, collagen vascular diseases, or medications. Hallmarks of this disease are elevated cardiac enzymes and myocardial damage that results in reduction in heart function.
Aortic dissection typically causes a sharp, tearing chest pain that radiates to the back. This diagnosis is unlikely in this patient.
Pneumonia. Although our patient did not have a cough and no crackles were heard on lung examination to suggest pneumonia, his fever, pleuritic chest pain, and leukocytosis with a left shift warrant a workup for it. A parapneumonic effusion could manifest with fevers and pleuritic chest pain. However, the acuity of the symptoms and the characteristic electrocardiographic changes and elevated cardiac enzymes are better explained by the other diagnoses, notably acute pericarditis.
ACUTE PERICARDITIS: WHAT IS THE CAUSE?
2. Which is the most common cause of acute pericarditis?
- Idiopathic
- Neoplasm
- Autoimmune
- Tuberculosis
Most (approximately 80%) of cases of acute pericarditis are idiopathic.6,7 In a study in 100 patients with acute pericarditis,6 a specific cause was identified in only 22. The most common identified cause was neoplasm, which was present in seven patients: four with lung cancer and one each with breast carcinoma, cystic duct adenocarcinoma, and cardiac angiosarcoma.
CASE CONTINUES: PERICARDIAL EFFUSION
The patient is admitted to the hospital for additional workup. His fever, myalgia, and chest pain persist, though the pain is less intense than before.
A chest roentgenogram and transthoracic echocardiogram are ordered and blood cultures are drawn.
The roentgenogram shows marked cardiomegaly, bilateral small pleural effusions, and minimal atelectatic changes in the lungs.
Echocardiography reveals a normal ejection fraction (60%) and a moderate-sized pericardial effusion without evidence of tamponade.
3. Which is the most common cause of pericardial effusion?
- Idiopathic
- Infection
- Malignancy
- Collagen vascular disease
Pericardial effusion is relatively common after acute pericarditis but also has many other possible causes. In a study of 204 patients with pericardial effusion,8 48% of cases were labeled as idiopathic. Of the remaining 52%, the most common specific diagnoses were infection (16%) and cancer (15%). Collagen vascular disease accounted for 8% of the cases and included systemic lupus erythematosus, rheumatoid arthritis, and scleroderma.
Although small pericardial effusions are common in pericarditis, larger pericardial effusions or failure to respond to therapy necessitates additional workup.2
In our patient, an extensive workup is initiated to look for bacterial, viral, fungal, and autoimmune causes of pericardial effusion, but the results of the workup are negative.
TREATING ACUTE PERICARDITIS
4. Which is the most appropriate treatment for acute pericarditis?
- Steroids
- A nonsteroidal anti-inflammatory drug (NSAID) or aspirin
- Opioids
- Colchicine
- Colchicine plus an NSAID or aspirin
An NSAID or aspirin is the basis of treatment for acute pericarditis and is very effective in relieving symptoms. Aspirin 2–4 g daily, indomethacin (Indocin) 75–225 mg daily, or ibuprofen (Motrin) 1,600–3,200 mg daily are prescribed most often; ibuprofen is preferred because it has a lower incidence of adverse effects than the others.9
Colchicine is recommended in addition to aspirin or NSAIDs for the treatment of acute pericarditis. Although in the past colchicine was reserved for recurrent pericarditis, the Colchicine for Acute Pericarditis (COPE) trial10 found it to be beneficial for first episodes of pericarditis as well.10 In this study, patients were randomized to receive conventional treatment with aspirin 800 mg every 6 or 8 hours or aspirin at the same dose combined with colchicine 0.5 to 1.0 mg daily. Colchicine showed significant benefit over conventional therapy, resulting in reduced rates of recurrence.
CASE CONTINUES: HEPATIC LESIONS ON MRI
Although aspirin and colchicine were started at the time of admission, our patient’s symptoms fail to improve. A suspicion remains that a neoplastic disorder could be the underlying cause of the presentation and could explain his chronic malaise, pericardial disease, and fever. In view of the liver hemangiomas reported previously on CT, we decide to evaluate the liver further with magnetic resonance imaging (MRI).
Since our patient’s symptoms have improved significantly during the past few days and his fever has resolved, biopsy is scheduled on an outpatient basis. Biopsy with ultrasonographic guidance is performed a week later and yields a pathologic diagnosis of hemangioma. The improvement, however, is short-lived, and his pain and dyspnea recur after 2 months. A follow-up echocardiogram is ordered.
A remarkable echocardiographic finding
The original echocardiogram that was performed a little over 2-1/2 months ago is re-reviewed. It very subtly suggests a complexity to the pericardial effusion in the area of the current mass, apparent only when the two studies are directly compared. Clearly, there has been interval development of a mass easily detectable by echocardiography. Although a small mass may have been obscured by the pericardial effusion in the original echocardiogram, the development of a mass of this size in such a short time suggests a rapidly growing tumor.
CARDIAC TUMORS
5. Which is the most common primary cardiac tumor?
- Myxoma
- Papillary fibroelastoma
- Sarcoma
- Lymphoma
Primary cardiac tumors are rare, with an incidence on autopsy series ranging between 0.0017% and 0.33%,11,12 making them far less common than metastases to the heart.
Myxomas are benign cardiac tumors and are the most common primary cardiac neoplasm. Approximately 80% of myxomas originate in the left atrium, typically presenting with one or more of the triad of intracardiac obstruction, systemic embolization, and constitutional symptoms.14
Cardiac papillary fibroelastomas, the second most common cardiac tumors, are benign and predominantly affect the cardiac valves.15
Only one-fourth of all cardiac tumors are malignant. Nearly all of these malignant tumors are sarcomas, with angiosarcoma being the most common morphologic type, accounting for 30% of primary cardiac sarcomas.13
Primary cardiac lymphomas are extremely rare and account for only 1.3% of all primary cardiac tumors.16
A DIAGNOSIS IS MADE
CARDIAC ANGIOSARCOMA
Cardiac angiosarcoma, the most common malignant primary cardiac tumor, has a predilection for the right atrium.13 These tumors tend to occur between the third and fifth decade of life and are three times more common in men than in women. Cardiac sarcomas proliferate rapidly and commonly extend into the pericardial space, causing pericardial effusion in up to one-fourth of patients.
Surgical resection is the treatment of choice, but due to the location and extent of involvement, complete resection is often difficult. Also, distant metastases are present at the time of diagnosis in 80% of cases, precluding a surgical cure.17 Adjuvant chemotherapy, radiotherapy, and even heart transplantation do not substantially improve the survival of these patients.18–20 Because no effective treatment is available, the prognosis is dismal, with a median survival of 6 to 12 months.
Our patient is discharged home to follow up with an oncologist and initiate chemotherapy.
Acknowledgment: We thank Lisa M. Yerian, MD, for interpreting the biopsy specimens described in this article.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.
- Ariyarajah V, Spodick DH. Acute pericarditis: diagnostic cues and common electrocardiographic manifestations. Cardiol Rev 2007; 15:24–30.
- LeWinter MM, Kabbani S. Pericardial disease. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1757–1781.
- Karjalainen J, Heikkila J. “Acute pericarditis”: myocardial enzyme release as evidence for myocarditis”. Am Heart J 1986; 111:546–552.
- Brandt RR, Filzmaier K, Hanrath P. Circulating cardiac troponin I in acute pericarditis. Am J Cardiol 2001; 87:1326–1328.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease: the present and the future. J Am Coll Cardiol 2006; 48:1–11.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol 1995; 75:378–382.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation 2007; 115:2739–2744.
- Levy PY, Corey R, Berger P, et al. Etiologic diagnosis of 204 pericardial effusions. Medicine (Baltimore) 2003; 82:385–391.
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med 2004; 351:2195–2202.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: Results of the COlchicine for acute PEricarditis (COPE) trial. Circulation 2005; 112:2012–2016.
- Heath D. Pathology of cardiac tumors. Am J Cardiol 1968; 21:315–327.
- Wold LE, Lie JT. Cardiac myxomas: a clinicopathologic profile. Am J Pathol 1980; 101:219–240.
- Sabatine MS, Colucci WS, Schoen FS. Primary tumors of the heart. In: Zipes DP, et al, editor. Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7th ed. Philadelphia: Elsevier/WB Saunders; 2005:1741–1757.
- Gowda RM, Khan IA, Nair CK, Mehta NJ, Vasavada BC, Sacchi TJ. Cardiac papillary fibroelastoma: a comprehensive analysis of 725 cases. Am Heart J 2003; 146:404–410.
- Glancy DL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol 1968; 21:413–419.
- Ceresoli GL, Ferreri AJ, Bucci E, Ripa C, Ponzoni M, Villa E. Primary cardiac lymphoma in immunocompetent patients: diagnostic and therapeutic management. Cancer 1997; 80:1497–1506.
- Bear PA, Moodie DS. Malignant primary cardiac tumors. The Cleveland Clinic experience, 1956 to 1986. Chest 1987; 92:860–862.
- Llombart–Cussac A, Pivot X, Contesso G, et al. Adjuvant chemotherapy for primary cardiac sarcomas: the IGR experience. Br J Cancer 1998; 78:1624–1628.
- Putnam JB, Sweeney MS, Colon R, Lanza LA, Frazier OH, Cooley DA. Primary cardiac sarcomas. Ann Thorac Surg 1991; 51:906–910.
- Conklin LD, Reardon MJ. Autotransplantation of the heart for primary cardiac malignancy: development and surgical technique. Tex Heart Inst J 2002; 29:105–108.