User login
Case Studies in Toxicology: The Perils of Playing Catch-up
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
Case
A 16-year-old girl, who recently emigrated from Haiti, was brought to the pediatric ED by her mother for evaluation of a 2-hour history of gastric discomfort. Upon arrival at the ED waiting area, the patient experienced a sudden onset of generalized tonic-clonic movement with altered sensorium, though she did not fall to the ground and was not injured. Vital signs from triage were: blood pressure, 110/76 mm Hg; heart rate, 112 beats/min; respiratory rate, 22 breaths/min; and temperature, 97°F. Oxygen saturation was 98% on room air.
The patient was immediately attached to a cardiac monitor, given oxygen via a face mask, and received airway suctioning. Despite receiving a total of 4 mg of lorazepam, the seizure continued. Physical examination revealed no signs of external injury, but the ongoing generalized status epilepticus made the examination difficult.
What are the causes of refractory seizures in an adolescent patient?
The differential diagnosis for pediatric patients presenting with refractory seizure is the same as that for adult patients and should include treatment noncompliance, infection, vascular event (eg, stroke, hemorrhage), trauma (eg, cerebral contusions), metabolic and electrolyte disturbances, anticonvulsant toxicity, and exposure to a convulsant toxin.
While certain drugs (eg, cocaine) may cause status epilepticus through a secondary effect such as ischemia or a bleed, some drugs can directly cause refractory seizures. A few drugs and toxins are responsible for the majority of such seizures: bupropion; carbon monoxide; diphenhydramine; ethanol (withdrawal); hypoglycemics; lead; theophylline; tramadol; and certain antibiotics, including cephalosporins, penicillins, quinolones, and, in particular, isoniazid (INH).1
Case Continuation
Upon further history-taking, the patient’s mother informed the ED staff that during a recent visit to a local clinic, her daughter tested positive on routine screening for tuberculosis and was given “some medications.” The patient’s mother further noted that her daughter was scheduled for a follow-up appointment at the same clinic later this morning. She believed the patient had taken “a few” of the prescribed pills at once to “catch-up” on missed doses prior to that appointment, and provided the ED staff with an empty bottle of INH that she had found in her daughter’s purse.
What are the signs and symptoms of acute isoniazid toxicity?
Isoniazid toxicity should be suspected in any patient who has access to INH—even if the drug was prescribed for someone other than the patient. Acute toxicity develops rapidly after the ingestion of supratherapeutic doses of INH and includes nausea, abdominal discomfort, vomiting, dizziness, and excessive fatigue or lethargy. Patients can present with tachycardia, stupor, agitation, mydriasis, increased anion gap metabolic acidosis, and encephalopathy.
Seizures occur due to an INH-induced functional pyridoxine deficiency. Isoniazid inhibits pyridoxine phosphokinase, the enzyme that converts pyridoxine (vitamin B6) to its physiologically active form, pyridoxal 5’-phosphate (PLP). Because the conversion of glutamate (an excitatory neurotransmitter) to gamma-aminobutyric acid (GABA; the body’s main inhibitory neurotransmitter) is dependent on PLP, an excess of glutamate and a deficiency of GABA occurs following INH overdose. The result is neuroexcitation, which manifests as generalized seizures in affected patients.
The most consequential effect of INH overdose, however, is the development of seizure refractory to conventional therapy, such as benzodiazepines. This occurs because benzodiazepines are indirect-acting GABA agonists, and require the presence of GABA to elicit their effect. Therefore, due to the impairment of GABA synthesis, benzodiazepines are limited or ineffective as anticonvulsants. Although INH doses in excess of 20 mg/kg may result in neuroexcitation, refractory seizures are uncommon with doses <70 mg/kg.
Complications of chronic INH use include hepatotoxicity, and patients will present with jaundice, hepatomegaly, and right upper quadrant pain and tenderness. Isoniazid must be discontinued rapidly in
How is acute isoniazid-induced seizure managed?
Management of patients with refractory seizure should initially include an assessment and management of the patient’s airway, breathing, and circulation. Although seizures induced by INH toxicity are often resistant to benzodiazepines, these agents remain the first-line therapy. For patients who fail to respond to a reasonable trial of benzodiazepines (eg, lorazepam 6 mg intravenously [IV]), pyridoxine should be administered.3 The recommended dose is 1 g pyridoxine per every 1 g of INH ingested—if the initial dose ingested is known—with a maximum dose of 5 g pyridoxine. If the initial dose of INH is not known, 70 mg/kg of pyridoxine, up to 5 g, is recommended. Repeated doses of pyridoxine can be administered if the seizure continues, up to a total dose of 10 g in an adult. At extremely high doses, pyridoxine itself can be neurotoxic, limiting the maximal antidotal dose.
Rapid initiation of pyridoxine is a challenge since typical stocks in most EDs are not in an adequate supply required for treatment. Additionally, a typical vial of pyridoxine contains 100 mg, highlighting the rare need to open dozens of vials for a single patient. Drawing up adequate doses of the IV formulation can be a challenge and time-consuming.
Regardless, the most reliable and rapid route of administration for pyridoxine is IV, at a rate of 0.5 to 1 g/min. Even if the seizure resolves prior to completion of the initial dose, the remaining doses should still be administered over a 4- to 6-hour period. Oral or (more likely) nasogastric administration of pyridoxine can be administered if the IV formulation is not available, but neither are optimal routes of delivery. Every effort should be made to stock pyridoxine in the antidote supply in the ED to avoid time delays involving finding, preparing, and administering the drug in these scenarios. Previous studies have found that most EDs are not prepared to handle pyridoxine replacement.4,5
Since benzodiazepines and barbiturates are GABA agonists with complementary mechanisms of actions to pyridoxine, they should be administered to potentiate the antiseizure effect of pyridoxine. If the seizure does not terminate, the use of propofol or general anesthesia may be required. Once the seizure is terminated, oral activated charcoal can be administered if the ingestion occurred within several hours of presentation. Given the rapid onset of effect of a large dose of INH, most patients will develop seizure shortly after exposure, limiting the benefits of both aggressive gastrointestinal decontamination and delayed activated charcoal. Charcoal also can be used for patients who overdose on INH but do not develop seizures.
Although the utility of a head computed tomography (CT) scan or laboratory studies is limited given the context of the exposure, these are generally obtained for patients with new-onset seizure. Since many patients with INH toxicity do not seize, such a patient may have a lower seizure threshold due to the existence of a subclinical cerebral lesion or metabolic abnormality.
Case Conclusion
The patient’s INH-induced refractory seizure was treated with pyridoxine. Her history suggested that she had ingested an unknown number of INH tablets within an hour. On this initial basis, an IV dose of 5,000 mg of pyridoxine was administered. The patient’s seizures terminated within 2 minutes of the infusion, and no additional doses of pyridoxine were required. Given the lack of concern for self-harm, an acetaminophen concentration was not obtained. A urine toxicology screen was negative for cocaine and amphetamines, and a CT scan of the head was negative for any abnormality. The patient was admitted to the pediatric intensive care unit for status epileptics and was discharged home on hospital day 2 after an uneventful stay.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.
1. Cock HR. Drug-induced status epilepticus. Epilepsy Behav. 2015;49:76-82. doi:10.1016/j.yebeh.2015.04.034.
2. Latent tuberculosis infection: a guide for primary health care providers. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/tb/publications/LTBI/treatment.htm. Updated August 5, 2016. Accessed December 13, 2016.
3. Howland MA. Antidotes in depth: pyridoxine. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR, eds. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2015:797-799.
4. Shah BR, Santucci K, Sinert R, Steiner P. Acute isoniazid neurotoxicity in an urban hospital. Pediatrics. 1995;95(5):700-704.
5. Santucci KA, Shah BR, Linakis JG. Acute isoniazid exposures and antidote availability. Pediatr Emerg Care. 1999;15(2):99-101.

He Huffed and He Puffed and He Got Frostbite
Case
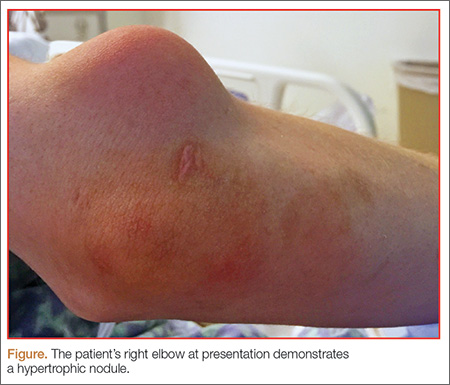
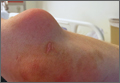
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
Case
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
Case
A 27-year-old man presented to an ED after experiencing a syncopal episode. His vital signs at presentation were normal. Physical examination was generally normal except that there were blisters on the patient’s abdomen, left hand, and right arm, as well as a hypertrophic nodule on the right elbow (Figure) and hard growths on the digits of the right hand. The patient stated the growths started 5 months ago and had been increasing in size. On further questioning, the patient admitted to “huffing” (ie, inhaling) at least six cans of pressurized dust-removal keyboard cleaning spray daily for the past 11 months.
Why do patients abuse keyboard cleaning spray?
The propellant used in certain liquefied compressed gas products is 1,1-difluoroethane (1,1-DFE), a fluorinated hydrocarbon. It is a member of a broad class of related compounds that are present in spray paints, glues, nail polish removers, fuels, hair sprays, and air-freshening products. These 1,1-DFE-containing products are abused for their rapid and short-acting central nervous system (CNS) depressant effects—not unlike that of ethanol. Typically, the vapor of a volatile hydrocarbon is inhaled directly from the open container (“sniffing”), from a bag (“bagging”), or from a soaked rag (huffing). Not only are such hydrocarbon-containing products easy to conceal, they are also highly accessible and inexpensive. Moreover, there are generally no direct legal consequences resulting from abuse of these substances.
All of the aforementioned factors make hydrocarbons a popular drug of abuse among adolescents. Approximately 75% of the population abusing hydrocarbons is younger than age 18 years, half of whom reported first use prior to age 13 years.1,2 Though inhalant abuse rarely continues into adulthood, 0.1% of individuals between the ages of 18 and 30 years report having an inhalant-use disorder.
Hydrocarbons and their halogenated derivatives are lipophilic compounds that are rapidly absorbed after inhalation and rapidly distributed to CNS and cardiac tissue. The brain concentration of 1,1-DFE likely peaks higher than concentrations in other organs and is cleared more rapidly.3 Hydrocarbons produce CNS depression secondary to multiple mechanisms, including gamma-aminobutyric acid agonism, dopamine modulation, and N-methyl-D-aspartate-receptor antagonism.4,5
What causes skin lesions on the abdomen and arms?
The lesions on the patient’s abdomen and extremities were consistent with frostbite. The liquefied compressed gas in computer-cleaning and related products is housed in a pressurized canister. The pressure is released when the spray nozzle is depressed; this causes the liquid to rapidly expand to a gas as it is released, resulting in a quick decrease in the temperature of the metal canister. This process, referred to as adiabatic cooling, demonstrates the first law of thermodynamics. The cold temperature of both the liquid and the canister can cause frostbite in the digits and other parts of the body with which the canister or liquid comes into contact.6
Why did the patient have syncope?
Halogenated hydrocarbons inhibit the cardiac delayed rectifier potassium channels involved in the repolarization of cardiac myocytes, causing a delay in repolarization that is manifested as prolongation of the QT interval on an electrocardiogram. This condition places patients at an increased risk of developing torsades de pointes (TdP).7 In most cases, TdP is self-terminating; however, if TdP persists, degeneration to ventricular fibrillation will result. Deaths caused in this fashion have been referred to as “sudden sniffing death syndrome,” and account for half of all hydrocarbon-related deaths.6,8 In addition to the cardiac effects, hydrocarbons are simple asphyxiants that act by displacing oxygen from inspired air, which also contributes to syncope.
It is important to note that epinephrine and other catecholamines increase the risk for dysrhythmias such as TdP in the setting of hydrocarbon abuse.9 For this reason, epinephrine should be used with caution in the setting of a hydrocarbon-induced arrhythmia. Beta-adrenergic antagonists such as esmolol and propranolol are preferable because they reduce the incidence of ectopia that may trigger TdP.10
What is the significance of the masses noted on the examination and radiograph?
Fluorosis is associated with abnormalities of skeletal and dental tissue. Skeletal fluorosis causes osteosclerosis of the axial skeleton, periosteal new bone formation, ligamentous and tendinous ossification, and osteophyte formation. Dental fluorosis causes a yellow/brown discoloration of the teeth with horizontal streaking (mottling), pitting, and chipping.11 Fluorosis is well-described in regions where water fluoride concentrations are high due to industrial exposure; from consumption of fluorinated wine or chronic overconsumption of tea (especially green or black tea); or from fluoridated toothpaste.12-14 More recently, fluorosis has been described in patients treated for an extended duration of time with voriconazole, a fluorinated antifungal agent.15 Unlike other hydrocarbon products, fluorinated hydrocarbons such as 1,1-DFE can significantly increase systemic fluoride concentrations with excessive use. Rapid skeletal fluorosis is not well described, but has been reported after chronic abuse of fluorinated hydrocarbons.16
How is fluorosis diagnosed and managed?
The lack of rapid laboratory testing available for serum, urine, and bone fluoride concentrations makes the initial diagnosis of fluorosis a clinical one. Imaging studies are generally highly suggestive of fluorosis and can be used to support the diagnosis. A dual energy X-ray absorptiometry scan of the spine, hip, femur, and distal portions of the radii can reveal elevated T-scores consistent with osteosclerosis.14 These findings, in conjunction with bone or joint pain, reduced range of motion, or kyphosis, should prompt clinicians to conduct further testing—even without a confirmed fluoride source. A serum fluoride (reference range, 0.2-3.2 mg/L) and 24-hour urine fluoride (reference range, 0.2-3.2 mg/dL) and creatinine evaluation can be used to diagnose fluorosis. However, a bone biopsy with quantitative bone ash fluoride analysis remains the gold standard for the diagnosis of skeletal fluorosis.16 Laboratory evaluation should also include an assessment of electrolytes, specifically calcium, 25-hydroxyvitamin D, and alkaline phosphatase. The differential diagnosis should include hemoglobinopathies, renal osteodystrophy, Paget disease, hypothyroidism, and skeletal metastases.16
Treatment of fluorosis is largely symptomatic and supportive, with identification and discontinuation of the fluoride source. Patients should be referred to an orthopedist for evaluation and management as needed. Evaluation by an endocrinologist should also be considered because patients may have chronic vitamin D and calcium deficiencies as a result of systemic fluorosis.
Case Conclusion
The patient’s laboratory assessment was notable for the following: alkaline phosphatase, 624 U/L (reference range, 44-147 IU/L); vitamin D, 10 ng/mL (reference range, 20-40 ng/mL); serum fluoride, 0.3 mg/L (reference range, 0.2-3.2 mg/L); urine fluoride, 52 mg/dL (0.2-3.2 mg/dL); and urine creatinine, 1 g/L (reference range, 0.3-3 g/L). Imaging studies noted periosteal bone formation on the lateral epicondyle of the distal right humerus, as well as similar osseous abnormalities in other locations. A bone biopsy was scheduled. The patient was treated with oral vitamin D and educated about the importance of discontinuing the huffing of all hydrocarbons.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
1. Williams JF, Storck M; American Academy of Pediatrics Committee on Substance Abuse; American Academy of Pediatrics Committee on Native American Child Health. Inhalant abuse. Pediatrics. 2007;119(5):1009-1017.
2. Wu LT, Pilowsky DJ, Schlenger WE. Inhalant abuse and dependence among adolescents in the United States. J Am Acad Child Adolesc Psychiatry. 2004;43(10):1206-1214.
3. Avella J, Kunaparaju N, Kumar S, Lehrer M, Zito SW, Barletta M. Uptake and distribution of the abused inhalant 1,1-difluoroethane in the rat. J Anal Toxicol. 2010;34(7):381-388.
4. Tormoehlen LM, Tekulve KJ, Nañagas KA. Hydrocarbon toxicity: A review. Clin Toxicol (Phila). 2014;52(5):479-489.
5. Duncan JR, Lawrence AJ. Conventional concepts and new perspectives for understanding the addictive properties of inhalants. J Pharmacol Sci. 2013;122(4):237-243.
6. Sakai K, Maruyama-Maebashi K, Takatsu A, et al. Sudden death involving inhalation of 1,1-difluoroethane (HFC-152a) with spray cleaner: three case reports. Forensic Sci Int. 2011;206(1-3):e58-e61.
7. Himmel HM. Mechanisms involved in cardiac sensitization by volatile anesthetics: general applicability to halogenated hydrocarbons? Crit Rev Toxicol. 2008;38(9):773-803.
8. Avella J, Wilson JC, Lehrer M. Fatal cardiac arrhythmia after repeated exposure to 1,1-difluoroethane (DFE). Am J Forensic Med Pathol. 2006;27(1):58-60.
9. Nelson LS. Toxicologic myocardial sensitization. J Toxicol Clin Toxicol. 2002;40(7):867-879.
10. Mortiz F, de La Chapelle A, Bauer F, Leroy JP, Goullé JP, Bonmarchand G. Esmolol in the treatment of severe arrhythmia after acute trichloroethylene poisoning. Intensive Care Med. 2000;26(2):256.
11. Majumdar KK. Health impact of supplying safe drinking water containing fluoride below permissible level on flourosis patients in a fluoride-endemic rural area of West Bengal. Indian J Public Health. 2011;55(4):303-308.
12. Kakumanu N, Rao SD. Images in clinical medicine. Skeletal fluorosis due to excessive tea drinking. N Engl J Med 2013;368(12):1140.
13. Soriano M, Manchón F. Radiological aspects of a new type of bone fluorosis, periostitis deformans. Radiology 1966;87(6):1089-1094.
14. Tamer MN, Kale Köroğlu B, Arslan C, et al. Osteosclerosis due to endemic fluorosis. Sci Total Environ. 2007;373(1):43-48.
15. Bucknor MD, Gross AJ, Link TM. Voriconazole-induced periostitis in two post-transplant patients. J Radiol Case Rep. 2013;7(8):10-17.
16. Cohen E, Hsu RY, Evangelista P, Aaron R, Rubin LE. Rapid-onset diffuse skeletal fluorosis from inhalant abuse: a case report. JBJS Case Connector. 2014;4(4):e108.
Case Studies In Toxicology: Withdrawal: Another Danger of Diversion
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?

Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?
Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?
Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.

Case Studies in Toxicology: The Perilous Pursuit of Perfection
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?

The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.

Case Studies in Toxicology: One Last Kick—Transverse Myelitis After an Overdose of Heroin via Insufflation

Case
A 17-year-old adolescent girl with a history of depression and opioid dependence, for which she was taking buprenorphine until 2 weeks earlier, presented to the ED via emergency medical services (EMS) after her father found her lying on the couch unresponsive and with shallow respirations. Naloxone was administered by EMS and her mental status improved.
At presentation, the patient admitted to insufflation of an unknown amount of heroin and ingestion of 2 mg of alprazolam earlier in the day. She denied any past or current use of intravenous (IV) drugs. During monitoring, she began to complain of numbness in her legs and an inability to urinate. Examination revealed paralysis and decreased sensation of her bilateral lower extremities to the midthigh, with decreased rectal tone. Because of the patient’s history of drug use and temporal association with the heroin overdose, both neurosurgery and toxicology services were consulted.
What can cause lower extremity paralysis in a drug user?
The differential diagnosis for the patient at this point included toxin-induced myelopathy, Guillain-Barré syndrome, hypokalemic periodic paralysis, spinal compression, epidural abscess, cerebrovascular accident, spinal lesion, and spinal artery dissection or infarction.
Although Guillain-Barré syndrome presents with ascending paralysis, there is usually an antecedent respiratory or gastrointestinal infection. While epidural abscess with spinal compression is associated with IV drug use and can present similarly, the patient in this case denied IV use. In the absence of any risk factors, cerebrovascular accident and spinal artery dissection were also unlikely.
Case Continuation
A bladder catheter was placed due to the patient’s inability to urinate, and approximately 1 L of urine output was retrieved. Immediate magnetic resonance imaging (MRI) demonstrated increased T2 signal intensity and expansion of the distal thoracic cord and conus without mass lesion, consistent with transverse myelitis (TM).
What is transverse myelitis and why does it occur?
Transverse myelitis is an inflammatory demyelinating disorder that focally affects the spinal cord, resulting in a specific pattern of motor, sensory, and autonomic dysfunction.1 Signs and symptoms include paresthesia, paralysis of the extremities, and loss of bladder and bowel control. The level of the spinal cord affected determines the clinical effects. Demyelination typically occurs at the thoracic segment, producing findings in the legs, as well as bladder and bowel dysfunction.
The exact cause of TM is unknown, but the inflammation may result from a viral complication or an abnormal immune response. Infectious viral agents suspected of causing TM include varicella zoster, herpes simplex, cytomegalovirus, Epstein-Barr, influenza, human immunodeficiency virus, hepatitis A, and rubella. It has also been postulated that an autoimmune reaction is responsible for the condition.
In some individuals, TM represents the first manifestation of an underlying demyelinating disorder such as multiple sclerosis or neuromyelitis optica. A diagnosis of TM is made through patient history, physical examination, and characteristic findings on neuroimaging, specifically MRI.
Heroin use has long been associated with the development of TM, and is usually associated with IV administration of the drug after a period of abstinence.2 This association strengthens the basis for an immunologic etiology—an initial sensitization and subsequent reexposure causing the effects of TM. There have also been cases of TM coexisting with rhabdomyolysis due to the patient being found in a contorted position.3 Another theory of the etiology of heroin-associated TM is a reaction to a possible adulterant or contaminant in the heroin.4
What is the treatment and prognosis of transverse myelitis?
Since there is no cure for TM, treatment is directed at reducing inflammation in the spinal cord. Initial therapy generally includes corticosteroids. In patients with a minimal response to corticosteroids, plasma exchange can be attempted. There are also limited data to suggest a beneficial role for the use of IV immunoglobulin.5 In addition to treatment, general supportive care must also be optimized, such as the use of prophylaxis for thrombophlebitis due to immobility and physical therapy, if possible.
The prognosis of patients with TM is variable, and up to two thirds of patients will have moderate-to-severe residual neurological disability.6 Recovery is slow, with most patients beginning to show improvement within the first 2 to 12 weeks from treatment and supportive care. The recovery process can continue for 2 years. However, if no improvement is made within the first 3 to 6 months, recovery is unlikely.7 Cases of heroin-associated TM may have a more favorable prognosis.8
A majority of individuals will only experience this clinical entity once, but there are rare causes of recurrent or relapsing TM.7 In these situations, a search for underlying demyelinating diseases should be performed.
Case Conclusion
The patient was immediately started on IV corticosteroids, but as there was no improvement after 5 days, plasmapheresis was performed. She received 5 cycles of plasmapheresis and a 5-day course of IV immunoglobulin but still without any improvement. A repeat MRI of the thoracic spine was performed and raised the possibility of cord infarct, but infectious or inflammatory myelitis remained within differential consideration. The patient continued to make minimal improvement with physical therapy and, after a 3-week hospital course, she was transferred to inpatient rehabilitation for further care. Over the next 2 months, the loss of sensation and motor ability of her legs did not improve, but she did regain control of her bowels and bladder.
Dr Regina is a medical toxicology fellow in the department of emergency medicine at North Shore Long Island Jewish Health System, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Pandit L. Transverse myelitis spectrum disorders. Neurol India. 2009;57(2):126-133.
- Richter RW, Rosenberg RN. Transverse myelitis associated with heroin addiction. JAMA. 1968;206(6):1255-1257.
- Sahni V, Garg D, Garg S, Agarwal SK, Singh NP. Unusual complications of heroin abuse: transverse myelitis, rhabdomyolysis, compartment syndrome, and ARF. Clin Toxicol (Phila). 2008;46(2):153-155.
- Schein PS, Yessayan L, Mayman CI. Acute transverse myelitis associated with intravenous opium. Neurology. 1971;21(1):101-102.
- Absoud M, Gadian J, Hellier J, et al. Protocol for a multicentre randomiSed controlled TRial of IntraVEnous immunoglobulin versus standard therapy for the treatment of transverse myelitis in adults and children (STRIVE). BMJ Open. 2015;5(5):e008312.
- West TW. Transverse myelitis--a review of the presentation, diagnosis, and initial management. Discov Med. 2013;16(88):167-177.
- Transverse myelitis fact sheet. National Institute of Neurological Disorders and Stroke. http://www.ninds.nih.gov/disorders/transversemyelitis/detail_transversemyelitis.htm. Updated June 24, 2015. Accessed September 2, 2015.
- McGuire JL, Beslow LA, Finkel RS, Zimmerman RA, Henretig FM. A teenager with focal weakness. Pediatr Emerg Care. 2008;24(12):875-879.
Case
A 17-year-old adolescent girl with a history of depression and opioid dependence, for which she was taking buprenorphine until 2 weeks earlier, presented to the ED via emergency medical services (EMS) after her father found her lying on the couch unresponsive and with shallow respirations. Naloxone was administered by EMS and her mental status improved.
At presentation, the patient admitted to insufflation of an unknown amount of heroin and ingestion of 2 mg of alprazolam earlier in the day. She denied any past or current use of intravenous (IV) drugs. During monitoring, she began to complain of numbness in her legs and an inability to urinate. Examination revealed paralysis and decreased sensation of her bilateral lower extremities to the midthigh, with decreased rectal tone. Because of the patient’s history of drug use and temporal association with the heroin overdose, both neurosurgery and toxicology services were consulted.
What can cause lower extremity paralysis in a drug user?
The differential diagnosis for the patient at this point included toxin-induced myelopathy, Guillain-Barré syndrome, hypokalemic periodic paralysis, spinal compression, epidural abscess, cerebrovascular accident, spinal lesion, and spinal artery dissection or infarction.
Although Guillain-Barré syndrome presents with ascending paralysis, there is usually an antecedent respiratory or gastrointestinal infection. While epidural abscess with spinal compression is associated with IV drug use and can present similarly, the patient in this case denied IV use. In the absence of any risk factors, cerebrovascular accident and spinal artery dissection were also unlikely.
Case Continuation
A bladder catheter was placed due to the patient’s inability to urinate, and approximately 1 L of urine output was retrieved. Immediate magnetic resonance imaging (MRI) demonstrated increased T2 signal intensity and expansion of the distal thoracic cord and conus without mass lesion, consistent with transverse myelitis (TM).
What is transverse myelitis and why does it occur?
Transverse myelitis is an inflammatory demyelinating disorder that focally affects the spinal cord, resulting in a specific pattern of motor, sensory, and autonomic dysfunction.1 Signs and symptoms include paresthesia, paralysis of the extremities, and loss of bladder and bowel control. The level of the spinal cord affected determines the clinical effects. Demyelination typically occurs at the thoracic segment, producing findings in the legs, as well as bladder and bowel dysfunction.
The exact cause of TM is unknown, but the inflammation may result from a viral complication or an abnormal immune response. Infectious viral agents suspected of causing TM include varicella zoster, herpes simplex, cytomegalovirus, Epstein-Barr, influenza, human immunodeficiency virus, hepatitis A, and rubella. It has also been postulated that an autoimmune reaction is responsible for the condition.
In some individuals, TM represents the first manifestation of an underlying demyelinating disorder such as multiple sclerosis or neuromyelitis optica. A diagnosis of TM is made through patient history, physical examination, and characteristic findings on neuroimaging, specifically MRI.
Heroin use has long been associated with the development of TM, and is usually associated with IV administration of the drug after a period of abstinence.2 This association strengthens the basis for an immunologic etiology—an initial sensitization and subsequent reexposure causing the effects of TM. There have also been cases of TM coexisting with rhabdomyolysis due to the patient being found in a contorted position.3 Another theory of the etiology of heroin-associated TM is a reaction to a possible adulterant or contaminant in the heroin.4
What is the treatment and prognosis of transverse myelitis?
Since there is no cure for TM, treatment is directed at reducing inflammation in the spinal cord. Initial therapy generally includes corticosteroids. In patients with a minimal response to corticosteroids, plasma exchange can be attempted. There are also limited data to suggest a beneficial role for the use of IV immunoglobulin.5 In addition to treatment, general supportive care must also be optimized, such as the use of prophylaxis for thrombophlebitis due to immobility and physical therapy, if possible.
The prognosis of patients with TM is variable, and up to two thirds of patients will have moderate-to-severe residual neurological disability.6 Recovery is slow, with most patients beginning to show improvement within the first 2 to 12 weeks from treatment and supportive care. The recovery process can continue for 2 years. However, if no improvement is made within the first 3 to 6 months, recovery is unlikely.7 Cases of heroin-associated TM may have a more favorable prognosis.8
A majority of individuals will only experience this clinical entity once, but there are rare causes of recurrent or relapsing TM.7 In these situations, a search for underlying demyelinating diseases should be performed.
Case Conclusion
The patient was immediately started on IV corticosteroids, but as there was no improvement after 5 days, plasmapheresis was performed. She received 5 cycles of plasmapheresis and a 5-day course of IV immunoglobulin but still without any improvement. A repeat MRI of the thoracic spine was performed and raised the possibility of cord infarct, but infectious or inflammatory myelitis remained within differential consideration. The patient continued to make minimal improvement with physical therapy and, after a 3-week hospital course, she was transferred to inpatient rehabilitation for further care. Over the next 2 months, the loss of sensation and motor ability of her legs did not improve, but she did regain control of her bowels and bladder.
Dr Regina is a medical toxicology fellow in the department of emergency medicine at North Shore Long Island Jewish Health System, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 17-year-old adolescent girl with a history of depression and opioid dependence, for which she was taking buprenorphine until 2 weeks earlier, presented to the ED via emergency medical services (EMS) after her father found her lying on the couch unresponsive and with shallow respirations. Naloxone was administered by EMS and her mental status improved.
At presentation, the patient admitted to insufflation of an unknown amount of heroin and ingestion of 2 mg of alprazolam earlier in the day. She denied any past or current use of intravenous (IV) drugs. During monitoring, she began to complain of numbness in her legs and an inability to urinate. Examination revealed paralysis and decreased sensation of her bilateral lower extremities to the midthigh, with decreased rectal tone. Because of the patient’s history of drug use and temporal association with the heroin overdose, both neurosurgery and toxicology services were consulted.
What can cause lower extremity paralysis in a drug user?
The differential diagnosis for the patient at this point included toxin-induced myelopathy, Guillain-Barré syndrome, hypokalemic periodic paralysis, spinal compression, epidural abscess, cerebrovascular accident, spinal lesion, and spinal artery dissection or infarction.
Although Guillain-Barré syndrome presents with ascending paralysis, there is usually an antecedent respiratory or gastrointestinal infection. While epidural abscess with spinal compression is associated with IV drug use and can present similarly, the patient in this case denied IV use. In the absence of any risk factors, cerebrovascular accident and spinal artery dissection were also unlikely.
Case Continuation
A bladder catheter was placed due to the patient’s inability to urinate, and approximately 1 L of urine output was retrieved. Immediate magnetic resonance imaging (MRI) demonstrated increased T2 signal intensity and expansion of the distal thoracic cord and conus without mass lesion, consistent with transverse myelitis (TM).
What is transverse myelitis and why does it occur?
Transverse myelitis is an inflammatory demyelinating disorder that focally affects the spinal cord, resulting in a specific pattern of motor, sensory, and autonomic dysfunction.1 Signs and symptoms include paresthesia, paralysis of the extremities, and loss of bladder and bowel control. The level of the spinal cord affected determines the clinical effects. Demyelination typically occurs at the thoracic segment, producing findings in the legs, as well as bladder and bowel dysfunction.
The exact cause of TM is unknown, but the inflammation may result from a viral complication or an abnormal immune response. Infectious viral agents suspected of causing TM include varicella zoster, herpes simplex, cytomegalovirus, Epstein-Barr, influenza, human immunodeficiency virus, hepatitis A, and rubella. It has also been postulated that an autoimmune reaction is responsible for the condition.
In some individuals, TM represents the first manifestation of an underlying demyelinating disorder such as multiple sclerosis or neuromyelitis optica. A diagnosis of TM is made through patient history, physical examination, and characteristic findings on neuroimaging, specifically MRI.
Heroin use has long been associated with the development of TM, and is usually associated with IV administration of the drug after a period of abstinence.2 This association strengthens the basis for an immunologic etiology—an initial sensitization and subsequent reexposure causing the effects of TM. There have also been cases of TM coexisting with rhabdomyolysis due to the patient being found in a contorted position.3 Another theory of the etiology of heroin-associated TM is a reaction to a possible adulterant or contaminant in the heroin.4
What is the treatment and prognosis of transverse myelitis?
Since there is no cure for TM, treatment is directed at reducing inflammation in the spinal cord. Initial therapy generally includes corticosteroids. In patients with a minimal response to corticosteroids, plasma exchange can be attempted. There are also limited data to suggest a beneficial role for the use of IV immunoglobulin.5 In addition to treatment, general supportive care must also be optimized, such as the use of prophylaxis for thrombophlebitis due to immobility and physical therapy, if possible.
The prognosis of patients with TM is variable, and up to two thirds of patients will have moderate-to-severe residual neurological disability.6 Recovery is slow, with most patients beginning to show improvement within the first 2 to 12 weeks from treatment and supportive care. The recovery process can continue for 2 years. However, if no improvement is made within the first 3 to 6 months, recovery is unlikely.7 Cases of heroin-associated TM may have a more favorable prognosis.8
A majority of individuals will only experience this clinical entity once, but there are rare causes of recurrent or relapsing TM.7 In these situations, a search for underlying demyelinating diseases should be performed.
Case Conclusion
The patient was immediately started on IV corticosteroids, but as there was no improvement after 5 days, plasmapheresis was performed. She received 5 cycles of plasmapheresis and a 5-day course of IV immunoglobulin but still without any improvement. A repeat MRI of the thoracic spine was performed and raised the possibility of cord infarct, but infectious or inflammatory myelitis remained within differential consideration. The patient continued to make minimal improvement with physical therapy and, after a 3-week hospital course, she was transferred to inpatient rehabilitation for further care. Over the next 2 months, the loss of sensation and motor ability of her legs did not improve, but she did regain control of her bowels and bladder.
Dr Regina is a medical toxicology fellow in the department of emergency medicine at North Shore Long Island Jewish Health System, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Pandit L. Transverse myelitis spectrum disorders. Neurol India. 2009;57(2):126-133.
- Richter RW, Rosenberg RN. Transverse myelitis associated with heroin addiction. JAMA. 1968;206(6):1255-1257.
- Sahni V, Garg D, Garg S, Agarwal SK, Singh NP. Unusual complications of heroin abuse: transverse myelitis, rhabdomyolysis, compartment syndrome, and ARF. Clin Toxicol (Phila). 2008;46(2):153-155.
- Schein PS, Yessayan L, Mayman CI. Acute transverse myelitis associated with intravenous opium. Neurology. 1971;21(1):101-102.
- Absoud M, Gadian J, Hellier J, et al. Protocol for a multicentre randomiSed controlled TRial of IntraVEnous immunoglobulin versus standard therapy for the treatment of transverse myelitis in adults and children (STRIVE). BMJ Open. 2015;5(5):e008312.
- West TW. Transverse myelitis--a review of the presentation, diagnosis, and initial management. Discov Med. 2013;16(88):167-177.
- Transverse myelitis fact sheet. National Institute of Neurological Disorders and Stroke. http://www.ninds.nih.gov/disorders/transversemyelitis/detail_transversemyelitis.htm. Updated June 24, 2015. Accessed September 2, 2015.
- McGuire JL, Beslow LA, Finkel RS, Zimmerman RA, Henretig FM. A teenager with focal weakness. Pediatr Emerg Care. 2008;24(12):875-879.
- Pandit L. Transverse myelitis spectrum disorders. Neurol India. 2009;57(2):126-133.
- Richter RW, Rosenberg RN. Transverse myelitis associated with heroin addiction. JAMA. 1968;206(6):1255-1257.
- Sahni V, Garg D, Garg S, Agarwal SK, Singh NP. Unusual complications of heroin abuse: transverse myelitis, rhabdomyolysis, compartment syndrome, and ARF. Clin Toxicol (Phila). 2008;46(2):153-155.
- Schein PS, Yessayan L, Mayman CI. Acute transverse myelitis associated with intravenous opium. Neurology. 1971;21(1):101-102.
- Absoud M, Gadian J, Hellier J, et al. Protocol for a multicentre randomiSed controlled TRial of IntraVEnous immunoglobulin versus standard therapy for the treatment of transverse myelitis in adults and children (STRIVE). BMJ Open. 2015;5(5):e008312.
- West TW. Transverse myelitis--a review of the presentation, diagnosis, and initial management. Discov Med. 2013;16(88):167-177.
- Transverse myelitis fact sheet. National Institute of Neurological Disorders and Stroke. http://www.ninds.nih.gov/disorders/transversemyelitis/detail_transversemyelitis.htm. Updated June 24, 2015. Accessed September 2, 2015.
- McGuire JL, Beslow LA, Finkel RS, Zimmerman RA, Henretig FM. A teenager with focal weakness. Pediatr Emerg Care. 2008;24(12):875-879.

Case Studies in Toxicology: Managing Missed Methadone
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.

The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.
The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
A 53-year-old woman presented to the ED after experiencing a fall. Her medical history was significant for chronic obstructive pulmonary disease, hepatitis, and a remote history of intravenous drug use, for which she had been maintained on methadone for the past 20 years. She reported that she had suffered several “fainting episodes” over the past month, and the morning prior to arrival, had sustained what she thought was a mechanical fall outside of the methadone program she attended. She complained of tenderness on her head but denied any other injuries.
The methadone program had referred the patient to the ED for evaluation, noting to the ED staff that her daily methadone dose of 185 mg had not been dispensed prior to transfer. During evaluation, the patient requested that the emergency physician (EP) provide the methadone dose since the clinic would close prior to her discharge from the ED.
How can requests for methadone be managed in the ED?
Methadone is a long-acting oral opioid that is used for both opioid replacement therapy and pain management. When used to reduce craving in opioid-dependent patients, methadone is administered daily through federally sanctioned methadone maintenance treatment (MMT) programs. Patients who consistently adhere to the required guidelines are given “take home” doses. When used for pain management, methadone is typically administered several times daily and may be prescribed by any provider with an appropriate DEA registration.
When given for MMT, methadone saturates the µ-opioid receptors and hinders their binding and agonism by other opioids such as heroin or oxycodone. Patients in MMT programs are started on a low initial dose and slowly titrated upward as tolerance to the adverse effects (eg, sedation) develop.
How are symptomatic patients with methadone withdrawal treated?
Most methadone programs have limited hours and require that patients who miss a dose wait until the following day to return to the program. This is typically without medical consequence because the high dose dispensed by these programs maintains a therapeutic blood concentration for far longer than the expected delay. Although the half-life of methadone exhibits wide interindividual variability, it generally ranges from 12 hours to more than 40 hours.1 Regardless, patients may feel anxious about potential opioid withdrawal, and this often leads them to access the ED for a missed dose.
The neuropsychiatric symptoms attending withdrawal may precede the objective signs of opioid withdrawal. Patients with objective signs of opioid withdrawal (eg, piloerection, vomiting, diarrhea, dilated pupils) may be sufficiently treated with supportive care alone, using antiemetics, hydration, and sometimes clonidine.
Administration of substitute opioids is problematic due to the patient’s underlying tolerance necessitating careful dose titration. Therefore, direct replacement of methadone in the ED remains controversial, and some EDs have strict policies prohibiting the administration of methadone to patients who have missed an MMT dose. Such policies, which are intended to discourage patients from using the ED as a convenience, may be appropriate given the generally benign—though uncomfortable—course of opioid withdrawal due to abstinence.
Other EDs provide replacement methadone for asymptomatic, treat-and-release patients confirmed to be enrolled in an MMT program when the time to the next dose is likely to be 24 hours or greater from the missed dose. Typically, a dose of no more than 10 mg orally or 10 mg intramuscularly (IM) is recommended, and patients should be advised that they will be receiving only a low dose to sufficient to prevent withdrawal—one that may not have the equivalent effects of the outpatient dose.
Whenever possible, a patient’s MMT program should be contacted and informed of the ED visit. For patients who display objective signs of withdrawal and who cannot be confirmed or who do not participate in an MMT program, 10 mg of methadone IM will prevent uncertainty of drug absorption in the setting of nausea or vomiting. All patients receiving oral methadone should be observed for 1 hour, and those receiving IM methadone should be observed for at least 90 minutes to assess for unexpected sedation.2
Patients encountering circumstances that prevent opioid access (eg, incarceration) and who are not in withdrawal but have gone without opioids for more than 5 days may have a loss of tolerance to their usual doses—whether the medication was obtained through an MMT program or illicitly. Harm-reduction strategies aimed at educating patients on the potential vulnerability to their familiar dosing regimens are warranted to avert inadvertent overdoses in chronic opioid users who are likely to resume illicit opoiod use.
Does this patient need syncope evaluation?
Further complicating the decision regarding ED dispensing of methadone are the effects of the drug on myocardial repolarization. Methadone affects conduction across the hERG potassium rectifier current and can prolong the QTc interval on the surface electrocardiogram (ECG), predisposing a patient to torsade de pointes (TdP). Although there is controversy regarding the role of ECG screening during the enrollment of patients in methadone maintenance clinics, doses above 60 mg, underlying myocardial disease, female sex, and electrolyte disturbances may increase the risk of QT prolongation and TdP.3
Whether there is value in obtaining a screening ECG in a patient receiving an initial dose of methadone in the ED is unclear, and this practice is controversial even among methadone clinics. However, some of the excess death in patients taking methadone may be explained by the dysrhythmogenic potential of methadone.4 An ECG therefore may elucidate a correctable cause in methadone patients presenting with syncope.
Administering methadone to patients with documented QT prolongation must weigh the risk of methadone’s conduction effects against the substantial risks of illicit opioid self-administration. For some patients at-risk for TdP, it may be preferable to use buprenorphine if possible, since it does not carry the same cardiac effects as methadone.1,5 Such therapy requires referral to a physician licensed to prescribe this medication.
How should admitted patients be managed?
While administration of methadone for withdrawal or maintenance therapy in the ED is acceptable, outpatient prescribing of methadone for these reasons is not legal, and only federally regulated clinics may engage in this practice. Hospitalized patients who are enrolled in an MMT program should have their daily methadone dose confirmed and continued—as long as the patient has not lost tolerance. Patients not participating in an MMT program can receive up to 3 days of methadone in the hospital, even if the practitioner is not registered to provide methadone.6 For these patients, it is recommended that the physician order a low dose of methadone and also consult with an addiction specialist to determine whether the patient should continue on MMT maintenance or undergo detoxification.
It is important to note that methadone may be prescribed for pain, but its use in the ED for this purpose is strongly discouraged, especially in patients who have never received methadone previously. For admitted patients requiring such potent opioid analgesia, consultation with a pain service or, when indicated, a palliative care/hospice specialist is warranted as the dosing intervals are different in each setting, and the risk of respiratory depression is high.
Case Conclusion
As requested by the MMT clinic, the patient was administered methadone 185 mg orally in the ED, though a dose of 10 mg would have been sufficient to prevent withdrawal. Unfortunately, the EP did not appreciate the relationship of the markedly prolonged QTc and the methadone, which should have prompted a dose reduction.
Evaluation of the patient’s electrolyte levels, which included magnesium and potassium, were normal. An ECG was repeated 24 hours later and revealed a persistent, but improved, QT interval at 505 ms. The remainder of the syncope workup was negative. Because the patient had no additional symptoms or events during her stay, she was discharged. At discharge, the EP followed up with the MMT clinic to discuss lowering the patient’s daily methadone dose, as well as close cardiology follow-up.
Dr Rao is the chief of the division of medical toxicology at New York Presbyterian Hospital/Weill Cornell Medical Center, New York. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.
- Chou R, Weimer MB, Dana T. Methadone overdose and cardiac arrhythmia potential: findings from a review of the evidence for an American Pain Society and College on Problems of Drug Dependence clinical practice guideline. J Pain. 2014;15(4):338-365.
- National Highway Traffic Safety Administration Web site. Methadone. http://www.nhtsa.gov/people/injury/research/job185drugs/methadone.htm. Accessed August 3, 2015.
- Martin JA, Campbell A, Killip T, et al; Substance Abuse and Mental Health Services Administration. QT interval screening in methadone maintenance treatment: report of a SAMHSA expert panel. J Addict Dis. 2011;30(4):283-306. Erratum in: J Addict Dis. 2012;31(1):91.
- Ray WA, Chung CP, Murray KT, Cooper WO, Hall K, Stein CM. Out-of-hospital mortality among patients receiving methadone for noncancer pain. JAMA Intern Med. 2015;175(3):420-427.
- Davis MP. Twelve reasons for considering buprenorphine as a frontline analgesic in the management of pain. J Support Oncol. 2012;10(6):209-219.
- US Government Printing Office. Federal Digital System. Administering or dispensing of narcotic drugs. Code of Federal Regulations. Title 21 CFR §1306.07. http://www.gpo.gov/fdsys/pkg/CFR-1998-title21-vol9/pdf/CFR-1998-title21-vol9-sec1306-07.pdf. Accessed August 4, 2015.

Mind the Gap
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications was brought to the ED with a two-week history of intermittent fever and poor oral intake. His current medications included sodium bromide (185 mg bid, orally) for his seizure disorder.
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were sodium, 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
WHAT DOES THE ANION GAP REPRESENT?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo and is accomplished by the presence of unmeasured anions (UA; eg, lactate and phosphate) and unmeasured cations (UC; eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus unmeasured anions must equal the sum of the measured plus unmeasured cations.
WHAT CAUSES A LOW OR NEGATIVE ANION GAP?
While most health care providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased concentration of unmeasured anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than that of the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of serum chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride.
However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
Continue for causes of the falsely elevated chloride >>
WHAT CAUSES THE FALSELY ELEVATED CHLORIDE?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride.
Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration; rather, it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
HOW ARE PATIENTS EXPOSED TO BROMIDE SALTS?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurologic and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide, as well as the brominated vegetable oils found in some soft drinks.5
Continue for how bromism is treated >>
HOW IS BROMISM TREATED?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10 to 12 days), but in patients with normal renal function, it is possible to reduce this half-life to approximately three days with hydration and diuresis with sodium chloride.3
Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
CASE CONCLUSION
The patient was admitted for observation and treated with IV sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide (185 mg bid, orally) since his seizures were refractory to other anticonvulsant medications.
REFERENCES
1. Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
2. Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
3. Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, et al. A negative anion gap as a clue to diagnose bromide intoxication. Nephron. 1995;69(3):311-313.
4. Yamamoto K, Kobayashi H, Kobayashi T, Murakami S. False hyperchloremia in bromism. J Anesth. 1991;5(1):88-91.
5. Ng YY, Lin WL, Chen TW. Spurious hyperchloremia and decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications was brought to the ED with a two-week history of intermittent fever and poor oral intake. His current medications included sodium bromide (185 mg bid, orally) for his seizure disorder.
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were sodium, 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
WHAT DOES THE ANION GAP REPRESENT?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo and is accomplished by the presence of unmeasured anions (UA; eg, lactate and phosphate) and unmeasured cations (UC; eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus unmeasured anions must equal the sum of the measured plus unmeasured cations.
WHAT CAUSES A LOW OR NEGATIVE ANION GAP?
While most health care providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased concentration of unmeasured anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than that of the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of serum chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride.
However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
Continue for causes of the falsely elevated chloride >>
WHAT CAUSES THE FALSELY ELEVATED CHLORIDE?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride.
Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration; rather, it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
HOW ARE PATIENTS EXPOSED TO BROMIDE SALTS?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurologic and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide, as well as the brominated vegetable oils found in some soft drinks.5
Continue for how bromism is treated >>
HOW IS BROMISM TREATED?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10 to 12 days), but in patients with normal renal function, it is possible to reduce this half-life to approximately three days with hydration and diuresis with sodium chloride.3
Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
CASE CONCLUSION
The patient was admitted for observation and treated with IV sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide (185 mg bid, orally) since his seizures were refractory to other anticonvulsant medications.
REFERENCES
1. Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
2. Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
3. Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, et al. A negative anion gap as a clue to diagnose bromide intoxication. Nephron. 1995;69(3):311-313.
4. Yamamoto K, Kobayashi H, Kobayashi T, Murakami S. False hyperchloremia in bromism. J Anesth. 1991;5(1):88-91.
5. Ng YY, Lin WL, Chen TW. Spurious hyperchloremia and decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications was brought to the ED with a two-week history of intermittent fever and poor oral intake. His current medications included sodium bromide (185 mg bid, orally) for his seizure disorder.
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were sodium, 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
WHAT DOES THE ANION GAP REPRESENT?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo and is accomplished by the presence of unmeasured anions (UA; eg, lactate and phosphate) and unmeasured cations (UC; eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus unmeasured anions must equal the sum of the measured plus unmeasured cations.
WHAT CAUSES A LOW OR NEGATIVE ANION GAP?
While most health care providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased concentration of unmeasured anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than that of the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of serum chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride.
However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
Continue for causes of the falsely elevated chloride >>
WHAT CAUSES THE FALSELY ELEVATED CHLORIDE?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride.
Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration; rather, it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
HOW ARE PATIENTS EXPOSED TO BROMIDE SALTS?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurologic and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide, as well as the brominated vegetable oils found in some soft drinks.5
Continue for how bromism is treated >>
HOW IS BROMISM TREATED?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10 to 12 days), but in patients with normal renal function, it is possible to reduce this half-life to approximately three days with hydration and diuresis with sodium chloride.3
Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
CASE CONCLUSION
The patient was admitted for observation and treated with IV sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide (185 mg bid, orally) since his seizures were refractory to other anticonvulsant medications.
REFERENCES
1. Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
2. Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
3. Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, et al. A negative anion gap as a clue to diagnose bromide intoxication. Nephron. 1995;69(3):311-313.
4. Yamamoto K, Kobayashi H, Kobayashi T, Murakami S. False hyperchloremia in bromism. J Anesth. 1991;5(1):88-91.
5. Ng YY, Lin WL, Chen TW. Spurious hyperchloremia and decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.
Case Studies in Toxicology: When Doing More for the Sake of Better Health Goes Wrong
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
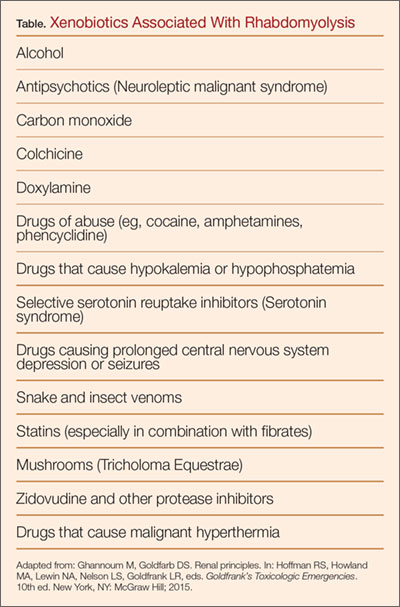
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 62-year-old man with a history of hypercholesterolemia and HIV infection presented to the ED for evaluation of diffuse myalgia and tea-colored urine. His medication history included lopinavir/ritonavir (Kaletra) and simvastatin. A week prior to presentation, the patient’s primary care physician had instructed him to increase his daily dose of simvastatin from 40 mg to 80 mg. The patient stated that he had taken simvastatin 80 mg daily for approximately 5 days and then, 2 days prior to presentation, had independently further increased the dose to 160 mg daily.
In the ED, the patient reported feeling fatigued. His initial vital signs were: blood pressure, 129/86 mm Hg; heart rate, 93 beats/minute; respiratory rate, 17 breaths/minute; and temperature, 98.5˚F. Oxygen saturation was 98% on room air. His physical examination was unremarkable. Initial laboratory testing revealed the following: creatine kinase (CK) 350,000 U/L; blood urea nitrogen, 27 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase (AST), 2,950 U/L; and alanine aminotransferase (ALT), 1,305 U/L.
What can cause tea-colored/cola-colored urine and myalgia?
Numerous medications can result in dark-colored urine. These include antimalarial drugs such as chloroquine and primaquine; antibiotics such as metronidazole or nitrofurantoin; and the muscle relaxant methocarbamol. Myalgia and tea-colored urine are the hallmarks of rhabdomyolysis. Rhabdomyolysis involves the destruction of myocytes, which can occur as a result of a long list of processes, including crush injuries, poor oxygenation or perfusion, hypermetabolic states, and direct (or indirect) toxin-mediated myocyte damage.1 The list of toxic substances that can cause rhabdomyolysis is extensive, and statins are one of the most common drug-induced causes (Table).
Simvastatin is one of seven currently available 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (ie, statins) that are commonly used to treat hypercholesterolemia. Because simvastatin is lipophilic, it can more readily cross cell membranes than nonlipophilic statins such as pravastatin. Simvastatin, therefore, has a propensity to disrupt the cellular integrity of myocytes and hepatocytes.What is the likely cause of this patient’s rhabdomyolysis?
At doses greater than 40 mg daily, simvastatin is associated with myalgia, myositis, and rhabdomyolysis. In December 2011, the US Food and Drug Administration (FDA) released a drug safety announcement recommending the originally approved maximum daily dose of simvastatin 80 mg be limited to patients who have already tolerated that dose for at least 12 months without evidence of muscular injury. The FDA further recommended no new patients be escalated to this dose. According to the FDA, patients taking 80 mg of simvastatin daily are also at increased risk of myopathy.
The metabolism of simvastatin, in addition to increased dosage of the drug, contributes to its potential for adverse effects. Of the seven available statins, only atorvastatin, lovastatin, and simvastatin are metabolized by the cytochrome P450 3A4 (CYP3A4). Lovastatin and simvastatin appear to have the highest potential for drug-drug interactions when coadministered with drugs that inhibit this enzyme (eg, ritonavir).2 The resulting elevation in blood concentration of simvastatin increases the risk of rhabdomyolysis. Other nonlipophilic statins, such as pravastatin, which are mostly eliminated unchanged in the urine and bile, would be preferable for patients taking CYP3A4 inhibitors.
How should patients with rhabdomyolysis be monitored?
Statins interfere with the myocyte’s ability to produce adenosine triphosphate, most likely by depleting coenzyme Q—one of the complexes found in the electron transport chain of the mitochondria. Under conditions of a high-energy requirement, myocytes incapable of producing sufficient energy ultimately fail and lyse, releasing cellular contents such as CK and myoglobin.1 The serum CK activity serves as a marker of muscle injury and should be monitored closely in patients with rhabdomyolysis. Although values above 5,000 U/L has been associated with renal injury,4 in healthy patients with access to hydration, renal injury is relatively uncommon with CK activities less than 50,000 U/L. Even though the prediction of renal failure is difficult, a validated nephrotoxicity prediction instrument using the patient’s age, gender, and initial laboratory data (serum creatinine, calcium, CK, phosphate, and bicarbonate) is available.5
Although the association between rhabdomyolysis and acute renal injury is well established, the mechanism remains unclear. Myoglobin from skeletal myocytes passes through the glomerulus without causing damage and is reabsorbed in the proximal renal tubular cell. Iron is subsequently released from the porphyrin ring and, in large concentrations, exceeds the binding capacity of the tissue ferritin. Because it is a transition metal, the free iron ion participates in oxidant stress reactions causing direct injury to the renal tubular cells.6 Furthermore, myoglobin also combines with renal tubular proteins, a process enhanced by an environment with lower pH, to form casts and cause renal tubular obstruction.
Patients with rhabdomyolysis may also be at risk for aminotransferase elevation, as occurred in the patient presented here. This elevation is most likely due to myocyte injury. In addition, potassium release due to myocyte destruction may cause life-threatening hyperkalemia, and phosphate liberation from these myocytes may cause hypocalcemia. Laboratory monitoring along with an electrocardiogram should be performed as required.
What is the treatment for rhabdomyolysis?
No adequate randomized controlled trials exist to guide the treatment of patients with rhabdomyolysis. As a result, recommendations for management come from retrospective observational studies, animal studies, case reports, and expert opinion.7
Once airway, breathing, and circulation have been addressed, patients with statin-induced rhabdomyolysis should be immediately treated with intravenous (IV) fluids to maintain renal perfusion, which helps to limit acute renal injury. Normal saline appears to be the most recommended fluid type, with a goal of maintaining a urine output of approximately 3 to 5 mL/kg/h.4,7
Some recommendations include the use of a sodium bicarbonate infusion to raise the urine pH, which may help limit the formation of renal casts from myoglobin. The data to support the benefit of sodium bicarbonate, however, is weak.3 A 2013 systematic review indicated that sodium bicarbonate should only be used to treat severe metabolic acidosis in patients with rhabdomyolysis.4
In addition to sodium bicarbonate, the use of diuretics is also discouraged by current recommendations. In patients with refractory electrolyte abnormalities or renal failure, hemodialysis may be required. Before disposition of a patient, his or her medication list should be reconciled to reflect statin discontinuation.
Case Conclusion
The patient received IV normal saline to maintain his urine output at 2 to 3 cc/kg/h. His repeat creatinine was 0.8 mg/dL and remained stable on repeat testing. His CK and AST concentrations trended down during his hospitalization. On hospital day 4, laboratory values were CK, less than 10,000 U/L; AST, 56 U/L; and ALT, 23 U/L. He had normal serum potassium levels and no dysrhythmia on electrocardiogram. His symptoms resolved on hospital day 2, and he was discharged on hospital day 4 with instructions to discontinue simvastatin.
Dr Fernandez is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
- Bench-to-bedside review: Rhabdomyolysis—an overview for clinicians. Crit Care. 2005;9(2):158-169.
- Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52(10):815-831.
- Brown CV, Rhee P, Chan L, Evans K, Demetriades D, Velmahos GC. Preventing renal failure in patients with rhabdomyolysis: do bicarbonate and mannitol make a difference? J Trauma. 2004;56(6):1191-1196.
- Scharman EJ, Troutman WG. Prevention of kidney injury following rhabdomyolysis: a systematic review. Ann Pharmacother. 2013;47(1):90-105.
- McMahon GM, Zeng X, Waikar SS. A risk prediction score for kidney failure or mortality in rhabdomyolysis. JAMA Intern Med. 2013;173(19):1821-1828.
- Visweswaran P, Guntupalli J. Rhabdomyolysis. Crit Care Clin. 1999;15(2):415-428, ix-x.
- Zimmerman JL, Shen MC. Rhabdomyolysis. Chest. 2013;144(3):1058-1065.
Case Studies in Toxicology: Babies and Booze—Pediatric Considerations in the Management of Ethanol Intoxication
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.

Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
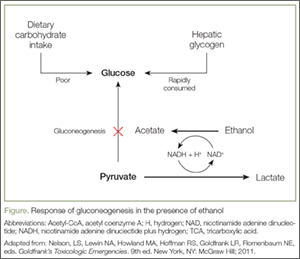
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.
Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
Case
A previously healthy 4-month-old girl was brought into the ED for concerns of alcohol ingestion. Reportedly, the infant’s father reconstituted 4 ounces of powdered formula using what he thought was water from an unmarked bottle in his refrigerator. He later realized that the bottle contained rum, although he still let the child finish the 4 ounces of formula in the hopes that she would vomit—which did not occur.
Upon arrival to the ED, the infant’s vital signs were: blood pressure, 100/61 mm Hg; heart rate, 155 beats/minute; respiratory rate, 36 breaths/minute; and temperature, normal. Oxygen saturation was 98% on room air. A rapid bedside blood glucose test was 89 mg/dL. The infant’s physical examination was unremarkable. She appeared active but hungry, had a strong cry, and had a developmentally appropriate gross neurological examination.
How does ethanol exposure in children typically occur?
Recent reports from the American Association of Poison Control Centers’ National Poison Data System demonstrate that ethanol exposures comprise 1% to 3% of total exposures in children aged ≤5 years.
The most common sources are ethanol-containing beverages, mouthwash, and cologne/perfume.1 Ethanol can also be found as a solvent for certain pediatric liquid medications (eg, ranitidine) or in flavor extracts (eg, vanilla extract, orange extract). Any clear alcohol (eg, vodka, gin, rum) stored in an accessible site, such as a refrigerator, may be mistaken for water. In many reports, a caregiver unintentionally used the alcohol to reconstitute formula; however, intentional provision of alcohol to toddlers, usually as a sedative, is a recurring concern.2
What are the clinical concerns in children with ethanol intoxication?
Under usual conditions, a normal serum glucose concentration is maintained from ingested carbohydrates and via glycogenolysis of hepatic glycogen stores. Such glycogen reserves can sustain normal blood glucose concentrations for several hours in adults but for a shorter period in children. Once glycogen is depleted, as is common after an overnight fast, glucose can be generated through gluconeogenesis.
However, in the presence of ethanol (Figure), the excessive reducing potential (ie, NADH) that results from ethanol metabolism shunts pyruvate away from the gluconeogenic pathway (toward lactate), inhibiting glucose production. Unlike adults, children and infants, who have relatively low glycogen reserves, are at significant risk for hypoglycemia following ethanol exposure. This represents the largest contributor to morbidity and mortality of children with ethanol intoxication.3 Patients with hypoglycemia can have a highly variable clinical presentation including agitation, seizures, focality, or coma.4
Case Continuation
Intravenous (IV) access was obtained, and the patient was placed on a dextrose-containing fluid at 1.5 times the maintenance flow rate. Pertinent laboratory studies revealed a serum glucose level of 90 mg/dL, normal electrolyte panel, and an initial blood alcohol concentration of 337 mg/dL (approximately 30 minutes postingestion).
How do children with ethanol intoxication present?
While there is some variation in clinical effects among nontolerant adults, acute ethanol intoxication with a serum concentration >250 mg/dL is frequently associated with stupor, respiratory depression, and hypotension. A concentration >400 mg/dL may be associated with coma or apnea. Although similar clinical effects are expected in adolescents and children, infants often have counterintuitive clinical findings.
To date, eight cases of significant infant ethanol exposure exist in the literature (age range, 29 days to 9 months; ethanol concentration, 183-524 mg/dL). Respiratory depression was absent in all cases.5-9 In all but two cases, the neurological examination revealed only subtle decreases in interaction or tone. The remaining two children were described as obtunded and flaccid (ethanol levels, 405 mg/dL and 524 mg/dL, respectively) and were intubated for airway protection despite normal respiratory rates.7,10
The incongruence between the clinical findings (both the neurological examination and respiratory effects) and the ethanol concentration is difficult to explain. It may be due to age-related neurological immaturity or a limited ability to perform the required detailed neurological examinations in children. In particular, the relatively preserved level of consciousness, despite an otherwise coma-inducing ethanol concentration, is unique to infants. Accordingly, there should be a low threshold to check ethanol concentrations in infants presenting with apparent life-threatening events, altered mental status, decreased tone, or unexplained hypoglycemia or hypothermia.
What is the estimated time to sobriety in infants?
Ethanol is eliminated via a hepatic enzymatic oxidation pathway that becomes saturated at low serum levels. In nontolerant adults, this results in a zero-order kinetic elimination pattern with an ethanol elimination rate of approximately 20 mg/dL per hour. Anecdotally, it had been thought that children clear ethanol at roughly double this rate via unclear mechanisms. However, a review of published kinetic data suggests the actual rate of clearance may not differ substantially from adults (range, 19-34 mg/dL per hour).5-7,10,11
Case Conclusion
The patient was transferred to a tertiary care pediatric hospital for continued management, where the markedly elevated serum ethanol concentration was confirmed. She was maintained on a dextrose-containing IV fluid and observed overnight without development of any complications. Serial serum ethanol concentrations were performed and complete clearance was achieved approximately 20 hours postingestion, suggesting a metabolic rate of 16 mg/dL per hour. The infant was discharged home with supervision by child protective services.
Dr Boroughf is a toxicology fellow, department of emergency medicine, Albert Einstein Medical Center, Philadelphia, Pennsylvania. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board. Dr Henretig is an attending toxicologist, department of emergency medicine, Children’s Hospital of Philadelphia, Pennsylvania.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila). 2013;51(10):949-1229.
- Wood JN, Pecker LH, Russo ME, Henretig F, Christian CW. Evaluation and referral for child maltreatment in pediatric poisoning victims. Child Abuse Negl. 2012;36(4):362-369.
- Lamminpää A. Alcohol intoxication in childhood and adolescence. Alcohol Alcohol. 1995;30(1):5-12.
- Malouf R, Brust JC. Hypoglycemia: causes, neurological manifestations, and outcome. Ann Neurol.1985;17(5):421-430.
- Chikava K, Lower DR, Frangiskakis SH, Sepulveda JL, Virji MA, Rao KN. Acute ethanol intoxication in a 7-month old-infant. Pediatr Dev Pathol. 2004;7(4):400-402.
- Ford JB, Wayment MT, Albertson TE, Owen KP, Radke JB, Sutter ME. Elimination kinetics of ethanol in a 5-week-old infant and a literature review of infant ethanol pharmacokinetics. Case Rep Med. 2013;2013:250716. doi:10.1155/2013/250716
- McCormick T, Levine M, Knox O, Claudius I. Ethanol ingestion in two infants under 2 months old: a previously unreported cause of ALTE. Pediatrics. 2013;131(2);e604-e607.
- Fong HF, Muller AA. An unexpected clinical course in a 29-day-old infant with ethanol exposure. Pediatr Emerg Care. 2014;30(2):111-113.
- Iyer SS, Haupt A, Henretig FM. Pick your poison: straight from the spring? Ped Emerg Care. 2009;25(3):194-196.
- Edmunds SM, Ajizian SJ, Liguori A. Acute obtundation in a 9-month-old patient: ethanol ingestion. Pediatr Emerg Care. 2014;30(10):739-741.
- Simon HK, Cox JM, Sucov A, Linakis JG. Serum ethanol clearance in intoxicated children and adolescents presenting to the ED. Acad Emerg Med. 1994;1(6):520-524.

Mind the Gap: Case Study in Toxicology
Case
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications, was presented to the ED with a 2-week history of intermittent fever and poor oral intake. His current medications included sodium bromide 185 mg orally twice daily for his seizure disorder.
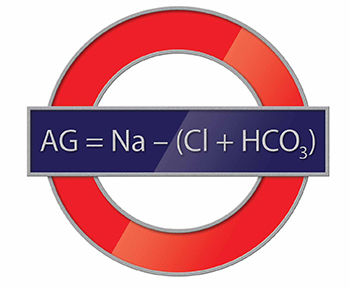
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were: sodium 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
What does the anion gap represent?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo, and is accomplished by the presence of unmeasured anions (UA) (eg, lactate and phosphate) and unmeasured cations (UC) (eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus the unmeasured anions must equal the sum of the measured plus unmeasured cations.
What causes a low or negative anion gap?
While most healthcare providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased Concentration of Unmeasured Anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium Cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of Serum Chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride. However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
What causes the falsely elevated chloride?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride. Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration, but rather it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
How are patients exposed to bromide salts?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders, but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurological and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide as well as the brominated vegetable oils found in some soft drinks.5
How is bromism treated?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10-12 days), and in patients with normal renal function, it is possible to reduce this half-life to approximately 3 days with hydration and diuresis with sodium chloride.3 Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
Case Conclusion
The patient was admitted for observation and treated with intravenous sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide 185 mg orally twice daily since his seizures were refractory to other anticonvulsant medications.
Dr Repplinger is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
- Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
- Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, Nimmannit S, Nilwarangkur S. A negative aniongap as a clue to diagnose bromide intoxication.Nephron. 1995;69(3):311-313.
- Yamamoto K, Kobayashi H, Kobayashi T, MurakamiS. False hyperchloremia in bromism. J Anesth.1991;5(1):88-91.
- Ng YY, Lin WL, Chen TW. Spurious hyperchloremiaand decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.
Case
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications, was presented to the ED with a 2-week history of intermittent fever and poor oral intake. His current medications included sodium bromide 185 mg orally twice daily for his seizure disorder.
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were: sodium 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
What does the anion gap represent?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo, and is accomplished by the presence of unmeasured anions (UA) (eg, lactate and phosphate) and unmeasured cations (UC) (eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus the unmeasured anions must equal the sum of the measured plus unmeasured cations.
What causes a low or negative anion gap?
While most healthcare providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased Concentration of Unmeasured Anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium Cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of Serum Chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride. However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
What causes the falsely elevated chloride?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride. Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration, but rather it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
How are patients exposed to bromide salts?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders, but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurological and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide as well as the brominated vegetable oils found in some soft drinks.5
How is bromism treated?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10-12 days), and in patients with normal renal function, it is possible to reduce this half-life to approximately 3 days with hydration and diuresis with sodium chloride.3 Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
Case Conclusion
The patient was admitted for observation and treated with intravenous sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide 185 mg orally twice daily since his seizures were refractory to other anticonvulsant medications.
Dr Repplinger is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
An 8-month-old boy with a history of hypotonia, developmental delay, and seizure disorder refractory to multiple anticonvulsant medications, was presented to the ED with a 2-week history of intermittent fever and poor oral intake. His current medications included sodium bromide 185 mg orally twice daily for his seizure disorder.
On physical examination, the boy appeared small for his age, with diffuse hypotonia and diminished reflexes. He was able to track with his eyes but was otherwise unresponsive. No rash was present. Results of initial laboratory studies were: sodium 144 mEq/L; potassium, 4.8 mEq/L; chloride, 179 mEq/L; bicarbonate, 21 mEq/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.1 mg/dL; and glucose, 63 mg/dL. His anion gap (AG) was −56.
What does the anion gap represent?
The AG is a valuable clinical calculation derived from the measured extracellular electrolytes and provides an index of acid-base status.1 Due to the necessity of electroneutrality, the sum of positive charges (cations) in the extracellular fluid must be balanced exactly with the sum of negative charges (anions). However, to routinely measure all of the cations and anions in the serum would be time-consuming and is also unnecessary. Because most clinical laboratories commonly only measure one relevant cation (sodium) and two anions (chloride and bicarbonate), the positive and negative sums are not completely balanced. The AG therefore refers to this difference (ie, AG = Na – [Cl + HCO3]).
Of course, electroneutrality exists in vivo, and is accomplished by the presence of unmeasured anions (UA) (eg, lactate and phosphate) and unmeasured cations (UC) (eg, potassium and calcium) not accounted for in the AG (ie, AG = UA – UC). In other words, the sum of measured plus the unmeasured anions must equal the sum of the measured plus unmeasured cations.
What causes a low or negative anion gap?
While most healthcare providers are well versed in the clinical significance of an elevated AG (eg, MUDPILES [methanol, uremia, diabetic ketoacidosis, propylene glycol or phenformin, iron or isoniazid, lactate, ethylene glycol, salicylates]), the meaning of a low or negative AG is underappreciated. There are several scenarios that could potentially yield a low or negative AG, including decreased concentration of UA, increased concentrations of nonsodium cations (UC), and overestimation of serum chloride.
Decreased Concentration of Unmeasured Anions. This most commonly occurs by two mechanisms: dilution of the extracellular fluid or hypoalbuminemia. The addition of water to the extracellular fluid will cause a proportionate dilution of all the measured electrolytes. Since the concentration of measured cations is higher than the measured anions, there is a small and relatively insignificant decrease in the AG.
Alternatively, hypoalbuminemia results in a low AG due to the change in UA; albumin is negatively charged. At physiologic pH, the overwhelming majority of serum proteins are anionic and counter-balanced by the positive charge of sodium. Albumin, the most abundant serum protein, accounts for approximately 75% of the normal AG. Hypoalbuminemic states, such as cirrhosis or nephrotic syndrome, can therefore cause low AG due to the retention of chloride to replace the lost negative charge. The albumin concentration can be corrected to calculate the AG.2
Nonsodium Cations. There are a number of clinical conditions that result in the retention of nonsodium cations. For example, the excess positively charged paraproteins associated with IgG myeloma raise the UC concentration, resulting in a low AG. Similarly, elevations of unmeasured cationic electrolytes, such as calcium and magnesium, may also result in a lower AG. Significant changes in AG, though, are caused only by profound (and often life-threatening) hypercalcemia or hypermagnesemia.
Overestimation of Serum Chloride. Overestimation of serum chloride most commonly occurs in the clinical scenario of bromide exposure. In normal physiologic conditions, chloride is the only halide present in the extracellular fluid. With intake of brominated products, chloride may be partially replaced by bromide. As there is greater renal tubular avidity for bromide, chronic ingestion of bromide results in a gradual rise in serum bromide concentrations with a proportional fall in chloride. However, and more importantly, bromide interferes with a number of laboratory techniques measuring chloride concentrations, resulting in a spuriously elevated chloride, or pseudohyperchloremia. Because the measured sodium and bicarbonate concentrations will remain unchanged, this falsely elevated chloride measurement will result in a negative AG.
What causes the falsely elevated chloride?
All of the current laboratory techniques for measurement of serum chloride concentration can potentially result in a falsely elevated value. However, the degree of pseudohyperchloremia will depend on the specific assay used for measurement. The ion-selective electrode method used by many common laboratory analyzers appears to have the greatest interference on chloride measurement in the presence of bromide. This is simply due to the molecular similarity of bromide and chloride. Conversely, the coulometry method, often used as a reference standard, has the least interference of current laboratory methods.3 This is because coulometry does not completely rely on molecular structure to measure concentration, but rather it measures the amount of energy produced or consumed in an electrolysis reaction. Iodide, another halide compound, has also been described as a cause of pseudohyperchloremia, whereas fluoride does not seem to have significant interference.4
How are patients exposed to bromide salts?
Bromide salts, specifically sodium bromide, are infrequently used to treat seizure disorders, but are generally reserved for patients with epilepsy refractory to other, less toxic anticonvulsant medications. During the era when bromide salts were more commonly used to treat epilepsy, bromide intoxication, or bromism, was frequently observed.
Bromism may manifest as a constellation of nonspecific neurological and psychiatric symptoms. These most commonly include headache, weakness, agitation, confusion, and hallucinations. In more severe cases of bromism, stupor and coma may occur.3,5
Although bromide salts are no longer commonly prescribed, a number of products still contain brominated ingredients. Symptoms of bromide intoxication can occur with chronic use of a cough syrup containing dextromethorphan hydrobromide as well as the brominated vegetable oils found in some soft drinks.5
How is bromism treated?
The treatment of bromism involves preventing further exposure to bromide and promoting bromide excretion. Bromide has a long half-life (10-12 days), and in patients with normal renal function, it is possible to reduce this half-life to approximately 3 days with hydration and diuresis with sodium chloride.3 Alternatively, in patients with impaired renal function or severe intoxication, hemodialysis has been used effectively.5
Case Conclusion
The patient was admitted for observation and treated with intravenous sodium chloride. After consultation with his neurologist, he was discharged home in the care of his parents, who were advised to continue him on sodium bromide 185 mg orally twice daily since his seizures were refractory to other anticonvulsant medications.
Dr Repplinger is a medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
- Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
- Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, Nimmannit S, Nilwarangkur S. A negative aniongap as a clue to diagnose bromide intoxication.Nephron. 1995;69(3):311-313.
- Yamamoto K, Kobayashi H, Kobayashi T, MurakamiS. False hyperchloremia in bromism. J Anesth.1991;5(1):88-91.
- Ng YY, Lin WL, Chen TW. Spurious hyperchloremiaand decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.
- Emmett M, Narins RG. Clinical use of the anion gap. Medicine (Baltimore). 1977;56(1):38-54.
- Figge J, Jabor A, Kazda A, Fencl V. Anion gap and hypoalbuminemia. Crit Care Med. 1998;26(11):1807-1810.
- Vasuyattakul S, Lertpattanasuwan N, Vareesangthip K, Nimmannit S, Nilwarangkur S. A negative aniongap as a clue to diagnose bromide intoxication.Nephron. 1995;69(3):311-313.
- Yamamoto K, Kobayashi H, Kobayashi T, MurakamiS. False hyperchloremia in bromism. J Anesth.1991;5(1):88-91.
- Ng YY, Lin WL, Chen TW. Spurious hyperchloremiaand decreased anion gap in a patient with dextromethorphan bromide. Am J Nephrol. 1992;12(4):268-270.