User login
More on treating chronic insomnia
In “Treating chronic insomnia: An alternating medication strategy” (
Leslie Citrome, MD, MPH
Valhalla, New York
1. Rosenberg R, Citrome L, Drake CL. Advances in the treatment of chronic insomnia: a narrative review of new nonpharmacologic and pharmacologic therapies. Neuropsychiatr Dis Treat. 2021;17:2549-2566.
2. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
3. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
4. Citrome L, Juday TR, Frech F, et al. Lemborexant for the treatment of insomnia: direct and indirect comparisons with other hypnotics using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2021;82:20m13795. doi:10.4088/JCP.20m13795
5. Citrome L, Juday TR, Lundwall C. Lemborexant and daridorexant for the treatment of insomnia: an indirect comparison using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2023;84(6):23m14851. doi:10.4088/JCP.23m14851
In “Treating chronic insomnia: An alternating medication strategy” (
Leslie Citrome, MD, MPH
Valhalla, New York
In “Treating chronic insomnia: An alternating medication strategy” (
Leslie Citrome, MD, MPH
Valhalla, New York
1. Rosenberg R, Citrome L, Drake CL. Advances in the treatment of chronic insomnia: a narrative review of new nonpharmacologic and pharmacologic therapies. Neuropsychiatr Dis Treat. 2021;17:2549-2566.
2. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
3. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
4. Citrome L, Juday TR, Frech F, et al. Lemborexant for the treatment of insomnia: direct and indirect comparisons with other hypnotics using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2021;82:20m13795. doi:10.4088/JCP.20m13795
5. Citrome L, Juday TR, Lundwall C. Lemborexant and daridorexant for the treatment of insomnia: an indirect comparison using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2023;84(6):23m14851. doi:10.4088/JCP.23m14851
1. Rosenberg R, Citrome L, Drake CL. Advances in the treatment of chronic insomnia: a narrative review of new nonpharmacologic and pharmacologic therapies. Neuropsychiatr Dis Treat. 2021;17:2549-2566.
2. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
3. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
4. Citrome L, Juday TR, Frech F, et al. Lemborexant for the treatment of insomnia: direct and indirect comparisons with other hypnotics using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2021;82:20m13795. doi:10.4088/JCP.20m13795
5. Citrome L, Juday TR, Lundwall C. Lemborexant and daridorexant for the treatment of insomnia: an indirect comparison using number needed to treat, number needed to harm, and likelihood to be helped or harmed. J Clin Psychiatry. 2023;84(6):23m14851. doi:10.4088/JCP.23m14851

Dexmedetomidine sublingual film for agitation
Approved by the FDA on April 5, 2022, dexmedetomidine sublingual film (Igalmi, manufactured and distributed by BioXcel Therapeutics, Inc., New Haven, CT USA) is indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder (Table).1,2 It is administered sublingually or buccally under the supervision of a health care provider. After administration, patients should have their vital signs and alertness assessed but there is no FDA Risk Evaluation and Mitigation Strategy (REMS) required for use. A limitation of use is that the safety and effectiveness of dexmedetomidine sublingual film has not been established beyond 24 hours from the first dose.2 There are no contraindications for use.2

Dexmedetomidine is a well-known efficacious alpha-2 adrenergic receptor agonist available since 1999 in an IV formulation indicated for sedation of initially intubated and mechanically ventilated patients in an ICU setting, and sedation of nonintubated patients prior to and/or during surgical and other procedures.3,4 The reformulation of dexmedetomidine as a sublingual film allows the broader use of this agent in psychiatric settings when managing agitation in patients with schizophrenia or bipolar disorder, and thus potentially avoiding the use of IM administration of antipsychotics and/or benzodiazepines. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.5
Dosing
Dexmedetomidine sublingual film is distributed commercially in the following strengths: 180 mcg and 120 mcg. It consists of a lightly mint-flavored, rectangular film containing 2 microdeposits of dexmedetomidine hydrochloride. Dosage strengths of 90 mcg and 60 mcg are available by cutting the 180 mcg or 120 mcg film in half
If agitation persists after the initial dose, up to 2 additional doses (90 mcg if the initial dose was 180 mcg, otherwise 60 mcg if the initial dose was 120, 90, or 60 mcg) may be given at least 2 hours apart. Assessment of vital signs, including orthostatic measurements, is required prior to the administration of any subsequent doses. Due to risk of hypotension, additional doses are not recommended in patients with systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, heart rate <60 beats per minute, or postural decrease in systolic blood pressure ≥20 mm Hg or in diastolic blood pressure ≥10 mm Hg.
Mechanism of action and pharmacodynamics
Dexmedetomidine is an alpha-2 adrenergic receptor agonist and the mechanism of action in the acute treatment of agitation is thought to be due to activation of presynaptic alpha-2 adrenergic receptors.2 Binding affinities (Ki values) are 4 to 6 nM at the alpha-2 adrenergic receptor subtypes.2
Dexmedetomidine exhibits concentration-dependent QT prolongation, with mean QTc increases from baseline from 6 msec (120 mcg single dose) to 11 msec (180 mcg plus 2 additional doses of 90 mcg 2 hours apart for a total of 3 doses).2 Placing the observation about QTc prolongation into clinical context, studies of IM administration of ziprasidone 20 mg and 30 mg and haloperidol 7.5 mg and 10 mg resulted in changes of the QTc interval of 4.6 msec and 6.0 msec, respectively, after 1 dose.6 After a second injection, these values were 12.8 msec and 14.7 msec, respectively.6
Clinical pharmacokinetics
The sublingual film formulation is absorbed orally, bypassing first-pass metabolism, and achieving higher dexmedetomidine bioavailability than ingested formulations.7 Exposure is dose-dependent, with dexmedetomidine being quantifiable in plasma after 5 to 20 minutes post dosing, and with a plasma half-life of 2 to 3 hours.2,8 Mean time for the film to dissolve in the mouth was approximately 6 to 8 minutes following sublingual administration, and 18 minutes following buccal administration.2 Absolute bioavailability was approximately 72% and 82% following sublingual and buccal administration, respectively.2 Mean maximal plasma concentrations of dexmedetomidine were reached approximately 2 hours after sublingual or buccal administration.2 Compared to drinking water at 2 hours post administration, early water intake (as early as 15 minutes post-dose) had minimal effects on the rate or extent of sublingual absorption but was not assessed after buccal administration.2 The average protein binding was 94% and was constant across the different plasma concentrations evaluated and similar in males and females, but significantly decreased in participants with hepatic impairment compared to healthy individuals.2 In contrast, the pharmacokinetic profile of dexmedetomidine is not significantly different in patients with creatinine clearance <30 mL/minute compared to those with normal renal function.2 Dexmedetomidine undergoes almost complete biotransformation to inactive metabolites via direct glucuronidation as well as cytochrome P450 (CYP) (primarily CYP2A6)–mediated metabolism.2 There is no evidence of any CYP–mediated drug interactions that are likely to be of clinical relevance.2
Continue to: Efficacy
Efficacy
The efficacy and tolerability of 120 mcg and 180 mcg doses of dexmedetomidine sublingual film was evaluated in 2 similarly designed, randomized, double-blind, placebo-controlled, Phase 3 trials in the treatment of acute agitation associated with schizophrenia, schizoaffective, or schizophreniform disorder9 and bipolar I or II disorder.10 These studies included a total of 758 adult patients age range 18 to 71 (mean age approximately 46.5), with about 59% male participants.2 In contrast to other agents approved by the FDA for treatment of agitation associated with bipolar disorder, dexmedetomidine sublingual film was assessed in patients regardless of polarity (manic, mixed features, or depressed).5 The primary efficacy measure for the dexmedetomidine sublingual film studies was the investigator-administered Positive and Negative Syndrome Scale-Excited Component (PANSS-EC), consisting of the following 5 items: excitement, tension, hostility, uncooperativeness, and poor impulse control.11 The items from the PANSS-EC are rated from 1 (not present) to 7 (extremely severe) and thus the total scores range from 5 to 35. For enrollment in the studies, patients had to be judged to be clinically agitated with a total PANSS-EC score ≥14, with at least 1 individual item score ≥4.2
After study medication administration, the PANSS-EC was assessed from 10 minutes through 24 hours, with the primary endpoint being at 2 hours post-dose. Patients with schizophrenia or bipolar disorder who were treated with dexmedetomidine sublingual film 120 mcg or 180 mcg had superior symptomatic improvements from baseline to 2 hours post-dose compared to placebo, with treatment effects beginning as early as 20 to 30 minutes post-dose (for patients with schizophrenia, dexmedetomidine was statistically significantly superior to placebo beginning at 20 minutes following dosing with the 180 mcg dose and 30 minutes after the 120 mcg dose; for patients with bipolar disorder, differences from placebo were statistically significant beginning at 20 minutes after treatment with both the 120 mcg and 180 mcg doses).2 Evaluation of effect size for dexmedetomidine vs placebo for PANSS-EC response at 2 hours (defined as ≥40% improvement from baseline) resulted in a number needed to treat (NNT) of 3 when combining both studies and both doses,12 comparing favorably with the NNT values observed for IM formulations of aripiprazole, haloperidol, lorazepam, olanzapine, and ziprasidone,13 and inhaled loxapine.14
Overall tolerability and safety
The highlights of the prescribing information contain warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence.2 Advice is provided to ensure that patients are alert and not experiencing orthostatic or symptomatic hypotension prior to resuming ambulation, a concern commonly raised when assessing potential treatments for agitation.15 Dexmedetomidine sublingual film should be avoided in patients with risk factors for prolonged QT interval, a precaution that was evident for the use of ziprasidone16 and where an effect is also noted with haloperidol.6 As per the prescribing information, the most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) are somnolence, oral paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. Rates of adverse reactions of somnolence (including fatigue and sluggishness) with dexmedetomidine 120 mcg or 180 mcg are almost the same (22% and 23%, respectively), and higher than the 6% observed with placebo.2 Other adverse reactions are substantially lower in frequency. These include oral paresthesia or oral hypoesthesia (6%, 7%, and 1%, for dexmedetomidine 120 mcg, 180 mcg, or placebo, respectively), dizziness (4%, 6%, 1%), hypotension (5%, 5%, 0%), orthostatic hypotension (3%, 5%, <1%), dry mouth (7%, 4%, 1%), nausea (2%, 3%, 2%), bradycardia (2%, 2%, 0%), and abdominal discomfort (0%, 2%, 1%).2
Regarding dose-dependent changes in blood pressure during the studies, 16%, 18%, and 9% of patients treated with 120 mcg, 180 mcg, and placebo, respectively, experienced orthostatic hypotension at 2 hours post dose. However, at 24 hours, none of the patients in the 180-mcg group experienced a systolic blood pressure ≤90 mm Hg with a decrease ≥20 mm Hg, compared with one patient (<1%) in the 120-mcg group and none in the placebo group.2
The prescribing information advises that concomitant use of dexmedetomidine sublingual film with anesthetics, sedatives, hypnotics, or opioids is likely to lead to enhanced CNS depressant effects, and that the prescriber should consider a reduction in dosage of dexmedetomidine or the concomitant anesthetic, sedative, hypnotic, or opioid.2
Summary
Dexmedetomidine sublingual film is an oral medication indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder. The recommended dose depends on severity of agitation, age, and the presence of hepatic impairment. A dose of 180 mcg is recommended for severe agitation and a dose of 120 mcg is recommended for mild or moderate agitation, with doses adjusted lower in the presence of hepatic impairment. There are no contraindications but there are warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence. Clinicians should monitor vital signs and alertness after administration to prevent falls and syncope; however, there is no FDA REMS required for use. The clinical trial evidence supporting the use of dexmedetomidine is robust, with evidence of a treatment effect as early as 20 minutes after administration. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.
Bottom Line
Dexmedetomidine sublingual film provides an opportunity to rethink the approach to the management of agitation and avoid the potentially unnecessary use of IM injections. Dexmedetomidine sublingual film acts rapidly and is simple to use.
Related Resources
- Dexmedetomidine sublingual film (Iglami) prescribing information. https://www.igalmihcp.com/igalmi-pi.pdf
Drug Brand Names
Aripiprazole • Abilify
Dexmedetomidine • Igalmi, Precedex
Haloperidol • Haldol
Lorazepam • Ativan
Loxapine inhaled • Adasuve
Olanzapine • Zyprexa
Ziprasidone • Geodon
1. US Food and Drug Administration. NDA 215390 Approval Letter. Accessed April 5, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215390Orig1s000ltr.pdf
2. Igalmi [package insert]. BioXcel Therapeutics, Inc; 2022.
3. Weerink MAS, Struys MMRF, Hannivoort LN, et al. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893-913. doi:10.1007/s40262-017-0507-7
4. Precedex [package insert]. Hospira, Inc; 2021.
5. Zeller SL, Citrome L. Managing agitation associated with schizophrenia and bipolar disorder in the emergency setting. West J Emerg Med. 2016;17(2):165-172. doi:10.5811/westjem.2015.12.28763
6. Miceli JJ, Tensfeldt TG, Shiovitz T, et al. Effects of high-dose ziprasidone and haloperidol on the QTc interval after intramuscular administration: a randomized, single-blind, parallel-group study in patients with schizophrenia or schizoaffective disorder. Clin Ther. 2010;32(3):472-491. doi:10.1016/j.clinthera.2010.03.003
7. Yocca F, DeVivo M, Seth S, et al. Dexmedetomidine—highly favorable pharmacokinetic and pharmacological features for a CNS therapeutic drug. Poster presented at: 58th Annual Meeting of the American College of Neuropsychopharmacology; December 8-11, 2019; Orlando, FL.
8. Adedoyin A, Preskorn S, Lathia CD. Pharmacokinetics of dexmedetomidine after a single sublingual dose of BXCL501 in patients with agitation associated with schizophrenia. Poster presented at: 23rd Annual Conference of the International Society for Bipolar Disorders; May 13-15, 2021. Virtual. Session 17.
9. Citrome LL, Lauriello J, Risinger R, et al. A novel rapidly effective treatment of agitation for schizophrenia with the oral dissolving film BXCL501. Poster presented at: American Psychiatric Association Annual Meeting; May 1-3, 2021. Virtual. Accessed November 11, 2021. https://www.psychiatry.org/File%20Library/Psychiatrists/Meetings/Annual-Meeting/2021/2021-APA-Annual-Meeting-Poster-Proceedings.pdf
10. Preskorn SH, Zeller S, Citrome L, et al. Effect of sublingual dexmedetomidine vs placebo on acute agitation associated with bipolar disorder: a randomized clinical trial. JAMA. 2022;327(8):727-736. doi:10.1001/jama.2022.0799
11. Montoya A, Valladares A, Lizán L, et al. Validation of the Excited Component of the Positive and Negative Syndrome Scale (PANSS-EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Qual Life Outcomes. 2011;9:18. doi:10.1186/1477-7525-9-18
12. Citrome L, Palko L, Hokett S, et al. Number needed to treat and number needed to harm from two phase 3 studies of BXCL501 for treating acute agitation in patients with schizophrenia and bipolar disorder. Poster presented at: Academy of Managed Care Pharmacy Nexus 2021; October 18-21, 2021; Denver, CO.
13. Citrome L. Comparison of intramuscular ziprasidone, olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885. doi:10.4088/jcp.v68n1207
14. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325. doi:10.1111/j.1742-1241.2011.02890.x
15. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of the American Association for Emergency Psychiatry project Beta psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34. doi:10.5811/westjem.2011.9.6866
16. Zimbroff DL, Allen MH, Battaglia J, et al. Best clinical practice with ziprasidone IM: update after 2 years of experience. CNS Spectr. 2005;10(9):1-15. doi:10.1017/s1092852900025487
Approved by the FDA on April 5, 2022, dexmedetomidine sublingual film (Igalmi, manufactured and distributed by BioXcel Therapeutics, Inc., New Haven, CT USA) is indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder (Table).1,2 It is administered sublingually or buccally under the supervision of a health care provider. After administration, patients should have their vital signs and alertness assessed but there is no FDA Risk Evaluation and Mitigation Strategy (REMS) required for use. A limitation of use is that the safety and effectiveness of dexmedetomidine sublingual film has not been established beyond 24 hours from the first dose.2 There are no contraindications for use.2
Dexmedetomidine is a well-known efficacious alpha-2 adrenergic receptor agonist available since 1999 in an IV formulation indicated for sedation of initially intubated and mechanically ventilated patients in an ICU setting, and sedation of nonintubated patients prior to and/or during surgical and other procedures.3,4 The reformulation of dexmedetomidine as a sublingual film allows the broader use of this agent in psychiatric settings when managing agitation in patients with schizophrenia or bipolar disorder, and thus potentially avoiding the use of IM administration of antipsychotics and/or benzodiazepines. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.5
Dosing
Dexmedetomidine sublingual film is distributed commercially in the following strengths: 180 mcg and 120 mcg. It consists of a lightly mint-flavored, rectangular film containing 2 microdeposits of dexmedetomidine hydrochloride. Dosage strengths of 90 mcg and 60 mcg are available by cutting the 180 mcg or 120 mcg film in half
If agitation persists after the initial dose, up to 2 additional doses (90 mcg if the initial dose was 180 mcg, otherwise 60 mcg if the initial dose was 120, 90, or 60 mcg) may be given at least 2 hours apart. Assessment of vital signs, including orthostatic measurements, is required prior to the administration of any subsequent doses. Due to risk of hypotension, additional doses are not recommended in patients with systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, heart rate <60 beats per minute, or postural decrease in systolic blood pressure ≥20 mm Hg or in diastolic blood pressure ≥10 mm Hg.
Mechanism of action and pharmacodynamics
Dexmedetomidine is an alpha-2 adrenergic receptor agonist and the mechanism of action in the acute treatment of agitation is thought to be due to activation of presynaptic alpha-2 adrenergic receptors.2 Binding affinities (Ki values) are 4 to 6 nM at the alpha-2 adrenergic receptor subtypes.2
Dexmedetomidine exhibits concentration-dependent QT prolongation, with mean QTc increases from baseline from 6 msec (120 mcg single dose) to 11 msec (180 mcg plus 2 additional doses of 90 mcg 2 hours apart for a total of 3 doses).2 Placing the observation about QTc prolongation into clinical context, studies of IM administration of ziprasidone 20 mg and 30 mg and haloperidol 7.5 mg and 10 mg resulted in changes of the QTc interval of 4.6 msec and 6.0 msec, respectively, after 1 dose.6 After a second injection, these values were 12.8 msec and 14.7 msec, respectively.6
Clinical pharmacokinetics
The sublingual film formulation is absorbed orally, bypassing first-pass metabolism, and achieving higher dexmedetomidine bioavailability than ingested formulations.7 Exposure is dose-dependent, with dexmedetomidine being quantifiable in plasma after 5 to 20 minutes post dosing, and with a plasma half-life of 2 to 3 hours.2,8 Mean time for the film to dissolve in the mouth was approximately 6 to 8 minutes following sublingual administration, and 18 minutes following buccal administration.2 Absolute bioavailability was approximately 72% and 82% following sublingual and buccal administration, respectively.2 Mean maximal plasma concentrations of dexmedetomidine were reached approximately 2 hours after sublingual or buccal administration.2 Compared to drinking water at 2 hours post administration, early water intake (as early as 15 minutes post-dose) had minimal effects on the rate or extent of sublingual absorption but was not assessed after buccal administration.2 The average protein binding was 94% and was constant across the different plasma concentrations evaluated and similar in males and females, but significantly decreased in participants with hepatic impairment compared to healthy individuals.2 In contrast, the pharmacokinetic profile of dexmedetomidine is not significantly different in patients with creatinine clearance <30 mL/minute compared to those with normal renal function.2 Dexmedetomidine undergoes almost complete biotransformation to inactive metabolites via direct glucuronidation as well as cytochrome P450 (CYP) (primarily CYP2A6)–mediated metabolism.2 There is no evidence of any CYP–mediated drug interactions that are likely to be of clinical relevance.2
Continue to: Efficacy
Efficacy
The efficacy and tolerability of 120 mcg and 180 mcg doses of dexmedetomidine sublingual film was evaluated in 2 similarly designed, randomized, double-blind, placebo-controlled, Phase 3 trials in the treatment of acute agitation associated with schizophrenia, schizoaffective, or schizophreniform disorder9 and bipolar I or II disorder.10 These studies included a total of 758 adult patients age range 18 to 71 (mean age approximately 46.5), with about 59% male participants.2 In contrast to other agents approved by the FDA for treatment of agitation associated with bipolar disorder, dexmedetomidine sublingual film was assessed in patients regardless of polarity (manic, mixed features, or depressed).5 The primary efficacy measure for the dexmedetomidine sublingual film studies was the investigator-administered Positive and Negative Syndrome Scale-Excited Component (PANSS-EC), consisting of the following 5 items: excitement, tension, hostility, uncooperativeness, and poor impulse control.11 The items from the PANSS-EC are rated from 1 (not present) to 7 (extremely severe) and thus the total scores range from 5 to 35. For enrollment in the studies, patients had to be judged to be clinically agitated with a total PANSS-EC score ≥14, with at least 1 individual item score ≥4.2
After study medication administration, the PANSS-EC was assessed from 10 minutes through 24 hours, with the primary endpoint being at 2 hours post-dose. Patients with schizophrenia or bipolar disorder who were treated with dexmedetomidine sublingual film 120 mcg or 180 mcg had superior symptomatic improvements from baseline to 2 hours post-dose compared to placebo, with treatment effects beginning as early as 20 to 30 minutes post-dose (for patients with schizophrenia, dexmedetomidine was statistically significantly superior to placebo beginning at 20 minutes following dosing with the 180 mcg dose and 30 minutes after the 120 mcg dose; for patients with bipolar disorder, differences from placebo were statistically significant beginning at 20 minutes after treatment with both the 120 mcg and 180 mcg doses).2 Evaluation of effect size for dexmedetomidine vs placebo for PANSS-EC response at 2 hours (defined as ≥40% improvement from baseline) resulted in a number needed to treat (NNT) of 3 when combining both studies and both doses,12 comparing favorably with the NNT values observed for IM formulations of aripiprazole, haloperidol, lorazepam, olanzapine, and ziprasidone,13 and inhaled loxapine.14
Overall tolerability and safety
The highlights of the prescribing information contain warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence.2 Advice is provided to ensure that patients are alert and not experiencing orthostatic or symptomatic hypotension prior to resuming ambulation, a concern commonly raised when assessing potential treatments for agitation.15 Dexmedetomidine sublingual film should be avoided in patients with risk factors for prolonged QT interval, a precaution that was evident for the use of ziprasidone16 and where an effect is also noted with haloperidol.6 As per the prescribing information, the most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) are somnolence, oral paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. Rates of adverse reactions of somnolence (including fatigue and sluggishness) with dexmedetomidine 120 mcg or 180 mcg are almost the same (22% and 23%, respectively), and higher than the 6% observed with placebo.2 Other adverse reactions are substantially lower in frequency. These include oral paresthesia or oral hypoesthesia (6%, 7%, and 1%, for dexmedetomidine 120 mcg, 180 mcg, or placebo, respectively), dizziness (4%, 6%, 1%), hypotension (5%, 5%, 0%), orthostatic hypotension (3%, 5%, <1%), dry mouth (7%, 4%, 1%), nausea (2%, 3%, 2%), bradycardia (2%, 2%, 0%), and abdominal discomfort (0%, 2%, 1%).2
Regarding dose-dependent changes in blood pressure during the studies, 16%, 18%, and 9% of patients treated with 120 mcg, 180 mcg, and placebo, respectively, experienced orthostatic hypotension at 2 hours post dose. However, at 24 hours, none of the patients in the 180-mcg group experienced a systolic blood pressure ≤90 mm Hg with a decrease ≥20 mm Hg, compared with one patient (<1%) in the 120-mcg group and none in the placebo group.2
The prescribing information advises that concomitant use of dexmedetomidine sublingual film with anesthetics, sedatives, hypnotics, or opioids is likely to lead to enhanced CNS depressant effects, and that the prescriber should consider a reduction in dosage of dexmedetomidine or the concomitant anesthetic, sedative, hypnotic, or opioid.2
Summary
Dexmedetomidine sublingual film is an oral medication indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder. The recommended dose depends on severity of agitation, age, and the presence of hepatic impairment. A dose of 180 mcg is recommended for severe agitation and a dose of 120 mcg is recommended for mild or moderate agitation, with doses adjusted lower in the presence of hepatic impairment. There are no contraindications but there are warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence. Clinicians should monitor vital signs and alertness after administration to prevent falls and syncope; however, there is no FDA REMS required for use. The clinical trial evidence supporting the use of dexmedetomidine is robust, with evidence of a treatment effect as early as 20 minutes after administration. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.
Bottom Line
Dexmedetomidine sublingual film provides an opportunity to rethink the approach to the management of agitation and avoid the potentially unnecessary use of IM injections. Dexmedetomidine sublingual film acts rapidly and is simple to use.
Related Resources
- Dexmedetomidine sublingual film (Iglami) prescribing information. https://www.igalmihcp.com/igalmi-pi.pdf
Drug Brand Names
Aripiprazole • Abilify
Dexmedetomidine • Igalmi, Precedex
Haloperidol • Haldol
Lorazepam • Ativan
Loxapine inhaled • Adasuve
Olanzapine • Zyprexa
Ziprasidone • Geodon
Approved by the FDA on April 5, 2022, dexmedetomidine sublingual film (Igalmi, manufactured and distributed by BioXcel Therapeutics, Inc., New Haven, CT USA) is indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder (Table).1,2 It is administered sublingually or buccally under the supervision of a health care provider. After administration, patients should have their vital signs and alertness assessed but there is no FDA Risk Evaluation and Mitigation Strategy (REMS) required for use. A limitation of use is that the safety and effectiveness of dexmedetomidine sublingual film has not been established beyond 24 hours from the first dose.2 There are no contraindications for use.2
Dexmedetomidine is a well-known efficacious alpha-2 adrenergic receptor agonist available since 1999 in an IV formulation indicated for sedation of initially intubated and mechanically ventilated patients in an ICU setting, and sedation of nonintubated patients prior to and/or during surgical and other procedures.3,4 The reformulation of dexmedetomidine as a sublingual film allows the broader use of this agent in psychiatric settings when managing agitation in patients with schizophrenia or bipolar disorder, and thus potentially avoiding the use of IM administration of antipsychotics and/or benzodiazepines. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.5
Dosing
Dexmedetomidine sublingual film is distributed commercially in the following strengths: 180 mcg and 120 mcg. It consists of a lightly mint-flavored, rectangular film containing 2 microdeposits of dexmedetomidine hydrochloride. Dosage strengths of 90 mcg and 60 mcg are available by cutting the 180 mcg or 120 mcg film in half
If agitation persists after the initial dose, up to 2 additional doses (90 mcg if the initial dose was 180 mcg, otherwise 60 mcg if the initial dose was 120, 90, or 60 mcg) may be given at least 2 hours apart. Assessment of vital signs, including orthostatic measurements, is required prior to the administration of any subsequent doses. Due to risk of hypotension, additional doses are not recommended in patients with systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, heart rate <60 beats per minute, or postural decrease in systolic blood pressure ≥20 mm Hg or in diastolic blood pressure ≥10 mm Hg.
Mechanism of action and pharmacodynamics
Dexmedetomidine is an alpha-2 adrenergic receptor agonist and the mechanism of action in the acute treatment of agitation is thought to be due to activation of presynaptic alpha-2 adrenergic receptors.2 Binding affinities (Ki values) are 4 to 6 nM at the alpha-2 adrenergic receptor subtypes.2
Dexmedetomidine exhibits concentration-dependent QT prolongation, with mean QTc increases from baseline from 6 msec (120 mcg single dose) to 11 msec (180 mcg plus 2 additional doses of 90 mcg 2 hours apart for a total of 3 doses).2 Placing the observation about QTc prolongation into clinical context, studies of IM administration of ziprasidone 20 mg and 30 mg and haloperidol 7.5 mg and 10 mg resulted in changes of the QTc interval of 4.6 msec and 6.0 msec, respectively, after 1 dose.6 After a second injection, these values were 12.8 msec and 14.7 msec, respectively.6
Clinical pharmacokinetics
The sublingual film formulation is absorbed orally, bypassing first-pass metabolism, and achieving higher dexmedetomidine bioavailability than ingested formulations.7 Exposure is dose-dependent, with dexmedetomidine being quantifiable in plasma after 5 to 20 minutes post dosing, and with a plasma half-life of 2 to 3 hours.2,8 Mean time for the film to dissolve in the mouth was approximately 6 to 8 minutes following sublingual administration, and 18 minutes following buccal administration.2 Absolute bioavailability was approximately 72% and 82% following sublingual and buccal administration, respectively.2 Mean maximal plasma concentrations of dexmedetomidine were reached approximately 2 hours after sublingual or buccal administration.2 Compared to drinking water at 2 hours post administration, early water intake (as early as 15 minutes post-dose) had minimal effects on the rate or extent of sublingual absorption but was not assessed after buccal administration.2 The average protein binding was 94% and was constant across the different plasma concentrations evaluated and similar in males and females, but significantly decreased in participants with hepatic impairment compared to healthy individuals.2 In contrast, the pharmacokinetic profile of dexmedetomidine is not significantly different in patients with creatinine clearance <30 mL/minute compared to those with normal renal function.2 Dexmedetomidine undergoes almost complete biotransformation to inactive metabolites via direct glucuronidation as well as cytochrome P450 (CYP) (primarily CYP2A6)–mediated metabolism.2 There is no evidence of any CYP–mediated drug interactions that are likely to be of clinical relevance.2
Continue to: Efficacy
Efficacy
The efficacy and tolerability of 120 mcg and 180 mcg doses of dexmedetomidine sublingual film was evaluated in 2 similarly designed, randomized, double-blind, placebo-controlled, Phase 3 trials in the treatment of acute agitation associated with schizophrenia, schizoaffective, or schizophreniform disorder9 and bipolar I or II disorder.10 These studies included a total of 758 adult patients age range 18 to 71 (mean age approximately 46.5), with about 59% male participants.2 In contrast to other agents approved by the FDA for treatment of agitation associated with bipolar disorder, dexmedetomidine sublingual film was assessed in patients regardless of polarity (manic, mixed features, or depressed).5 The primary efficacy measure for the dexmedetomidine sublingual film studies was the investigator-administered Positive and Negative Syndrome Scale-Excited Component (PANSS-EC), consisting of the following 5 items: excitement, tension, hostility, uncooperativeness, and poor impulse control.11 The items from the PANSS-EC are rated from 1 (not present) to 7 (extremely severe) and thus the total scores range from 5 to 35. For enrollment in the studies, patients had to be judged to be clinically agitated with a total PANSS-EC score ≥14, with at least 1 individual item score ≥4.2
After study medication administration, the PANSS-EC was assessed from 10 minutes through 24 hours, with the primary endpoint being at 2 hours post-dose. Patients with schizophrenia or bipolar disorder who were treated with dexmedetomidine sublingual film 120 mcg or 180 mcg had superior symptomatic improvements from baseline to 2 hours post-dose compared to placebo, with treatment effects beginning as early as 20 to 30 minutes post-dose (for patients with schizophrenia, dexmedetomidine was statistically significantly superior to placebo beginning at 20 minutes following dosing with the 180 mcg dose and 30 minutes after the 120 mcg dose; for patients with bipolar disorder, differences from placebo were statistically significant beginning at 20 minutes after treatment with both the 120 mcg and 180 mcg doses).2 Evaluation of effect size for dexmedetomidine vs placebo for PANSS-EC response at 2 hours (defined as ≥40% improvement from baseline) resulted in a number needed to treat (NNT) of 3 when combining both studies and both doses,12 comparing favorably with the NNT values observed for IM formulations of aripiprazole, haloperidol, lorazepam, olanzapine, and ziprasidone,13 and inhaled loxapine.14
Overall tolerability and safety
The highlights of the prescribing information contain warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence.2 Advice is provided to ensure that patients are alert and not experiencing orthostatic or symptomatic hypotension prior to resuming ambulation, a concern commonly raised when assessing potential treatments for agitation.15 Dexmedetomidine sublingual film should be avoided in patients with risk factors for prolonged QT interval, a precaution that was evident for the use of ziprasidone16 and where an effect is also noted with haloperidol.6 As per the prescribing information, the most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) are somnolence, oral paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. Rates of adverse reactions of somnolence (including fatigue and sluggishness) with dexmedetomidine 120 mcg or 180 mcg are almost the same (22% and 23%, respectively), and higher than the 6% observed with placebo.2 Other adverse reactions are substantially lower in frequency. These include oral paresthesia or oral hypoesthesia (6%, 7%, and 1%, for dexmedetomidine 120 mcg, 180 mcg, or placebo, respectively), dizziness (4%, 6%, 1%), hypotension (5%, 5%, 0%), orthostatic hypotension (3%, 5%, <1%), dry mouth (7%, 4%, 1%), nausea (2%, 3%, 2%), bradycardia (2%, 2%, 0%), and abdominal discomfort (0%, 2%, 1%).2
Regarding dose-dependent changes in blood pressure during the studies, 16%, 18%, and 9% of patients treated with 120 mcg, 180 mcg, and placebo, respectively, experienced orthostatic hypotension at 2 hours post dose. However, at 24 hours, none of the patients in the 180-mcg group experienced a systolic blood pressure ≤90 mm Hg with a decrease ≥20 mm Hg, compared with one patient (<1%) in the 120-mcg group and none in the placebo group.2
The prescribing information advises that concomitant use of dexmedetomidine sublingual film with anesthetics, sedatives, hypnotics, or opioids is likely to lead to enhanced CNS depressant effects, and that the prescriber should consider a reduction in dosage of dexmedetomidine or the concomitant anesthetic, sedative, hypnotic, or opioid.2
Summary
Dexmedetomidine sublingual film is an oral medication indicated in adults for the acute treatment of agitation associated with schizophrenia or bipolar I or II disorder. The recommended dose depends on severity of agitation, age, and the presence of hepatic impairment. A dose of 180 mcg is recommended for severe agitation and a dose of 120 mcg is recommended for mild or moderate agitation, with doses adjusted lower in the presence of hepatic impairment. There are no contraindications but there are warnings and precautions regarding hypotension/orthostatic hypotension/bradycardia, QT interval prolongation, and somnolence. Clinicians should monitor vital signs and alertness after administration to prevent falls and syncope; however, there is no FDA REMS required for use. The clinical trial evidence supporting the use of dexmedetomidine is robust, with evidence of a treatment effect as early as 20 minutes after administration. Noninvasive formulations, although requiring cooperation from patients, have the potential to improve overall patient experience, thereby improving future cooperation between patients and health care professionals.
Bottom Line
Dexmedetomidine sublingual film provides an opportunity to rethink the approach to the management of agitation and avoid the potentially unnecessary use of IM injections. Dexmedetomidine sublingual film acts rapidly and is simple to use.
Related Resources
- Dexmedetomidine sublingual film (Iglami) prescribing information. https://www.igalmihcp.com/igalmi-pi.pdf
Drug Brand Names
Aripiprazole • Abilify
Dexmedetomidine • Igalmi, Precedex
Haloperidol • Haldol
Lorazepam • Ativan
Loxapine inhaled • Adasuve
Olanzapine • Zyprexa
Ziprasidone • Geodon
1. US Food and Drug Administration. NDA 215390 Approval Letter. Accessed April 5, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215390Orig1s000ltr.pdf
2. Igalmi [package insert]. BioXcel Therapeutics, Inc; 2022.
3. Weerink MAS, Struys MMRF, Hannivoort LN, et al. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893-913. doi:10.1007/s40262-017-0507-7
4. Precedex [package insert]. Hospira, Inc; 2021.
5. Zeller SL, Citrome L. Managing agitation associated with schizophrenia and bipolar disorder in the emergency setting. West J Emerg Med. 2016;17(2):165-172. doi:10.5811/westjem.2015.12.28763
6. Miceli JJ, Tensfeldt TG, Shiovitz T, et al. Effects of high-dose ziprasidone and haloperidol on the QTc interval after intramuscular administration: a randomized, single-blind, parallel-group study in patients with schizophrenia or schizoaffective disorder. Clin Ther. 2010;32(3):472-491. doi:10.1016/j.clinthera.2010.03.003
7. Yocca F, DeVivo M, Seth S, et al. Dexmedetomidine—highly favorable pharmacokinetic and pharmacological features for a CNS therapeutic drug. Poster presented at: 58th Annual Meeting of the American College of Neuropsychopharmacology; December 8-11, 2019; Orlando, FL.
8. Adedoyin A, Preskorn S, Lathia CD. Pharmacokinetics of dexmedetomidine after a single sublingual dose of BXCL501 in patients with agitation associated with schizophrenia. Poster presented at: 23rd Annual Conference of the International Society for Bipolar Disorders; May 13-15, 2021. Virtual. Session 17.
9. Citrome LL, Lauriello J, Risinger R, et al. A novel rapidly effective treatment of agitation for schizophrenia with the oral dissolving film BXCL501. Poster presented at: American Psychiatric Association Annual Meeting; May 1-3, 2021. Virtual. Accessed November 11, 2021. https://www.psychiatry.org/File%20Library/Psychiatrists/Meetings/Annual-Meeting/2021/2021-APA-Annual-Meeting-Poster-Proceedings.pdf
10. Preskorn SH, Zeller S, Citrome L, et al. Effect of sublingual dexmedetomidine vs placebo on acute agitation associated with bipolar disorder: a randomized clinical trial. JAMA. 2022;327(8):727-736. doi:10.1001/jama.2022.0799
11. Montoya A, Valladares A, Lizán L, et al. Validation of the Excited Component of the Positive and Negative Syndrome Scale (PANSS-EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Qual Life Outcomes. 2011;9:18. doi:10.1186/1477-7525-9-18
12. Citrome L, Palko L, Hokett S, et al. Number needed to treat and number needed to harm from two phase 3 studies of BXCL501 for treating acute agitation in patients with schizophrenia and bipolar disorder. Poster presented at: Academy of Managed Care Pharmacy Nexus 2021; October 18-21, 2021; Denver, CO.
13. Citrome L. Comparison of intramuscular ziprasidone, olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885. doi:10.4088/jcp.v68n1207
14. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325. doi:10.1111/j.1742-1241.2011.02890.x
15. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of the American Association for Emergency Psychiatry project Beta psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34. doi:10.5811/westjem.2011.9.6866
16. Zimbroff DL, Allen MH, Battaglia J, et al. Best clinical practice with ziprasidone IM: update after 2 years of experience. CNS Spectr. 2005;10(9):1-15. doi:10.1017/s1092852900025487
1. US Food and Drug Administration. NDA 215390 Approval Letter. Accessed April 5, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215390Orig1s000ltr.pdf
2. Igalmi [package insert]. BioXcel Therapeutics, Inc; 2022.
3. Weerink MAS, Struys MMRF, Hannivoort LN, et al. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893-913. doi:10.1007/s40262-017-0507-7
4. Precedex [package insert]. Hospira, Inc; 2021.
5. Zeller SL, Citrome L. Managing agitation associated with schizophrenia and bipolar disorder in the emergency setting. West J Emerg Med. 2016;17(2):165-172. doi:10.5811/westjem.2015.12.28763
6. Miceli JJ, Tensfeldt TG, Shiovitz T, et al. Effects of high-dose ziprasidone and haloperidol on the QTc interval after intramuscular administration: a randomized, single-blind, parallel-group study in patients with schizophrenia or schizoaffective disorder. Clin Ther. 2010;32(3):472-491. doi:10.1016/j.clinthera.2010.03.003
7. Yocca F, DeVivo M, Seth S, et al. Dexmedetomidine—highly favorable pharmacokinetic and pharmacological features for a CNS therapeutic drug. Poster presented at: 58th Annual Meeting of the American College of Neuropsychopharmacology; December 8-11, 2019; Orlando, FL.
8. Adedoyin A, Preskorn S, Lathia CD. Pharmacokinetics of dexmedetomidine after a single sublingual dose of BXCL501 in patients with agitation associated with schizophrenia. Poster presented at: 23rd Annual Conference of the International Society for Bipolar Disorders; May 13-15, 2021. Virtual. Session 17.
9. Citrome LL, Lauriello J, Risinger R, et al. A novel rapidly effective treatment of agitation for schizophrenia with the oral dissolving film BXCL501. Poster presented at: American Psychiatric Association Annual Meeting; May 1-3, 2021. Virtual. Accessed November 11, 2021. https://www.psychiatry.org/File%20Library/Psychiatrists/Meetings/Annual-Meeting/2021/2021-APA-Annual-Meeting-Poster-Proceedings.pdf
10. Preskorn SH, Zeller S, Citrome L, et al. Effect of sublingual dexmedetomidine vs placebo on acute agitation associated with bipolar disorder: a randomized clinical trial. JAMA. 2022;327(8):727-736. doi:10.1001/jama.2022.0799
11. Montoya A, Valladares A, Lizán L, et al. Validation of the Excited Component of the Positive and Negative Syndrome Scale (PANSS-EC) in a naturalistic sample of 278 patients with acute psychosis and agitation in a psychiatric emergency room. Health Qual Life Outcomes. 2011;9:18. doi:10.1186/1477-7525-9-18
12. Citrome L, Palko L, Hokett S, et al. Number needed to treat and number needed to harm from two phase 3 studies of BXCL501 for treating acute agitation in patients with schizophrenia and bipolar disorder. Poster presented at: Academy of Managed Care Pharmacy Nexus 2021; October 18-21, 2021; Denver, CO.
13. Citrome L. Comparison of intramuscular ziprasidone, olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885. doi:10.4088/jcp.v68n1207
14. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325. doi:10.1111/j.1742-1241.2011.02890.x
15. Wilson MP, Pepper D, Currier GW, et al. The psychopharmacology of agitation: consensus statement of the American Association for Emergency Psychiatry project Beta psychopharmacology workgroup. West J Emerg Med. 2012;13(1):26-34. doi:10.5811/westjem.2011.9.6866
16. Zimbroff DL, Allen MH, Battaglia J, et al. Best clinical practice with ziprasidone IM: update after 2 years of experience. CNS Spectr. 2005;10(9):1-15. doi:10.1017/s1092852900025487

Evidence-based medicine: It’s not a cookbook!
The term evidence-based medicine (EBM) has been derided by some as “cookbook medicine.” To others, EBM conjures up the efforts of describing interventions in terms of comparative effectiveness, drowning us in a deluge of “evidence-based” publications. The moniker has also been hijacked by companies to name their Health Economics and Outcomes research divisions. The spirit behind EBM is getting lost. EBM is not just about the evidence; it is about how we use it.1
In this commentary, we describe the concept of EBM and discuss teaching EBM to medical students and residents, its role in continuing medical education, and how it may be applied to practice, using a case scenario as a guide.
What is evidence-based medicine?
Sackett et al2 summed it best in an editorial published in the BMJ in 1996, where he emphasized decision-making in the care of individual patients. When making clinical decisions, using the best evidence available makes sense, but so does integrating individual clinical expertise and considering the individual patient’s preferences. Sackett et al2 warns about practice becoming tyrannized by evidence: “even excellent external evidence may be inapplicable to or inappropriate for an individual patient.” Clearly, EBM is not cookbook medicine.
Figure 13 illustrates EBM as the confluence of clinical judgment, relevant scientific evidence, and patients’ values and preferences. The results from a clinical trial are only one part of the equation. As practitioners, we have the advantage of detailed knowledge about the patient, and our decisions are not “one size fits all.” Prior information about the patient dictates how we apply the evidence that supports potential interventions.

The concept of EBM was born out of necessity to bring scientific principles into the heart of medicine. As outlined by Sackett,4 the practice of EBM is a process of lifelong, self-directed learning in which caring for our own patients creates the need for clinically important information about diagnosis, prognosis, therapy, and other clinical and health care issues. Through EBM, we:
- convert these information needs into answerable questions
- track down, with maximum efficiency, the best evidence with which to answer questions (whether from clinical examination, diagnostic laboratory results, research evidence, or other sources)
- critically appraise that evidence for its validity (closeness to the truth) and usefulness (clinical applicability)
- integrate this appraisal with our clinical expertise and apply it in practice
- evaluate our performance.
Over the years, the original aim of EBM as a self-directed method for clinicians to practice high-quality medicine was morphed by some into a tool of enforced standardization and a boilerplate approach to managing costs across systems of care. As a result, the term EBM has been criticized because of:
- its reliance on empiricism
- a narrow definition of evidence
- a lack of evidence of efficacy
- its limited usefulness for individual patients
- threats to the autonomy of the doctor-patient relationship.
These 5 categories are associated with severe drawbacks when used for individual patient care.5 In addition to problems with applying standardized population research to a specific patient with a specific set of symptoms, medications, genetic variations, and unique environment, it can take years for clinicians to change their practices to incorporate new information.6
Continue to: Evidence that is too narrow...
Evidence that is too narrow in scope may not be useful. Single-molecule pharmaceutical clinical trials have erroneously become a synonym of EBM. Such studies do not reflect complex, real-life situations. Based on such studies, FDA product labeling can be inadequate in its guidance, particularly when faced with complex comorbidities. The standard comparison of active treatment to placebo is also seen as EBM, narrowing its scope and deflecting from clinical medicine when physicians measure one treatment’s success against another vs measuring real treatments against shams. Real-life treatment choice is frequently based on considering adverse effects as important to consider as therapeutic efficacy; however, this concept is outside of the common (mis)understanding of EBM.
Conflicting and ever-changing data and the push to replace clinical thinking with general dogmas trivializes medical practice and endangers treatment outcomes. This would not happen to the extent we see now if EBM was again seen as a guide and general direction rather than a blanket, distorted requirement to follow rigid recommendations for specific patients.
Insurance companies have driven a change in the understanding of EBM by using the FDA label as an excuse to deny, delay, and/or refuse to pay for treatments that are not explicitly and narrowly on-label. Dependence on on-label treatments is even more challenging in specialty medicine because primary care clinicians generally have tried the conventional approaches before referring patients to a specialist. However, insurance denials rarely differentiate between practice settings.
Medicolegal issues have cemented the present situation when clinically valid “off-label” treatments may be a reasonable consideration for patients but can place health care practitioners in jeopardy. The distorted EBM doctrine has become a justification for legal actions against clinicians who practice individualized medicine.
Concision bias (selectively focusing on information, losing nuance) and selection bias (patients in clinical trials who do not reflect real-life patients) have become an impediment to progress and EBM as originally intended.
Continue to: Training medical students and residents
Training medical students and residents
Although there is some variation in how EBM is taught to medical students and residents,7,8 the expectation is that such education occurs. The Accreditation Council for Graduate Medical Education requirements for a residency program state that “the program must advance residents’ knowledge and practice of the scholarly approach to evidence-based patient care.”9 The topic has been part of the American Society of Clinical Psychopharmacology Model Psychopharmacology Curriculum, but only in an optional lecture.10 The formal teaching of EBM includes how to find relevant biomedical publications for the clinical issues at hand, understand the different hierarchies of evidence, interpret results in terms of effect size, and apply this knowledge in the care of patients. This 5-step process is illustrated in Figure 28. See Related Resources for 3 books that provide a scholarly yet clinically relevant approach to EBM.

Continuing medical education
Most
Practical applications
There are common clinical scenarios where evidence is ignored, or where it is overvalued. For example, the treatment of bipolar depression can be made worse with the use of antidepressants.14 Does this mean that antidepressants should never be used? What about patient history and preference? What if the approved agents fail to relieve symptoms or are not well tolerated? Available FDA-approved choices may not always be suitable.15 The Table illustrates some of these scenarios.

1. Citrome L. Evidence-based medicine: it’s not just about the evidence. Int J Clin Pract. 2011;65(6):634-635.
2. Sackett DL, Rosenberg WM, Gray JA, et al. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71.
3. Citrome L. Think Bayesian, think smarter! Int J Clin Pract. 2019;73(4):e13351. doi.org/10.1111/ijcp.13351
4. Sackett DL. Evidence-based medicine. Semin Perinatol. 1997;21(1):3-5.
5. Cohen AM, Stavri PZ, Hersh WR. A categorization and analysis of the criticisms of evidence-based medicine. Int J Med Inform. 2004;73(1):35-43.
6. Dutton DB. Worse than the disease: pitfalls of medical progress. Cambridge University Press; 1988.
7. Maggio LA. Educating physicians in evidence based medicine: current practices and curricular strategies. Perspect Med Educ. 2016;5(6):358-361.
8. Citrome L, Ketter TA. Teaching the philosophy and tools of evidence-based medicine: misunderstandings and solutions. Int J Clin Pract. 2009;63(3):353-359.
9. Accreditation Council for Graduate Medical Education. ACGME Common Program Requirements (Residency). Revised February 3, 2020. Accessed March 30, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
10. Citrome L, Ellison JM. Show me the evidence! Understanding the philosophy of evidence-based medicine and interpreting clinical trials. In: Glick ID, Macaluso M (Chair, Co-chair). ASCP model psychopharmacology curriculum for training directors and teachers of psychopharmacology in psychiatric residency programs, 10th ed. American Society of Clinical Psychopharmacology; 2019.
11. Citrome L. Interpreting and applying the CATIE results: with CATIE, context is key, when sorting out Phases 1, 1A, 1B, 2E, and 2T. Psychiatry (Edgmont). 2007;4(10):23-29.
12. Citrome L, Stroup TS. Schizophrenia, clinical antipsychotic trials of intervention effectiveness (CATIE) and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-940. doi: 10.1111/j.1742-1241.2006.01044.x
13. Citrome L. Dissecting clinical trials with ‘number needed to treat’. Current Psychiatry. 2007;6(3):66-71.
14. Goldberg JF, Freeman MP, Balon R, et al. The American Society of Clinical Psychopharmacology survey of psychopharmacologists’ practice patterns for the treatment of mood disorders. Depress Anxiety. 2015;32(8):605-613.
15. Citrome L. Food and Drug Administration-approved treatments for acute bipolar depression: what we have and what we need. J Clin Psychopharmacol. 2020;40(4):334-338.
The term evidence-based medicine (EBM) has been derided by some as “cookbook medicine.” To others, EBM conjures up the efforts of describing interventions in terms of comparative effectiveness, drowning us in a deluge of “evidence-based” publications. The moniker has also been hijacked by companies to name their Health Economics and Outcomes research divisions. The spirit behind EBM is getting lost. EBM is not just about the evidence; it is about how we use it.1
In this commentary, we describe the concept of EBM and discuss teaching EBM to medical students and residents, its role in continuing medical education, and how it may be applied to practice, using a case scenario as a guide.
What is evidence-based medicine?
Sackett et al2 summed it best in an editorial published in the BMJ in 1996, where he emphasized decision-making in the care of individual patients. When making clinical decisions, using the best evidence available makes sense, but so does integrating individual clinical expertise and considering the individual patient’s preferences. Sackett et al2 warns about practice becoming tyrannized by evidence: “even excellent external evidence may be inapplicable to or inappropriate for an individual patient.” Clearly, EBM is not cookbook medicine.
Figure 13 illustrates EBM as the confluence of clinical judgment, relevant scientific evidence, and patients’ values and preferences. The results from a clinical trial are only one part of the equation. As practitioners, we have the advantage of detailed knowledge about the patient, and our decisions are not “one size fits all.” Prior information about the patient dictates how we apply the evidence that supports potential interventions.
The concept of EBM was born out of necessity to bring scientific principles into the heart of medicine. As outlined by Sackett,4 the practice of EBM is a process of lifelong, self-directed learning in which caring for our own patients creates the need for clinically important information about diagnosis, prognosis, therapy, and other clinical and health care issues. Through EBM, we:
- convert these information needs into answerable questions
- track down, with maximum efficiency, the best evidence with which to answer questions (whether from clinical examination, diagnostic laboratory results, research evidence, or other sources)
- critically appraise that evidence for its validity (closeness to the truth) and usefulness (clinical applicability)
- integrate this appraisal with our clinical expertise and apply it in practice
- evaluate our performance.
Over the years, the original aim of EBM as a self-directed method for clinicians to practice high-quality medicine was morphed by some into a tool of enforced standardization and a boilerplate approach to managing costs across systems of care. As a result, the term EBM has been criticized because of:
- its reliance on empiricism
- a narrow definition of evidence
- a lack of evidence of efficacy
- its limited usefulness for individual patients
- threats to the autonomy of the doctor-patient relationship.
These 5 categories are associated with severe drawbacks when used for individual patient care.5 In addition to problems with applying standardized population research to a specific patient with a specific set of symptoms, medications, genetic variations, and unique environment, it can take years for clinicians to change their practices to incorporate new information.6
Continue to: Evidence that is too narrow...
Evidence that is too narrow in scope may not be useful. Single-molecule pharmaceutical clinical trials have erroneously become a synonym of EBM. Such studies do not reflect complex, real-life situations. Based on such studies, FDA product labeling can be inadequate in its guidance, particularly when faced with complex comorbidities. The standard comparison of active treatment to placebo is also seen as EBM, narrowing its scope and deflecting from clinical medicine when physicians measure one treatment’s success against another vs measuring real treatments against shams. Real-life treatment choice is frequently based on considering adverse effects as important to consider as therapeutic efficacy; however, this concept is outside of the common (mis)understanding of EBM.
Conflicting and ever-changing data and the push to replace clinical thinking with general dogmas trivializes medical practice and endangers treatment outcomes. This would not happen to the extent we see now if EBM was again seen as a guide and general direction rather than a blanket, distorted requirement to follow rigid recommendations for specific patients.
Insurance companies have driven a change in the understanding of EBM by using the FDA label as an excuse to deny, delay, and/or refuse to pay for treatments that are not explicitly and narrowly on-label. Dependence on on-label treatments is even more challenging in specialty medicine because primary care clinicians generally have tried the conventional approaches before referring patients to a specialist. However, insurance denials rarely differentiate between practice settings.
Medicolegal issues have cemented the present situation when clinically valid “off-label” treatments may be a reasonable consideration for patients but can place health care practitioners in jeopardy. The distorted EBM doctrine has become a justification for legal actions against clinicians who practice individualized medicine.
Concision bias (selectively focusing on information, losing nuance) and selection bias (patients in clinical trials who do not reflect real-life patients) have become an impediment to progress and EBM as originally intended.
Continue to: Training medical students and residents
Training medical students and residents
Although there is some variation in how EBM is taught to medical students and residents,7,8 the expectation is that such education occurs. The Accreditation Council for Graduate Medical Education requirements for a residency program state that “the program must advance residents’ knowledge and practice of the scholarly approach to evidence-based patient care.”9 The topic has been part of the American Society of Clinical Psychopharmacology Model Psychopharmacology Curriculum, but only in an optional lecture.10 The formal teaching of EBM includes how to find relevant biomedical publications for the clinical issues at hand, understand the different hierarchies of evidence, interpret results in terms of effect size, and apply this knowledge in the care of patients. This 5-step process is illustrated in Figure 28. See Related Resources for 3 books that provide a scholarly yet clinically relevant approach to EBM.
Continuing medical education
Most
Practical applications
There are common clinical scenarios where evidence is ignored, or where it is overvalued. For example, the treatment of bipolar depression can be made worse with the use of antidepressants.14 Does this mean that antidepressants should never be used? What about patient history and preference? What if the approved agents fail to relieve symptoms or are not well tolerated? Available FDA-approved choices may not always be suitable.15 The Table illustrates some of these scenarios.
The term evidence-based medicine (EBM) has been derided by some as “cookbook medicine.” To others, EBM conjures up the efforts of describing interventions in terms of comparative effectiveness, drowning us in a deluge of “evidence-based” publications. The moniker has also been hijacked by companies to name their Health Economics and Outcomes research divisions. The spirit behind EBM is getting lost. EBM is not just about the evidence; it is about how we use it.1
In this commentary, we describe the concept of EBM and discuss teaching EBM to medical students and residents, its role in continuing medical education, and how it may be applied to practice, using a case scenario as a guide.
What is evidence-based medicine?
Sackett et al2 summed it best in an editorial published in the BMJ in 1996, where he emphasized decision-making in the care of individual patients. When making clinical decisions, using the best evidence available makes sense, but so does integrating individual clinical expertise and considering the individual patient’s preferences. Sackett et al2 warns about practice becoming tyrannized by evidence: “even excellent external evidence may be inapplicable to or inappropriate for an individual patient.” Clearly, EBM is not cookbook medicine.
Figure 13 illustrates EBM as the confluence of clinical judgment, relevant scientific evidence, and patients’ values and preferences. The results from a clinical trial are only one part of the equation. As practitioners, we have the advantage of detailed knowledge about the patient, and our decisions are not “one size fits all.” Prior information about the patient dictates how we apply the evidence that supports potential interventions.
The concept of EBM was born out of necessity to bring scientific principles into the heart of medicine. As outlined by Sackett,4 the practice of EBM is a process of lifelong, self-directed learning in which caring for our own patients creates the need for clinically important information about diagnosis, prognosis, therapy, and other clinical and health care issues. Through EBM, we:
- convert these information needs into answerable questions
- track down, with maximum efficiency, the best evidence with which to answer questions (whether from clinical examination, diagnostic laboratory results, research evidence, or other sources)
- critically appraise that evidence for its validity (closeness to the truth) and usefulness (clinical applicability)
- integrate this appraisal with our clinical expertise and apply it in practice
- evaluate our performance.
Over the years, the original aim of EBM as a self-directed method for clinicians to practice high-quality medicine was morphed by some into a tool of enforced standardization and a boilerplate approach to managing costs across systems of care. As a result, the term EBM has been criticized because of:
- its reliance on empiricism
- a narrow definition of evidence
- a lack of evidence of efficacy
- its limited usefulness for individual patients
- threats to the autonomy of the doctor-patient relationship.
These 5 categories are associated with severe drawbacks when used for individual patient care.5 In addition to problems with applying standardized population research to a specific patient with a specific set of symptoms, medications, genetic variations, and unique environment, it can take years for clinicians to change their practices to incorporate new information.6
Continue to: Evidence that is too narrow...
Evidence that is too narrow in scope may not be useful. Single-molecule pharmaceutical clinical trials have erroneously become a synonym of EBM. Such studies do not reflect complex, real-life situations. Based on such studies, FDA product labeling can be inadequate in its guidance, particularly when faced with complex comorbidities. The standard comparison of active treatment to placebo is also seen as EBM, narrowing its scope and deflecting from clinical medicine when physicians measure one treatment’s success against another vs measuring real treatments against shams. Real-life treatment choice is frequently based on considering adverse effects as important to consider as therapeutic efficacy; however, this concept is outside of the common (mis)understanding of EBM.
Conflicting and ever-changing data and the push to replace clinical thinking with general dogmas trivializes medical practice and endangers treatment outcomes. This would not happen to the extent we see now if EBM was again seen as a guide and general direction rather than a blanket, distorted requirement to follow rigid recommendations for specific patients.
Insurance companies have driven a change in the understanding of EBM by using the FDA label as an excuse to deny, delay, and/or refuse to pay for treatments that are not explicitly and narrowly on-label. Dependence on on-label treatments is even more challenging in specialty medicine because primary care clinicians generally have tried the conventional approaches before referring patients to a specialist. However, insurance denials rarely differentiate between practice settings.
Medicolegal issues have cemented the present situation when clinically valid “off-label” treatments may be a reasonable consideration for patients but can place health care practitioners in jeopardy. The distorted EBM doctrine has become a justification for legal actions against clinicians who practice individualized medicine.
Concision bias (selectively focusing on information, losing nuance) and selection bias (patients in clinical trials who do not reflect real-life patients) have become an impediment to progress and EBM as originally intended.
Continue to: Training medical students and residents
Training medical students and residents
Although there is some variation in how EBM is taught to medical students and residents,7,8 the expectation is that such education occurs. The Accreditation Council for Graduate Medical Education requirements for a residency program state that “the program must advance residents’ knowledge and practice of the scholarly approach to evidence-based patient care.”9 The topic has been part of the American Society of Clinical Psychopharmacology Model Psychopharmacology Curriculum, but only in an optional lecture.10 The formal teaching of EBM includes how to find relevant biomedical publications for the clinical issues at hand, understand the different hierarchies of evidence, interpret results in terms of effect size, and apply this knowledge in the care of patients. This 5-step process is illustrated in Figure 28. See Related Resources for 3 books that provide a scholarly yet clinically relevant approach to EBM.
Continuing medical education
Most
Practical applications
There are common clinical scenarios where evidence is ignored, or where it is overvalued. For example, the treatment of bipolar depression can be made worse with the use of antidepressants.14 Does this mean that antidepressants should never be used? What about patient history and preference? What if the approved agents fail to relieve symptoms or are not well tolerated? Available FDA-approved choices may not always be suitable.15 The Table illustrates some of these scenarios.
1. Citrome L. Evidence-based medicine: it’s not just about the evidence. Int J Clin Pract. 2011;65(6):634-635.
2. Sackett DL, Rosenberg WM, Gray JA, et al. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71.
3. Citrome L. Think Bayesian, think smarter! Int J Clin Pract. 2019;73(4):e13351. doi.org/10.1111/ijcp.13351
4. Sackett DL. Evidence-based medicine. Semin Perinatol. 1997;21(1):3-5.
5. Cohen AM, Stavri PZ, Hersh WR. A categorization and analysis of the criticisms of evidence-based medicine. Int J Med Inform. 2004;73(1):35-43.
6. Dutton DB. Worse than the disease: pitfalls of medical progress. Cambridge University Press; 1988.
7. Maggio LA. Educating physicians in evidence based medicine: current practices and curricular strategies. Perspect Med Educ. 2016;5(6):358-361.
8. Citrome L, Ketter TA. Teaching the philosophy and tools of evidence-based medicine: misunderstandings and solutions. Int J Clin Pract. 2009;63(3):353-359.
9. Accreditation Council for Graduate Medical Education. ACGME Common Program Requirements (Residency). Revised February 3, 2020. Accessed March 30, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
10. Citrome L, Ellison JM. Show me the evidence! Understanding the philosophy of evidence-based medicine and interpreting clinical trials. In: Glick ID, Macaluso M (Chair, Co-chair). ASCP model psychopharmacology curriculum for training directors and teachers of psychopharmacology in psychiatric residency programs, 10th ed. American Society of Clinical Psychopharmacology; 2019.
11. Citrome L. Interpreting and applying the CATIE results: with CATIE, context is key, when sorting out Phases 1, 1A, 1B, 2E, and 2T. Psychiatry (Edgmont). 2007;4(10):23-29.
12. Citrome L, Stroup TS. Schizophrenia, clinical antipsychotic trials of intervention effectiveness (CATIE) and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-940. doi: 10.1111/j.1742-1241.2006.01044.x
13. Citrome L. Dissecting clinical trials with ‘number needed to treat’. Current Psychiatry. 2007;6(3):66-71.
14. Goldberg JF, Freeman MP, Balon R, et al. The American Society of Clinical Psychopharmacology survey of psychopharmacologists’ practice patterns for the treatment of mood disorders. Depress Anxiety. 2015;32(8):605-613.
15. Citrome L. Food and Drug Administration-approved treatments for acute bipolar depression: what we have and what we need. J Clin Psychopharmacol. 2020;40(4):334-338.
1. Citrome L. Evidence-based medicine: it’s not just about the evidence. Int J Clin Pract. 2011;65(6):634-635.
2. Sackett DL, Rosenberg WM, Gray JA, et al. Evidence based medicine: what it is and what it isn’t. BMJ. 1996;312(7023):71.
3. Citrome L. Think Bayesian, think smarter! Int J Clin Pract. 2019;73(4):e13351. doi.org/10.1111/ijcp.13351
4. Sackett DL. Evidence-based medicine. Semin Perinatol. 1997;21(1):3-5.
5. Cohen AM, Stavri PZ, Hersh WR. A categorization and analysis of the criticisms of evidence-based medicine. Int J Med Inform. 2004;73(1):35-43.
6. Dutton DB. Worse than the disease: pitfalls of medical progress. Cambridge University Press; 1988.
7. Maggio LA. Educating physicians in evidence based medicine: current practices and curricular strategies. Perspect Med Educ. 2016;5(6):358-361.
8. Citrome L, Ketter TA. Teaching the philosophy and tools of evidence-based medicine: misunderstandings and solutions. Int J Clin Pract. 2009;63(3):353-359.
9. Accreditation Council for Graduate Medical Education. ACGME Common Program Requirements (Residency). Revised February 3, 2020. Accessed March 30, 2021. https://www.acgme.org/Portals/0/PFAssets/ProgramRequirements/CPRResidency2020.pdf
10. Citrome L, Ellison JM. Show me the evidence! Understanding the philosophy of evidence-based medicine and interpreting clinical trials. In: Glick ID, Macaluso M (Chair, Co-chair). ASCP model psychopharmacology curriculum for training directors and teachers of psychopharmacology in psychiatric residency programs, 10th ed. American Society of Clinical Psychopharmacology; 2019.
11. Citrome L. Interpreting and applying the CATIE results: with CATIE, context is key, when sorting out Phases 1, 1A, 1B, 2E, and 2T. Psychiatry (Edgmont). 2007;4(10):23-29.
12. Citrome L, Stroup TS. Schizophrenia, clinical antipsychotic trials of intervention effectiveness (CATIE) and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-940. doi: 10.1111/j.1742-1241.2006.01044.x
13. Citrome L. Dissecting clinical trials with ‘number needed to treat’. Current Psychiatry. 2007;6(3):66-71.
14. Goldberg JF, Freeman MP, Balon R, et al. The American Society of Clinical Psychopharmacology survey of psychopharmacologists’ practice patterns for the treatment of mood disorders. Depress Anxiety. 2015;32(8):605-613.
15. Citrome L. Food and Drug Administration-approved treatments for acute bipolar depression: what we have and what we need. J Clin Psychopharmacol. 2020;40(4):334-338.

Asenapine transdermal system for schizophrenia

The asenapine transdermal system is available in 3 patch sizes: 20, 30, and 40 cm2, which deliver 3.8, 5.7, and 7.6 mg/24 hours of asenapine, respectively.3 Based on the average exposure (area under the plasma concentration curve [AUC]) of asenapine, 3.8 mg/24 hours corresponds to 5 mg twice daily of sublingual asenapine, and 7.6 mg/24 hours corresponds to 10 mg twice daily of sublingual asenapine.3 The “in-between” dose strength of 5.7 mg/24 hours would correspond to exposure to a total of 15 mg/d of sublingual asenapine. The recommended starting dose for asenapine transdermal system is 3.8 mg/24 hours. The dosage may be increased to 5.7 mg/24 hours or 7.6 mg/24 hours, as needed, after 1 week. The safety of doses above 7.6 mg/24 hours has not been evaluated in clinical studies. Asenapine transdermal system is applied once daily and should be worn for 24 hours only, with only 1 patch at any time. Application sites include the upper arm, upper back, abdomen, and hip. A different application site of clean, dry, intact skin should be selected each time a new patch is applied. Although showering is permitted, the use of asenapine transdermal system during swimming or taking a bath has not been evaluated. Of note, prolonged application of heat over an asenapine transdermal system increases plasma concentrations of asenapine, and thus application of external heat sources (eg, heating pads) over the patch should be avoided.
How it works
Product labeling notes that asenapine is an atypical antipsychotic, and that its efficacy in schizophrenia could be mediated through a combination of antagonist activity at dopamine D2 and serotonin 5-HT2A receptors.3 The pharmacodynamic profile of asenapine is complex5 and receptor-binding assays performed using cloned human serotonin, norepinephrine, dopamine, histamine, and muscarinic receptors demonstrated picomolar affinity (extremely high) for 5-HT2C and 5-HT2A receptors, subnanomolar affinity (very high) for 5-HT7, 5-HT2B, 5-HT6, and D3 receptors, and nanomolar affinity (high) for D2 receptors, as well as histamine H1, D4, a1-adrenergic, a2-adrenergic, D1, 5-HT5, 5-HT1A, 5-HT1B, and histamine H2 receptors. Activity of asenapine is that of antagonism at these receptors. Asenapine has no appreciable affinity for muscarinic cholinergic receptors.
The asenapine receptor-binding “fingerprint” differs from that of other antipsychotics. Some of these receptor affinities are of special interest in terms of potential efficacy for pro-cognitive effects and amelioration of abnormal mood.5,9 In terms of tolerability, a relative absence of affinity to muscarinic receptors would predict a low risk for anticholinergic adverse effects, but antagonism at histamine H1 and at a1-adrenergic receptors, either alone or in combination, may cause sedation, and blockade of H1 receptors would also predict weight gain.9 Antagonism of a1-adrenergic receptors can be associated with orthostatic hypotension and neurally mediated reflex bradycardia.9
Clinical pharmacokinetics
Three open-label, randomized, phase 1 studies were conducted to assess the relative bioavailability of asenapine transdermal system vs sublingual asenapine.10 These included single- and multiple-dose studies and clinical trials that examined the effects of different application sites and ethnic groups, and the effect of external heat on medication absorption. Studies were conducted in healthy individuals, except for the multiple-dose study, which was performed in adults with schizophrenia. The AUC for asenapine transdermal system was within the range of that of equivalent doses of sublingual asenapine, but peak exposure (maximum concentration) was significantly lower. As already noted, the AUC of the asenapine patch for 3.8 mg/24 hours and 7.6 mg/24 hours corresponds to sublingual asenapine 5 mg and 10 mg twice daily, respectively. Maximum asenapine concentrations are typically reached between 12 and 24 hours, with sustained concentrations during the 24-hour wear time.3 On average, approximately 60% of the available asenapine is released from the transdermal system over 24 hours. Steady-state plasma concentrations for asenapine transdermal system were achieved approximately 72 hours after the first application and, in contrast to sublingual asenapine, the peak-trough fluctuations were small (peak-to-trough ratio is 1.5 for asenapine transdermal system compared with >3 for sublingual asenapine). Dose-proportionality at steady state was evident for asenapine transdermal system. This is in contrast to sublingual asenapine, where exposure increases 1.7-fold with a 2-fold increase in dose.4,5 Following patch removal, the apparent elimination half-life is approximately 30 hours.3 The pharmacokinetics of the patch did not vary with regards to the application site (upper arm, upper back, abdomen, or hip area), and the pharmacokinetic profile was similar across the ethnic groups that participated in the study. Direct exposure to external heat did increase both the rate and extent of absorption, so external heat sources should be avoided.3
Efficacy
The efficacy profile for asenapine transdermal system would be expected to mirror that for sublingual asenapine.6,7 In addition to data supporting the use of asenapine as administered sublingually, a phase 3 study specifically assessed efficacy and safety of asenapine transdermal system in adults with schizophrenia.11,12 This study was conducted in the United States and 4 other countries at a total of 59 study sites, and 616 patients with acutely exacerbated schizophrenia were enrolled. After a 3- to 14-day screening/single-blind run-in washout period, participants entered a 6-week inpatient double-blind period. Randomization was 1:1:1 to asenapine transdermal system 3.8 mg/24 hours, 7.6 mg/24 hours, or a placebo patch. Each of the patch doses demonstrated significant improvement vs placebo at Week 6 for the primary (change in Positive and Negative Syndrome Scale [PANSS] total score) and key secondary (change in Clinical Global Impression-Severity of Illness) endpoints. Response at endpoint, as defined by a ≥30% improvement from baseline PANSS total score, or by a Clinical Global Impression–Improvement score of 1 (very much improved) or 2 (much improved), was also assessed. For either definition of response, both doses of asenapine transdermal system were superior to placebo, with number needed to treat (NNT) (Box) values <10 for the 3.8 mg/24 hours dose (Table 2). These effect sizes are similar to what is known about sublingual asenapine as determined in a meta-analysis performed by the manufacturer and using individual patient data.13
Box
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies, you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we reviewing a result that may be statistically significant but irrelevant for day-today patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—two tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between two interventions. If we see a therapeutic difference once every 100 patients (NNT of 100), the difference between the treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 7 patients being treated with an intervention vs another (NNT of 7), the result will likely influence dayto-day practice.
How to calculate NNT (or NNH):
What is the NNT for an outcome for drug A vs drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates: .75 -.55 = .20
- NNT: 1/.20 = 5
A rule of thumb: NNT values for a medication vs placebo <10 usually denote a medication we use on a regular basis to treat patients.
a Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71. Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82. Additional information can be found in Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411 (free to access at onlinelibrary.wiley.com/doi/full/10.1111/ijcp.12142)
Overall tolerability and safety
The systemic safety and tolerability profile for asenapine transdermal system would be expected to be similar to that for sublingual asenapine, unless there are adverse events that are related to high peak plasma concentrations or large differences between peak and trough plasma concentrations.6 Nonsystemic local application site adverse events would, of course, differ between sublingual vs transdermal administration.

Continue to: Use of asenapine transdermal system...
Use of asenapine transdermal system avoids the dysgeusia and oral hypoesthesia that can be observed with sublingual asenapine4,6; however, dermal effects need to be considered (see Dermal safety). The most commonly observed adverse reactions (incidence ≥5% and at least twice that for placebo) for asenapine transdermal system are extrapyramidal disorder, application site reaction, and weight gain.3 For sublingual asenapine for adults with schizophrenia, the list includes akathisia, oral hypoesthesia, and somnolence.4 These adverse events can be further described using the metric of number needed to harm (NNH) as shown in Table 3.3,4,11,12,14 Of note, extrapyramidal disorder and weight gain appear to be dose-related for asenapine transdermal system. Akathisia appears to be dose-related for sublingual asenapine but not for asenapine transdermal system. Somnolence appears to be associated with sublingual asenapine but not necessarily with asenapine transdermal system.

For sublingual asenapine, the additional indications (bipolar I disorder as acute monotherapy treatment of manic or mixed episodes in adults and pediatric patients age 10 to 17, adjunctive treatment to lithium or valproate in adults, and maintenance monotherapy treatment in adults) have varying commonly encountered adverse reactions.4 Both transdermal asenapine system and sublingual asenapine are contraindicated in patients with severe hepatic impairment (Child-Pugh C) and those with known hypersensitivity to asenapine or to any components in the formulation. Both formulations carry similar warnings in their prescribing information regarding increased mortality in older patients with dementia-related psychosis, cerebrovascular adverse reactions in older patients with dementia-related psychosis, neuroleptic malignant syndrome, tardive dyskinesia, metabolic changes, orthostatic hypotension, leukopenia (and neutropenia and agranulocytosis), QT prolongation, seizures, and potential for cognitive and motor impairment.
Adverse events leading to discontinuation of study treatment in the asenapine transdermal system pivotal trial occurred in 4.9%, 7.8%, and 6.8% of participants in the 3.8 mg/24 hour, 7.6 mg/24 hour, and placebo groups, respectively.11
Dermal safety
In the pivotal efficacy study,11 the incidence of adverse events at patch application sites was higher in the active groups vs placebo (Table 33,4,11,12,14). The most frequently reported patch application site reactions were erythema and pruritus, occurring in approximately 10% and 4% in the active treatment arms vs 1.5% and 1.9% for placebo, respectively. With the exception of 1 adverse event of severe application site erythema during Week 2 (participant received 7.6 mg/24 hour, erythema resolved without intervention, and the patient continued the study), all other patch application site events were mild or moderate in severity. Rates of discontinuation due to application site reactions or skin disorders were ≤0.5% across all groups. In the pharmacokinetic studies,10 no patches were removed because of unacceptable irritation.
Why Rx?
Asenapine transdermal system is the first antipsychotic “patch” FDA-approved for the treatment of adults with schizophrenia. Asenapine has been available since 2009 as a sublingual formulation administered twice daily. The pharmacokinetic profile of the once-daily transdermal system demonstrates dose-proportional kinetics and sustained delivery of asenapine with a low peak-to-trough plasma level ratio. Three dosage strengths (3.8, 5.7, and 7.6 mg/24 hours) are available, corresponding to blood levels attained with sublingual asenapine exposures of 10, 15, and 20 mg/d, respectively. Application sites are rotated daily and include the upper arms, upper back, abdomen, or hip. Dysgeusia and hypoesthesia of the tongue are avoided with the use of the patch, and there are no food or drink restrictions. Attention will be needed in case of dermal reactions, similar to that observed with other medication patches.
Bottom Line
The asenapine transdermal drug delivery system appears to be efficacious and reasonably well tolerated. The treatment of schizophrenia is complex and requires individualized choices in order to optimize outcomes. A patch may be the preferred formulation for selected patients, and caregivers will have the ability to visually check if the medication is being used.
Related Resource
- Hisamitsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
Drug Brand Names
Asenapine sublingual • Saphris
Asenapine transdermal system • Secuado
Lithium • Eskalith, Lithobid
Valproate • Depakote
1. Noven. US FDA approves SECUADO® (asenapine) transdermal system, the first-and-only transdermal patch for the treatment of adults with schizophrenia. October 15, 2019. Accessed January 15, 2021. https://www.noven.com/wp-content/uploads/2020/04/PR101519.pdf
2. US Food and Drug Administration. Center for Drug Evaluation and Research. Approval Package for: APPLICATION NUMBER: 212268Orig1s000. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000Approv.pdf
3. Hisam itsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
4. Allergan USA, Inc. SAPHRIS® (asenapine) sublingual tablets prescribing information. February 2017. Accessed January 15, 2021. https://media.allergan.com/actavis/actavis/media/allergan-pdf-documents/product-prescribing/Final_labeling_text_SAPHRIS-clean-02-2017.pdf
5. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
6. Citrome L. Asenapine review, part II: clinical efficacy, safety and tolerability. Expert Opin Drug Saf. 2014;13(6):803-830.
7. Citrome L. Chapter 31: Asenapine. In: Schatzberg AF, Nemeroff CB, eds. The American Psychiatric Association Publishing Textbook of Psychopharmacology, 5th ed. American Psychiatric Association Publishing; 2017:797-808.
8. Citrome L, Zeni CM, Correll CU. Patches: established and emerging transdermal treatments in psychiatry. J Clin Psychiatry. 2019;80(4):18nr12554. doi: 10.4088/JCP.18nr12554
9. Shayegan DK, Stahl SM. Atypical antipsychotics: matching receptor profile to individual patient’s clinical profile. CNS Spectr. 2004;9(10 suppl 11):6-14.
10. Castelli M, Suzuki K, Komaroff M, et al. Pharmacokinetic profile of asenapine transdermal system HP-3070: The first antipsychotic patch in the US. Poster presented virtually at the American Society for Clinical Psychopharmacology (ASCP) 2020 Annual Meeting, May 29-30, 2020. https://www.psychiatrist.com/ascpcorner/Documents/ascp2020/3_ASCP%20Poster%20Abstracts%202020-JCP.pdf
11. Citrome L, Walling DP, Zeni CM, et al. Efficacy and safety of HP-3070, an asenapine transdermal system, in patients with schizophrenia: a phase 3, randomized, placebo-controlled study. J Clin Psychiatry. 2020;82(1):20m13602. doi: 10.4088/JCP.20m13602
12. US Food and Drug Administration. Drug Approval Package: SECAUDO. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000TOC.cfm
13. Szegedi A, Verweij P, van Duijnhoven W, et al. Meta-analyses of the efficacy of asenapine for acute schizophrenia: comparisons with placebo and other antipsychotics. J Clin Psychiatry. 2012;73(12):1533-1540.
14. Citrome L. Asenapine for schizophrenia and bipolar disorder: a review of the efficacy and safety profile for this newly approved sublingually absorbed second-generation antipsychotic. Int J Clin Pract. 2009;63(12):1762-1784.
The asenapine transdermal system is available in 3 patch sizes: 20, 30, and 40 cm2, which deliver 3.8, 5.7, and 7.6 mg/24 hours of asenapine, respectively.3 Based on the average exposure (area under the plasma concentration curve [AUC]) of asenapine, 3.8 mg/24 hours corresponds to 5 mg twice daily of sublingual asenapine, and 7.6 mg/24 hours corresponds to 10 mg twice daily of sublingual asenapine.3 The “in-between” dose strength of 5.7 mg/24 hours would correspond to exposure to a total of 15 mg/d of sublingual asenapine. The recommended starting dose for asenapine transdermal system is 3.8 mg/24 hours. The dosage may be increased to 5.7 mg/24 hours or 7.6 mg/24 hours, as needed, after 1 week. The safety of doses above 7.6 mg/24 hours has not been evaluated in clinical studies. Asenapine transdermal system is applied once daily and should be worn for 24 hours only, with only 1 patch at any time. Application sites include the upper arm, upper back, abdomen, and hip. A different application site of clean, dry, intact skin should be selected each time a new patch is applied. Although showering is permitted, the use of asenapine transdermal system during swimming or taking a bath has not been evaluated. Of note, prolonged application of heat over an asenapine transdermal system increases plasma concentrations of asenapine, and thus application of external heat sources (eg, heating pads) over the patch should be avoided.
How it works
Product labeling notes that asenapine is an atypical antipsychotic, and that its efficacy in schizophrenia could be mediated through a combination of antagonist activity at dopamine D2 and serotonin 5-HT2A receptors.3 The pharmacodynamic profile of asenapine is complex5 and receptor-binding assays performed using cloned human serotonin, norepinephrine, dopamine, histamine, and muscarinic receptors demonstrated picomolar affinity (extremely high) for 5-HT2C and 5-HT2A receptors, subnanomolar affinity (very high) for 5-HT7, 5-HT2B, 5-HT6, and D3 receptors, and nanomolar affinity (high) for D2 receptors, as well as histamine H1, D4, a1-adrenergic, a2-adrenergic, D1, 5-HT5, 5-HT1A, 5-HT1B, and histamine H2 receptors. Activity of asenapine is that of antagonism at these receptors. Asenapine has no appreciable affinity for muscarinic cholinergic receptors.
The asenapine receptor-binding “fingerprint” differs from that of other antipsychotics. Some of these receptor affinities are of special interest in terms of potential efficacy for pro-cognitive effects and amelioration of abnormal mood.5,9 In terms of tolerability, a relative absence of affinity to muscarinic receptors would predict a low risk for anticholinergic adverse effects, but antagonism at histamine H1 and at a1-adrenergic receptors, either alone or in combination, may cause sedation, and blockade of H1 receptors would also predict weight gain.9 Antagonism of a1-adrenergic receptors can be associated with orthostatic hypotension and neurally mediated reflex bradycardia.9
Clinical pharmacokinetics
Three open-label, randomized, phase 1 studies were conducted to assess the relative bioavailability of asenapine transdermal system vs sublingual asenapine.10 These included single- and multiple-dose studies and clinical trials that examined the effects of different application sites and ethnic groups, and the effect of external heat on medication absorption. Studies were conducted in healthy individuals, except for the multiple-dose study, which was performed in adults with schizophrenia. The AUC for asenapine transdermal system was within the range of that of equivalent doses of sublingual asenapine, but peak exposure (maximum concentration) was significantly lower. As already noted, the AUC of the asenapine patch for 3.8 mg/24 hours and 7.6 mg/24 hours corresponds to sublingual asenapine 5 mg and 10 mg twice daily, respectively. Maximum asenapine concentrations are typically reached between 12 and 24 hours, with sustained concentrations during the 24-hour wear time.3 On average, approximately 60% of the available asenapine is released from the transdermal system over 24 hours. Steady-state plasma concentrations for asenapine transdermal system were achieved approximately 72 hours after the first application and, in contrast to sublingual asenapine, the peak-trough fluctuations were small (peak-to-trough ratio is 1.5 for asenapine transdermal system compared with >3 for sublingual asenapine). Dose-proportionality at steady state was evident for asenapine transdermal system. This is in contrast to sublingual asenapine, where exposure increases 1.7-fold with a 2-fold increase in dose.4,5 Following patch removal, the apparent elimination half-life is approximately 30 hours.3 The pharmacokinetics of the patch did not vary with regards to the application site (upper arm, upper back, abdomen, or hip area), and the pharmacokinetic profile was similar across the ethnic groups that participated in the study. Direct exposure to external heat did increase both the rate and extent of absorption, so external heat sources should be avoided.3
Efficacy
The efficacy profile for asenapine transdermal system would be expected to mirror that for sublingual asenapine.6,7 In addition to data supporting the use of asenapine as administered sublingually, a phase 3 study specifically assessed efficacy and safety of asenapine transdermal system in adults with schizophrenia.11,12 This study was conducted in the United States and 4 other countries at a total of 59 study sites, and 616 patients with acutely exacerbated schizophrenia were enrolled. After a 3- to 14-day screening/single-blind run-in washout period, participants entered a 6-week inpatient double-blind period. Randomization was 1:1:1 to asenapine transdermal system 3.8 mg/24 hours, 7.6 mg/24 hours, or a placebo patch. Each of the patch doses demonstrated significant improvement vs placebo at Week 6 for the primary (change in Positive and Negative Syndrome Scale [PANSS] total score) and key secondary (change in Clinical Global Impression-Severity of Illness) endpoints. Response at endpoint, as defined by a ≥30% improvement from baseline PANSS total score, or by a Clinical Global Impression–Improvement score of 1 (very much improved) or 2 (much improved), was also assessed. For either definition of response, both doses of asenapine transdermal system were superior to placebo, with number needed to treat (NNT) (Box) values <10 for the 3.8 mg/24 hours dose (Table 2). These effect sizes are similar to what is known about sublingual asenapine as determined in a meta-analysis performed by the manufacturer and using individual patient data.13
Box
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies, you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we reviewing a result that may be statistically significant but irrelevant for day-today patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—two tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between two interventions. If we see a therapeutic difference once every 100 patients (NNT of 100), the difference between the treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 7 patients being treated with an intervention vs another (NNT of 7), the result will likely influence dayto-day practice.
How to calculate NNT (or NNH):
What is the NNT for an outcome for drug A vs drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates: .75 -.55 = .20
- NNT: 1/.20 = 5
A rule of thumb: NNT values for a medication vs placebo <10 usually denote a medication we use on a regular basis to treat patients.
a Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71. Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82. Additional information can be found in Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411 (free to access at onlinelibrary.wiley.com/doi/full/10.1111/ijcp.12142)
Overall tolerability and safety
The systemic safety and tolerability profile for asenapine transdermal system would be expected to be similar to that for sublingual asenapine, unless there are adverse events that are related to high peak plasma concentrations or large differences between peak and trough plasma concentrations.6 Nonsystemic local application site adverse events would, of course, differ between sublingual vs transdermal administration.
Continue to: Use of asenapine transdermal system...
Use of asenapine transdermal system avoids the dysgeusia and oral hypoesthesia that can be observed with sublingual asenapine4,6; however, dermal effects need to be considered (see Dermal safety). The most commonly observed adverse reactions (incidence ≥5% and at least twice that for placebo) for asenapine transdermal system are extrapyramidal disorder, application site reaction, and weight gain.3 For sublingual asenapine for adults with schizophrenia, the list includes akathisia, oral hypoesthesia, and somnolence.4 These adverse events can be further described using the metric of number needed to harm (NNH) as shown in Table 3.3,4,11,12,14 Of note, extrapyramidal disorder and weight gain appear to be dose-related for asenapine transdermal system. Akathisia appears to be dose-related for sublingual asenapine but not for asenapine transdermal system. Somnolence appears to be associated with sublingual asenapine but not necessarily with asenapine transdermal system.
For sublingual asenapine, the additional indications (bipolar I disorder as acute monotherapy treatment of manic or mixed episodes in adults and pediatric patients age 10 to 17, adjunctive treatment to lithium or valproate in adults, and maintenance monotherapy treatment in adults) have varying commonly encountered adverse reactions.4 Both transdermal asenapine system and sublingual asenapine are contraindicated in patients with severe hepatic impairment (Child-Pugh C) and those with known hypersensitivity to asenapine or to any components in the formulation. Both formulations carry similar warnings in their prescribing information regarding increased mortality in older patients with dementia-related psychosis, cerebrovascular adverse reactions in older patients with dementia-related psychosis, neuroleptic malignant syndrome, tardive dyskinesia, metabolic changes, orthostatic hypotension, leukopenia (and neutropenia and agranulocytosis), QT prolongation, seizures, and potential for cognitive and motor impairment.
Adverse events leading to discontinuation of study treatment in the asenapine transdermal system pivotal trial occurred in 4.9%, 7.8%, and 6.8% of participants in the 3.8 mg/24 hour, 7.6 mg/24 hour, and placebo groups, respectively.11
Dermal safety
In the pivotal efficacy study,11 the incidence of adverse events at patch application sites was higher in the active groups vs placebo (Table 33,4,11,12,14). The most frequently reported patch application site reactions were erythema and pruritus, occurring in approximately 10% and 4% in the active treatment arms vs 1.5% and 1.9% for placebo, respectively. With the exception of 1 adverse event of severe application site erythema during Week 2 (participant received 7.6 mg/24 hour, erythema resolved without intervention, and the patient continued the study), all other patch application site events were mild or moderate in severity. Rates of discontinuation due to application site reactions or skin disorders were ≤0.5% across all groups. In the pharmacokinetic studies,10 no patches were removed because of unacceptable irritation.
Why Rx?
Asenapine transdermal system is the first antipsychotic “patch” FDA-approved for the treatment of adults with schizophrenia. Asenapine has been available since 2009 as a sublingual formulation administered twice daily. The pharmacokinetic profile of the once-daily transdermal system demonstrates dose-proportional kinetics and sustained delivery of asenapine with a low peak-to-trough plasma level ratio. Three dosage strengths (3.8, 5.7, and 7.6 mg/24 hours) are available, corresponding to blood levels attained with sublingual asenapine exposures of 10, 15, and 20 mg/d, respectively. Application sites are rotated daily and include the upper arms, upper back, abdomen, or hip. Dysgeusia and hypoesthesia of the tongue are avoided with the use of the patch, and there are no food or drink restrictions. Attention will be needed in case of dermal reactions, similar to that observed with other medication patches.
Bottom Line
The asenapine transdermal drug delivery system appears to be efficacious and reasonably well tolerated. The treatment of schizophrenia is complex and requires individualized choices in order to optimize outcomes. A patch may be the preferred formulation for selected patients, and caregivers will have the ability to visually check if the medication is being used.
Related Resource
- Hisamitsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
Drug Brand Names
Asenapine sublingual • Saphris
Asenapine transdermal system • Secuado
Lithium • Eskalith, Lithobid
Valproate • Depakote
The asenapine transdermal system is available in 3 patch sizes: 20, 30, and 40 cm2, which deliver 3.8, 5.7, and 7.6 mg/24 hours of asenapine, respectively.3 Based on the average exposure (area under the plasma concentration curve [AUC]) of asenapine, 3.8 mg/24 hours corresponds to 5 mg twice daily of sublingual asenapine, and 7.6 mg/24 hours corresponds to 10 mg twice daily of sublingual asenapine.3 The “in-between” dose strength of 5.7 mg/24 hours would correspond to exposure to a total of 15 mg/d of sublingual asenapine. The recommended starting dose for asenapine transdermal system is 3.8 mg/24 hours. The dosage may be increased to 5.7 mg/24 hours or 7.6 mg/24 hours, as needed, after 1 week. The safety of doses above 7.6 mg/24 hours has not been evaluated in clinical studies. Asenapine transdermal system is applied once daily and should be worn for 24 hours only, with only 1 patch at any time. Application sites include the upper arm, upper back, abdomen, and hip. A different application site of clean, dry, intact skin should be selected each time a new patch is applied. Although showering is permitted, the use of asenapine transdermal system during swimming or taking a bath has not been evaluated. Of note, prolonged application of heat over an asenapine transdermal system increases plasma concentrations of asenapine, and thus application of external heat sources (eg, heating pads) over the patch should be avoided.
How it works
Product labeling notes that asenapine is an atypical antipsychotic, and that its efficacy in schizophrenia could be mediated through a combination of antagonist activity at dopamine D2 and serotonin 5-HT2A receptors.3 The pharmacodynamic profile of asenapine is complex5 and receptor-binding assays performed using cloned human serotonin, norepinephrine, dopamine, histamine, and muscarinic receptors demonstrated picomolar affinity (extremely high) for 5-HT2C and 5-HT2A receptors, subnanomolar affinity (very high) for 5-HT7, 5-HT2B, 5-HT6, and D3 receptors, and nanomolar affinity (high) for D2 receptors, as well as histamine H1, D4, a1-adrenergic, a2-adrenergic, D1, 5-HT5, 5-HT1A, 5-HT1B, and histamine H2 receptors. Activity of asenapine is that of antagonism at these receptors. Asenapine has no appreciable affinity for muscarinic cholinergic receptors.
The asenapine receptor-binding “fingerprint” differs from that of other antipsychotics. Some of these receptor affinities are of special interest in terms of potential efficacy for pro-cognitive effects and amelioration of abnormal mood.5,9 In terms of tolerability, a relative absence of affinity to muscarinic receptors would predict a low risk for anticholinergic adverse effects, but antagonism at histamine H1 and at a1-adrenergic receptors, either alone or in combination, may cause sedation, and blockade of H1 receptors would also predict weight gain.9 Antagonism of a1-adrenergic receptors can be associated with orthostatic hypotension and neurally mediated reflex bradycardia.9
Clinical pharmacokinetics
Three open-label, randomized, phase 1 studies were conducted to assess the relative bioavailability of asenapine transdermal system vs sublingual asenapine.10 These included single- and multiple-dose studies and clinical trials that examined the effects of different application sites and ethnic groups, and the effect of external heat on medication absorption. Studies were conducted in healthy individuals, except for the multiple-dose study, which was performed in adults with schizophrenia. The AUC for asenapine transdermal system was within the range of that of equivalent doses of sublingual asenapine, but peak exposure (maximum concentration) was significantly lower. As already noted, the AUC of the asenapine patch for 3.8 mg/24 hours and 7.6 mg/24 hours corresponds to sublingual asenapine 5 mg and 10 mg twice daily, respectively. Maximum asenapine concentrations are typically reached between 12 and 24 hours, with sustained concentrations during the 24-hour wear time.3 On average, approximately 60% of the available asenapine is released from the transdermal system over 24 hours. Steady-state plasma concentrations for asenapine transdermal system were achieved approximately 72 hours after the first application and, in contrast to sublingual asenapine, the peak-trough fluctuations were small (peak-to-trough ratio is 1.5 for asenapine transdermal system compared with >3 for sublingual asenapine). Dose-proportionality at steady state was evident for asenapine transdermal system. This is in contrast to sublingual asenapine, where exposure increases 1.7-fold with a 2-fold increase in dose.4,5 Following patch removal, the apparent elimination half-life is approximately 30 hours.3 The pharmacokinetics of the patch did not vary with regards to the application site (upper arm, upper back, abdomen, or hip area), and the pharmacokinetic profile was similar across the ethnic groups that participated in the study. Direct exposure to external heat did increase both the rate and extent of absorption, so external heat sources should be avoided.3
Efficacy
The efficacy profile for asenapine transdermal system would be expected to mirror that for sublingual asenapine.6,7 In addition to data supporting the use of asenapine as administered sublingually, a phase 3 study specifically assessed efficacy and safety of asenapine transdermal system in adults with schizophrenia.11,12 This study was conducted in the United States and 4 other countries at a total of 59 study sites, and 616 patients with acutely exacerbated schizophrenia were enrolled. After a 3- to 14-day screening/single-blind run-in washout period, participants entered a 6-week inpatient double-blind period. Randomization was 1:1:1 to asenapine transdermal system 3.8 mg/24 hours, 7.6 mg/24 hours, or a placebo patch. Each of the patch doses demonstrated significant improvement vs placebo at Week 6 for the primary (change in Positive and Negative Syndrome Scale [PANSS] total score) and key secondary (change in Clinical Global Impression-Severity of Illness) endpoints. Response at endpoint, as defined by a ≥30% improvement from baseline PANSS total score, or by a Clinical Global Impression–Improvement score of 1 (very much improved) or 2 (much improved), was also assessed. For either definition of response, both doses of asenapine transdermal system were superior to placebo, with number needed to treat (NNT) (Box) values <10 for the 3.8 mg/24 hours dose (Table 2). These effect sizes are similar to what is known about sublingual asenapine as determined in a meta-analysis performed by the manufacturer and using individual patient data.13
Box
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies, you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we reviewing a result that may be statistically significant but irrelevant for day-today patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—two tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between two interventions. If we see a therapeutic difference once every 100 patients (NNT of 100), the difference between the treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 7 patients being treated with an intervention vs another (NNT of 7), the result will likely influence dayto-day practice.
How to calculate NNT (or NNH):
What is the NNT for an outcome for drug A vs drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates: .75 -.55 = .20
- NNT: 1/.20 = 5
A rule of thumb: NNT values for a medication vs placebo <10 usually denote a medication we use on a regular basis to treat patients.
a Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71. Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82. Additional information can be found in Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411 (free to access at onlinelibrary.wiley.com/doi/full/10.1111/ijcp.12142)
Overall tolerability and safety
The systemic safety and tolerability profile for asenapine transdermal system would be expected to be similar to that for sublingual asenapine, unless there are adverse events that are related to high peak plasma concentrations or large differences between peak and trough plasma concentrations.6 Nonsystemic local application site adverse events would, of course, differ between sublingual vs transdermal administration.
Continue to: Use of asenapine transdermal system...
Use of asenapine transdermal system avoids the dysgeusia and oral hypoesthesia that can be observed with sublingual asenapine4,6; however, dermal effects need to be considered (see Dermal safety). The most commonly observed adverse reactions (incidence ≥5% and at least twice that for placebo) for asenapine transdermal system are extrapyramidal disorder, application site reaction, and weight gain.3 For sublingual asenapine for adults with schizophrenia, the list includes akathisia, oral hypoesthesia, and somnolence.4 These adverse events can be further described using the metric of number needed to harm (NNH) as shown in Table 3.3,4,11,12,14 Of note, extrapyramidal disorder and weight gain appear to be dose-related for asenapine transdermal system. Akathisia appears to be dose-related for sublingual asenapine but not for asenapine transdermal system. Somnolence appears to be associated with sublingual asenapine but not necessarily with asenapine transdermal system.
For sublingual asenapine, the additional indications (bipolar I disorder as acute monotherapy treatment of manic or mixed episodes in adults and pediatric patients age 10 to 17, adjunctive treatment to lithium or valproate in adults, and maintenance monotherapy treatment in adults) have varying commonly encountered adverse reactions.4 Both transdermal asenapine system and sublingual asenapine are contraindicated in patients with severe hepatic impairment (Child-Pugh C) and those with known hypersensitivity to asenapine or to any components in the formulation. Both formulations carry similar warnings in their prescribing information regarding increased mortality in older patients with dementia-related psychosis, cerebrovascular adverse reactions in older patients with dementia-related psychosis, neuroleptic malignant syndrome, tardive dyskinesia, metabolic changes, orthostatic hypotension, leukopenia (and neutropenia and agranulocytosis), QT prolongation, seizures, and potential for cognitive and motor impairment.
Adverse events leading to discontinuation of study treatment in the asenapine transdermal system pivotal trial occurred in 4.9%, 7.8%, and 6.8% of participants in the 3.8 mg/24 hour, 7.6 mg/24 hour, and placebo groups, respectively.11
Dermal safety
In the pivotal efficacy study,11 the incidence of adverse events at patch application sites was higher in the active groups vs placebo (Table 33,4,11,12,14). The most frequently reported patch application site reactions were erythema and pruritus, occurring in approximately 10% and 4% in the active treatment arms vs 1.5% and 1.9% for placebo, respectively. With the exception of 1 adverse event of severe application site erythema during Week 2 (participant received 7.6 mg/24 hour, erythema resolved without intervention, and the patient continued the study), all other patch application site events were mild or moderate in severity. Rates of discontinuation due to application site reactions or skin disorders were ≤0.5% across all groups. In the pharmacokinetic studies,10 no patches were removed because of unacceptable irritation.
Why Rx?
Asenapine transdermal system is the first antipsychotic “patch” FDA-approved for the treatment of adults with schizophrenia. Asenapine has been available since 2009 as a sublingual formulation administered twice daily. The pharmacokinetic profile of the once-daily transdermal system demonstrates dose-proportional kinetics and sustained delivery of asenapine with a low peak-to-trough plasma level ratio. Three dosage strengths (3.8, 5.7, and 7.6 mg/24 hours) are available, corresponding to blood levels attained with sublingual asenapine exposures of 10, 15, and 20 mg/d, respectively. Application sites are rotated daily and include the upper arms, upper back, abdomen, or hip. Dysgeusia and hypoesthesia of the tongue are avoided with the use of the patch, and there are no food or drink restrictions. Attention will be needed in case of dermal reactions, similar to that observed with other medication patches.
Bottom Line
The asenapine transdermal drug delivery system appears to be efficacious and reasonably well tolerated. The treatment of schizophrenia is complex and requires individualized choices in order to optimize outcomes. A patch may be the preferred formulation for selected patients, and caregivers will have the ability to visually check if the medication is being used.
Related Resource
- Hisamitsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
Drug Brand Names
Asenapine sublingual • Saphris
Asenapine transdermal system • Secuado
Lithium • Eskalith, Lithobid
Valproate • Depakote
1. Noven. US FDA approves SECUADO® (asenapine) transdermal system, the first-and-only transdermal patch for the treatment of adults with schizophrenia. October 15, 2019. Accessed January 15, 2021. https://www.noven.com/wp-content/uploads/2020/04/PR101519.pdf
2. US Food and Drug Administration. Center for Drug Evaluation and Research. Approval Package for: APPLICATION NUMBER: 212268Orig1s000. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000Approv.pdf
3. Hisam itsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
4. Allergan USA, Inc. SAPHRIS® (asenapine) sublingual tablets prescribing information. February 2017. Accessed January 15, 2021. https://media.allergan.com/actavis/actavis/media/allergan-pdf-documents/product-prescribing/Final_labeling_text_SAPHRIS-clean-02-2017.pdf
5. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
6. Citrome L. Asenapine review, part II: clinical efficacy, safety and tolerability. Expert Opin Drug Saf. 2014;13(6):803-830.
7. Citrome L. Chapter 31: Asenapine. In: Schatzberg AF, Nemeroff CB, eds. The American Psychiatric Association Publishing Textbook of Psychopharmacology, 5th ed. American Psychiatric Association Publishing; 2017:797-808.
8. Citrome L, Zeni CM, Correll CU. Patches: established and emerging transdermal treatments in psychiatry. J Clin Psychiatry. 2019;80(4):18nr12554. doi: 10.4088/JCP.18nr12554
9. Shayegan DK, Stahl SM. Atypical antipsychotics: matching receptor profile to individual patient’s clinical profile. CNS Spectr. 2004;9(10 suppl 11):6-14.
10. Castelli M, Suzuki K, Komaroff M, et al. Pharmacokinetic profile of asenapine transdermal system HP-3070: The first antipsychotic patch in the US. Poster presented virtually at the American Society for Clinical Psychopharmacology (ASCP) 2020 Annual Meeting, May 29-30, 2020. https://www.psychiatrist.com/ascpcorner/Documents/ascp2020/3_ASCP%20Poster%20Abstracts%202020-JCP.pdf
11. Citrome L, Walling DP, Zeni CM, et al. Efficacy and safety of HP-3070, an asenapine transdermal system, in patients with schizophrenia: a phase 3, randomized, placebo-controlled study. J Clin Psychiatry. 2020;82(1):20m13602. doi: 10.4088/JCP.20m13602
12. US Food and Drug Administration. Drug Approval Package: SECAUDO. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000TOC.cfm
13. Szegedi A, Verweij P, van Duijnhoven W, et al. Meta-analyses of the efficacy of asenapine for acute schizophrenia: comparisons with placebo and other antipsychotics. J Clin Psychiatry. 2012;73(12):1533-1540.
14. Citrome L. Asenapine for schizophrenia and bipolar disorder: a review of the efficacy and safety profile for this newly approved sublingually absorbed second-generation antipsychotic. Int J Clin Pract. 2009;63(12):1762-1784.
1. Noven. US FDA approves SECUADO® (asenapine) transdermal system, the first-and-only transdermal patch for the treatment of adults with schizophrenia. October 15, 2019. Accessed January 15, 2021. https://www.noven.com/wp-content/uploads/2020/04/PR101519.pdf
2. US Food and Drug Administration. Center for Drug Evaluation and Research. Approval Package for: APPLICATION NUMBER: 212268Orig1s000. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000Approv.pdf
3. Hisam itsu Pharmaceutical Co., Inc. SECUADO® (asenapine) transdermal system prescribing information. October 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212268s000lbl.pdf
4. Allergan USA, Inc. SAPHRIS® (asenapine) sublingual tablets prescribing information. February 2017. Accessed January 15, 2021. https://media.allergan.com/actavis/actavis/media/allergan-pdf-documents/product-prescribing/Final_labeling_text_SAPHRIS-clean-02-2017.pdf
5. Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893-903.
6. Citrome L. Asenapine review, part II: clinical efficacy, safety and tolerability. Expert Opin Drug Saf. 2014;13(6):803-830.
7. Citrome L. Chapter 31: Asenapine. In: Schatzberg AF, Nemeroff CB, eds. The American Psychiatric Association Publishing Textbook of Psychopharmacology, 5th ed. American Psychiatric Association Publishing; 2017:797-808.
8. Citrome L, Zeni CM, Correll CU. Patches: established and emerging transdermal treatments in psychiatry. J Clin Psychiatry. 2019;80(4):18nr12554. doi: 10.4088/JCP.18nr12554
9. Shayegan DK, Stahl SM. Atypical antipsychotics: matching receptor profile to individual patient’s clinical profile. CNS Spectr. 2004;9(10 suppl 11):6-14.
10. Castelli M, Suzuki K, Komaroff M, et al. Pharmacokinetic profile of asenapine transdermal system HP-3070: The first antipsychotic patch in the US. Poster presented virtually at the American Society for Clinical Psychopharmacology (ASCP) 2020 Annual Meeting, May 29-30, 2020. https://www.psychiatrist.com/ascpcorner/Documents/ascp2020/3_ASCP%20Poster%20Abstracts%202020-JCP.pdf
11. Citrome L, Walling DP, Zeni CM, et al. Efficacy and safety of HP-3070, an asenapine transdermal system, in patients with schizophrenia: a phase 3, randomized, placebo-controlled study. J Clin Psychiatry. 2020;82(1):20m13602. doi: 10.4088/JCP.20m13602
12. US Food and Drug Administration. Drug Approval Package: SECAUDO. October 11, 2019. Accessed January 15, 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212268Orig1s000TOC.cfm
13. Szegedi A, Verweij P, van Duijnhoven W, et al. Meta-analyses of the efficacy of asenapine for acute schizophrenia: comparisons with placebo and other antipsychotics. J Clin Psychiatry. 2012;73(12):1533-1540.
14. Citrome L. Asenapine for schizophrenia and bipolar disorder: a review of the efficacy and safety profile for this newly approved sublingually absorbed second-generation antipsychotic. Int J Clin Pract. 2009;63(12):1762-1784.

Aripiprazole, brexpiprazole, and cariprazine: Not all the same
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
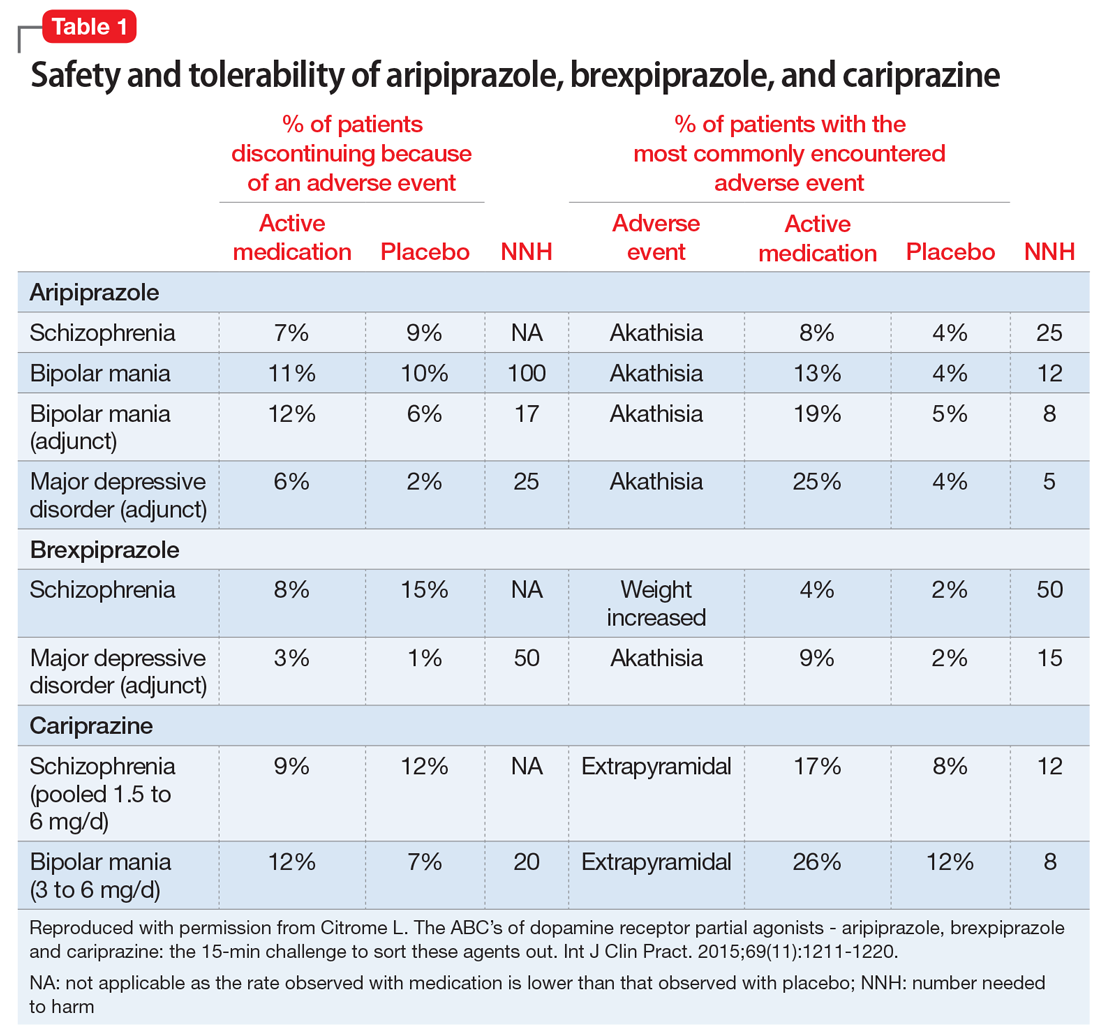
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.

Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
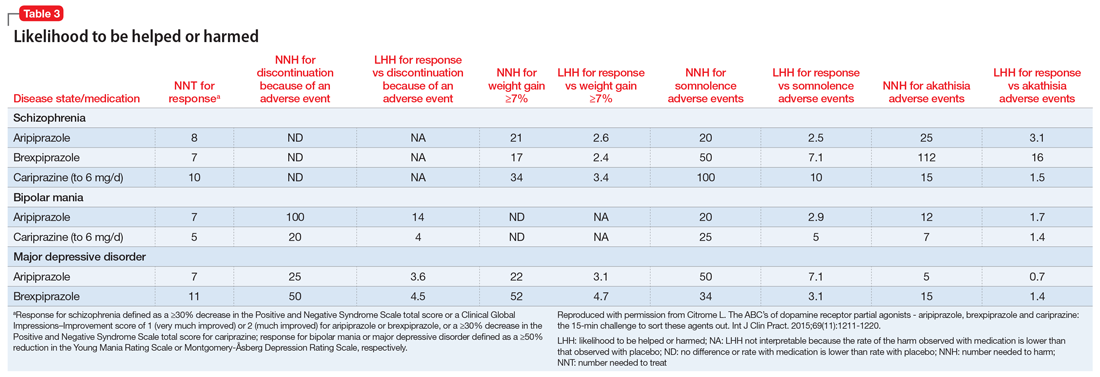
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
Aripiprazole, brexpiprazole, and cariprazine are dopamine receptor partial agonists, and on the surface, they appear similar. However, there are key differences in terms of available indications, formulations, pharmacodynamics, pharmacokinetics, dosing, drug interactions, tolerability, and other factors related to successful use.1 This review will cover the main points that the knowledgeable clinician will need to be mindful of when prescribing these agents.
Aripiprazole
Aripiprazole was launched in the United States in 20022 as the first dopamine receptor partial agonist approved for the treatment of schizophrenia; it later received additional indications for adults with manic or mixed episodes associated with bipolar I disorder and the maintenance treatment of bipolar I disorder, as well as for the adjunctive treatment of major depressive disorder (MDD). Pediatric indications include schizophrenia, acute treatment of manic or mixed episodes associated with bipolar I disorder, irritability associated with autistic disorder, and Tourette’s disorder.
Several formulations also became available, including a short-acting injection indicated for agitation associated with schizophrenia or bipolar mania, and oral disintegrating tablets and an oral solution that could substitute for the regular tablet. Presently the medication has gone “generic,” and not all formulations are being manufactured. The long-acting formulations of aripiprazole (aripiprazole monohydrate and aripiprazole lauroxil) are considered different products, each with its own product insert, with indications that are more limited in scope than for the oral forms.3,4
Although dopamine D2 receptor partial agonism is a relevant mechanism of action, partial agonist activity at serotonin 5-HT1A receptors and antagonist activity at 5-HT2A receptors also play a role.2 Actions at receptors other than dopamine D2, serotonin 5-HT1A, and serotonin 5-HT2A may explain some of the other clinical effects of aripiprazole. In terms of binding, aripiprazole has very high binding affinities (Ki) to dopamine D2 (0.34 nM), dopamine D3 (0.8 nM), and serotonin 5-HT2B (0.36 nM) receptors, and high binding affinities to serotonin 5-HT1A (1.7 nM) and serotonin 5-HT2A (3.4 nM) receptors.
Dosage recommendations for adults with schizophrenia suggest a starting and maintenance dose of 10 to 15 mg/d.2 Although the maximum dose is 30 mg/d, there is no evidence that doses >15 mg/d are superior to lower doses.5 In adolescents with schizophrenia, the product label recommends a starting dose of 2 mg/d, a maintenance dose of 10 mg/d, and a maximum dose of 30 mg/d. Recommendations for dosing in bipolar mania are similar. Dosing for the other indications is lower.
Efficacy in schizophrenia can be quantified using number needed to treat (NNT) for response vs placebo. The NNT answers the question “How many patients need to be randomized to aripiprazole vs placebo before expecting to encounter one additional responder?”6 From the 4 positive pivotal short-term acute schizophrenia trials for aripiprazole in adults,7-10 using the definition of response as a ≥30% decrease in the Positive and Negative Syndrome Scale (PANSS) total score or a Clinical Global Impressions–Improvement (CGI-I) score of 1 (very much improved) or 2 (much improved), and pooling the data for aripiprazole doses 10 to 30 mg/d, response rates were 38% for aripiprazole vs 24% for placebo, resulting in a NNT of 8 (95% confidence interval [CI] 6 to 13).
From the 4 positive pivotal short-term acute bipolar mania trials for aripiprazole monotherapy in adults11-14 using the definition of response as a ≥50% decrease in the Young Mania Rating Scale (YMRS) total score, and pooling the data for aripiprazole doses 15 to 30 mg/d, response rates were 47% for aripiprazole vs 31% for placebo, resulting in a NNT of 7 (95% CI 5 to 11).1 Similar results were observed in the adjunctive aripiprazole acute bipolar mania trial15 where the NNT for response was also 7.1
Continue to: From the 2 positive pivotal short-term...
From the 2 positive pivotal short-term acute MDD trials for aripiprazole,16,17 using the definition of response as a ≥50% decrease in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, and pooling the data (aripiprazole flexibly dosed 2 to 20 mg/d, with a median dose of 10 mg/d), response rates were 33% for aripiprazole vs 20% for placebo, resulting in a NNT of 8 (95% CI 6 to 17). After including a third trial not described in product labeling,18 the NNT became a more robust 7 (95% CI 5 to 11).1
The most commonly encountered adverse events (incidence ≥5% and at least twice the rate of placebo) in the pivotal trials were akathisia (schizophrenia); akathisia, sedation, restlessness, tremor, and extrapyramidal disorder (bipolar mania, monotherapy); akathisia, insomnia, and extrapyramidal disorder (bipolar mania, adjunctive therapy); akathisia, restlessness, insomnia, constipation, fatigue, and blurred vision (MDD); and nausea (short-acting IM formulation). Table 11 summarizes the tolerability information regarding rate of discontinuation due to adverse events (an overall indicator of tolerability), and the incidence of the most common adverse event, together with the calculated number needed to harm (NNH). Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability; for the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability for these indications.
Brexpiprazole
Brexpiprazole was launched in the United States in 2015 for 2 indications: schizophrenia and the adjunctive treatment of MDD, both in adults.19 In terms of binding, brexpiprazole has very high binding affinities to serotonin 5-HT1A (0.12 nM), adrenergic α1B (0.17 nM), dopamine D2 (0.30 nM), serotonin 5-HT2A (0.47 nM), and adrenergic α2C (0.59 nM) receptors, and high binding affinities to dopamine D3 (1.1 nM), serotonin 5-HT2B (1.9 nM), adrenergic α1D (2.6 nM), serotonin 5-HT7 (3.7 nM), and adrenergic α1A (3.8 nM) receptors.
The 1-mg/d starting dose for brexpiprazole is lower than the recommended dose range of 2 to 4 mg/d for schizophrenia or the recommended dose of 2 mg/d for MDD.19 Thus brexpiprazole requires titration. The recommended rate of titration depends on the disease state being treated. For schizophrenia, the recommended titration schedule is to increase the dose to 2 mg/d on Day 5 through Day 7, then to 4 mg/d (the maximum recommended dose) on Day 8 based on the patient’s clinical response and tolerability. For MDD, there is the option of starting at 0.5 mg/d and the titration process is slower, with dosage increases occurring at weekly intervals, and with a maximum dose of 3 mg/d.
Using the identical definition of response in persons with schizophrenia as for the aripiprazole data described above, pooling together all the available data for the recommended target dose of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 studies listed in the product label,20,21 the percentage of responders was 46%, compared with 31% for the pooled placebo groups, yielding a NNT of 7 (95% CI 5 to 12).22
Continue to: For MDD...
For MDD, using the definition of response as a ≥50% decrease in MADRS total score, and pooling the results for brexpiprazole 1, 2, and 3 mg/d from the 2 pivotal trials,23,24 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI 8 to 26).22 Including the 1.5-mg/d dose arm and the placebo arm from the phase II study for which results are also available but not included in product labelling, the NNT becomes a slightly more robust 11 (95% CI 8 to 20).22 Although the magnitude of the NNT effect size is stronger for aripiprazole than for brexpiprazole, the 95% CIs do overlap.
The most commonly encountered adverse event in the short-term trials in schizophrenia (incidence ≥4% and at least twice the rate of placebo) was increased weight. The most commonly encountered adverse events in the short-term trials in MDD (incidence ≥5% and at least twice the rate of placebo) were increased weight and akathisia. Rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability, and for MDD the NNH vs placebo on discontinuation because of an adverse event was 50, representing reasonable overall tolerability for this indication as well (Table 11).
Cariprazine
Cariprazine was launched in the United States in 2015 for 2 indications: schizophrenia, and the acute treatment of manic or mixed episodes associated with bipolar I disorder, both in adults.25 In terms of binding, cariprazine has very high binding affinities to dopamine D3 (0.085 nM), dopamine D2L (0.49 nM), serotonin 5-HT2B (0.58 nM), and dopamine D2S (0.69 nM) receptors, and high binding affinity to serotonin 5-HT1A (2.6 nM) receptors. Cariprazine forms 2 major metabolites, desmethyl cariprazine and didesmethyl cariprazine, that have in vitro receptor binding profiles similar to the parent drug. This latter metabolite, didesmethyl cariprazine, has a half-life of 1 to 3 weeks, and is the active moiety responsible for the majority of cariprazine’s effect when in steady state. Thus, following discontinuation of cariprazine, the decline in plasma concentrations of active drug will be slow.
The starting dose for cariprazine for schizophrenia, 1.5 mg/d, can be therapeutic. The dosage can be increased to 3 mg/d on Day 2. Depending upon clinical response and tolerability, further dose adjustments can be made in 1.5-mg or 3-mg increments to a maximum dose of 6 mg/d. For the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d; this can be done on Day 2. Cariprazine has been tested in clinical trials at higher doses; however, doses that exceed 6 mg/d did not confer significant additional benefit.25
A more conservative definition of response was used in the reporting of the cariprazine acute schizophrenia studies. This was simply a ≥30% decrease in the PANSS total score, and did not include the option of including patients who scored a 1 or 2 on the CGI-I. For pooled doses of cariprazine 1.5 to 6 mg/d,26-28 the percentage of responders was 31%, compared with 21% for the pooled placebo groups, yielding a NNT of 10 (95% CI 7 to 18).1 Although the magnitude of the NNT effect size is weaker for cariprazine than the other dopamine receptor partial agonists, the 95% CI overlaps with that of aripiprazole and brexpiprazole. An appropriately designed head-to-head trial would be necessary to directly test noninferiority.
Continue to: Pooling the data...
Pooling the data from the 3 pivotal short-term acute bipolar mania trials for cariprazine monotherapy in adults29-31 and using the definition of response as a ≥50% decrease in the YMRS total score for the recommended target dose of 3 to 6 mg/d, the percentage of responders was 57%, compared with 36% for the pooled placebo groups, yielding a NNT of 5 (95% CI 4 to 8).1 The magnitude of the NNT effect size is stronger for cariprazine than for aripiprazole, but the 95% CIs overlap.
The most commonly encountered adverse events in the short-term trials (incidence ≥5% and at least twice the rate of placebo) were extrapyramidal symptoms and akathisia (schizophrenia); and extrapyramidal symptoms, akathisia, dyspepsia, vomiting, somnolence, and restlessness (bipolar mania). In the schizophrenia studies, rates of discontinuation because of an adverse event were not higher for active medication vs placebo, suggesting excellent overall tolerability, and for bipolar disorder the NNH vs placebo on discontinuation because of an adverse event was 20, representing reasonable overall tolerability for this indication as well (Table 1).
Differences to consider
Indications. Although all 3 medications are approved for the treatment of schizophrenia, both aripiprazole and brexpiprazole are also approved for adjunctive treatment of MDD, and both aripiprazole and cariprazine are also approved for acute treatment of manic or mixed episodes associated with bipolar I disorder. In addition, aripiprazole is approved for a number of different disease states in pediatric patients. Aripiprazole has also been approved in a number of different formulations (oral and IM), but brexpiprazole and cariprazine are presently available only as oral pills (tablets for brexpiprazole, capsules for cariprazine).
Contraindications. All 3 agents are contraindicated in patients with a known hypersensitivity reaction to the product. All 3 also have a “black-box” warning for increased mortality in geriatric patients with dementia-related psychosis, a warning that is found in all antipsychotic medication labels. Additional black-box warnings are included regarding suicidality in the product labels of aripiprazole and brexpiprazole by virtue of their approval for the treatment of MDD.
Pharmacodynamics. All 3 agents describe a similar mechanism of action in their respective product labels: “efficacy … could be mediated through a combination of partial agonist activity at central dopamine D2 and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors.”2,19,25
Continue to: However, binding affinities differ...
However, binding affinities differ substantially among the agents (for example, cariprazine has only moderate binding affinity at serotonin 5-HT2A receptors [18.8 nM]), and differences also exist in terms of intrinsic activity at the receptors where partial agonism is operative. Compared with aripiprazole, brexpiprazole has lower intrinsic activity at the dopamine D2 receptor (and thus is expected to cause less akathisia), and has an approximately 10-fold higher affinity for serotonin 5-HT1A and 5-HT2A receptors, also potentially enhancing tolerability and perhaps anxiolytic activity.32,33 When cariprazine was compared with aripiprazole in functional assays for dopamine D2 and D3 receptors, similar D2 and higher D3 antagonist-partial agonist affinity and a 3- to 10-fold greater D3 vs D2 selectivity was observed for cariprazine.34 Whether specifically targeting the dopamine D3 receptor over the dopamine D2 receptor is clinically advantageous remains unknown, but in preclinical studies, dopamine D3–preferring agents may exert pro-cognitive effects.35-37 All 3 agents have only moderate binding affinities to histamine H1 receptors, thus sedation should not be prominent for any of them. None of the 3 agents have appreciable binding at muscarinic receptors, thus adverse effects related to antimuscarinic activity should not be present as well.
Schizophrenia is a heterogenous disorder. We know from clinical practice that patients respond differently to specific antipsychotics. Having different pharmacodynamic “fingerprints” to choose from allows for flexibility in treatment. Moreover, dopamine receptor partial agonists provide an alternative to the array of dopamine receptor antagonists, such as the other second-generation antipsychotics and all first-generation antipsychotics.
Dosing. Although all 3 agents are dosed once daily, only for aripiprazole is the recommended starting dose the same as the recommended maintenance dose in adults with schizophrenia or bipolar mania. Although the starting dose for cariprazine for schizophrenia can be therapeutic (1.5 mg/d), for the treatment of bipolar mania, cariprazine will need to be titrated from the starting dose of 1.5 mg/d to the recommended target dose range of 3 to 6 mg/d.
Half-life. Aripiprazole and brexpiprazole share a similar elimination half-life: approximately 75 hours and 94 hours for aripiprazole and its active metabolite dehydro-aripiprazole, respectively, and 91 hours and 86 hours for brexpiprazole and its major metabolite, DM-3411 (inactive), respectively. Cariprazine is strikingly different, with an elimination half-life of 2 to 4 days, and approximately 1 to 3 weeks for its active metabolite didesmethyl cariprazine.
Drug interactions. Both aripiprazole and brexpiprazole are metabolized via cytochrome P450 (CYP) 2D6 and CYP3A4, and thus the dose may need to be adjusted in the presence of CYP2D6 inhibitors or CYP3A4 inhibitors/inducers; with inhibitors, the dose is decreased by half or more, and with inducers, the dose is doubled. In contrast, cariprazine is primarily metabolized by CYP3A4 and thus potential drug–drug interactions are primarily focused on CYP3A4 inhibitors (decrease cariprazine dose by half) and inducers (co-prescribing of cariprazine with a CYP3A4 inducer is not recommended).
Continue to: Tolerability
Tolerability. For all 3 agents, rates of discontinuation because of an adverse event were not higher for active medication vs placebo for the schizophrenia studies, suggesting excellent overall tolerability.2,19,25 For the other disease states, NNH values ranged from 17 (adjunctive use of aripiprazole for bipolar mania) to 100 (aripiprazole monotherapy for bipolar mania), representing reasonable overall tolerability. For the most commonly encountered adverse event for each medication, the NNH values ranged from 5 (akathisia for aripiprazole for adjunctive use in MDD) to 50 (increased weight for brexpiprazole for schizophrenia). Of special interest are the adverse events of weight gain ≥7% from baseline, somnolence adverse events, and akathisia adverse events; the NNH values vs placebo for these are listed in Table 21. Pragmatically, NNH values <10 are likely to be more clinically relevant. For aripiprazole, brexpiprazole, and cariprazine for the treatment of schizophrenia, none of the NNH values for weight gain, somnolence, or akathisia were <10; however, this was not the case for the mood disorders, where in general, akathisia was more frequently observed for each of the agents. For the indication of schizophrenia, the rank order for propensity for weight gain appears to be brexpiprazole > aripiprazole > cariprazine, the propensity for somnolence aripiprazole > brexpiprazole > cariprazine, and the propensity for akathisia cariprazine > aripiprazole > brexpiprazole; however, this is by indirect comparison, and appropriately designed head-to-head clinical trials will be necessary in order to accurately assess these potential differences.
Because of the partial agonist activity at the dopamine D2 receptor, aripiprazole, brexpiprazole, and cariprazine are less likely to cause hyperprolactinemia than other first-line first- or second-generation antipsychotics. Other differentiating features of the dopamine receptor partial agonists compared with other choices include a relative lack of effect on the QT interval.38 In general, as predicted by their relatively lower binding affinities to histamine H1 receptors, the dopamine receptor partial agonists are not especially sedating.39
Likelihood to be helped or harmed
The concept of likelihood to be helped or harmed (LHH) can be useful to assess benefit vs risk, provided you select a relevant harm to contrast with the expected benefit.40 Table 31 provides the NNT for response, NNH for discontinuation because of an adverse event (where applicable), the NNHs for weight gain ≥7%, somnolence adverse events, and akathisia adverse events, together with the calculated LHH (where applicable). With the exception of aripiprazole for the treatment of MDD when comparing response vs akathisia, all LHH values are >1.0, and thus the benefit (response) would be encountered more often than the harm. When LHH values are ≥10, this can be interpreted that one would encounter a response at least 10 times more often than the adverse event of interest. This was observed for brexpiprazole for the treatment of schizophrenia when comparing response vs akathisia, for cariprazine for schizophrenia when comparing response vs somnolence, for aripiprazole for bipolar mania when comparing response vs discontinuation because of an adverse event, and for cariprazine for bipolar mania when comparing response vs somnolence.
Beyond acute studies
When treating patients with schizophrenia, delaying time to relapse is a main goal. In placebo-controlled randomized withdrawal studies of oral aripiprazole, brexpiprazole, and cariprazine in patients with schizophrenia, observed relapse rates vs placebo were reported, allowing the calculation of NNT vs placebo for the avoidance of relapse.41-44 These NNT values were similar and ranged from 4 to 5. For aripiprazole, relapse rates vs placebo in the 26-week study were 34% vs 57%, resulting in a NNT of 5 (95% CI 3 to 9); brexpiprazole, 52-week study, 13.5% vs 38.5%, NNT of 4 (95% CI 3 to 8); and cariprazine, 72-week study, 25% vs 47.5%, NNT of 5 (95% CI 3 to 11). In addition, cariprazine, 4.5 mg/d, has been directly compared with risperidone, 4 mg/d, in a 26-week double-blind study in non-geriatric adult patients with schizophrenia and predominant negative symptoms for at least 6 months.45 Cariprazine was superior to risperidone on the PANSS–Negative Factor Score, and response to treatment (decrease ≥20% in PANSS–Negative Factor Score) was achieved by more patients treated with cariprazine by 26 weeks than those treated with risperidone (69% vs 58%, NNT 9 [95% CI 5 to 44]).
Caveats
The harms discussed in this article are primarily from acute studies and do not reflect effects that can take time to develop, such as tardive dyskinesia, the long-term accumulation of body weight, and the development of insulin resistance/type 2 diabetes mellitus.40 The data presented are from carefully conducted registration trials that enrolled subjects who fulfilled restrictive inclusion/exclusion criteria. Such patients may differ from those encountered in routine clinical practice. Keep in mind that adverse events may differ in terms of impact and may not be clinically relevant if the adverse event is mild, time-limited, or easily managed. Moreover, different patients carry different propensities to experience different adverse events or to achieve a therapeutic response.
Continue to: Bottom Line
Bottom Line
Although aripiprazole, brexpiprazole, and cariprazine are all dopamine receptor partial agonists with demonstrated efficacy in psychiatric disorders, they differ in terms of available formulations, indications, pharmacodynamics, pharmacokinetics, titration requirements, and tolerability. Careful consideration of these factors can increase the likelihood of successful treatment.
Related Resources
- Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407-411.
- U.S. Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf.
Drug Brand Names
Aripiprazole • Abilify
Aripiprazole lauroxil • Aristada
Aripiprazole monohydrate • Abilify Maintena
Brexpiprazole • Rexulti
Cariprazine • Vraylar
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.
1. C
2. Otsuka. Abilify (aripiprazole) tablets, ABILIFY DISCMELT (aripiprazole) orally disintegrating tablets, ABILIFY (aripiprazole) oral solution, Abilify (aripiprazole) injection for intramuscular use only. Prescribing information. http://www.otsuka-us.com/Documents/Abilify.PI.pdf. Revised February 2018. Accessed March 14, 2018.
3. Citrome L. Aripiprazole long-acting injectable formulations for schizophrenia: aripiprazole monohydrate and aripiprazole lauroxil. Expert Rev Clin Pharmacol. 2016;9(2):169-186.
4. Citrome L. Long-acting injectable antipsychotics update: lengthening the dosing interval and expanding the diagnostic indications. Expert Rev Neurother. 2017;17(10):1029-1043.
5. Mace S, Taylor D. Aripiprazole: dose-response relationship in schizophrenia and schizoaffective disorder. CNS Drugs. 2009;23(9):773-780.
6. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
7. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry. 2002;63(9):763-771.
8. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2003;60(7):681-690.
9. McEvoy JP, Daniel DG, Carson WH Jr, et al. A randomized, double-blind, placebo-controlled, study of the efficacy and safety of aripiprazole 10, 15 or 20 mg/day for the treatment of patients with acute exacerbations of schizophrenia. J Psychiatr Res. 2007;41(11):895-905.
10. Cutler AJ, Marcus RN, Hardy SA, et al. The efficacy and safety of lower doses of aripiprazole for the treatment of patients with acute exacerbation of schizophrenia. CNS Spectr. 2006;11(9):691-702.
11. Sachs G, Sanchez R, Marcus R, et al; Aripiprazole Study Group. Aripiprazole in the treatment of acute manic or mixed episodes in patients with bipolar I disorder: a 3-week placebo-controlled study. J Psychopharmacol. 2006;20(4):536-546.
12. Keck PE Jr, Marcus R, Tourkodimitris S, et al; Aripiprazole Study Group. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651-1658.
13. Keck PE, Orsulak PJ, Cutler AJ, et al; CN138-135 Study Group. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1-3):36-49.
14. Young AH, Oren DA, Lowy A, et al. Aripiprazole monotherapy in acute mania: 12-week randomised placebo- and haloperidol-controlled study. Br J Psychiatry. 2009;194(1):40-48.
15. Vieta E, T’joen C, McQuade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316-1325.
16. Marcus RN, McQuade RD, Carson WH, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a second multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychopharmacol. 2008;28(2):156-165.
17. Berman RM, Marcus RN, Swanink R, et al. The efficacy and safety of aripiprazole as adjunctive therapy in major depressive disorder: a multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2007;68(6):843-853.
18. Berman RM, Fava M, Thase ME, et al. Aripiprazole augmentation in major depressive disorder: a double-blind, placebo-controlled study in patients with inadequate response to antidepressants. CNS Spectr. 2009;14(4):197-206.
19. Otsuka. Rexulti (brexpiprazole) tablets, for oral use. Prescribing information. http://www.otsuka-us.com/Products/Documents/Rexulti.PI.pdf. Revised February 2018. Accessed March 14, 2018.
20. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
21. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
22. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
23. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants. J Clin Psychiatry. 2015;76(9):1224-1231.
24. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76(9):1232-1240.
25. Allergan. Vraylar (cariprazine) capsules, for oral use. Prescribing information. https://www.allergan.com/assets/pdf/vraylar_pi. Revised November 2017. Accessed March 14, 2018.
26. Durgam S, Starace A, Li D, et al. An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res. 2014;152(2-3):450-457.
27. Durgam S, Cutler AJ, Lu K, et al. Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry. 2015;76(12):e1574-e1582.
28. Kane JM, Zukin S, Wang Y, et al. Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol. 2015;35(4):367-373.
29. Calabrese JR, Keck PE Jr, Starace A, et al. Efficacy and safety of low- and high-dose cariprazine in acute and mixed mania associated with bipolar I disorder: a double-blind, placebo-controlled study. J Clin Psychiatry. 2015;76(3):284-292.
30. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
31. Sachs GS, Greenberg WM, Starace A, et al. Cariprazine in the treatment of acute mania in bipolar I disorder: a double-blind, placebo-controlled, phase III trial. J Affect Disord. 2015;174:296-302.
32. Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15(10):1219-1229.
33. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
34. Kiss B, Horváth A, Némethy Z, et al. Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther. 2010;333(1):328-340.
35. Zimnisky R, Chang G, Gyertyán I, et al. Cariprazine, a dopamine D3-receptor-preferring partial agonist, blocks phencyclidine-induced impairments of working memory, attention set-shifting, and recognition memory in the mouse. Psychopharmacology (Berl). 2013; 226(1):91-100.
36. Neill JC, Grayson B, Kiss B, et al. Effects of cariprazine, a novel antipsychotic, on cognitive deficit and negative symptoms in a rodent model of schizophrenia symptomatology. Eur Neuropsychopharmacol. 2016;26(1):3-14.
37. Gyertyán I, Kiss B, Sághy K, et al. Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int. 2011;59(6):925-935.
38. Leucht S, Leucht C, Huhn M, et al. Sixty years of placebo-controlled antipsychotic drug trials in acute schizophrenia: systematic review, Bayesian meta-analysis, and meta-regression of efficacy predictors. Am J Psychiatry. 2017;174(10):927-942.
39. Citrome L. Activating and sedating adverse effects of second-generation antipsychotics in the treatment of schizophrenia and major depressive disorder: absolute risk increase and number needed to harm. J Clin Psychopharmacol. 2017;37(2):138-147.
40. Citrome L, Kantrowitz J. Antipsychotics for the treatment of schizophrenia: likelihood to be helped or harmed, understanding proximal and distal benefits and risks. Expert Rev Neurother. 2008;8(7):1079-1091.
41. Pigott TA, Carson WH, Saha AR, et al; Aripiprazole Study Group. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry. 2003;64(9):1048-1056.
42. Fleischhacker WW, Hobart M, Ouyang J, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Int J Neuropsychopharmacol. 2016;20(1):11-21.
43. Durgam S, Earley W, Li R, et al. Long-term cariprazine treatment for the prevention of relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled trial. Schizophr Res. 2016;176(2-3):264-271.
44. Citrome L. Schizophrenia relapse, patient considerations, and potential role of lurasidone. Patient Prefer Adherence. 2016;10:1529-1537.
45. Németh G, Laszlovszky I, Czobor P, et al. Cariprazine versus risperidone monotherapy for treatment of predominant negative symptoms in patients with schizophrenia: a randomised, double-blind, controlled trial. Lancet. 2017;389(10074):1103-1113.

Aripiprazole, brexpiprazole, and cariprazine

Brexpiprazole for schizophrenia and as adjunct for major depressive disorder
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
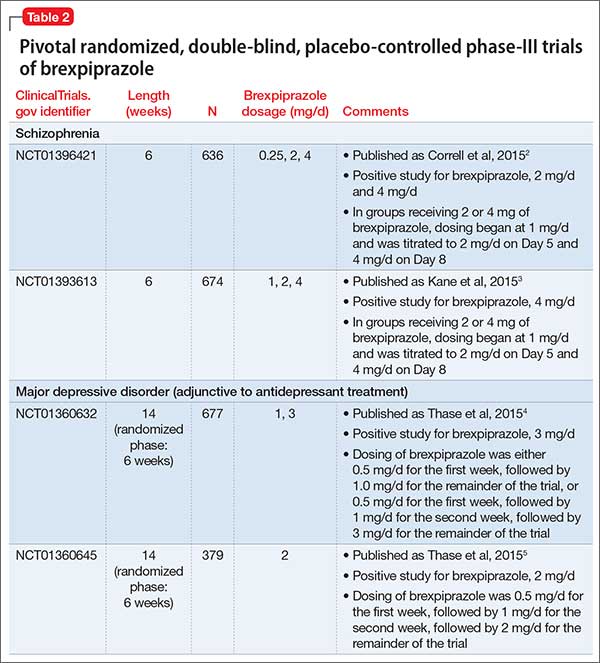
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
Brexpiprazole, FDA-approved in July 2015 to treat schizophrenia and as an adjunct for major depressive disorder (MDD) (Table 1), has shown efficacy in 2 phase-III acute trials for each indication.1-6 Although brexpiprazole is a dopamine D2 partial agonist, it differs from aripiprazole, the other available D2 partial agonist, because it is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors,7 which could mean better tolerability.
Clinical implications
Schizophrenia is heterogeneous, and individual response and tolerability to antipsychotics vary greatly8; therefore, new drug options are useful. For MDD, before the availability of brexpiprazole, only 3 antipsychotics were FDA-approved for adjunctive use with antidepressant therapy9; brexpiprazole represents another agent for patients whose depressive symptoms persist after standard antidepressant treatment.
Variables that limit the use of antipsychotics include extrapyramidal symptoms (EPS), akathisia, sedation/somnolence, weight gain, metabolic abnormalities, and hyperprolactinemia. If post-marketing studies and clinical experience confirm that brexpiprazole has an overall favorable side-effect profile regarding these tolerability obstacles, brexpiprazole would potentially have advantages over some other available agents, including aripiprazole.
How it works
In addition to a subnanomolar binding affinity (Ki < 1 nM) to dopamine D2 receptors as a partial agonist, brexpiprazole also exhibits similar binding affinities for serotonin 5-HT1A (partial agonist), 5-HT2A (antagonist), and adrenergic α1B (antagonist) and α2C (antagonist) receptors.7
Brexpiprazole also has high affinity (Ki < 5 nM) for dopamine D3 (partial ago nist), serotonin 5-HT2B (antagonist), and 5-HT7 (antagonist), and at adrenergic α1A (antagonist) and α1D (antagonist) receptors. Brexpiprazole has moderate affinity for histamine H1 receptors (Ki = 19 nM, antagonist), and low affinity for muscarinic M1 receptors (Ki > 1000 nM, antagonist).
Brexpiprazole’s pharmacodynamic profile differs from other available antipsychotics, including aripiprazole. Whether this translates to meaningful differences in efficacy and tolerability will depend on the outcomes of specifically designed clinical trials as well as “real-world” experience. Animal models have suggested amelioration of schizophrenia-like behavior, depression-like behavior, and anxiety-like behavior with brexipiprazole.6
Pharmacokinetics
At 91 hours, brexpiprazole’s half-life is relatively long; a steady-state concentration therefore is attained in approximately 2 weeks.1 In the phase-III clinical trials, brexpiprazole was titrated to target dosages, and therefore the product label recommends the same. Brexpiprazole can be administered with or without food.
In a study of brexpiprazole excretion, after a single oral dose of [14C]-labeled brexpiprazole, approximately 25% and 46% of the administered radioactivity was recovered in urine and feces, respectively. Less than 1% of unchanged brexpiprazole was excreted in the urine, and approximately 14% of the oral dose was recovered unchanged in the feces.
Exposure, as measured by maximum concentration and area under the concentration curve, is dose proportional.
Metabolism of brexpiprazole is mediated principally by cytochrome P450 (CYP) 3A4 and CYP2D6. Based on in vitro data, brexpiprazole shows little or no inhibition of CYP450 isozymes.
Efficacy
FDA approval for brexpiprazole for schizophrenia and for adjunctive use in MDD was based on 4 phase-III pivotal acute clinical trials conducted in adults, 2 studies each for each disorder.1-6 These studies are described in Table 2.2-5
Schizophrenia. The primary outcome measure for the acute schizophrenia trials was change on the Positive and Negative Syndrome Scale (PANSS) total scores from baseline to 6-week endpoint. Statistically significant reductions in PANSS total score were observed for brexpiprazole dosages of 2 mg/d and 4 mg/d in one study,2 and 4 mg/d in another study.3 Responder rates also were measured, with response defined as a reduction of ≥30% from baseline in PANSS total score or a Clinical Global Impressions-Improvement score of 1 (very much improved) or 2 (much improved).2,3 Pooling together the available data for the recommended target dosage of brexpiprazole for schizophrenia (2 to 4 mg/d) from the 2 phase-III studies, 45.5% of patients responded to the drug, compared with 31% for the pooled placebo groups, yielding a number needed to treat (NNT) of 7 (95% CI, 5-12).6
Although not described in product labeling, a phase-III 52-week maintenance study demonstrated brexpiprazole’s efficacy in preventing exacerbation of psychotic symptoms and impending relapse in patients with schizophrenia.10 Time from randomization to exacerbation of psychotic symptoms or impending relapse showed a beneficial effect with brexpiprazole compared with placebo (log-rank test: hazard ratio = 0.292, P < .0001). Significantly fewer patients in the brexpiprazole group relapsed compared with placebo (13.5% vs 38.5%, P < .0001), resulting in a NNT of 4 (95% CI, 3-8).
Major depressive disorder. The primary outcome measure for the acute MDD studies was change in Montgomery-Åsberg Depression Rating Scale (MADRS) scores from baseline to 6-week endpoint of the randomized treatment phase. All patients were required to have a history of inadequate response to 1 to 3 treatment trials of standard antidepressants for their current depressive episode. In addition, patients entered the randomized phase only if they had an inadequate response to antidepressant therapy during an 8-week prospective treatment trial of standard antidepressant treatment plus single-blind placebo.
Participants who responded adequately to the antidepressant in the prospective single-blind phase were not randomized, but instead continued on antidepressant treatment plus single-blind placebo for 6 weeks.
The phase-III studies showed positive results for brexpiprazole, 2 mg/d and 3 mg/d, with change in MADRS from baseline to endpoint superior to that observed with placebo.4,5
When examining treatment response, defined as a reduction of ≥50% in MADRS total score from baseline, NNT vs placebo for response were 12 at all dosages tested, however, NNT vs placebo for remission (defined as MADRS total score ≤10 and ≥50% improvement from baseline) ranged from 17 to 31 and were not statistically significant.6 When the results for brexpiprazole, 1 mg/d, 2 mg/d, and 3 mg/d, from the 2 phase-III trials are pooled together, 23.2% of the patients receiving brexpiprazole were responders, vs 14.5% for placebo, yielding a NNT of 12 (95% CI, 8-26); 14.4% of the brexpiprazole-treated patients met remission criteria, vs 9.6% for placebo, resulting in a NNT of 21 (95% CI, 12-138).6
Tolerability
Overall tolerability can be evaluated by examining the percentage of patients who discontinued the clinical trials because of an adverse event (AE). In the acute schizophrenia double-blind trials for the recommended dosage range of 2 to 4 mg, the discontinuation rates were lower overall for patients receiving brexpiprazole compared with placebo.2,3 In the acute MDD trials, 32.6% of brexpiprazole-treated patients and 10.7% of placebo-treated patients discontinued because of AEs,4,5 yielding a number needed to harm (NNH) of 53 (95% CI, 30-235).6
The most commonly encountered AEs for MDD (incidence ≥5% and at least twice the rate for placebo) were akathisia (8.6% vs 1.7% for brexpiprazole vs placebo, and dose-related) and weight gain (6.7% vs 1.9%),1 with NNH values of 15 (95% CI, 11-23), and 22 (95% CI, 15-42), respectively.6 The most commonly encountered AE for schizophrenia (incidence ≥4% and at least twice the rate for placebo) was weight gain (4% vs 2%),1 with a NNH of 50 (95% CI, 26-1773).6
Of note, rates of akathisia in the schizophrenia trials were 5.5% for brexpiprazole and 4.6% for placebo,1 yielding a non-statistically significant NNH of 112.6 In a 6-week exploratory study,11 the incidence of EPS-related AEs including akathisia was lower for brexpiprazole-treated patients (14.1%) compared with those receiving aripiprazole (30.3%), for a NNT advantage for brexpiprazole of 7 (not statistically significant).
Short-term weight gain appears modest; however, outliers with an increase of ≥7% of body weight were evident in open-label long-term safety studies.1,6 Effects on glucose and lipids were small. Minimal effects on prolactin were observed, and no clinically relevant effects on the QT interval were evident.
Contraindications
The only absolute contraindication for brexpiprazole is known hypersensitivity to brexpiprazole or any of its components. Reactions have included rash, facial swelling, urticaria, and anaphylaxis.1
As with all antipsychotics and antipsychotics with an indication for a depressive disorder:
• there is a bolded boxed warning in the product label regarding increased mortality in geriatric patients with dementia-related psychosis. Brexpiprazole is not approved for treating patients with dementia-related psychosis
• there is a bolded boxed warning in the product label about suicidal thoughts and behaviors in patients age ≤24. The safety and efficacy of brexpiprazole have not been established in pediatric patients.
Dosing
Schizophrenia. The recommended starting dosage for brexpiprazole for schizophrenia is 1 mg/d on Days 1 to 4. Brexpiprazole can be titrated to 2 mg/d on Day 5 through Day 7, then to 4 mg/d on Day 8 based on the patient’s response and ability to tolerate the medication. The recommended target dosage is 2 to 4 mg/d with a maximum recommended daily dosage of 4 mg.
Major depressive disorder. The recommended starting dosage for brexpiprazole as adjunctive treatment for MDD is 0.5 mg or 1 mg/d. Brexpiprazole can be titrated to 1 mg/d, then up to the target dosage of 2 mg/d, with dosage increases occurring at weekly intervals based on the patient’s clinical response and ability to tolerate the agent, with a maximum recommended dosage of 3 mg/d.
Other considerations. For patients with moderate to severe hepatic impairment, or moderate, severe, or end-stage renal impairment, the maximum recommended dosage is 3 mg/d for patients with schizophrenia, and 2 mg/d for patients with MDD.
In general, dosage adjustments are recommended in patients who are known CYP2D6 poor metabolizers and in those taking concomitant CYP3A4 inhibitors or CYP2D6 inhibitors or strong CYP3A4 inducers1:
• for strong CYP2D6 or CYP3A4 inhibitors, administer one-half the usual dosage
• for strong/moderate CYP2D6 with strong/moderate CYP3A4 inhibitors, administer a one-quarter of the usual dosage
• for known CYP2D6 poor metabolizers taking strong/moderate CYP3A4 inhibitors, also administer a one-quarter of the usual dosage
• for strong CYP3A4 inducers, double the usual dosage and further adjust based on clinical response.
In clinical trials for MDD, brexpiprazole dosage was not adjusted for strong CYP2D6 inhibitors (eg, paroxetine, fluoxetine). Therefore, CYP considerations are already factored into general dosing recommendations and brexpiprazole could be administered without dosage adjustment in patients with MDD; however, under these circumstances, it would be prudent to start brexpiprazole at 0.5 mg, which, although “on-label,” represents a low starting dosage. (Whenever 2 drugs are co-administered and 1 agent has the ability to disturb the metabolism of the other, using smaller increments to the target dosage and possibly waiting longer between dosage adjustments could help avoid potential drug–drug interactions.)
No dosage adjustment for brexpiprazole is required on the basis of sex, race or ethnicity, or smoking status. Although clinical studies did not include patients age ≥65, the product label recommends that in general, dose selection for a geriatric patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
Bottom Line
Brexpiprazole, an atypical antipsychotic, is FDA-approved for schizophrenia and as an adjunct to antidepressants in major depressive disorder. For both indications, brexpiprazole demonstrated positive results compared with placebo in phase-III trials. Brexpiprazole is more potent at serotonin 5-HT1A and 5-HT2A receptors and displays less intrinsic activity at D2 receptors than aripiprazole, which could mean that the drug may be better-tolerated.
Related Resources
• Citrome L. Brexpiprazole: a new dopamine D2 receptor partial agonist for the treatment of schizophrenia and major depressive disorder. Drugs Today (Barc). 2015;51(7):397-414.
• Citrome L, Stensbøl TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. In press.
Drug Brand Names
Aripiprazole • Abilify
Brexpiprazole • Rexulti
Fluoxetine • Prozac
Paroxetine • Paxil
Disclosure
Dr. Citrome is a consultant to Alexza Pharmaceuticals, Alkermes, Allergan, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly and Company, Forum Pharmaceuticals, Genentech, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Medivation, Mylan, Novartis, Noven, Otsuka, Pfizer, Reckitt Benckiser, Reviva, Shire, Sunovion, Takeda, Teva, and Valeant Pharmaceuticals; and is a speaker for Allergan, AstraZeneca, Janssen, Jazz Pharmaceuticals, Lundbeck, Merck, Novartis, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Teva.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.
1. Rexulti [package insert]. Rockville, MD: Otsuka; 2015.
2. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
3. Kane JM, Skuban A, Ouyang J, et al. A multicenter, randomized, double-blind, controlled phase 3 trial of fixed-dose brexpiprazole for the treatment of adults with acute schizophrenia. Schizophr Res. 2015;164(1-3):127-135.
4. Thase ME, Youakim JM, Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/ JCP.14m09689.
5. Thase ME, Youakim JM, Skuban A, et al. Efficacy and safety of adjunctive brexpiprazole 2 mg in major depressive disorder: a phase 3, randomized, placebo-controlled study in patients with inadequate response to antidepressants [published online August 4, 2015]. J Clin Psychiatry. doi: 10.4088/JCP.14m09688.
6. Citrome L. Brexpiprazole for schizophrenia and as adjunct for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antipsychotic—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2015;69(9):978-997.
7. Maeda K, Sugino H, Akazawa H, et al. Brexpiprazole I: in vitro and in vivo characterization of a novel serotonin-dopamine activity modulator. J Pharmacol Exp Ther. 2014;350(3):589-604.
8. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917-1928.
9. Citrome L. Adjunctive aripiprazole, olanzapine, or quetiapine for major depressive disorder: an analysis of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Postgrad Med. 2010;122(4):39-48.
10. Hobart M, Ouyang J, Forbes A, et al. Efficacy and safety of brexpiprazole (OPC-34712) as maintenance treatment in adults with schizophrenia: a randomized, double-blind, placebo-controlled study. Poster presented at: the American Society of Clinical Psychopharmacology Annual Meeting; June 22 to 25, 2015; Miami, FL.
11. Citrome L, Ota A, Nagamizu K, Perry P, et al. The effect of brexpiprazole (OPC‐34712) versus aripiprazole in adult patients with acute schizophrenia: an exploratory study. Poster presented at: the Society of Biological Psychiatry Annual Scientific Meeting and Convention; May 15, 2015; Toronto, Ontario, Canada.
Inhaled loxapine for agitation
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
How to best help patients with residual schizophrenia symptoms
Treatment-resistant schizophrenia: What can we do about it?

Discuss this article at www.facebook.com/CurrentPsychiatry
Patients with treatment-resistant schizophrenia can be broadly defined to include any persons with residual symptoms that cause distress or impairment despite several treatment attempts. Unfortunately, this definition may include most of our patients with schizophrenia.
Clinical trial data on treatment-resistant schizophrenia can be contradictory, leaving “N of 1” empirical treatment trials for individual patients as the current state of the art. This article presents data from clinical trials for pharmacologic and nonpharmacologic options and offers recommendations to try to help our treatment-resistant patients.
Defining treatment resistance
Research reports regarding treatment-resistant or treatment-refractory schizophrenia have relied on operational criteria such as that found in the pivotal study for clozapine1:
- at least 3 periods of treatment in the preceding 5 years with neuroleptic agents from at least 2 different chemical classes at dosages equivalent to ≥1000 mg/d of chlorpromazine for 6 weeks, each without significant symptomatic relief, and
- no period of good functioning within the preceding 5 years.1 In that study, patients also underwent a prospective treatment trial with what we now know are high doses of haloperidol (up to 60 mg/d or higher) and benztropine mesylate (6 mg/d) for a period of 6 weeks to confirm lack of drug responsiveness. Other studies have more relaxed criteria, such as:
- persistent positive symptoms—hallucinations, delusions, or marked thought disorder—after at least 6 contiguous weeks of past or present treatment, with ≥1 typical antipsychotics at doses of ≥600 mg/d in chlorpromazine equivalents
- a poor level of functioning over the past 2 years, as defined by the lack of competitive employment or enrollment in an academic or vocational program and not having age-expected interpersonal relations with someone outside the biologic family with whom ongoing regular contacts were maintained.2
In this study, no prospective period of treatment to confirm lack of drug responsiveness was required.
The most clinically relevant definition of treatment resistance depends on the patient’s individual circumstances. For some patients, targeting positive symptoms is a high priority; for others it may be negative and cognitive symptoms; for others, it may be excitement. Moreover, families may complain of symptoms or behavior that are of little or no concern to your patient.
Although we desire treatment response and remission for our patients, definitions for remission and functional recovery are in flux. Proposed criteria define symptomatic remission as 6-month maintenance of simultaneous ratings of mild or less on delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behavior, and negative symptoms.3,4 Emsley et al4 note that reported remission rates vary widely across studies (17% to 88%) and that patients in remission do better than their non-remitted counterparts in several other outcome domains. Also, patients move in and out of remission over time. Predictors of remission include:
- early treatment response
- baseline symptom severity
- subjective well-being.4
Recovery is a more complex construct than remission and includes social outcomes. Although recovery lacks a standard definition, it is the implied goal of treatment. Anything short of recovery can be viewed as inadequate. If we set the bar at this height, many or most of the patients we treat for schizophrenia could be considered treatment-resistant.
Confounding factors
Before concluding that a patient is treatment-resistant, address medication adherence and possible substance use. Partial or nonadherence with antipsychotic treatment is common—approximately one-half of patients are nonadherent5—and associated with relapse and re-hospitalization.6 In addition, an estimated one-half of all individuals with schizophrenia also use substances.7
Be aware of the optimal dose for any particular antipsychotic and factors that can interfere with achieving adequate plasma levels. This means acknowledging that dosing ranges established during registration studies may not reflect the needs of day-to-day clinical practice.8 Pharmacokinetic interactions with other medications, such as carbamazepine or rifampin, can induce liver enzymes and result in subtherapeutic antipsychotic levels. Cigarette smoking also may have this effect. Lowered clozapine or olanzapine plasma levels have been observed in patients who resume smoking after being discharged from a non-smoking inpatient environment. Some antipsychotics, such as ziprasidone and lurasidone, must be taken with food in order to have sufficient bioavailability.9
What does a patient want?
Patients with schizophrenia often have limited insight into their psychotic symptoms.10 Savvy clinicians will attempt to leverage a patient’s insight into ancillary symptoms—such as impaired sleep, anxiety, and dysphoria—to encourage a therapeutic alliance and therefore adherence. If patients feel their concerns are not addressed, they may consider treatment inadequate even though the intensity of their hallucinations and delusions may have decreased.
Which antipsychotic is best?
Meta-analyses of randomized controlled trials (RCTs) of antipsychotic treatment for schizophrenia found that, although individual response will vary, clozapine generally has better efficacy that other antipsychotics.11-13 Olanzapine, risperidone, and amisulpride (which is not available in the United States) appear to be more efficacious than first-generation antipsychotics. Other second-generation antipsychotics do not consistently show greater efficacy than first-generation antipsychotics, although their tolerability profiles vary greatly.11-13
Antipsychotic monotherapy. More than 25 RCTs have focused on antipsychotic monotherapy for treatment-resistant patients; for a bibliography of these studies, click here. For the most part, clozapine has consistently demonstrated superiority over comparators. Because not all patients with schizophrenia can tolerate clozapine or are willing to have their blood monitored as required, other second-generation antipsychotics have been suggested as possible substitutes. Olanzapine has established superior efficacy to first-generation antipsychotics11-13 and perhaps comparable efficacy to clozapine in some studies.2,14-17 Risperidone appeared to be comparable to clozapine in some studies,18,19 whereas clozapine’s superiority was evident in others.14,20,21 Although an RCT found comparable efficacy for ziprasidone vs clozapine,22 patients enrolled in this study may not have been treatment-resistant regarding efficacy but instead could not tolerate prior treatments. Enrolling patients on the basis of poor efficacy and/or poor tolerability to their prior antipsychotic regimen also has complicated the interpretation of studies comparing olanzapine with clozapine16 and risperidone with clozapine.18
Antipsychotic combinations. Combinations of antipsychotics are used commonly when treating chronic schizophrenia.23 Of the approximately 20 RCTs of antipsychotic combination therapy, most tested clozapine combined with other second-generation antipsychotics, such as risperidone. For a bibliography of these studies, click here. Only 5 studies support a combination approach (Table 1).
Table 1
Antipsychotic combinations: Few studies support efficacy
| Study | Design | Patients | Results |
|---|---|---|---|
| Shiloh et al, 1997a | 10-week, double-blind, placebo-controlled | 28 patients nonresponsive to typical antipsychotics and partially responsive to clozapine received add-on sulpiride,* 600 mg/d, or placebo | The sulpiride group showed improvements in positive and negative symptoms |
| Josiassen et al, 2005b | 12-week, randomized, double-blind, placebo-controlled | 40 schizophrenia patients unresponsive or partially responsive to clozapine randomized to clozapine + placebo or clozapine + risperidone, 6 mg/d | Mean BPRS total and positive symptom subscale scores reduced in both groups but reductions were greater in the clozapine/risperidone group; reduction in SANS also was observed in the clozapine/risperidone group |
| Genç et al, 2007c | 8-week, randomized, single-blind | 56 treatment-resistant schizophrenia patients randomly assigned to clozapine + amisulpride* or clozapine + quetiapine | Both groups improved at week 8 as measured by BPRS, SANS, SAPS, and CGI; however, patients receiving amisulpride showed greater improvement |
| Muscatello et al, 2011d | 24-week, randomized, double-blind, placebo-controlled | 31 treatment-resistant schizophrenia patients receiving clozapine randomized to receive adjunctive aripiprazole or placebo | Aripiprazole showed beneficial effect on positive and general psychopathologic symptomatology, but no significant effects on executive cognitive function |
| Takahashi et al, 1999e | 8-week, randomized, single-blind, crossover | 10 neuroleptic-treated patients received add-on risperidone and mosapramine* | Both additions resulted in significant, yet modest, improvement; no significant difference in PANSS between risperidone and mosapramine |
| *Not available in the United States BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression; PANSS: Positive and Negative Syndrome Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry. 1997;171:569-573. b. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136. c. Genç Y, Taner E, Candansayar S. Comparison of clozapine-amisulpride and clozapine-quetiapine combinations for patients with schizophrenia who are partially responsive to clozapine: a single-blind randomized study. Adv Ther. 2007;24(1):1-13. d. Muscatello MR, Bruno A, Pandolfo G, et al. Effect of aripiprazole augmentation of clozapine in schizophrenia: a double-blind, placebo-controlled study. Schizophr Res. 2011;127(1-3):93-99. e. Takahashi N, Terao T, Oga T, et al. Comparison of risperidone and mosapramine addition to neuroleptic treatment in chronic schizophrenia. Neuropsychobiology. 1999;39(2):81-85. | |||
What about augmentation?
Adjunctive non-antipsychotics also are commonly used when treating patients with chronic schizophrenia. For example, lithium and anticonvulsants are used in approximately one-half of all inpatients with schizophrenia in facilities operated by the State of New York Office of Mental Health.24,25 The evidence base for these agents as adjuncts to antipsychotics generally is weak.26 Specifically, early reports of benefit with adjunctive lithium have been negated by later studies. Similarly, large trials of adjunctive valproate and lamotrigine have failed to replicate early and promising efficacy signals from smaller trials, although the larger studies did not specifically target treatment-resistant schizophrenia.
Among mood stabilizers, lamotrigine may be the most promising for treatment-resistant schizophrenia. In a meta-analysis of clinical trials examining schizophrenia patients receiving clozapine (N=161) who were randomized to receive adjunctive lamotrigine or adjunctive placebo, lamotrigine was superior to placebo in total score for psychosis symptoms and scores for positive and negative symptoms.27
More than 125 published RCTs have studied a wide variety of adjunctive agents other than lithium or anticonvulsants for treating persistent schizophrenia symptoms (Table 2).
Only some of the approximately 40 RCTs regarding adjunctive antidepressants in patients with chronic schizophrenia focused on patients with ongoing depressive symptoms. For a bibliography of these studies, click here. In a meta-analysis measuring improvement of negative symptoms from 23 trials (N=819),28 the effect size was moderate in favor of antidepressants. Subgroup analysis revealed significant responses for fluoxetine, trazodone, and ritanserin.
More than 50 RCTs have focused on augmenting medications for cognitive dysfunction in chronic schizophrenia. Unfortunately, agents used to treat Alzheimer’s disease have shown disappointing results when tested in patients with schizophrenia, as have agents prescribed for attention-deficit/hyperactivity disorder (methylphenidate, guanfacine, atomoxetine) or agents used to promote alertness (modafinil and armodafinil).
Medications that act on glutamate receptors may offer another potential solution, although not in combination with clozapine.29
Other agents that require further study where ≥2 positive studies have been reported (with ≤2 negative studies) include celecoxib, neurosteroids and hormones, purinergic agents, serotonin 5-HT1A receptor agonists, and serotonin 5-HT3 receptor antagonists.
Table 2
Agents studied as adjuncts to antipsychotics
| Acetylsalicylic acid and nonsteroidal anti-inflammatory agents |
| Anticonvulsants and lithium |
| Antidepressants |
| Antiglucocorticoids |
| Agents used to treat attention-deficit/hyperactivity disorder |
| Beta blockers |
| Cholinesterase inhibitors and other agents used to treat Alzheimer’s disease |
| Experimental agents that act on glutamate receptors |
| GABAA receptor drugs |
| Neurosteroids and hormones |
| Omega-3 fatty acids |
| Opioid system agents |
| Peptides |
| Purinergic agents |
| Serotonin 5-HT1A receptor agonists |
| Serotonin 5-HT3 receptor antagonists |
| Wakefulness promoting agents |
Therapeutic neuromodulation
More than 10 RCTs of repetitive transcranial magnetic stimulation (rTMS) in patients with refractory symptoms of schizophrenia have been published; the results were mixed. For a bibliography of these studies, click here. In a meta-analysis of 9 trials (n=213),30 prefrontal rTMS for treating negative symptoms demonstrated a small-to-medium effect size. In another meta-analysis31 of all prospective studies of rTMS for negative symptoms and for auditory hallucinations and overall positive symptoms in refractory schizophrenia, the effect sizes showed moderate effects.
Fewer controlled trials are available for electroconvulsive therapy,32,33 but its use with clozapine appears encouraging.34
Psychological and behavioral intervention. Cognitive-behavioral therapy, although labor-intensive, can be helpful even in patients considered treatment-resistant (Table 3). These interventions generally are provided together with pharmacotherapy.
Complementary and alternative therapies. Patients and their families may ask about complementary and alternative therapies, particularly when conventional approaches have not been successful. A meta-analysis of 6 studies (n=828)35 that reviewed adjunctive use of ginkgo in patients with chronic schizophrenia found statistically significant moderate improvement in total and negative symptoms. Negative reports also are available, including a 5-month study of adjunctive megavitamins that did not demonstrate any benefits.36 In a review of 13 RCTs of acupuncture for schizophrenia, Lee et al found the overall methodological quality was too low to draw firm conclusions.37
Table 3
Cognitive-behavioral therapy for schizophrenia
| Study | Design | Patients | Results |
|---|---|---|---|
| Pinto et al, 1999a | 6-month, randomized controlled | 37 treatment-resistant schizophrenia patients were randomized to CBT plus social skills training or supportive therapy | Both groups showed statistically significant improvement on the BPRS, SAPS, and SANS; however, patients in the CBT group had lower BPRS and SAPS scores. No difference on SANS scores |
| Barretto et al, 2009b | 21-week, controlled (nonrandom-ized) | Patients refractory to clozapine were placed in a CBT or befriending control group | The CBT group showed significant improvement in PANSS total score and general psychopathology subscale score, as well as an improvement of QLS; improvement persisted at 6-month follow-up |
| BPRS: Brief Psychiatric Rating Scale; CBT: cognitive-behavioral therapy; PANSS: Positive and Negative Syndrome Scale; QLS: Quality of Life Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Pinto A, La Pia S, Mennella R, et al. Cognitive-behavioral therapy and clozapine for clients with treatment-refractory schizophrenia. Psychiatr Serv. 1999;50(7):901-904. b. Barretto EM, Kayo M, Avrichir BS, et al. A preliminary controlled trial of cognitive behavioral therapy in clozapine-resistant schizophrenia. J Nerv Ment Dis. 2009;197(11):865-868. | |||
Clinical recommendations
Before declaring a patient with schizophrenia as treatment-resistant, ensure that an adequate trial of medication did take place. This includes consideration of adequate dosing and pharmacokinetic issues. Awareness of potential substance use and/or partial adherence or nonadherence also is critical because these factors can impact treatment response.
When prescribing for a treatment-resistant schizophrenia patient, identify specific target symptoms to better inform medication selection—especially for symptoms that the patient feels are important. For example, consider an antidepressant for patients who have negative or depressive symptoms. Also take into account other patient-centered concerns, such as tolerability issues that may have interfered with adherence and response in the past.
Clozapine remains the medication of choice for treatment-resistant schizophrenia. Despite dozens of RCTs of potential adjunctive agents for treatment-resistant schizophrenia, no single approach has consistently shown efficacy in reducing symptoms, improving cognition, or increasing a patient’s level of function. Individual response can vary, and our search for the “outlier” who does respond to an adjunctive agent can explain our use of these strategies in clinical practice.
Related Resources
- Cochrane Database of Systematic Reviews. www.cochrane.org/reviews. This database contains reviews of additional therapeutic options for patients with treatment-resistant schizophrenia. As of February 23, 2011, 157 reviews were available.
- Citrome L. Treatment-refractory schizophrenia: What it is and what’s been done about it. Neuropsychiatry. 2011. Epub ahead of print.
- Citrome L. Clozapine for schizophrenia. Life-threatening or life-saving treatment? Current Psychiatry. 2009;8(12):56-63.
Drug Brand Names
- Aripiprazole • Abilify
- Armodafinil • Nuvigil
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Carbamazepine • Tegretol
- Celecoxib • Celebrex
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Guanfacine • Tenex
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Lurasidone • Latuda
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Rifampin • Rifadin
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproate (Divalproex) • Depakote, Depakote ER
- Ziprasidone • Geodon
Disclosure
No writing assistance or external financial support was used for this article. Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Avanir Pharmaceuticals, Azur Pharma Inc., Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Merck, Novartis, Pfizer Inc., Sunovion, Valeant Pharmaceuticals, and Vanda Pharmaceuticals.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry. 2002;159(2):255-262.
3. Andreasen NC, Carpenter WT Jr, Kane JM, et al. Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry. 2005;162(3):441-449.
4. Emsley R, Chiliza B, Asmal L, et al. The concepts of remission and recovery in schizophrenia. Curr Opin Psychiatry. 2011;24(2):114-121.
5. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
6. Robinson DG, Woerner MG, Delman HM, et al. Pharmacological treatments for first-episode schizophrenia. Schizophr Bull. 2005;31(3):705-722.
7. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
8. Citrome L, Volavka J. Optimal dosing of atypical antipsychotics in adults: a review of the current evidence. Harv Rev Psychiatry. 2002;10(5):280-291.
9. Citrome L. Iloperidone asenapine and lurasidone. A brief overview of three new second-generation antipsychotics. Postgrad Med. 2011;123(2):153-162.
10. Lincoln TM, Lüllmann E, Rief W. Correlates and long-term consequences of poor insight in patients with schizophrenia. A systematic review. Schizophr Bull. 2007;33(6):1324-1342.
11. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31-41.
12. Leucht S, Komossa K, Rummel-Kluge C, et al. A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry. 2009;166(2):152-163.
13. Leucht S, Arbter D, Engel RR, et al. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14(4):429-447.
14. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
15. Tollefson GD, Birkett MA, Kiesler GM, et al. Double-blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine. Biol Psychiatry. 2001;49(1):52-63.
16. Bitter I, Dossenbach MR, Brook S, et al. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
17. Meltzer HY, Bobo WV, Roy A, et al. A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia. J Clin Psychiatry. 2008;69(2):274-285.
18. Bondolfi G, Dufour H, Patris M, et al. Risperidone versus clozapine in treatment-resistant chronic schizophrenia: a randomized double-blind study. Am J Psychiatry. 1998;155(4):499-504.
19. Wahlbeck K, Cheine M, Tuisku K, et al. Risperidone versus clozapine in treatment-resistant schizophrenia: a randomized pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(6):911-922.
20. Breier AF, Malhotra AK, Su TP, et al. Clozapine and risperidone in chronic schizophrenia: effects on symptoms, parkinsonian side effects, and neuroendocrine response. Am J Psychiatry. 1999;156(2):294-298.
21. Azorin JM, Spiegel R, Remington G, et al. A double-blind comparative study of clozapine and risperidone in the management of severe chronic schizophrenia. Am J Psychiatry. 2001;158(8):1305-1313.
22. Sacchetti E, Galluzzo A, Valsecchi P, et al. Ziprasidone vs clozapine in schizophrenia patients refractory to multiple antipsychotic treatments: the MOZART study. Schizophr Res. 2009;113(1):112-121.
23. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf. 2003;12(1):41-48.
24. Citrome L, Levine J, Allingham B. Changes in use of valproate and other mood stabilizers for patients with schizophrenia from 1994 to 1998. Psychiatr Serv. 2000;51(5):634-638.
25. Citrome L, Jaffe A, Levine J, et al. Use of mood stabilizers among patients with schizophrenia, 1994-2001. Psychiatr Serv. 2002;53(10):1212.-
26. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
27. Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2009;109(1-3):10-14.
28. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
29. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
30. Dlabac-de Lange JJ, Knegtering R, Aleman A, et al. Repetitive transcranial magnetic stimulation for negative symptoms of schizophrenia: review and meta-analysis. J Clin Psychiatry. 2010;71(4):411-418.
31. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
32. Chanpattana W, Chakrabhand ML, Sackeim HA, et al. Continuation ECT in treatment-resistant schizophrenia: a controlled study. J ECT. 1999;15(3):178-192.
33. Goswami U, Kumar U, Singh B. Efficacy of electroconvulsive therapy in treatment resistant schizophrenia: a double-blind study. Indian J Psychiatry. 2003;45(1):26-29.
34. Braga RJ, Petrides G. The combined use of electroconvulsive therapy and antipsychotics in patients with schizophrenia. J ECT. 2005;21(2):75-83.
35. Singh V, Singh SP, Chan K. Review and meta-analysis of usage of ginkgo as an adjunct therapy in chronic schizophrenia. Int J Neuropsychopharmacol. 2010;13(2):257-271.
36. Vaughan K, McConaghy N. Megavitamin and dietary treatment in schizophrenia: a randomised controlled trial. Aust N Z J Psychiatry. 1999;33(1):84-88.
37. Lee MS, Shin BC, Ronan P, et al. Acupuncture for schizophrenia: a systematic review and meta-analysis. Int J Clin Pract. 2009;63(11):1622-1633.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients with treatment-resistant schizophrenia can be broadly defined to include any persons with residual symptoms that cause distress or impairment despite several treatment attempts. Unfortunately, this definition may include most of our patients with schizophrenia.
Clinical trial data on treatment-resistant schizophrenia can be contradictory, leaving “N of 1” empirical treatment trials for individual patients as the current state of the art. This article presents data from clinical trials for pharmacologic and nonpharmacologic options and offers recommendations to try to help our treatment-resistant patients.
Defining treatment resistance
Research reports regarding treatment-resistant or treatment-refractory schizophrenia have relied on operational criteria such as that found in the pivotal study for clozapine1:
- at least 3 periods of treatment in the preceding 5 years with neuroleptic agents from at least 2 different chemical classes at dosages equivalent to ≥1000 mg/d of chlorpromazine for 6 weeks, each without significant symptomatic relief, and
- no period of good functioning within the preceding 5 years.1 In that study, patients also underwent a prospective treatment trial with what we now know are high doses of haloperidol (up to 60 mg/d or higher) and benztropine mesylate (6 mg/d) for a period of 6 weeks to confirm lack of drug responsiveness. Other studies have more relaxed criteria, such as:
- persistent positive symptoms—hallucinations, delusions, or marked thought disorder—after at least 6 contiguous weeks of past or present treatment, with ≥1 typical antipsychotics at doses of ≥600 mg/d in chlorpromazine equivalents
- a poor level of functioning over the past 2 years, as defined by the lack of competitive employment or enrollment in an academic or vocational program and not having age-expected interpersonal relations with someone outside the biologic family with whom ongoing regular contacts were maintained.2
In this study, no prospective period of treatment to confirm lack of drug responsiveness was required.
The most clinically relevant definition of treatment resistance depends on the patient’s individual circumstances. For some patients, targeting positive symptoms is a high priority; for others it may be negative and cognitive symptoms; for others, it may be excitement. Moreover, families may complain of symptoms or behavior that are of little or no concern to your patient.
Although we desire treatment response and remission for our patients, definitions for remission and functional recovery are in flux. Proposed criteria define symptomatic remission as 6-month maintenance of simultaneous ratings of mild or less on delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behavior, and negative symptoms.3,4 Emsley et al4 note that reported remission rates vary widely across studies (17% to 88%) and that patients in remission do better than their non-remitted counterparts in several other outcome domains. Also, patients move in and out of remission over time. Predictors of remission include:
- early treatment response
- baseline symptom severity
- subjective well-being.4
Recovery is a more complex construct than remission and includes social outcomes. Although recovery lacks a standard definition, it is the implied goal of treatment. Anything short of recovery can be viewed as inadequate. If we set the bar at this height, many or most of the patients we treat for schizophrenia could be considered treatment-resistant.
Confounding factors
Before concluding that a patient is treatment-resistant, address medication adherence and possible substance use. Partial or nonadherence with antipsychotic treatment is common—approximately one-half of patients are nonadherent5—and associated with relapse and re-hospitalization.6 In addition, an estimated one-half of all individuals with schizophrenia also use substances.7
Be aware of the optimal dose for any particular antipsychotic and factors that can interfere with achieving adequate plasma levels. This means acknowledging that dosing ranges established during registration studies may not reflect the needs of day-to-day clinical practice.8 Pharmacokinetic interactions with other medications, such as carbamazepine or rifampin, can induce liver enzymes and result in subtherapeutic antipsychotic levels. Cigarette smoking also may have this effect. Lowered clozapine or olanzapine plasma levels have been observed in patients who resume smoking after being discharged from a non-smoking inpatient environment. Some antipsychotics, such as ziprasidone and lurasidone, must be taken with food in order to have sufficient bioavailability.9
What does a patient want?
Patients with schizophrenia often have limited insight into their psychotic symptoms.10 Savvy clinicians will attempt to leverage a patient’s insight into ancillary symptoms—such as impaired sleep, anxiety, and dysphoria—to encourage a therapeutic alliance and therefore adherence. If patients feel their concerns are not addressed, they may consider treatment inadequate even though the intensity of their hallucinations and delusions may have decreased.
Which antipsychotic is best?
Meta-analyses of randomized controlled trials (RCTs) of antipsychotic treatment for schizophrenia found that, although individual response will vary, clozapine generally has better efficacy that other antipsychotics.11-13 Olanzapine, risperidone, and amisulpride (which is not available in the United States) appear to be more efficacious than first-generation antipsychotics. Other second-generation antipsychotics do not consistently show greater efficacy than first-generation antipsychotics, although their tolerability profiles vary greatly.11-13
Antipsychotic monotherapy. More than 25 RCTs have focused on antipsychotic monotherapy for treatment-resistant patients; for a bibliography of these studies, click here. For the most part, clozapine has consistently demonstrated superiority over comparators. Because not all patients with schizophrenia can tolerate clozapine or are willing to have their blood monitored as required, other second-generation antipsychotics have been suggested as possible substitutes. Olanzapine has established superior efficacy to first-generation antipsychotics11-13 and perhaps comparable efficacy to clozapine in some studies.2,14-17 Risperidone appeared to be comparable to clozapine in some studies,18,19 whereas clozapine’s superiority was evident in others.14,20,21 Although an RCT found comparable efficacy for ziprasidone vs clozapine,22 patients enrolled in this study may not have been treatment-resistant regarding efficacy but instead could not tolerate prior treatments. Enrolling patients on the basis of poor efficacy and/or poor tolerability to their prior antipsychotic regimen also has complicated the interpretation of studies comparing olanzapine with clozapine16 and risperidone with clozapine.18
Antipsychotic combinations. Combinations of antipsychotics are used commonly when treating chronic schizophrenia.23 Of the approximately 20 RCTs of antipsychotic combination therapy, most tested clozapine combined with other second-generation antipsychotics, such as risperidone. For a bibliography of these studies, click here. Only 5 studies support a combination approach (Table 1).
Table 1
Antipsychotic combinations: Few studies support efficacy
| Study | Design | Patients | Results |
|---|---|---|---|
| Shiloh et al, 1997a | 10-week, double-blind, placebo-controlled | 28 patients nonresponsive to typical antipsychotics and partially responsive to clozapine received add-on sulpiride,* 600 mg/d, or placebo | The sulpiride group showed improvements in positive and negative symptoms |
| Josiassen et al, 2005b | 12-week, randomized, double-blind, placebo-controlled | 40 schizophrenia patients unresponsive or partially responsive to clozapine randomized to clozapine + placebo or clozapine + risperidone, 6 mg/d | Mean BPRS total and positive symptom subscale scores reduced in both groups but reductions were greater in the clozapine/risperidone group; reduction in SANS also was observed in the clozapine/risperidone group |
| Genç et al, 2007c | 8-week, randomized, single-blind | 56 treatment-resistant schizophrenia patients randomly assigned to clozapine + amisulpride* or clozapine + quetiapine | Both groups improved at week 8 as measured by BPRS, SANS, SAPS, and CGI; however, patients receiving amisulpride showed greater improvement |
| Muscatello et al, 2011d | 24-week, randomized, double-blind, placebo-controlled | 31 treatment-resistant schizophrenia patients receiving clozapine randomized to receive adjunctive aripiprazole or placebo | Aripiprazole showed beneficial effect on positive and general psychopathologic symptomatology, but no significant effects on executive cognitive function |
| Takahashi et al, 1999e | 8-week, randomized, single-blind, crossover | 10 neuroleptic-treated patients received add-on risperidone and mosapramine* | Both additions resulted in significant, yet modest, improvement; no significant difference in PANSS between risperidone and mosapramine |
| *Not available in the United States BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression; PANSS: Positive and Negative Syndrome Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry. 1997;171:569-573. b. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136. c. Genç Y, Taner E, Candansayar S. Comparison of clozapine-amisulpride and clozapine-quetiapine combinations for patients with schizophrenia who are partially responsive to clozapine: a single-blind randomized study. Adv Ther. 2007;24(1):1-13. d. Muscatello MR, Bruno A, Pandolfo G, et al. Effect of aripiprazole augmentation of clozapine in schizophrenia: a double-blind, placebo-controlled study. Schizophr Res. 2011;127(1-3):93-99. e. Takahashi N, Terao T, Oga T, et al. Comparison of risperidone and mosapramine addition to neuroleptic treatment in chronic schizophrenia. Neuropsychobiology. 1999;39(2):81-85. | |||
What about augmentation?
Adjunctive non-antipsychotics also are commonly used when treating patients with chronic schizophrenia. For example, lithium and anticonvulsants are used in approximately one-half of all inpatients with schizophrenia in facilities operated by the State of New York Office of Mental Health.24,25 The evidence base for these agents as adjuncts to antipsychotics generally is weak.26 Specifically, early reports of benefit with adjunctive lithium have been negated by later studies. Similarly, large trials of adjunctive valproate and lamotrigine have failed to replicate early and promising efficacy signals from smaller trials, although the larger studies did not specifically target treatment-resistant schizophrenia.
Among mood stabilizers, lamotrigine may be the most promising for treatment-resistant schizophrenia. In a meta-analysis of clinical trials examining schizophrenia patients receiving clozapine (N=161) who were randomized to receive adjunctive lamotrigine or adjunctive placebo, lamotrigine was superior to placebo in total score for psychosis symptoms and scores for positive and negative symptoms.27
More than 125 published RCTs have studied a wide variety of adjunctive agents other than lithium or anticonvulsants for treating persistent schizophrenia symptoms (Table 2).
Only some of the approximately 40 RCTs regarding adjunctive antidepressants in patients with chronic schizophrenia focused on patients with ongoing depressive symptoms. For a bibliography of these studies, click here. In a meta-analysis measuring improvement of negative symptoms from 23 trials (N=819),28 the effect size was moderate in favor of antidepressants. Subgroup analysis revealed significant responses for fluoxetine, trazodone, and ritanserin.
More than 50 RCTs have focused on augmenting medications for cognitive dysfunction in chronic schizophrenia. Unfortunately, agents used to treat Alzheimer’s disease have shown disappointing results when tested in patients with schizophrenia, as have agents prescribed for attention-deficit/hyperactivity disorder (methylphenidate, guanfacine, atomoxetine) or agents used to promote alertness (modafinil and armodafinil).
Medications that act on glutamate receptors may offer another potential solution, although not in combination with clozapine.29
Other agents that require further study where ≥2 positive studies have been reported (with ≤2 negative studies) include celecoxib, neurosteroids and hormones, purinergic agents, serotonin 5-HT1A receptor agonists, and serotonin 5-HT3 receptor antagonists.
Table 2
Agents studied as adjuncts to antipsychotics
| Acetylsalicylic acid and nonsteroidal anti-inflammatory agents |
| Anticonvulsants and lithium |
| Antidepressants |
| Antiglucocorticoids |
| Agents used to treat attention-deficit/hyperactivity disorder |
| Beta blockers |
| Cholinesterase inhibitors and other agents used to treat Alzheimer’s disease |
| Experimental agents that act on glutamate receptors |
| GABAA receptor drugs |
| Neurosteroids and hormones |
| Omega-3 fatty acids |
| Opioid system agents |
| Peptides |
| Purinergic agents |
| Serotonin 5-HT1A receptor agonists |
| Serotonin 5-HT3 receptor antagonists |
| Wakefulness promoting agents |
Therapeutic neuromodulation
More than 10 RCTs of repetitive transcranial magnetic stimulation (rTMS) in patients with refractory symptoms of schizophrenia have been published; the results were mixed. For a bibliography of these studies, click here. In a meta-analysis of 9 trials (n=213),30 prefrontal rTMS for treating negative symptoms demonstrated a small-to-medium effect size. In another meta-analysis31 of all prospective studies of rTMS for negative symptoms and for auditory hallucinations and overall positive symptoms in refractory schizophrenia, the effect sizes showed moderate effects.
Fewer controlled trials are available for electroconvulsive therapy,32,33 but its use with clozapine appears encouraging.34
Psychological and behavioral intervention. Cognitive-behavioral therapy, although labor-intensive, can be helpful even in patients considered treatment-resistant (Table 3). These interventions generally are provided together with pharmacotherapy.
Complementary and alternative therapies. Patients and their families may ask about complementary and alternative therapies, particularly when conventional approaches have not been successful. A meta-analysis of 6 studies (n=828)35 that reviewed adjunctive use of ginkgo in patients with chronic schizophrenia found statistically significant moderate improvement in total and negative symptoms. Negative reports also are available, including a 5-month study of adjunctive megavitamins that did not demonstrate any benefits.36 In a review of 13 RCTs of acupuncture for schizophrenia, Lee et al found the overall methodological quality was too low to draw firm conclusions.37
Table 3
Cognitive-behavioral therapy for schizophrenia
| Study | Design | Patients | Results |
|---|---|---|---|
| Pinto et al, 1999a | 6-month, randomized controlled | 37 treatment-resistant schizophrenia patients were randomized to CBT plus social skills training or supportive therapy | Both groups showed statistically significant improvement on the BPRS, SAPS, and SANS; however, patients in the CBT group had lower BPRS and SAPS scores. No difference on SANS scores |
| Barretto et al, 2009b | 21-week, controlled (nonrandom-ized) | Patients refractory to clozapine were placed in a CBT or befriending control group | The CBT group showed significant improvement in PANSS total score and general psychopathology subscale score, as well as an improvement of QLS; improvement persisted at 6-month follow-up |
| BPRS: Brief Psychiatric Rating Scale; CBT: cognitive-behavioral therapy; PANSS: Positive and Negative Syndrome Scale; QLS: Quality of Life Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Pinto A, La Pia S, Mennella R, et al. Cognitive-behavioral therapy and clozapine for clients with treatment-refractory schizophrenia. Psychiatr Serv. 1999;50(7):901-904. b. Barretto EM, Kayo M, Avrichir BS, et al. A preliminary controlled trial of cognitive behavioral therapy in clozapine-resistant schizophrenia. J Nerv Ment Dis. 2009;197(11):865-868. | |||
Clinical recommendations
Before declaring a patient with schizophrenia as treatment-resistant, ensure that an adequate trial of medication did take place. This includes consideration of adequate dosing and pharmacokinetic issues. Awareness of potential substance use and/or partial adherence or nonadherence also is critical because these factors can impact treatment response.
When prescribing for a treatment-resistant schizophrenia patient, identify specific target symptoms to better inform medication selection—especially for symptoms that the patient feels are important. For example, consider an antidepressant for patients who have negative or depressive symptoms. Also take into account other patient-centered concerns, such as tolerability issues that may have interfered with adherence and response in the past.
Clozapine remains the medication of choice for treatment-resistant schizophrenia. Despite dozens of RCTs of potential adjunctive agents for treatment-resistant schizophrenia, no single approach has consistently shown efficacy in reducing symptoms, improving cognition, or increasing a patient’s level of function. Individual response can vary, and our search for the “outlier” who does respond to an adjunctive agent can explain our use of these strategies in clinical practice.
Related Resources
- Cochrane Database of Systematic Reviews. www.cochrane.org/reviews. This database contains reviews of additional therapeutic options for patients with treatment-resistant schizophrenia. As of February 23, 2011, 157 reviews were available.
- Citrome L. Treatment-refractory schizophrenia: What it is and what’s been done about it. Neuropsychiatry. 2011. Epub ahead of print.
- Citrome L. Clozapine for schizophrenia. Life-threatening or life-saving treatment? Current Psychiatry. 2009;8(12):56-63.
Drug Brand Names
- Aripiprazole • Abilify
- Armodafinil • Nuvigil
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Carbamazepine • Tegretol
- Celecoxib • Celebrex
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Guanfacine • Tenex
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Lurasidone • Latuda
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Rifampin • Rifadin
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproate (Divalproex) • Depakote, Depakote ER
- Ziprasidone • Geodon
Disclosure
No writing assistance or external financial support was used for this article. Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Avanir Pharmaceuticals, Azur Pharma Inc., Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Merck, Novartis, Pfizer Inc., Sunovion, Valeant Pharmaceuticals, and Vanda Pharmaceuticals.
Discuss this article at www.facebook.com/CurrentPsychiatry
Patients with treatment-resistant schizophrenia can be broadly defined to include any persons with residual symptoms that cause distress or impairment despite several treatment attempts. Unfortunately, this definition may include most of our patients with schizophrenia.
Clinical trial data on treatment-resistant schizophrenia can be contradictory, leaving “N of 1” empirical treatment trials for individual patients as the current state of the art. This article presents data from clinical trials for pharmacologic and nonpharmacologic options and offers recommendations to try to help our treatment-resistant patients.
Defining treatment resistance
Research reports regarding treatment-resistant or treatment-refractory schizophrenia have relied on operational criteria such as that found in the pivotal study for clozapine1:
- at least 3 periods of treatment in the preceding 5 years with neuroleptic agents from at least 2 different chemical classes at dosages equivalent to ≥1000 mg/d of chlorpromazine for 6 weeks, each without significant symptomatic relief, and
- no period of good functioning within the preceding 5 years.1 In that study, patients also underwent a prospective treatment trial with what we now know are high doses of haloperidol (up to 60 mg/d or higher) and benztropine mesylate (6 mg/d) for a period of 6 weeks to confirm lack of drug responsiveness. Other studies have more relaxed criteria, such as:
- persistent positive symptoms—hallucinations, delusions, or marked thought disorder—after at least 6 contiguous weeks of past or present treatment, with ≥1 typical antipsychotics at doses of ≥600 mg/d in chlorpromazine equivalents
- a poor level of functioning over the past 2 years, as defined by the lack of competitive employment or enrollment in an academic or vocational program and not having age-expected interpersonal relations with someone outside the biologic family with whom ongoing regular contacts were maintained.2
In this study, no prospective period of treatment to confirm lack of drug responsiveness was required.
The most clinically relevant definition of treatment resistance depends on the patient’s individual circumstances. For some patients, targeting positive symptoms is a high priority; for others it may be negative and cognitive symptoms; for others, it may be excitement. Moreover, families may complain of symptoms or behavior that are of little or no concern to your patient.
Although we desire treatment response and remission for our patients, definitions for remission and functional recovery are in flux. Proposed criteria define symptomatic remission as 6-month maintenance of simultaneous ratings of mild or less on delusions, hallucinations, disorganized speech, grossly disorganized or catatonic behavior, and negative symptoms.3,4 Emsley et al4 note that reported remission rates vary widely across studies (17% to 88%) and that patients in remission do better than their non-remitted counterparts in several other outcome domains. Also, patients move in and out of remission over time. Predictors of remission include:
- early treatment response
- baseline symptom severity
- subjective well-being.4
Recovery is a more complex construct than remission and includes social outcomes. Although recovery lacks a standard definition, it is the implied goal of treatment. Anything short of recovery can be viewed as inadequate. If we set the bar at this height, many or most of the patients we treat for schizophrenia could be considered treatment-resistant.
Confounding factors
Before concluding that a patient is treatment-resistant, address medication adherence and possible substance use. Partial or nonadherence with antipsychotic treatment is common—approximately one-half of patients are nonadherent5—and associated with relapse and re-hospitalization.6 In addition, an estimated one-half of all individuals with schizophrenia also use substances.7
Be aware of the optimal dose for any particular antipsychotic and factors that can interfere with achieving adequate plasma levels. This means acknowledging that dosing ranges established during registration studies may not reflect the needs of day-to-day clinical practice.8 Pharmacokinetic interactions with other medications, such as carbamazepine or rifampin, can induce liver enzymes and result in subtherapeutic antipsychotic levels. Cigarette smoking also may have this effect. Lowered clozapine or olanzapine plasma levels have been observed in patients who resume smoking after being discharged from a non-smoking inpatient environment. Some antipsychotics, such as ziprasidone and lurasidone, must be taken with food in order to have sufficient bioavailability.9
What does a patient want?
Patients with schizophrenia often have limited insight into their psychotic symptoms.10 Savvy clinicians will attempt to leverage a patient’s insight into ancillary symptoms—such as impaired sleep, anxiety, and dysphoria—to encourage a therapeutic alliance and therefore adherence. If patients feel their concerns are not addressed, they may consider treatment inadequate even though the intensity of their hallucinations and delusions may have decreased.
Which antipsychotic is best?
Meta-analyses of randomized controlled trials (RCTs) of antipsychotic treatment for schizophrenia found that, although individual response will vary, clozapine generally has better efficacy that other antipsychotics.11-13 Olanzapine, risperidone, and amisulpride (which is not available in the United States) appear to be more efficacious than first-generation antipsychotics. Other second-generation antipsychotics do not consistently show greater efficacy than first-generation antipsychotics, although their tolerability profiles vary greatly.11-13
Antipsychotic monotherapy. More than 25 RCTs have focused on antipsychotic monotherapy for treatment-resistant patients; for a bibliography of these studies, click here. For the most part, clozapine has consistently demonstrated superiority over comparators. Because not all patients with schizophrenia can tolerate clozapine or are willing to have their blood monitored as required, other second-generation antipsychotics have been suggested as possible substitutes. Olanzapine has established superior efficacy to first-generation antipsychotics11-13 and perhaps comparable efficacy to clozapine in some studies.2,14-17 Risperidone appeared to be comparable to clozapine in some studies,18,19 whereas clozapine’s superiority was evident in others.14,20,21 Although an RCT found comparable efficacy for ziprasidone vs clozapine,22 patients enrolled in this study may not have been treatment-resistant regarding efficacy but instead could not tolerate prior treatments. Enrolling patients on the basis of poor efficacy and/or poor tolerability to their prior antipsychotic regimen also has complicated the interpretation of studies comparing olanzapine with clozapine16 and risperidone with clozapine.18
Antipsychotic combinations. Combinations of antipsychotics are used commonly when treating chronic schizophrenia.23 Of the approximately 20 RCTs of antipsychotic combination therapy, most tested clozapine combined with other second-generation antipsychotics, such as risperidone. For a bibliography of these studies, click here. Only 5 studies support a combination approach (Table 1).
Table 1
Antipsychotic combinations: Few studies support efficacy
| Study | Design | Patients | Results |
|---|---|---|---|
| Shiloh et al, 1997a | 10-week, double-blind, placebo-controlled | 28 patients nonresponsive to typical antipsychotics and partially responsive to clozapine received add-on sulpiride,* 600 mg/d, or placebo | The sulpiride group showed improvements in positive and negative symptoms |
| Josiassen et al, 2005b | 12-week, randomized, double-blind, placebo-controlled | 40 schizophrenia patients unresponsive or partially responsive to clozapine randomized to clozapine + placebo or clozapine + risperidone, 6 mg/d | Mean BPRS total and positive symptom subscale scores reduced in both groups but reductions were greater in the clozapine/risperidone group; reduction in SANS also was observed in the clozapine/risperidone group |
| Genç et al, 2007c | 8-week, randomized, single-blind | 56 treatment-resistant schizophrenia patients randomly assigned to clozapine + amisulpride* or clozapine + quetiapine | Both groups improved at week 8 as measured by BPRS, SANS, SAPS, and CGI; however, patients receiving amisulpride showed greater improvement |
| Muscatello et al, 2011d | 24-week, randomized, double-blind, placebo-controlled | 31 treatment-resistant schizophrenia patients receiving clozapine randomized to receive adjunctive aripiprazole or placebo | Aripiprazole showed beneficial effect on positive and general psychopathologic symptomatology, but no significant effects on executive cognitive function |
| Takahashi et al, 1999e | 8-week, randomized, single-blind, crossover | 10 neuroleptic-treated patients received add-on risperidone and mosapramine* | Both additions resulted in significant, yet modest, improvement; no significant difference in PANSS between risperidone and mosapramine |
| *Not available in the United States BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression; PANSS: Positive and Negative Syndrome Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry. 1997;171:569-573. b. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136. c. Genç Y, Taner E, Candansayar S. Comparison of clozapine-amisulpride and clozapine-quetiapine combinations for patients with schizophrenia who are partially responsive to clozapine: a single-blind randomized study. Adv Ther. 2007;24(1):1-13. d. Muscatello MR, Bruno A, Pandolfo G, et al. Effect of aripiprazole augmentation of clozapine in schizophrenia: a double-blind, placebo-controlled study. Schizophr Res. 2011;127(1-3):93-99. e. Takahashi N, Terao T, Oga T, et al. Comparison of risperidone and mosapramine addition to neuroleptic treatment in chronic schizophrenia. Neuropsychobiology. 1999;39(2):81-85. | |||
What about augmentation?
Adjunctive non-antipsychotics also are commonly used when treating patients with chronic schizophrenia. For example, lithium and anticonvulsants are used in approximately one-half of all inpatients with schizophrenia in facilities operated by the State of New York Office of Mental Health.24,25 The evidence base for these agents as adjuncts to antipsychotics generally is weak.26 Specifically, early reports of benefit with adjunctive lithium have been negated by later studies. Similarly, large trials of adjunctive valproate and lamotrigine have failed to replicate early and promising efficacy signals from smaller trials, although the larger studies did not specifically target treatment-resistant schizophrenia.
Among mood stabilizers, lamotrigine may be the most promising for treatment-resistant schizophrenia. In a meta-analysis of clinical trials examining schizophrenia patients receiving clozapine (N=161) who were randomized to receive adjunctive lamotrigine or adjunctive placebo, lamotrigine was superior to placebo in total score for psychosis symptoms and scores for positive and negative symptoms.27
More than 125 published RCTs have studied a wide variety of adjunctive agents other than lithium or anticonvulsants for treating persistent schizophrenia symptoms (Table 2).
Only some of the approximately 40 RCTs regarding adjunctive antidepressants in patients with chronic schizophrenia focused on patients with ongoing depressive symptoms. For a bibliography of these studies, click here. In a meta-analysis measuring improvement of negative symptoms from 23 trials (N=819),28 the effect size was moderate in favor of antidepressants. Subgroup analysis revealed significant responses for fluoxetine, trazodone, and ritanserin.
More than 50 RCTs have focused on augmenting medications for cognitive dysfunction in chronic schizophrenia. Unfortunately, agents used to treat Alzheimer’s disease have shown disappointing results when tested in patients with schizophrenia, as have agents prescribed for attention-deficit/hyperactivity disorder (methylphenidate, guanfacine, atomoxetine) or agents used to promote alertness (modafinil and armodafinil).
Medications that act on glutamate receptors may offer another potential solution, although not in combination with clozapine.29
Other agents that require further study where ≥2 positive studies have been reported (with ≤2 negative studies) include celecoxib, neurosteroids and hormones, purinergic agents, serotonin 5-HT1A receptor agonists, and serotonin 5-HT3 receptor antagonists.
Table 2
Agents studied as adjuncts to antipsychotics
| Acetylsalicylic acid and nonsteroidal anti-inflammatory agents |
| Anticonvulsants and lithium |
| Antidepressants |
| Antiglucocorticoids |
| Agents used to treat attention-deficit/hyperactivity disorder |
| Beta blockers |
| Cholinesterase inhibitors and other agents used to treat Alzheimer’s disease |
| Experimental agents that act on glutamate receptors |
| GABAA receptor drugs |
| Neurosteroids and hormones |
| Omega-3 fatty acids |
| Opioid system agents |
| Peptides |
| Purinergic agents |
| Serotonin 5-HT1A receptor agonists |
| Serotonin 5-HT3 receptor antagonists |
| Wakefulness promoting agents |
Therapeutic neuromodulation
More than 10 RCTs of repetitive transcranial magnetic stimulation (rTMS) in patients with refractory symptoms of schizophrenia have been published; the results were mixed. For a bibliography of these studies, click here. In a meta-analysis of 9 trials (n=213),30 prefrontal rTMS for treating negative symptoms demonstrated a small-to-medium effect size. In another meta-analysis31 of all prospective studies of rTMS for negative symptoms and for auditory hallucinations and overall positive symptoms in refractory schizophrenia, the effect sizes showed moderate effects.
Fewer controlled trials are available for electroconvulsive therapy,32,33 but its use with clozapine appears encouraging.34
Psychological and behavioral intervention. Cognitive-behavioral therapy, although labor-intensive, can be helpful even in patients considered treatment-resistant (Table 3). These interventions generally are provided together with pharmacotherapy.
Complementary and alternative therapies. Patients and their families may ask about complementary and alternative therapies, particularly when conventional approaches have not been successful. A meta-analysis of 6 studies (n=828)35 that reviewed adjunctive use of ginkgo in patients with chronic schizophrenia found statistically significant moderate improvement in total and negative symptoms. Negative reports also are available, including a 5-month study of adjunctive megavitamins that did not demonstrate any benefits.36 In a review of 13 RCTs of acupuncture for schizophrenia, Lee et al found the overall methodological quality was too low to draw firm conclusions.37
Table 3
Cognitive-behavioral therapy for schizophrenia
| Study | Design | Patients | Results |
|---|---|---|---|
| Pinto et al, 1999a | 6-month, randomized controlled | 37 treatment-resistant schizophrenia patients were randomized to CBT plus social skills training or supportive therapy | Both groups showed statistically significant improvement on the BPRS, SAPS, and SANS; however, patients in the CBT group had lower BPRS and SAPS scores. No difference on SANS scores |
| Barretto et al, 2009b | 21-week, controlled (nonrandom-ized) | Patients refractory to clozapine were placed in a CBT or befriending control group | The CBT group showed significant improvement in PANSS total score and general psychopathology subscale score, as well as an improvement of QLS; improvement persisted at 6-month follow-up |
| BPRS: Brief Psychiatric Rating Scale; CBT: cognitive-behavioral therapy; PANSS: Positive and Negative Syndrome Scale; QLS: Quality of Life Scale; SANS: Scale for the Assessment of Negative Symptoms; SAPS: Scale for the Assessment of Positive Symptoms | |||
| Source: References a. Pinto A, La Pia S, Mennella R, et al. Cognitive-behavioral therapy and clozapine for clients with treatment-refractory schizophrenia. Psychiatr Serv. 1999;50(7):901-904. b. Barretto EM, Kayo M, Avrichir BS, et al. A preliminary controlled trial of cognitive behavioral therapy in clozapine-resistant schizophrenia. J Nerv Ment Dis. 2009;197(11):865-868. | |||
Clinical recommendations
Before declaring a patient with schizophrenia as treatment-resistant, ensure that an adequate trial of medication did take place. This includes consideration of adequate dosing and pharmacokinetic issues. Awareness of potential substance use and/or partial adherence or nonadherence also is critical because these factors can impact treatment response.
When prescribing for a treatment-resistant schizophrenia patient, identify specific target symptoms to better inform medication selection—especially for symptoms that the patient feels are important. For example, consider an antidepressant for patients who have negative or depressive symptoms. Also take into account other patient-centered concerns, such as tolerability issues that may have interfered with adherence and response in the past.
Clozapine remains the medication of choice for treatment-resistant schizophrenia. Despite dozens of RCTs of potential adjunctive agents for treatment-resistant schizophrenia, no single approach has consistently shown efficacy in reducing symptoms, improving cognition, or increasing a patient’s level of function. Individual response can vary, and our search for the “outlier” who does respond to an adjunctive agent can explain our use of these strategies in clinical practice.
Related Resources
- Cochrane Database of Systematic Reviews. www.cochrane.org/reviews. This database contains reviews of additional therapeutic options for patients with treatment-resistant schizophrenia. As of February 23, 2011, 157 reviews were available.
- Citrome L. Treatment-refractory schizophrenia: What it is and what’s been done about it. Neuropsychiatry. 2011. Epub ahead of print.
- Citrome L. Clozapine for schizophrenia. Life-threatening or life-saving treatment? Current Psychiatry. 2009;8(12):56-63.
Drug Brand Names
- Aripiprazole • Abilify
- Armodafinil • Nuvigil
- Atomoxetine • Strattera
- Benztropine mesylate • Cogentin
- Carbamazepine • Tegretol
- Celecoxib • Celebrex
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Fluoxetine • Prozac
- Guanfacine • Tenex
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Lurasidone • Latuda
- Methylphenidate • Ritalin, Methylin, others
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Rifampin • Rifadin
- Risperidone • Risperdal
- Trazodone • Desyrel, Oleptro
- Valproate (Divalproex) • Depakote, Depakote ER
- Ziprasidone • Geodon
Disclosure
No writing assistance or external financial support was used for this article. Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Avanir Pharmaceuticals, Azur Pharma Inc., Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Merck, Novartis, Pfizer Inc., Sunovion, Valeant Pharmaceuticals, and Vanda Pharmaceuticals.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry. 2002;159(2):255-262.
3. Andreasen NC, Carpenter WT Jr, Kane JM, et al. Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry. 2005;162(3):441-449.
4. Emsley R, Chiliza B, Asmal L, et al. The concepts of remission and recovery in schizophrenia. Curr Opin Psychiatry. 2011;24(2):114-121.
5. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
6. Robinson DG, Woerner MG, Delman HM, et al. Pharmacological treatments for first-episode schizophrenia. Schizophr Bull. 2005;31(3):705-722.
7. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
8. Citrome L, Volavka J. Optimal dosing of atypical antipsychotics in adults: a review of the current evidence. Harv Rev Psychiatry. 2002;10(5):280-291.
9. Citrome L. Iloperidone asenapine and lurasidone. A brief overview of three new second-generation antipsychotics. Postgrad Med. 2011;123(2):153-162.
10. Lincoln TM, Lüllmann E, Rief W. Correlates and long-term consequences of poor insight in patients with schizophrenia. A systematic review. Schizophr Bull. 2007;33(6):1324-1342.
11. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31-41.
12. Leucht S, Komossa K, Rummel-Kluge C, et al. A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry. 2009;166(2):152-163.
13. Leucht S, Arbter D, Engel RR, et al. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14(4):429-447.
14. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
15. Tollefson GD, Birkett MA, Kiesler GM, et al. Double-blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine. Biol Psychiatry. 2001;49(1):52-63.
16. Bitter I, Dossenbach MR, Brook S, et al. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
17. Meltzer HY, Bobo WV, Roy A, et al. A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia. J Clin Psychiatry. 2008;69(2):274-285.
18. Bondolfi G, Dufour H, Patris M, et al. Risperidone versus clozapine in treatment-resistant chronic schizophrenia: a randomized double-blind study. Am J Psychiatry. 1998;155(4):499-504.
19. Wahlbeck K, Cheine M, Tuisku K, et al. Risperidone versus clozapine in treatment-resistant schizophrenia: a randomized pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(6):911-922.
20. Breier AF, Malhotra AK, Su TP, et al. Clozapine and risperidone in chronic schizophrenia: effects on symptoms, parkinsonian side effects, and neuroendocrine response. Am J Psychiatry. 1999;156(2):294-298.
21. Azorin JM, Spiegel R, Remington G, et al. A double-blind comparative study of clozapine and risperidone in the management of severe chronic schizophrenia. Am J Psychiatry. 2001;158(8):1305-1313.
22. Sacchetti E, Galluzzo A, Valsecchi P, et al. Ziprasidone vs clozapine in schizophrenia patients refractory to multiple antipsychotic treatments: the MOZART study. Schizophr Res. 2009;113(1):112-121.
23. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf. 2003;12(1):41-48.
24. Citrome L, Levine J, Allingham B. Changes in use of valproate and other mood stabilizers for patients with schizophrenia from 1994 to 1998. Psychiatr Serv. 2000;51(5):634-638.
25. Citrome L, Jaffe A, Levine J, et al. Use of mood stabilizers among patients with schizophrenia, 1994-2001. Psychiatr Serv. 2002;53(10):1212.-
26. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
27. Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2009;109(1-3):10-14.
28. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
29. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
30. Dlabac-de Lange JJ, Knegtering R, Aleman A, et al. Repetitive transcranial magnetic stimulation for negative symptoms of schizophrenia: review and meta-analysis. J Clin Psychiatry. 2010;71(4):411-418.
31. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
32. Chanpattana W, Chakrabhand ML, Sackeim HA, et al. Continuation ECT in treatment-resistant schizophrenia: a controlled study. J ECT. 1999;15(3):178-192.
33. Goswami U, Kumar U, Singh B. Efficacy of electroconvulsive therapy in treatment resistant schizophrenia: a double-blind study. Indian J Psychiatry. 2003;45(1):26-29.
34. Braga RJ, Petrides G. The combined use of electroconvulsive therapy and antipsychotics in patients with schizophrenia. J ECT. 2005;21(2):75-83.
35. Singh V, Singh SP, Chan K. Review and meta-analysis of usage of ginkgo as an adjunct therapy in chronic schizophrenia. Int J Neuropsychopharmacol. 2010;13(2):257-271.
36. Vaughan K, McConaghy N. Megavitamin and dietary treatment in schizophrenia: a randomised controlled trial. Aust N Z J Psychiatry. 1999;33(1):84-88.
37. Lee MS, Shin BC, Ronan P, et al. Acupuncture for schizophrenia: a systematic review and meta-analysis. Int J Clin Pract. 2009;63(11):1622-1633.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Volavka J, Czobor P, Sheitman B, et al. Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry. 2002;159(2):255-262.
3. Andreasen NC, Carpenter WT Jr, Kane JM, et al. Remission in schizophrenia: proposed criteria and rationale for consensus. Am J Psychiatry. 2005;162(3):441-449.
4. Emsley R, Chiliza B, Asmal L, et al. The concepts of remission and recovery in schizophrenia. Curr Opin Psychiatry. 2011;24(2):114-121.
5. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
6. Robinson DG, Woerner MG, Delman HM, et al. Pharmacological treatments for first-episode schizophrenia. Schizophr Bull. 2005;31(3):705-722.
7. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
8. Citrome L, Volavka J. Optimal dosing of atypical antipsychotics in adults: a review of the current evidence. Harv Rev Psychiatry. 2002;10(5):280-291.
9. Citrome L. Iloperidone asenapine and lurasidone. A brief overview of three new second-generation antipsychotics. Postgrad Med. 2011;123(2):153-162.
10. Lincoln TM, Lüllmann E, Rief W. Correlates and long-term consequences of poor insight in patients with schizophrenia. A systematic review. Schizophr Bull. 2007;33(6):1324-1342.
11. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31-41.
12. Leucht S, Komossa K, Rummel-Kluge C, et al. A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry. 2009;166(2):152-163.
13. Leucht S, Arbter D, Engel RR, et al. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14(4):429-447.
14. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
15. Tollefson GD, Birkett MA, Kiesler GM, et al. Double-blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine. Biol Psychiatry. 2001;49(1):52-63.
16. Bitter I, Dossenbach MR, Brook S, et al. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
17. Meltzer HY, Bobo WV, Roy A, et al. A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia. J Clin Psychiatry. 2008;69(2):274-285.
18. Bondolfi G, Dufour H, Patris M, et al. Risperidone versus clozapine in treatment-resistant chronic schizophrenia: a randomized double-blind study. Am J Psychiatry. 1998;155(4):499-504.
19. Wahlbeck K, Cheine M, Tuisku K, et al. Risperidone versus clozapine in treatment-resistant schizophrenia: a randomized pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(6):911-922.
20. Breier AF, Malhotra AK, Su TP, et al. Clozapine and risperidone in chronic schizophrenia: effects on symptoms, parkinsonian side effects, and neuroendocrine response. Am J Psychiatry. 1999;156(2):294-298.
21. Azorin JM, Spiegel R, Remington G, et al. A double-blind comparative study of clozapine and risperidone in the management of severe chronic schizophrenia. Am J Psychiatry. 2001;158(8):1305-1313.
22. Sacchetti E, Galluzzo A, Valsecchi P, et al. Ziprasidone vs clozapine in schizophrenia patients refractory to multiple antipsychotic treatments: the MOZART study. Schizophr Res. 2009;113(1):112-121.
23. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf. 2003;12(1):41-48.
24. Citrome L, Levine J, Allingham B. Changes in use of valproate and other mood stabilizers for patients with schizophrenia from 1994 to 1998. Psychiatr Serv. 2000;51(5):634-638.
25. Citrome L, Jaffe A, Levine J, et al. Use of mood stabilizers among patients with schizophrenia, 1994-2001. Psychiatr Serv. 2002;53(10):1212.-
26. Citrome L. Adjunctive lithium and anticonvulsants for the treatment of schizophrenia: what is the evidence? Expert Rev Neurother. 2009;9(1):55-71.
27. Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2009;109(1-3):10-14.
28. Singh SP, Singh V, Kar N, et al. Efficacy of antidepressants in treating the negative symptoms of chronic schizophrenia: meta-analysis. Br J Psychiatry. 2010;197(3):174-179.
29. Kantrowitz JT, Javitt DC. Thinking glutamatergically: changing concepts of schizophrenia based upon changing neurochemical models. Clin Schizophr Relat Psychoses. 2010;4(3):189-200.
30. Dlabac-de Lange JJ, Knegtering R, Aleman A, et al. Repetitive transcranial magnetic stimulation for negative symptoms of schizophrenia: review and meta-analysis. J Clin Psychiatry. 2010;71(4):411-418.
31. Freitas C, Fregni F, Pascual-Leone A. Meta-analysis of the effects of repetitive transcranial magnetic stimulation (rTMS) on negative and positive symptoms in schizophrenia. Schizophr Res. 2009;108(1-3):11-24.
32. Chanpattana W, Chakrabhand ML, Sackeim HA, et al. Continuation ECT in treatment-resistant schizophrenia: a controlled study. J ECT. 1999;15(3):178-192.
33. Goswami U, Kumar U, Singh B. Efficacy of electroconvulsive therapy in treatment resistant schizophrenia: a double-blind study. Indian J Psychiatry. 2003;45(1):26-29.
34. Braga RJ, Petrides G. The combined use of electroconvulsive therapy and antipsychotics in patients with schizophrenia. J ECT. 2005;21(2):75-83.
35. Singh V, Singh SP, Chan K. Review and meta-analysis of usage of ginkgo as an adjunct therapy in chronic schizophrenia. Int J Neuropsychopharmacol. 2010;13(2):257-271.
36. Vaughan K, McConaghy N. Megavitamin and dietary treatment in schizophrenia: a randomised controlled trial. Aust N Z J Psychiatry. 1999;33(1):84-88.
37. Lee MS, Shin BC, Ronan P, et al. Acupuncture for schizophrenia: a systematic review and meta-analysis. Int J Clin Pract. 2009;63(11):1622-1633.