User login
Best Practices for Capturing Clinical and Dermoscopic Images With Smartphone Photography
Best Practices for Capturing Clinical and Dermoscopic Images With Smartphone Photography
PRACTICE GAP
Photography is an essential tool in modern dermatologic practice, aiding in the evaluation, documentation, and monitoring of nevi, skin cancers, and other cutaneous pathologies.1 With the rapid technologic advancement of smartphone cameras, high-quality clinical and dermoscopic images have become increasingly easy to attain; however, best practices for optimizing smartphone photography are limited in the medical literature. We have collated a series of recommendations to help fill this knowledge gap.
A search of PubMed articles indexed for MEDLINE was conducted using the terms clinical imaging AND smartphone, clinical photography AND smartphone, dermatology AND photography, dermatology AND imaging, dermoscopy AND photography, and dermoscopy AND imaging. We also consulted with Elizabeth Seiverling, MD (Annville, Pennsylvania) and Jennifer Stein, MD (New York, New York)—both renowned experts in the fields of dermatology, dermoscopy, and medical photography—via email and video meetings conducted during the period from June 1, 2022, through August 20, 2022. Our goal in creating this guide is to facilitate standardized yet simple ways to integrate smartphone photography into current dermatologic practice.
THE TECHNIQUE
Clinical Photography
Clinical images should be captured in a space with ample indirect natural light, such as a patient examination room with frosted or draped windows, ensuring patient privacy is maintained.1,2 The smartphone’s flash can be used if natural lighting is insufficient, but caution should be exercised when photographing patients with darker skin types, as the flash may create an undesired glare. To combat this, consider using a small clip-on light-emitting diode ring light positioned at a 45° angle for more uniform lighting and reduced glare (eFigures 1 and 2).2 This additional light source helps to distribute light evenly across the patient’s skin, enhancing detail visibility, minimizing harsh shadows, and ensuring a more accurate representation of skin pigmentation.2

When a magnified image is required (eg, to capture suspicious lesions with unique and detailed findings such as irregular borders or atypical pigmentation), use the smartphone’s digital zoom function rather than physically moving the camera lens closer to the subject. Moving the camera too close can cause proximity distortion, artificially enlarging objects close to the lens and degrading the quality of the image.1,2 Unnecessary camera features such as portrait mode, live focus, and filters should be turned off to maintain image accuracy. It also is important to avoid excessive manual adjustments to exposure and brightness settings.1,2 The tap-to-focus feature that is integrated into many smartphone cameras can be utilized to ensure the capture of sharp, focused images. After verifying the image preview on the smartphone display, take the photograph. Immediately review the captured image to ensure it is clear and well lit and accurately depicts the area of interest, including its color, texture, and any relevant details, without glare or distortion. If the image does not meet these criteria, promptly reattempt to achieve the desired quality.
Dermoscopic Photography
Dermoscopy, which enables magnified examination of skin lesions, is increasingly being utilized in dermatology. While traditional dermoscopic photography requires specialized equipment, such as large single-lens reflex cameras with dedicated dermoscopic lens attachments, smartphone cameras now can be used to obtain dermoscopic images of reasonable quality.3,4 Adhering to specific practices can help to optimize the quality of dermoscopic images obtained via this technique.
Before capturing an image, it is essential to prepare both the lesion and the surrounding skin. Ensure the area is cleaned thoroughly and trim any hairs that may obscure the image. Apply an interface fluid such as rubbing alcohol or ultrasonography gel to improve image clarity by reducing surface tension and reflections, minimizing glare, and ensuring even light transmission throughout the lesion.5 As recommended for clinical photography, images should be captured in a space with ample indirect light. For best results, we recommend utilizing the primary photo capture option instead of portrait or panoramic mode or additional settings. It is crucial to disable features such as live focus, filters, night mode, and flash, as they may alter image accuracy; however, use of the tap-to-focus feature or manual settings adjustment is encouraged to ensure a high-resolution photograph.
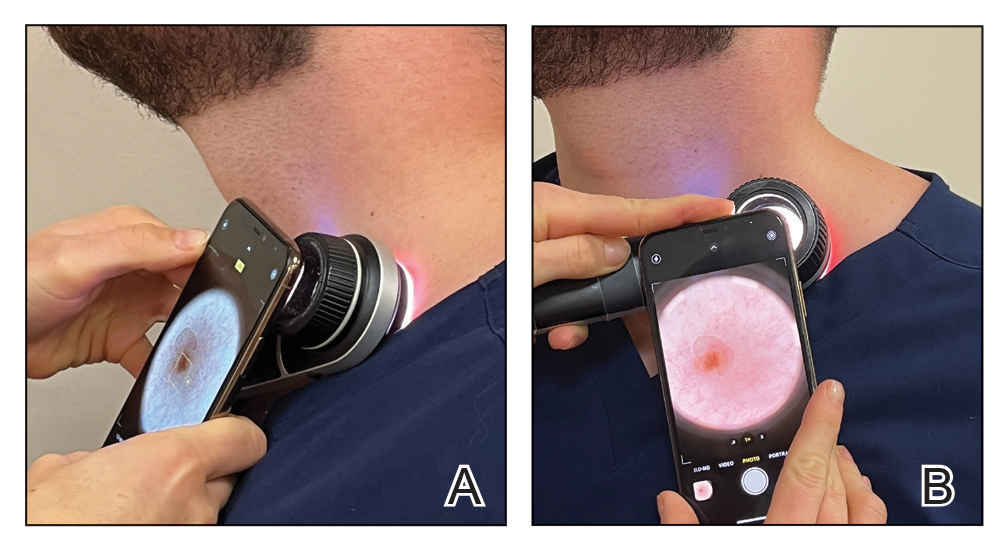
Once these smartphone settings have been verified, position the dermatoscope directly over the lesion of interest. Next, place the smartphone camera lens directly against the eyepiece of the dermatoscope (Figure). Center the lesion in the field of view on the screen. Most smartphones enable adjustment to the image magnification on the photo capture screen. A single tap on the screen should populate the zoom options (eg, ×0.5, ×1, ×3) and allow for adjustment. For the majority of dermoscopic photographs, we recommend standard ×1 magnification, as it typically provides a clear and accurate representation of the lesion without introducing the possibility of image distortion. To obtain a close-up image, use the smartphone’s digital zoom function prior to taking the photograph rather than zooming in on the image after it has been captured; however, to minimize proximity distortion and maintain optimal image quality, avoid exceeding the halfway point on the camera’s zoom dial. After verifying the image preview on the smartphone display, capture the photograph. Immediate review is recommended to allow for prompt reattempt at capturing the image if needed.
PRACTICE IMPLICATIONS
The inherent convenience and accessibility offered by smartphone photography further solidifies its status as a valuable tool in modern dermatologic practice. By adhering to the best practices outlined in this guide, dermatologists can utilize smartphones to capture high-quality clinical and dermoscopic images that support accurate diagnosis and enhance patient care. This approach helps streamline workflows, enhance consistency in image quality, and standardize image capture across different settings and providers.
Additionally, smartphone photography can enhance both education and telemedicine by enabling physicians to easily share high-quality images with colleagues for virtual consultations, second opinions, and collaborative diagnoses. This sharing of images fosters learning opportunities, supports knowledge exchange, and allows for real-time feedback—all of which can improve clinical decision-making. Moreover, it broadens access to dermatologic expertise, strengthens communication between health care providers, and facilitates timely decision-making. As a result, patients benefit from more efficient, accurate, and collaborative care.
- Muraco L. Improved medical photography: key tips for creating images of lasting value. JAMA Dermatol. 2020;156:121-123. doi:10.1001 /jamadermatol.2019.3849
- Alvarado SM, Flessland P, Grant-Kels JM, et al. Practical strategies for improving clinical photography of dark skin. J Am Acad Dermatol. 2022;86:E21-E23. doi:10.1016/j.jaad.2021.09.001
- Pagliarello C, Feliciani C, Fantini C, et al. Use of the dermoscope as a smartphone close-up lens and LED annular macro ring flash. J Am Acad Dermatol. 2016;75:E27–E28. doi:10.1016/j.jaad .2015.12.04
- Zuo KJ, Guo D, Rao J. Mobile teledermatology: a promising future in clinical practice. J Cutan Med Surg. 2013;17:387-391. doi:10.2310/7750.2013.13030
- Gewirtzman AJ, Saurat J-H, Braun RP. An evaluation of dermscopy fluids and application techniques. Br J Dermatol. 2003;149:59-63. doi:10.1046/j.1365-2133.2003.05366.x
PRACTICE GAP
Photography is an essential tool in modern dermatologic practice, aiding in the evaluation, documentation, and monitoring of nevi, skin cancers, and other cutaneous pathologies.1 With the rapid technologic advancement of smartphone cameras, high-quality clinical and dermoscopic images have become increasingly easy to attain; however, best practices for optimizing smartphone photography are limited in the medical literature. We have collated a series of recommendations to help fill this knowledge gap.
A search of PubMed articles indexed for MEDLINE was conducted using the terms clinical imaging AND smartphone, clinical photography AND smartphone, dermatology AND photography, dermatology AND imaging, dermoscopy AND photography, and dermoscopy AND imaging. We also consulted with Elizabeth Seiverling, MD (Annville, Pennsylvania) and Jennifer Stein, MD (New York, New York)—both renowned experts in the fields of dermatology, dermoscopy, and medical photography—via email and video meetings conducted during the period from June 1, 2022, through August 20, 2022. Our goal in creating this guide is to facilitate standardized yet simple ways to integrate smartphone photography into current dermatologic practice.
THE TECHNIQUE
Clinical Photography
Clinical images should be captured in a space with ample indirect natural light, such as a patient examination room with frosted or draped windows, ensuring patient privacy is maintained.1,2 The smartphone’s flash can be used if natural lighting is insufficient, but caution should be exercised when photographing patients with darker skin types, as the flash may create an undesired glare. To combat this, consider using a small clip-on light-emitting diode ring light positioned at a 45° angle for more uniform lighting and reduced glare (eFigures 1 and 2).2 This additional light source helps to distribute light evenly across the patient’s skin, enhancing detail visibility, minimizing harsh shadows, and ensuring a more accurate representation of skin pigmentation.2
When a magnified image is required (eg, to capture suspicious lesions with unique and detailed findings such as irregular borders or atypical pigmentation), use the smartphone’s digital zoom function rather than physically moving the camera lens closer to the subject. Moving the camera too close can cause proximity distortion, artificially enlarging objects close to the lens and degrading the quality of the image.1,2 Unnecessary camera features such as portrait mode, live focus, and filters should be turned off to maintain image accuracy. It also is important to avoid excessive manual adjustments to exposure and brightness settings.1,2 The tap-to-focus feature that is integrated into many smartphone cameras can be utilized to ensure the capture of sharp, focused images. After verifying the image preview on the smartphone display, take the photograph. Immediately review the captured image to ensure it is clear and well lit and accurately depicts the area of interest, including its color, texture, and any relevant details, without glare or distortion. If the image does not meet these criteria, promptly reattempt to achieve the desired quality.
Dermoscopic Photography
Dermoscopy, which enables magnified examination of skin lesions, is increasingly being utilized in dermatology. While traditional dermoscopic photography requires specialized equipment, such as large single-lens reflex cameras with dedicated dermoscopic lens attachments, smartphone cameras now can be used to obtain dermoscopic images of reasonable quality.3,4 Adhering to specific practices can help to optimize the quality of dermoscopic images obtained via this technique.
Before capturing an image, it is essential to prepare both the lesion and the surrounding skin. Ensure the area is cleaned thoroughly and trim any hairs that may obscure the image. Apply an interface fluid such as rubbing alcohol or ultrasonography gel to improve image clarity by reducing surface tension and reflections, minimizing glare, and ensuring even light transmission throughout the lesion.5 As recommended for clinical photography, images should be captured in a space with ample indirect light. For best results, we recommend utilizing the primary photo capture option instead of portrait or panoramic mode or additional settings. It is crucial to disable features such as live focus, filters, night mode, and flash, as they may alter image accuracy; however, use of the tap-to-focus feature or manual settings adjustment is encouraged to ensure a high-resolution photograph.
Once these smartphone settings have been verified, position the dermatoscope directly over the lesion of interest. Next, place the smartphone camera lens directly against the eyepiece of the dermatoscope (Figure). Center the lesion in the field of view on the screen. Most smartphones enable adjustment to the image magnification on the photo capture screen. A single tap on the screen should populate the zoom options (eg, ×0.5, ×1, ×3) and allow for adjustment. For the majority of dermoscopic photographs, we recommend standard ×1 magnification, as it typically provides a clear and accurate representation of the lesion without introducing the possibility of image distortion. To obtain a close-up image, use the smartphone’s digital zoom function prior to taking the photograph rather than zooming in on the image after it has been captured; however, to minimize proximity distortion and maintain optimal image quality, avoid exceeding the halfway point on the camera’s zoom dial. After verifying the image preview on the smartphone display, capture the photograph. Immediate review is recommended to allow for prompt reattempt at capturing the image if needed.
PRACTICE IMPLICATIONS
The inherent convenience and accessibility offered by smartphone photography further solidifies its status as a valuable tool in modern dermatologic practice. By adhering to the best practices outlined in this guide, dermatologists can utilize smartphones to capture high-quality clinical and dermoscopic images that support accurate diagnosis and enhance patient care. This approach helps streamline workflows, enhance consistency in image quality, and standardize image capture across different settings and providers.
Additionally, smartphone photography can enhance both education and telemedicine by enabling physicians to easily share high-quality images with colleagues for virtual consultations, second opinions, and collaborative diagnoses. This sharing of images fosters learning opportunities, supports knowledge exchange, and allows for real-time feedback—all of which can improve clinical decision-making. Moreover, it broadens access to dermatologic expertise, strengthens communication between health care providers, and facilitates timely decision-making. As a result, patients benefit from more efficient, accurate, and collaborative care.
PRACTICE GAP
Photography is an essential tool in modern dermatologic practice, aiding in the evaluation, documentation, and monitoring of nevi, skin cancers, and other cutaneous pathologies.1 With the rapid technologic advancement of smartphone cameras, high-quality clinical and dermoscopic images have become increasingly easy to attain; however, best practices for optimizing smartphone photography are limited in the medical literature. We have collated a series of recommendations to help fill this knowledge gap.
A search of PubMed articles indexed for MEDLINE was conducted using the terms clinical imaging AND smartphone, clinical photography AND smartphone, dermatology AND photography, dermatology AND imaging, dermoscopy AND photography, and dermoscopy AND imaging. We also consulted with Elizabeth Seiverling, MD (Annville, Pennsylvania) and Jennifer Stein, MD (New York, New York)—both renowned experts in the fields of dermatology, dermoscopy, and medical photography—via email and video meetings conducted during the period from June 1, 2022, through August 20, 2022. Our goal in creating this guide is to facilitate standardized yet simple ways to integrate smartphone photography into current dermatologic practice.
THE TECHNIQUE
Clinical Photography
Clinical images should be captured in a space with ample indirect natural light, such as a patient examination room with frosted or draped windows, ensuring patient privacy is maintained.1,2 The smartphone’s flash can be used if natural lighting is insufficient, but caution should be exercised when photographing patients with darker skin types, as the flash may create an undesired glare. To combat this, consider using a small clip-on light-emitting diode ring light positioned at a 45° angle for more uniform lighting and reduced glare (eFigures 1 and 2).2 This additional light source helps to distribute light evenly across the patient’s skin, enhancing detail visibility, minimizing harsh shadows, and ensuring a more accurate representation of skin pigmentation.2
When a magnified image is required (eg, to capture suspicious lesions with unique and detailed findings such as irregular borders or atypical pigmentation), use the smartphone’s digital zoom function rather than physically moving the camera lens closer to the subject. Moving the camera too close can cause proximity distortion, artificially enlarging objects close to the lens and degrading the quality of the image.1,2 Unnecessary camera features such as portrait mode, live focus, and filters should be turned off to maintain image accuracy. It also is important to avoid excessive manual adjustments to exposure and brightness settings.1,2 The tap-to-focus feature that is integrated into many smartphone cameras can be utilized to ensure the capture of sharp, focused images. After verifying the image preview on the smartphone display, take the photograph. Immediately review the captured image to ensure it is clear and well lit and accurately depicts the area of interest, including its color, texture, and any relevant details, without glare or distortion. If the image does not meet these criteria, promptly reattempt to achieve the desired quality.
Dermoscopic Photography
Dermoscopy, which enables magnified examination of skin lesions, is increasingly being utilized in dermatology. While traditional dermoscopic photography requires specialized equipment, such as large single-lens reflex cameras with dedicated dermoscopic lens attachments, smartphone cameras now can be used to obtain dermoscopic images of reasonable quality.3,4 Adhering to specific practices can help to optimize the quality of dermoscopic images obtained via this technique.
Before capturing an image, it is essential to prepare both the lesion and the surrounding skin. Ensure the area is cleaned thoroughly and trim any hairs that may obscure the image. Apply an interface fluid such as rubbing alcohol or ultrasonography gel to improve image clarity by reducing surface tension and reflections, minimizing glare, and ensuring even light transmission throughout the lesion.5 As recommended for clinical photography, images should be captured in a space with ample indirect light. For best results, we recommend utilizing the primary photo capture option instead of portrait or panoramic mode or additional settings. It is crucial to disable features such as live focus, filters, night mode, and flash, as they may alter image accuracy; however, use of the tap-to-focus feature or manual settings adjustment is encouraged to ensure a high-resolution photograph.
Once these smartphone settings have been verified, position the dermatoscope directly over the lesion of interest. Next, place the smartphone camera lens directly against the eyepiece of the dermatoscope (Figure). Center the lesion in the field of view on the screen. Most smartphones enable adjustment to the image magnification on the photo capture screen. A single tap on the screen should populate the zoom options (eg, ×0.5, ×1, ×3) and allow for adjustment. For the majority of dermoscopic photographs, we recommend standard ×1 magnification, as it typically provides a clear and accurate representation of the lesion without introducing the possibility of image distortion. To obtain a close-up image, use the smartphone’s digital zoom function prior to taking the photograph rather than zooming in on the image after it has been captured; however, to minimize proximity distortion and maintain optimal image quality, avoid exceeding the halfway point on the camera’s zoom dial. After verifying the image preview on the smartphone display, capture the photograph. Immediate review is recommended to allow for prompt reattempt at capturing the image if needed.
PRACTICE IMPLICATIONS
The inherent convenience and accessibility offered by smartphone photography further solidifies its status as a valuable tool in modern dermatologic practice. By adhering to the best practices outlined in this guide, dermatologists can utilize smartphones to capture high-quality clinical and dermoscopic images that support accurate diagnosis and enhance patient care. This approach helps streamline workflows, enhance consistency in image quality, and standardize image capture across different settings and providers.
Additionally, smartphone photography can enhance both education and telemedicine by enabling physicians to easily share high-quality images with colleagues for virtual consultations, second opinions, and collaborative diagnoses. This sharing of images fosters learning opportunities, supports knowledge exchange, and allows for real-time feedback—all of which can improve clinical decision-making. Moreover, it broadens access to dermatologic expertise, strengthens communication between health care providers, and facilitates timely decision-making. As a result, patients benefit from more efficient, accurate, and collaborative care.
- Muraco L. Improved medical photography: key tips for creating images of lasting value. JAMA Dermatol. 2020;156:121-123. doi:10.1001 /jamadermatol.2019.3849
- Alvarado SM, Flessland P, Grant-Kels JM, et al. Practical strategies for improving clinical photography of dark skin. J Am Acad Dermatol. 2022;86:E21-E23. doi:10.1016/j.jaad.2021.09.001
- Pagliarello C, Feliciani C, Fantini C, et al. Use of the dermoscope as a smartphone close-up lens and LED annular macro ring flash. J Am Acad Dermatol. 2016;75:E27–E28. doi:10.1016/j.jaad .2015.12.04
- Zuo KJ, Guo D, Rao J. Mobile teledermatology: a promising future in clinical practice. J Cutan Med Surg. 2013;17:387-391. doi:10.2310/7750.2013.13030
- Gewirtzman AJ, Saurat J-H, Braun RP. An evaluation of dermscopy fluids and application techniques. Br J Dermatol. 2003;149:59-63. doi:10.1046/j.1365-2133.2003.05366.x
- Muraco L. Improved medical photography: key tips for creating images of lasting value. JAMA Dermatol. 2020;156:121-123. doi:10.1001 /jamadermatol.2019.3849
- Alvarado SM, Flessland P, Grant-Kels JM, et al. Practical strategies for improving clinical photography of dark skin. J Am Acad Dermatol. 2022;86:E21-E23. doi:10.1016/j.jaad.2021.09.001
- Pagliarello C, Feliciani C, Fantini C, et al. Use of the dermoscope as a smartphone close-up lens and LED annular macro ring flash. J Am Acad Dermatol. 2016;75:E27–E28. doi:10.1016/j.jaad .2015.12.04
- Zuo KJ, Guo D, Rao J. Mobile teledermatology: a promising future in clinical practice. J Cutan Med Surg. 2013;17:387-391. doi:10.2310/7750.2013.13030
- Gewirtzman AJ, Saurat J-H, Braun RP. An evaluation of dermscopy fluids and application techniques. Br J Dermatol. 2003;149:59-63. doi:10.1046/j.1365-2133.2003.05366.x
Best Practices for Capturing Clinical and Dermoscopic Images With Smartphone Photography
Best Practices for Capturing Clinical and Dermoscopic Images With Smartphone Photography

Diffuse Pruritic Eruption in an Immunocompromised Patient
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
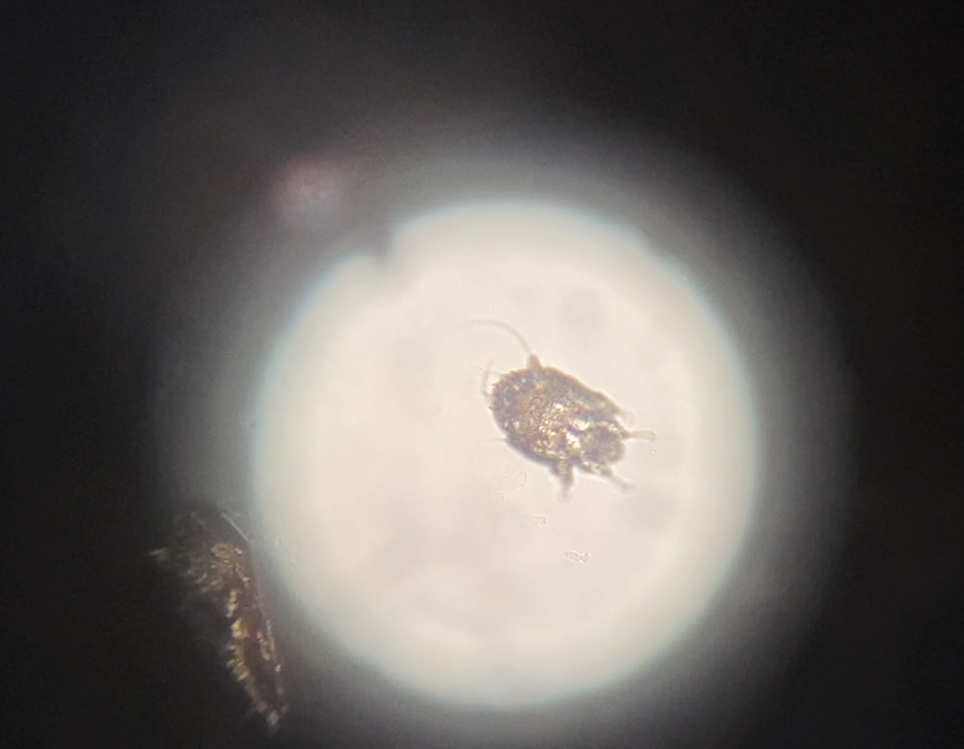
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
The Diagnosis: Scabies Infestation
Direct microscopy revealed the presence of a live scabies mite and numerous eggs (Figure), confirming the diagnosis of a scabies infestation. Scabies, caused by the Sarcoptes scabiei var hominis mite, characteristically presents in adults as pruritic hyperkeratotic plaques of the interdigital web spaces of the hands, flexor surfaces of the wrists and elbows, axillae, male genitalia, and breasts; however, an atypical presentation is common in immunocompromised or immunosuppressed individuals, such as our patient. In children, the palms, soles, and head (ie, face, scalp, neck) are common sites of involvement. Although dermatologists generally are familiar with severe atypical presentations such as Norwegian crusted scabies or bullous scabies, it is important that they are aware of other atypical presentations, such as the diffuse papulonodular variant observed in our patient.1 As such, a low threshold of suspicion for scabies infestations should be employed in immunocompromised patients with new-onset pruritic eruptions.
Direct microscopy is widely accepted as the gold standard for the diagnosis of scabies infestations; it is a fast and low-cost diagnostic tool. However, this technique displays variable sensitivity in clinical practice, requiring experience and a skilled hand.1,2 Other more sensitive diagnostic options for suspected scabies infestations include histopathology, serology, and molecular-based techniques such as DNA isolation and polymerase chain reaction. Although these tests do demonstrate greater sensitivity, they also are more invasive, time intensive, and costly.2 Therefore, they typically are not the first choice for a suspected scabies infestation. Dermoscopy has emerged as another tool to aid in the diagnosis of a suspected scabies infestation, enabling visualization of scaly burrows, eggs, and live mites. Classically, findings resembling a delta wing with contrail are seen on dermoscopic examination. The delta wing represents the brown triangular structure of the pigmented scabies mite head and anterior legs; the contrail is the lighter linear structures streaming behind the scabies mite (similar to visible vapor streams occurring behind flying jets), representing the burrow of the mite.
Although treatment of scabies infestations typically can be accomplished with permethrin cream 5%, the diffuse nature of our patient’s lesions in combination with his immunocompromised state made oral therapy a more appropriate choice. Based on Centers for Disease Control and Prevention recommendations, the patient received 2 doses of oral weight-based ivermectin (200 μg/kg per dose) administered 1 week apart.1,3 The initial dose at day 1 serves to eliminate any scabies mites that are present, while the second dose 1 week later eliminates any residual eggs. Our patient experienced complete resolution of the symptoms following this treatment regimen.
It was important to differentiate our patient’s scabies infestation from other intensely pruritic conditions and morphologic mimics including papular urticaria, lichenoid drug eruptions, tinea corporis, and prurigo nodularis. Papular urticaria is an intensely pruritic hypersensitivity reaction to insect bites that commonly affects the extremities or other exposed areas. Visible puncta may be present.4 Our patient’s lesion distribution involved areas covered by clothing, no puncta were present, and he had no history of a recent arthropod assault, making the diagnosis of papular urticaria less likely.
Lichenoid drug eruptions classically present with symmetric, diffuse, pruritic, violaceous, scaling papules and plaques that present 2 to 3 months after exposure to an offending agent.5 Our patient’s eruption was papulonodular with no violaceous plaques, and he did not report changes to his medications, making a lichenoid drug eruption less likely.
Tinea corporis is another intensely pruritic condition that should be considered, especially in immunocompromised patients. It is caused by dermatophytes and classically presents as erythematous pruritic plaques with an annular, advancing, scaling border.6 Although immunocompromised patients may display extensive involvement, our patient’s lesions were papulonodular with no annular morphology or scale, rendering tinea corporis less likely.
Prurigo nodularis is a chronic condition characterized by pruritic, violaceous, dome-shaped, smooth or crusted nodules secondary to repeated scratching or pressure. Although prurigo nodules can develop as a secondary change due to chronic excoriations in scabies infestations, prurigo nodules usually do not develop in areas such as the midline of the back that are not easily reached by the fingernails,7 which made prurigo nodularis less likely in our patient.
This case describes a unique papulonodular variant of scabies presenting in an immunocompromised cancer patient. Timely recognition and diagnosis of atypical scabies infestations can decrease morbidity and improve the quality of life of these patients.
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
- Chandler DJ, Fuller LC. A review of scabies: an infestation more than skin deep. Dermatology. 2019;235:79-90. doi:10.1159/000495290
- Siddig EE, Hay R. Laboratory-based diagnosis of scabies: a review of the current status. Trans R Soc Trop Med Hyg. 2022;116:4-9. doi:10.1093/trstmh/trab049
- Centers for Disease Control and Prevention. Parasites—scabies. medications. Accessed September 19, 2023. https://www.cdc.gov/parasites/ scabies/health_professionals/meds.html
- Örnek S, Zuberbier T, Kocatürk E. Annular urticarial lesions. Clin Dermatol. 2022;40:480-504. doi:10.1016/j.clindermatol .2021.12.010
- Cheraghlou S, Levy LL. Fixed drug eruptions, bullous drug eruptions, and lichenoid drug eruptions. Clin Dermatol. 2020;38:679-692. doi:10.1016/j.clindermatol.2020.06.010
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9:2020-5-6. doi:10.7573/dic.2020-5-6
- Kwon CD, Khanna R, Williams KA, et al. Diagnostic workup and evaluation of patients with prurigo nodularis. Medicines (Basel). 2019;6:97. doi:10.3390/medicines6040097
A 54-year-old man presented to our dermatology clinic for evaluation of a widespread intensely pruritic rash of 4 weeks’ duration. Calamine lotion and oral hydroxyzine provided minimal relief. He was being treated for a myeloproliferative disorder with immunosuppressive therapy consisting of a combination of cladribine, low-dose cytarabine, and fedratinib. Physical examination revealed multiple excoriated papules and indurated nodules on the extensor and flexor surfaces of the arms and legs (top), chest, midline of the back (bottom), and groin. No lesions were noted on the volar aspect of the patient’s wrists or interdigital spaces, and no central puncta or scales were present. He denied any preceding arthropod bites, trauma, new environmental exposures, or changes to his medications. Scrapings from several representative lesions were obtained for mineral oil preparation and microscopic evaluation.


Cerebral Cavernous Malformations Associated With Cutaneous Angiokeratomas and Hemangiomas
Case Report
A 66-year-old non-Hispanic white man with a history of adult-onset seizures, renal cell carcinoma, and nephrolithiasis presented to the dermatology clinic with numerous dark red, dry, scaling lesions on the lower legs, abdomen, back, thighs, and scrotum that bled easily. The lesions had developed 2 to 4 years prior to presentation. The patient also noted several dark blue lesions on the bilateral arms that had been present for several years.
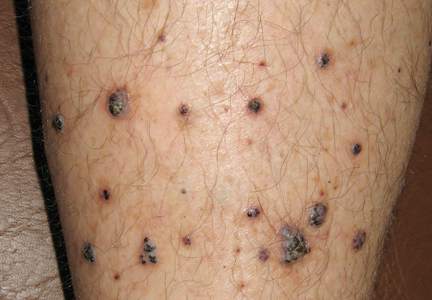
Physical examination revealed multiple black, keratotic papules on the bilateral lower legs (Figure 1), with similar lesions on the lower abdomen and lower back. Numerous dark red, scaling papules were present on the thighs and scrotum, and firm, 1- to 3-cm, dark blue nodules were noted on the bilateral arms (Figure 2) and right side of the forehead. Biopsy of papules from the lower right leg and back demonstrated angiokeratomas on pathologic review (Figure 3). Four separate biopsies taken from the left forearm demonstrated lobular and cavernous hemangiomas (Figure 4).
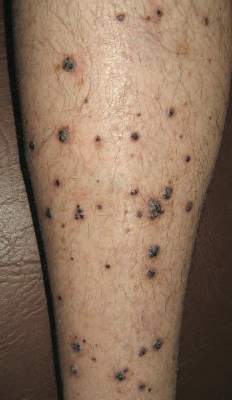
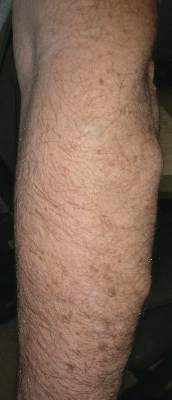
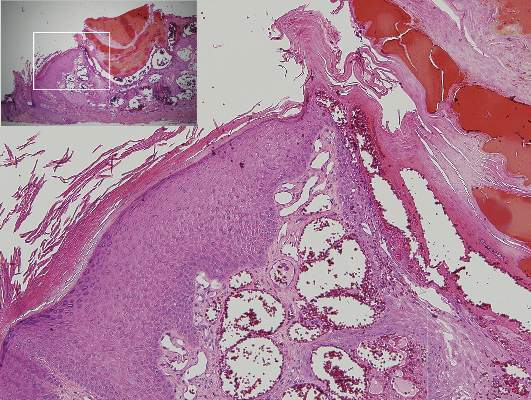
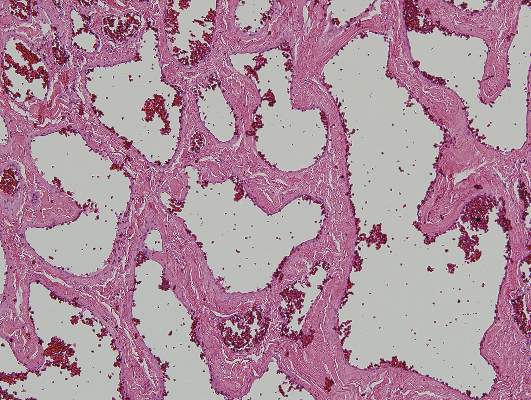
The patient’s medical history was remarkable for neurologic symptoms including adult-onset seizures at the age of 42 years. Recent magnetic resonance imaging of the head showed multiple areas of heterogeneous signal intensity with T2 hypointense rims consistent with cerebral cavernous malformations (CCMs) involving both cerebral hemispheres, the posterior cranial fossa, and the brainstem (Figure 5). Of note, the patient’s mother had been diagnosed with epilepsy in her 40s, but no other family history of a clinically or radiographically diagnosed neurologic disorder was reported.
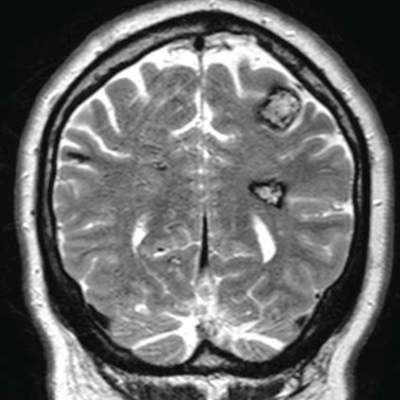
This presentation of numerous CCMs and cutaneous vascular lesions prompted genetic testing for mutations in the 3 CCM genes. Bidirectional sequencing from a blood specimen confirmed a 1267C→T substitution resulting in an Arg423X nonsense mutation in the KRIT1, ankyrin repeat containing gene (also known as CCM1) on chromosome 7, which is a published mutation.1 No treatment of the cutaneous lesions was administered. The patient continued to be treated by a neurologist for the seizure disorder after genetic testing and counseling was complete.
Comment
Cerebral cavernous malformations represent collections of dilated, capillarylike vessels without intervening brain parenchyma,2 which can occur as a sporadic or autosomal-dominant condition with variable prevalence. Imaging and autopsy studies suggest CCMs may be present in 0.5% of the general population, with the incidence of clinically symptomatic disease being much lower.3,4 While sporadic cases are usually associated with the presence of a single CCM, hereditary cases generally display multiple CCMs5-7; studies indicate that most patients who present with symptomatic CCMs are in the second to fifth decades of life.3,8-10 The most common clinical manifestations of CCMs are headaches, seizures, and focal neurologic deficits due to cerebral hemorrhages.11
Three autosomal-dominant forms of familial CCMs are associated with several different heterozygous mutations in the CCM1 gene, which encodes Krev interaction trapped protein 1; the cerebral cavernous malformation 2 gene (CCM2), which encodes the protein malcavernin; and the programmed cell death 10 gene (PDCD10)(also known as CCM3), which encodes the PDCD10 protein.12-14 Although the precise pathophysiologic mechanisms linking these mutations with the resultant clinical phenotypes have yet to be fully elucidated, these genes are thought to play roles in angiogenesis as well as vascular maintenance and remodeling.11 Among non-Hispanic white kindreds with familial CCMs, 40% to 72% of kindreds were linked to mutations in CCM1, 20% were linked to mutations in CCM2, and 8% to 40% were linked to mutations in CCM3, depending on the series.13,15 The prevalence of symptomatic disease among gene carriers for kindreds linked to CCM1, CCM2, and CCM3 was 88%, 100%, and 63%, respectively.13 A specific founder mutation in CCM1 is thought to be responsible for virtually all cases of familial CCMs in Hispanic American individuals16 and up to 50% of this patient population are thought to have the familial form of the condition.5 As more is learned about the role of CCM gene mutations in the pathogenesis of CCMs, understanding how different mutations with different degrees of prevalence affect different patient populations will be important to help guide testing and perhaps treatment in the future.
Extracerebral manifestations of familial CCMs include retinal and cutaneous involvement. The incidence of retinal cavernomas has been estimated at 5% in familial CCM patients.17 The most commonly reported cutaneous finding (with an incidence of 3% to 5% reported in large case series) is hyperkeratotic cutaneous capillary-venous malformations (HCCVMs), which are composed of dilated capillaries and blood-filled, venouslike channels extending into the dermis and hypodermis with an overlying hyperkeratotic epidermis.7,18-20 Although the prevalence for CCM1-associated CCMs is reported to be higher than that for the HCCVMs in families affected by both cerebral and cutaneous lesions, all patients manifesting HCCVMs also have CCMs.18-20 Perhaps the cutaneous lesions may serve as markers of neurologic involvement in affected families. Other reports have shown an association between CCMs and cutaneous cavernous hemangiomas,21 capillary malformations,20 venous malformations,20,22 and cherry angiomas,21,23 although the relevance of cherry angiomas is unclear given their high prevalence in the general population.
There are few reports of patients with CCMs having angiokeratomas, including a 77-year-old Hispanic man with a solitary angiokeratoma, cutaneous venous malformations, multiple CCMs, and demonstration of the 2105C→T CCM1 founder mutation, which is commonly found in Hispanic patients24; a Chinese man with a CCM1 mutation and purple-black, raised, angiomalike skin lesions (similar to those seen in our patient) that which were not biopsied25; and a patient with multiple eruptive angiokeratomas who was found to have numerous CCMs before the availability of genetic testing.26 In our patient, the presentation of eruptive angiokeratomas prompted consideration of genetic testing and subsequent confirmation of a CCM1 mutation. Knowledge of the association between familial CCMs and cutaneous vascular lesions may be used to identify patients with clinically silent, undetected CCMs who may be at risk of developing future neurologic disorders or who may benefit from appropriate genetic counseling.
Conclusion
Familial CCMs are associated with extracerebral manifestations that include retinal cavernomas along with a variety of cutaneous manifestations. We present a case of a patient with adult-onset seizures, eruptive angiokeratomas, and cutaneous hemangiomas in the setting of numerous CCMs with confirmation of CCM1 mutation on genetic testing. We hope to raise awareness of the cutaneous findings associated with CCMs and the availability of genetic testing for CCM gene mutations so that patients with multiple cutaneous vascular lesions, including eruptive angiokeratomas, can be screened and tested for genetic mutations and neurologic involvement when appropriate.
1. Cavé-Riant F, Denier C, Labauge P, et al. Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with cerebral cavernous malformations. Eur J Hum Genet. 2002;10:733-740.
2. Simard JM, Garcia-Bengochea F, Ballinger WE Jr, et al. Cavernous angioma: a review of 126 collected and 12 new clinical cases. Neurosurgery. 1986;18:162-172.
3. Del Curling O Jr, Kelly DL Jr, Elster AD, et al. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702-708.
4. Otten P, Pizzolato GP, Rilliet B, et al. 131 cases of cavernous angioma (cavernomas) of the cns, discovered by retrospective analysis of 24,535 autopsies [article in French]. Neurochirurgie. 1989;35:82-83, 128-131.
5. Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. incidence and familial occurrence. N Eng J Med. 1988;319:343-347.
6. Labauge P, Laberge S, Brunereau L, et al. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Société Française de Neurochirurgie. Lancet. 1998;352:1892-1897.
7. Denier C, Labauge P, Brunereau L, et al. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol. 2004;55:213-220.
8. Moriarity JL, Wetzel M, Clatterbuck RE, et al. The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery. 1999;44:1166-1171.
9. Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709-714.
10. Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
11. Cavalcanti DD, Kalani MY, Martirosyan NL, et al. Cerebral cavernous malformations: from genes to proteins to disease. J Neurosurg. 2012;116:122-132.
12. Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a KREV-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325-2333.
13. Craig HD, Gunel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851-1858.
14. Bergametti F, Denier C, Labauge P, et al; Société Française de Neurochirurgie. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42-51.
15. Denier C, Labauge P, Bergametti F, et al; Brain Vascular Malformation Consortium (BVMC) Study. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550-556.
16. Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Eng J Med. 1996;334:946-951.
17. Labauge P, Krivosic V, Denier C, et al. Frequency of retinal cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. Arch Ophthalmol. 2006;124:885-886.
18. Eerola I, Plate KH, Spiegel R, et al. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351-1355.
19. Labauge P, Enjolras O, Bonerandi JJ, et al. An association between autosomal dominant cerebral cavernomas and a distinctive hyperkeratotic cutaneous vascular malformation in 4 families. Ann Neurol. 1999;45:250-254.
20. Sirvente J, Enjolras O, Wassef M, et al. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. 2009;23:1066-1072.
21. Filling-Katz MR, Levin SW, Patronas NJ, et al. Terminal transverse limb defects associated with familial cavernous angiomatosis. Am J Med Genet. 1992;42:346-351.
22. Toll A, Parera E, Giménez-Arnau AM, et al. Cutaneous venous malformations in familial cerebral cavernomatosis caused by KRIT1 gene mutations. Dermatology. 2009;218:307-313.
23. Clatterbuck RE, Rigamonti D. Cherry angiomas associated with familial cerebral cavernous malformations: case illustration. J Neurosurg. 2002;96:964.
24. Zlotoff BJ, Bang RH, Padilla RS, et al. Cutaneous angiokeratoma and venous malformations in a Hispanic-American patient with cerebral cavernous malformations. Br J Dermatol. 2007;157:210-212.
25. Chen DH, Lipe HP, Qin Z, et al. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurol Sci. 2002;196:91-96.
26. Ostlere L, Hart Y, Misch KJ. Cutaneous and cerebral haemangiomas associated with eruptive angiokeratomas. Br J Dermatol. 1996;135:98-101.
Case Report
A 66-year-old non-Hispanic white man with a history of adult-onset seizures, renal cell carcinoma, and nephrolithiasis presented to the dermatology clinic with numerous dark red, dry, scaling lesions on the lower legs, abdomen, back, thighs, and scrotum that bled easily. The lesions had developed 2 to 4 years prior to presentation. The patient also noted several dark blue lesions on the bilateral arms that had been present for several years.
Physical examination revealed multiple black, keratotic papules on the bilateral lower legs (Figure 1), with similar lesions on the lower abdomen and lower back. Numerous dark red, scaling papules were present on the thighs and scrotum, and firm, 1- to 3-cm, dark blue nodules were noted on the bilateral arms (Figure 2) and right side of the forehead. Biopsy of papules from the lower right leg and back demonstrated angiokeratomas on pathologic review (Figure 3). Four separate biopsies taken from the left forearm demonstrated lobular and cavernous hemangiomas (Figure 4).
The patient’s medical history was remarkable for neurologic symptoms including adult-onset seizures at the age of 42 years. Recent magnetic resonance imaging of the head showed multiple areas of heterogeneous signal intensity with T2 hypointense rims consistent with cerebral cavernous malformations (CCMs) involving both cerebral hemispheres, the posterior cranial fossa, and the brainstem (Figure 5). Of note, the patient’s mother had been diagnosed with epilepsy in her 40s, but no other family history of a clinically or radiographically diagnosed neurologic disorder was reported.
This presentation of numerous CCMs and cutaneous vascular lesions prompted genetic testing for mutations in the 3 CCM genes. Bidirectional sequencing from a blood specimen confirmed a 1267C→T substitution resulting in an Arg423X nonsense mutation in the KRIT1, ankyrin repeat containing gene (also known as CCM1) on chromosome 7, which is a published mutation.1 No treatment of the cutaneous lesions was administered. The patient continued to be treated by a neurologist for the seizure disorder after genetic testing and counseling was complete.
Comment
Cerebral cavernous malformations represent collections of dilated, capillarylike vessels without intervening brain parenchyma,2 which can occur as a sporadic or autosomal-dominant condition with variable prevalence. Imaging and autopsy studies suggest CCMs may be present in 0.5% of the general population, with the incidence of clinically symptomatic disease being much lower.3,4 While sporadic cases are usually associated with the presence of a single CCM, hereditary cases generally display multiple CCMs5-7; studies indicate that most patients who present with symptomatic CCMs are in the second to fifth decades of life.3,8-10 The most common clinical manifestations of CCMs are headaches, seizures, and focal neurologic deficits due to cerebral hemorrhages.11
Three autosomal-dominant forms of familial CCMs are associated with several different heterozygous mutations in the CCM1 gene, which encodes Krev interaction trapped protein 1; the cerebral cavernous malformation 2 gene (CCM2), which encodes the protein malcavernin; and the programmed cell death 10 gene (PDCD10)(also known as CCM3), which encodes the PDCD10 protein.12-14 Although the precise pathophysiologic mechanisms linking these mutations with the resultant clinical phenotypes have yet to be fully elucidated, these genes are thought to play roles in angiogenesis as well as vascular maintenance and remodeling.11 Among non-Hispanic white kindreds with familial CCMs, 40% to 72% of kindreds were linked to mutations in CCM1, 20% were linked to mutations in CCM2, and 8% to 40% were linked to mutations in CCM3, depending on the series.13,15 The prevalence of symptomatic disease among gene carriers for kindreds linked to CCM1, CCM2, and CCM3 was 88%, 100%, and 63%, respectively.13 A specific founder mutation in CCM1 is thought to be responsible for virtually all cases of familial CCMs in Hispanic American individuals16 and up to 50% of this patient population are thought to have the familial form of the condition.5 As more is learned about the role of CCM gene mutations in the pathogenesis of CCMs, understanding how different mutations with different degrees of prevalence affect different patient populations will be important to help guide testing and perhaps treatment in the future.
Extracerebral manifestations of familial CCMs include retinal and cutaneous involvement. The incidence of retinal cavernomas has been estimated at 5% in familial CCM patients.17 The most commonly reported cutaneous finding (with an incidence of 3% to 5% reported in large case series) is hyperkeratotic cutaneous capillary-venous malformations (HCCVMs), which are composed of dilated capillaries and blood-filled, venouslike channels extending into the dermis and hypodermis with an overlying hyperkeratotic epidermis.7,18-20 Although the prevalence for CCM1-associated CCMs is reported to be higher than that for the HCCVMs in families affected by both cerebral and cutaneous lesions, all patients manifesting HCCVMs also have CCMs.18-20 Perhaps the cutaneous lesions may serve as markers of neurologic involvement in affected families. Other reports have shown an association between CCMs and cutaneous cavernous hemangiomas,21 capillary malformations,20 venous malformations,20,22 and cherry angiomas,21,23 although the relevance of cherry angiomas is unclear given their high prevalence in the general population.
There are few reports of patients with CCMs having angiokeratomas, including a 77-year-old Hispanic man with a solitary angiokeratoma, cutaneous venous malformations, multiple CCMs, and demonstration of the 2105C→T CCM1 founder mutation, which is commonly found in Hispanic patients24; a Chinese man with a CCM1 mutation and purple-black, raised, angiomalike skin lesions (similar to those seen in our patient) that which were not biopsied25; and a patient with multiple eruptive angiokeratomas who was found to have numerous CCMs before the availability of genetic testing.26 In our patient, the presentation of eruptive angiokeratomas prompted consideration of genetic testing and subsequent confirmation of a CCM1 mutation. Knowledge of the association between familial CCMs and cutaneous vascular lesions may be used to identify patients with clinically silent, undetected CCMs who may be at risk of developing future neurologic disorders or who may benefit from appropriate genetic counseling.
Conclusion
Familial CCMs are associated with extracerebral manifestations that include retinal cavernomas along with a variety of cutaneous manifestations. We present a case of a patient with adult-onset seizures, eruptive angiokeratomas, and cutaneous hemangiomas in the setting of numerous CCMs with confirmation of CCM1 mutation on genetic testing. We hope to raise awareness of the cutaneous findings associated with CCMs and the availability of genetic testing for CCM gene mutations so that patients with multiple cutaneous vascular lesions, including eruptive angiokeratomas, can be screened and tested for genetic mutations and neurologic involvement when appropriate.
Case Report
A 66-year-old non-Hispanic white man with a history of adult-onset seizures, renal cell carcinoma, and nephrolithiasis presented to the dermatology clinic with numerous dark red, dry, scaling lesions on the lower legs, abdomen, back, thighs, and scrotum that bled easily. The lesions had developed 2 to 4 years prior to presentation. The patient also noted several dark blue lesions on the bilateral arms that had been present for several years.
Physical examination revealed multiple black, keratotic papules on the bilateral lower legs (Figure 1), with similar lesions on the lower abdomen and lower back. Numerous dark red, scaling papules were present on the thighs and scrotum, and firm, 1- to 3-cm, dark blue nodules were noted on the bilateral arms (Figure 2) and right side of the forehead. Biopsy of papules from the lower right leg and back demonstrated angiokeratomas on pathologic review (Figure 3). Four separate biopsies taken from the left forearm demonstrated lobular and cavernous hemangiomas (Figure 4).
The patient’s medical history was remarkable for neurologic symptoms including adult-onset seizures at the age of 42 years. Recent magnetic resonance imaging of the head showed multiple areas of heterogeneous signal intensity with T2 hypointense rims consistent with cerebral cavernous malformations (CCMs) involving both cerebral hemispheres, the posterior cranial fossa, and the brainstem (Figure 5). Of note, the patient’s mother had been diagnosed with epilepsy in her 40s, but no other family history of a clinically or radiographically diagnosed neurologic disorder was reported.
This presentation of numerous CCMs and cutaneous vascular lesions prompted genetic testing for mutations in the 3 CCM genes. Bidirectional sequencing from a blood specimen confirmed a 1267C→T substitution resulting in an Arg423X nonsense mutation in the KRIT1, ankyrin repeat containing gene (also known as CCM1) on chromosome 7, which is a published mutation.1 No treatment of the cutaneous lesions was administered. The patient continued to be treated by a neurologist for the seizure disorder after genetic testing and counseling was complete.
Comment
Cerebral cavernous malformations represent collections of dilated, capillarylike vessels without intervening brain parenchyma,2 which can occur as a sporadic or autosomal-dominant condition with variable prevalence. Imaging and autopsy studies suggest CCMs may be present in 0.5% of the general population, with the incidence of clinically symptomatic disease being much lower.3,4 While sporadic cases are usually associated with the presence of a single CCM, hereditary cases generally display multiple CCMs5-7; studies indicate that most patients who present with symptomatic CCMs are in the second to fifth decades of life.3,8-10 The most common clinical manifestations of CCMs are headaches, seizures, and focal neurologic deficits due to cerebral hemorrhages.11
Three autosomal-dominant forms of familial CCMs are associated with several different heterozygous mutations in the CCM1 gene, which encodes Krev interaction trapped protein 1; the cerebral cavernous malformation 2 gene (CCM2), which encodes the protein malcavernin; and the programmed cell death 10 gene (PDCD10)(also known as CCM3), which encodes the PDCD10 protein.12-14 Although the precise pathophysiologic mechanisms linking these mutations with the resultant clinical phenotypes have yet to be fully elucidated, these genes are thought to play roles in angiogenesis as well as vascular maintenance and remodeling.11 Among non-Hispanic white kindreds with familial CCMs, 40% to 72% of kindreds were linked to mutations in CCM1, 20% were linked to mutations in CCM2, and 8% to 40% were linked to mutations in CCM3, depending on the series.13,15 The prevalence of symptomatic disease among gene carriers for kindreds linked to CCM1, CCM2, and CCM3 was 88%, 100%, and 63%, respectively.13 A specific founder mutation in CCM1 is thought to be responsible for virtually all cases of familial CCMs in Hispanic American individuals16 and up to 50% of this patient population are thought to have the familial form of the condition.5 As more is learned about the role of CCM gene mutations in the pathogenesis of CCMs, understanding how different mutations with different degrees of prevalence affect different patient populations will be important to help guide testing and perhaps treatment in the future.
Extracerebral manifestations of familial CCMs include retinal and cutaneous involvement. The incidence of retinal cavernomas has been estimated at 5% in familial CCM patients.17 The most commonly reported cutaneous finding (with an incidence of 3% to 5% reported in large case series) is hyperkeratotic cutaneous capillary-venous malformations (HCCVMs), which are composed of dilated capillaries and blood-filled, venouslike channels extending into the dermis and hypodermis with an overlying hyperkeratotic epidermis.7,18-20 Although the prevalence for CCM1-associated CCMs is reported to be higher than that for the HCCVMs in families affected by both cerebral and cutaneous lesions, all patients manifesting HCCVMs also have CCMs.18-20 Perhaps the cutaneous lesions may serve as markers of neurologic involvement in affected families. Other reports have shown an association between CCMs and cutaneous cavernous hemangiomas,21 capillary malformations,20 venous malformations,20,22 and cherry angiomas,21,23 although the relevance of cherry angiomas is unclear given their high prevalence in the general population.
There are few reports of patients with CCMs having angiokeratomas, including a 77-year-old Hispanic man with a solitary angiokeratoma, cutaneous venous malformations, multiple CCMs, and demonstration of the 2105C→T CCM1 founder mutation, which is commonly found in Hispanic patients24; a Chinese man with a CCM1 mutation and purple-black, raised, angiomalike skin lesions (similar to those seen in our patient) that which were not biopsied25; and a patient with multiple eruptive angiokeratomas who was found to have numerous CCMs before the availability of genetic testing.26 In our patient, the presentation of eruptive angiokeratomas prompted consideration of genetic testing and subsequent confirmation of a CCM1 mutation. Knowledge of the association between familial CCMs and cutaneous vascular lesions may be used to identify patients with clinically silent, undetected CCMs who may be at risk of developing future neurologic disorders or who may benefit from appropriate genetic counseling.
Conclusion
Familial CCMs are associated with extracerebral manifestations that include retinal cavernomas along with a variety of cutaneous manifestations. We present a case of a patient with adult-onset seizures, eruptive angiokeratomas, and cutaneous hemangiomas in the setting of numerous CCMs with confirmation of CCM1 mutation on genetic testing. We hope to raise awareness of the cutaneous findings associated with CCMs and the availability of genetic testing for CCM gene mutations so that patients with multiple cutaneous vascular lesions, including eruptive angiokeratomas, can be screened and tested for genetic mutations and neurologic involvement when appropriate.
1. Cavé-Riant F, Denier C, Labauge P, et al. Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with cerebral cavernous malformations. Eur J Hum Genet. 2002;10:733-740.
2. Simard JM, Garcia-Bengochea F, Ballinger WE Jr, et al. Cavernous angioma: a review of 126 collected and 12 new clinical cases. Neurosurgery. 1986;18:162-172.
3. Del Curling O Jr, Kelly DL Jr, Elster AD, et al. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702-708.
4. Otten P, Pizzolato GP, Rilliet B, et al. 131 cases of cavernous angioma (cavernomas) of the cns, discovered by retrospective analysis of 24,535 autopsies [article in French]. Neurochirurgie. 1989;35:82-83, 128-131.
5. Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. incidence and familial occurrence. N Eng J Med. 1988;319:343-347.
6. Labauge P, Laberge S, Brunereau L, et al. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Société Française de Neurochirurgie. Lancet. 1998;352:1892-1897.
7. Denier C, Labauge P, Brunereau L, et al. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol. 2004;55:213-220.
8. Moriarity JL, Wetzel M, Clatterbuck RE, et al. The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery. 1999;44:1166-1171.
9. Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709-714.
10. Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
11. Cavalcanti DD, Kalani MY, Martirosyan NL, et al. Cerebral cavernous malformations: from genes to proteins to disease. J Neurosurg. 2012;116:122-132.
12. Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a KREV-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325-2333.
13. Craig HD, Gunel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851-1858.
14. Bergametti F, Denier C, Labauge P, et al; Société Française de Neurochirurgie. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42-51.
15. Denier C, Labauge P, Bergametti F, et al; Brain Vascular Malformation Consortium (BVMC) Study. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550-556.
16. Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Eng J Med. 1996;334:946-951.
17. Labauge P, Krivosic V, Denier C, et al. Frequency of retinal cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. Arch Ophthalmol. 2006;124:885-886.
18. Eerola I, Plate KH, Spiegel R, et al. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351-1355.
19. Labauge P, Enjolras O, Bonerandi JJ, et al. An association between autosomal dominant cerebral cavernomas and a distinctive hyperkeratotic cutaneous vascular malformation in 4 families. Ann Neurol. 1999;45:250-254.
20. Sirvente J, Enjolras O, Wassef M, et al. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. 2009;23:1066-1072.
21. Filling-Katz MR, Levin SW, Patronas NJ, et al. Terminal transverse limb defects associated with familial cavernous angiomatosis. Am J Med Genet. 1992;42:346-351.
22. Toll A, Parera E, Giménez-Arnau AM, et al. Cutaneous venous malformations in familial cerebral cavernomatosis caused by KRIT1 gene mutations. Dermatology. 2009;218:307-313.
23. Clatterbuck RE, Rigamonti D. Cherry angiomas associated with familial cerebral cavernous malformations: case illustration. J Neurosurg. 2002;96:964.
24. Zlotoff BJ, Bang RH, Padilla RS, et al. Cutaneous angiokeratoma and venous malformations in a Hispanic-American patient with cerebral cavernous malformations. Br J Dermatol. 2007;157:210-212.
25. Chen DH, Lipe HP, Qin Z, et al. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurol Sci. 2002;196:91-96.
26. Ostlere L, Hart Y, Misch KJ. Cutaneous and cerebral haemangiomas associated with eruptive angiokeratomas. Br J Dermatol. 1996;135:98-101.
1. Cavé-Riant F, Denier C, Labauge P, et al. Spectrum and expression analysis of KRIT1 mutations in 121 consecutive and unrelated patients with cerebral cavernous malformations. Eur J Hum Genet. 2002;10:733-740.
2. Simard JM, Garcia-Bengochea F, Ballinger WE Jr, et al. Cavernous angioma: a review of 126 collected and 12 new clinical cases. Neurosurgery. 1986;18:162-172.
3. Del Curling O Jr, Kelly DL Jr, Elster AD, et al. An analysis of the natural history of cavernous angiomas. J Neurosurg. 1991;75:702-708.
4. Otten P, Pizzolato GP, Rilliet B, et al. 131 cases of cavernous angioma (cavernomas) of the cns, discovered by retrospective analysis of 24,535 autopsies [article in French]. Neurochirurgie. 1989;35:82-83, 128-131.
5. Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations. incidence and familial occurrence. N Eng J Med. 1988;319:343-347.
6. Labauge P, Laberge S, Brunereau L, et al. Hereditary cerebral cavernous angiomas: clinical and genetic features in 57 French families. Société Française de Neurochirurgie. Lancet. 1998;352:1892-1897.
7. Denier C, Labauge P, Brunereau L, et al. Clinical features of cerebral cavernous malformations patients with KRIT1 mutations. Ann Neurol. 2004;55:213-220.
8. Moriarity JL, Wetzel M, Clatterbuck RE, et al. The natural history of cavernous malformations: a prospective study of 68 patients. Neurosurgery. 1999;44:1166-1171.
9. Robinson JR, Awad IA, Little JR. Natural history of the cavernous angioma. J Neurosurg. 1991;75:709-714.
10. Zabramski JM, Wascher TM, Spetzler RF, et al. The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg. 1994;80:422-432.
11. Cavalcanti DD, Kalani MY, Martirosyan NL, et al. Cerebral cavernous malformations: from genes to proteins to disease. J Neurosurg. 2012;116:122-132.
12. Sahoo T, Johnson EW, Thomas JW, et al. Mutations in the gene encoding KRIT1, a KREV-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325-2333.
13. Craig HD, Gunel M, Cepeda O, et al. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851-1858.
14. Bergametti F, Denier C, Labauge P, et al; Société Française de Neurochirurgie. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42-51.
15. Denier C, Labauge P, Bergametti F, et al; Brain Vascular Malformation Consortium (BVMC) Study. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550-556.
16. Gunel M, Awad IA, Finberg K, et al. A founder mutation as a cause of cerebral cavernous malformation in Hispanic Americans. N Eng J Med. 1996;334:946-951.
17. Labauge P, Krivosic V, Denier C, et al. Frequency of retinal cavernomas in 60 patients with familial cerebral cavernomas: a clinical and genetic study. Arch Ophthalmol. 2006;124:885-886.
18. Eerola I, Plate KH, Spiegel R, et al. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351-1355.
19. Labauge P, Enjolras O, Bonerandi JJ, et al. An association between autosomal dominant cerebral cavernomas and a distinctive hyperkeratotic cutaneous vascular malformation in 4 families. Ann Neurol. 1999;45:250-254.
20. Sirvente J, Enjolras O, Wassef M, et al. Frequency and phenotypes of cutaneous vascular malformations in a consecutive series of 417 patients with familial cerebral cavernous malformations. J Eur Acad Dermatol Venereol. 2009;23:1066-1072.
21. Filling-Katz MR, Levin SW, Patronas NJ, et al. Terminal transverse limb defects associated with familial cavernous angiomatosis. Am J Med Genet. 1992;42:346-351.
22. Toll A, Parera E, Giménez-Arnau AM, et al. Cutaneous venous malformations in familial cerebral cavernomatosis caused by KRIT1 gene mutations. Dermatology. 2009;218:307-313.
23. Clatterbuck RE, Rigamonti D. Cherry angiomas associated with familial cerebral cavernous malformations: case illustration. J Neurosurg. 2002;96:964.
24. Zlotoff BJ, Bang RH, Padilla RS, et al. Cutaneous angiokeratoma and venous malformations in a Hispanic-American patient with cerebral cavernous malformations. Br J Dermatol. 2007;157:210-212.
25. Chen DH, Lipe HP, Qin Z, et al. Cerebral cavernous malformation: novel mutation in a Chinese family and evidence for heterogeneity. J Neurol Sci. 2002;196:91-96.
26. Ostlere L, Hart Y, Misch KJ. Cutaneous and cerebral haemangiomas associated with eruptive angiokeratomas. Br J Dermatol. 1996;135:98-101.
Practice Points
- Familial cerebral cavernous malformations (CCMs) are associated with extra-cerebral manifestations that include retinal cavernomas as well as a variety of cutaneous manifestations.
- Our patient presented with adult-onset eruptive angiokeratomas, cutaneous hemangiomas, and adult-onset seizures in the setting of numerous CCMs with confirmation of CCM1 mutation.
- Dermatologists should be aware of the availability of genetic testing for CCM gene mutations and neurologic screening in patients presenting with multiple cutaneous vascular lesions, including eruptive angiokeratomas.