User login
Left Out in the Cold
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
© 2020 Society of Hospital Medicine
Missed Opportunities for Treatment of Opioid Use Disorder in the Hospital Setting: Updating an Outdated Policy
THE PROBLEM AND THE ROLE OF THE HOSPITALIST
Opioid use disorder (OUD) is a common, underrecognized, undertreated, and deadly medical condition. Although the focus of addressing the opioid epidemic has been centered in the outpatient setting, hospitalists play an important—and often underutilized—role in identifying OUD, initiating treatment, and assisting with linkage to longitudinal care after discharge.
Over the past 20 years, the annual rate of hospital discharges documenting OUD has quadrupled.1 During 2010-2016, the annual discharge rate for heroin overdoses increased by 23%.1 Although the total number of hospitalizations in the United States remained stable from 2002 to 2012, the number of admissions for opioid abuse or dependence increased from 301,707 to 520,275. More than 500,000 hospital admissions per year (1% of total nationwide hospitalizations) are now due primarily to OUD.2
Injection opioid use increases the risk of endocarditis, osteomyelitis, septic arthritis, and epidural abscesses, conditions that often prolong hospitalizations and frequently lead to readmissions. Admissions for OUD-related infections are rising at a startling rate. Between 2002 and 2012, the number of admissions for infections associated with OUDs had increased from 3,421 to 6,535.2 In addition to providing the opportunity to diagnose OUD, hospitalizations offer an ideal time to engage patients in OUD treatment and linkage to outpatient care.
Although we uniformly offer patients antibiotic treatment for acute infection, hospitalists should consistently incorporate treatment of OUD to address the root cause of these admissions. As infection is but one sequelae of the underlying disease of addiction, treating without medications for OUD (MOUD) would be akin to treating a diabetic foot ulcer with antibiotics and not providing medications to improve glycemic control. Omitting such addiction treatment can contribute to treatment failure and worse health outcomes. Among patients with endocarditis and an associated valve repair, those who continue injection drug use have a 10 times higher risk of death or reoperation between 90 and 180 days after repair than those not engaged in drug use.3
Despite data demonstrating the significant benefit and the minimal harm of MOUD, significant gaps remain in providing MOUD and linking patients from the hospital to community care.1,4 Hospital encounters are missed opportunities to provide life-saving MOUD treatment; the majority of patients with OUD do not receive evidence-based treatment while inpatient.5 Rosenthal et al. found that of 102 patients admitted with injection drug use-associated infective endocarditis from 2004 to 2014, only 8% received MOUD, and approximately half had a documentation of substance use treatment in their discharge worksheet.4 In Massachusetts, among individuals who experienced a nonfatal opioid overdose and had interaction with healthcare services, only 26% were on MOUD one year later.6 Based on our experience, a substantial proportion of patients with OUD do not seek or have access to medical care, acute care settings offer a critical opportunity to engage them in treatment for their addiction.
WHY SHOULD HOSPITALISTS INITIATE BUPRENORPHINE?
First, buprenorphine effectively treats withdrawal symptoms. Buprenorphine and methadone are superior to other medications in treating symptoms of withdrawal.7 If withdrawal symptoms are treated, patients are less likely to leave against medical advice8 and are more likely to complete treatment.
Second, MOUD is the standard of care for treating OUD.9 Medications include the full agonist methadone, the partial agonist buprenorphine, and the long-acting antagonist naltrexone. Although all these drugs are effective and legal to initiate for inpatients,6 this perspective focuses on buprenorphine in an effort to draw attention to associated policy barriers. Buprenorphine is the only MOUD that can be offered as office-based therapy by providers in the outpatient setting. Meta-analyses show that MOUD is associated with lower rates of mortality, illicit opioid use, HIV transmission, and violent crime and arrest.9
Third, MOUD treatment, rather than just referral, leads to higher long-term treatment success.10 When initiating buprenorphine in the hospital, treatment retention rates at one month were double that of referral alone. Six months after discharge, patients were five times more likely to remain engaged in treatment compared with those who received a detoxification protocol only.
Fourth, buprenorphine is not only effective, but it is also safe and has low risks of misuse. Because buprenorphine is a partial agonist, it has both a ceiling effect on respiratory depression (decreasing potential lethality) and on euphoria (decreasing the likelihood of misuse). Among individuals who took nonprescribed buprenorphine on the street, less than 7% reported taking it for any attempt at euphoria. Instead, people with OUD most often use nonprescribed or diverted buprenorphine to treat withdrawal symptoms.11
Fifth, buprenorphine treatment is associated with fewer hospital readmissions.12
Finally, initiating OUD treatment is feasible in the hospital setting. Any hospitalist can legally prescribe buprenorphine to treat opioid withdrawal for hospitalized patients admitted for medical or surgical reasons. A waiver is necessary only for prescribing at the time of hospital discharge for use in non-inpatient settings of care.
A POLICY BARRIER: THE X WAIVER
The United States Congress passed the Drug Addiction Treatment Act (DATA) of 2000, which codified the X waiver, in response to the growing opioid crisis. Only those providers with the DATA X waiver can write buprenorphine prescriptions to be filled in an outpatient pharmacy. To obtain an X waiver, physicians must complete an 8-hour course, whereas physician assistants and nurse practitioners must complete a 24-hour course. This training far exceeds any required training to prescribe opioids for pain.
Unfortunately, the X waiver requirement obstructs hospitalists from initiating buprenorphine in the inpatient setting in the following ways: (1) hospitalists often choose not to initiate chronic buprenorphine treatment if they lack the X waiver that would allow them to write the discharge prescription and/or (2) they are unable to identify a waivered provider in the community to continue the prescription. Unfortunately, only 6% of all medical practitioners are waivered to prescribe buprenorphine; greater than 40% of US counties are “buprenorphine deserts,” with no providers waivered to prescribe buprenorphine.13
To address the opioid crisis, we must rethink our current policies. The Department of Health and Human Services should eliminate the X waiver and allow any licensed physician, nurse practitioner, or physician assistant to prescribe buprenorphine.14 Recent American Medical Association Opioid Task Force recommendations have called to “remove… inappropriate administrative burdens or barriers that delay or deny care for FDA-approved medications used as part of medication-assisted treatment for OUD.”15 Legislation to remove the X wavier has been proposed in the United States.16
The removal of a buprenorphine waiver requirement has had success in other settings. The French deregulation of buprenorphine was associated with a reduction in opioid overdose deaths by 79%. Similar success in the United States would save an estimated 30,000 lives yearly.14 Removing the X waiver is an important step in empowering hospitalists to initiate MOUD for individuals in the hospital setting. Moreover, it opens the door to more outpatient primary care providers serving as community linkages for long-term addiction care.
NOT A PANACEA
Without the X waiver, the associated OUD training will no longer be required. This could have unintended consequences. For example, if hospitalists order buprenorphine while opioids remain active, precipitated withdrawal may ensue. Crucially, the current literature does not indicate that the required X waiver training improves knowledge, patient care, or outcomes.17 Nevertheless, MOUD and addiction training may help reduce knowledge gaps and empower providers to engage in productive conversations surrounding addiction. This highlights the crucial role of physician organizations, such as the Society of Hospital Medicine, in educating hospitalists about MOUD. (This organization, among others, has developed robust MOUD training.18)
It is also important to acknowledge that the waiver is only one obstacle. Other barriers have been identified in initiating buprenorphine, including access to treatment after discharge, access to social work support, and lack of EMR order sets, among others.19 Professional societies, hospitals, and hospitalists need to help address these barriers through ancillary support staff, quality improvement initiatives, and improved inpatient treatment of withdrawal with MOUD. This can be done successfully; one study found that 82% of hospitalized patients who engaged in a new transitional opioid program subsequently presented to outpatient opioid treatment.20 Novel interventions must be part of a hospital-wide approach to optimizing improved longitudinal treatment for patients suffering from addiction.
CONCLUSION
Hospitalization is an ideal opportunity for clinicians to diagnose and treat OUD in a population that often has not sought, or has fallen out of, addiction treatment. Hospitalists can and should initiate buprenorphine in appropriate inpatients and plan for their transition to chronic care. Eliminating the waiver in combination with designing innovative educational opportunities and systems approaches to provide better linkages to outpatient OUD treatment is needed to combat the opioid crisis. To enable more hospitalists to successfully initiate long-term buprenorphine therapy—and to enable more outpatient providers to continue prescriptions—we must eliminate the X waiver.
Disclosures
Dr. Wilson received honorarium from the American Society of Addiction Medicine for teaching and creating
1. Peterson C, Xu L, Florence C, Mack KA. Opioid-related US hospital discharges by type, 1993–2016. J Subst Abuse Treat. 2019;103:9-13. https://doi.org/10.1016/j.jsat.2019.05.003.
2. Ronan MV, Herzig SJ. Hospitalizations related to opioid abuse/dependence and associated serious infections increased sharply, 2002-12. Health Aff. 2016;35(5):832-837. https://doi.org/10.1377/hlthaff.2015.1424.
3. Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg. 2015;100(3):875-882. https://doi.org/10.1016/j.athoracsur.2015.03.019.
4. Rosenthal ES, Karchmer AW, Theisen-Toupal J, Castillo RA, Rowley CF. Suboptimal addiction interventions for patients hospitalized with injection drug use-associated infective endocarditis. Am J Med. 2016;129(5):481-485. https://doi.org/10.1016/j.amjmed.2015.09.024.
5. Winetsky D, Weinrieb RM, Perrone J. Expanding treatment opportunities for hospitalized patients with opioid use disorders. J Hosp Med. 2017;13(1):62-64. https://doi.org/10.12788/jhm.2861.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Gowing L, Ali R, White JM, Mbewe D. Buprenorphine for managing opioid withdrawal. Cochrane Database Syst Rev. 2017;(2):CD002025. https://doi.org/10.1002/14651858.CD002025.pub5.
8. Ti L, Ti L. Leaving the hospital against medical advice among people who use illicit drugs: a systematic review. Am J Public Health. 2015;105(12):e53-e59. https://doi.org/10.2105/AJPH.2015.302885.
9. Schuckit MA. Treatment of opioid-use disorders. N Engl J Med. 2016;375(4):357-368. https://doi.org/10.1056/NEJMra1604339.
10. Liebschutz JM, Crooks D, Herman D, et al. Buprenorphine treatment for hospitalized, opioid-dependent patients. JAMA Intern Med. 2014;174(8):1369. https://doi.org/10.1001/jamainternmed.2014.2556.
11. Cicero TJ, Ellis MS, Surratt HL, Kurtz SP. Factors contributing to the rise of buprenorphine misuse: 2008-2013. Drug Alcohol Depend. 2014;142:98-104. https://doi.org/10.1016/j.drugalcdep.2014.06.005.
12. Moreno JL, Wakeman SE, Duprey MS, Roberts RJ, Jacobson JS, Devlin JW. Predictors for 30-day and 90-day hospital readmission among patients with opioid use disorder. J Addict Med. 2019;13(4):306-313. https://doi.org/10.1097/ADM.0000000000000499.
13. Andrilla CHA, Moore TE, Patterson DG, Larson EH. Geographic distribution of providers with a dea waiver to prescribe buprenorphine for the treatment of opioid use disorder: a 5-year update. J Rural Heal. 2019;35(1):108-112. https://doi.org/10.1111/jrh.12307.
14. Fiscella K, Wakeman SE, Beletsky L. Buprenorphine deregulation and mainstreaming treatment for opioid use disorder: x the x waiver. JAMA Psychiatry. 2019;76(3):229-230. https://doi.org/10.1001/jamapsychiatry.2018.3685.
15. American Medical Association Opioid Task Force. AMA Opioid Task Force recommendations offer roadmap to policymakers | American Medical Association. https://www.ama-assn.org/press-center/press-releases/ama-opioid-task-force-recommendations-offer-roadmap-policymakers. Accessed June 14, 2019.
16. Tonko P. H.R.2482: Mainstreaming Addiction Treatment Act of 2019. House Of Representatives (116th Congress); 2019. https://www.congress.gov/bill/116th-congress/house-bill/2482. Accessed July 10, 2019.
17. Frank JW, Wakeman SE, Gordon AJ. No end to the crisis without an end to the waiver. Subst Abus. 2018;39(3):263-265. https://doi.org/10.1080/08897077.2018.1543382
18. Society of Hospital Medicine. Clinical Topics: Opioid Safety. https://www.hospitalmedicine.org/clinical-topics/opioid-safety/. Accessed October 24, 2019.
19. Lowenstein M, Kilaru A, Perrone J, et al. Barriers and facilitators for emergency department initiation of buprenorphine: a physician survey. Am J Emerg Med. 2019;37(9):1787-1790. https://doi.org/10.1016/j.ajem.2019.02.025.
20. Shanahan CW, Beers D, Alford DP, Brigandi E, Samet JH. A transitional opioid program to engage hospitalized drug users. J Gen Intern Med. 2010;25(8):803-808. https://doi.org/10.1007/s11606-010-1311-3.
THE PROBLEM AND THE ROLE OF THE HOSPITALIST
Opioid use disorder (OUD) is a common, underrecognized, undertreated, and deadly medical condition. Although the focus of addressing the opioid epidemic has been centered in the outpatient setting, hospitalists play an important—and often underutilized—role in identifying OUD, initiating treatment, and assisting with linkage to longitudinal care after discharge.
Over the past 20 years, the annual rate of hospital discharges documenting OUD has quadrupled.1 During 2010-2016, the annual discharge rate for heroin overdoses increased by 23%.1 Although the total number of hospitalizations in the United States remained stable from 2002 to 2012, the number of admissions for opioid abuse or dependence increased from 301,707 to 520,275. More than 500,000 hospital admissions per year (1% of total nationwide hospitalizations) are now due primarily to OUD.2
Injection opioid use increases the risk of endocarditis, osteomyelitis, septic arthritis, and epidural abscesses, conditions that often prolong hospitalizations and frequently lead to readmissions. Admissions for OUD-related infections are rising at a startling rate. Between 2002 and 2012, the number of admissions for infections associated with OUDs had increased from 3,421 to 6,535.2 In addition to providing the opportunity to diagnose OUD, hospitalizations offer an ideal time to engage patients in OUD treatment and linkage to outpatient care.
Although we uniformly offer patients antibiotic treatment for acute infection, hospitalists should consistently incorporate treatment of OUD to address the root cause of these admissions. As infection is but one sequelae of the underlying disease of addiction, treating without medications for OUD (MOUD) would be akin to treating a diabetic foot ulcer with antibiotics and not providing medications to improve glycemic control. Omitting such addiction treatment can contribute to treatment failure and worse health outcomes. Among patients with endocarditis and an associated valve repair, those who continue injection drug use have a 10 times higher risk of death or reoperation between 90 and 180 days after repair than those not engaged in drug use.3
Despite data demonstrating the significant benefit and the minimal harm of MOUD, significant gaps remain in providing MOUD and linking patients from the hospital to community care.1,4 Hospital encounters are missed opportunities to provide life-saving MOUD treatment; the majority of patients with OUD do not receive evidence-based treatment while inpatient.5 Rosenthal et al. found that of 102 patients admitted with injection drug use-associated infective endocarditis from 2004 to 2014, only 8% received MOUD, and approximately half had a documentation of substance use treatment in their discharge worksheet.4 In Massachusetts, among individuals who experienced a nonfatal opioid overdose and had interaction with healthcare services, only 26% were on MOUD one year later.6 Based on our experience, a substantial proportion of patients with OUD do not seek or have access to medical care, acute care settings offer a critical opportunity to engage them in treatment for their addiction.
WHY SHOULD HOSPITALISTS INITIATE BUPRENORPHINE?
First, buprenorphine effectively treats withdrawal symptoms. Buprenorphine and methadone are superior to other medications in treating symptoms of withdrawal.7 If withdrawal symptoms are treated, patients are less likely to leave against medical advice8 and are more likely to complete treatment.
Second, MOUD is the standard of care for treating OUD.9 Medications include the full agonist methadone, the partial agonist buprenorphine, and the long-acting antagonist naltrexone. Although all these drugs are effective and legal to initiate for inpatients,6 this perspective focuses on buprenorphine in an effort to draw attention to associated policy barriers. Buprenorphine is the only MOUD that can be offered as office-based therapy by providers in the outpatient setting. Meta-analyses show that MOUD is associated with lower rates of mortality, illicit opioid use, HIV transmission, and violent crime and arrest.9
Third, MOUD treatment, rather than just referral, leads to higher long-term treatment success.10 When initiating buprenorphine in the hospital, treatment retention rates at one month were double that of referral alone. Six months after discharge, patients were five times more likely to remain engaged in treatment compared with those who received a detoxification protocol only.
Fourth, buprenorphine is not only effective, but it is also safe and has low risks of misuse. Because buprenorphine is a partial agonist, it has both a ceiling effect on respiratory depression (decreasing potential lethality) and on euphoria (decreasing the likelihood of misuse). Among individuals who took nonprescribed buprenorphine on the street, less than 7% reported taking it for any attempt at euphoria. Instead, people with OUD most often use nonprescribed or diverted buprenorphine to treat withdrawal symptoms.11
Fifth, buprenorphine treatment is associated with fewer hospital readmissions.12
Finally, initiating OUD treatment is feasible in the hospital setting. Any hospitalist can legally prescribe buprenorphine to treat opioid withdrawal for hospitalized patients admitted for medical or surgical reasons. A waiver is necessary only for prescribing at the time of hospital discharge for use in non-inpatient settings of care.
A POLICY BARRIER: THE X WAIVER
The United States Congress passed the Drug Addiction Treatment Act (DATA) of 2000, which codified the X waiver, in response to the growing opioid crisis. Only those providers with the DATA X waiver can write buprenorphine prescriptions to be filled in an outpatient pharmacy. To obtain an X waiver, physicians must complete an 8-hour course, whereas physician assistants and nurse practitioners must complete a 24-hour course. This training far exceeds any required training to prescribe opioids for pain.
Unfortunately, the X waiver requirement obstructs hospitalists from initiating buprenorphine in the inpatient setting in the following ways: (1) hospitalists often choose not to initiate chronic buprenorphine treatment if they lack the X waiver that would allow them to write the discharge prescription and/or (2) they are unable to identify a waivered provider in the community to continue the prescription. Unfortunately, only 6% of all medical practitioners are waivered to prescribe buprenorphine; greater than 40% of US counties are “buprenorphine deserts,” with no providers waivered to prescribe buprenorphine.13
To address the opioid crisis, we must rethink our current policies. The Department of Health and Human Services should eliminate the X waiver and allow any licensed physician, nurse practitioner, or physician assistant to prescribe buprenorphine.14 Recent American Medical Association Opioid Task Force recommendations have called to “remove… inappropriate administrative burdens or barriers that delay or deny care for FDA-approved medications used as part of medication-assisted treatment for OUD.”15 Legislation to remove the X wavier has been proposed in the United States.16
The removal of a buprenorphine waiver requirement has had success in other settings. The French deregulation of buprenorphine was associated with a reduction in opioid overdose deaths by 79%. Similar success in the United States would save an estimated 30,000 lives yearly.14 Removing the X waiver is an important step in empowering hospitalists to initiate MOUD for individuals in the hospital setting. Moreover, it opens the door to more outpatient primary care providers serving as community linkages for long-term addiction care.
NOT A PANACEA
Without the X waiver, the associated OUD training will no longer be required. This could have unintended consequences. For example, if hospitalists order buprenorphine while opioids remain active, precipitated withdrawal may ensue. Crucially, the current literature does not indicate that the required X waiver training improves knowledge, patient care, or outcomes.17 Nevertheless, MOUD and addiction training may help reduce knowledge gaps and empower providers to engage in productive conversations surrounding addiction. This highlights the crucial role of physician organizations, such as the Society of Hospital Medicine, in educating hospitalists about MOUD. (This organization, among others, has developed robust MOUD training.18)
It is also important to acknowledge that the waiver is only one obstacle. Other barriers have been identified in initiating buprenorphine, including access to treatment after discharge, access to social work support, and lack of EMR order sets, among others.19 Professional societies, hospitals, and hospitalists need to help address these barriers through ancillary support staff, quality improvement initiatives, and improved inpatient treatment of withdrawal with MOUD. This can be done successfully; one study found that 82% of hospitalized patients who engaged in a new transitional opioid program subsequently presented to outpatient opioid treatment.20 Novel interventions must be part of a hospital-wide approach to optimizing improved longitudinal treatment for patients suffering from addiction.
CONCLUSION
Hospitalization is an ideal opportunity for clinicians to diagnose and treat OUD in a population that often has not sought, or has fallen out of, addiction treatment. Hospitalists can and should initiate buprenorphine in appropriate inpatients and plan for their transition to chronic care. Eliminating the waiver in combination with designing innovative educational opportunities and systems approaches to provide better linkages to outpatient OUD treatment is needed to combat the opioid crisis. To enable more hospitalists to successfully initiate long-term buprenorphine therapy—and to enable more outpatient providers to continue prescriptions—we must eliminate the X waiver.
Disclosures
Dr. Wilson received honorarium from the American Society of Addiction Medicine for teaching and creating
THE PROBLEM AND THE ROLE OF THE HOSPITALIST
Opioid use disorder (OUD) is a common, underrecognized, undertreated, and deadly medical condition. Although the focus of addressing the opioid epidemic has been centered in the outpatient setting, hospitalists play an important—and often underutilized—role in identifying OUD, initiating treatment, and assisting with linkage to longitudinal care after discharge.
Over the past 20 years, the annual rate of hospital discharges documenting OUD has quadrupled.1 During 2010-2016, the annual discharge rate for heroin overdoses increased by 23%.1 Although the total number of hospitalizations in the United States remained stable from 2002 to 2012, the number of admissions for opioid abuse or dependence increased from 301,707 to 520,275. More than 500,000 hospital admissions per year (1% of total nationwide hospitalizations) are now due primarily to OUD.2
Injection opioid use increases the risk of endocarditis, osteomyelitis, septic arthritis, and epidural abscesses, conditions that often prolong hospitalizations and frequently lead to readmissions. Admissions for OUD-related infections are rising at a startling rate. Between 2002 and 2012, the number of admissions for infections associated with OUDs had increased from 3,421 to 6,535.2 In addition to providing the opportunity to diagnose OUD, hospitalizations offer an ideal time to engage patients in OUD treatment and linkage to outpatient care.
Although we uniformly offer patients antibiotic treatment for acute infection, hospitalists should consistently incorporate treatment of OUD to address the root cause of these admissions. As infection is but one sequelae of the underlying disease of addiction, treating without medications for OUD (MOUD) would be akin to treating a diabetic foot ulcer with antibiotics and not providing medications to improve glycemic control. Omitting such addiction treatment can contribute to treatment failure and worse health outcomes. Among patients with endocarditis and an associated valve repair, those who continue injection drug use have a 10 times higher risk of death or reoperation between 90 and 180 days after repair than those not engaged in drug use.3
Despite data demonstrating the significant benefit and the minimal harm of MOUD, significant gaps remain in providing MOUD and linking patients from the hospital to community care.1,4 Hospital encounters are missed opportunities to provide life-saving MOUD treatment; the majority of patients with OUD do not receive evidence-based treatment while inpatient.5 Rosenthal et al. found that of 102 patients admitted with injection drug use-associated infective endocarditis from 2004 to 2014, only 8% received MOUD, and approximately half had a documentation of substance use treatment in their discharge worksheet.4 In Massachusetts, among individuals who experienced a nonfatal opioid overdose and had interaction with healthcare services, only 26% were on MOUD one year later.6 Based on our experience, a substantial proportion of patients with OUD do not seek or have access to medical care, acute care settings offer a critical opportunity to engage them in treatment for their addiction.
WHY SHOULD HOSPITALISTS INITIATE BUPRENORPHINE?
First, buprenorphine effectively treats withdrawal symptoms. Buprenorphine and methadone are superior to other medications in treating symptoms of withdrawal.7 If withdrawal symptoms are treated, patients are less likely to leave against medical advice8 and are more likely to complete treatment.
Second, MOUD is the standard of care for treating OUD.9 Medications include the full agonist methadone, the partial agonist buprenorphine, and the long-acting antagonist naltrexone. Although all these drugs are effective and legal to initiate for inpatients,6 this perspective focuses on buprenorphine in an effort to draw attention to associated policy barriers. Buprenorphine is the only MOUD that can be offered as office-based therapy by providers in the outpatient setting. Meta-analyses show that MOUD is associated with lower rates of mortality, illicit opioid use, HIV transmission, and violent crime and arrest.9
Third, MOUD treatment, rather than just referral, leads to higher long-term treatment success.10 When initiating buprenorphine in the hospital, treatment retention rates at one month were double that of referral alone. Six months after discharge, patients were five times more likely to remain engaged in treatment compared with those who received a detoxification protocol only.
Fourth, buprenorphine is not only effective, but it is also safe and has low risks of misuse. Because buprenorphine is a partial agonist, it has both a ceiling effect on respiratory depression (decreasing potential lethality) and on euphoria (decreasing the likelihood of misuse). Among individuals who took nonprescribed buprenorphine on the street, less than 7% reported taking it for any attempt at euphoria. Instead, people with OUD most often use nonprescribed or diverted buprenorphine to treat withdrawal symptoms.11
Fifth, buprenorphine treatment is associated with fewer hospital readmissions.12
Finally, initiating OUD treatment is feasible in the hospital setting. Any hospitalist can legally prescribe buprenorphine to treat opioid withdrawal for hospitalized patients admitted for medical or surgical reasons. A waiver is necessary only for prescribing at the time of hospital discharge for use in non-inpatient settings of care.
A POLICY BARRIER: THE X WAIVER
The United States Congress passed the Drug Addiction Treatment Act (DATA) of 2000, which codified the X waiver, in response to the growing opioid crisis. Only those providers with the DATA X waiver can write buprenorphine prescriptions to be filled in an outpatient pharmacy. To obtain an X waiver, physicians must complete an 8-hour course, whereas physician assistants and nurse practitioners must complete a 24-hour course. This training far exceeds any required training to prescribe opioids for pain.
Unfortunately, the X waiver requirement obstructs hospitalists from initiating buprenorphine in the inpatient setting in the following ways: (1) hospitalists often choose not to initiate chronic buprenorphine treatment if they lack the X waiver that would allow them to write the discharge prescription and/or (2) they are unable to identify a waivered provider in the community to continue the prescription. Unfortunately, only 6% of all medical practitioners are waivered to prescribe buprenorphine; greater than 40% of US counties are “buprenorphine deserts,” with no providers waivered to prescribe buprenorphine.13
To address the opioid crisis, we must rethink our current policies. The Department of Health and Human Services should eliminate the X waiver and allow any licensed physician, nurse practitioner, or physician assistant to prescribe buprenorphine.14 Recent American Medical Association Opioid Task Force recommendations have called to “remove… inappropriate administrative burdens or barriers that delay or deny care for FDA-approved medications used as part of medication-assisted treatment for OUD.”15 Legislation to remove the X wavier has been proposed in the United States.16
The removal of a buprenorphine waiver requirement has had success in other settings. The French deregulation of buprenorphine was associated with a reduction in opioid overdose deaths by 79%. Similar success in the United States would save an estimated 30,000 lives yearly.14 Removing the X waiver is an important step in empowering hospitalists to initiate MOUD for individuals in the hospital setting. Moreover, it opens the door to more outpatient primary care providers serving as community linkages for long-term addiction care.
NOT A PANACEA
Without the X waiver, the associated OUD training will no longer be required. This could have unintended consequences. For example, if hospitalists order buprenorphine while opioids remain active, precipitated withdrawal may ensue. Crucially, the current literature does not indicate that the required X waiver training improves knowledge, patient care, or outcomes.17 Nevertheless, MOUD and addiction training may help reduce knowledge gaps and empower providers to engage in productive conversations surrounding addiction. This highlights the crucial role of physician organizations, such as the Society of Hospital Medicine, in educating hospitalists about MOUD. (This organization, among others, has developed robust MOUD training.18)
It is also important to acknowledge that the waiver is only one obstacle. Other barriers have been identified in initiating buprenorphine, including access to treatment after discharge, access to social work support, and lack of EMR order sets, among others.19 Professional societies, hospitals, and hospitalists need to help address these barriers through ancillary support staff, quality improvement initiatives, and improved inpatient treatment of withdrawal with MOUD. This can be done successfully; one study found that 82% of hospitalized patients who engaged in a new transitional opioid program subsequently presented to outpatient opioid treatment.20 Novel interventions must be part of a hospital-wide approach to optimizing improved longitudinal treatment for patients suffering from addiction.
CONCLUSION
Hospitalization is an ideal opportunity for clinicians to diagnose and treat OUD in a population that often has not sought, or has fallen out of, addiction treatment. Hospitalists can and should initiate buprenorphine in appropriate inpatients and plan for their transition to chronic care. Eliminating the waiver in combination with designing innovative educational opportunities and systems approaches to provide better linkages to outpatient OUD treatment is needed to combat the opioid crisis. To enable more hospitalists to successfully initiate long-term buprenorphine therapy—and to enable more outpatient providers to continue prescriptions—we must eliminate the X waiver.
Disclosures
Dr. Wilson received honorarium from the American Society of Addiction Medicine for teaching and creating
1. Peterson C, Xu L, Florence C, Mack KA. Opioid-related US hospital discharges by type, 1993–2016. J Subst Abuse Treat. 2019;103:9-13. https://doi.org/10.1016/j.jsat.2019.05.003.
2. Ronan MV, Herzig SJ. Hospitalizations related to opioid abuse/dependence and associated serious infections increased sharply, 2002-12. Health Aff. 2016;35(5):832-837. https://doi.org/10.1377/hlthaff.2015.1424.
3. Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg. 2015;100(3):875-882. https://doi.org/10.1016/j.athoracsur.2015.03.019.
4. Rosenthal ES, Karchmer AW, Theisen-Toupal J, Castillo RA, Rowley CF. Suboptimal addiction interventions for patients hospitalized with injection drug use-associated infective endocarditis. Am J Med. 2016;129(5):481-485. https://doi.org/10.1016/j.amjmed.2015.09.024.
5. Winetsky D, Weinrieb RM, Perrone J. Expanding treatment opportunities for hospitalized patients with opioid use disorders. J Hosp Med. 2017;13(1):62-64. https://doi.org/10.12788/jhm.2861.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Gowing L, Ali R, White JM, Mbewe D. Buprenorphine for managing opioid withdrawal. Cochrane Database Syst Rev. 2017;(2):CD002025. https://doi.org/10.1002/14651858.CD002025.pub5.
8. Ti L, Ti L. Leaving the hospital against medical advice among people who use illicit drugs: a systematic review. Am J Public Health. 2015;105(12):e53-e59. https://doi.org/10.2105/AJPH.2015.302885.
9. Schuckit MA. Treatment of opioid-use disorders. N Engl J Med. 2016;375(4):357-368. https://doi.org/10.1056/NEJMra1604339.
10. Liebschutz JM, Crooks D, Herman D, et al. Buprenorphine treatment for hospitalized, opioid-dependent patients. JAMA Intern Med. 2014;174(8):1369. https://doi.org/10.1001/jamainternmed.2014.2556.
11. Cicero TJ, Ellis MS, Surratt HL, Kurtz SP. Factors contributing to the rise of buprenorphine misuse: 2008-2013. Drug Alcohol Depend. 2014;142:98-104. https://doi.org/10.1016/j.drugalcdep.2014.06.005.
12. Moreno JL, Wakeman SE, Duprey MS, Roberts RJ, Jacobson JS, Devlin JW. Predictors for 30-day and 90-day hospital readmission among patients with opioid use disorder. J Addict Med. 2019;13(4):306-313. https://doi.org/10.1097/ADM.0000000000000499.
13. Andrilla CHA, Moore TE, Patterson DG, Larson EH. Geographic distribution of providers with a dea waiver to prescribe buprenorphine for the treatment of opioid use disorder: a 5-year update. J Rural Heal. 2019;35(1):108-112. https://doi.org/10.1111/jrh.12307.
14. Fiscella K, Wakeman SE, Beletsky L. Buprenorphine deregulation and mainstreaming treatment for opioid use disorder: x the x waiver. JAMA Psychiatry. 2019;76(3):229-230. https://doi.org/10.1001/jamapsychiatry.2018.3685.
15. American Medical Association Opioid Task Force. AMA Opioid Task Force recommendations offer roadmap to policymakers | American Medical Association. https://www.ama-assn.org/press-center/press-releases/ama-opioid-task-force-recommendations-offer-roadmap-policymakers. Accessed June 14, 2019.
16. Tonko P. H.R.2482: Mainstreaming Addiction Treatment Act of 2019. House Of Representatives (116th Congress); 2019. https://www.congress.gov/bill/116th-congress/house-bill/2482. Accessed July 10, 2019.
17. Frank JW, Wakeman SE, Gordon AJ. No end to the crisis without an end to the waiver. Subst Abus. 2018;39(3):263-265. https://doi.org/10.1080/08897077.2018.1543382
18. Society of Hospital Medicine. Clinical Topics: Opioid Safety. https://www.hospitalmedicine.org/clinical-topics/opioid-safety/. Accessed October 24, 2019.
19. Lowenstein M, Kilaru A, Perrone J, et al. Barriers and facilitators for emergency department initiation of buprenorphine: a physician survey. Am J Emerg Med. 2019;37(9):1787-1790. https://doi.org/10.1016/j.ajem.2019.02.025.
20. Shanahan CW, Beers D, Alford DP, Brigandi E, Samet JH. A transitional opioid program to engage hospitalized drug users. J Gen Intern Med. 2010;25(8):803-808. https://doi.org/10.1007/s11606-010-1311-3.
1. Peterson C, Xu L, Florence C, Mack KA. Opioid-related US hospital discharges by type, 1993–2016. J Subst Abuse Treat. 2019;103:9-13. https://doi.org/10.1016/j.jsat.2019.05.003.
2. Ronan MV, Herzig SJ. Hospitalizations related to opioid abuse/dependence and associated serious infections increased sharply, 2002-12. Health Aff. 2016;35(5):832-837. https://doi.org/10.1377/hlthaff.2015.1424.
3. Shrestha NK, Jue J, Hussain ST, et al. Injection drug use and outcomes after surgical intervention for infective endocarditis. Ann Thorac Surg. 2015;100(3):875-882. https://doi.org/10.1016/j.athoracsur.2015.03.019.
4. Rosenthal ES, Karchmer AW, Theisen-Toupal J, Castillo RA, Rowley CF. Suboptimal addiction interventions for patients hospitalized with injection drug use-associated infective endocarditis. Am J Med. 2016;129(5):481-485. https://doi.org/10.1016/j.amjmed.2015.09.024.
5. Winetsky D, Weinrieb RM, Perrone J. Expanding treatment opportunities for hospitalized patients with opioid use disorders. J Hosp Med. 2017;13(1):62-64. https://doi.org/10.12788/jhm.2861.
6. Larochelle MR, Bernson D, Land T, et al. Medication for opioid use disorder after nonfatal opioid overdose and association with mortality: a cohort study. Ann Intern Med. 2018;169(3):137-145. https://doi.org/10.7326/M17-3107.
7. Gowing L, Ali R, White JM, Mbewe D. Buprenorphine for managing opioid withdrawal. Cochrane Database Syst Rev. 2017;(2):CD002025. https://doi.org/10.1002/14651858.CD002025.pub5.
8. Ti L, Ti L. Leaving the hospital against medical advice among people who use illicit drugs: a systematic review. Am J Public Health. 2015;105(12):e53-e59. https://doi.org/10.2105/AJPH.2015.302885.
9. Schuckit MA. Treatment of opioid-use disorders. N Engl J Med. 2016;375(4):357-368. https://doi.org/10.1056/NEJMra1604339.
10. Liebschutz JM, Crooks D, Herman D, et al. Buprenorphine treatment for hospitalized, opioid-dependent patients. JAMA Intern Med. 2014;174(8):1369. https://doi.org/10.1001/jamainternmed.2014.2556.
11. Cicero TJ, Ellis MS, Surratt HL, Kurtz SP. Factors contributing to the rise of buprenorphine misuse: 2008-2013. Drug Alcohol Depend. 2014;142:98-104. https://doi.org/10.1016/j.drugalcdep.2014.06.005.
12. Moreno JL, Wakeman SE, Duprey MS, Roberts RJ, Jacobson JS, Devlin JW. Predictors for 30-day and 90-day hospital readmission among patients with opioid use disorder. J Addict Med. 2019;13(4):306-313. https://doi.org/10.1097/ADM.0000000000000499.
13. Andrilla CHA, Moore TE, Patterson DG, Larson EH. Geographic distribution of providers with a dea waiver to prescribe buprenorphine for the treatment of opioid use disorder: a 5-year update. J Rural Heal. 2019;35(1):108-112. https://doi.org/10.1111/jrh.12307.
14. Fiscella K, Wakeman SE, Beletsky L. Buprenorphine deregulation and mainstreaming treatment for opioid use disorder: x the x waiver. JAMA Psychiatry. 2019;76(3):229-230. https://doi.org/10.1001/jamapsychiatry.2018.3685.
15. American Medical Association Opioid Task Force. AMA Opioid Task Force recommendations offer roadmap to policymakers | American Medical Association. https://www.ama-assn.org/press-center/press-releases/ama-opioid-task-force-recommendations-offer-roadmap-policymakers. Accessed June 14, 2019.
16. Tonko P. H.R.2482: Mainstreaming Addiction Treatment Act of 2019. House Of Representatives (116th Congress); 2019. https://www.congress.gov/bill/116th-congress/house-bill/2482. Accessed July 10, 2019.
17. Frank JW, Wakeman SE, Gordon AJ. No end to the crisis without an end to the waiver. Subst Abus. 2018;39(3):263-265. https://doi.org/10.1080/08897077.2018.1543382
18. Society of Hospital Medicine. Clinical Topics: Opioid Safety. https://www.hospitalmedicine.org/clinical-topics/opioid-safety/. Accessed October 24, 2019.
19. Lowenstein M, Kilaru A, Perrone J, et al. Barriers and facilitators for emergency department initiation of buprenorphine: a physician survey. Am J Emerg Med. 2019;37(9):1787-1790. https://doi.org/10.1016/j.ajem.2019.02.025.
20. Shanahan CW, Beers D, Alford DP, Brigandi E, Samet JH. A transitional opioid program to engage hospitalized drug users. J Gen Intern Med. 2010;25(8):803-808. https://doi.org/10.1007/s11606-010-1311-3.
© 2019 Society of Hospital Medicine
Things We Do for No Reason™: Lumbar Punctures in Low-Risk Febrile Infants with Bronchiolitis

CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.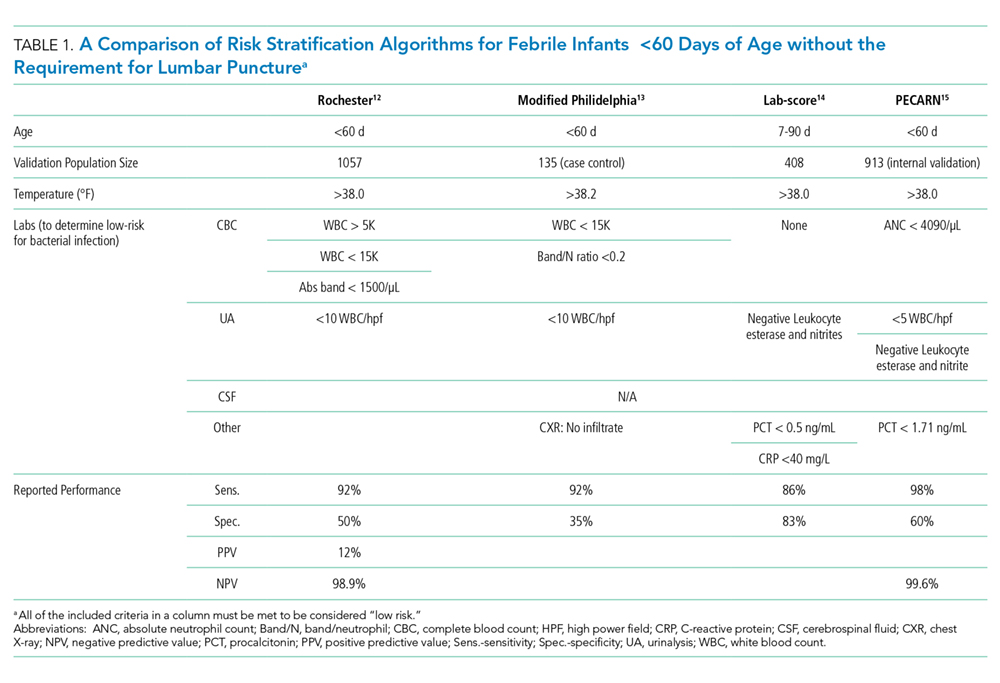
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing TWDFNR@hospitalmedicine.org.
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing TWDFNR@hospitalmedicine.org.
CLINICAL SCENARIO
A 22-day-old full-term previously healthy male infant was evaluated in the emergency department (ED). The patient’s mother reported a three-day history of nasal congestion, cough and labored breathing, decreased oral intake, and subjective fever.
In the ED, the patient was found to have a rectal temperature of 101.3 °F (38.3 °C), heart rate of 112 beats per minute, and a respiratory rate of 54 breaths per minute, with subcostal retractions and diffuse expiratory wheezing. His appearance was otherwise unremarkable. His evaluation in the ED included a normal complete blood count (CBC) with differential, a normal urinalysis, and a chest radiograph with diffuse peribronchial thickening. Blood and catheterized urine cultures were also collected. The patient’s provider informs the parents that a lumbar puncture (LP) would be performed to rule out bacterial meningitis. Is it necessary for this patient to receive an LP?
INTRODUCTION
Fever in an infant <90 days old is a common clinical presentation.1 Because a newborn’s immune system is still developing, there is a heightened concern for bacterial infections in this age group. These include bloodstream infections, meningitis, pneumonia, urinary tract infections (UTIs), skin/soft tissue infections, and osteoarticular infections. Bacterial infections collectively account for approximately 10% of illness in young febrile infants <90 days.2 Of these, UTIs are the most common. The most recent literature has narrowed the focus on infants <60 days old as the risk of serious infection is inversely correlated with age. Meningitis accounts for 1% of infections or less in children <60 days of age who present with a fever.3
Frequently, the evaluation of fever in young infants leads to cerebrospinal fluid (CSF) collection and hospitalization.4 Among febrile infants, current practice patterns regarding LPs vary across institutions.5 Some clinical practice guidelines recommend universal CSF testing for all febrile infants ≤56 days old.6
Bronchiolitis is also a common presentation. Up to 90% of children are infected with respiratory syncytial virus, the most common viral cause of bronchiolitis, within the first two years of life.7 Fever may be a presenting symptom in infants with bronchiolitis and one study found approximately 11% of febrile infants less than 90 days old met clinical criteria for bronchiolitis.8
WHY YOU MIGHT THINK LUMBAR PUNCTURE IN FEBRILE INFANTS WITH BRONCHIOLITIS IS HELPFUL
While clinical guidelines for bronchiolitis are well established,7 the evaluation and management of fever in an infant <90 days old remains a challenge because of concern for missing a bloodstream infection or meningitis. Meningitis can devastate an infant neurologically.9 Signs and symptoms of bacterial meningitis in infants are not specific, including the physical exam.10 Blood cultures are only concomitantly positive in 62% of cases of culture-confirmed bacterial meningitis.11
Several risk stratification algorithms exist to evaluate the likelihood of bacterial infections in febrile infants (Table). Two of the most common criteria—the Boston and Philadelphia—were validated using CSF cell count data. Other algorithms do not require an LP.12-15 All of the fever criteria algorithms have several limitations including lack of robust validation studies, under-powered methodologies (particularly for meningitis), and different inclusion criteria.2 Even with these risk stratification algorithms, some providers may continue to feel more comfortable obtaining CSF due to fear of missing meningitis in well-appearing, low-risk infants.
WHY LUMBAR PUNCTURE IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS IS NOT NECESSARY
Bacterial meningitis, even in young infants, is rare. A recent meta-analysis estimated the general prevalence of meningitis in febrile neonates (regardless of risk stratification or bronchiolitis symptoms) in their first and second months of life were 1.2% (95% CI, 0.8%-1.9%) and 0.4% (95% CI, 0.2%-1.0%), respectively.3
Febrile infant risk stratification algorithms have high negative predictive values (NPVs) in ruling out meningitis. The Rochester criteria, which does not utilize CSF, has an NPV of greater than 98%.12 A recent Pediatric Emergency Care Applied Research Network Clinical Prediction Rule has an NPV of 99.9% among febrile infants <60 days, using only absolute neutrophil count, urinalysis, and procalcitonin.15
Among the patients that are already a low risk, concomitant viral infections further decrease the pretest probability. Febrile infants with lab-confirmed respiratory viral infections are at lower risk for serious bacterial infections.16,17 Multiple retrospective and prospective observational studies have demonstrated that low-risk patients with bronchiolitis symptoms are extremely unlikely to have bacterial meningitis.8,18-22 A systematic review of 1749 febrile patients under 90 days of age with clinical bronchiolitis demonstrated no cases of meningitis.23 Many of these studies included infants aged <28 days. Though the total number of neonates (<28 days) in all studies is somewhat unclear, it suggests that the cut-off to avoid an LP may be even lower.
Recent literature has advocated outpatient observation without an LP for low-risk infants as a cost-effective management tool,24 and this is particularly true in patients with concomitant viral bronchiolitis.
Based on the latest data confirming the low prevalence of meningitis among all infants,3 the ability to identify low-risk infants based on risk stratification algorithms (Table), and the decreased prevalence of meningitis in patients with clinical bronchiolitis,23 low-risk infants with bronchiolitis seem to have minimal, if any, risk of meningitis. Therefore, low-risk infants with bronchiolitis do not warrant an LP.
Importantly, LPs are not risk neutral. Their benefit versus harm should be weighed every time they are considered. Approximately 19% of LP attempts in infants under 90 days old are either traumatic or unsuccessful.25 Infants aged 28 to 60 days with traumatic or unsuccessful LPs are more frequently hospitalized.25 Increased hospitalizations are associated with higher costs.4 The majority of positive CSF cultures are deemed to be “contaminants” (87% in one study26), but the positive result still leads to unnecessary further evaluation, hospitalization, repeated invasive procedures, and family distress.27 These data further support refraining from pursuing an LP in low-risk infants with bronchiolitis.
WHY LUMBAR PUNCTURE MIGHT BE HELPFUL IN CERTAIN CIRCUMSTANCES
If the patient is not low risk based on criteria or does not have clinical bronchiolitis, consider performing an LP. A recent study demonstrated a 0.4% incidence of bacterial meningitis in febrile infants with viral co-infection,29 though it is not determined if the patients presented with symptoms of bronchiolitis or were risk-stratified using the algorithms discussed.
In the studies looking at viral infections in febrile infants, each has important exclusion criteria including prematurity, comorbidities, and recent antibiotic administration.23 For these patients, an LP may be warranted (though the evidence is lacking). In addition, in very young infants (less than seven-14 days old), viral infections may be less common than in older infants, resulting in a desire to rule out bacterial infections more thoroughly in this population.
WHAT YOU SHOULD DO INSTEAD: AVOID AN LP IN LOW-RISK FEBRILE INFANTS WITH BRONCHIOLITIS
For low-risk febrile infants with signs of bronchiolitis, evaluation for bacterial meningitis is not necessary. The low prevalence of meningitis in this age range along with the even lower likelihood of meningitis when bronchiolitis is identified suggests that the procedure is unnecessary. Moreover, the risks associated with LP—including trauma, hospitalization, costs, and family stress—likely outweigh the benefits of CSF analysis.
RECOMMENDATIONS
- In febrile infants, determine the risk of serious bacterial infections using published algorithms (Table) before considering lumbar puncture.
- In low-risk febrile infants with typical bronchiolitis, evaluation for bacterial meningitis with an LP is not necessary.
CONCLUSION
Infants under 90 days of age often present to care with fever. While there is a concern for missing bacterial meningitis, the prevalence of such an infection in infants is very low. Moreover, in low-risk patients that present with typical bronchiolitis symptoms, the prevalence is effectively zero. LP practices vary by institution and can be associated with risks. In low-risk infants with typical bronchiolitis symptoms, an LP is one of the Things We Do for No Reason™.
Do you think this is a low-value practice? Is this truly a “Thing We Do for No Reason™”? Share what you do in your practice and join in the conversation online by retweeting it on Twitter (#TWDFNR) and liking it on Facebook. We invite you to propose ideas for other “Things We Do for No Reason™” topics by emailing TWDFNR@hospitalmedicine.org.
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
1. Cioffredi L-A, Jhaveri R. Evaluation and management of febrile children. JAMA Pediatr. 2016;170(8):794. https://doi.org/10.1001/jamapediatrics.2016.0596.
2. Huppler AR, Eickhoff JC, Wald ER. Performance of low-risk criteria in the evaluation of young infants with fever: review of the literature. Pediatrics. 2010;125(2):228-233. https://doi.org/10.1542/peds.2009-1070.
3. Biondi EA, Lee B, Ralston SL, et al. Prevalence of bacteremia and bacterial meningitis in febrile neonates and infants in the second month of life a systematic review and meta-analysis + supplemental content. JAMA Netw Open. 2019;2(3):190874. https://doi.org/10.1001/jamanetworkopen.2019.0874.
4. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
5. Aronson PL, Thurm C, Alpern ER, et al. Variation in care of the febrile young infant <90 days in us pediatric emergency departments. Pediatrics. 2014;134(4):667-677. https://doi.org/10.1542/peds.2014-1382.
6. Aronson PL, Thurm C, Williams DJ, et al. Association of clinical practice guidelines with emergency department management of febrile infants ≤56 days of age. J Hosp Med. 2015;10(6):358-365. https://doi.org/10.1002/jhm.2329.
7. Mendonca EA, Meissner HC, Gadomski AM, et al. Clinical practice guideline: the diagnosis, management, and prevention of bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. https://doi.org/10.1542/peds.2014-2742.
8. Melendez E, Harper MB. Utility of sepsis evaluation in infants 90 days of age or younger with fever and clinical bronchiolitis. Pediatr Infect Dis J. 2003;22(12):1053-1056. https://doi.org/10.1097/01.inf.0000101296.68993.4d.
9. Pruitt CM, Neuman MI, Shah SS, et al. Factors associated with adverse outcomes among febrile young infants with invasive bacterial infections. J. Pediatr. 2018;204:177-182. https://doi.org/10.1016/j.jpeds.2018.08.066.
10. Casper TC, Mahajan PV., Tzimenatos L, et al. The Yale Observation Scale Score and the risk of serious bacterial infections in febrile infants. Pediatrics. 2017;140(1):e20170695. https://doi.org/10.1542/peds.2017-0695.
11. Garges HP. Neonatal meningitis: what is the correlation among cerebrospinal fluid cultures, blood cultures, and cerebrospinal fluid parameters? Pediatrics. 2006;117(4):1094-1100. https://doi.org/10.1542/peds.2005-1132.
12. Jaskiewicz JA, McCarthy CA, Richardson AC, et al. Febrile infants at low risk for serious bacterial infection-an appraisal of the Rochester criteria and implications for management. Pediatrics. 1994;94(3):390-396. http://www.ncbi.nlm.nih.gov/pubmed/8065869. Accessed March 23, 2019.
13. Aronson P, Wang M, Shapiro E, et al. Risk stratification of febrile infants ≤60 days old without routine lumbar puncture. Pediatrics. 2018;142(6):e20181879. https://doi.org/10.1542/peds.2018-1879.
14. Galetto-Lacour A, Zamora SA, Andreola B, et al. Validation of a laboratory risk index score for the identification of severe bacterial infection in children with fever without source. Arch Dis Child. 2010;95(12):968-973. https://doi.org/10.1136/adc.2009.176800.
15. Kuppermann N, Dayan PS, Levine DA, et al. A clinical prediction rule to identify febrile infants 60 days and younger at low risk for serious bacterial infections. JAMA Pediatr. 2019;173(4):342. https://doi.org/10.1001/jamapediatrics.2018.5501.
16. Byington CL, Enriquez FR, Hoff C, et al. Serious bacterial infections in febrile infants 1 to 90 days old with and without viral infections. Pediatrics. 2004;113(6):1662-1666. https://doi.org/10.1542/peds.113.6.1662.
17. Cioffredi LA, Jhaveri R. Evaluation and management of febrile children: a review. JAMA Pediatr. 2016;170(8):794-800. https://doi.org/10.1001/jamapediatrics.2016.0596.
18. Dayan PS, Roskind CG, Levine DA, Kuppermann N. Controversies in the management of children with bronchiolitis. Clin Pediatr Emerg Med. 2004;5(1):41-53. https://doi.org/10.1016/j.cpem.2003.11.001.
19. Oray-Schrom P, Phoenix C, St. Martin D, Amoateng-Adjepong Y. Sepsis workup in febrile infants 0-90 days of age with respiratory syncytial virus infection. Pediatr Emerg Care. 2003;19(5):314-319. https://doi.org/10.1097/01.pec.0000092576.40174.28.
20. Purcell K, Fergie J. Concurrent serious bacterial infections in 2396 infants and children hospitalized with respiratory syncytial virus lower respiratory tract infections. Arch Pediatr Adolesc Med. 2002;156(4):322-324. https://doi.org/10.1001/archpedi.156.4.322.
21. Purcell K, Fergie J. Concurrent serious bacterial infections in 912 infants and children hospitalized for treatment of respiratory syncytial virus lower respiratory tract infection. Pediatr Infect Dis J. 2004;23(3):267-269. https://doi.org/10.1097/01.inf.0000116759.21252.29.
22. Yarden-Bilavsky H, Ashkenazi-Hoffnung L, Livni G, Amir J, Bilavsky E. Month-by-month age analysis of the risk for serious bacterial infections in febrile infants with bronchiolitis. Clin Pediatr (Phila). 2011;50(11):1052-1056. https://doi.org/10.1177/0009922811412949.
23. Ralston S, Hill V, Waters A. Occult serious bacterial infection in infants younger than 60 to 90 days with bronchiolitis: a systematic review. Arch Pediatr Adolesc Med. 2011;165(10):951-956. https://doi.org/10.1001/archpediatrics.2011.155.
24. Lee TJ, Aronson PL. To spinal tap or not to spinal tap, that is the question. Hosp Pediatr. 2018;8(4):236-238. https://doi.org/10.1542/hpeds.2017-0207.
25. Pingree EW, Kimia AA, Nigrovic LE. The effect of traumatic lumbar puncture on hospitalization rate for febrile infants 28 to 60 days of age. Acad Emerg Med. 2015;22(2):240-243. https://doi.org/10.1111/acem.12582.
26. Leazer R, Erickson N, Paulson J, et al. epidemiology of cerebrospinal fluid cultures and time to detection in term infants. Pediatrics. 2017;139(5):e20163268. https://doi.org/10.1542/peds.2016-3268.
27. Paxton RD, Byington CL. An examination of the unintended consequences of the rule-out sepsis evaluation: a parental perspective. Clin Pediatr (Phila). 2001;40(2):71-77. https://doi.org/10.1177/000992280104000202.
28. Mahajan P, Br owne LR, Levine DA, et al. Risk of bacterial coinfections in febrile infants 60 days old and younger with documented viral infections. J Pediatr. 2018;203:86-91.e2. https://doi.org/10.1016/j.jpeds.2018.07.073.
© 2019 Society of Hospital Medicine
The Basement Flight
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.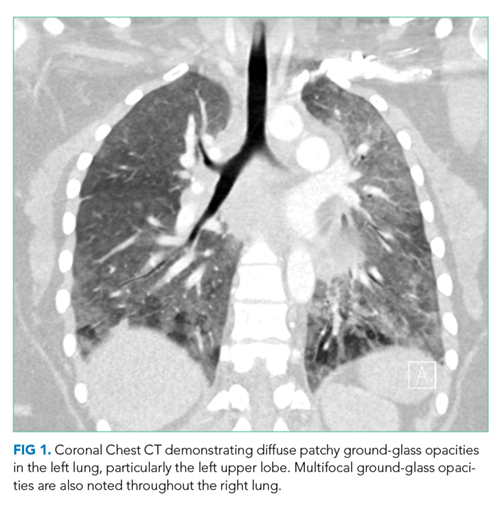
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).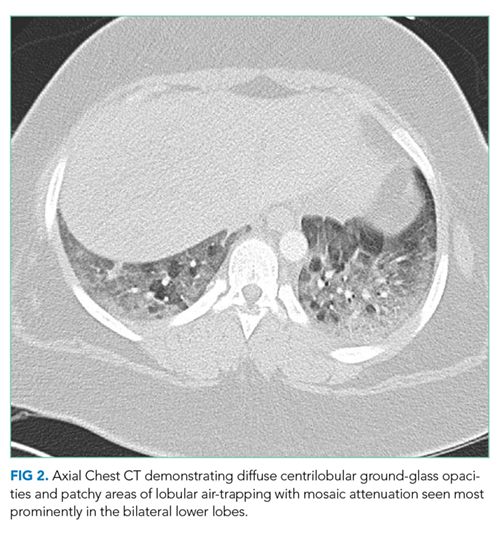
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.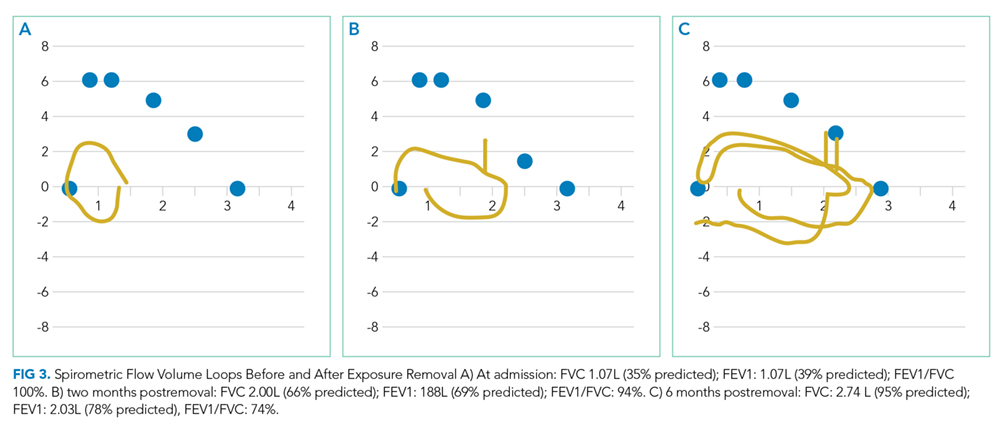
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
© 2019 Society of Hospital Medicine