User login
Christopher Palmer has been an associate editor at MDedge News since 2017. When he's not tidying grammar, he writes short pieces about breaking FDA announcements and approvals, as well as journal articles. He proudly holds a BA in English and philosophy. Follow him on Twitter @cmacmpalm.
Some HCV medications associated with serious liver injury

Many of the affected patients had signs or symptoms of moderate to severe liver impairment (Child-Pugh class B or C), and given that these medications – glecaprevir/pibrentasvir (Mavyret), elbasvir/grazoprevir (Zepatier), and sofosbuvir/velpatasvir/voxilaprevir (Vosevi) – are not indicated for such patients, they should not have been prescribed in the first place, the FDA noted in the drug safety communication. Some cases had other preexisting risk factors, such as liver cancer, alcohol abuse, or serious medical illnesses associated with liver problems.
In most cases, impairment or decompensation occurred within the first 4 weeks of starting treatment, and symptoms resolved or new-onset worsening of liver function improved after stopping. These medicines have been widely used and, among patients with no or mild liver impairment, have been shown to be safe and effective.
Health care professionals should continue prescribing these medicines as indicated; they should assess patients at baseline for severity of liver disease and other risk factors and closely monitor these patients after for signs and symptoms of worsening liver function. Patients should be aware that the risk of injury is rare and continue taking prescribed medicines; if they develop fatigue, weakness, loss of appetite, nausea and vomiting, yellow eyes or skin, or light-colored stools, they should talk with their health care professional but should continue taking the medications in question until instructed to do otherwise.
The full communication is available on the FDA website and includes more facts about these drugs and information for patients and health care professionals.
Many of the affected patients had signs or symptoms of moderate to severe liver impairment (Child-Pugh class B or C), and given that these medications – glecaprevir/pibrentasvir (Mavyret), elbasvir/grazoprevir (Zepatier), and sofosbuvir/velpatasvir/voxilaprevir (Vosevi) – are not indicated for such patients, they should not have been prescribed in the first place, the FDA noted in the drug safety communication. Some cases had other preexisting risk factors, such as liver cancer, alcohol abuse, or serious medical illnesses associated with liver problems.
In most cases, impairment or decompensation occurred within the first 4 weeks of starting treatment, and symptoms resolved or new-onset worsening of liver function improved after stopping. These medicines have been widely used and, among patients with no or mild liver impairment, have been shown to be safe and effective.
Health care professionals should continue prescribing these medicines as indicated; they should assess patients at baseline for severity of liver disease and other risk factors and closely monitor these patients after for signs and symptoms of worsening liver function. Patients should be aware that the risk of injury is rare and continue taking prescribed medicines; if they develop fatigue, weakness, loss of appetite, nausea and vomiting, yellow eyes or skin, or light-colored stools, they should talk with their health care professional but should continue taking the medications in question until instructed to do otherwise.
The full communication is available on the FDA website and includes more facts about these drugs and information for patients and health care professionals.
Many of the affected patients had signs or symptoms of moderate to severe liver impairment (Child-Pugh class B or C), and given that these medications – glecaprevir/pibrentasvir (Mavyret), elbasvir/grazoprevir (Zepatier), and sofosbuvir/velpatasvir/voxilaprevir (Vosevi) – are not indicated for such patients, they should not have been prescribed in the first place, the FDA noted in the drug safety communication. Some cases had other preexisting risk factors, such as liver cancer, alcohol abuse, or serious medical illnesses associated with liver problems.
In most cases, impairment or decompensation occurred within the first 4 weeks of starting treatment, and symptoms resolved or new-onset worsening of liver function improved after stopping. These medicines have been widely used and, among patients with no or mild liver impairment, have been shown to be safe and effective.
Health care professionals should continue prescribing these medicines as indicated; they should assess patients at baseline for severity of liver disease and other risk factors and closely monitor these patients after for signs and symptoms of worsening liver function. Patients should be aware that the risk of injury is rare and continue taking prescribed medicines; if they develop fatigue, weakness, loss of appetite, nausea and vomiting, yellow eyes or skin, or light-colored stools, they should talk with their health care professional but should continue taking the medications in question until instructed to do otherwise.
The full communication is available on the FDA website and includes more facts about these drugs and information for patients and health care professionals.
Farxiga gets Fast Track status from FDA
The Food and Drug Administration has given Fast Track designation to the development of dapagliflozin (Farxiga) to delay progression of renal failure and to prevent cardiovascular and renal death in patients with chronic kidney disease with and without type 2 diabetes, according to a release from AstraZeneca.
The Fast Track designation is meant to accelerate the development and review process for the treatment of serious conditions that have unmet therapeutic needs.
Dapagliflozin, an oral daily sodium-glucose transporter 2 inhibitor, is approved both as a monotherapy and a component of combination therapy for the improvement of glycemic control in patients with type 2 diabetes, according to the release. It is given as an adjunct to diet and exercise, and has also shown additional benefits of weight loss and reduction in blood pressure.
A phase 3, randomized, placebo-controlled trial, DAPA-CVD (NCT03036150), is currently underway to evaluate the drug’s efficacy specifically in terms of renal outcomes and cardiovascular mortality in patients with chronic kidney disease, with and without type 2 diabetes. Participants receive once-daily dapagliflozin or placebo in addition to standard care.
Taking dapagliflozin carries risks of hypotension, renal impairment, hypoglycemia, and other concerns. The most common adverse reactions (5% or greater incidence) include female genital mycotic infections, nasopharyngitis, and urinary tract infections. Full prescribing information can be found on the agency’s website.
The Food and Drug Administration has given Fast Track designation to the development of dapagliflozin (Farxiga) to delay progression of renal failure and to prevent cardiovascular and renal death in patients with chronic kidney disease with and without type 2 diabetes, according to a release from AstraZeneca.
The Fast Track designation is meant to accelerate the development and review process for the treatment of serious conditions that have unmet therapeutic needs.
Dapagliflozin, an oral daily sodium-glucose transporter 2 inhibitor, is approved both as a monotherapy and a component of combination therapy for the improvement of glycemic control in patients with type 2 diabetes, according to the release. It is given as an adjunct to diet and exercise, and has also shown additional benefits of weight loss and reduction in blood pressure.
A phase 3, randomized, placebo-controlled trial, DAPA-CVD (NCT03036150), is currently underway to evaluate the drug’s efficacy specifically in terms of renal outcomes and cardiovascular mortality in patients with chronic kidney disease, with and without type 2 diabetes. Participants receive once-daily dapagliflozin or placebo in addition to standard care.
Taking dapagliflozin carries risks of hypotension, renal impairment, hypoglycemia, and other concerns. The most common adverse reactions (5% or greater incidence) include female genital mycotic infections, nasopharyngitis, and urinary tract infections. Full prescribing information can be found on the agency’s website.
The Food and Drug Administration has given Fast Track designation to the development of dapagliflozin (Farxiga) to delay progression of renal failure and to prevent cardiovascular and renal death in patients with chronic kidney disease with and without type 2 diabetes, according to a release from AstraZeneca.
The Fast Track designation is meant to accelerate the development and review process for the treatment of serious conditions that have unmet therapeutic needs.
Dapagliflozin, an oral daily sodium-glucose transporter 2 inhibitor, is approved both as a monotherapy and a component of combination therapy for the improvement of glycemic control in patients with type 2 diabetes, according to the release. It is given as an adjunct to diet and exercise, and has also shown additional benefits of weight loss and reduction in blood pressure.
A phase 3, randomized, placebo-controlled trial, DAPA-CVD (NCT03036150), is currently underway to evaluate the drug’s efficacy specifically in terms of renal outcomes and cardiovascular mortality in patients with chronic kidney disease, with and without type 2 diabetes. Participants receive once-daily dapagliflozin or placebo in addition to standard care.
Taking dapagliflozin carries risks of hypotension, renal impairment, hypoglycemia, and other concerns. The most common adverse reactions (5% or greater incidence) include female genital mycotic infections, nasopharyngitis, and urinary tract infections. Full prescribing information can be found on the agency’s website.
Class I recall issued for Sapien 3 balloon
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.

Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.
Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
The Food and Drug Administration has issued a class I correction recall for Edward Lifescience’s Sapien 3 balloon system, which is used to deploy transcatheter aortic valve replacements, according to a release from the agency. A class I recall is the most serious the agency issues and indicates risk of severe injury or even death.
Rather than indicate removal of the device from market, this recall provides details on how to use the device cautiously and safely and instructs physicians on proper technique to safely retract the delivery system into the sheath in cases of a suspected balloon burst. Failure to observe these recommendations can result in vascular injury, bleeding, or surgical intervention.
These recommendations and instructions reiterate those issued in an Urgent Field Safety Notice that was provided by Edward Lifesciences on July 9, 2019. Edward Lifesciences could not be reached for comment.
FDA approves Wakix for excessive daytime sleepiness
The Food and Drug Administration has approved pitolisant (Wakix) for excessive daytime sleepiness among patients with narcolepsy, according to a release from the drug’s developer.
Approval of this once-daily, selective histamine 3–receptor antagonist/inverse agonist was based on a pair of multicenter, randomized, double-blind, placebo-controlled studies that included a total of 261 patients. Patients in both studies experienced statistically significant improvements in excessive daytime sleepiness according to Epworth Sleepiness Scale scores.
Rates of adverse advents at or greater than 5% and more than double that of placebo included insomnia (6%), nausea (6%), and anxiety (5%). Patients with severe liver disease should not use pitolisant. Pitolisant has not been evaluated in patients under 18 years of age, and patients who are pregnant or planning to become pregnant are encouraged to enroll in a pregnancy exposure registry.
Full prescribing information, including contraindications and warnings, can be found on the FDA website.
The Food and Drug Administration has approved pitolisant (Wakix) for excessive daytime sleepiness among patients with narcolepsy, according to a release from the drug’s developer.
Approval of this once-daily, selective histamine 3–receptor antagonist/inverse agonist was based on a pair of multicenter, randomized, double-blind, placebo-controlled studies that included a total of 261 patients. Patients in both studies experienced statistically significant improvements in excessive daytime sleepiness according to Epworth Sleepiness Scale scores.
Rates of adverse advents at or greater than 5% and more than double that of placebo included insomnia (6%), nausea (6%), and anxiety (5%). Patients with severe liver disease should not use pitolisant. Pitolisant has not been evaluated in patients under 18 years of age, and patients who are pregnant or planning to become pregnant are encouraged to enroll in a pregnancy exposure registry.
Full prescribing information, including contraindications and warnings, can be found on the FDA website.
The Food and Drug Administration has approved pitolisant (Wakix) for excessive daytime sleepiness among patients with narcolepsy, according to a release from the drug’s developer.
Approval of this once-daily, selective histamine 3–receptor antagonist/inverse agonist was based on a pair of multicenter, randomized, double-blind, placebo-controlled studies that included a total of 261 patients. Patients in both studies experienced statistically significant improvements in excessive daytime sleepiness according to Epworth Sleepiness Scale scores.
Rates of adverse advents at or greater than 5% and more than double that of placebo included insomnia (6%), nausea (6%), and anxiety (5%). Patients with severe liver disease should not use pitolisant. Pitolisant has not been evaluated in patients under 18 years of age, and patients who are pregnant or planning to become pregnant are encouraged to enroll in a pregnancy exposure registry.
Full prescribing information, including contraindications and warnings, can be found on the FDA website.
FCC backs designating 988 as suicide prevention hotline
A Federal Communications Commission report recommends that 988 be designated as a nationwide suicide and mental health crisis support hotline, because a three-digit number “could be more effective” than the current 10-digit number, according to an FCC release.
The report was prepared by the FCC’s Wireline Competition Bureau and Office of Economics and Analytics at the direction of the National Suicide Hotline Improvement Act of 2018. The current service, known as the National Suicide Prevention Lifeline, uses the 10-digit number of 800-273-8255 (TALK). The report noted that the hotline has proved effective: The service answered 2.2 million calls in 2018 and various assessments have shown significant reductions in hopelessness and suicidal ideation among callers.
However, based on data and conclusions from some of those assessments, the report determined “that the Lifeline could be more effective in preventing suicides and providing crisis intervention if it were accessible via a simple, easy-to-remember, 3-digit dialing code.” The report examined the feasibility of three-digit options, including N11 options such as 211 and 511, but based on an analysis by the North American Numbering Council, it found that “technical and operational concerns related to the 988 code could be more easily and quickly addressed and resolved than any re-education efforts related to repurposing a N11 code.”
Suicide and mental health crises have been on the rise, according to numbers from the Centers for Disease Control and Prevention cited by the FCC report. Among the 50 states, 49 saw increases during 1999-2016, and more than half saw increases greater than 20%. The report describes how some groups, such as veterans and LGBTQ youth, are at especially high risk:
A Federal Communications Commission report recommends that 988 be designated as a nationwide suicide and mental health crisis support hotline, because a three-digit number “could be more effective” than the current 10-digit number, according to an FCC release.
The report was prepared by the FCC’s Wireline Competition Bureau and Office of Economics and Analytics at the direction of the National Suicide Hotline Improvement Act of 2018. The current service, known as the National Suicide Prevention Lifeline, uses the 10-digit number of 800-273-8255 (TALK). The report noted that the hotline has proved effective: The service answered 2.2 million calls in 2018 and various assessments have shown significant reductions in hopelessness and suicidal ideation among callers.
However, based on data and conclusions from some of those assessments, the report determined “that the Lifeline could be more effective in preventing suicides and providing crisis intervention if it were accessible via a simple, easy-to-remember, 3-digit dialing code.” The report examined the feasibility of three-digit options, including N11 options such as 211 and 511, but based on an analysis by the North American Numbering Council, it found that “technical and operational concerns related to the 988 code could be more easily and quickly addressed and resolved than any re-education efforts related to repurposing a N11 code.”
Suicide and mental health crises have been on the rise, according to numbers from the Centers for Disease Control and Prevention cited by the FCC report. Among the 50 states, 49 saw increases during 1999-2016, and more than half saw increases greater than 20%. The report describes how some groups, such as veterans and LGBTQ youth, are at especially high risk:
A Federal Communications Commission report recommends that 988 be designated as a nationwide suicide and mental health crisis support hotline, because a three-digit number “could be more effective” than the current 10-digit number, according to an FCC release.
The report was prepared by the FCC’s Wireline Competition Bureau and Office of Economics and Analytics at the direction of the National Suicide Hotline Improvement Act of 2018. The current service, known as the National Suicide Prevention Lifeline, uses the 10-digit number of 800-273-8255 (TALK). The report noted that the hotline has proved effective: The service answered 2.2 million calls in 2018 and various assessments have shown significant reductions in hopelessness and suicidal ideation among callers.
However, based on data and conclusions from some of those assessments, the report determined “that the Lifeline could be more effective in preventing suicides and providing crisis intervention if it were accessible via a simple, easy-to-remember, 3-digit dialing code.” The report examined the feasibility of three-digit options, including N11 options such as 211 and 511, but based on an analysis by the North American Numbering Council, it found that “technical and operational concerns related to the 988 code could be more easily and quickly addressed and resolved than any re-education efforts related to repurposing a N11 code.”
Suicide and mental health crises have been on the rise, according to numbers from the Centers for Disease Control and Prevention cited by the FCC report. Among the 50 states, 49 saw increases during 1999-2016, and more than half saw increases greater than 20%. The report describes how some groups, such as veterans and LGBTQ youth, are at especially high risk:
FDA approves upadacitinib for rheumatoid arthritis
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
according to a release from its developer. The indication specifies that patients must have shown inadequate response or intolerance to methotrexate.
The approval is based on the SELECT program, which included 4,400 patients across five studies that tested the oral Janus kinase inhibitor in various settings, such as alone or with methotrexate. All primary and secondary endpoints were met in these trials. For example, among patients with inadequate response to methotrexate in one study, 68% of those treated with 15-mg upadacitinib monotherapy achieved 20% improvement in American College of Rheumatology response criteria (ACR20) at week 14 versus 41% of those who had continued on methotrexate. In another study of patients with in adequate response to methotrexate, 71% of those treated with upadacitinib/methotrexate combination therapy achieved ACR20 at week 12 versus 36% of those receiving placebo and methotrexate. Compared with other treatments, better rates of clinical remission and radiographic inhibition were seen with upadacitinib-based therapies, too.
Upadacitinib carries a boxed warning – the most serious warning the FDA issues – for risks of serious infection, malignancy, and thrombosis; these serious risks should be weighed against treatment benefits in any patients with increased risk for these problems or currently experiencing any of them. Concomitant use with other JAK inhibitors, biologic DMARDs, or with potent immunosuppressants is not recommended; its use also is not recommended for patients with severe hepatic impairment. The most common adverse reactions are upper respiratory tract infection, nausea, cough, and pyrexia. Live vaccines should be avoided with patients taking this drug, and patients who are breastfeeding should be advised against use of it.
More prescribing information can be found in the drug’s label, which can be found on the FDA’s website.
Upadacitinib is also being evaluated for treatment of other immune-mediated diseases.
Nebraska issues SUNucate-based guidance for schools
Nebraska’s Department of Education recommended in a guidance that children be allowed to possess and use sunscreen products in school and at school-sponsored events, according to an Aug. 2 release from the American Society for Dermatologic Surgery.
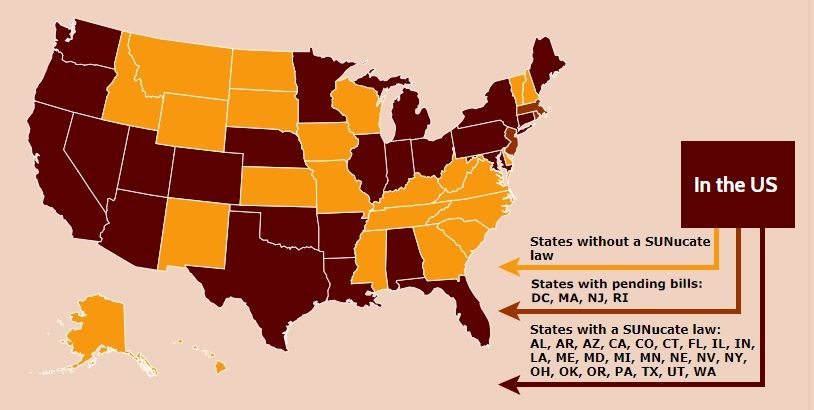
The department’s guidance is based on model legislation developed by the SUNucate Coalition, which was created by the American Society for Dermatologic Surgery Association and “works to address barriers to sunscreen use in school and camps and promote sun-safe behavior.” The coalition was created in 2016 because of reports that some U.S. schools had banned sunscreen products as part of broader medication bans because of these products’ classification as an over-the-counter medication.
Twenty-three other states have moved to lift such bans; these states were joined by Arkansas, Connecticut, Illinois, Maine, Minnesota, Nevada, and now Nebraska in 2019 alone. The District of Columbia, Massachusetts, New Jersey, and Rhode Island are expected to follow suit.
Nebraska’s Department of Education recommended in a guidance that children be allowed to possess and use sunscreen products in school and at school-sponsored events, according to an Aug. 2 release from the American Society for Dermatologic Surgery.
The department’s guidance is based on model legislation developed by the SUNucate Coalition, which was created by the American Society for Dermatologic Surgery Association and “works to address barriers to sunscreen use in school and camps and promote sun-safe behavior.” The coalition was created in 2016 because of reports that some U.S. schools had banned sunscreen products as part of broader medication bans because of these products’ classification as an over-the-counter medication.
Twenty-three other states have moved to lift such bans; these states were joined by Arkansas, Connecticut, Illinois, Maine, Minnesota, Nevada, and now Nebraska in 2019 alone. The District of Columbia, Massachusetts, New Jersey, and Rhode Island are expected to follow suit.
Nebraska’s Department of Education recommended in a guidance that children be allowed to possess and use sunscreen products in school and at school-sponsored events, according to an Aug. 2 release from the American Society for Dermatologic Surgery.
The department’s guidance is based on model legislation developed by the SUNucate Coalition, which was created by the American Society for Dermatologic Surgery Association and “works to address barriers to sunscreen use in school and camps and promote sun-safe behavior.” The coalition was created in 2016 because of reports that some U.S. schools had banned sunscreen products as part of broader medication bans because of these products’ classification as an over-the-counter medication.
Twenty-three other states have moved to lift such bans; these states were joined by Arkansas, Connecticut, Illinois, Maine, Minnesota, Nevada, and now Nebraska in 2019 alone. The District of Columbia, Massachusetts, New Jersey, and Rhode Island are expected to follow suit.
Evinacumab shows promise for HoFH in top line results
(HoFH), according to a release from the company developing the drug.
LDL cholesterol levels were 255 mg/dL on average for patients at the outset of the trial despite treatment with other lipid-lowering therapies; however, combining this drug with lipid-lowering therapies including maximally-tolerated statins, PCSK9 inhibitors, and LDL apheresis, reduced LDL cholesterol by an average of 49% by week 24 relative to treatment with lipid-lowering therapies alone (P less than .0001). Furthermore, 47% of patients taking evinacumab achieved LDL cholesterol levels under 100 mg/dL by that time point versus 23% of those taking lipid-lowering therapies only.
Treatment with evinacumab showed lowering effects as early as the first assessment at 2 weeks, and these effects were maintained.
HoFH is an inherited, rare, but serious condition estimated to affect 1,300 people in the United States; it can lead to early atherosclerotic disease, and even teenagers with this genetic disorder can suffer cardiac events. Further details and data from the trial, called ELIPSE HoFH, will be reported at a future medical meeting, and will be submitted to the Food and Drug Administration for consideration.
Evinacumab is an investigational, fully-human, monoclonal antibody that specifically binds to angiopoietin-like protein 3 (ANGPTL3), which acts as an inhibitor of lipoprotein lipase and endothelial lipase, and appears to play a central role in lipoprotein metabolism.
Evinacumab was granted breakthrough therapy designation for treatment of HoFH by the FDA in 2017, which entails an expedited review and development process for this drug because preliminary results have suggested it could have a substantial effect on a life-threatening or serious condition.
The company’s full release can be read on its website.
(HoFH), according to a release from the company developing the drug.
LDL cholesterol levels were 255 mg/dL on average for patients at the outset of the trial despite treatment with other lipid-lowering therapies; however, combining this drug with lipid-lowering therapies including maximally-tolerated statins, PCSK9 inhibitors, and LDL apheresis, reduced LDL cholesterol by an average of 49% by week 24 relative to treatment with lipid-lowering therapies alone (P less than .0001). Furthermore, 47% of patients taking evinacumab achieved LDL cholesterol levels under 100 mg/dL by that time point versus 23% of those taking lipid-lowering therapies only.
Treatment with evinacumab showed lowering effects as early as the first assessment at 2 weeks, and these effects were maintained.
HoFH is an inherited, rare, but serious condition estimated to affect 1,300 people in the United States; it can lead to early atherosclerotic disease, and even teenagers with this genetic disorder can suffer cardiac events. Further details and data from the trial, called ELIPSE HoFH, will be reported at a future medical meeting, and will be submitted to the Food and Drug Administration for consideration.
Evinacumab is an investigational, fully-human, monoclonal antibody that specifically binds to angiopoietin-like protein 3 (ANGPTL3), which acts as an inhibitor of lipoprotein lipase and endothelial lipase, and appears to play a central role in lipoprotein metabolism.
Evinacumab was granted breakthrough therapy designation for treatment of HoFH by the FDA in 2017, which entails an expedited review and development process for this drug because preliminary results have suggested it could have a substantial effect on a life-threatening or serious condition.
The company’s full release can be read on its website.
(HoFH), according to a release from the company developing the drug.
LDL cholesterol levels were 255 mg/dL on average for patients at the outset of the trial despite treatment with other lipid-lowering therapies; however, combining this drug with lipid-lowering therapies including maximally-tolerated statins, PCSK9 inhibitors, and LDL apheresis, reduced LDL cholesterol by an average of 49% by week 24 relative to treatment with lipid-lowering therapies alone (P less than .0001). Furthermore, 47% of patients taking evinacumab achieved LDL cholesterol levels under 100 mg/dL by that time point versus 23% of those taking lipid-lowering therapies only.
Treatment with evinacumab showed lowering effects as early as the first assessment at 2 weeks, and these effects were maintained.
HoFH is an inherited, rare, but serious condition estimated to affect 1,300 people in the United States; it can lead to early atherosclerotic disease, and even teenagers with this genetic disorder can suffer cardiac events. Further details and data from the trial, called ELIPSE HoFH, will be reported at a future medical meeting, and will be submitted to the Food and Drug Administration for consideration.
Evinacumab is an investigational, fully-human, monoclonal antibody that specifically binds to angiopoietin-like protein 3 (ANGPTL3), which acts as an inhibitor of lipoprotein lipase and endothelial lipase, and appears to play a central role in lipoprotein metabolism.
Evinacumab was granted breakthrough therapy designation for treatment of HoFH by the FDA in 2017, which entails an expedited review and development process for this drug because preliminary results have suggested it could have a substantial effect on a life-threatening or serious condition.
The company’s full release can be read on its website.
FDA update: Higher late mortality with paclitaxel-coated devices
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
Paclitaxel-coated devices, which are used to treat peripheral artery disease (PAD), appear to have a nearly 60% higher mortality risk than uncoated devices, according to a letter to health care providers from the Food and Drug Administration.
This letter updates details about long-term follow-up data and panel conclusions reviewed by the Food and Drug Administration, as well as recommendations from the agency regarding these devices. On Jan. 17, 2019, the FDA notified providers regarding an apparent increased late mortality risk seen with paclitaxel-eluting stents and paclitaxel-coated balloons placed in the femoropopliteal artery in patients with PAD. The agency issued an update March 15.
In a public meeting June 19-20, the Circulatory System Devices Panel of the Medical Devices Advisory Committee discussed long-term follow-up data that demonstrated a 57% relative increase in mortality among PAD patients treated with paclitaxel-coated devices when compared with those receiving uncoated devices. The panel concluded that the late mortality signal was real and warranted further study and action, a conclusion with which the FDA has concurred.
Among other recommendations issued by the FDA, health care professionals should continue to closely monitor patients who’ve already received the devices and fully discuss the risks and benefits of these devices with patients. The FDA has decided that, given the demonstrated short-term benefits of these devices, clinical studies may continue and should collect long-term safety and effectiveness data.
The magnitude of this late mortality signal should be interpreted with caution, the FDA noted in the update, because of the wide confidence intervals (although the relative risk was 1.57, the 95% confidence interval was 1.16-2.13, which translates to 16%-113% higher relative risk), pooling studies of different devices that weren’t meant to be combined, missing data, and other reasons.
The full letter, including more detailed data and the full list of recommendations, is available on the FDA’s website.
FDA accepts dasotraline NDA for binge-eating disorder
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.
Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.
Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.
The Food and Drug Administration has accepted the new drug application for dasotraline for the treatment of moderate to severe binge-eating disorder, the drug’s developer, Sunovion, announced July 30.
Dasotraline, a dopamine and norepinephrine reuptake inhibitor, demonstrated significant efficacy in a pair of 12-week, randomized, placebo-controlled studies (SEP360-221 and SEP360-321). The drug also was found to be well tolerated by patients with binge-eating disorder (BED), both in those studies and in a long-term safety study that followed patients for up to a year (SEP360-322).
The medication – characterized by an extended half-life – is to be taken once a day. The most common adverse events reported by patients who took dasotraline include insomnia, dry mouth, decreased appetite, anxiety, nausea, and decreased weight.
BED is more common than any other eating disorder, with an estimated lifetime prevalence among U.S. adults of 1.25% for women and 0.42% for men (CNS Spectr. 2019 Jun 14. doi: 10.1017/S109285291900103). The condition also might run in families. BED often is comorbid with other psychiatric and behavioral disorders, such as depression, substance use, and PTSD, noted Antony Loebel, MD, president and CEO of Sunovion, in a press release. He also said BED often is underrecognized and undertreated.
Meta-analytic reviews show that cognitive-behavioral therapy is considered first-line treatment for BED. However, limited access to such psychological treatments makes the development of medication options such as dasotraline important.
Last year, the agency rejected a new drug application for dasotraline for the treatment of ADHD, citing a need for additional data.