User login
EADV: Patients Often Unaware of Infliximab Infusion Risk
GOTHENBURG, SWEDEN - More than one-fifth of infliximab patients who had a temporary medical situation contraindicating administration of the biologic presented for their scheduled infusion unaware treatment would be inappropriate at that time.
The finding from the RemiTRAC Infusion Registry – a large, prospective, observational Canadian registry of patients on infliximab for any indication – underscores the importance of reviewing the full list of contraindications to infliximab with every patient prior to each infusion, said Dr. Warren J. Winkelman said at the annual congress of the European Academy of Dermatology and Venereology. This is the key to minimizing infusion reactions.
The most common medical conditions RemiTRAC participants mistakenly perceived as not being a problem were flulike symptoms and upper respiratory infections, despite the fact patients were informed that these symptoms increased the risk of adverse events as part of standard pretreatment education.
The lesson provided by RemiTRAC is that it is particularly important to inquire about flulike symptoms and colds before administering an infliximab infusion, according to Dr. Winkelman of the University of Toronto.
He reported on 718 patients on infliximab for any indication at more than a dozen Canadian sites participating in the RemiTRAC Infusion Registry. The indication for the antitumor necrosis factor (TNF)–alpha agent was rheumatoid arthritis in 51% of patients, ankylosing spondylitis in 17%, Crohn’s disease in 11%, psoriasis in 9%, psoriatic arthritis in 6%, and ulcerative colitis in 4%.
Participants in the registry received a mean of 15 infliximab infusions during more than 900 patient-years of exposure to the biologic agent. Of 8,051 scheduled infusions, 6% were not administered because of clinical reasons. Sixty-two percent of these cancelled infusions were scotched because of infections, mainly upper respiratory infections or flulike symptoms. Another 9% of cancelled treatments were the result of scheduled surgery.
In 22% of instances of cancelled infusion, patients came to the clinic for their infusion unaware that their health status at that particular time was inappropriate for administration of anti-TNF therapy, according to Dr. Winkelman.
The overall incidence of infusion reactions was 2.1%. The rate was 2.6% in patients being treated for rheumatoid arthritis, 1.9% in those with psoriasis, 0.7% in those with psoriatic arthritis, and 1.3% in patients with ankylosing spondylitis.
The RemiTRAC Infusion Registry was supported by Schering-Plough Canada, which is now part of Merck. Dr. Winkelman disclosed serving as a consultant to the company.
GOTHENBURG, SWEDEN - More than one-fifth of infliximab patients who had a temporary medical situation contraindicating administration of the biologic presented for their scheduled infusion unaware treatment would be inappropriate at that time.
The finding from the RemiTRAC Infusion Registry – a large, prospective, observational Canadian registry of patients on infliximab for any indication – underscores the importance of reviewing the full list of contraindications to infliximab with every patient prior to each infusion, said Dr. Warren J. Winkelman said at the annual congress of the European Academy of Dermatology and Venereology. This is the key to minimizing infusion reactions.
The most common medical conditions RemiTRAC participants mistakenly perceived as not being a problem were flulike symptoms and upper respiratory infections, despite the fact patients were informed that these symptoms increased the risk of adverse events as part of standard pretreatment education.
The lesson provided by RemiTRAC is that it is particularly important to inquire about flulike symptoms and colds before administering an infliximab infusion, according to Dr. Winkelman of the University of Toronto.
He reported on 718 patients on infliximab for any indication at more than a dozen Canadian sites participating in the RemiTRAC Infusion Registry. The indication for the antitumor necrosis factor (TNF)–alpha agent was rheumatoid arthritis in 51% of patients, ankylosing spondylitis in 17%, Crohn’s disease in 11%, psoriasis in 9%, psoriatic arthritis in 6%, and ulcerative colitis in 4%.
Participants in the registry received a mean of 15 infliximab infusions during more than 900 patient-years of exposure to the biologic agent. Of 8,051 scheduled infusions, 6% were not administered because of clinical reasons. Sixty-two percent of these cancelled infusions were scotched because of infections, mainly upper respiratory infections or flulike symptoms. Another 9% of cancelled treatments were the result of scheduled surgery.
In 22% of instances of cancelled infusion, patients came to the clinic for their infusion unaware that their health status at that particular time was inappropriate for administration of anti-TNF therapy, according to Dr. Winkelman.
The overall incidence of infusion reactions was 2.1%. The rate was 2.6% in patients being treated for rheumatoid arthritis, 1.9% in those with psoriasis, 0.7% in those with psoriatic arthritis, and 1.3% in patients with ankylosing spondylitis.
The RemiTRAC Infusion Registry was supported by Schering-Plough Canada, which is now part of Merck. Dr. Winkelman disclosed serving as a consultant to the company.
GOTHENBURG, SWEDEN - More than one-fifth of infliximab patients who had a temporary medical situation contraindicating administration of the biologic presented for their scheduled infusion unaware treatment would be inappropriate at that time.
The finding from the RemiTRAC Infusion Registry – a large, prospective, observational Canadian registry of patients on infliximab for any indication – underscores the importance of reviewing the full list of contraindications to infliximab with every patient prior to each infusion, said Dr. Warren J. Winkelman said at the annual congress of the European Academy of Dermatology and Venereology. This is the key to minimizing infusion reactions.
The most common medical conditions RemiTRAC participants mistakenly perceived as not being a problem were flulike symptoms and upper respiratory infections, despite the fact patients were informed that these symptoms increased the risk of adverse events as part of standard pretreatment education.
The lesson provided by RemiTRAC is that it is particularly important to inquire about flulike symptoms and colds before administering an infliximab infusion, according to Dr. Winkelman of the University of Toronto.
He reported on 718 patients on infliximab for any indication at more than a dozen Canadian sites participating in the RemiTRAC Infusion Registry. The indication for the antitumor necrosis factor (TNF)–alpha agent was rheumatoid arthritis in 51% of patients, ankylosing spondylitis in 17%, Crohn’s disease in 11%, psoriasis in 9%, psoriatic arthritis in 6%, and ulcerative colitis in 4%.
Participants in the registry received a mean of 15 infliximab infusions during more than 900 patient-years of exposure to the biologic agent. Of 8,051 scheduled infusions, 6% were not administered because of clinical reasons. Sixty-two percent of these cancelled infusions were scotched because of infections, mainly upper respiratory infections or flulike symptoms. Another 9% of cancelled treatments were the result of scheduled surgery.
In 22% of instances of cancelled infusion, patients came to the clinic for their infusion unaware that their health status at that particular time was inappropriate for administration of anti-TNF therapy, according to Dr. Winkelman.
The overall incidence of infusion reactions was 2.1%. The rate was 2.6% in patients being treated for rheumatoid arthritis, 1.9% in those with psoriasis, 0.7% in those with psoriatic arthritis, and 1.3% in patients with ankylosing spondylitis.
The RemiTRAC Infusion Registry was supported by Schering-Plough Canada, which is now part of Merck. Dr. Winkelman disclosed serving as a consultant to the company.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: In 22% of instances of cancelled infusion, patients came to the clinic for their infusion unaware that their health status at that particular time was inappropriate for administration of anti-TNF therapy.
Data Source: 718 patients on infliximab for any indication at more than a dozen Canadian sites participating in the RemiTRAC Infusion Registry
Disclosures: The RemiTRAC Infusion Registry was supported by Schering-Plough Canada, which is now part of Merck. Dr. Winkelman disclosed serving as a consultant to the company.
Expert Panel: Switch Methotrexate Nonresponders to Subcutaneous Form
GOTHENBURG, Sweden - A psoriasis patient should not be considered a methotrexate nonresponder before a subcutaneous dose is tried, an expert panel from the International Psoriasis Council said at the annual congress of the European Academy of Dermatology and Venereology.
"Improvement in psoriasis with methotrexate is dependent on the blood level. And we know that absorption of methotrexate in the gastrointestinal tract is incomplete and quite variable from person to person, especially at higher doses. You can turn a patient into a responder by switching from oral to subcutaneous administration," said Dr. Knud Kragballe, professor of dermatology at Aarhus (Denmark) University Hospital.
Methotrexate is a dermatologic tradition, and most dermatologists were trained to administer the drug orally, said Dr. Kragballe. The oral form is easier to use than injectable methotrexate, and a logical starting place; however, some patients who do not get sufficient benefit because oral methotrexate’s bioavailability is about 30% less than that of the subcutaneous form, he added.
The switch is a straightforward matter. The subcutaneous dose is equivalent to the oral dose. If a patient is not showing sufficient benefit, for example, on 20 mg of oral methotrexate once weekly, try 20 mg subcutaneously once weekly, he said.
The widespread dermatologic reluctance to use subcutaneous methotrexate is partly a matter of training, but is also a function of the plethora of biologic agents available, according to Dr. Kragballe.
"I think it’s important to utilize the full potential of any drug before switching to another drug. We have too many cases dismissed as methotrexate nonresponders when the problem is the drug is not being used at the proper dose and route of administration," he said.
An audience show of hands indicated only a minority of attendees at the International Psoriasis Council–sponsored meet-the-experts session now utilize subcutaneous methotrexate. But the subcutaneous route is not the hassle many dermatologists imagine, according to Dr. Wolfram Sterry.

"We switched 3 or 4 years ago in my department, and now we do only subcutaneous methotrexate. That way you know 10 mg is 10 mg, and that’s it. I haven’t yet found a patient who is uncomfortable with subcutaneous administration," said Dr. Sterry, professor and chairman of the department of dermatology and allergy at Charité University Hospital, Berlin.
Dr. Craig Leonardi said that he, like Dr. Kragballe, typically starts patients on oral methotrexate because it is easier, and that’s how he was trained, but he doesn’t hesitate to shift to the subcutaneous form.
In the United States, another reason to favor subcutaneous methotrexate, besides better bioavailability, is that the cost is only about one-tenth that of tablets.

"If a patient comes in who doesn’t have insurance and really can’t afford to pay for medications, subcutaneous methotrexate is the most cost-effective way to get the job done," said Dr. Leonardi a dermatologist and psoriasis specialist at St. Louis University.
International Psoriasis Council President, Dr. Alan Menter, commented that even though dermatologists have been using methotrexate for 40 years, the pharmacokinetics of the drug is still unclear.
Dr. Menter and his coworkers at Baylor University Medical Center, Dallas, where he is chief of dermatology, have been investigating a novel form of aminopterin – an antineoplastic drug closely related to methotrexate – which in preliminary phase I and II studies offered significant advantages over methotrexate: more predictable oral absorption, less uptake in cerebrospinal fluid, and less hepatotoxicity.

Speaking of hepatotoxicity, Dr. Menter said he and his colleagues have not conducted a liver biopsy in years, even though they have patients who have been on methotrexate for 2 decades.
"I think our gastroenterologists can now scan livers and look at early fibrosis much better than we can," Dr. Menter said.
Panelists noted there is no consensus on how to give folic acid to patients on methotrexate. Dr. Leonardi has patients take 1 mg daily. He thinks it is easier for patients to follow a daily routine.
Dr. Kragballe, in contrast, gives folic acid once per week 2 days after the methotrexate. "But there’s no science behind it," he noted. The important thing is that every psoriasis patient on methotrexate be on folic acid.
All of the International Psoriasis Council panelists disclosed serving as consultants, speakers, and investigators for numerous companies that market psoriasis therapies.
GOTHENBURG, Sweden - A psoriasis patient should not be considered a methotrexate nonresponder before a subcutaneous dose is tried, an expert panel from the International Psoriasis Council said at the annual congress of the European Academy of Dermatology and Venereology.
"Improvement in psoriasis with methotrexate is dependent on the blood level. And we know that absorption of methotrexate in the gastrointestinal tract is incomplete and quite variable from person to person, especially at higher doses. You can turn a patient into a responder by switching from oral to subcutaneous administration," said Dr. Knud Kragballe, professor of dermatology at Aarhus (Denmark) University Hospital.
Methotrexate is a dermatologic tradition, and most dermatologists were trained to administer the drug orally, said Dr. Kragballe. The oral form is easier to use than injectable methotrexate, and a logical starting place; however, some patients who do not get sufficient benefit because oral methotrexate’s bioavailability is about 30% less than that of the subcutaneous form, he added.
The switch is a straightforward matter. The subcutaneous dose is equivalent to the oral dose. If a patient is not showing sufficient benefit, for example, on 20 mg of oral methotrexate once weekly, try 20 mg subcutaneously once weekly, he said.
The widespread dermatologic reluctance to use subcutaneous methotrexate is partly a matter of training, but is also a function of the plethora of biologic agents available, according to Dr. Kragballe.
"I think it’s important to utilize the full potential of any drug before switching to another drug. We have too many cases dismissed as methotrexate nonresponders when the problem is the drug is not being used at the proper dose and route of administration," he said.
An audience show of hands indicated only a minority of attendees at the International Psoriasis Council–sponsored meet-the-experts session now utilize subcutaneous methotrexate. But the subcutaneous route is not the hassle many dermatologists imagine, according to Dr. Wolfram Sterry.
"We switched 3 or 4 years ago in my department, and now we do only subcutaneous methotrexate. That way you know 10 mg is 10 mg, and that’s it. I haven’t yet found a patient who is uncomfortable with subcutaneous administration," said Dr. Sterry, professor and chairman of the department of dermatology and allergy at Charité University Hospital, Berlin.
Dr. Craig Leonardi said that he, like Dr. Kragballe, typically starts patients on oral methotrexate because it is easier, and that’s how he was trained, but he doesn’t hesitate to shift to the subcutaneous form.
In the United States, another reason to favor subcutaneous methotrexate, besides better bioavailability, is that the cost is only about one-tenth that of tablets.
"If a patient comes in who doesn’t have insurance and really can’t afford to pay for medications, subcutaneous methotrexate is the most cost-effective way to get the job done," said Dr. Leonardi a dermatologist and psoriasis specialist at St. Louis University.
International Psoriasis Council President, Dr. Alan Menter, commented that even though dermatologists have been using methotrexate for 40 years, the pharmacokinetics of the drug is still unclear.
Dr. Menter and his coworkers at Baylor University Medical Center, Dallas, where he is chief of dermatology, have been investigating a novel form of aminopterin – an antineoplastic drug closely related to methotrexate – which in preliminary phase I and II studies offered significant advantages over methotrexate: more predictable oral absorption, less uptake in cerebrospinal fluid, and less hepatotoxicity.
Speaking of hepatotoxicity, Dr. Menter said he and his colleagues have not conducted a liver biopsy in years, even though they have patients who have been on methotrexate for 2 decades.
"I think our gastroenterologists can now scan livers and look at early fibrosis much better than we can," Dr. Menter said.
Panelists noted there is no consensus on how to give folic acid to patients on methotrexate. Dr. Leonardi has patients take 1 mg daily. He thinks it is easier for patients to follow a daily routine.
Dr. Kragballe, in contrast, gives folic acid once per week 2 days after the methotrexate. "But there’s no science behind it," he noted. The important thing is that every psoriasis patient on methotrexate be on folic acid.
All of the International Psoriasis Council panelists disclosed serving as consultants, speakers, and investigators for numerous companies that market psoriasis therapies.
GOTHENBURG, Sweden - A psoriasis patient should not be considered a methotrexate nonresponder before a subcutaneous dose is tried, an expert panel from the International Psoriasis Council said at the annual congress of the European Academy of Dermatology and Venereology.
"Improvement in psoriasis with methotrexate is dependent on the blood level. And we know that absorption of methotrexate in the gastrointestinal tract is incomplete and quite variable from person to person, especially at higher doses. You can turn a patient into a responder by switching from oral to subcutaneous administration," said Dr. Knud Kragballe, professor of dermatology at Aarhus (Denmark) University Hospital.
Methotrexate is a dermatologic tradition, and most dermatologists were trained to administer the drug orally, said Dr. Kragballe. The oral form is easier to use than injectable methotrexate, and a logical starting place; however, some patients who do not get sufficient benefit because oral methotrexate’s bioavailability is about 30% less than that of the subcutaneous form, he added.
The switch is a straightforward matter. The subcutaneous dose is equivalent to the oral dose. If a patient is not showing sufficient benefit, for example, on 20 mg of oral methotrexate once weekly, try 20 mg subcutaneously once weekly, he said.
The widespread dermatologic reluctance to use subcutaneous methotrexate is partly a matter of training, but is also a function of the plethora of biologic agents available, according to Dr. Kragballe.
"I think it’s important to utilize the full potential of any drug before switching to another drug. We have too many cases dismissed as methotrexate nonresponders when the problem is the drug is not being used at the proper dose and route of administration," he said.
An audience show of hands indicated only a minority of attendees at the International Psoriasis Council–sponsored meet-the-experts session now utilize subcutaneous methotrexate. But the subcutaneous route is not the hassle many dermatologists imagine, according to Dr. Wolfram Sterry.
"We switched 3 or 4 years ago in my department, and now we do only subcutaneous methotrexate. That way you know 10 mg is 10 mg, and that’s it. I haven’t yet found a patient who is uncomfortable with subcutaneous administration," said Dr. Sterry, professor and chairman of the department of dermatology and allergy at Charité University Hospital, Berlin.
Dr. Craig Leonardi said that he, like Dr. Kragballe, typically starts patients on oral methotrexate because it is easier, and that’s how he was trained, but he doesn’t hesitate to shift to the subcutaneous form.
In the United States, another reason to favor subcutaneous methotrexate, besides better bioavailability, is that the cost is only about one-tenth that of tablets.
"If a patient comes in who doesn’t have insurance and really can’t afford to pay for medications, subcutaneous methotrexate is the most cost-effective way to get the job done," said Dr. Leonardi a dermatologist and psoriasis specialist at St. Louis University.
International Psoriasis Council President, Dr. Alan Menter, commented that even though dermatologists have been using methotrexate for 40 years, the pharmacokinetics of the drug is still unclear.
Dr. Menter and his coworkers at Baylor University Medical Center, Dallas, where he is chief of dermatology, have been investigating a novel form of aminopterin – an antineoplastic drug closely related to methotrexate – which in preliminary phase I and II studies offered significant advantages over methotrexate: more predictable oral absorption, less uptake in cerebrospinal fluid, and less hepatotoxicity.
Speaking of hepatotoxicity, Dr. Menter said he and his colleagues have not conducted a liver biopsy in years, even though they have patients who have been on methotrexate for 2 decades.
"I think our gastroenterologists can now scan livers and look at early fibrosis much better than we can," Dr. Menter said.
Panelists noted there is no consensus on how to give folic acid to patients on methotrexate. Dr. Leonardi has patients take 1 mg daily. He thinks it is easier for patients to follow a daily routine.
Dr. Kragballe, in contrast, gives folic acid once per week 2 days after the methotrexate. "But there’s no science behind it," he noted. The important thing is that every psoriasis patient on methotrexate be on folic acid.
All of the International Psoriasis Council panelists disclosed serving as consultants, speakers, and investigators for numerous companies that market psoriasis therapies.
EXPERT ANALYSIS FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
AMG 827 Trial Participants Show Improvement of Chronic Plaque Psoriasis
GOTHENBURG, SWEDEN – Blockade of the receptor for the proinflammatory cytokine interleukin-17 using a novel fully human IgG2 monoclonal antibody has shown promise as a treatment for chronic plaque psoriasis.
In a small, phase I, double-blind randomized study, a single dose of AMG 827 was well tolerated and led to rapid clinical improvement accompanied by reversal of the molecular, cellular, and histologic hallmarks of the disease, Dr. David A. Martin reported at the annual congress of the European Academy of Dermatology and Venereology.
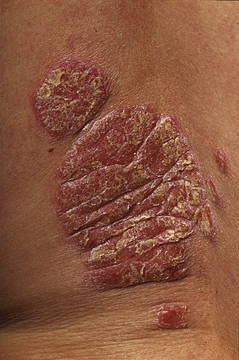
Based upon these findings, a phase II study of AMG 827 in patients with plaque psoriasis is underway, added Dr. Martin, an employee of the drug’s manufacturer and study sponsor, Amgen (Seattle).
He reported on 20 psoriasis patients who were randomized 4:4:1 to a single IV dose of AMG 827 at either 350 mg or 700 mg or to placebo. Biopsies were obtained from lesional and nonlesional skin at baseline and from lesional skin on days 8, 15, and 43.
By day 15 the 16 patients who received AMG 827 showed roughly a mean 60% improvement over baseline in terms of Psoriasis Area and Severity Index (PASI) scores. By day 43 the group treated with the 700-mg dose showed continued gains, with an 85% improvement in PASI scores over baseline. A PASI 75, or a 75% improvement over baseline, was achieved by day 43 in five of eight patients in the 350-mg group, seven of eight who got 700 mg of AMG 827, and none of four placebo-treated controls.
Physician’s Global Assessment ratings of psoriasis as "clear" or "minimal" were attained by all eight patients in the 700-mg group, five of eight who received 350 mg, and none of four controls.
At baseline, roughly 10,000 gene expression sequences were abnormally up- or downregulated in lesional skin. By day 8, this was reduced to about 2,000 abnormally expressed gene sequences. By day 15, the abnormal gene expression signature in the 700-mg group had dropped to roughly 500 abnormal sequences, nearly the same as in nonlesional skin at baseline.
The low level of abnormal gene expression was maintained in the 700-mg group out to day 43. Meanwhile, the abnormal gene expression signature in the 350-mg group improved from 2,000 sequences on day 8 to about 1,000 on day 43, Dr. Martin said.
Histopathologic findings in lesional skin included significant reductions in epidermal thickness, keratin-16 mRNA levels, Ki-67 cell counts, and the number of infiltrating leukocyte subsets by day 15, with further improvements noted by day 43.
AMG 827 is also under study for the treatment of rheumatoid arthritis and other inflammatory diseases. Interleukin-17 is known to activate local expression of a range of cytokines, metalloproteinases, chemokines, and antimicrobial peptides.
GOTHENBURG, SWEDEN – Blockade of the receptor for the proinflammatory cytokine interleukin-17 using a novel fully human IgG2 monoclonal antibody has shown promise as a treatment for chronic plaque psoriasis.
In a small, phase I, double-blind randomized study, a single dose of AMG 827 was well tolerated and led to rapid clinical improvement accompanied by reversal of the molecular, cellular, and histologic hallmarks of the disease, Dr. David A. Martin reported at the annual congress of the European Academy of Dermatology and Venereology.
Based upon these findings, a phase II study of AMG 827 in patients with plaque psoriasis is underway, added Dr. Martin, an employee of the drug’s manufacturer and study sponsor, Amgen (Seattle).
He reported on 20 psoriasis patients who were randomized 4:4:1 to a single IV dose of AMG 827 at either 350 mg or 700 mg or to placebo. Biopsies were obtained from lesional and nonlesional skin at baseline and from lesional skin on days 8, 15, and 43.
By day 15 the 16 patients who received AMG 827 showed roughly a mean 60% improvement over baseline in terms of Psoriasis Area and Severity Index (PASI) scores. By day 43 the group treated with the 700-mg dose showed continued gains, with an 85% improvement in PASI scores over baseline. A PASI 75, or a 75% improvement over baseline, was achieved by day 43 in five of eight patients in the 350-mg group, seven of eight who got 700 mg of AMG 827, and none of four placebo-treated controls.
Physician’s Global Assessment ratings of psoriasis as "clear" or "minimal" were attained by all eight patients in the 700-mg group, five of eight who received 350 mg, and none of four controls.
At baseline, roughly 10,000 gene expression sequences were abnormally up- or downregulated in lesional skin. By day 8, this was reduced to about 2,000 abnormally expressed gene sequences. By day 15, the abnormal gene expression signature in the 700-mg group had dropped to roughly 500 abnormal sequences, nearly the same as in nonlesional skin at baseline.
The low level of abnormal gene expression was maintained in the 700-mg group out to day 43. Meanwhile, the abnormal gene expression signature in the 350-mg group improved from 2,000 sequences on day 8 to about 1,000 on day 43, Dr. Martin said.
Histopathologic findings in lesional skin included significant reductions in epidermal thickness, keratin-16 mRNA levels, Ki-67 cell counts, and the number of infiltrating leukocyte subsets by day 15, with further improvements noted by day 43.
AMG 827 is also under study for the treatment of rheumatoid arthritis and other inflammatory diseases. Interleukin-17 is known to activate local expression of a range of cytokines, metalloproteinases, chemokines, and antimicrobial peptides.
GOTHENBURG, SWEDEN – Blockade of the receptor for the proinflammatory cytokine interleukin-17 using a novel fully human IgG2 monoclonal antibody has shown promise as a treatment for chronic plaque psoriasis.
In a small, phase I, double-blind randomized study, a single dose of AMG 827 was well tolerated and led to rapid clinical improvement accompanied by reversal of the molecular, cellular, and histologic hallmarks of the disease, Dr. David A. Martin reported at the annual congress of the European Academy of Dermatology and Venereology.
Based upon these findings, a phase II study of AMG 827 in patients with plaque psoriasis is underway, added Dr. Martin, an employee of the drug’s manufacturer and study sponsor, Amgen (Seattle).
He reported on 20 psoriasis patients who were randomized 4:4:1 to a single IV dose of AMG 827 at either 350 mg or 700 mg or to placebo. Biopsies were obtained from lesional and nonlesional skin at baseline and from lesional skin on days 8, 15, and 43.
By day 15 the 16 patients who received AMG 827 showed roughly a mean 60% improvement over baseline in terms of Psoriasis Area and Severity Index (PASI) scores. By day 43 the group treated with the 700-mg dose showed continued gains, with an 85% improvement in PASI scores over baseline. A PASI 75, or a 75% improvement over baseline, was achieved by day 43 in five of eight patients in the 350-mg group, seven of eight who got 700 mg of AMG 827, and none of four placebo-treated controls.
Physician’s Global Assessment ratings of psoriasis as "clear" or "minimal" were attained by all eight patients in the 700-mg group, five of eight who received 350 mg, and none of four controls.
At baseline, roughly 10,000 gene expression sequences were abnormally up- or downregulated in lesional skin. By day 8, this was reduced to about 2,000 abnormally expressed gene sequences. By day 15, the abnormal gene expression signature in the 700-mg group had dropped to roughly 500 abnormal sequences, nearly the same as in nonlesional skin at baseline.
The low level of abnormal gene expression was maintained in the 700-mg group out to day 43. Meanwhile, the abnormal gene expression signature in the 350-mg group improved from 2,000 sequences on day 8 to about 1,000 on day 43, Dr. Martin said.
Histopathologic findings in lesional skin included significant reductions in epidermal thickness, keratin-16 mRNA levels, Ki-67 cell counts, and the number of infiltrating leukocyte subsets by day 15, with further improvements noted by day 43.
AMG 827 is also under study for the treatment of rheumatoid arthritis and other inflammatory diseases. Interleukin-17 is known to activate local expression of a range of cytokines, metalloproteinases, chemokines, and antimicrobial peptides.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: PASI 75 was achieved by day 43 in five of eight patients who received 350 mg AMG 827, in seven of eight who got 700 mg, and none of four placebo-treated controls.
Data Source: A phase II study of AMG 827 of 20 patients with plaque psoriasis who were randomized 4:4:1 to a single intravenous dose of AMG 827 at either 350 mg or 700 mg or to placebo.
Disclosures: Dr. Martin is an employee of the AMG 827 manufacturer and study sponsor, Amgen (Seattle).
EADV: Study Points to Optimal Etanercept Regimen for Psoriasis, PsA
GOTHENBURG, Sweden - In patients with both plaque psoriasis and psoriatic arthritis, initial treatment with etanercept at 50 mg twice weekly resulted in faster clearance of skin lesions than 50 mg once weekly.
The twice-weekly regimen, however, offered no advantage for treatment of joint and tendon symptoms. These were the key findings of the Psoriasis Randomized Etanercept Study in Subjects with Psoriatic Arthritis (PRESTA), a collaborative study that paired dermatologists and rheumatologists in an effort to determine how to optimize the use of etanercept to treat both skin and rheumatologic symptoms in these dual-diagnosis patients.

The PRESTA results will enable physicians to individualize treatment of such patients with a degree of confidence not before possible, Dr. Wolfram Sterry, one of the study investigators, said at the annual congress of the European Academy of Dermatology and Venereology.
PRESTA findings suggest that patients with more severe cutaneous disease are best off with an initial 12 weeks of etanercept (Enbrel) at 50 mg twice weekly before stepping down to 50 mg once weekly, while 50 mg/week from the outset is sufficient in patients with milder skin involvement, explained Dr. Sterry, professor and chair of the department of dermatology and allergy at Charité University Hospital, Berlin.
PRESTA also showed the ideal dosing regimen for psoriatic arthritis to be closer to that employed in rheumatoid arthritis than for that used in psoriasis, he added.
PRESTA was a 24-week, multicenter, randomized, double-blind clinical trial in 752 patients with both psoriasis evaluated by dermatologists and psoriatic arthritis evaluated by rheumatologists. Participants were assigned to subcutaneous etanercept at either 50 mg once or twice weekly for 12 weeks, after which everyone received open-label etanercept at 50 mg/week for a further 12 weeks.
The primary efficacy end point was the number of patients achieving “clear” or “almost clear” on the Physician’s Global Assessment of psoriasis at 12 weeks. This outcome was obtained in 46% (176 of 379) of patients on twice-weekly therapy, compared with 32% (119 of 373) of patients on once-weekly treatment, a significant difference, said Dr. Sterry. In addition, the twice-weekly etanercept group showed significantly greater improvement from baseline to week 12 in the Psoriasis Area and Severity Index: 71%, compared with 62% for once-weekly therapy.
In contrast, both groups were equally likely to meet American College of Rheumatology (ACR) criteria for treatment response. At week 12, an ACR 20% response was achieved by 66% of patients on twice-weekly and 61% on once-weekly etanercept. An ACR 50% response was documented in 45% of those on twice-weekly and 41% on once-weekly etanercept. An ACR 70% response occurred in 20% on twice-weekly and 22% on once-weekly therapy, he noted.
By week 24, 66% of patients who initially got twice-weekly etanercept had an ACR 20% response and 35% had an ACR 70% response, as did 61% and 37%, respectively, of patients on 50 mg once weekly for the full 24 weeks.
Of note, by 24 weeks both patient groups achieved comparable improvements in their skin disease. Patients who initially received twice-weekly etanercept had a 78% improvement from baseline in the psoriasis area and severity index, while those on once-weekly therapy all along had a 74% improvement.
Another noteworthy finding, said Dr. Sterry, is that the study showed definitively, for the first time, that etanercept significantly improves two clinically important extra-articular manifestations of psoriatic arthritis: dactylitis and enthesis. These disease manifestations were present at baseline in 42% and 38% of patients, respectively. By week 24, dactylitis scores had improved by a mean of 85%, compared with baseline in both study arms, while enthesis scores improved by 81%.
There were no significant differences in the safety profiles between the two study arms.
The prevalence of psoriatic arthritis among psoriasis patients is estimated to be up to 30%, noted Dr. Sterry. The arthritis component typically does not present until about a decade after onset of the skin disease. This was true in PRESTA: Participants had a mean 18.9-year duration of psoriasis, with psoriatic arthritis appearing 11.9 years after the skin disease.
Dr. Sterry disclosed that he has received consulting fees from Wyeth, which sponsored PRESTA, as well as from Abbott, Schering-Plough, and Janssen-Cilag.
GOTHENBURG, Sweden - In patients with both plaque psoriasis and psoriatic arthritis, initial treatment with etanercept at 50 mg twice weekly resulted in faster clearance of skin lesions than 50 mg once weekly.
The twice-weekly regimen, however, offered no advantage for treatment of joint and tendon symptoms. These were the key findings of the Psoriasis Randomized Etanercept Study in Subjects with Psoriatic Arthritis (PRESTA), a collaborative study that paired dermatologists and rheumatologists in an effort to determine how to optimize the use of etanercept to treat both skin and rheumatologic symptoms in these dual-diagnosis patients.
The PRESTA results will enable physicians to individualize treatment of such patients with a degree of confidence not before possible, Dr. Wolfram Sterry, one of the study investigators, said at the annual congress of the European Academy of Dermatology and Venereology.
PRESTA findings suggest that patients with more severe cutaneous disease are best off with an initial 12 weeks of etanercept (Enbrel) at 50 mg twice weekly before stepping down to 50 mg once weekly, while 50 mg/week from the outset is sufficient in patients with milder skin involvement, explained Dr. Sterry, professor and chair of the department of dermatology and allergy at Charité University Hospital, Berlin.
PRESTA also showed the ideal dosing regimen for psoriatic arthritis to be closer to that employed in rheumatoid arthritis than for that used in psoriasis, he added.
PRESTA was a 24-week, multicenter, randomized, double-blind clinical trial in 752 patients with both psoriasis evaluated by dermatologists and psoriatic arthritis evaluated by rheumatologists. Participants were assigned to subcutaneous etanercept at either 50 mg once or twice weekly for 12 weeks, after which everyone received open-label etanercept at 50 mg/week for a further 12 weeks.
The primary efficacy end point was the number of patients achieving “clear” or “almost clear” on the Physician’s Global Assessment of psoriasis at 12 weeks. This outcome was obtained in 46% (176 of 379) of patients on twice-weekly therapy, compared with 32% (119 of 373) of patients on once-weekly treatment, a significant difference, said Dr. Sterry. In addition, the twice-weekly etanercept group showed significantly greater improvement from baseline to week 12 in the Psoriasis Area and Severity Index: 71%, compared with 62% for once-weekly therapy.
In contrast, both groups were equally likely to meet American College of Rheumatology (ACR) criteria for treatment response. At week 12, an ACR 20% response was achieved by 66% of patients on twice-weekly and 61% on once-weekly etanercept. An ACR 50% response was documented in 45% of those on twice-weekly and 41% on once-weekly etanercept. An ACR 70% response occurred in 20% on twice-weekly and 22% on once-weekly therapy, he noted.
By week 24, 66% of patients who initially got twice-weekly etanercept had an ACR 20% response and 35% had an ACR 70% response, as did 61% and 37%, respectively, of patients on 50 mg once weekly for the full 24 weeks.
Of note, by 24 weeks both patient groups achieved comparable improvements in their skin disease. Patients who initially received twice-weekly etanercept had a 78% improvement from baseline in the psoriasis area and severity index, while those on once-weekly therapy all along had a 74% improvement.
Another noteworthy finding, said Dr. Sterry, is that the study showed definitively, for the first time, that etanercept significantly improves two clinically important extra-articular manifestations of psoriatic arthritis: dactylitis and enthesis. These disease manifestations were present at baseline in 42% and 38% of patients, respectively. By week 24, dactylitis scores had improved by a mean of 85%, compared with baseline in both study arms, while enthesis scores improved by 81%.
There were no significant differences in the safety profiles between the two study arms.
The prevalence of psoriatic arthritis among psoriasis patients is estimated to be up to 30%, noted Dr. Sterry. The arthritis component typically does not present until about a decade after onset of the skin disease. This was true in PRESTA: Participants had a mean 18.9-year duration of psoriasis, with psoriatic arthritis appearing 11.9 years after the skin disease.
Dr. Sterry disclosed that he has received consulting fees from Wyeth, which sponsored PRESTA, as well as from Abbott, Schering-Plough, and Janssen-Cilag.
GOTHENBURG, Sweden - In patients with both plaque psoriasis and psoriatic arthritis, initial treatment with etanercept at 50 mg twice weekly resulted in faster clearance of skin lesions than 50 mg once weekly.
The twice-weekly regimen, however, offered no advantage for treatment of joint and tendon symptoms. These were the key findings of the Psoriasis Randomized Etanercept Study in Subjects with Psoriatic Arthritis (PRESTA), a collaborative study that paired dermatologists and rheumatologists in an effort to determine how to optimize the use of etanercept to treat both skin and rheumatologic symptoms in these dual-diagnosis patients.
The PRESTA results will enable physicians to individualize treatment of such patients with a degree of confidence not before possible, Dr. Wolfram Sterry, one of the study investigators, said at the annual congress of the European Academy of Dermatology and Venereology.
PRESTA findings suggest that patients with more severe cutaneous disease are best off with an initial 12 weeks of etanercept (Enbrel) at 50 mg twice weekly before stepping down to 50 mg once weekly, while 50 mg/week from the outset is sufficient in patients with milder skin involvement, explained Dr. Sterry, professor and chair of the department of dermatology and allergy at Charité University Hospital, Berlin.
PRESTA also showed the ideal dosing regimen for psoriatic arthritis to be closer to that employed in rheumatoid arthritis than for that used in psoriasis, he added.
PRESTA was a 24-week, multicenter, randomized, double-blind clinical trial in 752 patients with both psoriasis evaluated by dermatologists and psoriatic arthritis evaluated by rheumatologists. Participants were assigned to subcutaneous etanercept at either 50 mg once or twice weekly for 12 weeks, after which everyone received open-label etanercept at 50 mg/week for a further 12 weeks.
The primary efficacy end point was the number of patients achieving “clear” or “almost clear” on the Physician’s Global Assessment of psoriasis at 12 weeks. This outcome was obtained in 46% (176 of 379) of patients on twice-weekly therapy, compared with 32% (119 of 373) of patients on once-weekly treatment, a significant difference, said Dr. Sterry. In addition, the twice-weekly etanercept group showed significantly greater improvement from baseline to week 12 in the Psoriasis Area and Severity Index: 71%, compared with 62% for once-weekly therapy.
In contrast, both groups were equally likely to meet American College of Rheumatology (ACR) criteria for treatment response. At week 12, an ACR 20% response was achieved by 66% of patients on twice-weekly and 61% on once-weekly etanercept. An ACR 50% response was documented in 45% of those on twice-weekly and 41% on once-weekly etanercept. An ACR 70% response occurred in 20% on twice-weekly and 22% on once-weekly therapy, he noted.
By week 24, 66% of patients who initially got twice-weekly etanercept had an ACR 20% response and 35% had an ACR 70% response, as did 61% and 37%, respectively, of patients on 50 mg once weekly for the full 24 weeks.
Of note, by 24 weeks both patient groups achieved comparable improvements in their skin disease. Patients who initially received twice-weekly etanercept had a 78% improvement from baseline in the psoriasis area and severity index, while those on once-weekly therapy all along had a 74% improvement.
Another noteworthy finding, said Dr. Sterry, is that the study showed definitively, for the first time, that etanercept significantly improves two clinically important extra-articular manifestations of psoriatic arthritis: dactylitis and enthesis. These disease manifestations were present at baseline in 42% and 38% of patients, respectively. By week 24, dactylitis scores had improved by a mean of 85%, compared with baseline in both study arms, while enthesis scores improved by 81%.
There were no significant differences in the safety profiles between the two study arms.
The prevalence of psoriatic arthritis among psoriasis patients is estimated to be up to 30%, noted Dr. Sterry. The arthritis component typically does not present until about a decade after onset of the skin disease. This was true in PRESTA: Participants had a mean 18.9-year duration of psoriasis, with psoriatic arthritis appearing 11.9 years after the skin disease.
Dr. Sterry disclosed that he has received consulting fees from Wyeth, which sponsored PRESTA, as well as from Abbott, Schering-Plough, and Janssen-Cilag.
Annual Congress of the European Academy of Dermatology and Venereology
Major Finding: After 24 weeks of etanercept therapy, dactylitis scores had improved by a mean of 85%, compared with baseline, while enthesis scores improved by 81%.
Data Source: A 24-week, multicenter, randomized, double-blind clinical trial of 752 patients with both psoriasis and psoriatic arthritis.
Disclosures: Dr. Sterry disclosed that he has received consulting fees from Wyeth, which sponsored PRESTA, as well as from Abbott, Schering-Plough, and Janssen-Cilag.
EADV: Implant Reduced Phototoxic Attacks in Erythropoietic Protoporphyria
GOTHENBURG, Sweden - A resorbable subcutaneous implant of afamelanotide significantly reduced painful phototoxic attacks in patients with erythropoietic protoporphyria in a year-long multinational clinical trial.
Afamelanotide, a first-in-class investigational chemical analog of alpha-melanocyte-stimulating hormone, is a melanocortin-1 agonist that activates melanocytes and increases melanin levels in the skin.
Treatment with the controlled-release afamelanotide implants was life changing for study participants, Dr. Elisabeth I. Minder noted at the annual congress of the European Academy of Dermatology and Venereology.
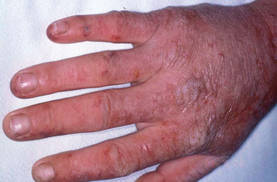
"You have to understand that EPP [erythropoietic protoporphyria] patients are conditioned early in life that sunlight is harmful: no exposure to sunlight, no painful attacks. Afamelanotide, compared to placebo, not only reduced phototoxic attacks, it also enabled patients to spend significantly more time outdoors. Anecdotally, patients were quite excited to explore new activities which were never possible in their lives previously," explained Dr. Minder of Triemli Hospital, Zurich.
EPP is a rare inherited disease characterized by severely painful, often intolerable phototoxic attacks lasting for days. An estimated 10,000 individuals are affected worldwide she said.
The afamelanotide implant (Scenesse) is also in clinical trials for more common diseases, including vitiligo, solar urticaria, and polymorphous light eruption, as well as for prevention of actinic keratoses and squamous cell carcinomas in organ transplant recipients.

The European phase III EPP trial involved 100 affected patients who were randomized double blind to a year's worth of alternating treatment cycles consisting of a subcutaneous implant of 16 mg of afamelanotide lasting for 60 days followed by 60 days of a placebo implant.
A total of 91 patients completed the study. The low dropout rate was testimony to how grateful participants were to finally have access to an affective therapy for their disorder, even if for only half of the study period, Dr. Minder said.
Daily pain diaries demonstrated that patients experienced significantly fewer days of severe, moderate, and mild pain during the periods in which they were being treated with afamelanotide. For example, patients spent one-third fewer days in severe pain and less than half as many days in moderate pain during the afamelanotide phase, compared with the placebo phase.
During the active-treatment periods, patients reported spending significantly more time outdoors exposed to the sun. More than half of patients reported spending greater than 6 hours per day outdoors while they were on afamelanotide, something that was unthinkable at baseline.
Only two adverse events were more frequent during active treatment: nausea, which occurred 1-2 days post-implant in 33% of patients when being treated with afamelanotide, compared with 18% of patients when being treated with placebo; and hot flushes, which were noted 1-2 days after afamelanotide implantation in 11% of patients, compared with just 1% with placebo.
Session chair Dr. Maurice Van Steensel of Maastricht (the Netherlands) University commented that a truly double-blind study is impossible with afamelanotide because the alpha-melanocyte-stimulating hormone analog causes tanning. For this very reason, he added, the tanning industry may be interested in this agent as a sunless tanning product.
The U.S. Food and Drug Administration announced in March that Clinuvel (afamelanotide’s manufacturer), could move forward with plans to conduct a phase II clinical trial for treating EPP patients. The confirmatory study is currently enrolling participants. The FDA also granted orphan drug status for the treatment for solar urticaria in late 2009.
In an earlier open-label pilot study, Dr. Minder and her associates showed that treatment with afamelanotide increased tolerance to exposure to artificial light under controlled conditions by 11-fold (N. Engl. J. Med. 2009;360:306-7).
The study was sponsored by Clinuvel Pharmaceuticals. Dr. Minder declared having no financial conflicts.
Having cared for patients with erythropoietic protoporphyria, as well as other forms of photosensitivity, I understand the need for effective therapies in this area. The results of the treatment in the study are impressive. However, I think further work needs to be done with this drug.
Vincent DeLeo, M.D. |

As pointed out by others, it is very difficult to run a completely blinded study with the drug since it does induce a visible tan so that both patients and evaluators are aware of the treatment group.
In addition, the end points studied are very subjective reports of pain.
More importantly, we should be concerned about the possibility of adverse effects of a drug which stimulates melanocytes.
It is imperative that long-term safety studies be done to ascertain the risk, if any, of this drug on the development or progression of malignant melanoma.
Dr. DeLeo is chairman of the department of dermatology at St. Luke's-Roosevelt Hospital Center and Beth Israel Medical Center in New York
Having cared for patients with erythropoietic protoporphyria, as well as other forms of photosensitivity, I understand the need for effective therapies in this area. The results of the treatment in the study are impressive. However, I think further work needs to be done with this drug.
Vincent DeLeo, M.D. |
As pointed out by others, it is very difficult to run a completely blinded study with the drug since it does induce a visible tan so that both patients and evaluators are aware of the treatment group.
In addition, the end points studied are very subjective reports of pain.
More importantly, we should be concerned about the possibility of adverse effects of a drug which stimulates melanocytes.
It is imperative that long-term safety studies be done to ascertain the risk, if any, of this drug on the development or progression of malignant melanoma.
Dr. DeLeo is chairman of the department of dermatology at St. Luke's-Roosevelt Hospital Center and Beth Israel Medical Center in New York
Having cared for patients with erythropoietic protoporphyria, as well as other forms of photosensitivity, I understand the need for effective therapies in this area. The results of the treatment in the study are impressive. However, I think further work needs to be done with this drug.
Vincent DeLeo, M.D. |
As pointed out by others, it is very difficult to run a completely blinded study with the drug since it does induce a visible tan so that both patients and evaluators are aware of the treatment group.
In addition, the end points studied are very subjective reports of pain.
More importantly, we should be concerned about the possibility of adverse effects of a drug which stimulates melanocytes.
It is imperative that long-term safety studies be done to ascertain the risk, if any, of this drug on the development or progression of malignant melanoma.
Dr. DeLeo is chairman of the department of dermatology at St. Luke's-Roosevelt Hospital Center and Beth Israel Medical Center in New York
GOTHENBURG, Sweden - A resorbable subcutaneous implant of afamelanotide significantly reduced painful phototoxic attacks in patients with erythropoietic protoporphyria in a year-long multinational clinical trial.
Afamelanotide, a first-in-class investigational chemical analog of alpha-melanocyte-stimulating hormone, is a melanocortin-1 agonist that activates melanocytes and increases melanin levels in the skin.
Treatment with the controlled-release afamelanotide implants was life changing for study participants, Dr. Elisabeth I. Minder noted at the annual congress of the European Academy of Dermatology and Venereology.
"You have to understand that EPP [erythropoietic protoporphyria] patients are conditioned early in life that sunlight is harmful: no exposure to sunlight, no painful attacks. Afamelanotide, compared to placebo, not only reduced phototoxic attacks, it also enabled patients to spend significantly more time outdoors. Anecdotally, patients were quite excited to explore new activities which were never possible in their lives previously," explained Dr. Minder of Triemli Hospital, Zurich.
EPP is a rare inherited disease characterized by severely painful, often intolerable phototoxic attacks lasting for days. An estimated 10,000 individuals are affected worldwide she said.
The afamelanotide implant (Scenesse) is also in clinical trials for more common diseases, including vitiligo, solar urticaria, and polymorphous light eruption, as well as for prevention of actinic keratoses and squamous cell carcinomas in organ transplant recipients.
The European phase III EPP trial involved 100 affected patients who were randomized double blind to a year's worth of alternating treatment cycles consisting of a subcutaneous implant of 16 mg of afamelanotide lasting for 60 days followed by 60 days of a placebo implant.
A total of 91 patients completed the study. The low dropout rate was testimony to how grateful participants were to finally have access to an affective therapy for their disorder, even if for only half of the study period, Dr. Minder said.
Daily pain diaries demonstrated that patients experienced significantly fewer days of severe, moderate, and mild pain during the periods in which they were being treated with afamelanotide. For example, patients spent one-third fewer days in severe pain and less than half as many days in moderate pain during the afamelanotide phase, compared with the placebo phase.
During the active-treatment periods, patients reported spending significantly more time outdoors exposed to the sun. More than half of patients reported spending greater than 6 hours per day outdoors while they were on afamelanotide, something that was unthinkable at baseline.
Only two adverse events were more frequent during active treatment: nausea, which occurred 1-2 days post-implant in 33% of patients when being treated with afamelanotide, compared with 18% of patients when being treated with placebo; and hot flushes, which were noted 1-2 days after afamelanotide implantation in 11% of patients, compared with just 1% with placebo.
Session chair Dr. Maurice Van Steensel of Maastricht (the Netherlands) University commented that a truly double-blind study is impossible with afamelanotide because the alpha-melanocyte-stimulating hormone analog causes tanning. For this very reason, he added, the tanning industry may be interested in this agent as a sunless tanning product.
The U.S. Food and Drug Administration announced in March that Clinuvel (afamelanotide’s manufacturer), could move forward with plans to conduct a phase II clinical trial for treating EPP patients. The confirmatory study is currently enrolling participants. The FDA also granted orphan drug status for the treatment for solar urticaria in late 2009.
In an earlier open-label pilot study, Dr. Minder and her associates showed that treatment with afamelanotide increased tolerance to exposure to artificial light under controlled conditions by 11-fold (N. Engl. J. Med. 2009;360:306-7).
The study was sponsored by Clinuvel Pharmaceuticals. Dr. Minder declared having no financial conflicts.
GOTHENBURG, Sweden - A resorbable subcutaneous implant of afamelanotide significantly reduced painful phototoxic attacks in patients with erythropoietic protoporphyria in a year-long multinational clinical trial.
Afamelanotide, a first-in-class investigational chemical analog of alpha-melanocyte-stimulating hormone, is a melanocortin-1 agonist that activates melanocytes and increases melanin levels in the skin.
Treatment with the controlled-release afamelanotide implants was life changing for study participants, Dr. Elisabeth I. Minder noted at the annual congress of the European Academy of Dermatology and Venereology.
"You have to understand that EPP [erythropoietic protoporphyria] patients are conditioned early in life that sunlight is harmful: no exposure to sunlight, no painful attacks. Afamelanotide, compared to placebo, not only reduced phototoxic attacks, it also enabled patients to spend significantly more time outdoors. Anecdotally, patients were quite excited to explore new activities which were never possible in their lives previously," explained Dr. Minder of Triemli Hospital, Zurich.
EPP is a rare inherited disease characterized by severely painful, often intolerable phototoxic attacks lasting for days. An estimated 10,000 individuals are affected worldwide she said.
The afamelanotide implant (Scenesse) is also in clinical trials for more common diseases, including vitiligo, solar urticaria, and polymorphous light eruption, as well as for prevention of actinic keratoses and squamous cell carcinomas in organ transplant recipients.
The European phase III EPP trial involved 100 affected patients who were randomized double blind to a year's worth of alternating treatment cycles consisting of a subcutaneous implant of 16 mg of afamelanotide lasting for 60 days followed by 60 days of a placebo implant.
A total of 91 patients completed the study. The low dropout rate was testimony to how grateful participants were to finally have access to an affective therapy for their disorder, even if for only half of the study period, Dr. Minder said.
Daily pain diaries demonstrated that patients experienced significantly fewer days of severe, moderate, and mild pain during the periods in which they were being treated with afamelanotide. For example, patients spent one-third fewer days in severe pain and less than half as many days in moderate pain during the afamelanotide phase, compared with the placebo phase.
During the active-treatment periods, patients reported spending significantly more time outdoors exposed to the sun. More than half of patients reported spending greater than 6 hours per day outdoors while they were on afamelanotide, something that was unthinkable at baseline.
Only two adverse events were more frequent during active treatment: nausea, which occurred 1-2 days post-implant in 33% of patients when being treated with afamelanotide, compared with 18% of patients when being treated with placebo; and hot flushes, which were noted 1-2 days after afamelanotide implantation in 11% of patients, compared with just 1% with placebo.
Session chair Dr. Maurice Van Steensel of Maastricht (the Netherlands) University commented that a truly double-blind study is impossible with afamelanotide because the alpha-melanocyte-stimulating hormone analog causes tanning. For this very reason, he added, the tanning industry may be interested in this agent as a sunless tanning product.
The U.S. Food and Drug Administration announced in March that Clinuvel (afamelanotide’s manufacturer), could move forward with plans to conduct a phase II clinical trial for treating EPP patients. The confirmatory study is currently enrolling participants. The FDA also granted orphan drug status for the treatment for solar urticaria in late 2009.
In an earlier open-label pilot study, Dr. Minder and her associates showed that treatment with afamelanotide increased tolerance to exposure to artificial light under controlled conditions by 11-fold (N. Engl. J. Med. 2009;360:306-7).
The study was sponsored by Clinuvel Pharmaceuticals. Dr. Minder declared having no financial conflicts.
From the Annual Congress of the European Academy of Dermatology and Venereology
Major Finding: During the active-treatment periods, more than half of
patients reported spending greater than 6 hours per day outdoors while
they were on afamelanotide.
Data Source: A European trial that involved 100 affected patients who were
randomized double blind to a year’s worth of alternating treatment
cycles consisting of a subcutaneous implant of 16 mg of afamelanotide
lasting for 60 days followed by 60 days of a placebo implant..
Disclosures: The study was sponsored by Clinuvel Pharmaceuticals. Dr. Minder declared having no financial conflicts.
Psoriatic Arthritis Patients May Be Predisposed to Depression
EDINBURGH - Depression is common, underdiagnosed, and undertreated in patients with psoriatic arthritis.
Psychiatric evaluation of 50 consecutive patients at the University of Glasgow psoriatic arthritis clinic indicated that 15 patients, or 30%, were depressed. Three were rated as severely depressed based on their scores on the Hospital Anxiety and Depression Scale (HADS), while 12 others had moderate depression, Dr. Rajeev Krishnadas reported at the International Congress of the Royal College of Psychiatrists.
This high prevalence of depression in psoriatic arthritis patients is consistent with reports in the dermatologic literature (Br. J. Dermatol. 2008;159:704-10).
Of note, none of the depressed Scottish psoriatic arthritis patients was on a therapeutic dose of an antidepressant, added Dr. Krishnadas of the Sackler Institute of Psychobiological Research, Southern General Hospital, Glasgow.
This study is part of a larger ongoing investigation looking at the relationship between systemic inflammation and depression in patients with rheumatoid arthritis or psoriatic arthritis. In this portion of the study, a positive association was noted between HADS scores and C-reactive protein levels in the psoriatic arthritis cohort, although it must be noted that CRP scores accounted for only 7% of the overall variance in HADS scores.
Not surprisingly, higher HADS scores were associated with worse quality of life as assessed by the Dermatology Quality of Life Questionnaire as well as with higher scores on a self-rated pain scale.
Of greater interest psychodynamically, a negative correlation was found between HADS scores and emotional intelligence as measured by the Trait Emotional Intelligence Questionnaire – Short Form. High trait emotional intelligence reflects greater awareness of one's own feelings as well as the feelings of others'. Individuals with high emotional intelligence are far better able to regulate their emotions than are those with lower trait emotional intelligence.
Patients who scored high in trait emotional intelligence had higher quality of life scores, lower CRP levels, and lower scores on the pain scale.
These findings are consistent with the hypothesis that a poor ability to be aware of and regulate one's emotions predisposes to depression in the presence of a chronic medical condition or other major stressor, according to Dr. Krishnadas.
He declared having no conflicts of interest.
EDINBURGH - Depression is common, underdiagnosed, and undertreated in patients with psoriatic arthritis.
Psychiatric evaluation of 50 consecutive patients at the University of Glasgow psoriatic arthritis clinic indicated that 15 patients, or 30%, were depressed. Three were rated as severely depressed based on their scores on the Hospital Anxiety and Depression Scale (HADS), while 12 others had moderate depression, Dr. Rajeev Krishnadas reported at the International Congress of the Royal College of Psychiatrists.
This high prevalence of depression in psoriatic arthritis patients is consistent with reports in the dermatologic literature (Br. J. Dermatol. 2008;159:704-10).
Of note, none of the depressed Scottish psoriatic arthritis patients was on a therapeutic dose of an antidepressant, added Dr. Krishnadas of the Sackler Institute of Psychobiological Research, Southern General Hospital, Glasgow.
This study is part of a larger ongoing investigation looking at the relationship between systemic inflammation and depression in patients with rheumatoid arthritis or psoriatic arthritis. In this portion of the study, a positive association was noted between HADS scores and C-reactive protein levels in the psoriatic arthritis cohort, although it must be noted that CRP scores accounted for only 7% of the overall variance in HADS scores.
Not surprisingly, higher HADS scores were associated with worse quality of life as assessed by the Dermatology Quality of Life Questionnaire as well as with higher scores on a self-rated pain scale.
Of greater interest psychodynamically, a negative correlation was found between HADS scores and emotional intelligence as measured by the Trait Emotional Intelligence Questionnaire – Short Form. High trait emotional intelligence reflects greater awareness of one's own feelings as well as the feelings of others'. Individuals with high emotional intelligence are far better able to regulate their emotions than are those with lower trait emotional intelligence.
Patients who scored high in trait emotional intelligence had higher quality of life scores, lower CRP levels, and lower scores on the pain scale.
These findings are consistent with the hypothesis that a poor ability to be aware of and regulate one's emotions predisposes to depression in the presence of a chronic medical condition or other major stressor, according to Dr. Krishnadas.
He declared having no conflicts of interest.
EDINBURGH - Depression is common, underdiagnosed, and undertreated in patients with psoriatic arthritis.
Psychiatric evaluation of 50 consecutive patients at the University of Glasgow psoriatic arthritis clinic indicated that 15 patients, or 30%, were depressed. Three were rated as severely depressed based on their scores on the Hospital Anxiety and Depression Scale (HADS), while 12 others had moderate depression, Dr. Rajeev Krishnadas reported at the International Congress of the Royal College of Psychiatrists.
This high prevalence of depression in psoriatic arthritis patients is consistent with reports in the dermatologic literature (Br. J. Dermatol. 2008;159:704-10).
Of note, none of the depressed Scottish psoriatic arthritis patients was on a therapeutic dose of an antidepressant, added Dr. Krishnadas of the Sackler Institute of Psychobiological Research, Southern General Hospital, Glasgow.
This study is part of a larger ongoing investigation looking at the relationship between systemic inflammation and depression in patients with rheumatoid arthritis or psoriatic arthritis. In this portion of the study, a positive association was noted between HADS scores and C-reactive protein levels in the psoriatic arthritis cohort, although it must be noted that CRP scores accounted for only 7% of the overall variance in HADS scores.
Not surprisingly, higher HADS scores were associated with worse quality of life as assessed by the Dermatology Quality of Life Questionnaire as well as with higher scores on a self-rated pain scale.
Of greater interest psychodynamically, a negative correlation was found between HADS scores and emotional intelligence as measured by the Trait Emotional Intelligence Questionnaire – Short Form. High trait emotional intelligence reflects greater awareness of one's own feelings as well as the feelings of others'. Individuals with high emotional intelligence are far better able to regulate their emotions than are those with lower trait emotional intelligence.
Patients who scored high in trait emotional intelligence had higher quality of life scores, lower CRP levels, and lower scores on the pain scale.
These findings are consistent with the hypothesis that a poor ability to be aware of and regulate one's emotions predisposes to depression in the presence of a chronic medical condition or other major stressor, according to Dr. Krishnadas.
He declared having no conflicts of interest.
from the annual International Congress of the Royal College of Psychiatrists
EADV: Cutaneous Lupus Rapidly Progresses to SLE, New Study Finds
GOTHENBURG, Sweden – Rapid progression of cutaneous lupus erythematosus to SLE occurs considerably more often than previously recognized, a comprehensive nationwide Swedish study found.
The population-based study, which included all 1,088 patients diagnosed with CLE in Sweden from 2005 to 2007, found that the risk of being diagnosed with SLE during the first year of CLE diagnosis was 12%. The 3-year rate of progression to SLE was 18%, Dr. Carina M. Grönhagen reported at the annual congress of the European Academy of Dermatology and Venereology.

Moreover, 24% of patients already carried the diagnosis of SLE at the time they were diagnosed with CLE, added Dr. Grönhagen, a dermatology resident at the Karolinska Institutet, Danderyd Hospital, Stockholm.
Patients with the subacute form of CLE had the highest rate of early progression to SLE, with a 3-year incidence of nearly 25%. Patients with subacute CLE accounted for 15.7% of the overall Swedish CLE cohort.
Discoid CLE accounted for 79.8% of all cases of CLE diagnosed in Sweden during the study period, while lupus panniculitis and other less common forms of local LE comprised the remainder.
Women with newly diagnosed CLE had a 3-year rate of progression to SLE of 20%, twice that of men. The incidence of CLE was threefold greater in Swedish women than men.
Patients with newly diagnosed CLE should be informed of the risk of early progression to SLE, Dr. Grönhagen said. They should be made aware of the systemic signs and symptoms, such as fevers and joint pains, so appropriate treatment for SLE is not delayed.
Her study relied upon Swedish National Patient Register data, which, since 2001, has included outpatient specialist care as well as inpatient care.
The incidence of CLE in Sweden, based on these data, is 4.0 per 100,000 population per year. The peak incidence of CLE in women was in the 65- to 74-year-old group, while the peak in men was at age 55-64. This pattern was skewed in the subset of patients with subacute CLE; their peak incidence was later, and there were far fewer cases diagnosed before age 45 than for discoid and other forms of CLE.
"One theory behind this is that the subacute form is drug induced to a larger extent than previously believed. An important known trigger for subacute CLE is antihypertensive drugs, which are mostly taken by people age 45 and older," Dr. Grönhagen said.
During her presentation at the meeting of the European Society of Cutaneous Lupus Erythematosus, held in conjunction with the EADV congress, audience members noted that there has been a dearth of population-based studies reporting the incidence of isolated CLE. They also commented on the large size of the population in Dr. Grönhagen’s study and the 3-year follow-up.
Audience members noted that the Swedish national study, since it included outpatients as well as inpatients, is relatively immune to selection bias. Although it was noted that the reported CLE incidence of 4.0/100,000 is up to 10-fold lower than cited in some other studies, Dr. Grönhagen countered that prior studies have been much smaller and less comprehensive.
Still, she said, the 4.0/100,000 incidence figure is probably an underestimate. It is likely that the mildest cases of discoid LE were never referred to a dermatologist or other specialist and, hence, weren't recorded in the outpatient portion of the national registry.
Dr. Grönhagen declared having no relevant financial interests.
GOTHENBURG, Sweden – Rapid progression of cutaneous lupus erythematosus to SLE occurs considerably more often than previously recognized, a comprehensive nationwide Swedish study found.
The population-based study, which included all 1,088 patients diagnosed with CLE in Sweden from 2005 to 2007, found that the risk of being diagnosed with SLE during the first year of CLE diagnosis was 12%. The 3-year rate of progression to SLE was 18%, Dr. Carina M. Grönhagen reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, 24% of patients already carried the diagnosis of SLE at the time they were diagnosed with CLE, added Dr. Grönhagen, a dermatology resident at the Karolinska Institutet, Danderyd Hospital, Stockholm.
Patients with the subacute form of CLE had the highest rate of early progression to SLE, with a 3-year incidence of nearly 25%. Patients with subacute CLE accounted for 15.7% of the overall Swedish CLE cohort.
Discoid CLE accounted for 79.8% of all cases of CLE diagnosed in Sweden during the study period, while lupus panniculitis and other less common forms of local LE comprised the remainder.
Women with newly diagnosed CLE had a 3-year rate of progression to SLE of 20%, twice that of men. The incidence of CLE was threefold greater in Swedish women than men.
Patients with newly diagnosed CLE should be informed of the risk of early progression to SLE, Dr. Grönhagen said. They should be made aware of the systemic signs and symptoms, such as fevers and joint pains, so appropriate treatment for SLE is not delayed.
Her study relied upon Swedish National Patient Register data, which, since 2001, has included outpatient specialist care as well as inpatient care.
The incidence of CLE in Sweden, based on these data, is 4.0 per 100,000 population per year. The peak incidence of CLE in women was in the 65- to 74-year-old group, while the peak in men was at age 55-64. This pattern was skewed in the subset of patients with subacute CLE; their peak incidence was later, and there were far fewer cases diagnosed before age 45 than for discoid and other forms of CLE.
"One theory behind this is that the subacute form is drug induced to a larger extent than previously believed. An important known trigger for subacute CLE is antihypertensive drugs, which are mostly taken by people age 45 and older," Dr. Grönhagen said.
During her presentation at the meeting of the European Society of Cutaneous Lupus Erythematosus, held in conjunction with the EADV congress, audience members noted that there has been a dearth of population-based studies reporting the incidence of isolated CLE. They also commented on the large size of the population in Dr. Grönhagen’s study and the 3-year follow-up.
Audience members noted that the Swedish national study, since it included outpatients as well as inpatients, is relatively immune to selection bias. Although it was noted that the reported CLE incidence of 4.0/100,000 is up to 10-fold lower than cited in some other studies, Dr. Grönhagen countered that prior studies have been much smaller and less comprehensive.
Still, she said, the 4.0/100,000 incidence figure is probably an underestimate. It is likely that the mildest cases of discoid LE were never referred to a dermatologist or other specialist and, hence, weren't recorded in the outpatient portion of the national registry.
Dr. Grönhagen declared having no relevant financial interests.
GOTHENBURG, Sweden – Rapid progression of cutaneous lupus erythematosus to SLE occurs considerably more often than previously recognized, a comprehensive nationwide Swedish study found.
The population-based study, which included all 1,088 patients diagnosed with CLE in Sweden from 2005 to 2007, found that the risk of being diagnosed with SLE during the first year of CLE diagnosis was 12%. The 3-year rate of progression to SLE was 18%, Dr. Carina M. Grönhagen reported at the annual congress of the European Academy of Dermatology and Venereology.
Moreover, 24% of patients already carried the diagnosis of SLE at the time they were diagnosed with CLE, added Dr. Grönhagen, a dermatology resident at the Karolinska Institutet, Danderyd Hospital, Stockholm.
Patients with the subacute form of CLE had the highest rate of early progression to SLE, with a 3-year incidence of nearly 25%. Patients with subacute CLE accounted for 15.7% of the overall Swedish CLE cohort.
Discoid CLE accounted for 79.8% of all cases of CLE diagnosed in Sweden during the study period, while lupus panniculitis and other less common forms of local LE comprised the remainder.
Women with newly diagnosed CLE had a 3-year rate of progression to SLE of 20%, twice that of men. The incidence of CLE was threefold greater in Swedish women than men.
Patients with newly diagnosed CLE should be informed of the risk of early progression to SLE, Dr. Grönhagen said. They should be made aware of the systemic signs and symptoms, such as fevers and joint pains, so appropriate treatment for SLE is not delayed.
Her study relied upon Swedish National Patient Register data, which, since 2001, has included outpatient specialist care as well as inpatient care.
The incidence of CLE in Sweden, based on these data, is 4.0 per 100,000 population per year. The peak incidence of CLE in women was in the 65- to 74-year-old group, while the peak in men was at age 55-64. This pattern was skewed in the subset of patients with subacute CLE; their peak incidence was later, and there were far fewer cases diagnosed before age 45 than for discoid and other forms of CLE.
"One theory behind this is that the subacute form is drug induced to a larger extent than previously believed. An important known trigger for subacute CLE is antihypertensive drugs, which are mostly taken by people age 45 and older," Dr. Grönhagen said.
During her presentation at the meeting of the European Society of Cutaneous Lupus Erythematosus, held in conjunction with the EADV congress, audience members noted that there has been a dearth of population-based studies reporting the incidence of isolated CLE. They also commented on the large size of the population in Dr. Grönhagen’s study and the 3-year follow-up.
Audience members noted that the Swedish national study, since it included outpatients as well as inpatients, is relatively immune to selection bias. Although it was noted that the reported CLE incidence of 4.0/100,000 is up to 10-fold lower than cited in some other studies, Dr. Grönhagen countered that prior studies have been much smaller and less comprehensive.
Still, she said, the 4.0/100,000 incidence figure is probably an underestimate. It is likely that the mildest cases of discoid LE were never referred to a dermatologist or other specialist and, hence, weren't recorded in the outpatient portion of the national registry.
Dr. Grönhagen declared having no relevant financial interests.
FROM THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
NICE Draft Nixes Golimumab for Psoriatic Arthritis
The clinical effectiveness agency for England and Wales said in draft guidance Oct. 6 that it would not recommend golimumab to treat psoriatic arthritis, saying that it was less effective than another drug in the same class, and that it may in practice be costlier than its manufacturer says.
In August, the National Institute for Health and Clinical Excellence recommended three similar drugs for adults with psoriatic arthritis who have not responded well to standard disease-modifying anti-rheumatic therapies, such as methotrexate and sulfasalazine. All three – adalimumab, etanercept and infliximab – are TNF-inhibitors, human monoclonal antibodies that inhibit the binding of tumor necrosis factor to its receptors.
Treatment with etanercerpt (Enbrel, Wyeth Pharmaceuticals) costs £9,295 ($14,782.50) per year assuming a 50 mg once-weekly dose (52 doses per year) or 25 mg twice-weekly dose (104 doses per year). Treatment with adalimumab (Humira, Abbot Laboratories) costs £9,295 ($14,782.50) per year, assuming a 40-mg dose given every two weeks (26 doses per year). Treatment with infliximab (Remicade, Schering-Plough) for an adult weighing 75 kg costs £10,910 ($17,352.16) per year based on infusions repeated every 8 weeks (average of 6.5 doses per year), according to a statement on psoriatic arthritis treatment issued by NICE in August.
Golimumab (Simponi, Schering-Plough/Centocor), also a TNF-inhibitor, is administered via monthly self-injections of 50 mg (or 100 mg for people weighing at least 100 kg). Treatment costs an estimated £9,295 (14,782.50) per year, on par with etanercept and adalimumab, for a normal-weight person. The NICE reviewers said, however, they believed a "significant" portion of patients would be eligible for the higher dose, which is double the cost, making cost-effectiveness estimates for golimumab less favorable.
The NICE reviewers said they did consider the drug's monthly dosing as an advantage to patients, compared with the other TNF inhibitors. They also determined, based on the results of one Phase III manufacturer-sponsored randomized controlled trial of 405 patients, that golimumab 50 mg was effective compared with placebo (Arthritis Rheum. 2009;60:976-86). However, a mixed treatment comparison presented by the manufacturer showed that it was not as effective as etanercept for the same patient group.
The reviewers also noted some safety concerns about golimumab, including a lack of long-term safety data; some adverse events reported in the randomized controlled trial (though the trial was not powered to detect statistically significant differences in adverse effect outcomes); and concerns related to its 12-day half life. For example, they said, "the longer retreatment interval with golimumab may result in longer periods of discomfort because of waning efficacy before retreatment," and in the case of an adverse event, "it would take longer for the treatment effect to wear off."
NICE's guidance on golimumab is not yet final; the agency is accepting comments until Oct. 27.
The clinical effectiveness agency for England and Wales said in draft guidance Oct. 6 that it would not recommend golimumab to treat psoriatic arthritis, saying that it was less effective than another drug in the same class, and that it may in practice be costlier than its manufacturer says.
In August, the National Institute for Health and Clinical Excellence recommended three similar drugs for adults with psoriatic arthritis who have not responded well to standard disease-modifying anti-rheumatic therapies, such as methotrexate and sulfasalazine. All three – adalimumab, etanercept and infliximab – are TNF-inhibitors, human monoclonal antibodies that inhibit the binding of tumor necrosis factor to its receptors.
Treatment with etanercerpt (Enbrel, Wyeth Pharmaceuticals) costs £9,295 ($14,782.50) per year assuming a 50 mg once-weekly dose (52 doses per year) or 25 mg twice-weekly dose (104 doses per year). Treatment with adalimumab (Humira, Abbot Laboratories) costs £9,295 ($14,782.50) per year, assuming a 40-mg dose given every two weeks (26 doses per year). Treatment with infliximab (Remicade, Schering-Plough) for an adult weighing 75 kg costs £10,910 ($17,352.16) per year based on infusions repeated every 8 weeks (average of 6.5 doses per year), according to a statement on psoriatic arthritis treatment issued by NICE in August.
Golimumab (Simponi, Schering-Plough/Centocor), also a TNF-inhibitor, is administered via monthly self-injections of 50 mg (or 100 mg for people weighing at least 100 kg). Treatment costs an estimated £9,295 (14,782.50) per year, on par with etanercept and adalimumab, for a normal-weight person. The NICE reviewers said, however, they believed a "significant" portion of patients would be eligible for the higher dose, which is double the cost, making cost-effectiveness estimates for golimumab less favorable.
The NICE reviewers said they did consider the drug's monthly dosing as an advantage to patients, compared with the other TNF inhibitors. They also determined, based on the results of one Phase III manufacturer-sponsored randomized controlled trial of 405 patients, that golimumab 50 mg was effective compared with placebo (Arthritis Rheum. 2009;60:976-86). However, a mixed treatment comparison presented by the manufacturer showed that it was not as effective as etanercept for the same patient group.
The reviewers also noted some safety concerns about golimumab, including a lack of long-term safety data; some adverse events reported in the randomized controlled trial (though the trial was not powered to detect statistically significant differences in adverse effect outcomes); and concerns related to its 12-day half life. For example, they said, "the longer retreatment interval with golimumab may result in longer periods of discomfort because of waning efficacy before retreatment," and in the case of an adverse event, "it would take longer for the treatment effect to wear off."
NICE's guidance on golimumab is not yet final; the agency is accepting comments until Oct. 27.
The clinical effectiveness agency for England and Wales said in draft guidance Oct. 6 that it would not recommend golimumab to treat psoriatic arthritis, saying that it was less effective than another drug in the same class, and that it may in practice be costlier than its manufacturer says.
In August, the National Institute for Health and Clinical Excellence recommended three similar drugs for adults with psoriatic arthritis who have not responded well to standard disease-modifying anti-rheumatic therapies, such as methotrexate and sulfasalazine. All three – adalimumab, etanercept and infliximab – are TNF-inhibitors, human monoclonal antibodies that inhibit the binding of tumor necrosis factor to its receptors.
Treatment with etanercerpt (Enbrel, Wyeth Pharmaceuticals) costs £9,295 ($14,782.50) per year assuming a 50 mg once-weekly dose (52 doses per year) or 25 mg twice-weekly dose (104 doses per year). Treatment with adalimumab (Humira, Abbot Laboratories) costs £9,295 ($14,782.50) per year, assuming a 40-mg dose given every two weeks (26 doses per year). Treatment with infliximab (Remicade, Schering-Plough) for an adult weighing 75 kg costs £10,910 ($17,352.16) per year based on infusions repeated every 8 weeks (average of 6.5 doses per year), according to a statement on psoriatic arthritis treatment issued by NICE in August.
Golimumab (Simponi, Schering-Plough/Centocor), also a TNF-inhibitor, is administered via monthly self-injections of 50 mg (or 100 mg for people weighing at least 100 kg). Treatment costs an estimated £9,295 (14,782.50) per year, on par with etanercept and adalimumab, for a normal-weight person. The NICE reviewers said, however, they believed a "significant" portion of patients would be eligible for the higher dose, which is double the cost, making cost-effectiveness estimates for golimumab less favorable.
The NICE reviewers said they did consider the drug's monthly dosing as an advantage to patients, compared with the other TNF inhibitors. They also determined, based on the results of one Phase III manufacturer-sponsored randomized controlled trial of 405 patients, that golimumab 50 mg was effective compared with placebo (Arthritis Rheum. 2009;60:976-86). However, a mixed treatment comparison presented by the manufacturer showed that it was not as effective as etanercept for the same patient group.
The reviewers also noted some safety concerns about golimumab, including a lack of long-term safety data; some adverse events reported in the randomized controlled trial (though the trial was not powered to detect statistically significant differences in adverse effect outcomes); and concerns related to its 12-day half life. For example, they said, "the longer retreatment interval with golimumab may result in longer periods of discomfort because of waning efficacy before retreatment," and in the case of an adverse event, "it would take longer for the treatment effect to wear off."
NICE's guidance on golimumab is not yet final; the agency is accepting comments until Oct. 27.
FDA Bans Marketing of Unapproved Forms of Colchicine
Companies that manufacture, distribute, and/or market unapproved formulations of the oral, single ingredient forms of the gout drug colchicine have been ordered to stop doing so, the Food and Drug Administration announced on Sept. 30.
The companies must stop manufacturing these products within 45 days of the announcement and stop shipping unapproved colchicine within 90 days of the announcement, according to the statement, which says that a small amount of unapproved colchicine will be available after these dates, until supplies are used up.
Oral colchicine products are among the drugs that have been on the market for so many years that they were never subjected to the current prescription drug approval process. The FDA has launched an initiative to target the manufacturers of colchicine and other older, unapproved drugs to conduct the necessary testing to meet current approval standards or take them off the market.
Once the older products are off the market, the only oral colchicine product that will be available is Colcrys, marketed by Mutual Pharmaceuticals/URL Pharma, which was approved in 2009, after the company met the current drug approval requirements. As a result, the drug’s label includes information on safety data, drug interactions, and dosing that is not available for the older unapproved versions, according to the FDA.
The FDA's approval of Colcrys and the plans to ban the marketing of the older products has been sharply criticized by some clinicians who treat patients with gout because Colcrys is significantly more expensive than the older, unapproved formulations.
The FDA statement notes that the manufacturer of Colcrys has established a patient assistance program for patients who can’t afford the brand-name drug and those on Medicare who don’t want the cost of the drug to contribute to their out-of-pocket Medicare Part D expenses. More information about that program is available at the Colcrys Web site or at Needy Meds, an umbrella organization that offers patient assistance, or by calling 888-811-8423. The company has said that the program will continue "at a minimum until there is FDA-approved generic competition" for Colcrys, according to the statement.
The agency is encouraging manufacturers of such products to pursue approval of their products or "to face the type of action announced today," Deborah Autor, director of the office of compliance at the FDA's Center for Drug Evaluation and Research (CDER), said in the statement.
The FDA ordered a halt to unapproved colchicine for injection products in 2008.
Companies that manufacture, distribute, and/or market unapproved formulations of the oral, single ingredient forms of the gout drug colchicine have been ordered to stop doing so, the Food and Drug Administration announced on Sept. 30.
The companies must stop manufacturing these products within 45 days of the announcement and stop shipping unapproved colchicine within 90 days of the announcement, according to the statement, which says that a small amount of unapproved colchicine will be available after these dates, until supplies are used up.
Oral colchicine products are among the drugs that have been on the market for so many years that they were never subjected to the current prescription drug approval process. The FDA has launched an initiative to target the manufacturers of colchicine and other older, unapproved drugs to conduct the necessary testing to meet current approval standards or take them off the market.
Once the older products are off the market, the only oral colchicine product that will be available is Colcrys, marketed by Mutual Pharmaceuticals/URL Pharma, which was approved in 2009, after the company met the current drug approval requirements. As a result, the drug’s label includes information on safety data, drug interactions, and dosing that is not available for the older unapproved versions, according to the FDA.
The FDA's approval of Colcrys and the plans to ban the marketing of the older products has been sharply criticized by some clinicians who treat patients with gout because Colcrys is significantly more expensive than the older, unapproved formulations.
The FDA statement notes that the manufacturer of Colcrys has established a patient assistance program for patients who can’t afford the brand-name drug and those on Medicare who don’t want the cost of the drug to contribute to their out-of-pocket Medicare Part D expenses. More information about that program is available at the Colcrys Web site or at Needy Meds, an umbrella organization that offers patient assistance, or by calling 888-811-8423. The company has said that the program will continue "at a minimum until there is FDA-approved generic competition" for Colcrys, according to the statement.
The agency is encouraging manufacturers of such products to pursue approval of their products or "to face the type of action announced today," Deborah Autor, director of the office of compliance at the FDA's Center for Drug Evaluation and Research (CDER), said in the statement.
The FDA ordered a halt to unapproved colchicine for injection products in 2008.
Companies that manufacture, distribute, and/or market unapproved formulations of the oral, single ingredient forms of the gout drug colchicine have been ordered to stop doing so, the Food and Drug Administration announced on Sept. 30.
The companies must stop manufacturing these products within 45 days of the announcement and stop shipping unapproved colchicine within 90 days of the announcement, according to the statement, which says that a small amount of unapproved colchicine will be available after these dates, until supplies are used up.
Oral colchicine products are among the drugs that have been on the market for so many years that they were never subjected to the current prescription drug approval process. The FDA has launched an initiative to target the manufacturers of colchicine and other older, unapproved drugs to conduct the necessary testing to meet current approval standards or take them off the market.
Once the older products are off the market, the only oral colchicine product that will be available is Colcrys, marketed by Mutual Pharmaceuticals/URL Pharma, which was approved in 2009, after the company met the current drug approval requirements. As a result, the drug’s label includes information on safety data, drug interactions, and dosing that is not available for the older unapproved versions, according to the FDA.
The FDA's approval of Colcrys and the plans to ban the marketing of the older products has been sharply criticized by some clinicians who treat patients with gout because Colcrys is significantly more expensive than the older, unapproved formulations.
The FDA statement notes that the manufacturer of Colcrys has established a patient assistance program for patients who can’t afford the brand-name drug and those on Medicare who don’t want the cost of the drug to contribute to their out-of-pocket Medicare Part D expenses. More information about that program is available at the Colcrys Web site or at Needy Meds, an umbrella organization that offers patient assistance, or by calling 888-811-8423. The company has said that the program will continue "at a minimum until there is FDA-approved generic competition" for Colcrys, according to the statement.
The agency is encouraging manufacturers of such products to pursue approval of their products or "to face the type of action announced today," Deborah Autor, director of the office of compliance at the FDA's Center for Drug Evaluation and Research (CDER), said in the statement.
The FDA ordered a halt to unapproved colchicine for injection products in 2008.