User login
Select biologics for dose escalation in psoriasis
SONOMA, CALIF.– Data support dosage escalation or intensification for patients who don’t respond to biologic therapy for psoriasis, but only for three of the four biologic agents available in the United States, Dr. April W. Armstrong said at the annual Coastal Dermatology Symposium.
It may make sense to increase the dose or shorten the intervals between doses in some patients with psoriasis who don’t respond to etanercept, adalimumab, or ustekinumab, she suggested.

For infliximab, however, increasing the standard 5-mg/kg dose to 10 mg/kg in nonresponders did not significantly improve efficacy in one randomized study of patients with moderate to severe psoriasis. Consider combination therapy instead of dose escalation in those patients, said Dr. Armstrong, director of the psoriasis program at the University of Colorado, Denver.
Most clinical trials of dose escalation for psoriasis target patients in whom conventional doses produce only a partial response, defined as a Psoriasis Area and Severity Index (PASI) score of 50-75, or no response, defined as a PASI score below 50, she said.
A conventional dosage of etanercept for psoriasis uses 50 mg twice weekly for 12 weeks, followed by 50 mg once weekly for maintenance, Dr. Armstrong said. In an open-label extension of a study of 912 patients with moderate to severe psoriasis who received 12 weeks of etanercept therapy, nonresponders who escalated the maintenance dosage to 50 mg twice weekly boosted the likelihood of achieving a PASI 75 from 33% at 12 weeks to 44% at 48 weeks and 43% at 72 weeks (J. Drugs Dermatol. 2010;9:928-37).
Safety profiles were similar between standard dosing and escalated or intensified dosing in studies of all four biologic therapies, Dr. Armstrong said at the symposium, jointly presented by the University of Louisville (Ky.) and the Global Academy for Medical Education.
In the etanercept study, rates of serious infections were 0.9 events per 100 patient-years on standard maintenance therapy and 1.9 events per 100 patient-years on the escalated dosage. Myocardial infarctions occurred in none of 321 patients on standard maintenance therapy and in 2 of 591 patients on the twice-weekly dosage.
The conventional dosage of adalimumab for psoriasis is 80 mg at the start of therapy, followed by 40 mg every other week starting at week 1. In an open-label extension of a study of 147 patients, those who did not achieve a PASI 50 by week 25 were allowed to escalate the maintenance therapy dosage to 40 mg weekly. By week 60, 64% of patients in the dose-escalation group achieved a PASI 75, compared with 56% of patients who had been on the standard regimen and 45% of patients who had started with placebo for 12 weeks and then went on the standard dosage (J. Am. Acad. Dermatol. 2006;55:598-606). The mean PASI score improved by 27% after 8 weeks on the escalated dose, Dr. Armstrong said.
Three malignancies and two cardiovascular events (one leading to death) developed in the escalated-dosage group, compared with two malignancies and a serious case of coccidiomycosis in the standard-therapy group, she noted.
The conventional dosage for ustekinumab is 45 mg in patients weighing up to 100 kg or 90 mg in heavier patients, administered in subcutaneous injections at weeks 0 and 4, then every 12 weeks thereafter. In the only head-to-head study comparing biologic therapies for psoriasis, ustekinumab was more effective than etanercept in the first 3 months, Dr. Armstrong noted. The 12-week Active Comparator (CNTO1275/Enbrel) Psoriasis Trial (ACCEPT) found that 74% of 347 patients on 90 mg of ustekinumab and 68% of 209 patients on 45 mg of ustekinumab achieved a PASI 75, compared with 57% of 347 patients on etanercept (N. Engl. J. Med. 2010;362:118-28).
In a separate study of 158 patients with psoriasis who only partially responded to standard ustekinumab therapy after 24 weeks, maintenance dose escalation to every 8 weeks instead of 12 significantly improved response rates in patients on 90 mg but not in those on 45 mg. By week 52, 69% of patients receiving 90 mg ustekinumab every 8 weeks achieved a PASI 75, compared with 33% of patients on ustekinumab 90 mg every 12 weeks (Lancet 2008;371:1675-84). In the patients on 45 mg ustekinumab, the proportions achieving PASI 75 by week 52 did not differ significantly between those receiving the drug every 8 or 12 weeks.
One cutaneous malignancy, one noncutaneous malignancy, and two other serious adverse events occurred in the intensified-therapy groups, compared with one serious infection and two other serious adverse events in the standard-therapy groups.
The conventional dosage of infliximab for psoriasis is 5 mg/kg at weeks 0, 2, and 6, then every 8 weeks. A study that randomized 33 patients with moderate to severe psoriasis to 10 weeks of therapy with placebo or 5 mg/kg or 10 mg/kg of infliximab at weeks 0, 2, and 6 found that 91% of patients on the escalated dose and 82% on the standard dose of infliximab achieved a Physician Global Assessment score of good, excellent, or clear, compared with 18% of patients on placebo (Lancet 2001;357:1842-7).
The proportions of patients achieving a PASI 75 were not significantly different between the standard-dosage group and the escalated-dosage group (82% vs. 73%, respectively), although both were significantly higher than in the placebo group (18%). “So, there’s not much difference if you give 5 or 10 mg/kg” of infliximab, Dr. Armstrong said.
One of 11 patients on 5 mg/kg infliximab developed a dental abscess, and 1 of 11 patients on 10 mg/kg infliximab developed pneumonia.
Dr. Armstrong reported financial associations with AbbVie, Amgen, Celgene, Lilly, Novartis, Merck, Pfizer, UCB, Modernizing Medicine, and Janssen. This publication and the Global Academy for Medical Education are owned by the same parent company.
On Twitter @sherryboschert
SONOMA, CALIF.– Data support dosage escalation or intensification for patients who don’t respond to biologic therapy for psoriasis, but only for three of the four biologic agents available in the United States, Dr. April W. Armstrong said at the annual Coastal Dermatology Symposium.
It may make sense to increase the dose or shorten the intervals between doses in some patients with psoriasis who don’t respond to etanercept, adalimumab, or ustekinumab, she suggested.
For infliximab, however, increasing the standard 5-mg/kg dose to 10 mg/kg in nonresponders did not significantly improve efficacy in one randomized study of patients with moderate to severe psoriasis. Consider combination therapy instead of dose escalation in those patients, said Dr. Armstrong, director of the psoriasis program at the University of Colorado, Denver.
Most clinical trials of dose escalation for psoriasis target patients in whom conventional doses produce only a partial response, defined as a Psoriasis Area and Severity Index (PASI) score of 50-75, or no response, defined as a PASI score below 50, she said.
A conventional dosage of etanercept for psoriasis uses 50 mg twice weekly for 12 weeks, followed by 50 mg once weekly for maintenance, Dr. Armstrong said. In an open-label extension of a study of 912 patients with moderate to severe psoriasis who received 12 weeks of etanercept therapy, nonresponders who escalated the maintenance dosage to 50 mg twice weekly boosted the likelihood of achieving a PASI 75 from 33% at 12 weeks to 44% at 48 weeks and 43% at 72 weeks (J. Drugs Dermatol. 2010;9:928-37).
Safety profiles were similar between standard dosing and escalated or intensified dosing in studies of all four biologic therapies, Dr. Armstrong said at the symposium, jointly presented by the University of Louisville (Ky.) and the Global Academy for Medical Education.
In the etanercept study, rates of serious infections were 0.9 events per 100 patient-years on standard maintenance therapy and 1.9 events per 100 patient-years on the escalated dosage. Myocardial infarctions occurred in none of 321 patients on standard maintenance therapy and in 2 of 591 patients on the twice-weekly dosage.
The conventional dosage of adalimumab for psoriasis is 80 mg at the start of therapy, followed by 40 mg every other week starting at week 1. In an open-label extension of a study of 147 patients, those who did not achieve a PASI 50 by week 25 were allowed to escalate the maintenance therapy dosage to 40 mg weekly. By week 60, 64% of patients in the dose-escalation group achieved a PASI 75, compared with 56% of patients who had been on the standard regimen and 45% of patients who had started with placebo for 12 weeks and then went on the standard dosage (J. Am. Acad. Dermatol. 2006;55:598-606). The mean PASI score improved by 27% after 8 weeks on the escalated dose, Dr. Armstrong said.
Three malignancies and two cardiovascular events (one leading to death) developed in the escalated-dosage group, compared with two malignancies and a serious case of coccidiomycosis in the standard-therapy group, she noted.
The conventional dosage for ustekinumab is 45 mg in patients weighing up to 100 kg or 90 mg in heavier patients, administered in subcutaneous injections at weeks 0 and 4, then every 12 weeks thereafter. In the only head-to-head study comparing biologic therapies for psoriasis, ustekinumab was more effective than etanercept in the first 3 months, Dr. Armstrong noted. The 12-week Active Comparator (CNTO1275/Enbrel) Psoriasis Trial (ACCEPT) found that 74% of 347 patients on 90 mg of ustekinumab and 68% of 209 patients on 45 mg of ustekinumab achieved a PASI 75, compared with 57% of 347 patients on etanercept (N. Engl. J. Med. 2010;362:118-28).
In a separate study of 158 patients with psoriasis who only partially responded to standard ustekinumab therapy after 24 weeks, maintenance dose escalation to every 8 weeks instead of 12 significantly improved response rates in patients on 90 mg but not in those on 45 mg. By week 52, 69% of patients receiving 90 mg ustekinumab every 8 weeks achieved a PASI 75, compared with 33% of patients on ustekinumab 90 mg every 12 weeks (Lancet 2008;371:1675-84). In the patients on 45 mg ustekinumab, the proportions achieving PASI 75 by week 52 did not differ significantly between those receiving the drug every 8 or 12 weeks.
One cutaneous malignancy, one noncutaneous malignancy, and two other serious adverse events occurred in the intensified-therapy groups, compared with one serious infection and two other serious adverse events in the standard-therapy groups.
The conventional dosage of infliximab for psoriasis is 5 mg/kg at weeks 0, 2, and 6, then every 8 weeks. A study that randomized 33 patients with moderate to severe psoriasis to 10 weeks of therapy with placebo or 5 mg/kg or 10 mg/kg of infliximab at weeks 0, 2, and 6 found that 91% of patients on the escalated dose and 82% on the standard dose of infliximab achieved a Physician Global Assessment score of good, excellent, or clear, compared with 18% of patients on placebo (Lancet 2001;357:1842-7).
The proportions of patients achieving a PASI 75 were not significantly different between the standard-dosage group and the escalated-dosage group (82% vs. 73%, respectively), although both were significantly higher than in the placebo group (18%). “So, there’s not much difference if you give 5 or 10 mg/kg” of infliximab, Dr. Armstrong said.
One of 11 patients on 5 mg/kg infliximab developed a dental abscess, and 1 of 11 patients on 10 mg/kg infliximab developed pneumonia.
Dr. Armstrong reported financial associations with AbbVie, Amgen, Celgene, Lilly, Novartis, Merck, Pfizer, UCB, Modernizing Medicine, and Janssen. This publication and the Global Academy for Medical Education are owned by the same parent company.
On Twitter @sherryboschert
SONOMA, CALIF.– Data support dosage escalation or intensification for patients who don’t respond to biologic therapy for psoriasis, but only for three of the four biologic agents available in the United States, Dr. April W. Armstrong said at the annual Coastal Dermatology Symposium.
It may make sense to increase the dose or shorten the intervals between doses in some patients with psoriasis who don’t respond to etanercept, adalimumab, or ustekinumab, she suggested.
For infliximab, however, increasing the standard 5-mg/kg dose to 10 mg/kg in nonresponders did not significantly improve efficacy in one randomized study of patients with moderate to severe psoriasis. Consider combination therapy instead of dose escalation in those patients, said Dr. Armstrong, director of the psoriasis program at the University of Colorado, Denver.
Most clinical trials of dose escalation for psoriasis target patients in whom conventional doses produce only a partial response, defined as a Psoriasis Area and Severity Index (PASI) score of 50-75, or no response, defined as a PASI score below 50, she said.
A conventional dosage of etanercept for psoriasis uses 50 mg twice weekly for 12 weeks, followed by 50 mg once weekly for maintenance, Dr. Armstrong said. In an open-label extension of a study of 912 patients with moderate to severe psoriasis who received 12 weeks of etanercept therapy, nonresponders who escalated the maintenance dosage to 50 mg twice weekly boosted the likelihood of achieving a PASI 75 from 33% at 12 weeks to 44% at 48 weeks and 43% at 72 weeks (J. Drugs Dermatol. 2010;9:928-37).
Safety profiles were similar between standard dosing and escalated or intensified dosing in studies of all four biologic therapies, Dr. Armstrong said at the symposium, jointly presented by the University of Louisville (Ky.) and the Global Academy for Medical Education.
In the etanercept study, rates of serious infections were 0.9 events per 100 patient-years on standard maintenance therapy and 1.9 events per 100 patient-years on the escalated dosage. Myocardial infarctions occurred in none of 321 patients on standard maintenance therapy and in 2 of 591 patients on the twice-weekly dosage.
The conventional dosage of adalimumab for psoriasis is 80 mg at the start of therapy, followed by 40 mg every other week starting at week 1. In an open-label extension of a study of 147 patients, those who did not achieve a PASI 50 by week 25 were allowed to escalate the maintenance therapy dosage to 40 mg weekly. By week 60, 64% of patients in the dose-escalation group achieved a PASI 75, compared with 56% of patients who had been on the standard regimen and 45% of patients who had started with placebo for 12 weeks and then went on the standard dosage (J. Am. Acad. Dermatol. 2006;55:598-606). The mean PASI score improved by 27% after 8 weeks on the escalated dose, Dr. Armstrong said.
Three malignancies and two cardiovascular events (one leading to death) developed in the escalated-dosage group, compared with two malignancies and a serious case of coccidiomycosis in the standard-therapy group, she noted.
The conventional dosage for ustekinumab is 45 mg in patients weighing up to 100 kg or 90 mg in heavier patients, administered in subcutaneous injections at weeks 0 and 4, then every 12 weeks thereafter. In the only head-to-head study comparing biologic therapies for psoriasis, ustekinumab was more effective than etanercept in the first 3 months, Dr. Armstrong noted. The 12-week Active Comparator (CNTO1275/Enbrel) Psoriasis Trial (ACCEPT) found that 74% of 347 patients on 90 mg of ustekinumab and 68% of 209 patients on 45 mg of ustekinumab achieved a PASI 75, compared with 57% of 347 patients on etanercept (N. Engl. J. Med. 2010;362:118-28).
In a separate study of 158 patients with psoriasis who only partially responded to standard ustekinumab therapy after 24 weeks, maintenance dose escalation to every 8 weeks instead of 12 significantly improved response rates in patients on 90 mg but not in those on 45 mg. By week 52, 69% of patients receiving 90 mg ustekinumab every 8 weeks achieved a PASI 75, compared with 33% of patients on ustekinumab 90 mg every 12 weeks (Lancet 2008;371:1675-84). In the patients on 45 mg ustekinumab, the proportions achieving PASI 75 by week 52 did not differ significantly between those receiving the drug every 8 or 12 weeks.
One cutaneous malignancy, one noncutaneous malignancy, and two other serious adverse events occurred in the intensified-therapy groups, compared with one serious infection and two other serious adverse events in the standard-therapy groups.
The conventional dosage of infliximab for psoriasis is 5 mg/kg at weeks 0, 2, and 6, then every 8 weeks. A study that randomized 33 patients with moderate to severe psoriasis to 10 weeks of therapy with placebo or 5 mg/kg or 10 mg/kg of infliximab at weeks 0, 2, and 6 found that 91% of patients on the escalated dose and 82% on the standard dose of infliximab achieved a Physician Global Assessment score of good, excellent, or clear, compared with 18% of patients on placebo (Lancet 2001;357:1842-7).
The proportions of patients achieving a PASI 75 were not significantly different between the standard-dosage group and the escalated-dosage group (82% vs. 73%, respectively), although both were significantly higher than in the placebo group (18%). “So, there’s not much difference if you give 5 or 10 mg/kg” of infliximab, Dr. Armstrong said.
One of 11 patients on 5 mg/kg infliximab developed a dental abscess, and 1 of 11 patients on 10 mg/kg infliximab developed pneumonia.
Dr. Armstrong reported financial associations with AbbVie, Amgen, Celgene, Lilly, Novartis, Merck, Pfizer, UCB, Modernizing Medicine, and Janssen. This publication and the Global Academy for Medical Education are owned by the same parent company.
On Twitter @sherryboschert
EXPERT ANALYSIS FROM THE COASTAL DERMATOLOGY SYMPOSIUM

Reducing Risks
Psoriasis is associated with multiple comorbidities, including cardiovascular diseases. With the advent of anti-inflammatory therapy, there has been much investigation into whether treatments for psoriasis may reduce the risk for cardiovascular events. In a Journal of the European Academy of Dermatology and Venereology article published online on October 10, Ahlehoff et al examined the rate of cardiovascular events—cardiovascular death, myocardial infarction, and stroke—in patients with severe psoriasis treated with systemic anti-inflammatory drugs.
Individual-level linkage of administrative registries was utilized to perform a longitudinal nationwide cohort study in Denmark. Time-dependent multivariable adjusted Cox regression was used to estimate hazard ratios (HRs) with 95% confidence intervals (CIs) of cardiovascular events associated with use of biological drugs, methotrexate, cyclosporine, retinoids, and other antipsoriatic therapies (ie, topical treatments, phototherapy, climate therapy).
The investigators included a total of 6902 patients (9662 treatment exposures) with a maximum follow-up of 5 years. Incidence rates per 1000 patient-years for cardiovascular events were highest for retinoids and other therapies (18.95 and 14.63, respectively) followed by methotrexate, cyclosporine, and biological drugs (6.28, 6.08, and 4.16, respectively). Relative to other therapies, methotrexate (HR, 0.53; 95% CI, 0.34-0.83) was associated with reduced risk for the composite end point. A comparable but nonsignificant protective effect was observed with biological drugs (HR, 0.58; 95% CI, 0.30-1.10), whereas no protective effect was apparent with cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) and retinoids (HR, 1.80; 95% CI, 1.03-2.96). Tumor necrosis factor inhibitors (HR, 0.46; 95% CI, 0.22-0.98) were linked to reduced event rates but the IL-12/IL-23 inhibitor ustekinumab (HR, 1.52; 95% CI, 0.47-4.94) was not.
The authors concluded that systemic anti-inflammatory treatment with methotrexate was associated with lower rates of cardiovascular events during long-term follow-up compared to patients treated with other antipsoriatic therapies.
What’s the issue?
This study is consistent with other investigations evaluating the cardioprotective benefit of therapies for psoriasis. The cardioprotective benefits of methotrexate and tumor necrosis factor inhibitors have been previously reported. Further investigation will help to elucidate the role of these drugs as well as newer therapies in the reduction of comorbidities. Does this study influence your perception of therapies for psoriasis?
Psoriasis is associated with multiple comorbidities, including cardiovascular diseases. With the advent of anti-inflammatory therapy, there has been much investigation into whether treatments for psoriasis may reduce the risk for cardiovascular events. In a Journal of the European Academy of Dermatology and Venereology article published online on October 10, Ahlehoff et al examined the rate of cardiovascular events—cardiovascular death, myocardial infarction, and stroke—in patients with severe psoriasis treated with systemic anti-inflammatory drugs.
Individual-level linkage of administrative registries was utilized to perform a longitudinal nationwide cohort study in Denmark. Time-dependent multivariable adjusted Cox regression was used to estimate hazard ratios (HRs) with 95% confidence intervals (CIs) of cardiovascular events associated with use of biological drugs, methotrexate, cyclosporine, retinoids, and other antipsoriatic therapies (ie, topical treatments, phototherapy, climate therapy).
The investigators included a total of 6902 patients (9662 treatment exposures) with a maximum follow-up of 5 years. Incidence rates per 1000 patient-years for cardiovascular events were highest for retinoids and other therapies (18.95 and 14.63, respectively) followed by methotrexate, cyclosporine, and biological drugs (6.28, 6.08, and 4.16, respectively). Relative to other therapies, methotrexate (HR, 0.53; 95% CI, 0.34-0.83) was associated with reduced risk for the composite end point. A comparable but nonsignificant protective effect was observed with biological drugs (HR, 0.58; 95% CI, 0.30-1.10), whereas no protective effect was apparent with cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) and retinoids (HR, 1.80; 95% CI, 1.03-2.96). Tumor necrosis factor inhibitors (HR, 0.46; 95% CI, 0.22-0.98) were linked to reduced event rates but the IL-12/IL-23 inhibitor ustekinumab (HR, 1.52; 95% CI, 0.47-4.94) was not.
The authors concluded that systemic anti-inflammatory treatment with methotrexate was associated with lower rates of cardiovascular events during long-term follow-up compared to patients treated with other antipsoriatic therapies.
What’s the issue?
This study is consistent with other investigations evaluating the cardioprotective benefit of therapies for psoriasis. The cardioprotective benefits of methotrexate and tumor necrosis factor inhibitors have been previously reported. Further investigation will help to elucidate the role of these drugs as well as newer therapies in the reduction of comorbidities. Does this study influence your perception of therapies for psoriasis?
Psoriasis is associated with multiple comorbidities, including cardiovascular diseases. With the advent of anti-inflammatory therapy, there has been much investigation into whether treatments for psoriasis may reduce the risk for cardiovascular events. In a Journal of the European Academy of Dermatology and Venereology article published online on October 10, Ahlehoff et al examined the rate of cardiovascular events—cardiovascular death, myocardial infarction, and stroke—in patients with severe psoriasis treated with systemic anti-inflammatory drugs.
Individual-level linkage of administrative registries was utilized to perform a longitudinal nationwide cohort study in Denmark. Time-dependent multivariable adjusted Cox regression was used to estimate hazard ratios (HRs) with 95% confidence intervals (CIs) of cardiovascular events associated with use of biological drugs, methotrexate, cyclosporine, retinoids, and other antipsoriatic therapies (ie, topical treatments, phototherapy, climate therapy).
The investigators included a total of 6902 patients (9662 treatment exposures) with a maximum follow-up of 5 years. Incidence rates per 1000 patient-years for cardiovascular events were highest for retinoids and other therapies (18.95 and 14.63, respectively) followed by methotrexate, cyclosporine, and biological drugs (6.28, 6.08, and 4.16, respectively). Relative to other therapies, methotrexate (HR, 0.53; 95% CI, 0.34-0.83) was associated with reduced risk for the composite end point. A comparable but nonsignificant protective effect was observed with biological drugs (HR, 0.58; 95% CI, 0.30-1.10), whereas no protective effect was apparent with cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) and retinoids (HR, 1.80; 95% CI, 1.03-2.96). Tumor necrosis factor inhibitors (HR, 0.46; 95% CI, 0.22-0.98) were linked to reduced event rates but the IL-12/IL-23 inhibitor ustekinumab (HR, 1.52; 95% CI, 0.47-4.94) was not.
The authors concluded that systemic anti-inflammatory treatment with methotrexate was associated with lower rates of cardiovascular events during long-term follow-up compared to patients treated with other antipsoriatic therapies.
What’s the issue?
This study is consistent with other investigations evaluating the cardioprotective benefit of therapies for psoriasis. The cardioprotective benefits of methotrexate and tumor necrosis factor inhibitors have been previously reported. Further investigation will help to elucidate the role of these drugs as well as newer therapies in the reduction of comorbidities. Does this study influence your perception of therapies for psoriasis?

FDA panel unanimously supports secukinumab approval for psoriasis
SILVER SPRING, MD. – The human monoclonal antibody secukinumab is expected to be the first biologic that targets interleukin-17A to be approved by the agency for treating psoriasis, with a Food and Drug Administration advisory panel’s unanimous support for the approval.
At a meeting on Oct. 20, the FDA’s Dermatologic and Ophthalmic Drugs Advisory committee voted 7-0 to recommend approval of secukinumab for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the indication proposed by Novartis Pharmaceuticals. Secukinumab “selectively binds and neutralizes” IL-17A, a proinflammatory cytokine “that is involved in normal inflammatory and immune responses and plays a key role in the pathogenesis of plaque psoriasis,” according to the company.
If approved, secukinumab would be the first IL-17A blocker available and would be available in a lyophilized formulation for reconstitution, and in a liquid formulation in a refilled syringe or autoinjector pen. The 300-mg dose was determined to be the best dose to achieve clear or almost clear skin, with a favorable safety profile that was comparable to that of the 150-mg dose, which was also studied in phase II and III studies, according to Novartis. The company has proposed a dose of 300 mg, administered with a subcutaneous injection, at 0, 1, 2, 3, and 4 weeks, followed by 300 mg once a month.
The panel agreed that the 300-mg dose was effective with an acceptable risk-benefit profile, but agreed that it would be useful to have the 150-mg dose, which was also effective in trials, be available as well. The panel also recommended that the 450-mg dose, which was not formally studied, be evaluated further in postmarketing studies, because it could be useful in nonresponders and for heavier patients weighing 90 kg (about 198 pounds) or more, because of evidence that the 300-mg dose was less effective in heavier patients.
Secukinumab has been studied in 10 phase II and III studies of almost 4,000 patients with psoriasis. In the two main phase III studies, the 150-mg and 300-mg doses were compared with placebo (and to a formulation of etanercept not available in the United States in one study); the primary endpoints were the Psoriasis Area and Severity Index (PASI) 75, a 75% improvement of psoriasis from baseline, and an Investigator Global Assessment (IGA) score of 0 or 1 (clear or almost clear skin).
In one study of 738 patients, PASI 75 scores at 12 weeks were achieved by almost 82% and almost 72% of those on the 300-mg and 150-mg doses respectively, vs. 4.5% of those on placebo. In the second study, PASI 75 scores at 12 weeks were achieved by 77% and 67% of those on the 300-mg and 150-mg doses respectively, vs. almost 5% of those on placebo. About 63%-65% of those on the 300-mg dose and about 51% of those on the 150-mg dose had an IGA of 0/1 at 12 weeks, vs. about 2.4%-2.8% of those on placebo. Differences in the responses were sustained at week 52, according to Novartis.
Concentration of the drug decreases as body weight increases, and in studies, weight had a significant effect on efficacy results. The proportion of patients who achieved the PASI 75 and IGA 0/1 endpoints was higher among those weighing under 90 kg than those weighing 90 kg or more at both the 150-mg and 300-mg doses tested.
Pooled data of phase III trial data were used to evaluate safety over 12 and 52 weeks. Serious infections, malignancies and serious cardiovascular events were low; there were no reports of reactivation of latent tuberculosis. No deaths were reported related to treatment. During the first 12 weeks of treatment, there was a higher rate of nonserious upper respiratory infections among the treated patients, and superficial Candida infections were more common among those on the 300-mg dose (1.2%) than among those on the 150-mg dose (0.4%) and placebo (0.3%). The FDA’s analysis of safety concluded that infection rates “tend to increase” as the concentration of secukinumab increases, but that most adverse events associated with greater exposure in patients weighing less than 90 kg were mild to moderate.
The panel agreed with the company’s plan to follow long-term safety in a postmarketing registry of patients with moderate to severe psoriasis, in at least 2,000 patients treated with secukinumab, 2,500 treated with other biologics, and 500 treated with other systemic medications. One panelist also recommended that long-term safety be evaluated in studies using large administrative databases for outcomes including malignancies and autoimmune events.
The FDA is expected to decide on approval by early 2015, according to Novartis, which plans to market secukinumab as Cosentyx if approved. The FDA usually follows the recommendations of its advisory panels. Members of these two panels had no conflicts to disclose; occasionally, panelists with a conflict are given a waiver, but not at this meeting. Secukinumab is not yet approved elsewhere, and is also under review in Europe. Phase III studies in patients with psoriatic arthritis are underway.
SILVER SPRING, MD. – The human monoclonal antibody secukinumab is expected to be the first biologic that targets interleukin-17A to be approved by the agency for treating psoriasis, with a Food and Drug Administration advisory panel’s unanimous support for the approval.
At a meeting on Oct. 20, the FDA’s Dermatologic and Ophthalmic Drugs Advisory committee voted 7-0 to recommend approval of secukinumab for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the indication proposed by Novartis Pharmaceuticals. Secukinumab “selectively binds and neutralizes” IL-17A, a proinflammatory cytokine “that is involved in normal inflammatory and immune responses and plays a key role in the pathogenesis of plaque psoriasis,” according to the company.
If approved, secukinumab would be the first IL-17A blocker available and would be available in a lyophilized formulation for reconstitution, and in a liquid formulation in a refilled syringe or autoinjector pen. The 300-mg dose was determined to be the best dose to achieve clear or almost clear skin, with a favorable safety profile that was comparable to that of the 150-mg dose, which was also studied in phase II and III studies, according to Novartis. The company has proposed a dose of 300 mg, administered with a subcutaneous injection, at 0, 1, 2, 3, and 4 weeks, followed by 300 mg once a month.
The panel agreed that the 300-mg dose was effective with an acceptable risk-benefit profile, but agreed that it would be useful to have the 150-mg dose, which was also effective in trials, be available as well. The panel also recommended that the 450-mg dose, which was not formally studied, be evaluated further in postmarketing studies, because it could be useful in nonresponders and for heavier patients weighing 90 kg (about 198 pounds) or more, because of evidence that the 300-mg dose was less effective in heavier patients.
Secukinumab has been studied in 10 phase II and III studies of almost 4,000 patients with psoriasis. In the two main phase III studies, the 150-mg and 300-mg doses were compared with placebo (and to a formulation of etanercept not available in the United States in one study); the primary endpoints were the Psoriasis Area and Severity Index (PASI) 75, a 75% improvement of psoriasis from baseline, and an Investigator Global Assessment (IGA) score of 0 or 1 (clear or almost clear skin).
In one study of 738 patients, PASI 75 scores at 12 weeks were achieved by almost 82% and almost 72% of those on the 300-mg and 150-mg doses respectively, vs. 4.5% of those on placebo. In the second study, PASI 75 scores at 12 weeks were achieved by 77% and 67% of those on the 300-mg and 150-mg doses respectively, vs. almost 5% of those on placebo. About 63%-65% of those on the 300-mg dose and about 51% of those on the 150-mg dose had an IGA of 0/1 at 12 weeks, vs. about 2.4%-2.8% of those on placebo. Differences in the responses were sustained at week 52, according to Novartis.
Concentration of the drug decreases as body weight increases, and in studies, weight had a significant effect on efficacy results. The proportion of patients who achieved the PASI 75 and IGA 0/1 endpoints was higher among those weighing under 90 kg than those weighing 90 kg or more at both the 150-mg and 300-mg doses tested.
Pooled data of phase III trial data were used to evaluate safety over 12 and 52 weeks. Serious infections, malignancies and serious cardiovascular events were low; there were no reports of reactivation of latent tuberculosis. No deaths were reported related to treatment. During the first 12 weeks of treatment, there was a higher rate of nonserious upper respiratory infections among the treated patients, and superficial Candida infections were more common among those on the 300-mg dose (1.2%) than among those on the 150-mg dose (0.4%) and placebo (0.3%). The FDA’s analysis of safety concluded that infection rates “tend to increase” as the concentration of secukinumab increases, but that most adverse events associated with greater exposure in patients weighing less than 90 kg were mild to moderate.
The panel agreed with the company’s plan to follow long-term safety in a postmarketing registry of patients with moderate to severe psoriasis, in at least 2,000 patients treated with secukinumab, 2,500 treated with other biologics, and 500 treated with other systemic medications. One panelist also recommended that long-term safety be evaluated in studies using large administrative databases for outcomes including malignancies and autoimmune events.
The FDA is expected to decide on approval by early 2015, according to Novartis, which plans to market secukinumab as Cosentyx if approved. The FDA usually follows the recommendations of its advisory panels. Members of these two panels had no conflicts to disclose; occasionally, panelists with a conflict are given a waiver, but not at this meeting. Secukinumab is not yet approved elsewhere, and is also under review in Europe. Phase III studies in patients with psoriatic arthritis are underway.
SILVER SPRING, MD. – The human monoclonal antibody secukinumab is expected to be the first biologic that targets interleukin-17A to be approved by the agency for treating psoriasis, with a Food and Drug Administration advisory panel’s unanimous support for the approval.
At a meeting on Oct. 20, the FDA’s Dermatologic and Ophthalmic Drugs Advisory committee voted 7-0 to recommend approval of secukinumab for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, the indication proposed by Novartis Pharmaceuticals. Secukinumab “selectively binds and neutralizes” IL-17A, a proinflammatory cytokine “that is involved in normal inflammatory and immune responses and plays a key role in the pathogenesis of plaque psoriasis,” according to the company.
If approved, secukinumab would be the first IL-17A blocker available and would be available in a lyophilized formulation for reconstitution, and in a liquid formulation in a refilled syringe or autoinjector pen. The 300-mg dose was determined to be the best dose to achieve clear or almost clear skin, with a favorable safety profile that was comparable to that of the 150-mg dose, which was also studied in phase II and III studies, according to Novartis. The company has proposed a dose of 300 mg, administered with a subcutaneous injection, at 0, 1, 2, 3, and 4 weeks, followed by 300 mg once a month.
The panel agreed that the 300-mg dose was effective with an acceptable risk-benefit profile, but agreed that it would be useful to have the 150-mg dose, which was also effective in trials, be available as well. The panel also recommended that the 450-mg dose, which was not formally studied, be evaluated further in postmarketing studies, because it could be useful in nonresponders and for heavier patients weighing 90 kg (about 198 pounds) or more, because of evidence that the 300-mg dose was less effective in heavier patients.
Secukinumab has been studied in 10 phase II and III studies of almost 4,000 patients with psoriasis. In the two main phase III studies, the 150-mg and 300-mg doses were compared with placebo (and to a formulation of etanercept not available in the United States in one study); the primary endpoints were the Psoriasis Area and Severity Index (PASI) 75, a 75% improvement of psoriasis from baseline, and an Investigator Global Assessment (IGA) score of 0 or 1 (clear or almost clear skin).
In one study of 738 patients, PASI 75 scores at 12 weeks were achieved by almost 82% and almost 72% of those on the 300-mg and 150-mg doses respectively, vs. 4.5% of those on placebo. In the second study, PASI 75 scores at 12 weeks were achieved by 77% and 67% of those on the 300-mg and 150-mg doses respectively, vs. almost 5% of those on placebo. About 63%-65% of those on the 300-mg dose and about 51% of those on the 150-mg dose had an IGA of 0/1 at 12 weeks, vs. about 2.4%-2.8% of those on placebo. Differences in the responses were sustained at week 52, according to Novartis.
Concentration of the drug decreases as body weight increases, and in studies, weight had a significant effect on efficacy results. The proportion of patients who achieved the PASI 75 and IGA 0/1 endpoints was higher among those weighing under 90 kg than those weighing 90 kg or more at both the 150-mg and 300-mg doses tested.
Pooled data of phase III trial data were used to evaluate safety over 12 and 52 weeks. Serious infections, malignancies and serious cardiovascular events were low; there were no reports of reactivation of latent tuberculosis. No deaths were reported related to treatment. During the first 12 weeks of treatment, there was a higher rate of nonserious upper respiratory infections among the treated patients, and superficial Candida infections were more common among those on the 300-mg dose (1.2%) than among those on the 150-mg dose (0.4%) and placebo (0.3%). The FDA’s analysis of safety concluded that infection rates “tend to increase” as the concentration of secukinumab increases, but that most adverse events associated with greater exposure in patients weighing less than 90 kg were mild to moderate.
The panel agreed with the company’s plan to follow long-term safety in a postmarketing registry of patients with moderate to severe psoriasis, in at least 2,000 patients treated with secukinumab, 2,500 treated with other biologics, and 500 treated with other systemic medications. One panelist also recommended that long-term safety be evaluated in studies using large administrative databases for outcomes including malignancies and autoimmune events.
The FDA is expected to decide on approval by early 2015, according to Novartis, which plans to market secukinumab as Cosentyx if approved. The FDA usually follows the recommendations of its advisory panels. Members of these two panels had no conflicts to disclose; occasionally, panelists with a conflict are given a waiver, but not at this meeting. Secukinumab is not yet approved elsewhere, and is also under review in Europe. Phase III studies in patients with psoriatic arthritis are underway.
AT AN FDA ADVISORY COMMITTEE MEETING
Secukinumab showed sustained efficacy in psoriasis
AMSTERDAM – The investigational biologic agent secukinumab continued to show strong efficacy through 52 weeks of treatment in a new secondary analysis of the pivotal phase III ERASURE trial.
For example, 60% of patients treated for moderate-to-severe chronic plaque psoriasis at the 300-mg dose of secukinumab continued to maintain a PASI 90 response through 52 weeks of therapy, Dr. Mark G. Lebwohl reported at the annual congress of the European Academy of Dermatology and Venereology.
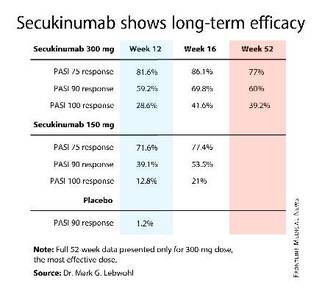
Secukinumab is a fully human monoclonal antibody directed against interleukin-17A. While the Food and Drug Administration requested that the primary outcome in ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) should be the degree of skin clearing at week 12, the efficacy actually peaked at 16 weeks and was then sustained with only modest tailoff through the remainder of the 52-week study (see graphic).
This is important new information, Dr. Lebwohl noted, because 12-week outcomes don’t provide a full picture regarding potent therapies. Psoriasis is a chronic disease requiring long-term therapy, and some biologic agents now marketed for psoriasis tend to show a loss of effect over time. Reassuringly, the long-term ERASURE data show that’s not the case for secukinumab, explained Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
ERASURE involved 738 patients randomized double-blind to secukinumab at either 150 mg or 300 mg, or to placebo. Following an initial loading-dose phase when the biologic was given subcutaneously once weekly for 5 weeks, it was then administered every 4 weeks for the remainder of the year-long trial.
The safety profile of secukinumab was similar to placebo with one exception: Upper respiratory tract infections were three- to fourfold more common in patients on the IL-17A inhibitor.
Novartis funded the ERASURE trial, whose primary results were recently published (N. Engl. J. Med. 2014;371:326-38). Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AMSTERDAM – The investigational biologic agent secukinumab continued to show strong efficacy through 52 weeks of treatment in a new secondary analysis of the pivotal phase III ERASURE trial.
For example, 60% of patients treated for moderate-to-severe chronic plaque psoriasis at the 300-mg dose of secukinumab continued to maintain a PASI 90 response through 52 weeks of therapy, Dr. Mark G. Lebwohl reported at the annual congress of the European Academy of Dermatology and Venereology.
Secukinumab is a fully human monoclonal antibody directed against interleukin-17A. While the Food and Drug Administration requested that the primary outcome in ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) should be the degree of skin clearing at week 12, the efficacy actually peaked at 16 weeks and was then sustained with only modest tailoff through the remainder of the 52-week study (see graphic).
This is important new information, Dr. Lebwohl noted, because 12-week outcomes don’t provide a full picture regarding potent therapies. Psoriasis is a chronic disease requiring long-term therapy, and some biologic agents now marketed for psoriasis tend to show a loss of effect over time. Reassuringly, the long-term ERASURE data show that’s not the case for secukinumab, explained Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
ERASURE involved 738 patients randomized double-blind to secukinumab at either 150 mg or 300 mg, or to placebo. Following an initial loading-dose phase when the biologic was given subcutaneously once weekly for 5 weeks, it was then administered every 4 weeks for the remainder of the year-long trial.
The safety profile of secukinumab was similar to placebo with one exception: Upper respiratory tract infections were three- to fourfold more common in patients on the IL-17A inhibitor.
Novartis funded the ERASURE trial, whose primary results were recently published (N. Engl. J. Med. 2014;371:326-38). Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AMSTERDAM – The investigational biologic agent secukinumab continued to show strong efficacy through 52 weeks of treatment in a new secondary analysis of the pivotal phase III ERASURE trial.
For example, 60% of patients treated for moderate-to-severe chronic plaque psoriasis at the 300-mg dose of secukinumab continued to maintain a PASI 90 response through 52 weeks of therapy, Dr. Mark G. Lebwohl reported at the annual congress of the European Academy of Dermatology and Venereology.
Secukinumab is a fully human monoclonal antibody directed against interleukin-17A. While the Food and Drug Administration requested that the primary outcome in ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) should be the degree of skin clearing at week 12, the efficacy actually peaked at 16 weeks and was then sustained with only modest tailoff through the remainder of the 52-week study (see graphic).
This is important new information, Dr. Lebwohl noted, because 12-week outcomes don’t provide a full picture regarding potent therapies. Psoriasis is a chronic disease requiring long-term therapy, and some biologic agents now marketed for psoriasis tend to show a loss of effect over time. Reassuringly, the long-term ERASURE data show that’s not the case for secukinumab, explained Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
ERASURE involved 738 patients randomized double-blind to secukinumab at either 150 mg or 300 mg, or to placebo. Following an initial loading-dose phase when the biologic was given subcutaneously once weekly for 5 weeks, it was then administered every 4 weeks for the remainder of the year-long trial.
The safety profile of secukinumab was similar to placebo with one exception: Upper respiratory tract infections were three- to fourfold more common in patients on the IL-17A inhibitor.
Novartis funded the ERASURE trial, whose primary results were recently published (N. Engl. J. Med. 2014;371:326-38). Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: Psoriasis patients’ initial strong clinical response to the interleukin-17A inhibitor secukinumab is sustained through 52 weeks of therapy.
Major finding: Sixty percent of patients with moderate-to-severe chronic plaque psoriasis had a PASI 90 response sustained through a full year of treatment.
Data source: The 52-week long, double-blind, multicenter ERASURE trial randomized 738 patients with moderate-to-severe chronic plaque psoriasis to secukinumab at 150 mg or 300 mg, or to placebo.
Disclosures: Novartis funded the study. Dr. Lebwohl reported serving as a consultant to the company.
In psoriasis, is pushing for PASI 90 really worthwhile?
AMSTERDAM – Does pushing for a PASI 90 response instead of settling for a PASI 75 matter to patients being treated for moderate-to-severe chronic plaque psoriasis?
You bet it does, Dr. Mark G. Lebwohl asserted at the annual congress of the European Academy of Dermatology and Venereology.

He presented a pooled analysis of data from two large pivotal phase III randomized trials of secukinumab for psoriasis. The primary endpoints in the analysis were how often and how soon patients who achieved a PASI 75 or PASI 90 response at 12 weeks reported obtaining a Dermatology Life Quality Index (DLQI) response, defined as a score of 0 or 1.
The answer: More patients who had a PASI 90 response (meaning almost clear at 12 weeks) had a DLQI response, and it occurred a full 4 weeks faster than in PASI 75 responders – at a median of 8 weeks, compared with 12 weeks, reported Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
Scores on the DLQI can range from 0, meaning no psoriasis-related impairment of the patient’s quality of life, up to 30. The average baseline score in this study population was 13.5, so a DLQI response dropping the score down to 0 or 1 represents a dramatic improvement in this patient-reported outcome.
Study participants completed the DLQI questionnaire at weeks 4, 8, 12, 24, 36, and again at week 52. The subjects’ mean baseline PASI score was 23.2.
The two double-blind, randomized, placebo-controlled clinical trials that formed the basis for this analysis were the recently published 52-week ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) and FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis) studies (N. Engl. J. Med. 2014;371:326-38), in which patients were assigned to secukinumab at a dose of either 150 mg or 300 mg, placebo, or in the case of FIXTURE, to etanercept. The PASI 75 and 90 response rates at 12 weeks were markedly higher at both doses of secukinumab than with etanercept.
Dr. Lebwohl’s pooled analysis was restricted to the 1,470 study participants in the two studies who were randomized to active therapy. A total of 612 patients achieved a PASI 90 response by week 12. Another 365 had a PASI 75 response. Fully 89% of PASI 90 responders also had a DLQI response maintained out to week 52, as did 77% of PASI 75 responders.
The key finding: The median time to a DLQI response in the PASI 90 responders was 8 weeks, compared with 12 weeks in the PASI 75 responders. Thus, patients with a PASI 90 response obtained virtually total relief from what had previously been a debilitating disease a full month sooner than PASI 75 responders. And that, as reported by the patients themselves, constitutes a meaningful advantage, Dr. Lebwohl stated.
Secukinumab is a fully human monoclonal antibody directed against a novel target: interleukin-17A. Novartis has filed for marketing approval of the biologic agent with an indication for psoriasis both with the Food and Drug Administration and European regulators. Secukinumab is also being developed as a treatment for psoriatic arthritis, rheumatoid arthritis, and ankylosing spondylitis.
Novartis funded the analysis. Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AMSTERDAM – Does pushing for a PASI 90 response instead of settling for a PASI 75 matter to patients being treated for moderate-to-severe chronic plaque psoriasis?
You bet it does, Dr. Mark G. Lebwohl asserted at the annual congress of the European Academy of Dermatology and Venereology.
He presented a pooled analysis of data from two large pivotal phase III randomized trials of secukinumab for psoriasis. The primary endpoints in the analysis were how often and how soon patients who achieved a PASI 75 or PASI 90 response at 12 weeks reported obtaining a Dermatology Life Quality Index (DLQI) response, defined as a score of 0 or 1.
The answer: More patients who had a PASI 90 response (meaning almost clear at 12 weeks) had a DLQI response, and it occurred a full 4 weeks faster than in PASI 75 responders – at a median of 8 weeks, compared with 12 weeks, reported Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
Scores on the DLQI can range from 0, meaning no psoriasis-related impairment of the patient’s quality of life, up to 30. The average baseline score in this study population was 13.5, so a DLQI response dropping the score down to 0 or 1 represents a dramatic improvement in this patient-reported outcome.
Study participants completed the DLQI questionnaire at weeks 4, 8, 12, 24, 36, and again at week 52. The subjects’ mean baseline PASI score was 23.2.
The two double-blind, randomized, placebo-controlled clinical trials that formed the basis for this analysis were the recently published 52-week ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) and FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis) studies (N. Engl. J. Med. 2014;371:326-38), in which patients were assigned to secukinumab at a dose of either 150 mg or 300 mg, placebo, or in the case of FIXTURE, to etanercept. The PASI 75 and 90 response rates at 12 weeks were markedly higher at both doses of secukinumab than with etanercept.
Dr. Lebwohl’s pooled analysis was restricted to the 1,470 study participants in the two studies who were randomized to active therapy. A total of 612 patients achieved a PASI 90 response by week 12. Another 365 had a PASI 75 response. Fully 89% of PASI 90 responders also had a DLQI response maintained out to week 52, as did 77% of PASI 75 responders.
The key finding: The median time to a DLQI response in the PASI 90 responders was 8 weeks, compared with 12 weeks in the PASI 75 responders. Thus, patients with a PASI 90 response obtained virtually total relief from what had previously been a debilitating disease a full month sooner than PASI 75 responders. And that, as reported by the patients themselves, constitutes a meaningful advantage, Dr. Lebwohl stated.
Secukinumab is a fully human monoclonal antibody directed against a novel target: interleukin-17A. Novartis has filed for marketing approval of the biologic agent with an indication for psoriasis both with the Food and Drug Administration and European regulators. Secukinumab is also being developed as a treatment for psoriatic arthritis, rheumatoid arthritis, and ankylosing spondylitis.
Novartis funded the analysis. Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AMSTERDAM – Does pushing for a PASI 90 response instead of settling for a PASI 75 matter to patients being treated for moderate-to-severe chronic plaque psoriasis?
You bet it does, Dr. Mark G. Lebwohl asserted at the annual congress of the European Academy of Dermatology and Venereology.
He presented a pooled analysis of data from two large pivotal phase III randomized trials of secukinumab for psoriasis. The primary endpoints in the analysis were how often and how soon patients who achieved a PASI 75 or PASI 90 response at 12 weeks reported obtaining a Dermatology Life Quality Index (DLQI) response, defined as a score of 0 or 1.
The answer: More patients who had a PASI 90 response (meaning almost clear at 12 weeks) had a DLQI response, and it occurred a full 4 weeks faster than in PASI 75 responders – at a median of 8 weeks, compared with 12 weeks, reported Dr. Lebwohl, professor and chairman of the department of dermatology at Mt. Sinai Medical Center in New York.
Scores on the DLQI can range from 0, meaning no psoriasis-related impairment of the patient’s quality of life, up to 30. The average baseline score in this study population was 13.5, so a DLQI response dropping the score down to 0 or 1 represents a dramatic improvement in this patient-reported outcome.
Study participants completed the DLQI questionnaire at weeks 4, 8, 12, 24, 36, and again at week 52. The subjects’ mean baseline PASI score was 23.2.
The two double-blind, randomized, placebo-controlled clinical trials that formed the basis for this analysis were the recently published 52-week ERASURE (Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis) and FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis) studies (N. Engl. J. Med. 2014;371:326-38), in which patients were assigned to secukinumab at a dose of either 150 mg or 300 mg, placebo, or in the case of FIXTURE, to etanercept. The PASI 75 and 90 response rates at 12 weeks were markedly higher at both doses of secukinumab than with etanercept.
Dr. Lebwohl’s pooled analysis was restricted to the 1,470 study participants in the two studies who were randomized to active therapy. A total of 612 patients achieved a PASI 90 response by week 12. Another 365 had a PASI 75 response. Fully 89% of PASI 90 responders also had a DLQI response maintained out to week 52, as did 77% of PASI 75 responders.
The key finding: The median time to a DLQI response in the PASI 90 responders was 8 weeks, compared with 12 weeks in the PASI 75 responders. Thus, patients with a PASI 90 response obtained virtually total relief from what had previously been a debilitating disease a full month sooner than PASI 75 responders. And that, as reported by the patients themselves, constitutes a meaningful advantage, Dr. Lebwohl stated.
Secukinumab is a fully human monoclonal antibody directed against a novel target: interleukin-17A. Novartis has filed for marketing approval of the biologic agent with an indication for psoriasis both with the Food and Drug Administration and European regulators. Secukinumab is also being developed as a treatment for psoriatic arthritis, rheumatoid arthritis, and ankylosing spondylitis.
Novartis funded the analysis. Dr. Lebwohl reported serving as a consultant to Novartis and more than a dozen other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: Psoriasis patients who obtain a PASI 90 response report the quality of life burden imposed by the disease is lifted a full month sooner than in those with a PASI 75 response.
Major finding: The median time to a patient-reported Dermatology Life Quality Index score of 0 or 1 on the 0-30 scale was 8 weeks in PASI 90 responders, compared with 12 weeks in PASI 75 responders.
Data source: A pooled analysis of the 1,470 patients with moderate-to-severe chronic plaque psoriasis who were assigned to active therapy with either secukinumab or etanercept in two pivotal phase III randomized trials.
Disclosures: Novartis funded the analysis. Dr. Lebwohl reported serving as a consultant to the company. <caps/>

Recent Findings About Cardiovascular Comorbidities
Psoriasis Patients Have a Higher Risk for Myocardial Infarction
To determine if psoriasis is associated with a higher risk for myocardial infarction (MI), Wu et al (J Dermatolog Treat. doi:10.3109/09546634.2014.952609) performed a retrospective cohort study of 50,865 control patients matched to 10,173 patients with mild psoriasis and 19,205 control patients matched to 3841 patients with severe psoriasis. Multivariate analysis revealed that patients with mild and severe psoriasis had a higher risk for MI compared to matched control patients.
Practice Point: Psoriasis is associated with a higher risk for MI compared to control patients.
>>Read more at Journal of Dermatological Treatment
Screen Children With Psoriasis for Cardiovascular Comorbidities
Most evidence of the cardiovascular effects on psoriasis patients has focused on adults. Torres et al (Eur J Dermatol. 2014;24:229-235) evaluated the prevalence of excess adiposity, cardiovascular risk factors, metabolic syndrome, and lipid profile in children with psoriasis (age range, 5–15 years) compared to a control group. Children with psoriasis had a higher prevalence and greater odds of excess adiposity compared to controls. A higher prevalence of metabolic syndrome also was observed in children with psoriasis compared to controls.
Practice Point: Cardiovascular comorbidities known to be associated with adult psoriasis also are observed in children with psoriasis, warranting the need to screen children with psoriasis and promote healthy lifestyle choices.
>>Read more at European Journal of Dermatology
Psoriasis Patients Have a Greater Risk for Heart Failure
Khalid et al (Eur J Heart Fail. 2014;16:743-748) investigated the risk for new-onset heart failure in a nationwide cohort of psoriasis patients. They found that the overall incidence rates of new-onset heart failure were higher for patients with mild and severe psoriasis. Compared with the reference population, the fully adjusted hazard ratios for new-onset heart failure were increased in patients with mild and severe psoriasis.
Practice Point: Psoriasis may be associated with a disease severity–dependent increased risk for new-onset heart failure.
>>Read more at European Journal of Heart Failure
Psoriasis Patients Have a Higher Risk for Myocardial Infarction
To determine if psoriasis is associated with a higher risk for myocardial infarction (MI), Wu et al (J Dermatolog Treat. doi:10.3109/09546634.2014.952609) performed a retrospective cohort study of 50,865 control patients matched to 10,173 patients with mild psoriasis and 19,205 control patients matched to 3841 patients with severe psoriasis. Multivariate analysis revealed that patients with mild and severe psoriasis had a higher risk for MI compared to matched control patients.
Practice Point: Psoriasis is associated with a higher risk for MI compared to control patients.
>>Read more at Journal of Dermatological Treatment
Screen Children With Psoriasis for Cardiovascular Comorbidities
Most evidence of the cardiovascular effects on psoriasis patients has focused on adults. Torres et al (Eur J Dermatol. 2014;24:229-235) evaluated the prevalence of excess adiposity, cardiovascular risk factors, metabolic syndrome, and lipid profile in children with psoriasis (age range, 5–15 years) compared to a control group. Children with psoriasis had a higher prevalence and greater odds of excess adiposity compared to controls. A higher prevalence of metabolic syndrome also was observed in children with psoriasis compared to controls.
Practice Point: Cardiovascular comorbidities known to be associated with adult psoriasis also are observed in children with psoriasis, warranting the need to screen children with psoriasis and promote healthy lifestyle choices.
>>Read more at European Journal of Dermatology
Psoriasis Patients Have a Greater Risk for Heart Failure
Khalid et al (Eur J Heart Fail. 2014;16:743-748) investigated the risk for new-onset heart failure in a nationwide cohort of psoriasis patients. They found that the overall incidence rates of new-onset heart failure were higher for patients with mild and severe psoriasis. Compared with the reference population, the fully adjusted hazard ratios for new-onset heart failure were increased in patients with mild and severe psoriasis.
Practice Point: Psoriasis may be associated with a disease severity–dependent increased risk for new-onset heart failure.
>>Read more at European Journal of Heart Failure
Psoriasis Patients Have a Higher Risk for Myocardial Infarction
To determine if psoriasis is associated with a higher risk for myocardial infarction (MI), Wu et al (J Dermatolog Treat. doi:10.3109/09546634.2014.952609) performed a retrospective cohort study of 50,865 control patients matched to 10,173 patients with mild psoriasis and 19,205 control patients matched to 3841 patients with severe psoriasis. Multivariate analysis revealed that patients with mild and severe psoriasis had a higher risk for MI compared to matched control patients.
Practice Point: Psoriasis is associated with a higher risk for MI compared to control patients.
>>Read more at Journal of Dermatological Treatment
Screen Children With Psoriasis for Cardiovascular Comorbidities
Most evidence of the cardiovascular effects on psoriasis patients has focused on adults. Torres et al (Eur J Dermatol. 2014;24:229-235) evaluated the prevalence of excess adiposity, cardiovascular risk factors, metabolic syndrome, and lipid profile in children with psoriasis (age range, 5–15 years) compared to a control group. Children with psoriasis had a higher prevalence and greater odds of excess adiposity compared to controls. A higher prevalence of metabolic syndrome also was observed in children with psoriasis compared to controls.
Practice Point: Cardiovascular comorbidities known to be associated with adult psoriasis also are observed in children with psoriasis, warranting the need to screen children with psoriasis and promote healthy lifestyle choices.
>>Read more at European Journal of Dermatology
Psoriasis Patients Have a Greater Risk for Heart Failure
Khalid et al (Eur J Heart Fail. 2014;16:743-748) investigated the risk for new-onset heart failure in a nationwide cohort of psoriasis patients. They found that the overall incidence rates of new-onset heart failure were higher for patients with mild and severe psoriasis. Compared with the reference population, the fully adjusted hazard ratios for new-onset heart failure were increased in patients with mild and severe psoriasis.
Practice Point: Psoriasis may be associated with a disease severity–dependent increased risk for new-onset heart failure.
>>Read more at European Journal of Heart Failure
At Last? Apremilast
In late September 2014, the US Food and Drug Administration approved the medication apremilast for treatment of moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy. It was previously approved for psoriatic arthritis in March 2014. Its mechanism includes selective inhibition of phosphodiesterase 4, resulting in increased intracellular cyclic adenosine monophosphate levels, indirectly mediating production of inflammatory mediators in many cell types, namely decreasing tumor necrosis factor α and IL-23 and increasing IL-10. Orally dosed at 30 mg twice daily, safety and efficacy was determined via 2 multicenter, randomized, double-blind, placebo-controlled trials—ESTEEM 1 and ESTEEM 2 (N=1257)—that highlighted a PASI-75 (psoriasis area severity index) in 30% of patients in the first 4 months and up to 88% of patients with PASI-75 in the first year (J Am Acad Dermatol. 2014;70(suppl 1):AB164). Additionally, according to results presented at a recent European Academy of Dermatology and Venereology meeting in early October 2014, pruritus and difficult areas such as the scalp, palmoplantar area, and nails showed significant improvement at week 16 (P<.0001). The most common side effects were diarrhea, nausea, upper respiratory infection, and headache, which occurred most often in the first 2 weeks of therapy. The medication does not require routine laboratory monitoring; however, because weight loss is possible, it is recommended that weight should be periodically checked. There are no contraindications aside from hypersensitivity to the drug itself, and caution should be taken in patients with unstable depression, suicidal ideation, or severe renal impairment. It is pregnancy category C.
What’s the issue?
Because it is administered orally; is dually indicated for plaque psoriasis and psoriatic arthritis; and does not require laboratory monitoring, alcohol consumption restrictions, category X classification, or immunosuppressive infection or cancer risk, the window-shopping appeal of this drug seems attractive compared to the veteran and contemporary pharmaceutical army of psoriasis therapy. However, based on the ESTEEM studies, meager apremilast PASI scores are not blowing us away like those of biologic medications. At a time when the evolution of medications for psoriasis seems like a revolving door for new products highlighting new mechanisms in new pathways in even newer cells in relationship to inflammation, how will this drug fit in?
In late September 2014, the US Food and Drug Administration approved the medication apremilast for treatment of moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy. It was previously approved for psoriatic arthritis in March 2014. Its mechanism includes selective inhibition of phosphodiesterase 4, resulting in increased intracellular cyclic adenosine monophosphate levels, indirectly mediating production of inflammatory mediators in many cell types, namely decreasing tumor necrosis factor α and IL-23 and increasing IL-10. Orally dosed at 30 mg twice daily, safety and efficacy was determined via 2 multicenter, randomized, double-blind, placebo-controlled trials—ESTEEM 1 and ESTEEM 2 (N=1257)—that highlighted a PASI-75 (psoriasis area severity index) in 30% of patients in the first 4 months and up to 88% of patients with PASI-75 in the first year (J Am Acad Dermatol. 2014;70(suppl 1):AB164). Additionally, according to results presented at a recent European Academy of Dermatology and Venereology meeting in early October 2014, pruritus and difficult areas such as the scalp, palmoplantar area, and nails showed significant improvement at week 16 (P<.0001). The most common side effects were diarrhea, nausea, upper respiratory infection, and headache, which occurred most often in the first 2 weeks of therapy. The medication does not require routine laboratory monitoring; however, because weight loss is possible, it is recommended that weight should be periodically checked. There are no contraindications aside from hypersensitivity to the drug itself, and caution should be taken in patients with unstable depression, suicidal ideation, or severe renal impairment. It is pregnancy category C.
What’s the issue?
Because it is administered orally; is dually indicated for plaque psoriasis and psoriatic arthritis; and does not require laboratory monitoring, alcohol consumption restrictions, category X classification, or immunosuppressive infection or cancer risk, the window-shopping appeal of this drug seems attractive compared to the veteran and contemporary pharmaceutical army of psoriasis therapy. However, based on the ESTEEM studies, meager apremilast PASI scores are not blowing us away like those of biologic medications. At a time when the evolution of medications for psoriasis seems like a revolving door for new products highlighting new mechanisms in new pathways in even newer cells in relationship to inflammation, how will this drug fit in?
In late September 2014, the US Food and Drug Administration approved the medication apremilast for treatment of moderate to severe plaque psoriasis in adults who are candidates for phototherapy or systemic therapy. It was previously approved for psoriatic arthritis in March 2014. Its mechanism includes selective inhibition of phosphodiesterase 4, resulting in increased intracellular cyclic adenosine monophosphate levels, indirectly mediating production of inflammatory mediators in many cell types, namely decreasing tumor necrosis factor α and IL-23 and increasing IL-10. Orally dosed at 30 mg twice daily, safety and efficacy was determined via 2 multicenter, randomized, double-blind, placebo-controlled trials—ESTEEM 1 and ESTEEM 2 (N=1257)—that highlighted a PASI-75 (psoriasis area severity index) in 30% of patients in the first 4 months and up to 88% of patients with PASI-75 in the first year (J Am Acad Dermatol. 2014;70(suppl 1):AB164). Additionally, according to results presented at a recent European Academy of Dermatology and Venereology meeting in early October 2014, pruritus and difficult areas such as the scalp, palmoplantar area, and nails showed significant improvement at week 16 (P<.0001). The most common side effects were diarrhea, nausea, upper respiratory infection, and headache, which occurred most often in the first 2 weeks of therapy. The medication does not require routine laboratory monitoring; however, because weight loss is possible, it is recommended that weight should be periodically checked. There are no contraindications aside from hypersensitivity to the drug itself, and caution should be taken in patients with unstable depression, suicidal ideation, or severe renal impairment. It is pregnancy category C.
What’s the issue?
Because it is administered orally; is dually indicated for plaque psoriasis and psoriatic arthritis; and does not require laboratory monitoring, alcohol consumption restrictions, category X classification, or immunosuppressive infection or cancer risk, the window-shopping appeal of this drug seems attractive compared to the veteran and contemporary pharmaceutical army of psoriasis therapy. However, based on the ESTEEM studies, meager apremilast PASI scores are not blowing us away like those of biologic medications. At a time when the evolution of medications for psoriasis seems like a revolving door for new products highlighting new mechanisms in new pathways in even newer cells in relationship to inflammation, how will this drug fit in?

Biosimilars poised to make biologics more routine
The biosimilar age for rheumatology has arrived, and experts expect wider use of previously expensive biologic drugs as biosimilar competition drives prices down and makes biologics more affordable.
An infliximab biosimilar became the first such agent to arrive onto the European market in the second half of 2013, and by the start of 2014, it was already having an impact by, for example, dropping the cost of infliximab by a third in Norway. Norway may have received the biggest biosimilar effect so far because it runs an annual auction for the best prices on competing drugs from manufacturers and then mandates Norwegian clinicians to prescribe the lowest-price option when starting a new therapy.
A U.S. version of this scenario may soon follow. In August 2014, Celltrion and Hospira, the two companies jointly producing and marketing biosimilar forms of infliximab under the names Remsima and Inflectra (in parts of eastern Europe and elsewhere), announced they had submitted a marketing application to the Food and Drug Administration. In the announcement, Celltrion officials said they expected Food and Drug Administration approval within a year. If that were to happen, and if Celltrion mounts a successful patent challenge, the Remsima form of infliximab could become one of the first biosimilars on the U.S. market. (In July, Sandoz – a subsidiary of Novartis – announced it submitted an FDA application for Zarzio, a biosimilar version of filgrastim that until now has been only available as Neupogen, a granulocyte colony–stimulating factor. Biosimilar filgrastim seems like the only contender that could edge out Remsima as the first biosimilar on the U.S. market.)
At least two more biosimilars – a third form of infliximab, and a new form of etanercept – may come next, although rheumatologists following the field caution that additional studies are needed on top of what was reported for these two biosimilars last June at the annual European Congress of Rheumatology.
Lower cost broadens use
With one rheumatology biosimilar already on several global formularies and others nearing that status, the next challenge is convincing clinicians that cut-rate biologics are safe and effective and patients can switch from the brand-name form to a biosimilar without adverse effects. Meanwhile, payers and patients are pressing for biosimilars to cut the high cost of biologic treatment. By making biologic drugs more affordable for more patients, introduction of biosimilars will change patient care, experts said.
“A decrease in price will change how biologics are used in U.S. patients. In the United States today, about half of rheumatoid arthritis (RA) patients receive a biologic,” a rate substantially below where it should be, said Dr. Vibeke Strand, a biopharmaceutical consultant and rheumatologist at Stanford (Calif.) University. “Biosimilars will have a very big impact,” she predicted.

“Clinicians are under a lot of pressure from pharmacies, hospitals, and managed-care organization to avoid expensive medications when possible. Starting a biologic will become easier,” when prices start falling. And adherence may also improve. “Part of why patients don’t take their biologics for more than 1-2 years is they can’t afford the copay. That may change” if prices drop, Dr. Strand said in an interview.
Before biosimilars became available, “countries with low GDPs [gross domestic products] had less access to biologics and more restrictions; richer countries had better access,” noted Dr. Tore K. Kvien, a rheumatologist and professor of rheumatology at the University of Oslo.

Greater affordability and access to biologics is the sole factor driving the biosimilar movement. “Cost is the only reason why you have biosimilars,” noted Dr. Bruce N. Cronstein, a rheumatologist and professor of medicine, pathology, and pharmacology at New York University. Aside from cost, there are, by definition, no meaningful differences between a biosimilar and the reference brand-name formulation.
Biosimilars, Dr. Cronstein said, “will be cheaper, but it won’t be like the difference with generic and brand-name statins. You won’t see a 90% price drop. Maybe we’ll see a 30% or 40% price cut, which is considerable. The biologics are all very expensive drugs. But biosimilars will not lead to anything like the savings with small generic molecules.”

While Dr. Cronstein noted that there are no guarantees regarding the timing of price changes, the extent of price cuts, or how competition might affect pricing of the brand-name alternative, the experience in Norway showed a quick, one-third price cut when Remsima became an option, Dr. Kvien said in an interview. For years, Norway has negotiated 1-year contracts for setting drug costs with manufacturers. Norwegian officials invite competitors to submit bids in an annual auction. Celltrion won the auction to make Remsima the infliximab of choice in Norway during 2014 for all six European Medicines Agency (EMA)-approved infliximab indications: rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, Crohn’s disease, and ulcerative colitis.
“My colleagues tell me that the one-third reduction in price [compared with Remicade's price in 2014] was larger than they expected,” Dr. Kvien said. A year of treatment for an RA patient receiving Remsima averages about 6,000 euro now, down from the roughly 9,000 euro current annual cost for Remicade.
Dr. Strand maintained that economic pressures play a role in how easily biosimilars gain regulatory approval. For example, EMA received pressure from Eastern European countries where biologics have been relatively unavailable because of their price, she said. In the United States, pressure on the FDA comes from payers and patients because biologic copays are so high. But despite pressure, Dr. Strand and others believe that EMA and the FDA have set reasonable approval standards for biosimilars that clinicians can rely on.
“I think for all intents and purposes if EMA decides something is a biosimilar then I definitely think it is,” Dr. Strand said. “There may be subtle differences, but nothing big.” In some cases reference, brand-name biologics themselves have undergone substantial changes from what they originally had been, as happened with Enbrel and Rituxan, she noted. “The FDA will not entertain even a small change in an amino acid sequence, and requires two immunogenicity studies.”
Concerns about safety
But biosimilars may be a special case, where even the regulatory stamp of approval may not fully convince some clinicians.
“There is still quite a bit of skepticism,” Dr. Strand said. “I think there will be less skepticism once more data show that biosimilars are safe. The biggest concern seems to be that if the FDA decides something is not just biosimilar but also equivalent then there might be substitutions by a pharmacist without knowledge of the health care provider. I think there is concern because people are still not sure what biosimilar means. It will take more data to convince some people.”
“I think that physicians will be convinced by the data showing these compounds act in a similar way. What physicians worry about is that while these drugs may look good in a trial there might be some subtle difference that increases drug resistance or anaphylaxis,” said Dr. Cronstein. “I think clinicians will accept biosimilars and use them, but worry that a safety signal may not be seen in clinical trials. I think the FDA is trying to figure out how to be sure that switching from one drug to another does not cause an adverse effect. That is the major potential downside people see.”
The safety of switching to a biosimilar was an important enough issue for the Norwegian government to sponsor a 2.5 million euro trial to address the question. The 500-patient study, slated to start in the fall of 2014, will randomize patients on stable Remicade treatment to either remain on Remicade or switch to Remsima. The NOR-SWITCH trial will include patients with any of the six indications for which Remsima received EMA approval.
Until trial results are available, anticipated to be in 2016, Norway does not sanction switching patients on a stable Remicade regimen to Remsima even though Remsima is the infliximab of choice for patients starting infliximab for the first time. Despite this, some Norwegian departments have already made the switch in some patients, said Dr. Kvien, who is lead investigator for NOR-SWITCH. “They say the scientific data available now are sufficient to show switching is safe, but I do not agree.” The willingness of some Norwegian clinicians to switch their patients now from infliximab to the biosimilar shows how eager they and patients in Norway are to save money with Remsima.
While biosimilars face challenges entering and gaining traction in the European and American markets, they also seem driven by an overwhelming inevitability.
“Biosimilars will absolutely be routine in 10-20 years. They will be accepted, but it will take time,” Dr. Cronstein predicted.
“Biosimilars are here, they are not going away, and as patents run out we’ll see more and more of them,” predicted Dr. Paul Emery, professor of rheumatology at Leeds (England) University, while speaking at the annual European Congress of Rheumatology last June in Paris.
“I expect biosimilars will be important. My hope is that their lower cost will improve access to these treatments and this will mean better treatment for more patients around the world,” Dr. Kvien said.
Dr. Strand has been a consultant to Hospira, Celltrion, Amgen, Pfizer, Epirus, Baxter, and Merck Serono. Dr. Kvien has been a consultant to AbbVie, Bristol-Myers Squibb, Hospira, Celltrion, Pfizer/Wyeth, Merck Serono, Merck Sharp & Dohme, Roche, UCB, Orion, and Takeda. Dr. Cronstein has been a consultant to Pfizer and Merck Serono. Dr. Emery has been a consultant to AbbVie, Bristol-Myers Squibb, Merck, Pfizer, Roche, and Takeda.
On Twitter @mitchelzoler
The biosimilar age for rheumatology has arrived, and experts expect wider use of previously expensive biologic drugs as biosimilar competition drives prices down and makes biologics more affordable.
An infliximab biosimilar became the first such agent to arrive onto the European market in the second half of 2013, and by the start of 2014, it was already having an impact by, for example, dropping the cost of infliximab by a third in Norway. Norway may have received the biggest biosimilar effect so far because it runs an annual auction for the best prices on competing drugs from manufacturers and then mandates Norwegian clinicians to prescribe the lowest-price option when starting a new therapy.
A U.S. version of this scenario may soon follow. In August 2014, Celltrion and Hospira, the two companies jointly producing and marketing biosimilar forms of infliximab under the names Remsima and Inflectra (in parts of eastern Europe and elsewhere), announced they had submitted a marketing application to the Food and Drug Administration. In the announcement, Celltrion officials said they expected Food and Drug Administration approval within a year. If that were to happen, and if Celltrion mounts a successful patent challenge, the Remsima form of infliximab could become one of the first biosimilars on the U.S. market. (In July, Sandoz – a subsidiary of Novartis – announced it submitted an FDA application for Zarzio, a biosimilar version of filgrastim that until now has been only available as Neupogen, a granulocyte colony–stimulating factor. Biosimilar filgrastim seems like the only contender that could edge out Remsima as the first biosimilar on the U.S. market.)
At least two more biosimilars – a third form of infliximab, and a new form of etanercept – may come next, although rheumatologists following the field caution that additional studies are needed on top of what was reported for these two biosimilars last June at the annual European Congress of Rheumatology.
Lower cost broadens use
With one rheumatology biosimilar already on several global formularies and others nearing that status, the next challenge is convincing clinicians that cut-rate biologics are safe and effective and patients can switch from the brand-name form to a biosimilar without adverse effects. Meanwhile, payers and patients are pressing for biosimilars to cut the high cost of biologic treatment. By making biologic drugs more affordable for more patients, introduction of biosimilars will change patient care, experts said.
“A decrease in price will change how biologics are used in U.S. patients. In the United States today, about half of rheumatoid arthritis (RA) patients receive a biologic,” a rate substantially below where it should be, said Dr. Vibeke Strand, a biopharmaceutical consultant and rheumatologist at Stanford (Calif.) University. “Biosimilars will have a very big impact,” she predicted.
“Clinicians are under a lot of pressure from pharmacies, hospitals, and managed-care organization to avoid expensive medications when possible. Starting a biologic will become easier,” when prices start falling. And adherence may also improve. “Part of why patients don’t take their biologics for more than 1-2 years is they can’t afford the copay. That may change” if prices drop, Dr. Strand said in an interview.
Before biosimilars became available, “countries with low GDPs [gross domestic products] had less access to biologics and more restrictions; richer countries had better access,” noted Dr. Tore K. Kvien, a rheumatologist and professor of rheumatology at the University of Oslo.
Greater affordability and access to biologics is the sole factor driving the biosimilar movement. “Cost is the only reason why you have biosimilars,” noted Dr. Bruce N. Cronstein, a rheumatologist and professor of medicine, pathology, and pharmacology at New York University. Aside from cost, there are, by definition, no meaningful differences between a biosimilar and the reference brand-name formulation.
Biosimilars, Dr. Cronstein said, “will be cheaper, but it won’t be like the difference with generic and brand-name statins. You won’t see a 90% price drop. Maybe we’ll see a 30% or 40% price cut, which is considerable. The biologics are all very expensive drugs. But biosimilars will not lead to anything like the savings with small generic molecules.”
While Dr. Cronstein noted that there are no guarantees regarding the timing of price changes, the extent of price cuts, or how competition might affect pricing of the brand-name alternative, the experience in Norway showed a quick, one-third price cut when Remsima became an option, Dr. Kvien said in an interview. For years, Norway has negotiated 1-year contracts for setting drug costs with manufacturers. Norwegian officials invite competitors to submit bids in an annual auction. Celltrion won the auction to make Remsima the infliximab of choice in Norway during 2014 for all six European Medicines Agency (EMA)-approved infliximab indications: rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, Crohn’s disease, and ulcerative colitis.
“My colleagues tell me that the one-third reduction in price [compared with Remicade's price in 2014] was larger than they expected,” Dr. Kvien said. A year of treatment for an RA patient receiving Remsima averages about 6,000 euro now, down from the roughly 9,000 euro current annual cost for Remicade.
Dr. Strand maintained that economic pressures play a role in how easily biosimilars gain regulatory approval. For example, EMA received pressure from Eastern European countries where biologics have been relatively unavailable because of their price, she said. In the United States, pressure on the FDA comes from payers and patients because biologic copays are so high. But despite pressure, Dr. Strand and others believe that EMA and the FDA have set reasonable approval standards for biosimilars that clinicians can rely on.
“I think for all intents and purposes if EMA decides something is a biosimilar then I definitely think it is,” Dr. Strand said. “There may be subtle differences, but nothing big.” In some cases reference, brand-name biologics themselves have undergone substantial changes from what they originally had been, as happened with Enbrel and Rituxan, she noted. “The FDA will not entertain even a small change in an amino acid sequence, and requires two immunogenicity studies.”
Concerns about safety
But biosimilars may be a special case, where even the regulatory stamp of approval may not fully convince some clinicians.
“There is still quite a bit of skepticism,” Dr. Strand said. “I think there will be less skepticism once more data show that biosimilars are safe. The biggest concern seems to be that if the FDA decides something is not just biosimilar but also equivalent then there might be substitutions by a pharmacist without knowledge of the health care provider. I think there is concern because people are still not sure what biosimilar means. It will take more data to convince some people.”
“I think that physicians will be convinced by the data showing these compounds act in a similar way. What physicians worry about is that while these drugs may look good in a trial there might be some subtle difference that increases drug resistance or anaphylaxis,” said Dr. Cronstein. “I think clinicians will accept biosimilars and use them, but worry that a safety signal may not be seen in clinical trials. I think the FDA is trying to figure out how to be sure that switching from one drug to another does not cause an adverse effect. That is the major potential downside people see.”
The safety of switching to a biosimilar was an important enough issue for the Norwegian government to sponsor a 2.5 million euro trial to address the question. The 500-patient study, slated to start in the fall of 2014, will randomize patients on stable Remicade treatment to either remain on Remicade or switch to Remsima. The NOR-SWITCH trial will include patients with any of the six indications for which Remsima received EMA approval.
Until trial results are available, anticipated to be in 2016, Norway does not sanction switching patients on a stable Remicade regimen to Remsima even though Remsima is the infliximab of choice for patients starting infliximab for the first time. Despite this, some Norwegian departments have already made the switch in some patients, said Dr. Kvien, who is lead investigator for NOR-SWITCH. “They say the scientific data available now are sufficient to show switching is safe, but I do not agree.” The willingness of some Norwegian clinicians to switch their patients now from infliximab to the biosimilar shows how eager they and patients in Norway are to save money with Remsima.
While biosimilars face challenges entering and gaining traction in the European and American markets, they also seem driven by an overwhelming inevitability.
“Biosimilars will absolutely be routine in 10-20 years. They will be accepted, but it will take time,” Dr. Cronstein predicted.
“Biosimilars are here, they are not going away, and as patents run out we’ll see more and more of them,” predicted Dr. Paul Emery, professor of rheumatology at Leeds (England) University, while speaking at the annual European Congress of Rheumatology last June in Paris.
“I expect biosimilars will be important. My hope is that their lower cost will improve access to these treatments and this will mean better treatment for more patients around the world,” Dr. Kvien said.
Dr. Strand has been a consultant to Hospira, Celltrion, Amgen, Pfizer, Epirus, Baxter, and Merck Serono. Dr. Kvien has been a consultant to AbbVie, Bristol-Myers Squibb, Hospira, Celltrion, Pfizer/Wyeth, Merck Serono, Merck Sharp & Dohme, Roche, UCB, Orion, and Takeda. Dr. Cronstein has been a consultant to Pfizer and Merck Serono. Dr. Emery has been a consultant to AbbVie, Bristol-Myers Squibb, Merck, Pfizer, Roche, and Takeda.
On Twitter @mitchelzoler
The biosimilar age for rheumatology has arrived, and experts expect wider use of previously expensive biologic drugs as biosimilar competition drives prices down and makes biologics more affordable.
An infliximab biosimilar became the first such agent to arrive onto the European market in the second half of 2013, and by the start of 2014, it was already having an impact by, for example, dropping the cost of infliximab by a third in Norway. Norway may have received the biggest biosimilar effect so far because it runs an annual auction for the best prices on competing drugs from manufacturers and then mandates Norwegian clinicians to prescribe the lowest-price option when starting a new therapy.
A U.S. version of this scenario may soon follow. In August 2014, Celltrion and Hospira, the two companies jointly producing and marketing biosimilar forms of infliximab under the names Remsima and Inflectra (in parts of eastern Europe and elsewhere), announced they had submitted a marketing application to the Food and Drug Administration. In the announcement, Celltrion officials said they expected Food and Drug Administration approval within a year. If that were to happen, and if Celltrion mounts a successful patent challenge, the Remsima form of infliximab could become one of the first biosimilars on the U.S. market. (In July, Sandoz – a subsidiary of Novartis – announced it submitted an FDA application for Zarzio, a biosimilar version of filgrastim that until now has been only available as Neupogen, a granulocyte colony–stimulating factor. Biosimilar filgrastim seems like the only contender that could edge out Remsima as the first biosimilar on the U.S. market.)
At least two more biosimilars – a third form of infliximab, and a new form of etanercept – may come next, although rheumatologists following the field caution that additional studies are needed on top of what was reported for these two biosimilars last June at the annual European Congress of Rheumatology.
Lower cost broadens use
With one rheumatology biosimilar already on several global formularies and others nearing that status, the next challenge is convincing clinicians that cut-rate biologics are safe and effective and patients can switch from the brand-name form to a biosimilar without adverse effects. Meanwhile, payers and patients are pressing for biosimilars to cut the high cost of biologic treatment. By making biologic drugs more affordable for more patients, introduction of biosimilars will change patient care, experts said.
“A decrease in price will change how biologics are used in U.S. patients. In the United States today, about half of rheumatoid arthritis (RA) patients receive a biologic,” a rate substantially below where it should be, said Dr. Vibeke Strand, a biopharmaceutical consultant and rheumatologist at Stanford (Calif.) University. “Biosimilars will have a very big impact,” she predicted.
“Clinicians are under a lot of pressure from pharmacies, hospitals, and managed-care organization to avoid expensive medications when possible. Starting a biologic will become easier,” when prices start falling. And adherence may also improve. “Part of why patients don’t take their biologics for more than 1-2 years is they can’t afford the copay. That may change” if prices drop, Dr. Strand said in an interview.
Before biosimilars became available, “countries with low GDPs [gross domestic products] had less access to biologics and more restrictions; richer countries had better access,” noted Dr. Tore K. Kvien, a rheumatologist and professor of rheumatology at the University of Oslo.
Greater affordability and access to biologics is the sole factor driving the biosimilar movement. “Cost is the only reason why you have biosimilars,” noted Dr. Bruce N. Cronstein, a rheumatologist and professor of medicine, pathology, and pharmacology at New York University. Aside from cost, there are, by definition, no meaningful differences between a biosimilar and the reference brand-name formulation.
Biosimilars, Dr. Cronstein said, “will be cheaper, but it won’t be like the difference with generic and brand-name statins. You won’t see a 90% price drop. Maybe we’ll see a 30% or 40% price cut, which is considerable. The biologics are all very expensive drugs. But biosimilars will not lead to anything like the savings with small generic molecules.”
While Dr. Cronstein noted that there are no guarantees regarding the timing of price changes, the extent of price cuts, or how competition might affect pricing of the brand-name alternative, the experience in Norway showed a quick, one-third price cut when Remsima became an option, Dr. Kvien said in an interview. For years, Norway has negotiated 1-year contracts for setting drug costs with manufacturers. Norwegian officials invite competitors to submit bids in an annual auction. Celltrion won the auction to make Remsima the infliximab of choice in Norway during 2014 for all six European Medicines Agency (EMA)-approved infliximab indications: rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, Crohn’s disease, and ulcerative colitis.
“My colleagues tell me that the one-third reduction in price [compared with Remicade's price in 2014] was larger than they expected,” Dr. Kvien said. A year of treatment for an RA patient receiving Remsima averages about 6,000 euro now, down from the roughly 9,000 euro current annual cost for Remicade.
Dr. Strand maintained that economic pressures play a role in how easily biosimilars gain regulatory approval. For example, EMA received pressure from Eastern European countries where biologics have been relatively unavailable because of their price, she said. In the United States, pressure on the FDA comes from payers and patients because biologic copays are so high. But despite pressure, Dr. Strand and others believe that EMA and the FDA have set reasonable approval standards for biosimilars that clinicians can rely on.
“I think for all intents and purposes if EMA decides something is a biosimilar then I definitely think it is,” Dr. Strand said. “There may be subtle differences, but nothing big.” In some cases reference, brand-name biologics themselves have undergone substantial changes from what they originally had been, as happened with Enbrel and Rituxan, she noted. “The FDA will not entertain even a small change in an amino acid sequence, and requires two immunogenicity studies.”
Concerns about safety
But biosimilars may be a special case, where even the regulatory stamp of approval may not fully convince some clinicians.
“There is still quite a bit of skepticism,” Dr. Strand said. “I think there will be less skepticism once more data show that biosimilars are safe. The biggest concern seems to be that if the FDA decides something is not just biosimilar but also equivalent then there might be substitutions by a pharmacist without knowledge of the health care provider. I think there is concern because people are still not sure what biosimilar means. It will take more data to convince some people.”
“I think that physicians will be convinced by the data showing these compounds act in a similar way. What physicians worry about is that while these drugs may look good in a trial there might be some subtle difference that increases drug resistance or anaphylaxis,” said Dr. Cronstein. “I think clinicians will accept biosimilars and use them, but worry that a safety signal may not be seen in clinical trials. I think the FDA is trying to figure out how to be sure that switching from one drug to another does not cause an adverse effect. That is the major potential downside people see.”
The safety of switching to a biosimilar was an important enough issue for the Norwegian government to sponsor a 2.5 million euro trial to address the question. The 500-patient study, slated to start in the fall of 2014, will randomize patients on stable Remicade treatment to either remain on Remicade or switch to Remsima. The NOR-SWITCH trial will include patients with any of the six indications for which Remsima received EMA approval.
Until trial results are available, anticipated to be in 2016, Norway does not sanction switching patients on a stable Remicade regimen to Remsima even though Remsima is the infliximab of choice for patients starting infliximab for the first time. Despite this, some Norwegian departments have already made the switch in some patients, said Dr. Kvien, who is lead investigator for NOR-SWITCH. “They say the scientific data available now are sufficient to show switching is safe, but I do not agree.” The willingness of some Norwegian clinicians to switch their patients now from infliximab to the biosimilar shows how eager they and patients in Norway are to save money with Remsima.
While biosimilars face challenges entering and gaining traction in the European and American markets, they also seem driven by an overwhelming inevitability.
“Biosimilars will absolutely be routine in 10-20 years. They will be accepted, but it will take time,” Dr. Cronstein predicted.
“Biosimilars are here, they are not going away, and as patents run out we’ll see more and more of them,” predicted Dr. Paul Emery, professor of rheumatology at Leeds (England) University, while speaking at the annual European Congress of Rheumatology last June in Paris.
“I expect biosimilars will be important. My hope is that their lower cost will improve access to these treatments and this will mean better treatment for more patients around the world,” Dr. Kvien said.
Dr. Strand has been a consultant to Hospira, Celltrion, Amgen, Pfizer, Epirus, Baxter, and Merck Serono. Dr. Kvien has been a consultant to AbbVie, Bristol-Myers Squibb, Hospira, Celltrion, Pfizer/Wyeth, Merck Serono, Merck Sharp & Dohme, Roche, UCB, Orion, and Takeda. Dr. Cronstein has been a consultant to Pfizer and Merck Serono. Dr. Emery has been a consultant to AbbVie, Bristol-Myers Squibb, Merck, Pfizer, Roche, and Takeda.
On Twitter @mitchelzoler

Psoriasis severity linked to uncontrolled hypertension
The more severe the psoriasis, the greater likelihood of uncontrolled hypertension, according to data from a population-based study. The findings were published online Oct. 15 in JAMA Dermatology.
In patients with diagnosed hypertension, those with moderate to severe psoriasis showed a positive dose-response relationship between their psoriasis activity and high blood pressure, wrote Dr. Junko Takeshita of the University of Pennsylvania, Philadelphia, and her associates (JAMA Dermatol. 2014 Oct. 15 [doi:10.1001/jamadermatol.2014.2094]).
The researchers compared a random sample of 1,322 adults aged 25-64 years with psoriasis and hypertension and 11,977 age- and practice-matched controls with hypertension. The data were taken from a population-based, cross-sectional study nested in a prospective cohort drawn from an electronic medical records database in the United Kingdom.
After investigators adjusted for age, sex, body mass index, smoking and alcohol use status, presence of comorbid conditions, and current use of antihypertensive medications and nonsteroidal anti-inflammatory drugs, they found psoriasis activity and uncontrolled hypertension correlated, with adjusted odds ratios of 0.97 for mild psoriasis,1.20 for moderate psoriasis, and 1.48 for severe psoriasis (P = .01). The likelihood of uncontrolled hypertension among psoriasis overall was also increased, but this increase was not statistically significant.
The study was limited by its cross-sectional design, which make the directionality of the two conditions hard to determine, the researchers noted.
However, the findings suggest that, among patients with hypertension, psoriasis “is independently associated with poorly controlled blood pressure,” and that more effective blood pressure management is warranted in psoriasis patients, especially those with more severe disease.
Dr. Takeshita reported receipt of payment for continuing medical education work related to psoriasis. Coauthor Dr. Joel Gelfand reported serving as a consultant for AbbVie, Amgen, Celgene, Eli Lilly, Janssen Biologics, and others. This study was supported in part by the National Heart, Lung, and Blood Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
On Twitter @whitneymcknight
The more severe the psoriasis, the greater likelihood of uncontrolled hypertension, according to data from a population-based study. The findings were published online Oct. 15 in JAMA Dermatology.
In patients with diagnosed hypertension, those with moderate to severe psoriasis showed a positive dose-response relationship between their psoriasis activity and high blood pressure, wrote Dr. Junko Takeshita of the University of Pennsylvania, Philadelphia, and her associates (JAMA Dermatol. 2014 Oct. 15 [doi:10.1001/jamadermatol.2014.2094]).
The researchers compared a random sample of 1,322 adults aged 25-64 years with psoriasis and hypertension and 11,977 age- and practice-matched controls with hypertension. The data were taken from a population-based, cross-sectional study nested in a prospective cohort drawn from an electronic medical records database in the United Kingdom.
After investigators adjusted for age, sex, body mass index, smoking and alcohol use status, presence of comorbid conditions, and current use of antihypertensive medications and nonsteroidal anti-inflammatory drugs, they found psoriasis activity and uncontrolled hypertension correlated, with adjusted odds ratios of 0.97 for mild psoriasis,1.20 for moderate psoriasis, and 1.48 for severe psoriasis (P = .01). The likelihood of uncontrolled hypertension among psoriasis overall was also increased, but this increase was not statistically significant.
The study was limited by its cross-sectional design, which make the directionality of the two conditions hard to determine, the researchers noted.
However, the findings suggest that, among patients with hypertension, psoriasis “is independently associated with poorly controlled blood pressure,” and that more effective blood pressure management is warranted in psoriasis patients, especially those with more severe disease.
Dr. Takeshita reported receipt of payment for continuing medical education work related to psoriasis. Coauthor Dr. Joel Gelfand reported serving as a consultant for AbbVie, Amgen, Celgene, Eli Lilly, Janssen Biologics, and others. This study was supported in part by the National Heart, Lung, and Blood Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
On Twitter @whitneymcknight
The more severe the psoriasis, the greater likelihood of uncontrolled hypertension, according to data from a population-based study. The findings were published online Oct. 15 in JAMA Dermatology.
In patients with diagnosed hypertension, those with moderate to severe psoriasis showed a positive dose-response relationship between their psoriasis activity and high blood pressure, wrote Dr. Junko Takeshita of the University of Pennsylvania, Philadelphia, and her associates (JAMA Dermatol. 2014 Oct. 15 [doi:10.1001/jamadermatol.2014.2094]).
The researchers compared a random sample of 1,322 adults aged 25-64 years with psoriasis and hypertension and 11,977 age- and practice-matched controls with hypertension. The data were taken from a population-based, cross-sectional study nested in a prospective cohort drawn from an electronic medical records database in the United Kingdom.
After investigators adjusted for age, sex, body mass index, smoking and alcohol use status, presence of comorbid conditions, and current use of antihypertensive medications and nonsteroidal anti-inflammatory drugs, they found psoriasis activity and uncontrolled hypertension correlated, with adjusted odds ratios of 0.97 for mild psoriasis,1.20 for moderate psoriasis, and 1.48 for severe psoriasis (P = .01). The likelihood of uncontrolled hypertension among psoriasis overall was also increased, but this increase was not statistically significant.
The study was limited by its cross-sectional design, which make the directionality of the two conditions hard to determine, the researchers noted.
However, the findings suggest that, among patients with hypertension, psoriasis “is independently associated with poorly controlled blood pressure,” and that more effective blood pressure management is warranted in psoriasis patients, especially those with more severe disease.
Dr. Takeshita reported receipt of payment for continuing medical education work related to psoriasis. Coauthor Dr. Joel Gelfand reported serving as a consultant for AbbVie, Amgen, Celgene, Eli Lilly, Janssen Biologics, and others. This study was supported in part by the National Heart, Lung, and Blood Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
On Twitter @whitneymcknight
Key clinical point: Patients with moderate to severe psoriasis may benefit from close hypertension management.
Major finding: Psoriasis severity had a significantly positive dose-response relationship with uncontrolled hypertension: adjusted odds ratio 0.97 for mild psoriasis, 1.20 for moderate psoriasis,and 1.48 for severe psoriasis, (P = .01).
Data source: Random sample of 1,322 adults between 25 and 64 years with psoriasis and hypertension and 11,977 age- and practice-matched controls taken from a population-based cross-sectional study nested in a prospective cohort drawn from an electronic medical records database.
Disclosures: Dr. Takeshita reported receipt of payment for continuing medical education work related to psoriasis. Coauthor Dr. Joel Gelfand reported serving as a consultant for AbbVie, Amgen, Celgene, Eli Lilly, Janssen Biologics, and others. This study was supported in part by the National Heart, Lung, and Blood Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Apremilast improves psoriasis with a side order of weight loss
AMSTERDAM – One in five psoriasis patients lost more than 5% of baseline body weight while on oral apremilast for 52 weeks in the phase III ESTEEM 1 and ESTEEM 2 trials.
Patients taking apremilast not only experienced marked improvement in their psoriasis, but also much-needed weight loss, Dr. Kristian Reich observed in presenting the ESTEEM trials analysis at the annual congress of the European Academy of Dermatology and Venereology.

“You could see the weight loss in these studies as a side effect. But I think many of us would think that, in a psoriasis population with a mean baseline weight greater than 90 kg, that’s a very good thing to have,” said Dr. Reich of Dermatologikum Hamburg (Germany).
The mechanism for this weight loss remains unclear, Dr. Reich said. The important point is that it proved completely unrelated to the diarrhea, nausea, and vomiting that are common, albeit mild and transient, side effects associated with apremilast, a phosphodiesterase 4 inhibitor. That is, patients who lost significant weight did not have an increased incidence of these GI adverse events. Nor was there any correlation between baseline body weight and weight loss on the drug, he added.
Apremilast (Otezla) was approved by the Food and Drug Administration earlier in 2014 for the treatment of psoriatic arthritis, and in late September for psoriasis, and is the only systemic psoriasis drug that requires no tuberculosis screening or laboratory monitoring. The drug also is being developed as a medication for rheumatoid arthritis and ankylosing spondylitis.
The prospective ESTEEM I and II trials totaled 1,250 patients with moderate to severe psoriasis and a mean baseline weight of 92.6 kg. They were initially randomized 2:1 to 16 weeks of double-blind apremilast at 30 mg twice daily or placebo. At 16 weeks, everyone was placed on apremilast 30 mg b.i.d. through week 32, when a randomized treatment withdrawal phase began that lasted through week 52.
The weight loss associated with apremilast was progressive. Patients continued to lose weight while taking the drug until approximately week 65 of therapy, when weight loss has plateaued in long-term extension studies.
At 16 weeks in the ESTEEM trials, patients on apremilast had a mean 1.51-kg weight loss compared to baseline, while the mean body weight in placebo-treated controls remained unchanged. Also at week 16, 13.7% of the apremilast group and 5.5% of controls demonstrated a greater than 5% weight reduction from baseline.
The 564 patients on apremilast for the full 52 weeks had a mean weight loss from baseline of 1.99 kg, and 19.2% of patients had a weight loss in excess of 5%.
“I would call a 2-kg weight loss over a 1-year period a moderate weight loss. But a greater than 5% weight loss is generally classified by experts as clinically meaningful,” Dr. Reich noted.
While weight loss increased with time on apremilast, the drug’s GI side effects peaked during the first 2 weeks of therapy and tailed off after the first month.
Weight loss in apremilast-treated patients did not lead to any overt medical sequelae. Only 0.2% of patients discontinued the drug because of weight loss.
The ESTEEM analysis showed no sign of any increased risks of major adverse cardiovascular events, malignancies, serious infections, depressive symptoms, or suicidality in apremilast-treated patients, according to Dr. Reich.
“This is a drug known for having a very good safety profile,” he said.
The ESTEEM trials were funded by Celgene, which markets apremilast. Dr. Reich reported receiving research grants from and serving as a consultant to the company.
AMSTERDAM – One in five psoriasis patients lost more than 5% of baseline body weight while on oral apremilast for 52 weeks in the phase III ESTEEM 1 and ESTEEM 2 trials.
Patients taking apremilast not only experienced marked improvement in their psoriasis, but also much-needed weight loss, Dr. Kristian Reich observed in presenting the ESTEEM trials analysis at the annual congress of the European Academy of Dermatology and Venereology.
“You could see the weight loss in these studies as a side effect. But I think many of us would think that, in a psoriasis population with a mean baseline weight greater than 90 kg, that’s a very good thing to have,” said Dr. Reich of Dermatologikum Hamburg (Germany).
The mechanism for this weight loss remains unclear, Dr. Reich said. The important point is that it proved completely unrelated to the diarrhea, nausea, and vomiting that are common, albeit mild and transient, side effects associated with apremilast, a phosphodiesterase 4 inhibitor. That is, patients who lost significant weight did not have an increased incidence of these GI adverse events. Nor was there any correlation between baseline body weight and weight loss on the drug, he added.
Apremilast (Otezla) was approved by the Food and Drug Administration earlier in 2014 for the treatment of psoriatic arthritis, and in late September for psoriasis, and is the only systemic psoriasis drug that requires no tuberculosis screening or laboratory monitoring. The drug also is being developed as a medication for rheumatoid arthritis and ankylosing spondylitis.
The prospective ESTEEM I and II trials totaled 1,250 patients with moderate to severe psoriasis and a mean baseline weight of 92.6 kg. They were initially randomized 2:1 to 16 weeks of double-blind apremilast at 30 mg twice daily or placebo. At 16 weeks, everyone was placed on apremilast 30 mg b.i.d. through week 32, when a randomized treatment withdrawal phase began that lasted through week 52.
The weight loss associated with apremilast was progressive. Patients continued to lose weight while taking the drug until approximately week 65 of therapy, when weight loss has plateaued in long-term extension studies.
At 16 weeks in the ESTEEM trials, patients on apremilast had a mean 1.51-kg weight loss compared to baseline, while the mean body weight in placebo-treated controls remained unchanged. Also at week 16, 13.7% of the apremilast group and 5.5% of controls demonstrated a greater than 5% weight reduction from baseline.
The 564 patients on apremilast for the full 52 weeks had a mean weight loss from baseline of 1.99 kg, and 19.2% of patients had a weight loss in excess of 5%.
“I would call a 2-kg weight loss over a 1-year period a moderate weight loss. But a greater than 5% weight loss is generally classified by experts as clinically meaningful,” Dr. Reich noted.
While weight loss increased with time on apremilast, the drug’s GI side effects peaked during the first 2 weeks of therapy and tailed off after the first month.
Weight loss in apremilast-treated patients did not lead to any overt medical sequelae. Only 0.2% of patients discontinued the drug because of weight loss.
The ESTEEM analysis showed no sign of any increased risks of major adverse cardiovascular events, malignancies, serious infections, depressive symptoms, or suicidality in apremilast-treated patients, according to Dr. Reich.
“This is a drug known for having a very good safety profile,” he said.
The ESTEEM trials were funded by Celgene, which markets apremilast. Dr. Reich reported receiving research grants from and serving as a consultant to the company.
AMSTERDAM – One in five psoriasis patients lost more than 5% of baseline body weight while on oral apremilast for 52 weeks in the phase III ESTEEM 1 and ESTEEM 2 trials.
Patients taking apremilast not only experienced marked improvement in their psoriasis, but also much-needed weight loss, Dr. Kristian Reich observed in presenting the ESTEEM trials analysis at the annual congress of the European Academy of Dermatology and Venereology.
“You could see the weight loss in these studies as a side effect. But I think many of us would think that, in a psoriasis population with a mean baseline weight greater than 90 kg, that’s a very good thing to have,” said Dr. Reich of Dermatologikum Hamburg (Germany).
The mechanism for this weight loss remains unclear, Dr. Reich said. The important point is that it proved completely unrelated to the diarrhea, nausea, and vomiting that are common, albeit mild and transient, side effects associated with apremilast, a phosphodiesterase 4 inhibitor. That is, patients who lost significant weight did not have an increased incidence of these GI adverse events. Nor was there any correlation between baseline body weight and weight loss on the drug, he added.
Apremilast (Otezla) was approved by the Food and Drug Administration earlier in 2014 for the treatment of psoriatic arthritis, and in late September for psoriasis, and is the only systemic psoriasis drug that requires no tuberculosis screening or laboratory monitoring. The drug also is being developed as a medication for rheumatoid arthritis and ankylosing spondylitis.
The prospective ESTEEM I and II trials totaled 1,250 patients with moderate to severe psoriasis and a mean baseline weight of 92.6 kg. They were initially randomized 2:1 to 16 weeks of double-blind apremilast at 30 mg twice daily or placebo. At 16 weeks, everyone was placed on apremilast 30 mg b.i.d. through week 32, when a randomized treatment withdrawal phase began that lasted through week 52.
The weight loss associated with apremilast was progressive. Patients continued to lose weight while taking the drug until approximately week 65 of therapy, when weight loss has plateaued in long-term extension studies.
At 16 weeks in the ESTEEM trials, patients on apremilast had a mean 1.51-kg weight loss compared to baseline, while the mean body weight in placebo-treated controls remained unchanged. Also at week 16, 13.7% of the apremilast group and 5.5% of controls demonstrated a greater than 5% weight reduction from baseline.
The 564 patients on apremilast for the full 52 weeks had a mean weight loss from baseline of 1.99 kg, and 19.2% of patients had a weight loss in excess of 5%.
“I would call a 2-kg weight loss over a 1-year period a moderate weight loss. But a greater than 5% weight loss is generally classified by experts as clinically meaningful,” Dr. Reich noted.
While weight loss increased with time on apremilast, the drug’s GI side effects peaked during the first 2 weeks of therapy and tailed off after the first month.
Weight loss in apremilast-treated patients did not lead to any overt medical sequelae. Only 0.2% of patients discontinued the drug because of weight loss.
The ESTEEM analysis showed no sign of any increased risks of major adverse cardiovascular events, malignancies, serious infections, depressive symptoms, or suicidality in apremilast-treated patients, according to Dr. Reich.
“This is a drug known for having a very good safety profile,” he said.
The ESTEEM trials were funded by Celgene, which markets apremilast. Dr. Reich reported receiving research grants from and serving as a consultant to the company.
AT THE EADV CONGRESS
Key clinical point: Oral apremilast promoted clinically significant weight reduction in addition to clinically meaningful improvement in skin disease for psoriasis patients.
Major finding: One in five psoriasis patients on apremilast for 52 weeks experienced more than a 5% reduction in weight with no untoward medical effects.
Data source: Analysis of 1,250 patients with moderate to severe psoriasis who participated in the randomized, double-blind, phase III ESTEEM 1 and ESTEEM 2 studies.
Disclosures: Dr. Reich disclosed ties to Celgene, which funded the ESTEEM trials.
