User login
Quizartinib-based combinations safe and effective for untreated MDS
Key clinical point: Quizartinib-based combinations were effective and well tolerated in both frontline and first salvage for patients with untreated myelodysplastic syndrome (MDS) and FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) acute myeloid leukemia (AML).
Major finding: Frontline quizartinib/azacitidine vs. quizartinib/low-dose cytarabine (LDAC) led to a higher proportion of patients achieve composite response (87% vs. 74%) and a longer overall survival (19.2 vs. 8.5 months; P = .036). Similarly, quizartinib/azacitidine vs. quizartinib/LDAC was associated with a higher proportion of patients with relapsed/refractory AML achieve composite response (64% vs. 29%) and a trend for longer overall survival (12.8 vs. 4 months; P = .053). Overall, therapy was well tolerated in both cohorts.
Study details: In this open-label phase I/II trial, patients with untreated MDS/AML or those receiving first-salvage treatment for FLT3-ITD AML were treated with a combination of quizartinib and either azacitidine or LDAC in frontline (n=34) or as first-salvage (n=39) therapy.
Disclosures: The study was supported in part by the Cancer Center Support Grant. The authors declared no competing financial interests.
Source: Swaminathan M et al. Haematologica. 2021 Apr 15. doi: 10.3324/haematol.2020.263392.
Key clinical point: Quizartinib-based combinations were effective and well tolerated in both frontline and first salvage for patients with untreated myelodysplastic syndrome (MDS) and FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) acute myeloid leukemia (AML).
Major finding: Frontline quizartinib/azacitidine vs. quizartinib/low-dose cytarabine (LDAC) led to a higher proportion of patients achieve composite response (87% vs. 74%) and a longer overall survival (19.2 vs. 8.5 months; P = .036). Similarly, quizartinib/azacitidine vs. quizartinib/LDAC was associated with a higher proportion of patients with relapsed/refractory AML achieve composite response (64% vs. 29%) and a trend for longer overall survival (12.8 vs. 4 months; P = .053). Overall, therapy was well tolerated in both cohorts.
Study details: In this open-label phase I/II trial, patients with untreated MDS/AML or those receiving first-salvage treatment for FLT3-ITD AML were treated with a combination of quizartinib and either azacitidine or LDAC in frontline (n=34) or as first-salvage (n=39) therapy.
Disclosures: The study was supported in part by the Cancer Center Support Grant. The authors declared no competing financial interests.
Source: Swaminathan M et al. Haematologica. 2021 Apr 15. doi: 10.3324/haematol.2020.263392.
Key clinical point: Quizartinib-based combinations were effective and well tolerated in both frontline and first salvage for patients with untreated myelodysplastic syndrome (MDS) and FMS-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) acute myeloid leukemia (AML).
Major finding: Frontline quizartinib/azacitidine vs. quizartinib/low-dose cytarabine (LDAC) led to a higher proportion of patients achieve composite response (87% vs. 74%) and a longer overall survival (19.2 vs. 8.5 months; P = .036). Similarly, quizartinib/azacitidine vs. quizartinib/LDAC was associated with a higher proportion of patients with relapsed/refractory AML achieve composite response (64% vs. 29%) and a trend for longer overall survival (12.8 vs. 4 months; P = .053). Overall, therapy was well tolerated in both cohorts.
Study details: In this open-label phase I/II trial, patients with untreated MDS/AML or those receiving first-salvage treatment for FLT3-ITD AML were treated with a combination of quizartinib and either azacitidine or LDAC in frontline (n=34) or as first-salvage (n=39) therapy.
Disclosures: The study was supported in part by the Cancer Center Support Grant. The authors declared no competing financial interests.
Source: Swaminathan M et al. Haematologica. 2021 Apr 15. doi: 10.3324/haematol.2020.263392.
Stanozolol: An effective alternative treatment for lower-risk MDS after epoetin alfa failure
Key clinical point: Stanozolol (STZ) monotherapy could be considered an alternative treatment in patients with lower-risk myelodysplastic syndrome (MDS) without del(5q) after the failure of epoetin alfa.
Major finding: Hematologic improvement-erythroid response (HI-E) and transfusion independence were achieved in 48.2% and 43.2% of patients, respectively. The estimated 5-year overall survival was higher in HI-E responders vs. nonresponders (88.6% vs. 33.8%). Most of the adverse events were manageable.
Study details: Findings are from a retrospective analysis of 56 patients with lower-risk MDS without del(5q) exclusively treated with STZ after failure of epoetin alfa as first-line treatment.
Disclosures: No source of funding was declared. The authors declared no competing interests.
Source: Qu WY et al. Ann Hematol. 2021 Apr 10. doi: 10.1007/s00277-021-04508-w.
Key clinical point: Stanozolol (STZ) monotherapy could be considered an alternative treatment in patients with lower-risk myelodysplastic syndrome (MDS) without del(5q) after the failure of epoetin alfa.
Major finding: Hematologic improvement-erythroid response (HI-E) and transfusion independence were achieved in 48.2% and 43.2% of patients, respectively. The estimated 5-year overall survival was higher in HI-E responders vs. nonresponders (88.6% vs. 33.8%). Most of the adverse events were manageable.
Study details: Findings are from a retrospective analysis of 56 patients with lower-risk MDS without del(5q) exclusively treated with STZ after failure of epoetin alfa as first-line treatment.
Disclosures: No source of funding was declared. The authors declared no competing interests.
Source: Qu WY et al. Ann Hematol. 2021 Apr 10. doi: 10.1007/s00277-021-04508-w.
Key clinical point: Stanozolol (STZ) monotherapy could be considered an alternative treatment in patients with lower-risk myelodysplastic syndrome (MDS) without del(5q) after the failure of epoetin alfa.
Major finding: Hematologic improvement-erythroid response (HI-E) and transfusion independence were achieved in 48.2% and 43.2% of patients, respectively. The estimated 5-year overall survival was higher in HI-E responders vs. nonresponders (88.6% vs. 33.8%). Most of the adverse events were manageable.
Study details: Findings are from a retrospective analysis of 56 patients with lower-risk MDS without del(5q) exclusively treated with STZ after failure of epoetin alfa as first-line treatment.
Disclosures: No source of funding was declared. The authors declared no competing interests.
Source: Qu WY et al. Ann Hematol. 2021 Apr 10. doi: 10.1007/s00277-021-04508-w.
Azacytidine-treated MDS patients at risk for invasive fungal infection
Key clinical point: Azacytidine-treated patients with myelodysplastic syndrome (MDS) are at significant risk for invasive fungal infection (IFI) with a corresponding higher risk for mortality, warranting mold-spectrum prophylaxis in these patients.
Major finding: Overall, 7.7% of patients developed IFI at a rate of 10.9% in patients who did not receive fungal prophylaxis. IFI was associated with a significantly higher risk for death (hazard ratio, 8.37; P less than .0001).
Study details: Findings are from a retrospective cohort study of 117 patients receiving 5-azacytidine for MDS and low blast count acute myeloid leukemia.
Disclosures: The study was funded by a Monash Haematology research grant. B Rogers and J Shortt declared being on advisory boards and receiving research grants, speaker, and consultation fees from various sources.
Source: Tey A et al. Eur J Haematol. 2021 Apr 7. doi: 10.1111/ejh.13631
Key clinical point: Azacytidine-treated patients with myelodysplastic syndrome (MDS) are at significant risk for invasive fungal infection (IFI) with a corresponding higher risk for mortality, warranting mold-spectrum prophylaxis in these patients.
Major finding: Overall, 7.7% of patients developed IFI at a rate of 10.9% in patients who did not receive fungal prophylaxis. IFI was associated with a significantly higher risk for death (hazard ratio, 8.37; P less than .0001).
Study details: Findings are from a retrospective cohort study of 117 patients receiving 5-azacytidine for MDS and low blast count acute myeloid leukemia.
Disclosures: The study was funded by a Monash Haematology research grant. B Rogers and J Shortt declared being on advisory boards and receiving research grants, speaker, and consultation fees from various sources.
Source: Tey A et al. Eur J Haematol. 2021 Apr 7. doi: 10.1111/ejh.13631
Key clinical point: Azacytidine-treated patients with myelodysplastic syndrome (MDS) are at significant risk for invasive fungal infection (IFI) with a corresponding higher risk for mortality, warranting mold-spectrum prophylaxis in these patients.
Major finding: Overall, 7.7% of patients developed IFI at a rate of 10.9% in patients who did not receive fungal prophylaxis. IFI was associated with a significantly higher risk for death (hazard ratio, 8.37; P less than .0001).
Study details: Findings are from a retrospective cohort study of 117 patients receiving 5-azacytidine for MDS and low blast count acute myeloid leukemia.
Disclosures: The study was funded by a Monash Haematology research grant. B Rogers and J Shortt declared being on advisory boards and receiving research grants, speaker, and consultation fees from various sources.
Source: Tey A et al. Eur J Haematol. 2021 Apr 7. doi: 10.1111/ejh.13631
MDS: Low lymphocyte-to-monocyte ratio predicts better outcomes
Key clinical point: Low lymphocyte-to-monocyte ratio (LMR) in patients with myelodysplastic syndrome (MDS) is associated with a favorable prognosis.
Major finding: LMR lesser than vs. greater than 5 was associated with a lower risk for leukemic transformation (median time not reached; P = .003) and better leukemia-free survival (median, 48 months vs. 21 months; P = .03).
Study details: Findings are from a retrospective study of 201 patients with a new diagnosis of MDS.
Disclosures: No source of funding was declared. The authors declared no potential conflicts of interest.
Source: Pénichoux J et al. Leuk Lymphoma. 2021 Apr 2. doi: 10.1080/10428194.2021.1907381
Key clinical point: Low lymphocyte-to-monocyte ratio (LMR) in patients with myelodysplastic syndrome (MDS) is associated with a favorable prognosis.
Major finding: LMR lesser than vs. greater than 5 was associated with a lower risk for leukemic transformation (median time not reached; P = .003) and better leukemia-free survival (median, 48 months vs. 21 months; P = .03).
Study details: Findings are from a retrospective study of 201 patients with a new diagnosis of MDS.
Disclosures: No source of funding was declared. The authors declared no potential conflicts of interest.
Source: Pénichoux J et al. Leuk Lymphoma. 2021 Apr 2. doi: 10.1080/10428194.2021.1907381
Key clinical point: Low lymphocyte-to-monocyte ratio (LMR) in patients with myelodysplastic syndrome (MDS) is associated with a favorable prognosis.
Major finding: LMR lesser than vs. greater than 5 was associated with a lower risk for leukemic transformation (median time not reached; P = .003) and better leukemia-free survival (median, 48 months vs. 21 months; P = .03).
Study details: Findings are from a retrospective study of 201 patients with a new diagnosis of MDS.
Disclosures: No source of funding was declared. The authors declared no potential conflicts of interest.
Source: Pénichoux J et al. Leuk Lymphoma. 2021 Apr 2. doi: 10.1080/10428194.2021.1907381
MDS-associated autoimmune manifestations predict poor prognosis
Key clinical point: Presence of autoimmune manifestations (AIMs) predicts poor prognosis irrespective of disease severity in patients with myelodysplastic syndrome (MDS).
Major finding: MDS-associated AIMs were identified in 20% of patients, with overall survival being shorter in patients with vs. without AIMs (P log-rank = .03). The prognosis was poor and comparable among patients with low-risk MDS and associated AIMs vs. those with high-risk MDS without AIMs (P log-rank = .9).
Study details: Findings are from a retrospective study of 61 patients with a new diagnosis of MDS.
Disclosures: This research was supported by Grant-in-Aid for Scientific Research(C). The authors declared no conflicts of interest.
Source: Arinobu Y et al. Medicine (Baltimore). 2021 Apr 2. doi: 10.1097/MD.0000000000025406
Key clinical point: Presence of autoimmune manifestations (AIMs) predicts poor prognosis irrespective of disease severity in patients with myelodysplastic syndrome (MDS).
Major finding: MDS-associated AIMs were identified in 20% of patients, with overall survival being shorter in patients with vs. without AIMs (P log-rank = .03). The prognosis was poor and comparable among patients with low-risk MDS and associated AIMs vs. those with high-risk MDS without AIMs (P log-rank = .9).
Study details: Findings are from a retrospective study of 61 patients with a new diagnosis of MDS.
Disclosures: This research was supported by Grant-in-Aid for Scientific Research(C). The authors declared no conflicts of interest.
Source: Arinobu Y et al. Medicine (Baltimore). 2021 Apr 2. doi: 10.1097/MD.0000000000025406
Key clinical point: Presence of autoimmune manifestations (AIMs) predicts poor prognosis irrespective of disease severity in patients with myelodysplastic syndrome (MDS).
Major finding: MDS-associated AIMs were identified in 20% of patients, with overall survival being shorter in patients with vs. without AIMs (P log-rank = .03). The prognosis was poor and comparable among patients with low-risk MDS and associated AIMs vs. those with high-risk MDS without AIMs (P log-rank = .9).
Study details: Findings are from a retrospective study of 61 patients with a new diagnosis of MDS.
Disclosures: This research was supported by Grant-in-Aid for Scientific Research(C). The authors declared no conflicts of interest.
Source: Arinobu Y et al. Medicine (Baltimore). 2021 Apr 2. doi: 10.1097/MD.0000000000025406
Azacitidine may allow bridging to salvage allo-HSCT after hematologic relapse
Key clinical point: Azacitidine treatment for hematological relapse of myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) is associated with poor outcomes; however, patients receiving subsequent allogeneic hematopoietic stem cell transplantation (allo-HSCT) may benefit from it.
Major finding: With a median follow-up of 4.7 and 13.6 months, the median overall survival (OS) was 5.9 (95% confidence interval [CI], 3.4-13) months and 9.5 (95% CI, 5.6-NA) months in patients receiving azacitidine as the first-line treatment of relapse and those receiving it after other treatment of relapse, respectively. In addition, the median OS was 11.6 (95% CI, 5.5-NA) months and not reached in patients who proceeded to salvage allo-HSCT in both groups, respectively.
Study details: This was a retrospective multicenter study of 31 patients with MDS or AML who had a hematological relapse after allo-HSCT and were treated with azacitidine.
Disclosures: This research did not receive any specific grant from funding agencies. The authors declared no conflicts of interest.
Source: Drozd-Sokołowska J et al. Eur J Haematol. 2021 Mar 25. doi: 10.1111/ejh.13628.
Key clinical point: Azacitidine treatment for hematological relapse of myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) is associated with poor outcomes; however, patients receiving subsequent allogeneic hematopoietic stem cell transplantation (allo-HSCT) may benefit from it.
Major finding: With a median follow-up of 4.7 and 13.6 months, the median overall survival (OS) was 5.9 (95% confidence interval [CI], 3.4-13) months and 9.5 (95% CI, 5.6-NA) months in patients receiving azacitidine as the first-line treatment of relapse and those receiving it after other treatment of relapse, respectively. In addition, the median OS was 11.6 (95% CI, 5.5-NA) months and not reached in patients who proceeded to salvage allo-HSCT in both groups, respectively.
Study details: This was a retrospective multicenter study of 31 patients with MDS or AML who had a hematological relapse after allo-HSCT and were treated with azacitidine.
Disclosures: This research did not receive any specific grant from funding agencies. The authors declared no conflicts of interest.
Source: Drozd-Sokołowska J et al. Eur J Haematol. 2021 Mar 25. doi: 10.1111/ejh.13628.
Key clinical point: Azacitidine treatment for hematological relapse of myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) is associated with poor outcomes; however, patients receiving subsequent allogeneic hematopoietic stem cell transplantation (allo-HSCT) may benefit from it.
Major finding: With a median follow-up of 4.7 and 13.6 months, the median overall survival (OS) was 5.9 (95% confidence interval [CI], 3.4-13) months and 9.5 (95% CI, 5.6-NA) months in patients receiving azacitidine as the first-line treatment of relapse and those receiving it after other treatment of relapse, respectively. In addition, the median OS was 11.6 (95% CI, 5.5-NA) months and not reached in patients who proceeded to salvage allo-HSCT in both groups, respectively.
Study details: This was a retrospective multicenter study of 31 patients with MDS or AML who had a hematological relapse after allo-HSCT and were treated with azacitidine.
Disclosures: This research did not receive any specific grant from funding agencies. The authors declared no conflicts of interest.
Source: Drozd-Sokołowska J et al. Eur J Haematol. 2021 Mar 25. doi: 10.1111/ejh.13628.
VEXAS: A novel rheumatologic, hematologic syndrome that’s making waves
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
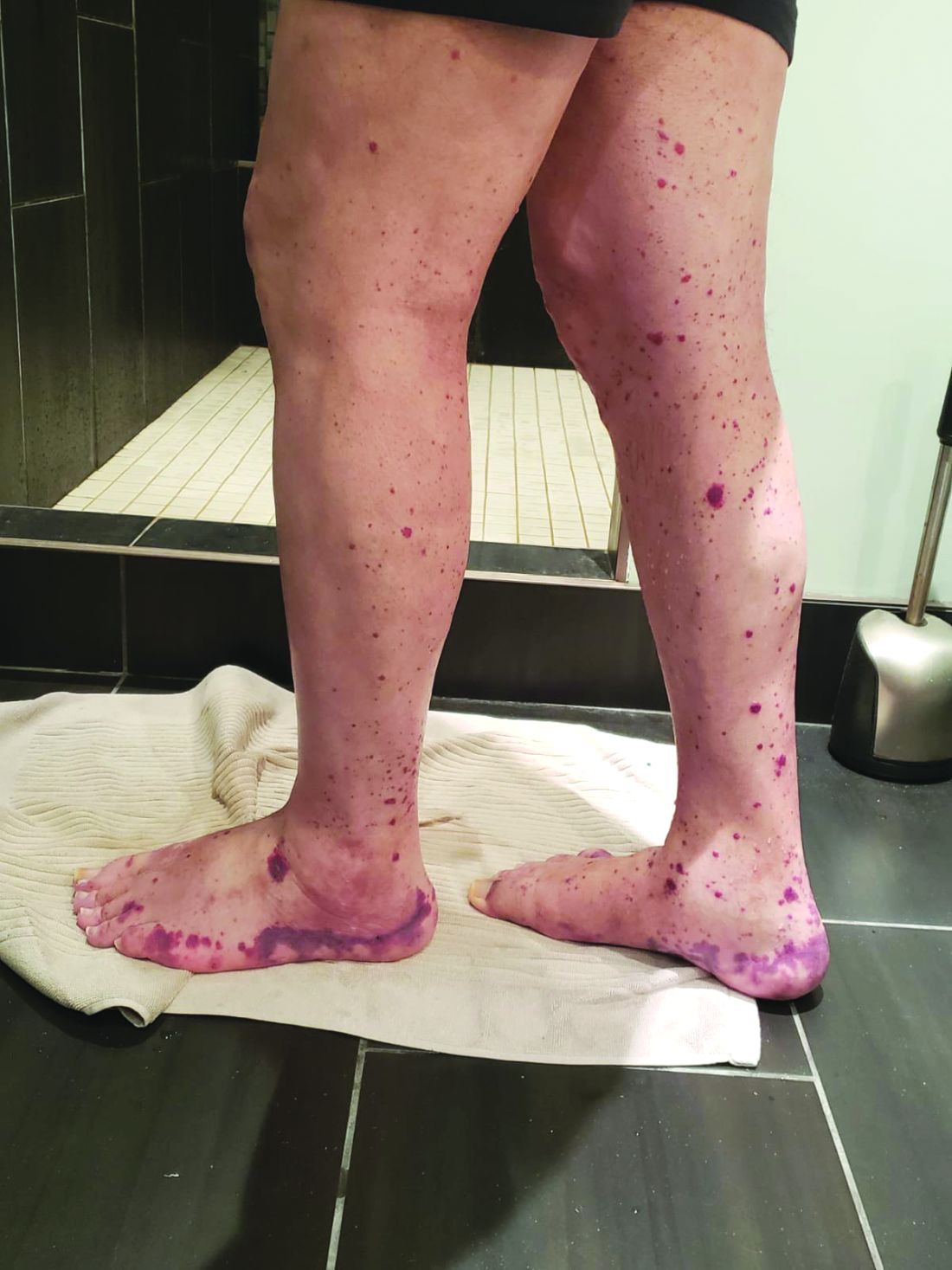
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
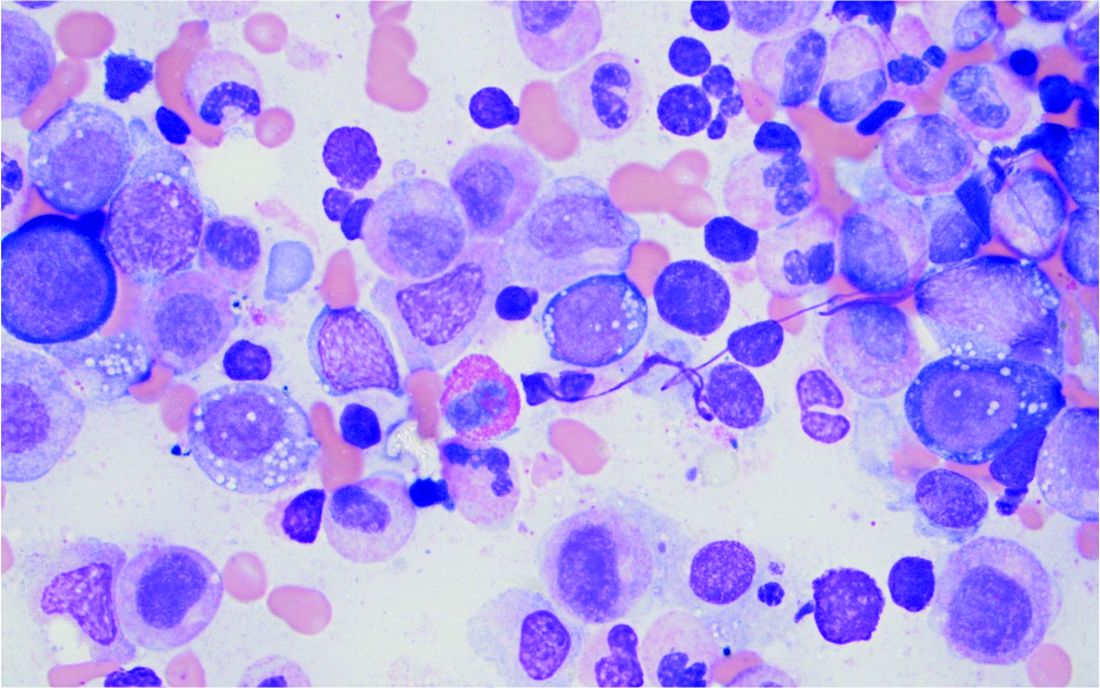
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers (david.beck@nih.gov).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Clinical Edge Commentary: MDS April 2021

The OS was similar between the two arms, although the analysis was underpowered. Nausea, diarrhea, and vomiting were the most frequently occurring treatment-emergent adverse events for oral azacitidine vs placebo (76% vs 23%, 68% vs 23%, 63% vs 9%). While oral azacitidine is not approved for MDS and trial was stopped early, it can provide meaningful reduction in RBC transfusions in low grade MDS, and further evaluation is needed in MDS.
Paroxysmal nocturnal hemoglobinuria (PNH) clones have been observed in bone marrow failure syndromes including MDS and aplastic anemia (AA). Fattizzo et al analyzed PNH clone size with clinical outcomes in MDS and AA in a cohort of 3085 patients tested for PNH at King’s College Hospital in London. 20.3% of MDS patients had presence of PNH clone; PNH cases occurred more in hypoplastic MDS, and lower risk IPSS MDS patients. The presence of PNH clone was a predictor of response to immunosuppressive therapy and to allogeneic stem cell transplant (84% vs 45%, p=0.01, 71% vs 57%, p=0.09), but not to azacitidine. MDS patients with PNH clone had lower rate of progression by IPSS and AML transformation, and higher OS accounting for MDS with excess blasts. Each 10% increase in clone size resulted in a 1% decrease in cumulative incidence of death. PNH testing is often done especially in cases with hypoplastic MDS; the underlying mechanism and role for monitoring PNH clones in MDS needs further investigation.
Outcomes in MDS patients after azacitidine therapy are generally poor. Clavio et al analyzed outcomes of 402 MDS patients post-azacitidine in the Italian MDS Registry. At azacitidine discontinuation, 20% of patients still derived response to azacitidine, 35% had primary resistance, and 44% had adaptive resistance, which was defined as loss of response or progression after achievement of a response. As expected, OS was longer for patients who stopped treatment due to planned allogeneic stem cell transplant (median OS not reached), compared to patients with primary resistance (median OS 4 months), or adaptive resistance (median OS 5 months), or patients intolerant or noncompliant to azacitidine (median OS 4 months, p=0.004). The North American MDS Consortium scoring system was evaluated in the study cohort; patients with high risk had worse OS than low risk (3 and 7 months, p<0.001). This retrospective analysis confirms the poor outlook of MDS patients after treatment of azacitidine, and effective therapy in this patient population is an unmet need.
The OS was similar between the two arms, although the analysis was underpowered. Nausea, diarrhea, and vomiting were the most frequently occurring treatment-emergent adverse events for oral azacitidine vs placebo (76% vs 23%, 68% vs 23%, 63% vs 9%). While oral azacitidine is not approved for MDS and trial was stopped early, it can provide meaningful reduction in RBC transfusions in low grade MDS, and further evaluation is needed in MDS.
Paroxysmal nocturnal hemoglobinuria (PNH) clones have been observed in bone marrow failure syndromes including MDS and aplastic anemia (AA). Fattizzo et al analyzed PNH clone size with clinical outcomes in MDS and AA in a cohort of 3085 patients tested for PNH at King’s College Hospital in London. 20.3% of MDS patients had presence of PNH clone; PNH cases occurred more in hypoplastic MDS, and lower risk IPSS MDS patients. The presence of PNH clone was a predictor of response to immunosuppressive therapy and to allogeneic stem cell transplant (84% vs 45%, p=0.01, 71% vs 57%, p=0.09), but not to azacitidine. MDS patients with PNH clone had lower rate of progression by IPSS and AML transformation, and higher OS accounting for MDS with excess blasts. Each 10% increase in clone size resulted in a 1% decrease in cumulative incidence of death. PNH testing is often done especially in cases with hypoplastic MDS; the underlying mechanism and role for monitoring PNH clones in MDS needs further investigation.
Outcomes in MDS patients after azacitidine therapy are generally poor. Clavio et al analyzed outcomes of 402 MDS patients post-azacitidine in the Italian MDS Registry. At azacitidine discontinuation, 20% of patients still derived response to azacitidine, 35% had primary resistance, and 44% had adaptive resistance, which was defined as loss of response or progression after achievement of a response. As expected, OS was longer for patients who stopped treatment due to planned allogeneic stem cell transplant (median OS not reached), compared to patients with primary resistance (median OS 4 months), or adaptive resistance (median OS 5 months), or patients intolerant or noncompliant to azacitidine (median OS 4 months, p=0.004). The North American MDS Consortium scoring system was evaluated in the study cohort; patients with high risk had worse OS than low risk (3 and 7 months, p<0.001). This retrospective analysis confirms the poor outlook of MDS patients after treatment of azacitidine, and effective therapy in this patient population is an unmet need.
The OS was similar between the two arms, although the analysis was underpowered. Nausea, diarrhea, and vomiting were the most frequently occurring treatment-emergent adverse events for oral azacitidine vs placebo (76% vs 23%, 68% vs 23%, 63% vs 9%). While oral azacitidine is not approved for MDS and trial was stopped early, it can provide meaningful reduction in RBC transfusions in low grade MDS, and further evaluation is needed in MDS.
Paroxysmal nocturnal hemoglobinuria (PNH) clones have been observed in bone marrow failure syndromes including MDS and aplastic anemia (AA). Fattizzo et al analyzed PNH clone size with clinical outcomes in MDS and AA in a cohort of 3085 patients tested for PNH at King’s College Hospital in London. 20.3% of MDS patients had presence of PNH clone; PNH cases occurred more in hypoplastic MDS, and lower risk IPSS MDS patients. The presence of PNH clone was a predictor of response to immunosuppressive therapy and to allogeneic stem cell transplant (84% vs 45%, p=0.01, 71% vs 57%, p=0.09), but not to azacitidine. MDS patients with PNH clone had lower rate of progression by IPSS and AML transformation, and higher OS accounting for MDS with excess blasts. Each 10% increase in clone size resulted in a 1% decrease in cumulative incidence of death. PNH testing is often done especially in cases with hypoplastic MDS; the underlying mechanism and role for monitoring PNH clones in MDS needs further investigation.
Outcomes in MDS patients after azacitidine therapy are generally poor. Clavio et al analyzed outcomes of 402 MDS patients post-azacitidine in the Italian MDS Registry. At azacitidine discontinuation, 20% of patients still derived response to azacitidine, 35% had primary resistance, and 44% had adaptive resistance, which was defined as loss of response or progression after achievement of a response. As expected, OS was longer for patients who stopped treatment due to planned allogeneic stem cell transplant (median OS not reached), compared to patients with primary resistance (median OS 4 months), or adaptive resistance (median OS 5 months), or patients intolerant or noncompliant to azacitidine (median OS 4 months, p=0.004). The North American MDS Consortium scoring system was evaluated in the study cohort; patients with high risk had worse OS than low risk (3 and 7 months, p<0.001). This retrospective analysis confirms the poor outlook of MDS patients after treatment of azacitidine, and effective therapy in this patient population is an unmet need.
High-risk MDS: Stanozolol improves PFS after effective induction therapy with decitabine
Key clinical point: Adding stanozolol to decitabine after effective decitabine treatment may improve progression-free survival (PFS) and reduce the severity of neutropenia in patients with high-risk myelodysplastic syndrome (MDS).
Major finding: PFS was significantly longer in the stanozolol group vs. stanozolol+decitabine group (15.0 vs. 9.0 months; hazard ratio [HR], 0.35; P = .0005). The proportion of patients with grade 3-4 neutropenia was lower in stanozolol group (76.2% vs. 95.1%; P = .039).
Study details: Findings are from a retrospective analysis of 62 patients with newly diagnosed high-risk MDS who achieved at least partial remission after 4 cycles of decitabine. For maintenance treatment, 21 patients received stanozolol and decitabine and 41 patients received decitabine alone.
Disclosures: This study was partly supported by the National Natural Science Foundation of China, the Natural Science Foundation of Tianjin China, Key Technology Research and Development Program of Tianjin China, Beijing Natural Science Foundation, and Chinese Academy of Medical Sciences innovation for medical sciences. The authors declared no conflicts of interest.
Source: Liu Y et al. Int J Hematol. 2021 Mar 1. doi: 10.1007/s12185-021-03115-9.
Key clinical point: Adding stanozolol to decitabine after effective decitabine treatment may improve progression-free survival (PFS) and reduce the severity of neutropenia in patients with high-risk myelodysplastic syndrome (MDS).
Major finding: PFS was significantly longer in the stanozolol group vs. stanozolol+decitabine group (15.0 vs. 9.0 months; hazard ratio [HR], 0.35; P = .0005). The proportion of patients with grade 3-4 neutropenia was lower in stanozolol group (76.2% vs. 95.1%; P = .039).
Study details: Findings are from a retrospective analysis of 62 patients with newly diagnosed high-risk MDS who achieved at least partial remission after 4 cycles of decitabine. For maintenance treatment, 21 patients received stanozolol and decitabine and 41 patients received decitabine alone.
Disclosures: This study was partly supported by the National Natural Science Foundation of China, the Natural Science Foundation of Tianjin China, Key Technology Research and Development Program of Tianjin China, Beijing Natural Science Foundation, and Chinese Academy of Medical Sciences innovation for medical sciences. The authors declared no conflicts of interest.
Source: Liu Y et al. Int J Hematol. 2021 Mar 1. doi: 10.1007/s12185-021-03115-9.
Key clinical point: Adding stanozolol to decitabine after effective decitabine treatment may improve progression-free survival (PFS) and reduce the severity of neutropenia in patients with high-risk myelodysplastic syndrome (MDS).
Major finding: PFS was significantly longer in the stanozolol group vs. stanozolol+decitabine group (15.0 vs. 9.0 months; hazard ratio [HR], 0.35; P = .0005). The proportion of patients with grade 3-4 neutropenia was lower in stanozolol group (76.2% vs. 95.1%; P = .039).
Study details: Findings are from a retrospective analysis of 62 patients with newly diagnosed high-risk MDS who achieved at least partial remission after 4 cycles of decitabine. For maintenance treatment, 21 patients received stanozolol and decitabine and 41 patients received decitabine alone.
Disclosures: This study was partly supported by the National Natural Science Foundation of China, the Natural Science Foundation of Tianjin China, Key Technology Research and Development Program of Tianjin China, Beijing Natural Science Foundation, and Chinese Academy of Medical Sciences innovation for medical sciences. The authors declared no conflicts of interest.
Source: Liu Y et al. Int J Hematol. 2021 Mar 1. doi: 10.1007/s12185-021-03115-9.
Comorbidities and prior malignancy negatively impact survival in MDS
Key clinical point: Over 13 years of observation reveals the presence of comorbidities, including a prior malignancy, in majority of patients with myelodysplastic syndrome (MDS). A combination of comorbidities was significantly associated with worse overall survival (OS).
Major finding: A comorbidity was reported in 67% of patients, of which 24.4% had a prior malignancy. Patients with Charlson Comorbidity Index score of 4 or higher vs. lower score had significantly impaired OS (P less than .01). Both therapy-related (hazard ratio [HR], 1.51) and secondary (HR, 1.58) vs. de novo MDS were associated with worse OS (P = .04).
Study details: Data come from an observational population-based study of 291 patients with newly diagnosed MDS.
Disclosures: No source of funding was declared. The lead author reported receiving financial support from Celgene for attending the MDS Foundation meeting in 2019. The remaining authors declared no competing financial interests.
Source: Rozema J et al. Blood Adv. 2021 Mar 3. doi: 10.1182/bloodadvances.2020003381.
Key clinical point: Over 13 years of observation reveals the presence of comorbidities, including a prior malignancy, in majority of patients with myelodysplastic syndrome (MDS). A combination of comorbidities was significantly associated with worse overall survival (OS).
Major finding: A comorbidity was reported in 67% of patients, of which 24.4% had a prior malignancy. Patients with Charlson Comorbidity Index score of 4 or higher vs. lower score had significantly impaired OS (P less than .01). Both therapy-related (hazard ratio [HR], 1.51) and secondary (HR, 1.58) vs. de novo MDS were associated with worse OS (P = .04).
Study details: Data come from an observational population-based study of 291 patients with newly diagnosed MDS.
Disclosures: No source of funding was declared. The lead author reported receiving financial support from Celgene for attending the MDS Foundation meeting in 2019. The remaining authors declared no competing financial interests.
Source: Rozema J et al. Blood Adv. 2021 Mar 3. doi: 10.1182/bloodadvances.2020003381.
Key clinical point: Over 13 years of observation reveals the presence of comorbidities, including a prior malignancy, in majority of patients with myelodysplastic syndrome (MDS). A combination of comorbidities was significantly associated with worse overall survival (OS).
Major finding: A comorbidity was reported in 67% of patients, of which 24.4% had a prior malignancy. Patients with Charlson Comorbidity Index score of 4 or higher vs. lower score had significantly impaired OS (P less than .01). Both therapy-related (hazard ratio [HR], 1.51) and secondary (HR, 1.58) vs. de novo MDS were associated with worse OS (P = .04).
Study details: Data come from an observational population-based study of 291 patients with newly diagnosed MDS.
Disclosures: No source of funding was declared. The lead author reported receiving financial support from Celgene for attending the MDS Foundation meeting in 2019. The remaining authors declared no competing financial interests.
Source: Rozema J et al. Blood Adv. 2021 Mar 3. doi: 10.1182/bloodadvances.2020003381.