User login
EMA recommends orphan designation for cancer vaccine

The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
The European Medicines Agency (EMA) has recommended orphan designation for the WT1 cancer vaccine galinpepimut-S as a treatment for patients
with acute myeloid leukemia (AML) and patients with malignant pleural mesothelioma (MPM).
The EMA’s opinion has been forwarded to the European Commission (EC), which makes the final decision.
The EC grants orphan designation to products intended to treat, prevent, or diagnose a life-threatening condition affecting up to 5 in 10,000 people in the European Union. The product must provide significant benefit to those affected by the condition.
Orphan designation from the EC provides companies with certain development incentives, including protocol assistance, a type of scientific advice specific for orphan drugs, and 10 years of market exclusivity once the drug is approved for use.
About the vaccine
The WT1 vaccine consists of 4 modified peptide chains that induce an innate immune response (CD4+/CD8+ T cells) against the WT1 antigen. The vaccine is administered in combination with an adjuvant and an immune modulator to improve the immune response to the target.
Based on the vaccine’s mechanism and the accumulating evidence of activity in mid-stage trials, researchers believe the WT1 vaccine may have the potential to complement currently available therapies by destroying residual tumor cells of cancers in remission and providing ongoing immune surveillance for recurrent tumors.
The WT1 vaccine could potentially target more than 20 cancers that overexpress WT1, many of which are associated with relapse rates of up to 80% or more, as seen in patients with AML and MPM.
The vaccine is being developed by SELLAS Life Sciences Group. The company said that, in a phase 1 study, AML patients treated with the vaccine had a median overall survival of more than 3 years.
In a phase 2 trial of the vaccine, adult AML patients had a median overall survival of around 4 years. Data from the phase 2 trial are scheduled to be presented at the 2016 ASCO Annual Meeting.
SELLAS said it expects to begin a phase 3 trial of the vaccine in AML patients later this year. ![]()
Therapy may improve haplo-HSCT in leukemia patients

Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Photo by Bill Branson
VALENCIA, SPAIN—The adjunct T-cell therapy BPX-501 can make haploidentical hematopoietic stem cell transplant (haplo-HSCT) an “attractive option” for pediatric patients with acute leukemia, according to a presentation at the 42nd Annual Meeting of the European Society for Blood and Marrow Transplantation (EBMT).
Acute leukemia patients who received BPX-501 after haplo-HSCT in a phase 1/2 trial tended to have favorable outcomes.
At a median follow-up of 7 months, 16 of the 17 patients were alive and disease-free.
There were several cases of graft-versus-host disease (GVHD), but nearly all of these resolved.
Franco Locatelli, MD, PhD, of Bambino Gesù Children’s Hospital in Rome, Italy, presented these results at the EBMT meeting as abstract WP16.*
The trial, known as BP-004, was sponsored by Bellicum Pharmaceuticals, the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The goal is to allow physicians to more safely perform haplo-HSCTs by giving patients BPX-501 to speed immune reconstitution and provide control over viral infections. But the technology is designed to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. The idea is that, if a patient develops severe GVHD, he can receive an infusion with the small molecule rimiducid. And this will trigger activation of the domain of caspase-9, which leads to selective apoptosis of the CaspaCIDe-containing cells.
About BP-004
In late 2014, Bellicum initiated BP-004, a phase 1/2 trial in children with leukemias, lymphomas, or orphan inherited blood disorders. The trial is being conducted in European and US pediatric transplant centers and is set to enroll up to 90 patients.
At the EBMT meeting, researchers reported results in 41 patients treated on this trial.
Dr Locatelli presented data on 17 patients with acute leukemias—13 with acute lymphoblastic leukemia and 4 with acute myeloid leukemia. Their median age at HSCT was 6.5 years (range, 0.9-16.1)
All of these patients received a T-cell-depleted haplo-HSCT without post-transplant GVHD prophylaxis. All were in complete remission at the time of transplant.
The patients received BPX-501 within 14 ± 4 days after haplo-HSCT. The phase 1 portion of the trial consisted of a classical 3+3 design, with 3 cohorts receiving escalating doses of BPX-501 cells—2.5 x 105, 5 x 105, and 1 x 106 cells/kg.
In the phase 2 portion, patients received 1 X 106 BPX-501 cells/kg. Rimiducid was only used in the event of uncontrollable GVHD.
Results
The median follow-up was 7 months (range, 1-15.6). The median time to platelet recovery was 11 days (range, 9-13), and the median time to neutrophil recovery was 17 days (range, 10-22).
Three patients developed skin-only acute GVHD, were treated with topical steroids, and the GVHD resolved. Two patients developed acute grade 3 GVHD, were treated with systemic steroids, and the GVHD resolved.
Two patients developed mild chronic GVHD, received systemic steroids, and the GVHD resolved. And 1 patient developed severe chronic GVHD, received systemic steroids and rimiducid, and the GVHD improved.
One patient relapsed. The estimated 1-year disease-free survival was 92.9%. Dr Locatelli noted that, although the follow-up is still limited, these results compare favorably to results in historical controls.
“These interim results continue to be very encouraging and indicate that a haploidentical transplant, with the addition of BPX-501-modified donor T cells, can be an attractive option for children in need of a transplant,” he said.
“Future studies will address the role of repeated infusions or higher numbers of BPX-501 cells in malignant patients with resistant disease.”
The BP-004 trial also included 24 patients with nonmalignant disorders. Results in these patients were presented at the EBMT meeting as abstract O007. ![]()
*Information in the abstract differs from that presented at the meeting.
Cyclophosphamide nets low rate of chronic GVHD after mobilized blood cell transplantation
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.

The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.
The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
High-dose cyclophosphamide is safe and effective when given as prophylaxis for chronic graft-versus-host disease (GVHD) to patients who have undergone transplantation of mobilized blood cells, finds a phase 2 trial reported in Blood.
Investigators led by Dr. Marco Mielcarek, medical director of the Adult Blood and Marrow Transplant Program and an oncologist at the Fred Hutchinson Cancer Research Center in Seattle, Washington, enrolled in the trial 43 patients with high-risk hematologic malignancies.
The patients underwent myeloablative conditioning followed by transplantation with growth factor–mobilized blood cells from related or unrelated donors, and were given high-dose cyclophosphamide on two early posttransplantation days.
Main results showed that the cumulative 1-year incidence of chronic GVHD was 16%, less than half of the roughly 35% seen historically with conventional immunosuppression.
Moreover, cyclophosphamide did not appear to compromise engraftment or control of the underlying malignancy. Only a single patient, one with an HLA-mismatched donor, had failure of primary engraftment; after amendment of the protocol to require HLA matching, there were no additional cases. Just 17% of patients experienced a recurrence of their malignancy by 2 years.
Taken together, the findings suggest that high-dose cyclophosphamide—as combined with two myeloablative conditioning options (to accommodate different malignancies) and with posttransplantation cyclosporine (to reduce the risk of acute GVHD)—may eliminate most of the drawbacks to using mobilized blood cells for transplantation, according to the investigators.
“If these findings are confirmed in future studies, HLA-matched mobilized blood cell transplantation may gain even greater acceptance and further replace marrow as a source of stem cells for most indications,” they maintain.
The patients studied had a median age of 43 years, and slightly more than half were in remission without minimal residual disease.
Blood cells were mobilized with granulocyte colony-stimulating factor (G-CSF). Overall, 28% of patients received grafts from related donors, while 72% received grafts from unrelated donors.
For pretransplant conditioning, patients received fludarabine and targeted busulfan, or total body irradiation with use of a minimum dose of 12 Gy.
The patients were given cyclophosphamide at 50 mg/kg per day on days 3 and 4 after transplantation. This was followed by cyclosporine starting on day 5.
The cumulative 1-year incidence of chronic GVHD as defined by National Institutes of Health criteria (i.e., that requiring systemic immunosuppressive therapy)—the trial’s primary endpoint—was 16%, which fell just short of the goal of 15% the investigators were aiming for (Blood. 2016;127:1502-8). Analyses failed to identify any predictors of this outcome.
Although the estimated cumulative incidence of grade 2 acute GVHD was high, at 77%, none of the patients developed grade 3 or 4 acute GVHD, according to the investigators, who disclosed that they had no competing financial interests.
The single patient who experienced failure of primary engraftment had familial myelodysplastic syndrome and had received a graft from an HLA A-antigen–mismatched unrelated donor.
The 2-year cumulative incidence of nonrelapse mortality was 14%, and the 2-year cumulative incidence of recurrent malignancy was 17%. Projected overall survival was 70%.
Among the 42 patients having at least a year of follow-up, 50% were alive and free of relapse without any systemic immunosuppression at 1 year after transplantation.
FROM BLOOD
Key clinical point: High-dose posttransplant cyclophosphamide is safe and effective for reducing the risk of chronic GVHD after mobilized blood cell transplantation.
Major finding: The cumulative 1-year incidence of chronic GVHD requiring immunosuppressive therapy was 16%.
Data source: A single-arm trial among 43 patients with high-risk hematologic malignancies undergoing growth factor–mobilized blood cell transplantation.
Disclosures: The authors disclosed that they have no competing financial interests.

Study implicates circular RNAs in leukemia, other cancers

Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
FDA grants product orphan designation for AML

Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Drug for conditioning AML patients for transplant gets orphan drug designation
A radioimmunotherapeutic drug for conditioning relapsed and refractory acute myeloid leukemia (AML) patients for a hematopoietic stem cell transplant has been granted orphan drug designation by the Food and Drug Administration.
Iomab-B is a radioimmunoconjugate consisting of the murine monoclonal antibody BC8 and an iodine-131 radioisotope. The Fred Hutchinson Cancer Research Center developed BC8 to target CD45, a panleukocytic antigen widely expressed on white blood cells. “When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow while avoiding effects of radiation on most healthy tissues,” says a statement from Actinium Pharmaceuticals, which would market the drug.
Iomab-B will be tested in a multicenter trial that will include 150 patients over age 55 with refractory and relapsed AML. “There has not been a new drug approved for relapsed and refractory AML patients over the age of 55 in decades and with Iomab-B being the only therapy of its kind, we are pleased to have achieved this important milestone,” Sandesh Seth, executive chairman of Actinium, said in the statement.
A radioimmunotherapeutic drug for conditioning relapsed and refractory acute myeloid leukemia (AML) patients for a hematopoietic stem cell transplant has been granted orphan drug designation by the Food and Drug Administration.
Iomab-B is a radioimmunoconjugate consisting of the murine monoclonal antibody BC8 and an iodine-131 radioisotope. The Fred Hutchinson Cancer Research Center developed BC8 to target CD45, a panleukocytic antigen widely expressed on white blood cells. “When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow while avoiding effects of radiation on most healthy tissues,” says a statement from Actinium Pharmaceuticals, which would market the drug.
Iomab-B will be tested in a multicenter trial that will include 150 patients over age 55 with refractory and relapsed AML. “There has not been a new drug approved for relapsed and refractory AML patients over the age of 55 in decades and with Iomab-B being the only therapy of its kind, we are pleased to have achieved this important milestone,” Sandesh Seth, executive chairman of Actinium, said in the statement.
A radioimmunotherapeutic drug for conditioning relapsed and refractory acute myeloid leukemia (AML) patients for a hematopoietic stem cell transplant has been granted orphan drug designation by the Food and Drug Administration.
Iomab-B is a radioimmunoconjugate consisting of the murine monoclonal antibody BC8 and an iodine-131 radioisotope. The Fred Hutchinson Cancer Research Center developed BC8 to target CD45, a panleukocytic antigen widely expressed on white blood cells. “When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow while avoiding effects of radiation on most healthy tissues,” says a statement from Actinium Pharmaceuticals, which would market the drug.
Iomab-B will be tested in a multicenter trial that will include 150 patients over age 55 with refractory and relapsed AML. “There has not been a new drug approved for relapsed and refractory AML patients over the age of 55 in decades and with Iomab-B being the only therapy of its kind, we are pleased to have achieved this important milestone,” Sandesh Seth, executive chairman of Actinium, said in the statement.
New drug approved for hepatic veno-occlusive disease
Defibrotide sodium has been approved to treat hepatic veno-occlusive disease (VOD) in patients with renal or pulmonary dysfunction following a hematopoietic stem cell transplantation, the Food and Drug Administration has announced.
The drug, which will be marketed as Defitelio by Jazz Pharmaceuticals, was tested in two prospective clinical trials and an expanded access study that included a total of 528 patients with hepatic VOD and multiorgan dysfunction following a transplantation. All patients received 6.25 mg/kg doses of the drug intravenously, every 6 hours until resolution of VOD. The percentages of patients surviving more than 100 days after receiving a stem cell transplantation in each of the studies were 38%, 44%, and 45%, respectively, according to a statement from the FDA.
Contraindications for taking the drug are concurrent use of anticoagulants or fibrinolytic therapies.
Hypotension, diarrhea, vomiting, nausea, and epistaxis are the most common adverse reactions to the drug.
Full prescribing information is available at the FDA website.
Defibrotide sodium has been approved to treat hepatic veno-occlusive disease (VOD) in patients with renal or pulmonary dysfunction following a hematopoietic stem cell transplantation, the Food and Drug Administration has announced.
The drug, which will be marketed as Defitelio by Jazz Pharmaceuticals, was tested in two prospective clinical trials and an expanded access study that included a total of 528 patients with hepatic VOD and multiorgan dysfunction following a transplantation. All patients received 6.25 mg/kg doses of the drug intravenously, every 6 hours until resolution of VOD. The percentages of patients surviving more than 100 days after receiving a stem cell transplantation in each of the studies were 38%, 44%, and 45%, respectively, according to a statement from the FDA.
Contraindications for taking the drug are concurrent use of anticoagulants or fibrinolytic therapies.
Hypotension, diarrhea, vomiting, nausea, and epistaxis are the most common adverse reactions to the drug.
Full prescribing information is available at the FDA website.
Defibrotide sodium has been approved to treat hepatic veno-occlusive disease (VOD) in patients with renal or pulmonary dysfunction following a hematopoietic stem cell transplantation, the Food and Drug Administration has announced.
The drug, which will be marketed as Defitelio by Jazz Pharmaceuticals, was tested in two prospective clinical trials and an expanded access study that included a total of 528 patients with hepatic VOD and multiorgan dysfunction following a transplantation. All patients received 6.25 mg/kg doses of the drug intravenously, every 6 hours until resolution of VOD. The percentages of patients surviving more than 100 days after receiving a stem cell transplantation in each of the studies were 38%, 44%, and 45%, respectively, according to a statement from the FDA.
Contraindications for taking the drug are concurrent use of anticoagulants or fibrinolytic therapies.
Hypotension, diarrhea, vomiting, nausea, and epistaxis are the most common adverse reactions to the drug.
Full prescribing information is available at the FDA website.
Mouse model replicates aggressive AML subtype

Researchers have developed a mouse model of an aggressive type of acute myeloid leukemia (AML) that, they believe, accurately replicates the human form of the disease.
The model replicates AML with co-occurring mutations in FLT3 and DNMT3A.
The researchers said they found that mice with Flt3-ITD and inducible deletion of Dnmt3a developed a rapidly lethal, completely penetrant, and transplantable AML of normal karyotype.
The team described this work in Cancer Discovery.
“Our goal was to create a model that was faithful to the human form of the disease that can be used for preclinical testing of potential cures,” said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“Previous models were slow, difficult to analyze, and did not accurately represent the human disease. This model is rapid, fully penetrant, and completely spontaneous. We hope that it will open the way for other researchers to join us in attacking this particularly lethal AML subtype.”
Dr Grimes and his colleagues said they were able to look at the disease in a new way with the help of a powerful new core facility utilizing analytical tools related to single-cell RNA sequencing. The team used complementary single-cell analyses to identify the core leukemia-causing stem cells of the tumor.
“Before, researchers were comparing the gene expression patterns of one AML subtype to either normal cells or other AML subtypes,” said study author Sara Meyer, PhD, a fellow in the Grimes lab.
“That approach made it difficult to tease out the specific impact of Dnmt3a mutation. Instead, we isolated the variables and studied only human and murine AML with Flt3 mutation. Comparing Flt3-mutant AML with and without Dnmt3a mutation allowed us to more finely identify those patterns that were specific to the Dnmt3a mutation.”
With that more detailed understanding, the researchers gained new insights into the contributions of the Dnmt3a mutation to the disease.
First, their work confirms suspicions that low-level Dnmt3a activity is cancer-causing. Moreover, they discovered that reduced Dnmt3a function allows genes that are normally expressed only at early development stages of blood cell formation to continue expression at later stages, leading to the development of AML.
The researchers also found that, in mouse tumor cells, rescuing expression of Dnmt3a reversed the leukemia phenotypes and gene expression. But they said more research is warranted to determine if rescuing normal levels of DNMT3A function is a viable method for treating human AML.
The team also identified several potential treatment targets that are unique to this type of AML. In future studies, they plan to proceed with testing potential therapies. ![]()
Researchers have developed a mouse model of an aggressive type of acute myeloid leukemia (AML) that, they believe, accurately replicates the human form of the disease.
The model replicates AML with co-occurring mutations in FLT3 and DNMT3A.
The researchers said they found that mice with Flt3-ITD and inducible deletion of Dnmt3a developed a rapidly lethal, completely penetrant, and transplantable AML of normal karyotype.
The team described this work in Cancer Discovery.
“Our goal was to create a model that was faithful to the human form of the disease that can be used for preclinical testing of potential cures,” said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“Previous models were slow, difficult to analyze, and did not accurately represent the human disease. This model is rapid, fully penetrant, and completely spontaneous. We hope that it will open the way for other researchers to join us in attacking this particularly lethal AML subtype.”
Dr Grimes and his colleagues said they were able to look at the disease in a new way with the help of a powerful new core facility utilizing analytical tools related to single-cell RNA sequencing. The team used complementary single-cell analyses to identify the core leukemia-causing stem cells of the tumor.
“Before, researchers were comparing the gene expression patterns of one AML subtype to either normal cells or other AML subtypes,” said study author Sara Meyer, PhD, a fellow in the Grimes lab.
“That approach made it difficult to tease out the specific impact of Dnmt3a mutation. Instead, we isolated the variables and studied only human and murine AML with Flt3 mutation. Comparing Flt3-mutant AML with and without Dnmt3a mutation allowed us to more finely identify those patterns that were specific to the Dnmt3a mutation.”
With that more detailed understanding, the researchers gained new insights into the contributions of the Dnmt3a mutation to the disease.
First, their work confirms suspicions that low-level Dnmt3a activity is cancer-causing. Moreover, they discovered that reduced Dnmt3a function allows genes that are normally expressed only at early development stages of blood cell formation to continue expression at later stages, leading to the development of AML.
The researchers also found that, in mouse tumor cells, rescuing expression of Dnmt3a reversed the leukemia phenotypes and gene expression. But they said more research is warranted to determine if rescuing normal levels of DNMT3A function is a viable method for treating human AML.
The team also identified several potential treatment targets that are unique to this type of AML. In future studies, they plan to proceed with testing potential therapies. ![]()
Researchers have developed a mouse model of an aggressive type of acute myeloid leukemia (AML) that, they believe, accurately replicates the human form of the disease.
The model replicates AML with co-occurring mutations in FLT3 and DNMT3A.
The researchers said they found that mice with Flt3-ITD and inducible deletion of Dnmt3a developed a rapidly lethal, completely penetrant, and transplantable AML of normal karyotype.
The team described this work in Cancer Discovery.
“Our goal was to create a model that was faithful to the human form of the disease that can be used for preclinical testing of potential cures,” said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
“Previous models were slow, difficult to analyze, and did not accurately represent the human disease. This model is rapid, fully penetrant, and completely spontaneous. We hope that it will open the way for other researchers to join us in attacking this particularly lethal AML subtype.”
Dr Grimes and his colleagues said they were able to look at the disease in a new way with the help of a powerful new core facility utilizing analytical tools related to single-cell RNA sequencing. The team used complementary single-cell analyses to identify the core leukemia-causing stem cells of the tumor.
“Before, researchers were comparing the gene expression patterns of one AML subtype to either normal cells or other AML subtypes,” said study author Sara Meyer, PhD, a fellow in the Grimes lab.
“That approach made it difficult to tease out the specific impact of Dnmt3a mutation. Instead, we isolated the variables and studied only human and murine AML with Flt3 mutation. Comparing Flt3-mutant AML with and without Dnmt3a mutation allowed us to more finely identify those patterns that were specific to the Dnmt3a mutation.”
With that more detailed understanding, the researchers gained new insights into the contributions of the Dnmt3a mutation to the disease.
First, their work confirms suspicions that low-level Dnmt3a activity is cancer-causing. Moreover, they discovered that reduced Dnmt3a function allows genes that are normally expressed only at early development stages of blood cell formation to continue expression at later stages, leading to the development of AML.
The researchers also found that, in mouse tumor cells, rescuing expression of Dnmt3a reversed the leukemia phenotypes and gene expression. But they said more research is warranted to determine if rescuing normal levels of DNMT3A function is a viable method for treating human AML.
The team also identified several potential treatment targets that are unique to this type of AML. In future studies, they plan to proceed with testing potential therapies. ![]()
PET probe could aid treatment for leukemia
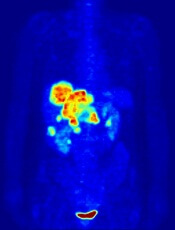
Image by Jens Langner
A PET probe known as [18F]CFA could be used to aid the treatment of leukemias and other cancers, according to research published in PNAS.
Investigators say [18F]CFA can detect the activity of deoxycytidine kinase (dCK) in humans more effectively than existing probes.
dCK is a rate-limiting enzyme in the cytosolic deoxyribonucleoside salvage pathway and is considered an important therapeutic and PET-imaging target in certain cancers.
Research has shown that dCK is highly expressed in acute leukemia cells and activated lymphocytes. And the enzyme plays an integral role in allowing drugs such as clofarabine, cytarabine, and fludarabine to treat certain leukemias.
“This enzyme is essential for the therapeutic activity of an entire class of anticancer drugs and even for some antiviral drugs,” said study author Caius Radu, MD, of the University of California, Los Angeles.
“It can take an inactive drug and activate it. If you trick a cancer cell or virus to activate the drug, it would be toxic for the cancer cell or viral genome.”
Until recently, PET technology was only able to clearly detect dCK in mice due to the metabolic instability of the available probes and cross-reactivity with a dCK-related enzyme in humans.
However, Dr Radu and his colleagues showed that [18F]CFA can clearly detect dCK in humans.
The team found that [18F]CFA accumulation in leukemia cells correlated with dCK expression, and they were able to inhibit [18F]CFA accumulation with a dCK inhibitor.
Experiments with [18F]CFA PET/CT in humans showed probe accumulation in tissues with high dCK expression, such as hematopoietic bone marrow and secondary lymphoid organs.
“We are able to clearly see tissues, including tumor tissues, with high dCK activity that we haven’t seen before in humans using any of the other probes previously developed for this enzyme,” Dr Radu said.
He added that, since activated immune cells increase their expression of dCK, [18F]CFA could also be used to monitor the effectiveness of immunotherapeutic interventions.
The investigators hope to begin clinical trials with [18F]CFA in the near future.
Dr Radu and his team invented [18F]CFA and its analogs, which were patented by the University of California and have been licensed to Sofie Biosciences, a company founded by Dr Radu and his team. The University of California also patented additional intellectual property for small-molecule dCK inhibitors. ![]()
Image by Jens Langner
A PET probe known as [18F]CFA could be used to aid the treatment of leukemias and other cancers, according to research published in PNAS.
Investigators say [18F]CFA can detect the activity of deoxycytidine kinase (dCK) in humans more effectively than existing probes.
dCK is a rate-limiting enzyme in the cytosolic deoxyribonucleoside salvage pathway and is considered an important therapeutic and PET-imaging target in certain cancers.
Research has shown that dCK is highly expressed in acute leukemia cells and activated lymphocytes. And the enzyme plays an integral role in allowing drugs such as clofarabine, cytarabine, and fludarabine to treat certain leukemias.
“This enzyme is essential for the therapeutic activity of an entire class of anticancer drugs and even for some antiviral drugs,” said study author Caius Radu, MD, of the University of California, Los Angeles.
“It can take an inactive drug and activate it. If you trick a cancer cell or virus to activate the drug, it would be toxic for the cancer cell or viral genome.”
Until recently, PET technology was only able to clearly detect dCK in mice due to the metabolic instability of the available probes and cross-reactivity with a dCK-related enzyme in humans.
However, Dr Radu and his colleagues showed that [18F]CFA can clearly detect dCK in humans.
The team found that [18F]CFA accumulation in leukemia cells correlated with dCK expression, and they were able to inhibit [18F]CFA accumulation with a dCK inhibitor.
Experiments with [18F]CFA PET/CT in humans showed probe accumulation in tissues with high dCK expression, such as hematopoietic bone marrow and secondary lymphoid organs.
“We are able to clearly see tissues, including tumor tissues, with high dCK activity that we haven’t seen before in humans using any of the other probes previously developed for this enzyme,” Dr Radu said.
He added that, since activated immune cells increase their expression of dCK, [18F]CFA could also be used to monitor the effectiveness of immunotherapeutic interventions.
The investigators hope to begin clinical trials with [18F]CFA in the near future.
Dr Radu and his team invented [18F]CFA and its analogs, which were patented by the University of California and have been licensed to Sofie Biosciences, a company founded by Dr Radu and his team. The University of California also patented additional intellectual property for small-molecule dCK inhibitors. ![]()
Image by Jens Langner
A PET probe known as [18F]CFA could be used to aid the treatment of leukemias and other cancers, according to research published in PNAS.
Investigators say [18F]CFA can detect the activity of deoxycytidine kinase (dCK) in humans more effectively than existing probes.
dCK is a rate-limiting enzyme in the cytosolic deoxyribonucleoside salvage pathway and is considered an important therapeutic and PET-imaging target in certain cancers.
Research has shown that dCK is highly expressed in acute leukemia cells and activated lymphocytes. And the enzyme plays an integral role in allowing drugs such as clofarabine, cytarabine, and fludarabine to treat certain leukemias.
“This enzyme is essential for the therapeutic activity of an entire class of anticancer drugs and even for some antiviral drugs,” said study author Caius Radu, MD, of the University of California, Los Angeles.
“It can take an inactive drug and activate it. If you trick a cancer cell or virus to activate the drug, it would be toxic for the cancer cell or viral genome.”
Until recently, PET technology was only able to clearly detect dCK in mice due to the metabolic instability of the available probes and cross-reactivity with a dCK-related enzyme in humans.
However, Dr Radu and his colleagues showed that [18F]CFA can clearly detect dCK in humans.
The team found that [18F]CFA accumulation in leukemia cells correlated with dCK expression, and they were able to inhibit [18F]CFA accumulation with a dCK inhibitor.
Experiments with [18F]CFA PET/CT in humans showed probe accumulation in tissues with high dCK expression, such as hematopoietic bone marrow and secondary lymphoid organs.
“We are able to clearly see tissues, including tumor tissues, with high dCK activity that we haven’t seen before in humans using any of the other probes previously developed for this enzyme,” Dr Radu said.
He added that, since activated immune cells increase their expression of dCK, [18F]CFA could also be used to monitor the effectiveness of immunotherapeutic interventions.
The investigators hope to begin clinical trials with [18F]CFA in the near future.
Dr Radu and his team invented [18F]CFA and its analogs, which were patented by the University of California and have been licensed to Sofie Biosciences, a company founded by Dr Radu and his team. The University of California also patented additional intellectual property for small-molecule dCK inhibitors. ![]()
High costs limit CML patients’ access to TKIs

Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said. ![]()
Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said. ![]()
Photo courtesy of the CDC
A new study suggests that cost-sharing policies in the US create a barrier to the treatment of chronic myeloid leukemia (CML).
Researchers examined Medicare claims data and found that “Part D” (prescription drug plan) co-insurance policies for “specialty drugs” seem to be reducing or delaying the use of tyrosine kinase inhibitors (TKIs) in patients with CML.
The team reported these findings in the American Journal of Managed Care.
“High out-of-pocket costs for specialty drugs appear to pose a very real barrier to treatment,” said study author Jalpa A. Doshi, PhD, of the Perelman School of Medicine at the University of Pennsylvania in Philadelphia.
While there is no standard definition for specialty drugs, the term typically refers to medications requiring special handling, administration, or monitoring. Most are aimed at treating chronic or life-threatening diseases.
Although specialty drugs typically tend to offer significant medical advances over non-specialty drugs, they are correspondingly more expensive. In 2014, such drugs accounted for less than 1% of prescriptions in the US but nearly a third of total prescription spending.
While insurers have been imposing higher cost-sharing requirements as part of their efforts to manage specialty drug spending, there has been limited information about the corresponding impact on patients.
“[I]t was particularly important to examine the extent to which the aggressive cost-sharing policies for specialty drugs seen under Medicare Part D, which are increasingly making their way into the private insurance market, adversely impact access to these treatments even for a condition like cancer,” Dr Doshi said.
So she and her colleagues examined the impact of specialty drug cost-sharing under the Medicare Part D prescription drug benefit on patients with CML. The team analyzed Medicare data on patients who were newly diagnosed with CML to examine whether and how quickly they initiated TKI treatment.
The researchers compared patients who were eligible for low-income subsidies and therefore faced nominal out-of-pocket costs to patients who faced average out-of-pocket costs of $2600 or more for their first 30-day TKI prescription fill.
Results showed that patients in the high-cost group were significantly less likely than the low-cost group to have a Part D claim for a TKI prescription within 6 months of their CML diagnosis. The rates were 45.3% and 66.9%, respectively (P<0.001).
Patients in the high cost-sharing group also took twice as long, on average, to initiate TKI treatment. The mean time to fill a TKI prescription was 50.9 days in the high-cost group and 23.7 days in the low-cost group (P<0.001).
“Medicare Part D was created to increase access to prescription drug treatment among beneficiaries, but our data suggest that current policies are interfering with that goal when it comes to specialty drugs,” Dr Doshi said.
She added that making Part D out-of-pocket costs more consistent and limiting them to more reasonable sums would help mitigate this negative impact.
Dr Doshi and her colleagues are now pursuing further studies of the impact of Part D cost-sharing policies in different disease areas. They hope to gain a better understanding of changes in drug access and of the long-range clinical outcomes and costs associated with any delays or interruptions in treatment.
“We need to know if the current aggressive cost-sharing arrangements have adverse long-term impacts on health and perhaps, paradoxically, increase overall spending due to complications of poorly controlled disease,” Dr Doshi said.