User login
Unleashing Our Immune Response to Quash Cancer
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
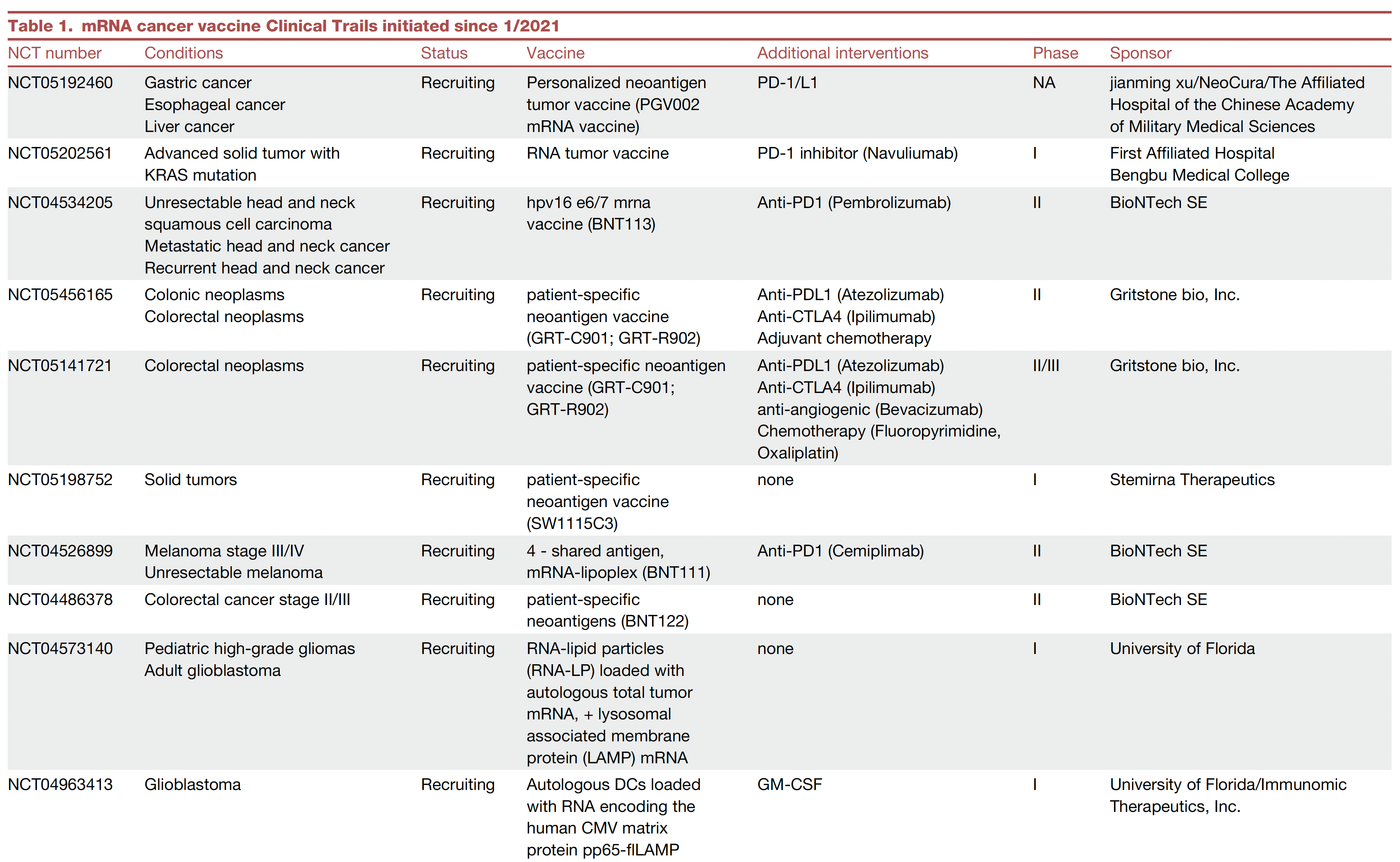
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
{kind=link}
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
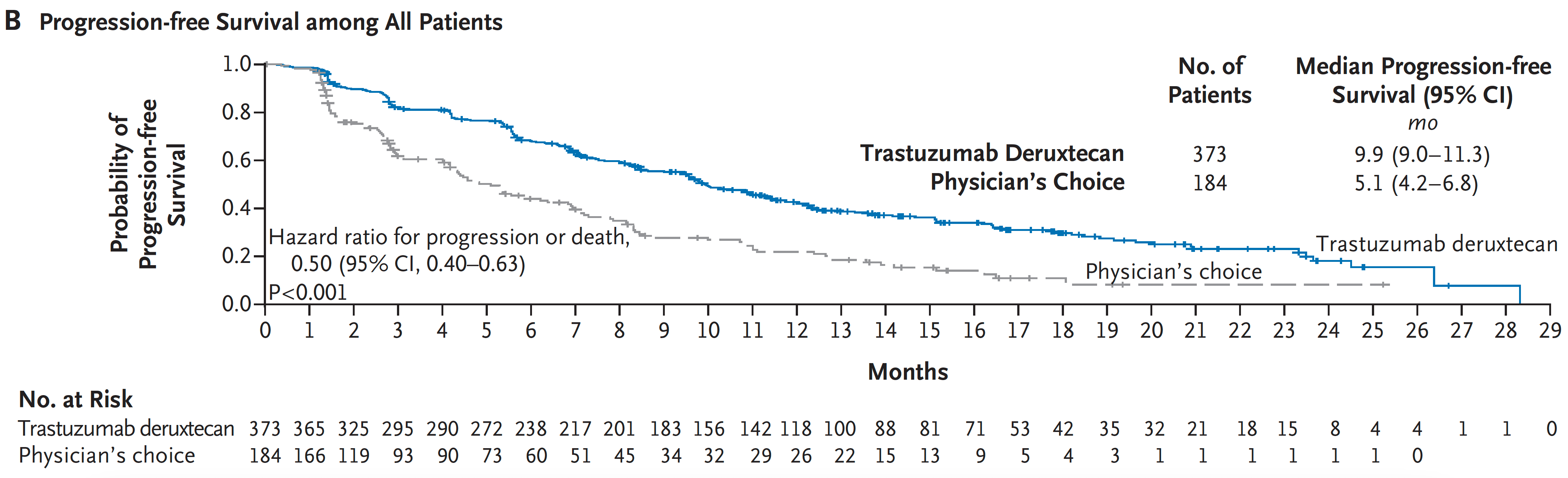
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
{kind=link}
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
This article was originally published on February 10 in Eric Topol’s substack “Ground Truths.”
It’s astounding how devious cancer cells and tumor tissue can be. This week in Science we learned how certain lung cancer cells can function like “Catch Me If You Can” — changing their driver mutation and cell identity to escape targeted therapy. This histologic transformation, as seen in an experimental model, is just one of so many cancer tricks that we are learning about.
Recently, as shown by single-cell sequencing, cancer cells can steal the mitochondria from T cells, a double whammy that turbocharges cancer cells with the hijacked fuel supply and, at the same time, dismantles the immune response.
Last week, we saw how tumor cells can release a virus-like protein that unleashes a vicious autoimmune response.
And then there’s the finding that cancer cell spread predominantly is occurring while we sleep.
As I previously reviewed, the ability for cancer cells to hijack neurons and neural circuits is now well established, no less their ability to reprogram neurons to become adrenergic and stimulate tumor progression, and interfere with the immune response. Stay tuned on that for a new Ground Truths podcast with Prof Michelle Monje, a leader in cancer neuroscience, which will post soon.
Add advancing age’s immunosenescence as yet another challenge to the long and growing list of formidable ways that cancer cells, and the tumor microenvironment, evade our immune response.
An Ever-Expanding Armamentarium
Immune Checkpoint Inhibitors
The field of immunotherapies took off with the immune checkpoint inhibitors, first approved by the FDA in 2011, that take the brakes off of T cells, with the programmed death-1 (PD-1), PD-ligand1, and anti-CTLA-4 monoclonal antibodies.
But we’re clearly learning they are not enough to prevail over cancer with common recurrences, only short term success in most patients, with some notable exceptions. Adding other immune response strategies, such as a vaccine, or antibody-drug conjugates, or engineered T cells, are showing improved chances for success.
Therapeutic Cancer Vaccines
There are many therapeutic cancer vaccines in the works, as reviewed in depth here.
Here’s a list of ongoing clinical trials of cancer vaccines. You’ll note most of these are on top of a checkpoint inhibitor and use personalized neoantigens (cancer cell surface proteins) derived from sequencing (whole-exome or whole genome, RNA-sequencing and HLA-profiling) the patient’s tumor.
An example of positive findings is with the combination of an mRNA-nanoparticle vaccine with up to 34 personalized neoantigens and pembrolizumab (Keytruda) vs pembrolizumab alone in advanced melanoma after resection, with improved outcomes at 3-year follow-up, cutting death or relapse rate in half.
Antibody-Drug Conjugates (ADC)
There is considerable excitement about antibody-drug conjugates (ADC) whereby a linker is used to attach a chemotherapy agent to the checkpoint inhibitor antibody, specifically targeting the cancer cell and facilitating entry of the chemotherapy into the cell. Akin to these are bispecific antibodies (BiTEs, binding to a tumor antigen and T cell receptor simultaneously), both of these conjugates acting as “biologic” or “guided” missiles.
A very good example of the potency of an ADC was seen in a “HER2-low” breast cancer randomized trial. The absence or very low expression or amplification of the HER2 receptor is common in breast cancer and successful treatment has been elusive. A randomized trial of an ADC (trastuzumab deruxtecan) compared to physician’s choice therapy demonstrated a marked success for progression-free survival in HER2-low patients, which was characterized as “unheard-of success” by media coverage.
This strategy is being used to target some of the most difficult cancer driver mutations such as TP53 and KRAS.
Oncolytic Viruses
Modifying viruses to infect the tumor and make it more visible to the immune system, potentiating anti-tumor responses, known as oncolytic viruses, have been proposed as a way to rev up the immune response for a long time but without positive Phase 3 clinical trials.
After decades of failure, a recent trial in refractory bladder cancer showed marked success, along with others, summarized here, now providing very encouraging results. It looks like oncolytic viruses are on a comeback path.
Engineering T Cells (Chimeric Antigen Receptor [CAR-T])
As I recently reviewed, there are over 500 ongoing clinical trials to build on the success of the first CAR-T approval for leukemia 7 years ago. I won’t go through that all again here, but to reiterate most of the success to date has been in “liquid” blood (leukemia and lymphoma) cancer tumors. This week in Nature is the discovery of a T cell cancer mutation, a gene fusion CARD11-PIK3R3, from a T cell lymphoma that can potentially be used to augment CAR-T efficacy. It has pronounced and prolonged effects in the experimental model. Instead of 1 million cells needed for treatment, even 20,000 were enough to melt the tumor. This is a noteworthy discovery since CAR-T work to date has largely not exploited such naturally occurring mutations, while instead concentrating on those seen in the patient’s set of key tumor mutations.
As currently conceived, CAR-T, and what is being referred to more broadly as adoptive cell therapies, involves removing T cells from the patient’s body and engineering their activation, then reintroducing them back to the patient. This is laborious, technically difficult, and very expensive. Recently, the idea of achieving all of this via an injection of virus that specifically infects T cells and inserts the genes needed, was advanced by two biotech companies with preclinical results, one in non-human primates.
Gearing up to meet the challenge of solid tumor CAR-T intervention, there’s more work using CRISPR genome editing of T cell receptors. A.I. is increasingly being exploited to process the data from sequencing and identify optimal neoantigens.
Instead of just CAR-T, we’re seeing the emergence of CAR-macrophage and CAR-natural killer (NK) cells strategies, and rapidly expanding potential combinations of all the strategies I’ve mentioned. No less, there’s been maturation of on-off suicide switches programmed in, to limit cytokine release and promote safety of these interventions. Overall, major side effects of immunotherapies are not only cytokine release syndromes, but also include interstitial pneumonitis and neurotoxicity.
Summary
Given the multitude of ways cancer cells and tumor tissue can evade our immune response, durably successful treatment remains a daunting challenge. But the ingenuity of so many different approaches to unleash our immune response, and their combinations, provides considerable hope that we’ll increasingly meet the challenge in the years ahead. We have clearly learned that combining different immunotherapy strategies will be essential for many patients with the most resilient solid tumors.
Of concern, as noted by a recent editorial in The Lancet, entitled “Cancer Research Equity: Innovations For The Many, Not The Few,” is that these individualized, sophisticated strategies are not scalable; they will have limited reach and benefit. The movement towards “off the shelf” CAR-T and inexpensive, orally active checkpoint inhibitors may help mitigate this issue.
Notwithstanding this important concern, we’re seeing an array of diverse and potent immunotherapy strategies that are providing highly encouraging results, engendering more excitement than we’ve seen in this space for some time. These should propel substantial improvements in outcomes for patients in the years ahead. It can’t happen soon enough.
Thanks for reading this edition of Ground Truths. If you found it informative, please share it with your colleagues.
Dr. Topol has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for Dexcom; Illumina; Molecular Stethoscope; Quest Diagnostics; Blue Cross Blue Shield Association. Received research grant from National Institutes of Health.
A version of this article appeared on Medscape.com.
Despite Few CNS Gene Therapies for Epilepsy, New Research Offers Hope
ORLANDO — Scientists have made major strides in gene therapy, and experts convened to share their insights on gene therapy development and challenges at the annual meeting of the American Epilepsy Society during a session called “Recent Advances Gene Therapies for the Epilepsies: A Preclinical Perspective.”
Four types of gene therapy
Suzanne Paradis, PhD, cofounder and president of Severin Therapeutics Inc., initiated the session, giving the audience an overview of the four types of gene therapy — the first being gene replacements, where a copy of the gene is added back. The second type of therapy, transcriptional enhancement, entails upregulating an endogenous copy of the gene.
“Both gene replacement and transcriptional enhancement can prove effective in treating monogenetic genetic disorders,” she said.
The third type is transcriptional enhancement, which upregulates an endogenous copy of the gene.
Generalizable gene therapies, the fourth type of gene therapy, involve adding a gene that bypasses either or both ictogenesis and seizure propagation.
As it stands, of the nearly 30 gene therapies currently marketed for neurological disorders, only four are indicated for central nervous system (CNS) disorders. Of the four currently approved by the FDA for seizures, onasemnogene abeparvovec-xioi (Zolgensma) is the only one that truly targets the CNS.
“Developing treatment that targets the CNS requires several important considerations,” Dr. Paradis said. “These include the right model system, appropriate delivery method, a product that can cross the blood-brain barrier (BBB) and target neurons, and the durability of transgene expression.”
Epilepsy May Be Amenable to Gene Therapy
To illustrate these principles, Meghan Eller, a PhD candidate at the University of Texas Southwestern in Dallas, shared research on potential new gene therapies that might one day become effective options in treating CNS diseases.
She spoke on viral-mediated gene delivery, specifically by employing adeno-associated virus (AAV) treatment in this arena.
“We capitalized on the ability of viruses to infect genetic materials,” she told the audience. “Viruses are naturally designed to infect cells and deliver genetic material.”
The viruses have three components that make them attractive. One of three viruses is typically used for this work — adenoviruses, lentiviruses, or AAV. The virus type used may be dictated by the gene of interest, meaning whether the gene is expressed, knocked down, or edited. Lastly, several regulatory elements are required; these are the promoter, polyadenylation signal, and the regulatory binding sites necessary for transcription.
“More recent technologies are CRISPR for gene editing, and with promoter, we can control the specific cell type in which gene will be expressed,” Ms. Eller explained.
Regulatory binding sites within a binding site allow regulation within an endogenous transgene.
“AAV genome is naturally single-stranded, but we can introduce a mutation to form a self-complementary cassette,” she said.
Using AAV as a vector for gene delivery has several advantages. First and foremost, it is easy to engineer. Moreover, it can infect dividing and non-dividing cells. It also exhibits long-lasting expression and has a low immune response. In addition, the AAV virion particle has demonstrated activity on cells found in numerous organs, including those of the lymph nodes, adrenal glands, kidneys, various muscle tissue, retinal cells, and digestive system as well as the CNS.
Yet, for all its benefits, the AAV comes with some limitations. For example, it carries as preexisting immunity and exhibits lost expression in dividing cells.
Another important drawback is its package size constraints, as many genes do not fall within its 2.4 kb self-complementary of 4.8 kb single-stranded packaging capacity.
For her research, Ms. Eller and colleagues took into account several considerations for therapy development. The appropriate route helps ensure the therapy reaches critical regions of the brain and that there is adequate expression in the periphery. The immune response becomes important regarding the body’s reaction to non-self proteins — a property, which, at times, can be modified based on dose. Thirdly, expression level and cell type expression can affect the therapy’s activity. In addition, a small amount of the vector will be incorporated into the host DNA.
The fact that AAV can cross the BBB allows for intravenous delivery; however, it limits brain transduction.
“Gene therapy may not be as effective if the delivery window is missed or there is significant neuron loss,” Ms. Eller said.
She stressed the importance of determining the minimal dose necessary for therapeutic benefit to minimize dose-related toxicity. She also distinguished when and why one might choose one type of gene therapy over another, using gene addition to help illustrate her point.
“Gene addition is the most important approach when there is a monogenic gene,” she said. “SLC13A5 and SLC6A1 are examples where gene addition is effective.”
Modulation of ion channels can help the delivery of therapeutic. Such is the case for NaV1.1 and Kv1.1. Finally, AAV can enhance the delivery of therapeutic proteins, as seen with Sema4D and neuropeptide Y.
Ms. Eller explained how the path to developing a gene therapy as an investigational new drug mirrors those historically traveled in conventional drug development to some extent. Preclinical studies offer proof of concept by determining efficacy, dosing, and toxicity in small animals such as mice. From there, studies progress to the pre-IND state by exploring pharmacology and clinical trial design while further investigating toxicity. FDA and regulatory approval require addressing safety concerns and establishing therapeutic benefit, at which point the therapy progresses to the fourth and final stage: clinical trials. During this stage, investigators monitor dosage and safety while evaluating efficacy.Optimal transgene expression regulation requires scientists to create an environment that gives rise to the perfect level of transgene expression. Otherwise, too little protein will result in no therapeutic benefit, while too much protein can become toxic.
Ms. Eller presented her work on investigating whether the reduction of Scn8a is therapeutic, given that epileptogenic Scn8a mutations increase neuronal firing. She treated both the control and Scn8a mice with antisense oligonucleotides (ASO), which depresses neuronal activity. Upon comparing the effects in ASO-treated mice to control, she found that long-term downregulation of Scn8a (50%) prevents seizures and increases survival — regardless of whether ASO therapy was initiated before or during seizure onset.
Additional studies exploring novel and potential gene therapies for epilepsy are on the horizon.
Dr. Paradis is an employee of Severin Therapeutics Inc. Ms Eller has no relevant disclosures.
ORLANDO — Scientists have made major strides in gene therapy, and experts convened to share their insights on gene therapy development and challenges at the annual meeting of the American Epilepsy Society during a session called “Recent Advances Gene Therapies for the Epilepsies: A Preclinical Perspective.”
Four types of gene therapy
Suzanne Paradis, PhD, cofounder and president of Severin Therapeutics Inc., initiated the session, giving the audience an overview of the four types of gene therapy — the first being gene replacements, where a copy of the gene is added back. The second type of therapy, transcriptional enhancement, entails upregulating an endogenous copy of the gene.
“Both gene replacement and transcriptional enhancement can prove effective in treating monogenetic genetic disorders,” she said.
The third type is transcriptional enhancement, which upregulates an endogenous copy of the gene.
Generalizable gene therapies, the fourth type of gene therapy, involve adding a gene that bypasses either or both ictogenesis and seizure propagation.
As it stands, of the nearly 30 gene therapies currently marketed for neurological disorders, only four are indicated for central nervous system (CNS) disorders. Of the four currently approved by the FDA for seizures, onasemnogene abeparvovec-xioi (Zolgensma) is the only one that truly targets the CNS.
“Developing treatment that targets the CNS requires several important considerations,” Dr. Paradis said. “These include the right model system, appropriate delivery method, a product that can cross the blood-brain barrier (BBB) and target neurons, and the durability of transgene expression.”
Epilepsy May Be Amenable to Gene Therapy
To illustrate these principles, Meghan Eller, a PhD candidate at the University of Texas Southwestern in Dallas, shared research on potential new gene therapies that might one day become effective options in treating CNS diseases.
She spoke on viral-mediated gene delivery, specifically by employing adeno-associated virus (AAV) treatment in this arena.
“We capitalized on the ability of viruses to infect genetic materials,” she told the audience. “Viruses are naturally designed to infect cells and deliver genetic material.”
The viruses have three components that make them attractive. One of three viruses is typically used for this work — adenoviruses, lentiviruses, or AAV. The virus type used may be dictated by the gene of interest, meaning whether the gene is expressed, knocked down, or edited. Lastly, several regulatory elements are required; these are the promoter, polyadenylation signal, and the regulatory binding sites necessary for transcription.
“More recent technologies are CRISPR for gene editing, and with promoter, we can control the specific cell type in which gene will be expressed,” Ms. Eller explained.
Regulatory binding sites within a binding site allow regulation within an endogenous transgene.
“AAV genome is naturally single-stranded, but we can introduce a mutation to form a self-complementary cassette,” she said.
Using AAV as a vector for gene delivery has several advantages. First and foremost, it is easy to engineer. Moreover, it can infect dividing and non-dividing cells. It also exhibits long-lasting expression and has a low immune response. In addition, the AAV virion particle has demonstrated activity on cells found in numerous organs, including those of the lymph nodes, adrenal glands, kidneys, various muscle tissue, retinal cells, and digestive system as well as the CNS.
Yet, for all its benefits, the AAV comes with some limitations. For example, it carries as preexisting immunity and exhibits lost expression in dividing cells.
Another important drawback is its package size constraints, as many genes do not fall within its 2.4 kb self-complementary of 4.8 kb single-stranded packaging capacity.
For her research, Ms. Eller and colleagues took into account several considerations for therapy development. The appropriate route helps ensure the therapy reaches critical regions of the brain and that there is adequate expression in the periphery. The immune response becomes important regarding the body’s reaction to non-self proteins — a property, which, at times, can be modified based on dose. Thirdly, expression level and cell type expression can affect the therapy’s activity. In addition, a small amount of the vector will be incorporated into the host DNA.
The fact that AAV can cross the BBB allows for intravenous delivery; however, it limits brain transduction.
“Gene therapy may not be as effective if the delivery window is missed or there is significant neuron loss,” Ms. Eller said.
She stressed the importance of determining the minimal dose necessary for therapeutic benefit to minimize dose-related toxicity. She also distinguished when and why one might choose one type of gene therapy over another, using gene addition to help illustrate her point.
“Gene addition is the most important approach when there is a monogenic gene,” she said. “SLC13A5 and SLC6A1 are examples where gene addition is effective.”
Modulation of ion channels can help the delivery of therapeutic. Such is the case for NaV1.1 and Kv1.1. Finally, AAV can enhance the delivery of therapeutic proteins, as seen with Sema4D and neuropeptide Y.
Ms. Eller explained how the path to developing a gene therapy as an investigational new drug mirrors those historically traveled in conventional drug development to some extent. Preclinical studies offer proof of concept by determining efficacy, dosing, and toxicity in small animals such as mice. From there, studies progress to the pre-IND state by exploring pharmacology and clinical trial design while further investigating toxicity. FDA and regulatory approval require addressing safety concerns and establishing therapeutic benefit, at which point the therapy progresses to the fourth and final stage: clinical trials. During this stage, investigators monitor dosage and safety while evaluating efficacy.Optimal transgene expression regulation requires scientists to create an environment that gives rise to the perfect level of transgene expression. Otherwise, too little protein will result in no therapeutic benefit, while too much protein can become toxic.
Ms. Eller presented her work on investigating whether the reduction of Scn8a is therapeutic, given that epileptogenic Scn8a mutations increase neuronal firing. She treated both the control and Scn8a mice with antisense oligonucleotides (ASO), which depresses neuronal activity. Upon comparing the effects in ASO-treated mice to control, she found that long-term downregulation of Scn8a (50%) prevents seizures and increases survival — regardless of whether ASO therapy was initiated before or during seizure onset.
Additional studies exploring novel and potential gene therapies for epilepsy are on the horizon.
Dr. Paradis is an employee of Severin Therapeutics Inc. Ms Eller has no relevant disclosures.
ORLANDO — Scientists have made major strides in gene therapy, and experts convened to share their insights on gene therapy development and challenges at the annual meeting of the American Epilepsy Society during a session called “Recent Advances Gene Therapies for the Epilepsies: A Preclinical Perspective.”
Four types of gene therapy
Suzanne Paradis, PhD, cofounder and president of Severin Therapeutics Inc., initiated the session, giving the audience an overview of the four types of gene therapy — the first being gene replacements, where a copy of the gene is added back. The second type of therapy, transcriptional enhancement, entails upregulating an endogenous copy of the gene.
“Both gene replacement and transcriptional enhancement can prove effective in treating monogenetic genetic disorders,” she said.
The third type is transcriptional enhancement, which upregulates an endogenous copy of the gene.
Generalizable gene therapies, the fourth type of gene therapy, involve adding a gene that bypasses either or both ictogenesis and seizure propagation.
As it stands, of the nearly 30 gene therapies currently marketed for neurological disorders, only four are indicated for central nervous system (CNS) disorders. Of the four currently approved by the FDA for seizures, onasemnogene abeparvovec-xioi (Zolgensma) is the only one that truly targets the CNS.
“Developing treatment that targets the CNS requires several important considerations,” Dr. Paradis said. “These include the right model system, appropriate delivery method, a product that can cross the blood-brain barrier (BBB) and target neurons, and the durability of transgene expression.”
Epilepsy May Be Amenable to Gene Therapy
To illustrate these principles, Meghan Eller, a PhD candidate at the University of Texas Southwestern in Dallas, shared research on potential new gene therapies that might one day become effective options in treating CNS diseases.
She spoke on viral-mediated gene delivery, specifically by employing adeno-associated virus (AAV) treatment in this arena.
“We capitalized on the ability of viruses to infect genetic materials,” she told the audience. “Viruses are naturally designed to infect cells and deliver genetic material.”
The viruses have three components that make them attractive. One of three viruses is typically used for this work — adenoviruses, lentiviruses, or AAV. The virus type used may be dictated by the gene of interest, meaning whether the gene is expressed, knocked down, or edited. Lastly, several regulatory elements are required; these are the promoter, polyadenylation signal, and the regulatory binding sites necessary for transcription.
“More recent technologies are CRISPR for gene editing, and with promoter, we can control the specific cell type in which gene will be expressed,” Ms. Eller explained.
Regulatory binding sites within a binding site allow regulation within an endogenous transgene.
“AAV genome is naturally single-stranded, but we can introduce a mutation to form a self-complementary cassette,” she said.
Using AAV as a vector for gene delivery has several advantages. First and foremost, it is easy to engineer. Moreover, it can infect dividing and non-dividing cells. It also exhibits long-lasting expression and has a low immune response. In addition, the AAV virion particle has demonstrated activity on cells found in numerous organs, including those of the lymph nodes, adrenal glands, kidneys, various muscle tissue, retinal cells, and digestive system as well as the CNS.
Yet, for all its benefits, the AAV comes with some limitations. For example, it carries as preexisting immunity and exhibits lost expression in dividing cells.
Another important drawback is its package size constraints, as many genes do not fall within its 2.4 kb self-complementary of 4.8 kb single-stranded packaging capacity.
For her research, Ms. Eller and colleagues took into account several considerations for therapy development. The appropriate route helps ensure the therapy reaches critical regions of the brain and that there is adequate expression in the periphery. The immune response becomes important regarding the body’s reaction to non-self proteins — a property, which, at times, can be modified based on dose. Thirdly, expression level and cell type expression can affect the therapy’s activity. In addition, a small amount of the vector will be incorporated into the host DNA.
The fact that AAV can cross the BBB allows for intravenous delivery; however, it limits brain transduction.
“Gene therapy may not be as effective if the delivery window is missed or there is significant neuron loss,” Ms. Eller said.
She stressed the importance of determining the minimal dose necessary for therapeutic benefit to minimize dose-related toxicity. She also distinguished when and why one might choose one type of gene therapy over another, using gene addition to help illustrate her point.
“Gene addition is the most important approach when there is a monogenic gene,” she said. “SLC13A5 and SLC6A1 are examples where gene addition is effective.”
Modulation of ion channels can help the delivery of therapeutic. Such is the case for NaV1.1 and Kv1.1. Finally, AAV can enhance the delivery of therapeutic proteins, as seen with Sema4D and neuropeptide Y.
Ms. Eller explained how the path to developing a gene therapy as an investigational new drug mirrors those historically traveled in conventional drug development to some extent. Preclinical studies offer proof of concept by determining efficacy, dosing, and toxicity in small animals such as mice. From there, studies progress to the pre-IND state by exploring pharmacology and clinical trial design while further investigating toxicity. FDA and regulatory approval require addressing safety concerns and establishing therapeutic benefit, at which point the therapy progresses to the fourth and final stage: clinical trials. During this stage, investigators monitor dosage and safety while evaluating efficacy.Optimal transgene expression regulation requires scientists to create an environment that gives rise to the perfect level of transgene expression. Otherwise, too little protein will result in no therapeutic benefit, while too much protein can become toxic.
Ms. Eller presented her work on investigating whether the reduction of Scn8a is therapeutic, given that epileptogenic Scn8a mutations increase neuronal firing. She treated both the control and Scn8a mice with antisense oligonucleotides (ASO), which depresses neuronal activity. Upon comparing the effects in ASO-treated mice to control, she found that long-term downregulation of Scn8a (50%) prevents seizures and increases survival — regardless of whether ASO therapy was initiated before or during seizure onset.
Additional studies exploring novel and potential gene therapies for epilepsy are on the horizon.
Dr. Paradis is an employee of Severin Therapeutics Inc. Ms Eller has no relevant disclosures.
FROM AES 2023
Sickle Cell CRISPR Gene Therapy May Offer Patients ‘Functional Cure’
One therapy — exagamglogene autotemcel or exa-cel (Casgevy) — is the first to use CRISPR gene-editing technology, and could “provide a one-time functional cure to patients with sickle cell disease,” said Haydar Frangoul, MD, of The Children’s Hospital at TriStar Centennial, Nashville, Tennessee.
Dr. Frangoul, who presented a recent interim analysis on the therapy at the American Society of Hematology (ASH) annual meeting earlier this month, reported that one infusion of exa-cel prompted rapid increases in total hemoglobin levels and almost completely eliminated a common and painful complication of sickle cell disease that can lead to irreversible organ damage, known as vaso-occlusive crisis.
Overall, the gene therapy led to “a rapid, robust, and durable increase in total hemoglobin to normal or near normal levels,” Dr. Frangoul said.
Exa-cel, from Vertex Pharmaceuticals and CRISPR Therapeutics, is a single-dose infusion containing a patient’s modified cells. First, a patient’s stem cells are harvested and then genetically modified to produce fetal hemoglobin.
The development of exa-cel was “grounded in human genetics, which show that fetal hemoglobin can substitute for sickle hemoglobin,” Dr. Frangoul explained. Patients receive these edited cells, which then help restore normal hemoglobin production.
The analysis showed that a one-time infusion of exa-cel following myeloablative conditioning prevented vaso-occlusive crisis in all but one patient with severe sickle cell disease. The therapy also prevented inpatient hospitalizations for vaso-occlusive crisis in all patients and led to sustained improvements in quality of life.
The results are “really striking,” said Sarah H. O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, who was not involved in the research. “The majority of our admissions on the hematology service are our patients with sickle cell. They’re uncomfortable, they’re in pain, they’re missing school, and they’re missing their activities,” which makes these interim findings quite “impactful.”
To examine the impact of exa-cel on vaso-occlusive crisis, the phase 3 trial included individuals aged 12 to 35 years with severe sickle cell disease and a history of at least two vaso-occlusive crises per year over the past 2 years.
Participants underwent cell CD34+ stem cell collection. These cells then underwent gene editing using CRISPR technology, explained Dr. Frangoul.
At the transplant center, patients received myeloablative conditioning chemotherapy with busulfan for 4 days before receiving an exa-cel infusion.
At the data cutoff in June 2023, 44 patients had been enrolled, of whom 30 were available for efficacy analysis. The mean age at screening was 22.1 years, and almost half (46.7%) were female. Prior to study recruitment, patients had a mean of 3.9 vaso-occlusive crises per year and a mean of 2.7 inpatient hospitalizations per year for severe vaso-occlusive crisis.
All but one patient (96.7%) met the primary endpoint of freedom from severe vaso-occlusive crisis for at least 12 consecutive months. The mean duration of freedom from vaso-occlusive crisis was 22.4 months, ranging from 14.8 months to 45.5 months. Moreover, 28 of the 29 patients who remained crisis-free at 12 months did not have a further vaso-occlusive crisis throughout the rest of the follow-up period.
Dr. Frangoul noted that results were similar for both adults and adolescents.
Exa-cel also led to a significant increase in freedom from inpatient hospitalizations, with 100% of patients achieving that goal, as well as early and sustained increases in both total and fetal hemoglobin levels, suggesting a “long-term meaningful benefit” from the therapy.
All 44 patients experienced adverse events related to myeloablative conditioning with busulfan, but only 29.5% had events linked to exa-cel. The most common adverse events overall were nausea (70.5%), stomatitis (63.6%), vomiting (56.8%), and febrile neutropenia (54.5%).
In a separate poster presented at ASH, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee, Dr. Frangoul, and colleagues reported that exa-cel also led to better health-related quality of life.
Patients showed “substantial improvements” in measures of quality of life, which included physical, emotional, social, and functional well-being as well as pain at a 6-month follow-up through year 2.
Typical outcomes studied in most trials are “emergency room visits and hospitalizations but what people may not appreciate as much is how much these patients are dealing with pain and discomfort at home,” Dr. O’Brien said. These recently reported quality-of-life metrics “are so key and really help us understand the impact” of this new therapy.
Dr. O’Brien noted, however, that “patients may be reluctant to undergo” this therapy because of the impact myeloablative conditioning has on fertility. That is why ongoing research on how stem cell transplants can be delivered “without impacting fertility is very important.”
It is “hard to know,” Dr. O’Brien explained, whether exa-cel will be a one-time treatment in practice, as many of the patients “already have end-organ damage from their disease.”
To that end, Dr. Frangoul noted that patients who complete the current trial can enroll in one that will include 13 years of additional follow-up.
Finally, Dr. O’Brien cautioned, gene therapies such as exa-cel “are only going to apply to a small segment of the population” — patients with the most severe form of the disease. That’s why “it’s important that we still prioritize hydroxyurea [and] multidisciplinary care for patients with sickle cell disease,” she said.
The study was sponsored by Vertex Pharmaceuticals in collaboration with CRISPR Therapeutics. Dr. Frangoul declared relationships with Editas Medicine, Rocket Pharmaceuticals, Jazz Pharmaceuticals, Vertex Pharmaceuticals, CRISPR Therapeutics, Bluebird Bio, and others. Dr. Sharma declared relationships with Vertex Pharmaceuticals, CRISPR Therapeutics, and others. Other authors declare numerous financial relationships.
A version of this article appeared on Medscape.com.
One therapy — exagamglogene autotemcel or exa-cel (Casgevy) — is the first to use CRISPR gene-editing technology, and could “provide a one-time functional cure to patients with sickle cell disease,” said Haydar Frangoul, MD, of The Children’s Hospital at TriStar Centennial, Nashville, Tennessee.
Dr. Frangoul, who presented a recent interim analysis on the therapy at the American Society of Hematology (ASH) annual meeting earlier this month, reported that one infusion of exa-cel prompted rapid increases in total hemoglobin levels and almost completely eliminated a common and painful complication of sickle cell disease that can lead to irreversible organ damage, known as vaso-occlusive crisis.
Overall, the gene therapy led to “a rapid, robust, and durable increase in total hemoglobin to normal or near normal levels,” Dr. Frangoul said.
Exa-cel, from Vertex Pharmaceuticals and CRISPR Therapeutics, is a single-dose infusion containing a patient’s modified cells. First, a patient’s stem cells are harvested and then genetically modified to produce fetal hemoglobin.
The development of exa-cel was “grounded in human genetics, which show that fetal hemoglobin can substitute for sickle hemoglobin,” Dr. Frangoul explained. Patients receive these edited cells, which then help restore normal hemoglobin production.
The analysis showed that a one-time infusion of exa-cel following myeloablative conditioning prevented vaso-occlusive crisis in all but one patient with severe sickle cell disease. The therapy also prevented inpatient hospitalizations for vaso-occlusive crisis in all patients and led to sustained improvements in quality of life.
The results are “really striking,” said Sarah H. O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, who was not involved in the research. “The majority of our admissions on the hematology service are our patients with sickle cell. They’re uncomfortable, they’re in pain, they’re missing school, and they’re missing their activities,” which makes these interim findings quite “impactful.”
To examine the impact of exa-cel on vaso-occlusive crisis, the phase 3 trial included individuals aged 12 to 35 years with severe sickle cell disease and a history of at least two vaso-occlusive crises per year over the past 2 years.
Participants underwent cell CD34+ stem cell collection. These cells then underwent gene editing using CRISPR technology, explained Dr. Frangoul.
At the transplant center, patients received myeloablative conditioning chemotherapy with busulfan for 4 days before receiving an exa-cel infusion.
At the data cutoff in June 2023, 44 patients had been enrolled, of whom 30 were available for efficacy analysis. The mean age at screening was 22.1 years, and almost half (46.7%) were female. Prior to study recruitment, patients had a mean of 3.9 vaso-occlusive crises per year and a mean of 2.7 inpatient hospitalizations per year for severe vaso-occlusive crisis.
All but one patient (96.7%) met the primary endpoint of freedom from severe vaso-occlusive crisis for at least 12 consecutive months. The mean duration of freedom from vaso-occlusive crisis was 22.4 months, ranging from 14.8 months to 45.5 months. Moreover, 28 of the 29 patients who remained crisis-free at 12 months did not have a further vaso-occlusive crisis throughout the rest of the follow-up period.
Dr. Frangoul noted that results were similar for both adults and adolescents.
Exa-cel also led to a significant increase in freedom from inpatient hospitalizations, with 100% of patients achieving that goal, as well as early and sustained increases in both total and fetal hemoglobin levels, suggesting a “long-term meaningful benefit” from the therapy.
All 44 patients experienced adverse events related to myeloablative conditioning with busulfan, but only 29.5% had events linked to exa-cel. The most common adverse events overall were nausea (70.5%), stomatitis (63.6%), vomiting (56.8%), and febrile neutropenia (54.5%).
In a separate poster presented at ASH, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee, Dr. Frangoul, and colleagues reported that exa-cel also led to better health-related quality of life.
Patients showed “substantial improvements” in measures of quality of life, which included physical, emotional, social, and functional well-being as well as pain at a 6-month follow-up through year 2.
Typical outcomes studied in most trials are “emergency room visits and hospitalizations but what people may not appreciate as much is how much these patients are dealing with pain and discomfort at home,” Dr. O’Brien said. These recently reported quality-of-life metrics “are so key and really help us understand the impact” of this new therapy.
Dr. O’Brien noted, however, that “patients may be reluctant to undergo” this therapy because of the impact myeloablative conditioning has on fertility. That is why ongoing research on how stem cell transplants can be delivered “without impacting fertility is very important.”
It is “hard to know,” Dr. O’Brien explained, whether exa-cel will be a one-time treatment in practice, as many of the patients “already have end-organ damage from their disease.”
To that end, Dr. Frangoul noted that patients who complete the current trial can enroll in one that will include 13 years of additional follow-up.
Finally, Dr. O’Brien cautioned, gene therapies such as exa-cel “are only going to apply to a small segment of the population” — patients with the most severe form of the disease. That’s why “it’s important that we still prioritize hydroxyurea [and] multidisciplinary care for patients with sickle cell disease,” she said.
The study was sponsored by Vertex Pharmaceuticals in collaboration with CRISPR Therapeutics. Dr. Frangoul declared relationships with Editas Medicine, Rocket Pharmaceuticals, Jazz Pharmaceuticals, Vertex Pharmaceuticals, CRISPR Therapeutics, Bluebird Bio, and others. Dr. Sharma declared relationships with Vertex Pharmaceuticals, CRISPR Therapeutics, and others. Other authors declare numerous financial relationships.
A version of this article appeared on Medscape.com.
One therapy — exagamglogene autotemcel or exa-cel (Casgevy) — is the first to use CRISPR gene-editing technology, and could “provide a one-time functional cure to patients with sickle cell disease,” said Haydar Frangoul, MD, of The Children’s Hospital at TriStar Centennial, Nashville, Tennessee.
Dr. Frangoul, who presented a recent interim analysis on the therapy at the American Society of Hematology (ASH) annual meeting earlier this month, reported that one infusion of exa-cel prompted rapid increases in total hemoglobin levels and almost completely eliminated a common and painful complication of sickle cell disease that can lead to irreversible organ damage, known as vaso-occlusive crisis.
Overall, the gene therapy led to “a rapid, robust, and durable increase in total hemoglobin to normal or near normal levels,” Dr. Frangoul said.
Exa-cel, from Vertex Pharmaceuticals and CRISPR Therapeutics, is a single-dose infusion containing a patient’s modified cells. First, a patient’s stem cells are harvested and then genetically modified to produce fetal hemoglobin.
The development of exa-cel was “grounded in human genetics, which show that fetal hemoglobin can substitute for sickle hemoglobin,” Dr. Frangoul explained. Patients receive these edited cells, which then help restore normal hemoglobin production.
The analysis showed that a one-time infusion of exa-cel following myeloablative conditioning prevented vaso-occlusive crisis in all but one patient with severe sickle cell disease. The therapy also prevented inpatient hospitalizations for vaso-occlusive crisis in all patients and led to sustained improvements in quality of life.
The results are “really striking,” said Sarah H. O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, who was not involved in the research. “The majority of our admissions on the hematology service are our patients with sickle cell. They’re uncomfortable, they’re in pain, they’re missing school, and they’re missing their activities,” which makes these interim findings quite “impactful.”
To examine the impact of exa-cel on vaso-occlusive crisis, the phase 3 trial included individuals aged 12 to 35 years with severe sickle cell disease and a history of at least two vaso-occlusive crises per year over the past 2 years.
Participants underwent cell CD34+ stem cell collection. These cells then underwent gene editing using CRISPR technology, explained Dr. Frangoul.
At the transplant center, patients received myeloablative conditioning chemotherapy with busulfan for 4 days before receiving an exa-cel infusion.
At the data cutoff in June 2023, 44 patients had been enrolled, of whom 30 were available for efficacy analysis. The mean age at screening was 22.1 years, and almost half (46.7%) were female. Prior to study recruitment, patients had a mean of 3.9 vaso-occlusive crises per year and a mean of 2.7 inpatient hospitalizations per year for severe vaso-occlusive crisis.
All but one patient (96.7%) met the primary endpoint of freedom from severe vaso-occlusive crisis for at least 12 consecutive months. The mean duration of freedom from vaso-occlusive crisis was 22.4 months, ranging from 14.8 months to 45.5 months. Moreover, 28 of the 29 patients who remained crisis-free at 12 months did not have a further vaso-occlusive crisis throughout the rest of the follow-up period.
Dr. Frangoul noted that results were similar for both adults and adolescents.
Exa-cel also led to a significant increase in freedom from inpatient hospitalizations, with 100% of patients achieving that goal, as well as early and sustained increases in both total and fetal hemoglobin levels, suggesting a “long-term meaningful benefit” from the therapy.
All 44 patients experienced adverse events related to myeloablative conditioning with busulfan, but only 29.5% had events linked to exa-cel. The most common adverse events overall were nausea (70.5%), stomatitis (63.6%), vomiting (56.8%), and febrile neutropenia (54.5%).
In a separate poster presented at ASH, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital in Memphis, Tennessee, Dr. Frangoul, and colleagues reported that exa-cel also led to better health-related quality of life.
Patients showed “substantial improvements” in measures of quality of life, which included physical, emotional, social, and functional well-being as well as pain at a 6-month follow-up through year 2.
Typical outcomes studied in most trials are “emergency room visits and hospitalizations but what people may not appreciate as much is how much these patients are dealing with pain and discomfort at home,” Dr. O’Brien said. These recently reported quality-of-life metrics “are so key and really help us understand the impact” of this new therapy.
Dr. O’Brien noted, however, that “patients may be reluctant to undergo” this therapy because of the impact myeloablative conditioning has on fertility. That is why ongoing research on how stem cell transplants can be delivered “without impacting fertility is very important.”
It is “hard to know,” Dr. O’Brien explained, whether exa-cel will be a one-time treatment in practice, as many of the patients “already have end-organ damage from their disease.”
To that end, Dr. Frangoul noted that patients who complete the current trial can enroll in one that will include 13 years of additional follow-up.
Finally, Dr. O’Brien cautioned, gene therapies such as exa-cel “are only going to apply to a small segment of the population” — patients with the most severe form of the disease. That’s why “it’s important that we still prioritize hydroxyurea [and] multidisciplinary care for patients with sickle cell disease,” she said.
The study was sponsored by Vertex Pharmaceuticals in collaboration with CRISPR Therapeutics. Dr. Frangoul declared relationships with Editas Medicine, Rocket Pharmaceuticals, Jazz Pharmaceuticals, Vertex Pharmaceuticals, CRISPR Therapeutics, Bluebird Bio, and others. Dr. Sharma declared relationships with Vertex Pharmaceuticals, CRISPR Therapeutics, and others. Other authors declare numerous financial relationships.
A version of this article appeared on Medscape.com.
FROM ASH 2023
Sickle Cell Gene Therapy ‘Truly Transformative’
.
More specifically, a single infusion of lovo-cel led to complete resolution of vaso-occlusive events in 88% of patients, with 94% achieving complete resolution of severe events. All 10 adolescents in the study achieved complete resolution of vaso-occlusive events. Most patients remained free of vaso-occlusive events at their last follow-up.
“This is a one-time, truly transformative treatment with lovo-cel,” lead author Julie Kanter, MD, director of the adult sickle cell clinic at the University of Alabama in Birmingham, said in a media briefing at the annual meeting of the American Society of Hematology. The gene therapy can essentially eliminate vaso-occlusive events in patients with sickle cell disease and lead to normal hemoglobin levels, Dr. Kanter added.
For “anybody who has rounded on the inpatient floor and taken care of adolescents admitted with a pain crisis multiple times a year,” seeing these results “is so compelling,” commented Sarah O’Brien, MD, a pediatric hematologist at Nationwide Children’s Hospital in Columbus, Ohio, who moderated the briefing but was not involved in the study.
One and Done
Sickle cell disease, a debilitating and potentially life-threatening blood disorder, affects an estimated 100,000 people in the US.
People with the condition have a mutation in hemoglobin, which causes red blood cells to develop an abnormal sickle shape. These sickled cells block the flow of blood, ultimately depriving tissues of oxygen and leading to organ damage and severe pain, known as vaso-occlusive events.
On Dec. 8, the U.S. Food and Drug Administration (FDA) approved lovo-cel for patients aged 12 years or older with severe sickle cell disease alongside another gene-editing therapy called exagamglogene autotemcel or exa-cel (Casgevy, Vertex Pharmaceuticals and Crispr Therapeutics). The two therapies use different gene-editing approaches — exa-cel is the first to use the gene-editing tool CRISPR while lovo-cel uses a lentiviral vector.
Both are one-time, single-dose cell-based gene therapies.
With lovo-cel, patients first undergo a transfusion regimen and myeloablative conditioning with busulfan to collect cells that can then be genetically modified. A patient’s harvested cells are modified with an anti-sickling version of hemoglobin A, HbAT87Q. Patients then receive an infusion of these edited cells and remain in the hospital during engraftment and reconstitution.
Dr. Kanter presented long-term follow-up data on 47 patients enrolled in phase 1/2 and phase 3 studies of lovo-cel.
All patients had stable HbAT87Q levels from 6 months to their last follow-up at a median of 35.5 months.
Most patients achieved a durable globin response through their final follow-up visit.
Among the 34 evaluable patients, 88% had complete resolution of vaso-occlusive events 6 to 18 months after their infusion, including all 10 adolescent patients. Almost all patients (94%) achieved complete resolution of serious vaso-occlusive events.
In the few patients who experienced posttreatment vaso-occlusive events, these individuals still achieved major reductions in hospital admissions and hospital days.
Among 20 patients followed for at least 3 years, more than half had clinically meaningful improvements in pain intensity, pain interference, and fatigue.
Most treatment-related adverse events occurred within 1 year of lovo-cel infusions and were primarily related to busulfan conditioning. No cases of veno-occlusive liver disease, graft failure, or graft vs host disease occurred, and patients did not have complications related to the viral vector. No patients who had a history of stroke prior to lovo-cel therapy experienced a post-therapy stroke.
One patient died at baseline from significant cardiopulmonary disease related to sickle cell disease, but the death was considered unrelated to lovo-cel therapy.
To see a one-time treatment that essentially eradicates vaso-occlusive events is “really unparalleled,” said Steven Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, who presented data on a different study at the briefing.
However, Dr. Kanter noted, “it’s important to highlight that many of these individuals come into this therapy with significant disease and end-organ complications, and this will be something we will really need to follow long-term to understand how much this therapy can stabilize or reverse these complications.”
The studies were funded by bluebird bio. Dr. Kanter disclosed honoraria from the company and consulting/advising activities and receipt of research funding from multiple other entities. Dr. O’Brien disclosed consultancy for AstraZeneca, honoraria from Pharmacosmos, and research funding from Bristol Myers Squibb. Dr. Pipe disclosed consulting activities from multiple companies, not including bluebird bio.
A version of this article appeared on Medscape.com.
.
More specifically, a single infusion of lovo-cel led to complete resolution of vaso-occlusive events in 88% of patients, with 94% achieving complete resolution of severe events. All 10 adolescents in the study achieved complete resolution of vaso-occlusive events. Most patients remained free of vaso-occlusive events at their last follow-up.
“This is a one-time, truly transformative treatment with lovo-cel,” lead author Julie Kanter, MD, director of the adult sickle cell clinic at the University of Alabama in Birmingham, said in a media briefing at the annual meeting of the American Society of Hematology. The gene therapy can essentially eliminate vaso-occlusive events in patients with sickle cell disease and lead to normal hemoglobin levels, Dr. Kanter added.
For “anybody who has rounded on the inpatient floor and taken care of adolescents admitted with a pain crisis multiple times a year,” seeing these results “is so compelling,” commented Sarah O’Brien, MD, a pediatric hematologist at Nationwide Children’s Hospital in Columbus, Ohio, who moderated the briefing but was not involved in the study.
One and Done
Sickle cell disease, a debilitating and potentially life-threatening blood disorder, affects an estimated 100,000 people in the US.
People with the condition have a mutation in hemoglobin, which causes red blood cells to develop an abnormal sickle shape. These sickled cells block the flow of blood, ultimately depriving tissues of oxygen and leading to organ damage and severe pain, known as vaso-occlusive events.
On Dec. 8, the U.S. Food and Drug Administration (FDA) approved lovo-cel for patients aged 12 years or older with severe sickle cell disease alongside another gene-editing therapy called exagamglogene autotemcel or exa-cel (Casgevy, Vertex Pharmaceuticals and Crispr Therapeutics). The two therapies use different gene-editing approaches — exa-cel is the first to use the gene-editing tool CRISPR while lovo-cel uses a lentiviral vector.
Both are one-time, single-dose cell-based gene therapies.
With lovo-cel, patients first undergo a transfusion regimen and myeloablative conditioning with busulfan to collect cells that can then be genetically modified. A patient’s harvested cells are modified with an anti-sickling version of hemoglobin A, HbAT87Q. Patients then receive an infusion of these edited cells and remain in the hospital during engraftment and reconstitution.
Dr. Kanter presented long-term follow-up data on 47 patients enrolled in phase 1/2 and phase 3 studies of lovo-cel.
All patients had stable HbAT87Q levels from 6 months to their last follow-up at a median of 35.5 months.
Most patients achieved a durable globin response through their final follow-up visit.
Among the 34 evaluable patients, 88% had complete resolution of vaso-occlusive events 6 to 18 months after their infusion, including all 10 adolescent patients. Almost all patients (94%) achieved complete resolution of serious vaso-occlusive events.
In the few patients who experienced posttreatment vaso-occlusive events, these individuals still achieved major reductions in hospital admissions and hospital days.
Among 20 patients followed for at least 3 years, more than half had clinically meaningful improvements in pain intensity, pain interference, and fatigue.
Most treatment-related adverse events occurred within 1 year of lovo-cel infusions and were primarily related to busulfan conditioning. No cases of veno-occlusive liver disease, graft failure, or graft vs host disease occurred, and patients did not have complications related to the viral vector. No patients who had a history of stroke prior to lovo-cel therapy experienced a post-therapy stroke.
One patient died at baseline from significant cardiopulmonary disease related to sickle cell disease, but the death was considered unrelated to lovo-cel therapy.
To see a one-time treatment that essentially eradicates vaso-occlusive events is “really unparalleled,” said Steven Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, who presented data on a different study at the briefing.
However, Dr. Kanter noted, “it’s important to highlight that many of these individuals come into this therapy with significant disease and end-organ complications, and this will be something we will really need to follow long-term to understand how much this therapy can stabilize or reverse these complications.”
The studies were funded by bluebird bio. Dr. Kanter disclosed honoraria from the company and consulting/advising activities and receipt of research funding from multiple other entities. Dr. O’Brien disclosed consultancy for AstraZeneca, honoraria from Pharmacosmos, and research funding from Bristol Myers Squibb. Dr. Pipe disclosed consulting activities from multiple companies, not including bluebird bio.
A version of this article appeared on Medscape.com.
.
More specifically, a single infusion of lovo-cel led to complete resolution of vaso-occlusive events in 88% of patients, with 94% achieving complete resolution of severe events. All 10 adolescents in the study achieved complete resolution of vaso-occlusive events. Most patients remained free of vaso-occlusive events at their last follow-up.
“This is a one-time, truly transformative treatment with lovo-cel,” lead author Julie Kanter, MD, director of the adult sickle cell clinic at the University of Alabama in Birmingham, said in a media briefing at the annual meeting of the American Society of Hematology. The gene therapy can essentially eliminate vaso-occlusive events in patients with sickle cell disease and lead to normal hemoglobin levels, Dr. Kanter added.
For “anybody who has rounded on the inpatient floor and taken care of adolescents admitted with a pain crisis multiple times a year,” seeing these results “is so compelling,” commented Sarah O’Brien, MD, a pediatric hematologist at Nationwide Children’s Hospital in Columbus, Ohio, who moderated the briefing but was not involved in the study.
One and Done
Sickle cell disease, a debilitating and potentially life-threatening blood disorder, affects an estimated 100,000 people in the US.
People with the condition have a mutation in hemoglobin, which causes red blood cells to develop an abnormal sickle shape. These sickled cells block the flow of blood, ultimately depriving tissues of oxygen and leading to organ damage and severe pain, known as vaso-occlusive events.
On Dec. 8, the U.S. Food and Drug Administration (FDA) approved lovo-cel for patients aged 12 years or older with severe sickle cell disease alongside another gene-editing therapy called exagamglogene autotemcel or exa-cel (Casgevy, Vertex Pharmaceuticals and Crispr Therapeutics). The two therapies use different gene-editing approaches — exa-cel is the first to use the gene-editing tool CRISPR while lovo-cel uses a lentiviral vector.
Both are one-time, single-dose cell-based gene therapies.
With lovo-cel, patients first undergo a transfusion regimen and myeloablative conditioning with busulfan to collect cells that can then be genetically modified. A patient’s harvested cells are modified with an anti-sickling version of hemoglobin A, HbAT87Q. Patients then receive an infusion of these edited cells and remain in the hospital during engraftment and reconstitution.
Dr. Kanter presented long-term follow-up data on 47 patients enrolled in phase 1/2 and phase 3 studies of lovo-cel.
All patients had stable HbAT87Q levels from 6 months to their last follow-up at a median of 35.5 months.
Most patients achieved a durable globin response through their final follow-up visit.
Among the 34 evaluable patients, 88% had complete resolution of vaso-occlusive events 6 to 18 months after their infusion, including all 10 adolescent patients. Almost all patients (94%) achieved complete resolution of serious vaso-occlusive events.
In the few patients who experienced posttreatment vaso-occlusive events, these individuals still achieved major reductions in hospital admissions and hospital days.
Among 20 patients followed for at least 3 years, more than half had clinically meaningful improvements in pain intensity, pain interference, and fatigue.
Most treatment-related adverse events occurred within 1 year of lovo-cel infusions and were primarily related to busulfan conditioning. No cases of veno-occlusive liver disease, graft failure, or graft vs host disease occurred, and patients did not have complications related to the viral vector. No patients who had a history of stroke prior to lovo-cel therapy experienced a post-therapy stroke.
One patient died at baseline from significant cardiopulmonary disease related to sickle cell disease, but the death was considered unrelated to lovo-cel therapy.
To see a one-time treatment that essentially eradicates vaso-occlusive events is “really unparalleled,” said Steven Pipe, MD, from the University of Michigan School of Medicine in Ann Arbor, who presented data on a different study at the briefing.
However, Dr. Kanter noted, “it’s important to highlight that many of these individuals come into this therapy with significant disease and end-organ complications, and this will be something we will really need to follow long-term to understand how much this therapy can stabilize or reverse these complications.”
The studies were funded by bluebird bio. Dr. Kanter disclosed honoraria from the company and consulting/advising activities and receipt of research funding from multiple other entities. Dr. O’Brien disclosed consultancy for AstraZeneca, honoraria from Pharmacosmos, and research funding from Bristol Myers Squibb. Dr. Pipe disclosed consulting activities from multiple companies, not including bluebird bio.
A version of this article appeared on Medscape.com.
FROM ASH 2023
This test may guide AML therapy for Black pediatric patients
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
.
The score, dubbed ACS10 and initially highlighted in a 2022 report, predicts how well patients will respond to cytarabine based on their genetic make-up, and has the potential to personalize treatment for Black pediatric patients, a group that often has worse outcomes than White patients.
In the current study, presented at the annual meeting of the American Society of Hematology (ASH) , Black patients with low ACS10 scores had significantly worse outcomes compared with those with high scores when initially treated with low-dose cytarabine, daunorubicin, and etoposide.
The difference in outcomes disappeared, however, for patients who received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine.
The genetic traits revealed by the test likely help explain why Black patients with AML typically fare worse on certain regimens, Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute, commented in an ASH press preview briefing.
This study also suggests that clinicians should perform testing for genetic variants and biomarkers that impact outcomes “instead of assuming that a certain dose should be given simply based on perceived or reported race or ethnicity,” said Dr. Dunbar, also secretary of ASH.
The ACS10 test, derived from a combination of 10 single nucleotide polymorphisms, is not yet available, but one could be developed to help guide treatment decisions for clinicians, especially those in developing countries where AML treatment can be very expensive, said study lead author Jatinder Lamba, PhD, MSc, of the University of Florida College of Pharmacy, Gainesville, at an ASH press briefing on Thursday.
Prior research shows that Black pediatric patients with AML often have worse outcomes than White patients. A recent study , for instance, found Black patients with AML, especially those aged 18 to 29 years, had a higher early death rate compared with White patients (16% vs 3%) and significantly lower 5-year overall survival rates (22% vs 51%). The authors of this study suggested that genetic differences between young Black and White patients could help explain the disparity.
In the new analysis, Dr. Lamba and colleagues explored how outcomes by race and cytarabine pharmacogenomics varied in pediatric patients with AML.
The study included 86 Black patients and 359 White patients with newly diagnosed AML treated on two multi-institutional clinical trials. The patients received one of three initial treatments that included cytarabine: high-dose or low-dose cytarabine, daunorubicin, and etoposide, or clofarabine and cytarabine.
Most Black patients in the analysis (73%) had low ACS10 scores compared with 30% of White patients.
Unlike other recent reports, this study found that Black and White patients had similar complete remission rates following two courses of induction therapy (92.6% vs 95%) as well as similar rates of minimal residual disease negativity after one course (55.8% vs 55.4%).
Event-free survival (EFS) and overall survival rates were also similar, with 5-year EFS estimates at 58.3% for Black patients and 58.2% for White patients and overall survival rates at 63.8% vs 69.4%, respectively (P = .24).
However, when separating outcomes by ACS10 scores, Black patients with low scores had significantly worse EFS following low-dose cytarabine, daunorubicin, and etoposide compared with those with high ACS10 scores. And when these patients received high-dose cytarabine, daunorubicin, and etoposide or clofarabine and cytarabine induction therapy instead, the differences went away.
Overall, Black patients demonstrated significantly better EFS following treatment with clofarabine and cytarabine compared with the low-dose cytarabine triple therapy (hazard ratio, 0.17; P = .01). After adjusting for cofounders, clofarabine and cytarabine induction was the best treatment for Black patients with low ACS10 scores (HR for EFS, 0.2).
“Our results suggest that pharmacogenomics differences between Black and White patients should be considered when tailoring induction regimens to improve outcomes of Black patients and bridge the racial disparity gap in AML treatment,” the researchers concluded.
In developing countries, especially in Africa, starting patients on high-dose cytarabine, daunorubicin, and etoposide can lead to better results “without increasing much of the economic burden” since this treatment is the cheapest, Dr. Lamba said. “At the same time, if the patients have high ACS10 score, you can reduce their economic burden by giving them standard dose” cytarabine, daunorubicin, and etoposide and achieve similar results.
No study funding was reported. Dr. Lamba reported no relevant financial relationships, and three other authors reported various disclosures. Disclosures for Dr. Dunbar were unavailable..
A version of this article appeared on Medscape.com.
FROM ASH 2023
FDA approves first 2 gene-editing therapies for sickle cell
These “milestone treatments” mark the first cell-based gene therapies for this debilitating and potentially life-threatening blood disorder that affects about 100,000 people in the United States.
The two therapies are exagamglogene autotemcel, or exa-cel (Casgevy; Vertex Pharmaceuticals and Crispr Therapeutics), and lovotibeglogene autotemcel, or lovo-cel (Lyfgenia; bluebird bio).
“The approval of the first gene therapies for [sickle cell disease] represents a tremendous step forward for the [sickle cell] community, which has been historically overlooked and underfunded,” said Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, in a statement from the American Society of Hematology, following the approval.
“We are excited to advance the field, especially for individuals whose lives have been severely disrupted by the disease, by approving two cell-based gene therapies today,” Nicole Verdun, MD, of the FDA’s Center for Biologics Evaluation and Research, added in an agency press release.
Sickle cell disease involves a mutation in hemoglobin, a protein in red blood cells that provides oxygen to tissues. The mutation leads red blood cells to develop a crescent or sickle shape, which can restrict blood flow and cause severe pain and organ damage, known as vaso-occlusive events or crises.
Treatment options prior to these approvals primarily included red blood transfusions and hydroxyurea alongside pain management. The only potential curative option has been allogeneic hematopoietic stem cell transplantation, but that comes with significant risks and most patients don’t have an appropriate donor.
Exa-cel
Exa-cel uses CRISPR gene-editing technology. Before the infusion, patients undergo myeloablative conditioning, which removes cells from the bone marrow. These cells are genetically modified to produce fetal hemoglobin. Patients then receive an infusion of the edited cells, which can help restore normal hemoglobin production.
The FDA approval was based on data from the pivotal CLIMB SCD-121 trial. In an October advisory committee meeting, the FDA highlighted trial data demonstrating that 29 of 31 patients reached the trial’s primary endpoint: freedom from severe vaso-occlusive crises over a 12-month period. In addition, 28 of these patients remained free of vaso-occlusive crises for almost 2 years.
The committee noted that one of the 31 patients died about 9 months after receiving an exa-cel infusion.
The cell-based gene therapy also increased both fetal and total hemoglobin, with total hemoglobin levels increasing to > 11 g/dL by month 3 and remaining at that level afterward. No patients experienced graft failure or rejection.
The most common side effects included low platelets and white blood cell counts, mouth sores, nausea, musculoskeletal pain, vomiting, and febrile neutropenia.
Exa-cel could “provide a one-time functional cure” for patients with severe sickle cell disease, according to Franco Locatelli, MD, of Sapienza University of Rome, who presented initial findings last year.
While the current approval is for patients with infusion-dependent sickle cell disease, exa-cel is also being evaluated in patients with another blood disorder, beta-thalassemia.
Lovo-cel
Lovo-cel, a cell-based gene therapy, uses a different technology — a lentiviral vector, or gene delivery vehicle — that can also genetically modify a patient’s blood stem cells.
Like exa-cel, lovo-cel is a one-time, single-dose infusion that contains the patient’s modified cells. Before the infusion, patients undergo myeloablative conditioning. The patient’s stem cells are then genetically modified to allow them to produce the most common form of hemoglobin, HbA
This approval was based on data from a single-arm, 24-month study in patients aged 12-50 years who had sickle cell disease and a history of vaso-occlusive events.
Overall, 88% of patients (28 of 32) achieved complete resolution of vaso-occlusive events 6-18 months after the infusion.
The most common side effects included stomatitis; febrile neutropenia; and low platelet, white blood cell, and red blood cell counts.
The FDA noted that hematologic cancer has occurred in patients treated with lovo-cel, and the label includes a black-box warning about the risk.
Dr. Brodsky noted, however, that “while these new gene therapies are potentially life-changing for individuals living with [sickle cell disease], they must be accessible to be effective.”
Access is a potential concern. Exa-cel and lovo-cel could cost about $2 million.
A version of this article appeared on Medscape.com.
These “milestone treatments” mark the first cell-based gene therapies for this debilitating and potentially life-threatening blood disorder that affects about 100,000 people in the United States.
The two therapies are exagamglogene autotemcel, or exa-cel (Casgevy; Vertex Pharmaceuticals and Crispr Therapeutics), and lovotibeglogene autotemcel, or lovo-cel (Lyfgenia; bluebird bio).
“The approval of the first gene therapies for [sickle cell disease] represents a tremendous step forward for the [sickle cell] community, which has been historically overlooked and underfunded,” said Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, in a statement from the American Society of Hematology, following the approval.
“We are excited to advance the field, especially for individuals whose lives have been severely disrupted by the disease, by approving two cell-based gene therapies today,” Nicole Verdun, MD, of the FDA’s Center for Biologics Evaluation and Research, added in an agency press release.
Sickle cell disease involves a mutation in hemoglobin, a protein in red blood cells that provides oxygen to tissues. The mutation leads red blood cells to develop a crescent or sickle shape, which can restrict blood flow and cause severe pain and organ damage, known as vaso-occlusive events or crises.
Treatment options prior to these approvals primarily included red blood transfusions and hydroxyurea alongside pain management. The only potential curative option has been allogeneic hematopoietic stem cell transplantation, but that comes with significant risks and most patients don’t have an appropriate donor.
Exa-cel
Exa-cel uses CRISPR gene-editing technology. Before the infusion, patients undergo myeloablative conditioning, which removes cells from the bone marrow. These cells are genetically modified to produce fetal hemoglobin. Patients then receive an infusion of the edited cells, which can help restore normal hemoglobin production.
The FDA approval was based on data from the pivotal CLIMB SCD-121 trial. In an October advisory committee meeting, the FDA highlighted trial data demonstrating that 29 of 31 patients reached the trial’s primary endpoint: freedom from severe vaso-occlusive crises over a 12-month period. In addition, 28 of these patients remained free of vaso-occlusive crises for almost 2 years.
The committee noted that one of the 31 patients died about 9 months after receiving an exa-cel infusion.
The cell-based gene therapy also increased both fetal and total hemoglobin, with total hemoglobin levels increasing to > 11 g/dL by month 3 and remaining at that level afterward. No patients experienced graft failure or rejection.
The most common side effects included low platelets and white blood cell counts, mouth sores, nausea, musculoskeletal pain, vomiting, and febrile neutropenia.
Exa-cel could “provide a one-time functional cure” for patients with severe sickle cell disease, according to Franco Locatelli, MD, of Sapienza University of Rome, who presented initial findings last year.
While the current approval is for patients with infusion-dependent sickle cell disease, exa-cel is also being evaluated in patients with another blood disorder, beta-thalassemia.
Lovo-cel
Lovo-cel, a cell-based gene therapy, uses a different technology — a lentiviral vector, or gene delivery vehicle — that can also genetically modify a patient’s blood stem cells.
Like exa-cel, lovo-cel is a one-time, single-dose infusion that contains the patient’s modified cells. Before the infusion, patients undergo myeloablative conditioning. The patient’s stem cells are then genetically modified to allow them to produce the most common form of hemoglobin, HbA
This approval was based on data from a single-arm, 24-month study in patients aged 12-50 years who had sickle cell disease and a history of vaso-occlusive events.
Overall, 88% of patients (28 of 32) achieved complete resolution of vaso-occlusive events 6-18 months after the infusion.
The most common side effects included stomatitis; febrile neutropenia; and low platelet, white blood cell, and red blood cell counts.
The FDA noted that hematologic cancer has occurred in patients treated with lovo-cel, and the label includes a black-box warning about the risk.
Dr. Brodsky noted, however, that “while these new gene therapies are potentially life-changing for individuals living with [sickle cell disease], they must be accessible to be effective.”
Access is a potential concern. Exa-cel and lovo-cel could cost about $2 million.
A version of this article appeared on Medscape.com.
These “milestone treatments” mark the first cell-based gene therapies for this debilitating and potentially life-threatening blood disorder that affects about 100,000 people in the United States.
The two therapies are exagamglogene autotemcel, or exa-cel (Casgevy; Vertex Pharmaceuticals and Crispr Therapeutics), and lovotibeglogene autotemcel, or lovo-cel (Lyfgenia; bluebird bio).
“The approval of the first gene therapies for [sickle cell disease] represents a tremendous step forward for the [sickle cell] community, which has been historically overlooked and underfunded,” said Robert A. Brodsky, MD, of Johns Hopkins University School of Medicine, in a statement from the American Society of Hematology, following the approval.
“We are excited to advance the field, especially for individuals whose lives have been severely disrupted by the disease, by approving two cell-based gene therapies today,” Nicole Verdun, MD, of the FDA’s Center for Biologics Evaluation and Research, added in an agency press release.
Sickle cell disease involves a mutation in hemoglobin, a protein in red blood cells that provides oxygen to tissues. The mutation leads red blood cells to develop a crescent or sickle shape, which can restrict blood flow and cause severe pain and organ damage, known as vaso-occlusive events or crises.
Treatment options prior to these approvals primarily included red blood transfusions and hydroxyurea alongside pain management. The only potential curative option has been allogeneic hematopoietic stem cell transplantation, but that comes with significant risks and most patients don’t have an appropriate donor.
Exa-cel
Exa-cel uses CRISPR gene-editing technology. Before the infusion, patients undergo myeloablative conditioning, which removes cells from the bone marrow. These cells are genetically modified to produce fetal hemoglobin. Patients then receive an infusion of the edited cells, which can help restore normal hemoglobin production.
The FDA approval was based on data from the pivotal CLIMB SCD-121 trial. In an October advisory committee meeting, the FDA highlighted trial data demonstrating that 29 of 31 patients reached the trial’s primary endpoint: freedom from severe vaso-occlusive crises over a 12-month period. In addition, 28 of these patients remained free of vaso-occlusive crises for almost 2 years.
The committee noted that one of the 31 patients died about 9 months after receiving an exa-cel infusion.
The cell-based gene therapy also increased both fetal and total hemoglobin, with total hemoglobin levels increasing to > 11 g/dL by month 3 and remaining at that level afterward. No patients experienced graft failure or rejection.
The most common side effects included low platelets and white blood cell counts, mouth sores, nausea, musculoskeletal pain, vomiting, and febrile neutropenia.
Exa-cel could “provide a one-time functional cure” for patients with severe sickle cell disease, according to Franco Locatelli, MD, of Sapienza University of Rome, who presented initial findings last year.
While the current approval is for patients with infusion-dependent sickle cell disease, exa-cel is also being evaluated in patients with another blood disorder, beta-thalassemia.
Lovo-cel
Lovo-cel, a cell-based gene therapy, uses a different technology — a lentiviral vector, or gene delivery vehicle — that can also genetically modify a patient’s blood stem cells.
Like exa-cel, lovo-cel is a one-time, single-dose infusion that contains the patient’s modified cells. Before the infusion, patients undergo myeloablative conditioning. The patient’s stem cells are then genetically modified to allow them to produce the most common form of hemoglobin, HbA
This approval was based on data from a single-arm, 24-month study in patients aged 12-50 years who had sickle cell disease and a history of vaso-occlusive events.
Overall, 88% of patients (28 of 32) achieved complete resolution of vaso-occlusive events 6-18 months after the infusion.
The most common side effects included stomatitis; febrile neutropenia; and low platelet, white blood cell, and red blood cell counts.
The FDA noted that hematologic cancer has occurred in patients treated with lovo-cel, and the label includes a black-box warning about the risk.
Dr. Brodsky noted, however, that “while these new gene therapies are potentially life-changing for individuals living with [sickle cell disease], they must be accessible to be effective.”
Access is a potential concern. Exa-cel and lovo-cel could cost about $2 million.
A version of this article appeared on Medscape.com.
Genetic therapies bring change to neurology clinics
PHOENIX – New therapies are on the horizon for genetic neuromuscular diseases, and this will raise both hopes for patients and challenges for neurologists. , according to Nicolas Madigan, MBBCh, PhD, who spoke at the 2023 annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine (AANEM).
“I think we will very soon be in a position to tell these patients that they might actually have a better treatment outcome with a genetic treatment than if they had a sporadic or inflammatory disorder,” said Dr. Madigan, who is an assistant professor of clinical research at Mayo Clinic, Rochester, N.Y.
To illustrate how genetic therapies are changing neurology practice, Dr. Madigan focused his talk on CMT neuropathy, which is the most common hereditary neuropathy and, as a result, has become a prime focus of gene therapy development. “In a city of about a million people, there will be 100-800 patients with one of these disorders,” said Dr. Madigan.
Case report illustrates a change in approach
There are more than 100 known genes that can contribute to CMT, but about 90% of patients harbor alterations in one of four genes: PMP22, GJB1, MFN2, and MPZ.
The trick is determining which patients are candidates for genetic testing, according to Dr. Madigan. He presented a case report of a 39-year-old woman who had experienced sensory symptoms for years, with a sudden exacerbation along with allodynia following COVID-19 vaccination. Her cerebrospinal fluid protein was high and outside electromyography indicated mild demyelinating neuropathy, consistent with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). After her insurance denied IVIG treatment, she received solumedrol, but her symptoms worsened and she was referred to Dr. Madigan.
After 6 months of methotrexate treatment, her sensory symptoms had not improved, and she was referred for genetic testing, which revealed a truncating mutation of the MPZ gene. “What I learned from this case really was that, in a young patient with conduction slowing, you might be considering CIDP. It might actually be better to do genetic testing first as opposed to starting inflammatory neuropathy type treatments with respect to cost – the genetic tests costs $300 versus tens of thousands of dollars for IVIG – and for [patient] welfare as well,” said Dr. Madigan.
Specifically, when clinical signs point to inherited neuropathy and there is conduction slowing, “the biggest bang for your buck might to be to go straight to PMP22 deletion or duplication testing and see if you can get a diagnosis. If that is negative or the clinical features are not as you might suspect, then, if you have other supportive features such as a very young age or there’s predominance of motor or sensory symptoms, you could test more broadly with a panel. If both of these are negative, then you could consider exome sequencing if the clinical phenotype really is consistent with that,” said Dr. Madigan.
The treatment landscape
With a diagnosis in hand, it’s possible to turn to treatment options, and the CMT landscape is promising. Dr. Madigan’s group recently reviewed 286 CMT clinical trials published between 1999 and 2022, 86% of which were interventions. Most were procedures based on carpal or cubital tunnel release, extracorporeal shockwave therapy, or nerve hydrodissection.
The small-molecule drug combination PXT3003 (Pharnext) – comprising baclofen, naltrexone, and sorbitol – downregulated PMP22 mRNA expression and led to improved myelination in animal models. It is currently being studied in a phase 2 clinical trial. Other approaches include supplements, stem cells, anesthetics, and various devices.
Genetic therapy is in the preclinical stage, including gene replacement using adeno-associated virus (AAV) vectors, gene silencing using antisense oligonucleotides or RNA interference, and gene editing using CRISPR-Cas 9 approaches.
Gene replacement strategies include delivering a normal copy of the gene, a supportive gene, or a gene that delays or reduces axon degeneration. Gene silencing targets PMP22, while CRISPR-Cas9 gene editing aims for PMP22 or neurofilament light polypeptide (NEFL) gene knockout.
The most clinically advanced AAV program delivers neurotrophin-3 via the viral vector to the target muscle, which has been demonstrated to improve symptoms in a mouse model using a muscle-specific promoter. A phase 1/2a trial will test the approach in three patients.
In the antisense space, chemical advances have improved the profile of the RNA, including modifications that influence inflammatory properties, stability, and targeting of specific tissues through conjugation to specific lipids, proteins, or antibodies. A 2018 study sponsored by DTxPharma showed that the formulation could improve outcomes and histologic myelination in a mouse model. In the wake of Novartis’s acquisition of the technology, Dr. Madigan anticipates that clinical trials will likely begin in 2024.
Finally, CRISPR-Cas9 targeting of a promoter region that leads to PMP22 transcription improved remyelination and electrophysiological parameters after injection into the sciatic nerve of mice.
A need for genetic counseling
Advances in testing and therapies represent exciting developments, but they also create a need for genetic counselors, according to Dr. Madigan. His clinic has two certified genetic counselors who meet with patients and discuss testing options, including risks and benefits to family members. The counselors also provide psychological support and assist in shared decision-making. They also handle testing paperwork, which eases the burden on physicians.
If the tests are negative, the genetic counselor informs the patient and lets them know of any additional testing required. In case of a positive test, the genetic counselor informs the patient, but the physician also makes contact to discuss clinical implications of the result. “I think it’s working extremely well, and I would encourage all practices to begin to explore those options moving forward,” said Dr. Madigan.
During the Q&A session after the talk, an audience member noted that genetic counselors are not covered by insurance, which places a financial burden on providers to hire them. He noted that his facility has a large clinical genomics department that was able to fund the two counselors, though they are both part-time. “It wasn’t easy. I think there was at least a year of trying to work out how to do it in terms of finding positions and negotiating, but I think once it’s accomplished it’s incredibly cost effective in terms of getting patients what they need from that perspective, and helping with the testing,” said Dr. Madigan.
Dr. Madigan reported no relevant financial disclosures.
PHOENIX – New therapies are on the horizon for genetic neuromuscular diseases, and this will raise both hopes for patients and challenges for neurologists. , according to Nicolas Madigan, MBBCh, PhD, who spoke at the 2023 annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine (AANEM).
“I think we will very soon be in a position to tell these patients that they might actually have a better treatment outcome with a genetic treatment than if they had a sporadic or inflammatory disorder,” said Dr. Madigan, who is an assistant professor of clinical research at Mayo Clinic, Rochester, N.Y.
To illustrate how genetic therapies are changing neurology practice, Dr. Madigan focused his talk on CMT neuropathy, which is the most common hereditary neuropathy and, as a result, has become a prime focus of gene therapy development. “In a city of about a million people, there will be 100-800 patients with one of these disorders,” said Dr. Madigan.
Case report illustrates a change in approach
There are more than 100 known genes that can contribute to CMT, but about 90% of patients harbor alterations in one of four genes: PMP22, GJB1, MFN2, and MPZ.
The trick is determining which patients are candidates for genetic testing, according to Dr. Madigan. He presented a case report of a 39-year-old woman who had experienced sensory symptoms for years, with a sudden exacerbation along with allodynia following COVID-19 vaccination. Her cerebrospinal fluid protein was high and outside electromyography indicated mild demyelinating neuropathy, consistent with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). After her insurance denied IVIG treatment, she received solumedrol, but her symptoms worsened and she was referred to Dr. Madigan.
After 6 months of methotrexate treatment, her sensory symptoms had not improved, and she was referred for genetic testing, which revealed a truncating mutation of the MPZ gene. “What I learned from this case really was that, in a young patient with conduction slowing, you might be considering CIDP. It might actually be better to do genetic testing first as opposed to starting inflammatory neuropathy type treatments with respect to cost – the genetic tests costs $300 versus tens of thousands of dollars for IVIG – and for [patient] welfare as well,” said Dr. Madigan.
Specifically, when clinical signs point to inherited neuropathy and there is conduction slowing, “the biggest bang for your buck might to be to go straight to PMP22 deletion or duplication testing and see if you can get a diagnosis. If that is negative or the clinical features are not as you might suspect, then, if you have other supportive features such as a very young age or there’s predominance of motor or sensory symptoms, you could test more broadly with a panel. If both of these are negative, then you could consider exome sequencing if the clinical phenotype really is consistent with that,” said Dr. Madigan.
The treatment landscape
With a diagnosis in hand, it’s possible to turn to treatment options, and the CMT landscape is promising. Dr. Madigan’s group recently reviewed 286 CMT clinical trials published between 1999 and 2022, 86% of which were interventions. Most were procedures based on carpal or cubital tunnel release, extracorporeal shockwave therapy, or nerve hydrodissection.
The small-molecule drug combination PXT3003 (Pharnext) – comprising baclofen, naltrexone, and sorbitol – downregulated PMP22 mRNA expression and led to improved myelination in animal models. It is currently being studied in a phase 2 clinical trial. Other approaches include supplements, stem cells, anesthetics, and various devices.
Genetic therapy is in the preclinical stage, including gene replacement using adeno-associated virus (AAV) vectors, gene silencing using antisense oligonucleotides or RNA interference, and gene editing using CRISPR-Cas 9 approaches.
Gene replacement strategies include delivering a normal copy of the gene, a supportive gene, or a gene that delays or reduces axon degeneration. Gene silencing targets PMP22, while CRISPR-Cas9 gene editing aims for PMP22 or neurofilament light polypeptide (NEFL) gene knockout.
The most clinically advanced AAV program delivers neurotrophin-3 via the viral vector to the target muscle, which has been demonstrated to improve symptoms in a mouse model using a muscle-specific promoter. A phase 1/2a trial will test the approach in three patients.
In the antisense space, chemical advances have improved the profile of the RNA, including modifications that influence inflammatory properties, stability, and targeting of specific tissues through conjugation to specific lipids, proteins, or antibodies. A 2018 study sponsored by DTxPharma showed that the formulation could improve outcomes and histologic myelination in a mouse model. In the wake of Novartis’s acquisition of the technology, Dr. Madigan anticipates that clinical trials will likely begin in 2024.
Finally, CRISPR-Cas9 targeting of a promoter region that leads to PMP22 transcription improved remyelination and electrophysiological parameters after injection into the sciatic nerve of mice.
A need for genetic counseling
Advances in testing and therapies represent exciting developments, but they also create a need for genetic counselors, according to Dr. Madigan. His clinic has two certified genetic counselors who meet with patients and discuss testing options, including risks and benefits to family members. The counselors also provide psychological support and assist in shared decision-making. They also handle testing paperwork, which eases the burden on physicians.
If the tests are negative, the genetic counselor informs the patient and lets them know of any additional testing required. In case of a positive test, the genetic counselor informs the patient, but the physician also makes contact to discuss clinical implications of the result. “I think it’s working extremely well, and I would encourage all practices to begin to explore those options moving forward,” said Dr. Madigan.
During the Q&A session after the talk, an audience member noted that genetic counselors are not covered by insurance, which places a financial burden on providers to hire them. He noted that his facility has a large clinical genomics department that was able to fund the two counselors, though they are both part-time. “It wasn’t easy. I think there was at least a year of trying to work out how to do it in terms of finding positions and negotiating, but I think once it’s accomplished it’s incredibly cost effective in terms of getting patients what they need from that perspective, and helping with the testing,” said Dr. Madigan.
Dr. Madigan reported no relevant financial disclosures.
PHOENIX – New therapies are on the horizon for genetic neuromuscular diseases, and this will raise both hopes for patients and challenges for neurologists. , according to Nicolas Madigan, MBBCh, PhD, who spoke at the 2023 annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine (AANEM).
“I think we will very soon be in a position to tell these patients that they might actually have a better treatment outcome with a genetic treatment than if they had a sporadic or inflammatory disorder,” said Dr. Madigan, who is an assistant professor of clinical research at Mayo Clinic, Rochester, N.Y.
To illustrate how genetic therapies are changing neurology practice, Dr. Madigan focused his talk on CMT neuropathy, which is the most common hereditary neuropathy and, as a result, has become a prime focus of gene therapy development. “In a city of about a million people, there will be 100-800 patients with one of these disorders,” said Dr. Madigan.
Case report illustrates a change in approach
There are more than 100 known genes that can contribute to CMT, but about 90% of patients harbor alterations in one of four genes: PMP22, GJB1, MFN2, and MPZ.
The trick is determining which patients are candidates for genetic testing, according to Dr. Madigan. He presented a case report of a 39-year-old woman who had experienced sensory symptoms for years, with a sudden exacerbation along with allodynia following COVID-19 vaccination. Her cerebrospinal fluid protein was high and outside electromyography indicated mild demyelinating neuropathy, consistent with chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). After her insurance denied IVIG treatment, she received solumedrol, but her symptoms worsened and she was referred to Dr. Madigan.
After 6 months of methotrexate treatment, her sensory symptoms had not improved, and she was referred for genetic testing, which revealed a truncating mutation of the MPZ gene. “What I learned from this case really was that, in a young patient with conduction slowing, you might be considering CIDP. It might actually be better to do genetic testing first as opposed to starting inflammatory neuropathy type treatments with respect to cost – the genetic tests costs $300 versus tens of thousands of dollars for IVIG – and for [patient] welfare as well,” said Dr. Madigan.
Specifically, when clinical signs point to inherited neuropathy and there is conduction slowing, “the biggest bang for your buck might to be to go straight to PMP22 deletion or duplication testing and see if you can get a diagnosis. If that is negative or the clinical features are not as you might suspect, then, if you have other supportive features such as a very young age or there’s predominance of motor or sensory symptoms, you could test more broadly with a panel. If both of these are negative, then you could consider exome sequencing if the clinical phenotype really is consistent with that,” said Dr. Madigan.
The treatment landscape
With a diagnosis in hand, it’s possible to turn to treatment options, and the CMT landscape is promising. Dr. Madigan’s group recently reviewed 286 CMT clinical trials published between 1999 and 2022, 86% of which were interventions. Most were procedures based on carpal or cubital tunnel release, extracorporeal shockwave therapy, or nerve hydrodissection.
The small-molecule drug combination PXT3003 (Pharnext) – comprising baclofen, naltrexone, and sorbitol – downregulated PMP22 mRNA expression and led to improved myelination in animal models. It is currently being studied in a phase 2 clinical trial. Other approaches include supplements, stem cells, anesthetics, and various devices.
Genetic therapy is in the preclinical stage, including gene replacement using adeno-associated virus (AAV) vectors, gene silencing using antisense oligonucleotides or RNA interference, and gene editing using CRISPR-Cas 9 approaches.
Gene replacement strategies include delivering a normal copy of the gene, a supportive gene, or a gene that delays or reduces axon degeneration. Gene silencing targets PMP22, while CRISPR-Cas9 gene editing aims for PMP22 or neurofilament light polypeptide (NEFL) gene knockout.
The most clinically advanced AAV program delivers neurotrophin-3 via the viral vector to the target muscle, which has been demonstrated to improve symptoms in a mouse model using a muscle-specific promoter. A phase 1/2a trial will test the approach in three patients.
In the antisense space, chemical advances have improved the profile of the RNA, including modifications that influence inflammatory properties, stability, and targeting of specific tissues through conjugation to specific lipids, proteins, or antibodies. A 2018 study sponsored by DTxPharma showed that the formulation could improve outcomes and histologic myelination in a mouse model. In the wake of Novartis’s acquisition of the technology, Dr. Madigan anticipates that clinical trials will likely begin in 2024.
Finally, CRISPR-Cas9 targeting of a promoter region that leads to PMP22 transcription improved remyelination and electrophysiological parameters after injection into the sciatic nerve of mice.
A need for genetic counseling
Advances in testing and therapies represent exciting developments, but they also create a need for genetic counselors, according to Dr. Madigan. His clinic has two certified genetic counselors who meet with patients and discuss testing options, including risks and benefits to family members. The counselors also provide psychological support and assist in shared decision-making. They also handle testing paperwork, which eases the burden on physicians.
If the tests are negative, the genetic counselor informs the patient and lets them know of any additional testing required. In case of a positive test, the genetic counselor informs the patient, but the physician also makes contact to discuss clinical implications of the result. “I think it’s working extremely well, and I would encourage all practices to begin to explore those options moving forward,” said Dr. Madigan.
During the Q&A session after the talk, an audience member noted that genetic counselors are not covered by insurance, which places a financial burden on providers to hire them. He noted that his facility has a large clinical genomics department that was able to fund the two counselors, though they are both part-time. “It wasn’t easy. I think there was at least a year of trying to work out how to do it in terms of finding positions and negotiating, but I think once it’s accomplished it’s incredibly cost effective in terms of getting patients what they need from that perspective, and helping with the testing,” said Dr. Madigan.
Dr. Madigan reported no relevant financial disclosures.
At AANEM 2023
Why genetic testing may be our best shot at progress in Parkinson’s disease
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
In 2017, Sanofi Genzyme launched a phase 2 clinical trial of a drug designed to target a specific genetic mutation in some patients with Parkinson’s disease. Researchers hoped the drug would slow or even stop disease progression.
Like many before it, the trial yielded disappointing results and the company shut it down in 2021. It was the latest in a string of unsuccessful clinical trials testing disease-modifying Parkinson’s disease drugs.
Although it failed, the Sanofi Genzyme study was different: It was the first to enroll patients with Parkinson’s disease who had a specific genotype and marked the earliest days of precision medicine and gene-specific drug development for the disease.
Once thought to play only a small role in a small number of patients with Parkinson’s disease,
“We’re about to enter this era of precision medicine for Parkinson’s disease, which makes genetic testing important,” said James Beck, PhD, senior vice president and chief scientific officer for the Parkinson’s Foundation.
“A number of companies have clinical trials or are in preparation for clinical trials to test some specific therapies that would depend upon people having a specific genetic mutation,” he said.
Today, at least four clinical trials of drugs that target specific Parkinson’s disease-related gene variants on LRRK2 and GBA are under way, and more are in the pipeline. Whether these drugs will be effective at modifying the course of the disease remains to be seen. First, the trials must enroll enough patients. And therein lies the challenge: Genetic testing isn’t part of routine Parkinson’s disease care and isn’t covered by most insurance policies. Most patients don’t know their genotype.
It’s a significant roadblock to the future of a precision medicine approach that is based on a patient’s individual genotype, which some experts argue offers the best shot at slowing disease progression.
“To enroll in clinical trials for precision drugs people with Parkinson’s disease have to be aware of their genetic status,” said Roy N. Alcalay, MD, chief of the movement disorders division at Tel Aviv Medical Center in Israel and part-time associate professor at Columbia University in New York. “How can a person with Parkinson’s and a LRRK2 mutation join a precision medicine trial for LRRK2 if she does not know she is a LRRK2 carrier?”
Free genetic testing
Previous studies have shown that some genetic variants increase the risk for Parkinson’s disease after exposure to environmental factors such as pesticides. Research has also shown that a patient’s genotype can predict survival time and that certain medications may prove more effective at slowing disease progression in patients with specific genotypes. All of this points to a significant role for genetics in a disorder that is rapidly increasing.
This makes expanding patient access to genetic testing even more important, Dr. Alcalay said, noting that it’s equally important that patients are informed of their genotype, something that doesn’t usually happen in blinded clinical trials.
To that end, Dr. Alcalay hopes a national genetics study he is leading will address access and need-to-know issues. PD GENEration, a project launched in 2019 by the Parkinson’s Foundation, offers patients free genetic testing for seven clinically relevant Parkinson’s disease-related genes.
Testing is done at home or in a nearby clinic and the results are shared with patients during a free genetic counseling session and with site investigators. Patient samples are stored in a genetic data bank that is open to researchers around the world.
“We surveyed clinical trialists in the Parkinson’s disease field prior to initiation of PD GENEration and estimated that over 90% of people with Parkinson’s disease prior to the effort were not aware of their genetic status,” Dr. Alcalay said.
“I think precision medicine in Parkinson’s disease will not happen without PD GENEration or similar efforts.”
‘Overwhelming’ patient interest
Participants in the study are screened for variants in seven genes known to be involved in Parkinson’s disease risk: GBA, LRRK2, PRKN, PINK1, SNCA, PARK7, and VPS35.
In less than 3 years, the study has already produced what is thought to be the largest genetic data bank of sequenced sets of Parkinson’s disease-risk genes made accessible to patients. Since the end of 2020, the first year of patient enrollment, the number of participants has increased from 676 to 10,515 and the number of participating clinical sites rose from 12 to 101.
The foundation has spent nearly $20 million on the project so far and plans to spend another $10 million to reach a goal of 15,000 patients. The study, which is funded by private donors, is so successful that the foundation has had to scale back enrollment.
“When we were at a peak, we had over 700 participants enrolling each month,” Dr. Beck said. Beginning in April, the program capped new sign-ups to 200 patients per month and created a waiting list for future enrollment. The waiting list is hundreds of patients long.
“The participants’ response to enroll in PD GENEration demonstrates there is an overwhelming interest by people with Parkinson’s disease to learn more about their genetic risk factors,” Dr. Alcalay said.
A research driver
Nearly 60% of participants enrolled so far are male and close to 80% are White. The average age is 69 years and 44% were diagnosed in the past 5 years. Close to 75% had never participated in a clinical trial.
Nearly 13% have tested positive for mutations on at least one of the seven target genes. Previous studies had suggested genetics were involved in only about 10% of cases.
The majority of those with positive results had early-onset Parkinson’s disease, high-risk ancestry, or a first-degree relative with the disease. However, 9% of people who tested positive weren’t in any of those categories.
Genetic information collected by the project is shared with the Global Parkinson’s Genetics Program (GP2), a resource program of the Aligning Science Across Parkinson’s initiative that is focused on the disease’s genetic architecture. Researchers around the world have access to GP2 data to study known gene variants and identify new ones.
PD GENEration participants can choose to be notified if they are carriers of gene variants discovered in the future.
“All DNA samples shared by participants are undergoing research-grade testing,” Dr. Beck said. “Not only do we want to be able to inform people with Parkinson’s disease about their genetic status, but we also want to be able to use this precious resource to further drive research into the genetics of Parkinson’s disease.”
Early success
Patient recruitment has long been one of the biggest challenges to any clinical trial’s success. Research suggests that 90% of all clinical trials fail to reach recruitment milestones in their allotted time frame and two-thirds of multicenter trials fold because too few patients sign up. Data from the Parkinson’s Foundation show that only about 1% of all patients with Parkinson’s disease participate in clinical trials.
Increasing those numbers is the primary goal of PD GENEration, Dr. Beck said. And there’s evidence it’s already paying off.
Earlier this year, one of the program’s participating clinical sites, Intermountain Health, in Salt Lake City, Utah, joined a phase 2 clinical trial of an experimental drug that targets a mutation on the GBA1 gene.
“One of the reasons we were able to participate was when we got the call about joining, we were able to say that we had patients with that specific gene mutation, and we could only say that because the patients had been genotyped through PD GENEration,” said Kathleen E. McKee, MD, director of movement disorders, associate medical director of neurosciences research, and PD GENEration principal investigator at Intermountain Health.
Since 2021, Dr. McKee has enrolled hundreds of patients in the foundation’s gene study and hopes to enroll even more. Few patients turn down the opportunity to participate, she added. Knowing their genotype has proven empowering for her patients, most of whom could not afford genetic testing on their own.
“Previously I would tell patients this is not going to change your immediate management,” Dr. McKee said. “Now I tell my patients that these trials are out there, it may actually change how I treat you and what I recommend.”
A version of this article appeared on Medscape.com.
DLBCL: Major new treatment breakthroughs
Significant breakthroughs have come in just the past few weeks and months, through the use of CAR T-cell and immunotherapies and with the approval in April by the Food and Drug Administration of polatuzumab for frontline DLBCL.
“Until the publishing of data from the POLARIX study (NCT03274492), which led to the approval of polatuzumab vedotin plus rituximab-cyclophosphamide, doxorubicin, and prednisone (pola + R-CHP), we had not had a breakthrough in frontline DLBCL therapies since the addition of rituximab 22 years ago,” said Dr. Charalambos Andreadis, MD, of the University of California at San Francisco’s Helen Diller Family Comprehensive Cancer Center.

“Pola + R-CHP is an improvement over the standard-of-care treatment, R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine, and prednisone), giving treatment naive patients an increase in PFS without an increase in side effects,” Dr. Andreadis said.
R-CHP-polatuzumab was approved only for patients with an International Prognostic Indices score between 2 and 5, leaving patients with IPI scores of 0 or 1 with the frontline standard of care (SoC) treatment of R-CHOP, which has a cure rate of between 60% and 70%.
“The highest likelihood of relapse is in the first year following treatment. After 2 years in remission, patients’ chance of relapsing is the same as the general populations’ chance of getting DLBCL for the first time. This is why even a slight increase in the progression-free survival rate with the addition of pola is so significant,” Dr. Andreadis noted.
Historically, patients with relapsed or refractory (RR) DLBCL who did not respond to R-CHOP or who experienced disease relapse less than a year after primary intervention were treated with alternative chemotherapy regimens, often followed by autologous stem cell transplants (ASCT). Randomized control studies have shown that CAR T-cell therapies yield higher success rates than chemotherapy and ASCT, leading to the SoC in RR patients being CAR-T cell therapy directly following failed primary treatment.
“There are many new CAR T-cell platforms in development, as well as novel combination strategies that aim to target critical genetic pathways,” Kieron Dunleavy, MD, professor of medicine at the Lombardi Comprehensive Cancer Center at Georgetown University Hospital, said in an interview. “While access to CAR T-cell therapies is becoming easier and more feasible in many centers, fast access continues to be an issue for many patients, often depending on geography and socioeconomic factors.”
Asked about the latest breakthroughs in treating DLBCL, Dr. Dunleavy said, “A significant proportion of patients with relapsed or refractory DLBCL do not have easy access to CAR T-cell therapies, so this needs to be addressed and improved. Sometimes the rapidity of clinical progression in DLBCL can make these therapies challenging to deliver, considering logistical issues like apheresis and insurance approvals, which are frequently complex. This highlights the need for alternative and ‘easier to deliver’ CAR-T cells and our continued prioritization of developing alternative effective agents for DLBCL.
“Currently, commercially approved CAR T-cells in DLBCL target the CD-19 marker on lymphoma cells but CAR T-cells targeting other and more than one antigen as well as alternative anti CD19 agents like loncastuximab and tafasitamab are similarly FDA approved and available for patients,” Dr. Dunleavy concluded.
Dr. Dunleavy is affiliated with the MedStar Georgetown Lymphoma group, where Rep. Raskin publicly announced that he had completed 4 months of chemotherapy treatment for DLBCL. On April 27, in an open letter to the U.S. public, he wrote that he rang the bell at MedStar to mark his preliminary diagnosis of being “in remission,” with a “90% prognosis of no relapse.”
Interviewed about the latest advances in treating DLBCL, Jason Westin, MD, associate professor of lymphoma and myeloma at the MD Anderson Cancer Center in Houston, said that even with improvements in overall survival possible with CAR T-cell therapies, “usually, a clinical trial should be considered strongly, as it is often the best option for patients, both in a newly diagnosed or in a relapsed setting, as they allow access to tomorrow’s breakthrough therapies today.”

Dr. Westin cited the example of bispecific T-cell engagers (BITE) as a promising therapy that is available to patients in clinical trials. These agents bind to one side to the lymphoma cell, but they also have a binding arm for T-cells, so they activate a patient’s own immune cells to kill lymphoma cells, in some cases offering a cure when CAR T-cell therapy has failed.
The first BITE to be approved, mosunetuzumab, is authorized only for the treatment of follicular lymphoma. However, data from a recent clinical study indicated that the agent yields complete responses in 24% of heavily pretreated patients with RR DLBCL.
Another BITE, glofitamab, was approved in Canada in March 2023 for use in RR DLBCL. Based on its high efficacy, it soon may be approved elsewhere.
Dr. Andreadis noted, “We are finally at a point where for both treatment naive and RR DLBCL patients, there are several promising options on the horizon that don’t involve ASCT. Furthermore, these breakthroughs reinforce each other, as there are studies in which therapies like BITE are being brought to the front line and pola to RR cases.”
The growing field of new frontline and RR DLBCL therapies lend credence to the optimism of specialists who treat DLBCL – and to the sanguine note that Congressman Raskin struck in published comments about his treatment for DLBCL.
Dr. Andreadis reported ties with BMS, Novartis, Roche, Genmab, Merck, Gilead, AbbVie, and J&J. Dr. Dunleavy disclosed relationships with ONO Pharmaceuticals, Kymera, Merck, Genentech, AstraZeneca, Amgen, ADC Therapeutics, MorphoSys and Incyte, Kite/Gilead, Cellectar. Dr. Westin reported ties with Kite/Gilead, BMS, Novartis, Genentech, AstraZeneca, Morphosys/Incyte, ADC Therapeutics, Kymera, Nurix, and MonteRosa.
Significant breakthroughs have come in just the past few weeks and months, through the use of CAR T-cell and immunotherapies and with the approval in April by the Food and Drug Administration of polatuzumab for frontline DLBCL.
“Until the publishing of data from the POLARIX study (NCT03274492), which led to the approval of polatuzumab vedotin plus rituximab-cyclophosphamide, doxorubicin, and prednisone (pola + R-CHP), we had not had a breakthrough in frontline DLBCL therapies since the addition of rituximab 22 years ago,” said Dr. Charalambos Andreadis, MD, of the University of California at San Francisco’s Helen Diller Family Comprehensive Cancer Center.
“Pola + R-CHP is an improvement over the standard-of-care treatment, R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine, and prednisone), giving treatment naive patients an increase in PFS without an increase in side effects,” Dr. Andreadis said.
R-CHP-polatuzumab was approved only for patients with an International Prognostic Indices score between 2 and 5, leaving patients with IPI scores of 0 or 1 with the frontline standard of care (SoC) treatment of R-CHOP, which has a cure rate of between 60% and 70%.
“The highest likelihood of relapse is in the first year following treatment. After 2 years in remission, patients’ chance of relapsing is the same as the general populations’ chance of getting DLBCL for the first time. This is why even a slight increase in the progression-free survival rate with the addition of pola is so significant,” Dr. Andreadis noted.
Historically, patients with relapsed or refractory (RR) DLBCL who did not respond to R-CHOP or who experienced disease relapse less than a year after primary intervention were treated with alternative chemotherapy regimens, often followed by autologous stem cell transplants (ASCT). Randomized control studies have shown that CAR T-cell therapies yield higher success rates than chemotherapy and ASCT, leading to the SoC in RR patients being CAR-T cell therapy directly following failed primary treatment.
“There are many new CAR T-cell platforms in development, as well as novel combination strategies that aim to target critical genetic pathways,” Kieron Dunleavy, MD, professor of medicine at the Lombardi Comprehensive Cancer Center at Georgetown University Hospital, said in an interview. “While access to CAR T-cell therapies is becoming easier and more feasible in many centers, fast access continues to be an issue for many patients, often depending on geography and socioeconomic factors.”
Asked about the latest breakthroughs in treating DLBCL, Dr. Dunleavy said, “A significant proportion of patients with relapsed or refractory DLBCL do not have easy access to CAR T-cell therapies, so this needs to be addressed and improved. Sometimes the rapidity of clinical progression in DLBCL can make these therapies challenging to deliver, considering logistical issues like apheresis and insurance approvals, which are frequently complex. This highlights the need for alternative and ‘easier to deliver’ CAR-T cells and our continued prioritization of developing alternative effective agents for DLBCL.
“Currently, commercially approved CAR T-cells in DLBCL target the CD-19 marker on lymphoma cells but CAR T-cells targeting other and more than one antigen as well as alternative anti CD19 agents like loncastuximab and tafasitamab are similarly FDA approved and available for patients,” Dr. Dunleavy concluded.
Dr. Dunleavy is affiliated with the MedStar Georgetown Lymphoma group, where Rep. Raskin publicly announced that he had completed 4 months of chemotherapy treatment for DLBCL. On April 27, in an open letter to the U.S. public, he wrote that he rang the bell at MedStar to mark his preliminary diagnosis of being “in remission,” with a “90% prognosis of no relapse.”
Interviewed about the latest advances in treating DLBCL, Jason Westin, MD, associate professor of lymphoma and myeloma at the MD Anderson Cancer Center in Houston, said that even with improvements in overall survival possible with CAR T-cell therapies, “usually, a clinical trial should be considered strongly, as it is often the best option for patients, both in a newly diagnosed or in a relapsed setting, as they allow access to tomorrow’s breakthrough therapies today.”
Dr. Westin cited the example of bispecific T-cell engagers (BITE) as a promising therapy that is available to patients in clinical trials. These agents bind to one side to the lymphoma cell, but they also have a binding arm for T-cells, so they activate a patient’s own immune cells to kill lymphoma cells, in some cases offering a cure when CAR T-cell therapy has failed.
The first BITE to be approved, mosunetuzumab, is authorized only for the treatment of follicular lymphoma. However, data from a recent clinical study indicated that the agent yields complete responses in 24% of heavily pretreated patients with RR DLBCL.
Another BITE, glofitamab, was approved in Canada in March 2023 for use in RR DLBCL. Based on its high efficacy, it soon may be approved elsewhere.
Dr. Andreadis noted, “We are finally at a point where for both treatment naive and RR DLBCL patients, there are several promising options on the horizon that don’t involve ASCT. Furthermore, these breakthroughs reinforce each other, as there are studies in which therapies like BITE are being brought to the front line and pola to RR cases.”
The growing field of new frontline and RR DLBCL therapies lend credence to the optimism of specialists who treat DLBCL – and to the sanguine note that Congressman Raskin struck in published comments about his treatment for DLBCL.
Dr. Andreadis reported ties with BMS, Novartis, Roche, Genmab, Merck, Gilead, AbbVie, and J&J. Dr. Dunleavy disclosed relationships with ONO Pharmaceuticals, Kymera, Merck, Genentech, AstraZeneca, Amgen, ADC Therapeutics, MorphoSys and Incyte, Kite/Gilead, Cellectar. Dr. Westin reported ties with Kite/Gilead, BMS, Novartis, Genentech, AstraZeneca, Morphosys/Incyte, ADC Therapeutics, Kymera, Nurix, and MonteRosa.
Significant breakthroughs have come in just the past few weeks and months, through the use of CAR T-cell and immunotherapies and with the approval in April by the Food and Drug Administration of polatuzumab for frontline DLBCL.
“Until the publishing of data from the POLARIX study (NCT03274492), which led to the approval of polatuzumab vedotin plus rituximab-cyclophosphamide, doxorubicin, and prednisone (pola + R-CHP), we had not had a breakthrough in frontline DLBCL therapies since the addition of rituximab 22 years ago,” said Dr. Charalambos Andreadis, MD, of the University of California at San Francisco’s Helen Diller Family Comprehensive Cancer Center.
“Pola + R-CHP is an improvement over the standard-of-care treatment, R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine, and prednisone), giving treatment naive patients an increase in PFS without an increase in side effects,” Dr. Andreadis said.
R-CHP-polatuzumab was approved only for patients with an International Prognostic Indices score between 2 and 5, leaving patients with IPI scores of 0 or 1 with the frontline standard of care (SoC) treatment of R-CHOP, which has a cure rate of between 60% and 70%.
“The highest likelihood of relapse is in the first year following treatment. After 2 years in remission, patients’ chance of relapsing is the same as the general populations’ chance of getting DLBCL for the first time. This is why even a slight increase in the progression-free survival rate with the addition of pola is so significant,” Dr. Andreadis noted.
Historically, patients with relapsed or refractory (RR) DLBCL who did not respond to R-CHOP or who experienced disease relapse less than a year after primary intervention were treated with alternative chemotherapy regimens, often followed by autologous stem cell transplants (ASCT). Randomized control studies have shown that CAR T-cell therapies yield higher success rates than chemotherapy and ASCT, leading to the SoC in RR patients being CAR-T cell therapy directly following failed primary treatment.
“There are many new CAR T-cell platforms in development, as well as novel combination strategies that aim to target critical genetic pathways,” Kieron Dunleavy, MD, professor of medicine at the Lombardi Comprehensive Cancer Center at Georgetown University Hospital, said in an interview. “While access to CAR T-cell therapies is becoming easier and more feasible in many centers, fast access continues to be an issue for many patients, often depending on geography and socioeconomic factors.”
Asked about the latest breakthroughs in treating DLBCL, Dr. Dunleavy said, “A significant proportion of patients with relapsed or refractory DLBCL do not have easy access to CAR T-cell therapies, so this needs to be addressed and improved. Sometimes the rapidity of clinical progression in DLBCL can make these therapies challenging to deliver, considering logistical issues like apheresis and insurance approvals, which are frequently complex. This highlights the need for alternative and ‘easier to deliver’ CAR-T cells and our continued prioritization of developing alternative effective agents for DLBCL.
“Currently, commercially approved CAR T-cells in DLBCL target the CD-19 marker on lymphoma cells but CAR T-cells targeting other and more than one antigen as well as alternative anti CD19 agents like loncastuximab and tafasitamab are similarly FDA approved and available for patients,” Dr. Dunleavy concluded.
Dr. Dunleavy is affiliated with the MedStar Georgetown Lymphoma group, where Rep. Raskin publicly announced that he had completed 4 months of chemotherapy treatment for DLBCL. On April 27, in an open letter to the U.S. public, he wrote that he rang the bell at MedStar to mark his preliminary diagnosis of being “in remission,” with a “90% prognosis of no relapse.”
Interviewed about the latest advances in treating DLBCL, Jason Westin, MD, associate professor of lymphoma and myeloma at the MD Anderson Cancer Center in Houston, said that even with improvements in overall survival possible with CAR T-cell therapies, “usually, a clinical trial should be considered strongly, as it is often the best option for patients, both in a newly diagnosed or in a relapsed setting, as they allow access to tomorrow’s breakthrough therapies today.”
Dr. Westin cited the example of bispecific T-cell engagers (BITE) as a promising therapy that is available to patients in clinical trials. These agents bind to one side to the lymphoma cell, but they also have a binding arm for T-cells, so they activate a patient’s own immune cells to kill lymphoma cells, in some cases offering a cure when CAR T-cell therapy has failed.
The first BITE to be approved, mosunetuzumab, is authorized only for the treatment of follicular lymphoma. However, data from a recent clinical study indicated that the agent yields complete responses in 24% of heavily pretreated patients with RR DLBCL.
Another BITE, glofitamab, was approved in Canada in March 2023 for use in RR DLBCL. Based on its high efficacy, it soon may be approved elsewhere.
Dr. Andreadis noted, “We are finally at a point where for both treatment naive and RR DLBCL patients, there are several promising options on the horizon that don’t involve ASCT. Furthermore, these breakthroughs reinforce each other, as there are studies in which therapies like BITE are being brought to the front line and pola to RR cases.”
The growing field of new frontline and RR DLBCL therapies lend credence to the optimism of specialists who treat DLBCL – and to the sanguine note that Congressman Raskin struck in published comments about his treatment for DLBCL.
Dr. Andreadis reported ties with BMS, Novartis, Roche, Genmab, Merck, Gilead, AbbVie, and J&J. Dr. Dunleavy disclosed relationships with ONO Pharmaceuticals, Kymera, Merck, Genentech, AstraZeneca, Amgen, ADC Therapeutics, MorphoSys and Incyte, Kite/Gilead, Cellectar. Dr. Westin reported ties with Kite/Gilead, BMS, Novartis, Genentech, AstraZeneca, Morphosys/Incyte, ADC Therapeutics, Kymera, Nurix, and MonteRosa.
Hemophilia A gene therapy under FDA review
Hemophilia A (a deficiency of clotting Factor X) is the most common form of the disease, accounting for about 85% of patients.
The other type is hemophilia B (deficiency of clotting Factor VIII), and a gene therapy for this form of the disease has recently been launched – etranacogene dezaparvovec (Hemgenix), at the enormous price tag of $3.5 million.
Both products are comprised of a one-off intravenous IV infusion that delivers a functional gene via an adeno-associated virus that instructs the body to make the missing clotting factor. The hope is that this one-off infusion will act as a ‘cure’ and that the individual will be freed from life-long prophylaxis and/or treatment.
The new clinical data on valoctocogene roxaparvovec, published online in the New England Journal of Medicine, show that the beneficial effects from the gene are largely durable at 2 years, but they are anticipated to fade with time.
Two years after the one-time infusion, there remained “a significant reduction in the annualized bleeding rates” among 132 men who, at baseline, had severe hemophilia A requiring ongoing factor VIII prophylaxis, said the investigators, led by hematologist Johnny Mahlangu, MBBCh, MMed, of the University of the Witwatersrand, Johannesburg, South Africa.
However, the team predicted that median factor VIII activity would decrease below 10% of normal by year 3 or 5 depending on measurement technique, which would still translate to mild disease with an annualized bleeding rate of less than 1 episode per year.
“Although valoctocogene roxaparvovec may not eliminate bleeding, it potentially provides more consistent protection than factor VIII prophylaxis with less treatment burden,” the team said.
New questions
Data from the study “will directly inform therapeutic decision-making” in Europe, where valoctocogene roxaparvovec is already conditionally approved, and the United States, where it is awaiting approval by the FDA, says Lindsey George, MD, a hematologist and gene therapy specialist at Children’s Hospital of Philadelphia, in an accompanying editorial.
The study speaks to an ongoing concern about the durability of gene therapy for hemophilia but also raises new questions, she said.
For instance, while some patients had normal Factor VIII production and activity at 2 years, activity had dropped substantially in others, including in six men who resumed prophylaxis. “The cause of the decrease in factor VIII expression is an unanswered question,” and despite an anticipated U.S. price tag of around $2.5 million per treatment, “it is not possible [at the moment] to predict where an individual patient may fall within this range,” she writes.
Also, some subjects had elevations in liver aminotransferase levels that lasted for several months, including 2 years after infusion in 29% of subjects. Elevations in liver aminotransferase levels were treated with immune suppression for a median of 33 weeks.
“This is a unique finding with an undefined cause and long-term safety implications,” Dr. George said.
Getting to the bottom of such issues will be necessary for hemophilia gene therapy to fulfill its promise as “a one-time, lifelong, disease-ameliorating” fix for the condition, she asserted.
Study details
The new report followed up on the initial trial in 134 men who were treated with a single infusion of 6 × 1013 vector genomes per kilogram of body weight.
Among the 132 subjects available for 2-year evaluation, median factor VIII activity was in the range of mild hemophilia (6%-49% of normal) with an 84.5% reduction in bleeding events from baseline.
More than 80% of participants had no bleeding events requiring treatment, and there was a 98% reduction from baseline in mean use of exogenous factor VIII.
Overall, at year 2, 4.5% of subjects had factor VIII activity consistent with severe hemophilia A; 9.1% had activity consistent with moderate disease; 59.8% had activity consistent with mild disease; and 26.5% had activity in the normal range above 40 IU/dL. The investigators estimated that the typical half-life of the transgene-derived factor VIII production system is 123 weeks.
Among the six men who resumed prophylaxis, most had fewer bleeding events than when they were on prophylaxis before the infusion, investigators noted.
All the subjects developed antibodies to the virus delivery vector, precluding retreatment.
The work was funded by valoctocogene roxaparvovec maker BioMarin Pharmaceuticals. Several investigators are employees. Others reported ties to BioMarin and other companies; Dr. Mahlangu, for instance, reported research grants from BioMarin, Roche, Novo Nordisk, Pfizer, and others. Dr. George reported a research grant from Asklepios Biopharmaceutical and having a patent licensed to the company. The full list of author disclosures can be found with the original article.
A version of this article first appeared on Medscape.com.
Hemophilia A (a deficiency of clotting Factor X) is the most common form of the disease, accounting for about 85% of patients.
The other type is hemophilia B (deficiency of clotting Factor VIII), and a gene therapy for this form of the disease has recently been launched – etranacogene dezaparvovec (Hemgenix), at the enormous price tag of $3.5 million.
Both products are comprised of a one-off intravenous IV infusion that delivers a functional gene via an adeno-associated virus that instructs the body to make the missing clotting factor. The hope is that this one-off infusion will act as a ‘cure’ and that the individual will be freed from life-long prophylaxis and/or treatment.
The new clinical data on valoctocogene roxaparvovec, published online in the New England Journal of Medicine, show that the beneficial effects from the gene are largely durable at 2 years, but they are anticipated to fade with time.
Two years after the one-time infusion, there remained “a significant reduction in the annualized bleeding rates” among 132 men who, at baseline, had severe hemophilia A requiring ongoing factor VIII prophylaxis, said the investigators, led by hematologist Johnny Mahlangu, MBBCh, MMed, of the University of the Witwatersrand, Johannesburg, South Africa.
However, the team predicted that median factor VIII activity would decrease below 10% of normal by year 3 or 5 depending on measurement technique, which would still translate to mild disease with an annualized bleeding rate of less than 1 episode per year.
“Although valoctocogene roxaparvovec may not eliminate bleeding, it potentially provides more consistent protection than factor VIII prophylaxis with less treatment burden,” the team said.
New questions
Data from the study “will directly inform therapeutic decision-making” in Europe, where valoctocogene roxaparvovec is already conditionally approved, and the United States, where it is awaiting approval by the FDA, says Lindsey George, MD, a hematologist and gene therapy specialist at Children’s Hospital of Philadelphia, in an accompanying editorial.
The study speaks to an ongoing concern about the durability of gene therapy for hemophilia but also raises new questions, she said.
For instance, while some patients had normal Factor VIII production and activity at 2 years, activity had dropped substantially in others, including in six men who resumed prophylaxis. “The cause of the decrease in factor VIII expression is an unanswered question,” and despite an anticipated U.S. price tag of around $2.5 million per treatment, “it is not possible [at the moment] to predict where an individual patient may fall within this range,” she writes.
Also, some subjects had elevations in liver aminotransferase levels that lasted for several months, including 2 years after infusion in 29% of subjects. Elevations in liver aminotransferase levels were treated with immune suppression for a median of 33 weeks.
“This is a unique finding with an undefined cause and long-term safety implications,” Dr. George said.
Getting to the bottom of such issues will be necessary for hemophilia gene therapy to fulfill its promise as “a one-time, lifelong, disease-ameliorating” fix for the condition, she asserted.
Study details
The new report followed up on the initial trial in 134 men who were treated with a single infusion of 6 × 1013 vector genomes per kilogram of body weight.
Among the 132 subjects available for 2-year evaluation, median factor VIII activity was in the range of mild hemophilia (6%-49% of normal) with an 84.5% reduction in bleeding events from baseline.
More than 80% of participants had no bleeding events requiring treatment, and there was a 98% reduction from baseline in mean use of exogenous factor VIII.
Overall, at year 2, 4.5% of subjects had factor VIII activity consistent with severe hemophilia A; 9.1% had activity consistent with moderate disease; 59.8% had activity consistent with mild disease; and 26.5% had activity in the normal range above 40 IU/dL. The investigators estimated that the typical half-life of the transgene-derived factor VIII production system is 123 weeks.
Among the six men who resumed prophylaxis, most had fewer bleeding events than when they were on prophylaxis before the infusion, investigators noted.
All the subjects developed antibodies to the virus delivery vector, precluding retreatment.
The work was funded by valoctocogene roxaparvovec maker BioMarin Pharmaceuticals. Several investigators are employees. Others reported ties to BioMarin and other companies; Dr. Mahlangu, for instance, reported research grants from BioMarin, Roche, Novo Nordisk, Pfizer, and others. Dr. George reported a research grant from Asklepios Biopharmaceutical and having a patent licensed to the company. The full list of author disclosures can be found with the original article.
A version of this article first appeared on Medscape.com.
Hemophilia A (a deficiency of clotting Factor X) is the most common form of the disease, accounting for about 85% of patients.
The other type is hemophilia B (deficiency of clotting Factor VIII), and a gene therapy for this form of the disease has recently been launched – etranacogene dezaparvovec (Hemgenix), at the enormous price tag of $3.5 million.
Both products are comprised of a one-off intravenous IV infusion that delivers a functional gene via an adeno-associated virus that instructs the body to make the missing clotting factor. The hope is that this one-off infusion will act as a ‘cure’ and that the individual will be freed from life-long prophylaxis and/or treatment.
The new clinical data on valoctocogene roxaparvovec, published online in the New England Journal of Medicine, show that the beneficial effects from the gene are largely durable at 2 years, but they are anticipated to fade with time.
Two years after the one-time infusion, there remained “a significant reduction in the annualized bleeding rates” among 132 men who, at baseline, had severe hemophilia A requiring ongoing factor VIII prophylaxis, said the investigators, led by hematologist Johnny Mahlangu, MBBCh, MMed, of the University of the Witwatersrand, Johannesburg, South Africa.
However, the team predicted that median factor VIII activity would decrease below 10% of normal by year 3 or 5 depending on measurement technique, which would still translate to mild disease with an annualized bleeding rate of less than 1 episode per year.
“Although valoctocogene roxaparvovec may not eliminate bleeding, it potentially provides more consistent protection than factor VIII prophylaxis with less treatment burden,” the team said.
New questions
Data from the study “will directly inform therapeutic decision-making” in Europe, where valoctocogene roxaparvovec is already conditionally approved, and the United States, where it is awaiting approval by the FDA, says Lindsey George, MD, a hematologist and gene therapy specialist at Children’s Hospital of Philadelphia, in an accompanying editorial.
The study speaks to an ongoing concern about the durability of gene therapy for hemophilia but also raises new questions, she said.
For instance, while some patients had normal Factor VIII production and activity at 2 years, activity had dropped substantially in others, including in six men who resumed prophylaxis. “The cause of the decrease in factor VIII expression is an unanswered question,” and despite an anticipated U.S. price tag of around $2.5 million per treatment, “it is not possible [at the moment] to predict where an individual patient may fall within this range,” she writes.
Also, some subjects had elevations in liver aminotransferase levels that lasted for several months, including 2 years after infusion in 29% of subjects. Elevations in liver aminotransferase levels were treated with immune suppression for a median of 33 weeks.
“This is a unique finding with an undefined cause and long-term safety implications,” Dr. George said.
Getting to the bottom of such issues will be necessary for hemophilia gene therapy to fulfill its promise as “a one-time, lifelong, disease-ameliorating” fix for the condition, she asserted.
Study details
The new report followed up on the initial trial in 134 men who were treated with a single infusion of 6 × 1013 vector genomes per kilogram of body weight.
Among the 132 subjects available for 2-year evaluation, median factor VIII activity was in the range of mild hemophilia (6%-49% of normal) with an 84.5% reduction in bleeding events from baseline.
More than 80% of participants had no bleeding events requiring treatment, and there was a 98% reduction from baseline in mean use of exogenous factor VIII.
Overall, at year 2, 4.5% of subjects had factor VIII activity consistent with severe hemophilia A; 9.1% had activity consistent with moderate disease; 59.8% had activity consistent with mild disease; and 26.5% had activity in the normal range above 40 IU/dL. The investigators estimated that the typical half-life of the transgene-derived factor VIII production system is 123 weeks.
Among the six men who resumed prophylaxis, most had fewer bleeding events than when they were on prophylaxis before the infusion, investigators noted.
All the subjects developed antibodies to the virus delivery vector, precluding retreatment.
The work was funded by valoctocogene roxaparvovec maker BioMarin Pharmaceuticals. Several investigators are employees. Others reported ties to BioMarin and other companies; Dr. Mahlangu, for instance, reported research grants from BioMarin, Roche, Novo Nordisk, Pfizer, and others. Dr. George reported a research grant from Asklepios Biopharmaceutical and having a patent licensed to the company. The full list of author disclosures can be found with the original article.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE