User login
How baseline death risk may impact transfusion outcome
![]()
Credit: UAB Hospital
A new study suggests the risks and benefits of red blood cell (RBC) transfusions can vary considerably for patients with trauma and major bleeding, depending on the patients’ risk of death at baseline.
Patients with the highest predicted risk of death on arrival at a trauma center received the greatest benefit from RBC transfusions.
But among patients with the lowest predicted risk of death at baseline, transfusion was associated with a higher risk of death post-treatment.
Pablo Perel, MD, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in PLOS Medicine.
To conduct this study, the team used data from the CRASH-2 trial, which assessed the effect of tranexamic acid in trauma patients. The trial included 20,127 patients with significant bleeding who were treated at 274 hospitals in 40 countries.
Dr Perel and his colleagues used that data to evaluate the association between receiving an RBC transfusion and death from all causes at 28 days post-trauma. The findings were stratified by predicted risk of death based on clinical observations on arrival at the trauma center.
The researchers found that patients with the greatest predicted risk of dying—greater than 50%—had a smaller chance of death from all causes if they were transfused than if they were not. The odds ratio (OR) was 0.59.
For patients whose predicted risk of death ranged from 21% to 50%, there was no significant difference in their chance of dying whether they were transfused or not. The OR was 0.92.
But for patients with a lower risk of death at baseline, transfusion was associated with an increased risk of death.
Patients with a 6% to 20% risk of death at baseline had an OR of 2.31 if they received a transfusion. And for patients whose initial risk of death was below 6%, the OR for death associated with transfusion was 5.40.
In absolute figures, compared to no transfusion, RBC transfusion was associated with 5.1 more deaths per 100 patients in the group with the lowest predicted risk of death but with 11.9 fewer deaths per 100 patients in the group with the highest predicted risk of death.
The researchers noted that, although these data suggest RBC transfusion could be harmful for patients whose predicted risk of death is low, this study was observational. So the team cannot confirm a causal link, and a randomized trial investigating the association is warranted. ![]()
![]()
Credit: UAB Hospital
A new study suggests the risks and benefits of red blood cell (RBC) transfusions can vary considerably for patients with trauma and major bleeding, depending on the patients’ risk of death at baseline.
Patients with the highest predicted risk of death on arrival at a trauma center received the greatest benefit from RBC transfusions.
But among patients with the lowest predicted risk of death at baseline, transfusion was associated with a higher risk of death post-treatment.
Pablo Perel, MD, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in PLOS Medicine.
To conduct this study, the team used data from the CRASH-2 trial, which assessed the effect of tranexamic acid in trauma patients. The trial included 20,127 patients with significant bleeding who were treated at 274 hospitals in 40 countries.
Dr Perel and his colleagues used that data to evaluate the association between receiving an RBC transfusion and death from all causes at 28 days post-trauma. The findings were stratified by predicted risk of death based on clinical observations on arrival at the trauma center.
The researchers found that patients with the greatest predicted risk of dying—greater than 50%—had a smaller chance of death from all causes if they were transfused than if they were not. The odds ratio (OR) was 0.59.
For patients whose predicted risk of death ranged from 21% to 50%, there was no significant difference in their chance of dying whether they were transfused or not. The OR was 0.92.
But for patients with a lower risk of death at baseline, transfusion was associated with an increased risk of death.
Patients with a 6% to 20% risk of death at baseline had an OR of 2.31 if they received a transfusion. And for patients whose initial risk of death was below 6%, the OR for death associated with transfusion was 5.40.
In absolute figures, compared to no transfusion, RBC transfusion was associated with 5.1 more deaths per 100 patients in the group with the lowest predicted risk of death but with 11.9 fewer deaths per 100 patients in the group with the highest predicted risk of death.
The researchers noted that, although these data suggest RBC transfusion could be harmful for patients whose predicted risk of death is low, this study was observational. So the team cannot confirm a causal link, and a randomized trial investigating the association is warranted. ![]()
![]()
Credit: UAB Hospital
A new study suggests the risks and benefits of red blood cell (RBC) transfusions can vary considerably for patients with trauma and major bleeding, depending on the patients’ risk of death at baseline.
Patients with the highest predicted risk of death on arrival at a trauma center received the greatest benefit from RBC transfusions.
But among patients with the lowest predicted risk of death at baseline, transfusion was associated with a higher risk of death post-treatment.
Pablo Perel, MD, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in PLOS Medicine.
To conduct this study, the team used data from the CRASH-2 trial, which assessed the effect of tranexamic acid in trauma patients. The trial included 20,127 patients with significant bleeding who were treated at 274 hospitals in 40 countries.
Dr Perel and his colleagues used that data to evaluate the association between receiving an RBC transfusion and death from all causes at 28 days post-trauma. The findings were stratified by predicted risk of death based on clinical observations on arrival at the trauma center.
The researchers found that patients with the greatest predicted risk of dying—greater than 50%—had a smaller chance of death from all causes if they were transfused than if they were not. The odds ratio (OR) was 0.59.
For patients whose predicted risk of death ranged from 21% to 50%, there was no significant difference in their chance of dying whether they were transfused or not. The OR was 0.92.
But for patients with a lower risk of death at baseline, transfusion was associated with an increased risk of death.
Patients with a 6% to 20% risk of death at baseline had an OR of 2.31 if they received a transfusion. And for patients whose initial risk of death was below 6%, the OR for death associated with transfusion was 5.40.
In absolute figures, compared to no transfusion, RBC transfusion was associated with 5.1 more deaths per 100 patients in the group with the lowest predicted risk of death but with 11.9 fewer deaths per 100 patients in the group with the highest predicted risk of death.
The researchers noted that, although these data suggest RBC transfusion could be harmful for patients whose predicted risk of death is low, this study was observational. So the team cannot confirm a causal link, and a randomized trial investigating the association is warranted. ![]()
Blood sterilization processes harmful to platelets
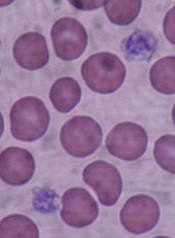
Some processes used to sterilize blood for transfusion are harmful to platelet function and could cause serious health issues in transfusion recipients, researchers say.
They found that some pathogen-reduction treatments impact platelets to the extent that they may be the cause of hemorrhages in recipients.
The pathogen reduction treatments “were developed more than 20 years ago, before we understood the importance of the genetic material contained in platelets,” explained study author Patrick Provost, PhD, of Université Laval and the CHU de Québec Research Center in Canada.
Platelets contain up to a third of the human genome in the form of ribonucleic acid (RNA), which enables them to synthesize over 1,000 proteins essential to the normal functioning of the human body.
The researchers studied the effects of 3 pathogen-reduction strategies—irradiation, riboflavin plus UVB light (Mirasol), and amotosalen plus UVA light (Intercept)—on platelet microRNAs, messenger RNAs (mRNAs), activation, and function.
They reported their findings in the journal Platelets.
The investigators collected 50 single-donor (apheresis) platelet concentrates (PCs) and subjected them to 5 treatments.
The control platelets were stored in donor plasma; additive solution platelets were stored in 65% storage solution and 35% donor plasma; the irradiation platelets were treated with 30Gy gamma irradiation and stored in donor plasma; the platelets treated with Mirasol were stored in donor plasma; and the platelets treated with Intercept were stored in the same solution as the additive solution group.
All treatments followed standard procedures or the manufacturer’s instructions.
After platelet isolation and RNA extraction, the investigators analyzed the levels of microRNA and mRNA levels of the platelets and assessed the impact of those levels on platelet activation and function.
MicroRNA profiles
They learned that platelets stored with additive solution or irradiation had significantly (P<0.05) reduced levels of one microRNA each, and only on day 7 of storage. Additive solution reduced the level of miR-223 and irradiation reduced the level of let-73.
Mirasol did not significantly reduce the level of any of the 11 tested micro RNAs.
And Intercept significantly reduced the level of 6 microRNAs on day 1, 1 microRNA on day 4, and 2 microRNAs on day 7. By day 7, let-7e was reduced by up to 70%.
The microRNA levels remained stable in the control sample for the entire 7-day storage period.
Platelet activation and function
Platelet counts in the Mirasol- and Intercept-treated platelets were significantly lower (P<0.001) on storage days 1, 4, and 7 compared with control platelets.
Pathogen-reduction treatments did not affect platelet microRNA synthesis, platelet microRNA function, nor did they induce the formation of cross-linked RNA adducts.
However, pathogen reduction caused platelet activation, which correlates with the observed reduction in platelet microRNAs.
The investigators measured CD62P expression, a marker of platelet activation, on the platelet surface. The additive solution platelets and Intercept-treated platelets, and to a lesser degree the irradiation group, had greater CD62P surface expression than the control group (P<0.05) on day 1.
The Mirasol group had similar activation to that of the control group.
On day 4, all treatment groups showed more activation than the control group (P<0.05). And on day 7, all groups had about the same activation level as the control group.
Pathogen reduction also impacted the aggregation response of platelets. Mirasol-treated platelets, which had the same aggregation response as that of controls on day 1, had no response on days 4 and 7.
And the aggregation response for Intercept-treated and additive solution platelets was already absent on day 1 and remained so on days 4 and 7.
Additive solution and Intercept also reduced platelet volume on day 1, which the investigators say could be explained by the platelet activation and release of microparticles induced by the treatments.
MicroRNA release
The investigators hypothesized that activated stored platelets could release microRNAs through microparticles in the supernatant. So they collected supernatant from each of the 5 groups and analyzed their total content of miR-223, which is one of the most abundant platelet microRNAs.
They discovered that the total amount of miR-223 was increased 30% to 86% in the microparticles released from additive solution and Intercept-treated platelets. They did not observe this increase in irradiation- or Mirasol-treated platelets compared to controls.
"The platelets end up depleted of RNA so, once transfused, they're unable to do what they normally would," Dr Provost said. Nevertheless, the clinical implications of the reduction in platelet activation and impaired platelet aggregation after Intercept treatment remain to be established.
The pathogen-reduction treatments are already marketed in some European countries, notably Switzerland, France, and Germany, and are under consideration in other countries, including Canada and the United States.
"In light of what we have demonstrated, the potentially harmful effects of these treatments should be carefully evaluated in the countries where they are not yet approved. It should also be re-evaluated in those countries where they are," Dr Provost said. ![]()
Some processes used to sterilize blood for transfusion are harmful to platelet function and could cause serious health issues in transfusion recipients, researchers say.
They found that some pathogen-reduction treatments impact platelets to the extent that they may be the cause of hemorrhages in recipients.
The pathogen reduction treatments “were developed more than 20 years ago, before we understood the importance of the genetic material contained in platelets,” explained study author Patrick Provost, PhD, of Université Laval and the CHU de Québec Research Center in Canada.
Platelets contain up to a third of the human genome in the form of ribonucleic acid (RNA), which enables them to synthesize over 1,000 proteins essential to the normal functioning of the human body.
The researchers studied the effects of 3 pathogen-reduction strategies—irradiation, riboflavin plus UVB light (Mirasol), and amotosalen plus UVA light (Intercept)—on platelet microRNAs, messenger RNAs (mRNAs), activation, and function.
They reported their findings in the journal Platelets.
The investigators collected 50 single-donor (apheresis) platelet concentrates (PCs) and subjected them to 5 treatments.
The control platelets were stored in donor plasma; additive solution platelets were stored in 65% storage solution and 35% donor plasma; the irradiation platelets were treated with 30Gy gamma irradiation and stored in donor plasma; the platelets treated with Mirasol were stored in donor plasma; and the platelets treated with Intercept were stored in the same solution as the additive solution group.
All treatments followed standard procedures or the manufacturer’s instructions.
After platelet isolation and RNA extraction, the investigators analyzed the levels of microRNA and mRNA levels of the platelets and assessed the impact of those levels on platelet activation and function.
MicroRNA profiles
They learned that platelets stored with additive solution or irradiation had significantly (P<0.05) reduced levels of one microRNA each, and only on day 7 of storage. Additive solution reduced the level of miR-223 and irradiation reduced the level of let-73.
Mirasol did not significantly reduce the level of any of the 11 tested micro RNAs.
And Intercept significantly reduced the level of 6 microRNAs on day 1, 1 microRNA on day 4, and 2 microRNAs on day 7. By day 7, let-7e was reduced by up to 70%.
The microRNA levels remained stable in the control sample for the entire 7-day storage period.
Platelet activation and function
Platelet counts in the Mirasol- and Intercept-treated platelets were significantly lower (P<0.001) on storage days 1, 4, and 7 compared with control platelets.
Pathogen-reduction treatments did not affect platelet microRNA synthesis, platelet microRNA function, nor did they induce the formation of cross-linked RNA adducts.
However, pathogen reduction caused platelet activation, which correlates with the observed reduction in platelet microRNAs.
The investigators measured CD62P expression, a marker of platelet activation, on the platelet surface. The additive solution platelets and Intercept-treated platelets, and to a lesser degree the irradiation group, had greater CD62P surface expression than the control group (P<0.05) on day 1.
The Mirasol group had similar activation to that of the control group.
On day 4, all treatment groups showed more activation than the control group (P<0.05). And on day 7, all groups had about the same activation level as the control group.
Pathogen reduction also impacted the aggregation response of platelets. Mirasol-treated platelets, which had the same aggregation response as that of controls on day 1, had no response on days 4 and 7.
And the aggregation response for Intercept-treated and additive solution platelets was already absent on day 1 and remained so on days 4 and 7.
Additive solution and Intercept also reduced platelet volume on day 1, which the investigators say could be explained by the platelet activation and release of microparticles induced by the treatments.
MicroRNA release
The investigators hypothesized that activated stored platelets could release microRNAs through microparticles in the supernatant. So they collected supernatant from each of the 5 groups and analyzed their total content of miR-223, which is one of the most abundant platelet microRNAs.
They discovered that the total amount of miR-223 was increased 30% to 86% in the microparticles released from additive solution and Intercept-treated platelets. They did not observe this increase in irradiation- or Mirasol-treated platelets compared to controls.
"The platelets end up depleted of RNA so, once transfused, they're unable to do what they normally would," Dr Provost said. Nevertheless, the clinical implications of the reduction in platelet activation and impaired platelet aggregation after Intercept treatment remain to be established.
The pathogen-reduction treatments are already marketed in some European countries, notably Switzerland, France, and Germany, and are under consideration in other countries, including Canada and the United States.
"In light of what we have demonstrated, the potentially harmful effects of these treatments should be carefully evaluated in the countries where they are not yet approved. It should also be re-evaluated in those countries where they are," Dr Provost said. ![]()
Some processes used to sterilize blood for transfusion are harmful to platelet function and could cause serious health issues in transfusion recipients, researchers say.
They found that some pathogen-reduction treatments impact platelets to the extent that they may be the cause of hemorrhages in recipients.
The pathogen reduction treatments “were developed more than 20 years ago, before we understood the importance of the genetic material contained in platelets,” explained study author Patrick Provost, PhD, of Université Laval and the CHU de Québec Research Center in Canada.
Platelets contain up to a third of the human genome in the form of ribonucleic acid (RNA), which enables them to synthesize over 1,000 proteins essential to the normal functioning of the human body.
The researchers studied the effects of 3 pathogen-reduction strategies—irradiation, riboflavin plus UVB light (Mirasol), and amotosalen plus UVA light (Intercept)—on platelet microRNAs, messenger RNAs (mRNAs), activation, and function.
They reported their findings in the journal Platelets.
The investigators collected 50 single-donor (apheresis) platelet concentrates (PCs) and subjected them to 5 treatments.
The control platelets were stored in donor plasma; additive solution platelets were stored in 65% storage solution and 35% donor plasma; the irradiation platelets were treated with 30Gy gamma irradiation and stored in donor plasma; the platelets treated with Mirasol were stored in donor plasma; and the platelets treated with Intercept were stored in the same solution as the additive solution group.
All treatments followed standard procedures or the manufacturer’s instructions.
After platelet isolation and RNA extraction, the investigators analyzed the levels of microRNA and mRNA levels of the platelets and assessed the impact of those levels on platelet activation and function.
MicroRNA profiles
They learned that platelets stored with additive solution or irradiation had significantly (P<0.05) reduced levels of one microRNA each, and only on day 7 of storage. Additive solution reduced the level of miR-223 and irradiation reduced the level of let-73.
Mirasol did not significantly reduce the level of any of the 11 tested micro RNAs.
And Intercept significantly reduced the level of 6 microRNAs on day 1, 1 microRNA on day 4, and 2 microRNAs on day 7. By day 7, let-7e was reduced by up to 70%.
The microRNA levels remained stable in the control sample for the entire 7-day storage period.
Platelet activation and function
Platelet counts in the Mirasol- and Intercept-treated platelets were significantly lower (P<0.001) on storage days 1, 4, and 7 compared with control platelets.
Pathogen-reduction treatments did not affect platelet microRNA synthesis, platelet microRNA function, nor did they induce the formation of cross-linked RNA adducts.
However, pathogen reduction caused platelet activation, which correlates with the observed reduction in platelet microRNAs.
The investigators measured CD62P expression, a marker of platelet activation, on the platelet surface. The additive solution platelets and Intercept-treated platelets, and to a lesser degree the irradiation group, had greater CD62P surface expression than the control group (P<0.05) on day 1.
The Mirasol group had similar activation to that of the control group.
On day 4, all treatment groups showed more activation than the control group (P<0.05). And on day 7, all groups had about the same activation level as the control group.
Pathogen reduction also impacted the aggregation response of platelets. Mirasol-treated platelets, which had the same aggregation response as that of controls on day 1, had no response on days 4 and 7.
And the aggregation response for Intercept-treated and additive solution platelets was already absent on day 1 and remained so on days 4 and 7.
Additive solution and Intercept also reduced platelet volume on day 1, which the investigators say could be explained by the platelet activation and release of microparticles induced by the treatments.
MicroRNA release
The investigators hypothesized that activated stored platelets could release microRNAs through microparticles in the supernatant. So they collected supernatant from each of the 5 groups and analyzed their total content of miR-223, which is one of the most abundant platelet microRNAs.
They discovered that the total amount of miR-223 was increased 30% to 86% in the microparticles released from additive solution and Intercept-treated platelets. They did not observe this increase in irradiation- or Mirasol-treated platelets compared to controls.
"The platelets end up depleted of RNA so, once transfused, they're unable to do what they normally would," Dr Provost said. Nevertheless, the clinical implications of the reduction in platelet activation and impaired platelet aggregation after Intercept treatment remain to be established.
The pathogen-reduction treatments are already marketed in some European countries, notably Switzerland, France, and Germany, and are under consideration in other countries, including Canada and the United States.
"In light of what we have demonstrated, the potentially harmful effects of these treatments should be carefully evaluated in the countries where they are not yet approved. It should also be re-evaluated in those countries where they are," Dr Provost said. ![]()
South Africa changes blood donation policy
![]()
Photo courtesy of UAB Hospital
The South African National Blood Service (SANBS) has lessened restrictions on blood donation for men who have sex with men (MSM) but placed a new restriction on heterosexual donors.
Now, all South Africans are prohibited from donating blood unless they have been celibate or in a monogamous sexual relationship for at least 6 months.
So any individual with a new sexual partner or multiple partners will not be allowed to donate blood, regardless of sexual orientation.
South Africa’s former policy was that MSM could only donate blood if they had been celibate for 6 months or longer. But heterosexual individuals who engaged in risky or casual sex were still allowed to donate.
This was because MSM were considered at high risk of contracting HIV. However, the South African HIV epidemic is primarily a heterosexual one, so the SANBS’s policy was thought by many to be discriminatory.
With the new policy, the question intended to identify MSM has been removed from the blood donor questionnaire. The new questionnaire addresses sexual risk in general, and any sexual act or contact with a new partner or partners during the preceding 6 months will be deemed a risk to the safety of the blood supply.
“It took us a while [to make this decision] because we didn’t have local facts that warranted changing our policy, although we knew South Africa was different from other countries in terms of risk of HIV,” said Vanessa Raju, SANBS Communications Manager.
“The policy wasn’t meant to be discriminatory, but it was seen as such. We then worked closely with the Department of Health and other organizations to reassess the situation.”
Blood donation policies perceived as discriminatory against MSM are in place in many countries.
In the US and Northern Ireland, for example, MSM are banned from donating blood for life. Canada lifted its lifetime ban last year but still requires that MSM be celibate for 5 years before donating.
Other countries have recently adopted similar policies. In England, Scotland, and Wales, MSM must be celibate for 12 months prior to donating blood. ![]()
![]()
Photo courtesy of UAB Hospital
The South African National Blood Service (SANBS) has lessened restrictions on blood donation for men who have sex with men (MSM) but placed a new restriction on heterosexual donors.
Now, all South Africans are prohibited from donating blood unless they have been celibate or in a monogamous sexual relationship for at least 6 months.
So any individual with a new sexual partner or multiple partners will not be allowed to donate blood, regardless of sexual orientation.
South Africa’s former policy was that MSM could only donate blood if they had been celibate for 6 months or longer. But heterosexual individuals who engaged in risky or casual sex were still allowed to donate.
This was because MSM were considered at high risk of contracting HIV. However, the South African HIV epidemic is primarily a heterosexual one, so the SANBS’s policy was thought by many to be discriminatory.
With the new policy, the question intended to identify MSM has been removed from the blood donor questionnaire. The new questionnaire addresses sexual risk in general, and any sexual act or contact with a new partner or partners during the preceding 6 months will be deemed a risk to the safety of the blood supply.
“It took us a while [to make this decision] because we didn’t have local facts that warranted changing our policy, although we knew South Africa was different from other countries in terms of risk of HIV,” said Vanessa Raju, SANBS Communications Manager.
“The policy wasn’t meant to be discriminatory, but it was seen as such. We then worked closely with the Department of Health and other organizations to reassess the situation.”
Blood donation policies perceived as discriminatory against MSM are in place in many countries.
In the US and Northern Ireland, for example, MSM are banned from donating blood for life. Canada lifted its lifetime ban last year but still requires that MSM be celibate for 5 years before donating.
Other countries have recently adopted similar policies. In England, Scotland, and Wales, MSM must be celibate for 12 months prior to donating blood. ![]()
![]()
Photo courtesy of UAB Hospital
The South African National Blood Service (SANBS) has lessened restrictions on blood donation for men who have sex with men (MSM) but placed a new restriction on heterosexual donors.
Now, all South Africans are prohibited from donating blood unless they have been celibate or in a monogamous sexual relationship for at least 6 months.
So any individual with a new sexual partner or multiple partners will not be allowed to donate blood, regardless of sexual orientation.
South Africa’s former policy was that MSM could only donate blood if they had been celibate for 6 months or longer. But heterosexual individuals who engaged in risky or casual sex were still allowed to donate.
This was because MSM were considered at high risk of contracting HIV. However, the South African HIV epidemic is primarily a heterosexual one, so the SANBS’s policy was thought by many to be discriminatory.
With the new policy, the question intended to identify MSM has been removed from the blood donor questionnaire. The new questionnaire addresses sexual risk in general, and any sexual act or contact with a new partner or partners during the preceding 6 months will be deemed a risk to the safety of the blood supply.
“It took us a while [to make this decision] because we didn’t have local facts that warranted changing our policy, although we knew South Africa was different from other countries in terms of risk of HIV,” said Vanessa Raju, SANBS Communications Manager.
“The policy wasn’t meant to be discriminatory, but it was seen as such. We then worked closely with the Department of Health and other organizations to reassess the situation.”
Blood donation policies perceived as discriminatory against MSM are in place in many countries.
In the US and Northern Ireland, for example, MSM are banned from donating blood for life. Canada lifted its lifetime ban last year but still requires that MSM be celibate for 5 years before donating.
Other countries have recently adopted similar policies. In England, Scotland, and Wales, MSM must be celibate for 12 months prior to donating blood. ![]()
FDA approves first molecular test for blood typing

Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Credit: Juan D. Alfonso
The US Food and Drug Administration (FDA) has approved the first molecular assay for determining blood compatibility prior to transfusion.
The Immucor PreciseType Human Erythrocyte Antigen (HEA) Molecular BeadChip Test can be used to determine donor and patient non-ABO/non-RhD red blood cell types.
The test provides an alternative to serological typing and may enhance patient care in certain situations, according to Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The Immucor PreciseType HEA Molecular BeadChip Test works by detecting genes that govern the expression of 36 antigens that can appear on the surface of red blood cells.
The test uses thousands of coded beads that bind with the genes coding for non-ABO red blood cell antigens that are present in a blood sample.
A light signal is generated from each bead that has captured a specific gene. Accompanying computer software decodes the light signals and reports which antigens are predicted to be present on the red cells, based on the genes detected.
Researchers conducted a study to compare the typing results of the PreciseType HEA Molecular BeadChip Test with licensed serological reagents and DNA sequencing. And the results demonstrated comparable performance between the methods.
The product was brought before the FDA’s Blood Products Advisory Committee on March 18, 2014. After reviewing the relevant information, the committee said the data provided reasonable assurance that the Immucor PreciseType HEA Molecular BeadChip Test is safe and effective for its intended use.
The test is manufactured by BioArray Solutions Ltd. of Warren, New Jersey. ![]()
Large-volume infusion pump recalled

Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
Credit: CDC
The medical technology company CareFusion has announced a Class I recall of its Alaris Pump model 8100, software version 9.1.18.
This large-volume infusion pump is used for the delivery of fluids, medicines, blood, and blood products.
Version 9.1.18 of the Alaris Pump model 8100 is being recalled due to the possibility of a software failure in which the pump module will not properly delay an infusion when the “Delay Until” option or “Multidose” feature is used.
There have been no reports of adverse events or deaths related to this malfunction, but it does pose risks. CareFusion has received 1 report where the device malfunctioned when the “Delay Until” option was selected.
The software failure also prevents the pump from properly delivering a multidose infusion under the following conditions:
- When the first dose is programmed to infuse when the system time is earlier than 7 pm and a subsequent dose is intended to infuse between 7 pm and 11:59 pm
- When the first dose is programmed to infuse when the system time is between 7 pm and 11:59 pm and a subsequent dose is intended to infuse between 12 am and 6:59 pm the next day.
If the infusion starts earlier or later than intended and is not immediately detected and stopped, serious injury or death could result. Therefore, healthcare professionals should not use the Alaris Pump module “Delay Until” option or the “Multidose” option.
However, CareFusion said it has identified the root cause of the issue and recommends that the previous Alaris Pump module software version 9.1.17 be installed to address this recall. The company said it will contact all affected customers to schedule the installation of software version 9.1.17.
As an interim guidance, customers may update their dataset to disable both “Delay” options (“Delay Until” and “Delay For”) and/or the “Multidose” option across all profiles to prevent the use of these features. These are shared configurations with the Alaris Syringe module and, if disabled, would prevent use of these features with the Alaris Syringe module as well.
For more information on this recall, see CareFusion’s recall notice, or contact the CareFusion Support Center at 888-562-6018 or SupportCenter@carefusion.com.
To report adverse reactions or quality problems associated with this product, visit the Food and Drug Administration’s MedWatch website. ![]()
Study reveals racial disparity in perioperative transfusion practices

Credit: Elise Amendola
Results of a large study showed that black patients were more likely than white patients to receive perioperative blood transfusions for 2 of 3 common surgical procedures.
Researchers evaluated transfusion practices in these 2 racial groups for coronary artery bypass surgery (CABG), total hip replacement (THR), and colectomy.
And they found that black patients undergoing CABG or THR had a significantly higher incidence of transfusion than white patients undergoing these procedures.
Feng Qian, PhD, of the University at Albany School of Public Health, and his colleagues reported these findings in BMC Health Services Research.
The team examined the use of perioperative red blood cell transfusion using patient data from the University Health System Consortium, a network of academic medical centers and affiliated hospitals. The data included hospitalizations occurring from 2009 to 2011.
The researchers’ final sample included 42,933 patients who underwent THR (37,888 white and 5045 black), 25,849 patients who underwent CABG (23,113 white and 2736 black), and 8255 patients who underwent colectomy (6861 white and 1394 black).
Black patients tended to be younger than white patients, with the overall age ranging from 48 to 73 years. Blacks were also less well-insured than whites and more likely to have comorbidities such as diabetes, renal failure, and anemia.
Dr Qian and his colleagues adjusted for these differences in their analysis, as well as for patient gender, admission status, and severity of illness.
The analysis revealed that black patients undergoing CABG had a 41% higher incidence of perioperative transfusion than white patients (P=0.002).
For THR, the incidence of transfusion was 39% higher among blacks than whites (P<0.001). And for colectomy, the incidence was 8% higher among blacks than whites (P=0.40).
The researchers then performed an analysis adjusted for the aforementioned factors as well as for hospital-fixed effects.
This revealed that black patients undergoing CABG had a 42% higher incidence of transfusion than whites (P<0.001). Blacks undergoing THR had a 43% higher incidence of transfusion (P<0.001). And blacks undergoing colectomy had a 1% higher incidence of transfusion (P=0.92)
The researchers noted that, although blood transfusion is widely employed in surgery, the practice is associated with adverse outcomes. So overuse of transfusions may pose serious health risks, specifically in black patients undergoing CABG and THR.
Dr Qian added that recognizing racial disparities related to the use of perioperative red blood cell transfusion may help reduce potentially unnecessary transfusions in minority patients. ![]()
Credit: Elise Amendola
Results of a large study showed that black patients were more likely than white patients to receive perioperative blood transfusions for 2 of 3 common surgical procedures.
Researchers evaluated transfusion practices in these 2 racial groups for coronary artery bypass surgery (CABG), total hip replacement (THR), and colectomy.
And they found that black patients undergoing CABG or THR had a significantly higher incidence of transfusion than white patients undergoing these procedures.
Feng Qian, PhD, of the University at Albany School of Public Health, and his colleagues reported these findings in BMC Health Services Research.
The team examined the use of perioperative red blood cell transfusion using patient data from the University Health System Consortium, a network of academic medical centers and affiliated hospitals. The data included hospitalizations occurring from 2009 to 2011.
The researchers’ final sample included 42,933 patients who underwent THR (37,888 white and 5045 black), 25,849 patients who underwent CABG (23,113 white and 2736 black), and 8255 patients who underwent colectomy (6861 white and 1394 black).
Black patients tended to be younger than white patients, with the overall age ranging from 48 to 73 years. Blacks were also less well-insured than whites and more likely to have comorbidities such as diabetes, renal failure, and anemia.
Dr Qian and his colleagues adjusted for these differences in their analysis, as well as for patient gender, admission status, and severity of illness.
The analysis revealed that black patients undergoing CABG had a 41% higher incidence of perioperative transfusion than white patients (P=0.002).
For THR, the incidence of transfusion was 39% higher among blacks than whites (P<0.001). And for colectomy, the incidence was 8% higher among blacks than whites (P=0.40).
The researchers then performed an analysis adjusted for the aforementioned factors as well as for hospital-fixed effects.
This revealed that black patients undergoing CABG had a 42% higher incidence of transfusion than whites (P<0.001). Blacks undergoing THR had a 43% higher incidence of transfusion (P<0.001). And blacks undergoing colectomy had a 1% higher incidence of transfusion (P=0.92)
The researchers noted that, although blood transfusion is widely employed in surgery, the practice is associated with adverse outcomes. So overuse of transfusions may pose serious health risks, specifically in black patients undergoing CABG and THR.
Dr Qian added that recognizing racial disparities related to the use of perioperative red blood cell transfusion may help reduce potentially unnecessary transfusions in minority patients. ![]()
Credit: Elise Amendola
Results of a large study showed that black patients were more likely than white patients to receive perioperative blood transfusions for 2 of 3 common surgical procedures.
Researchers evaluated transfusion practices in these 2 racial groups for coronary artery bypass surgery (CABG), total hip replacement (THR), and colectomy.
And they found that black patients undergoing CABG or THR had a significantly higher incidence of transfusion than white patients undergoing these procedures.
Feng Qian, PhD, of the University at Albany School of Public Health, and his colleagues reported these findings in BMC Health Services Research.
The team examined the use of perioperative red blood cell transfusion using patient data from the University Health System Consortium, a network of academic medical centers and affiliated hospitals. The data included hospitalizations occurring from 2009 to 2011.
The researchers’ final sample included 42,933 patients who underwent THR (37,888 white and 5045 black), 25,849 patients who underwent CABG (23,113 white and 2736 black), and 8255 patients who underwent colectomy (6861 white and 1394 black).
Black patients tended to be younger than white patients, with the overall age ranging from 48 to 73 years. Blacks were also less well-insured than whites and more likely to have comorbidities such as diabetes, renal failure, and anemia.
Dr Qian and his colleagues adjusted for these differences in their analysis, as well as for patient gender, admission status, and severity of illness.
The analysis revealed that black patients undergoing CABG had a 41% higher incidence of perioperative transfusion than white patients (P=0.002).
For THR, the incidence of transfusion was 39% higher among blacks than whites (P<0.001). And for colectomy, the incidence was 8% higher among blacks than whites (P=0.40).
The researchers then performed an analysis adjusted for the aforementioned factors as well as for hospital-fixed effects.
This revealed that black patients undergoing CABG had a 42% higher incidence of transfusion than whites (P<0.001). Blacks undergoing THR had a 43% higher incidence of transfusion (P<0.001). And blacks undergoing colectomy had a 1% higher incidence of transfusion (P=0.92)
The researchers noted that, although blood transfusion is widely employed in surgery, the practice is associated with adverse outcomes. So overuse of transfusions may pose serious health risks, specifically in black patients undergoing CABG and THR.
Dr Qian added that recognizing racial disparities related to the use of perioperative red blood cell transfusion may help reduce potentially unnecessary transfusions in minority patients. ![]()
Recycled RBCs prove more functional than banked ones
![]()
Credit: UAB Hospital
Reinfusing the blood a patient loses during cardiopulmonary bypass surgery confers benefits over transfusing the patient with banked blood, results of a small study suggest.
Investigators noted that both the surgery and red blood cell (RBC) storage are associated with changes in RBCs that can adversely affect oxygen delivery.
However, their study revealed minimal effects on RBC structure and function among patients who received their own recycled blood during surgery.
On the other hand, patients who received banked RBCs along with their own blood experienced a dose-dependent decrease in RBC cell membrane deformability that could persist beyond 3 days.
Steven Frank, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Anesthesia & Analgesia.
The team studied 32 patients undergoing cardiopulmonary bypass, categorizing them by their transfusion status: those who received their own RBCs (n=12), those who received their own blood plus fewer than 5 units of banked blood (n=10), and those who received their own RBCs plus 5 or more units of stored blood (n=10).
All patients had blood samples drawn before, during, and for 3 days after surgery. The investigators examined samples for blood cell membrane stiffness and flexibility.
In patients who received only their own recycled blood, their cells behaved normally right away, as if they had never been outside the body.
But the more banked blood a patient received, the less flexible their entire population of RBCs. Three days after surgery, RBCs in the group that received the largest number of transfused units still had not recovered their full function.
“We now have more evidence that fresh blood cells are of a higher quality than what comes from a blood bank,” Dr Frank said.
“If banked blood, which is stored for up to 6 weeks, is now shown to be of a lower quality, it makes more sense to use recycled blood that has only been outside the body for 1 or 2 hours. It’s always been the case that patients feel better about getting their own blood, and recycling is also more cost-effective.”
The investigators used a cell saver machine to collect the material a patient lost during surgery. They then rinsed away the unneeded fat and tissue, centrifuged and separated the red cells, and returned them to the patient.
Dr Frank and his colleagues noted that disposable parts of the cell saver, which can be used to process multiple units of blood, cost around $120, compared to $240 for each unit of banked blood. Additionally, recycling blood reduces a patient’s risk of contracting infections and experiencing transfusion-related adverse reactions.
Dr Frank pointed out, however, that cell saver machines are not appropriate for all operations, and not all hospitals have access to round-the-clock perfusionists to run them. For heart surgeries, a perfusionist is already in the operating room to run the heart-lung bypass machine.
Dr Frank also noted that many operations are considered to be a low risk for blood loss, in which case, the cell saver is unnecessary. Nevertheless, he advocates wider use of recycled blood.
“In any patient where you expect to give 1 unit of red blood cells or more, it’s cost-effective and beneficial to recycle,” he said. ![]()
![]()
Credit: UAB Hospital
Reinfusing the blood a patient loses during cardiopulmonary bypass surgery confers benefits over transfusing the patient with banked blood, results of a small study suggest.
Investigators noted that both the surgery and red blood cell (RBC) storage are associated with changes in RBCs that can adversely affect oxygen delivery.
However, their study revealed minimal effects on RBC structure and function among patients who received their own recycled blood during surgery.
On the other hand, patients who received banked RBCs along with their own blood experienced a dose-dependent decrease in RBC cell membrane deformability that could persist beyond 3 days.
Steven Frank, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Anesthesia & Analgesia.
The team studied 32 patients undergoing cardiopulmonary bypass, categorizing them by their transfusion status: those who received their own RBCs (n=12), those who received their own blood plus fewer than 5 units of banked blood (n=10), and those who received their own RBCs plus 5 or more units of stored blood (n=10).
All patients had blood samples drawn before, during, and for 3 days after surgery. The investigators examined samples for blood cell membrane stiffness and flexibility.
In patients who received only their own recycled blood, their cells behaved normally right away, as if they had never been outside the body.
But the more banked blood a patient received, the less flexible their entire population of RBCs. Three days after surgery, RBCs in the group that received the largest number of transfused units still had not recovered their full function.
“We now have more evidence that fresh blood cells are of a higher quality than what comes from a blood bank,” Dr Frank said.
“If banked blood, which is stored for up to 6 weeks, is now shown to be of a lower quality, it makes more sense to use recycled blood that has only been outside the body for 1 or 2 hours. It’s always been the case that patients feel better about getting their own blood, and recycling is also more cost-effective.”
The investigators used a cell saver machine to collect the material a patient lost during surgery. They then rinsed away the unneeded fat and tissue, centrifuged and separated the red cells, and returned them to the patient.
Dr Frank and his colleagues noted that disposable parts of the cell saver, which can be used to process multiple units of blood, cost around $120, compared to $240 for each unit of banked blood. Additionally, recycling blood reduces a patient’s risk of contracting infections and experiencing transfusion-related adverse reactions.
Dr Frank pointed out, however, that cell saver machines are not appropriate for all operations, and not all hospitals have access to round-the-clock perfusionists to run them. For heart surgeries, a perfusionist is already in the operating room to run the heart-lung bypass machine.
Dr Frank also noted that many operations are considered to be a low risk for blood loss, in which case, the cell saver is unnecessary. Nevertheless, he advocates wider use of recycled blood.
“In any patient where you expect to give 1 unit of red blood cells or more, it’s cost-effective and beneficial to recycle,” he said. ![]()
![]()
Credit: UAB Hospital
Reinfusing the blood a patient loses during cardiopulmonary bypass surgery confers benefits over transfusing the patient with banked blood, results of a small study suggest.
Investigators noted that both the surgery and red blood cell (RBC) storage are associated with changes in RBCs that can adversely affect oxygen delivery.
However, their study revealed minimal effects on RBC structure and function among patients who received their own recycled blood during surgery.
On the other hand, patients who received banked RBCs along with their own blood experienced a dose-dependent decrease in RBC cell membrane deformability that could persist beyond 3 days.
Steven Frank, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland, and his colleagues reported these findings in Anesthesia & Analgesia.
The team studied 32 patients undergoing cardiopulmonary bypass, categorizing them by their transfusion status: those who received their own RBCs (n=12), those who received their own blood plus fewer than 5 units of banked blood (n=10), and those who received their own RBCs plus 5 or more units of stored blood (n=10).
All patients had blood samples drawn before, during, and for 3 days after surgery. The investigators examined samples for blood cell membrane stiffness and flexibility.
In patients who received only their own recycled blood, their cells behaved normally right away, as if they had never been outside the body.
But the more banked blood a patient received, the less flexible their entire population of RBCs. Three days after surgery, RBCs in the group that received the largest number of transfused units still had not recovered their full function.
“We now have more evidence that fresh blood cells are of a higher quality than what comes from a blood bank,” Dr Frank said.
“If banked blood, which is stored for up to 6 weeks, is now shown to be of a lower quality, it makes more sense to use recycled blood that has only been outside the body for 1 or 2 hours. It’s always been the case that patients feel better about getting their own blood, and recycling is also more cost-effective.”
The investigators used a cell saver machine to collect the material a patient lost during surgery. They then rinsed away the unneeded fat and tissue, centrifuged and separated the red cells, and returned them to the patient.
Dr Frank and his colleagues noted that disposable parts of the cell saver, which can be used to process multiple units of blood, cost around $120, compared to $240 for each unit of banked blood. Additionally, recycling blood reduces a patient’s risk of contracting infections and experiencing transfusion-related adverse reactions.
Dr Frank pointed out, however, that cell saver machines are not appropriate for all operations, and not all hospitals have access to round-the-clock perfusionists to run them. For heart surgeries, a perfusionist is already in the operating room to run the heart-lung bypass machine.
Dr Frank also noted that many operations are considered to be a low risk for blood loss, in which case, the cell saver is unnecessary. Nevertheless, he advocates wider use of recycled blood.
“In any patient where you expect to give 1 unit of red blood cells or more, it’s cost-effective and beneficial to recycle,” he said. ![]()
Hospira issues Class I recall of infusion pumps

Credit: Daniel Gay
Hospira, Inc., has issued a Class I recall of Abbott Acclaim infusion pumps and Hospira Acclaim Encore infusion pumps, after receiving reports of broken door assemblies on these products.
If a door assembly breaks, the door may not close properly and an over-infusion or a delay of therapy may occur.
If the door cannot be closed, the pump cannot be used, and this can result in a delay of therapy.
Use of these products may cause serious adverse events, including death.
These pumps are used to deliver hydration fluids, drugs, blood and blood fractions, intravenous nutritionals, and enteral nutritionals.
The affected Abbott Acclaim Infusion Pumps, list Number 12032, were manufactured from February 1998 to November 1998 and distributed from September 1998 through February 2004.
The affected Hospira Acclaim Encore infusion pumps, list Number 12237, were manufactured from February 1997 to February 2010 and distributed from July 1999 through November 2013.
Hospira is recommending that users inspect each Hospira/Abbott Acclaim Encore infusion pump for door handle cracks prior to programming a therapy, by taking the following steps:
1. After inserting the tubing (with the roller clamp closed) and closing the door handle against the infusion pump, check that the door is fully closed.
If a pump has a door that does not close properly and a gap or separation exists between the completely closed door and the pump itself, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT). If the door closes correctly, proceed to Step 2.
2. If the door closes correctly and a gap or separation does not exist between the completely closed door and the pump itself, check that there is no free flow activity in the drip chamber of the administration set by opening the roller clamp.
If free flow is detected, close the roller clamp, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT).
3. If no issues are found through steps 1 and 2, the pump is acceptable for use. However, healthcare professionals should still ensure that anyone in their facility who might use these products is made aware of this safety notification and the recommended actions.
Healthcare professionals and patients can report adverse events or side effects related to the use of these products to the FDA’s MedWatch Program. ![]()
Credit: Daniel Gay
Hospira, Inc., has issued a Class I recall of Abbott Acclaim infusion pumps and Hospira Acclaim Encore infusion pumps, after receiving reports of broken door assemblies on these products.
If a door assembly breaks, the door may not close properly and an over-infusion or a delay of therapy may occur.
If the door cannot be closed, the pump cannot be used, and this can result in a delay of therapy.
Use of these products may cause serious adverse events, including death.
These pumps are used to deliver hydration fluids, drugs, blood and blood fractions, intravenous nutritionals, and enteral nutritionals.
The affected Abbott Acclaim Infusion Pumps, list Number 12032, were manufactured from February 1998 to November 1998 and distributed from September 1998 through February 2004.
The affected Hospira Acclaim Encore infusion pumps, list Number 12237, were manufactured from February 1997 to February 2010 and distributed from July 1999 through November 2013.
Hospira is recommending that users inspect each Hospira/Abbott Acclaim Encore infusion pump for door handle cracks prior to programming a therapy, by taking the following steps:
1. After inserting the tubing (with the roller clamp closed) and closing the door handle against the infusion pump, check that the door is fully closed.
If a pump has a door that does not close properly and a gap or separation exists between the completely closed door and the pump itself, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT). If the door closes correctly, proceed to Step 2.
2. If the door closes correctly and a gap or separation does not exist between the completely closed door and the pump itself, check that there is no free flow activity in the drip chamber of the administration set by opening the roller clamp.
If free flow is detected, close the roller clamp, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT).
3. If no issues are found through steps 1 and 2, the pump is acceptable for use. However, healthcare professionals should still ensure that anyone in their facility who might use these products is made aware of this safety notification and the recommended actions.
Healthcare professionals and patients can report adverse events or side effects related to the use of these products to the FDA’s MedWatch Program. ![]()
Credit: Daniel Gay
Hospira, Inc., has issued a Class I recall of Abbott Acclaim infusion pumps and Hospira Acclaim Encore infusion pumps, after receiving reports of broken door assemblies on these products.
If a door assembly breaks, the door may not close properly and an over-infusion or a delay of therapy may occur.
If the door cannot be closed, the pump cannot be used, and this can result in a delay of therapy.
Use of these products may cause serious adverse events, including death.
These pumps are used to deliver hydration fluids, drugs, blood and blood fractions, intravenous nutritionals, and enteral nutritionals.
The affected Abbott Acclaim Infusion Pumps, list Number 12032, were manufactured from February 1998 to November 1998 and distributed from September 1998 through February 2004.
The affected Hospira Acclaim Encore infusion pumps, list Number 12237, were manufactured from February 1997 to February 2010 and distributed from July 1999 through November 2013.
Hospira is recommending that users inspect each Hospira/Abbott Acclaim Encore infusion pump for door handle cracks prior to programming a therapy, by taking the following steps:
1. After inserting the tubing (with the roller clamp closed) and closing the door handle against the infusion pump, check that the door is fully closed.
If a pump has a door that does not close properly and a gap or separation exists between the completely closed door and the pump itself, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT). If the door closes correctly, proceed to Step 2.
2. If the door closes correctly and a gap or separation does not exist between the completely closed door and the pump itself, check that there is no free flow activity in the drip chamber of the administration set by opening the roller clamp.
If free flow is detected, close the roller clamp, remove the pump from clinical service, and call Hospira at 1-800-441-4100 (M-F, 8am-5pm, CT).
3. If no issues are found through steps 1 and 2, the pump is acceptable for use. However, healthcare professionals should still ensure that anyone in their facility who might use these products is made aware of this safety notification and the recommended actions.
Healthcare professionals and patients can report adverse events or side effects related to the use of these products to the FDA’s MedWatch Program. ![]()
Hospira announces device correction for infusion pump docking station
Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing ProductComplaintsPP@hospira.com.
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program. ![]()
Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing ProductComplaintsPP@hospira.com.
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program. ![]()
Credit: CDC
Hospira, Inc., has announced a medical device correction for the GemStar Docking Station (list number 13075), used in conjunction with the GemStar infusion pump.
The correction follows customer reports of 2 malfunctions that may occur with the docking station.
The company is not recalling the product but is notifying US customers of the potential malfunctions and providing instructions for overriding these errors.
The errors could potentially cause delays or interruptions in therapy. And this might result in serious adverse events or death, but there have been no such events reported to date.
Potential malfunctions
The GemStar Docking Station is an accessory to the GemStar infusion pump (sold separately) and provides an alternate power source to the GemStar pump.
When the docking station is used in conjunction with a GemStar Phase 3 pump (List 13000, 13100 or 13150), there is a risk that the GemStar Phase 3 pump may fail to power up while connected to the docking station.
When a GemStar Phase 3 (List 13000, 13100 or 13150) or GemStar Phase 4 pump (List 13086, 13087 or 13088) is used in conjunction with both a docking station and an external battery pack accessory (List 13073), the GemStar pump may display error code 11/003 and give an audible alarm, indicating excessive input voltage from the external sources.
If the GemStar pump detects what is perceived to be more than 3.6 volts, as measured on the external voltage input, the pump will stop the infusion. This will trigger an audible alarm, and the device will display alarm code 11/003.
If a GemStar fails to power up or the 11/003 error code stops an infusion, a patient’s therapy might be delayed or interrupted. This could result in significant injury or death, although there have been no reports of death or serious injury associated with these malfunctions to date.
The products impacted by these issues have been in distribution since February 2002.
Responding to/preventing malfunctions
Hospira is advising that healthcare professionals weigh the risk/benefit to patients associated with the use of the docking station when administering critical therapies. Clinicians should consider the use of an alternative pump, particularly in patients for whom a delay or interruption of therapy could result in serious injury or death.
However, the company says there is no need to return the GemStar Docking Station at this time. Instead, Hospira recommends that users take the following actions.
To avoid a failure to power up, turn on the pump before connecting it with the docking station. This will prevent the failure to power up.
To mitigate the potential for an 11/003 error code, remove the external battery pack accessory (List 13073) from the docking station and pump prior to installing the pump in the docking station.
In addition, clinicians should stop using a docking station in conjunction with an external battery pack accessory (List 13073). Contact Hospira to discuss an appropriate alternative option.
Docking station users who experience a failure to power up or an 11/003 error code should report the issue to Hospira by calling 1-800-441-4100 (M-F, 8am-5pm CT) or emailing ProductComplaintsPP@hospira.com.
For additional assistance or to obtain a copy of the Urgent Medical Device Correction letter and/or a reply form, contact Stericycle at 1-866-792-5451 (M-F, 8am-5pm ET).
On May 1, 2013, Hospira announced that it would begin the process of retiring the GemStar family of infusion devices in accordance with the company’s global device strategy. As of July 31, 2015, Hospira will consider the products within the GemStar Infusion System family retired and will no longer support them.
Adverse reactions or quality problems related to the GemStar Docking Station can be reported to the US Food and Drug Administration’s MedWatch Program.
Baxter issues Class I recall of infusion pumps

of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program.
of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program.
of chemotherapy drugs
Credit: Bill Branson
Baxter Healthcare Corporation is recalling some of its infusion pumps after receiving more than 3500 reports of the pumps malfunctioning.
According to the US Food and Drug Administration (FDA), the malfunctioning pumps have resulted in 9 severe adverse events but no deaths.
This Class I recall includes Sigma Spectrum Infusion Pumps with Master Drug Library Model No. 35700BAX and 35700ABB.
The pumps were made between July 1, 2005, and January 15, 2014. They were distributed between February 20, 2013, and January 15, 2014.
The Sigma Spectrum infusion pumps are intended to deliver controlled amounts of medicines, blood, blood products, and other intravenous fluids.
The FDA said there have been more than 3500 reports of these pumps malfunctioning—specifically, reports of System Error 322 “Link Switch Error (low).” This error occurs when the pump detects that the door is open even though it is closed. A System Error 322 may lead to an interruption or delay in therapy.
When this error occurs, the Sigma Spectrum infusion pump stops the infusion, an alarm sounds, and a light flashes (a visual “322” alarm). This requires a clinician to reset the alarm, reprogram the pump, and confirm the infusion is running properly.
The use of affected pumps may cause serious adverse health consequences, including death; hence, the Class I recall.
Customers who encounter a System Error 322 should turn off the pump by pressing the ON/OFF key, then turn the pump back on by pressing the ON/OFF key to clear the alarm.
Clinicians will need to reprogram the infusion after the pump is turned back on. If the alarm cannot be cleared using these instructions, the device should be removed from use and sent to the facility’s biomedical engineering department.
If the System Error 322 reoccurs, the pump may need to be inspected and serviced by Baxter Healthcare. To contact Baxter, call 1-800-356-3454 (choose option 1) Monday through Friday, 7 am to 7 pm, Eastern Time.
Adverse reactions or quality problems related to these pumps can be reported to the FDA’s MedWatch Program.