User login
Cutaneous reaction to AEDs? Think autoimmune epilepsy
BANGKOK – Cutaneous reactions to antiepileptic drugs in patients with chronic epilepsy suggest increased likelihood of an autoimmune element to their seizure disorder, Fernando Cendes, MD, PhD, reported at the International Epilepsy Congress.

“My recommendation based on our findings is that if you have a patient who has a history of skin reactions to AEDs [antiepileptic drugs], or who has psychosis, or who has a very strange response to antiepileptic medication – meaning that at some points they are refractory and at other points they are very well controlled – I think those patients are probably at risk for having an autoantibody,” he said at the congress sponsored by the International League Against Epilepsy.
Screening for autoantibodies in such patients is appropriate. However, there’s a caveat: “The thing is, we don’t have evidence that treating these autoantibodies with immunotherapy will have any benefit on seizure control in these patients. We don’t have that data yet, but we are looking into it,” according to Dr. Cendes, professor of neurology at the State University of Campinas (Brazil).
He presented a study of 221 consecutive adults with severe chronic refractory epilepsy as evidenced by a mean disease duration of nearly 29 years, with an average of 5.93 seizures per month. A total of 77% had a structural etiology for their epilepsy, in most cases hippocampal sclerosis. In 19% of patients, the etiology was unknown. Overall, 95% of subjects had focal epilepsy, and the remainder had generalized epilepsy. All underwent serum testing for a variety of antibodies against neuronal surface antigens that have been implicated in encephalitis, seizures, and/or psychosis. Those who tested positive then underwent confirmatory testing of their cerebrospinal fluid.
The impetus for this study, the neurologist explained, is that although it’s now well established that seizures are a common clinical expression of acute- and subacute-phase autoimmune encephalitis marked by neuronal autoantibodies, little is known about the relationship between chronic epilepsy and such antibodies.
Only five Brazilian patients with chronic epilepsy, or 2.2%, tested positive for autoantibodies, all of whom had mesial temporal lobe epilepsy with hippocampal sclerosis. This suggests a possible autoimmune etiology for hippocampal sclerosis. Three of the five patients had anti-N-methyl-D-aspartate receptor antibodies (anti-NMDA) and two had antiglutamic acid decarboxylate antibodies (anti-GAD). No one was positive for anti–leucine-rich glioma-inactivated 1 antibodies (anti-LGI1), anti–contactin-associated proteinlike 2 (anti-caspr2), anti-glutamate receptor antibodies (anti-AMPAr), or anti–gamma-aminobutyric acid receptor antibodies (anti-GABAr).
The autoantibody-negative and the much smaller autoantibody-positive groups didn’t differ significantly in terms of demographics, seizure frequency, disease duration, drug resistance, cognitive impairment, comorbid autoimmune conditions, or history of status epilepticus. Indeed, only two between-group differences were found: fluctuation in seizure control was an issue in 10.6% of autoantibody-negative and 40% of autoantibody-positive patients, and cutaneous adverse reactions to antiepileptic drugs were noted in 10.6% of antibody-negative and 60% of antibody-positive patients. Psychiatric comorbidities were present in 49.5% of autoantibody-negative patients as compared with 80% – that is, four of five – who were autoantibody-positive, a trend that didn’t achieve statistical significance.
Asked if he thinks the autoantibodies found in a small subset of patients with chronic epilepsy were a cause or an effect of repeated seizures for so long, Dr. Cendes replied, “That’s a very interesting question, and I don’t have an answer, actually. But if seizures trigger development of these antibodies – and remember, this population we’re talking about had many, many seizures over the years – I would expect antibodies to be more frequent than the figure we found.”
He reported having no financial conflicts regarding his study.
SOURCE: Watanabe N et al. IEC 2019, Abstract P004.
BANGKOK – Cutaneous reactions to antiepileptic drugs in patients with chronic epilepsy suggest increased likelihood of an autoimmune element to their seizure disorder, Fernando Cendes, MD, PhD, reported at the International Epilepsy Congress.
“My recommendation based on our findings is that if you have a patient who has a history of skin reactions to AEDs [antiepileptic drugs], or who has psychosis, or who has a very strange response to antiepileptic medication – meaning that at some points they are refractory and at other points they are very well controlled – I think those patients are probably at risk for having an autoantibody,” he said at the congress sponsored by the International League Against Epilepsy.
Screening for autoantibodies in such patients is appropriate. However, there’s a caveat: “The thing is, we don’t have evidence that treating these autoantibodies with immunotherapy will have any benefit on seizure control in these patients. We don’t have that data yet, but we are looking into it,” according to Dr. Cendes, professor of neurology at the State University of Campinas (Brazil).
He presented a study of 221 consecutive adults with severe chronic refractory epilepsy as evidenced by a mean disease duration of nearly 29 years, with an average of 5.93 seizures per month. A total of 77% had a structural etiology for their epilepsy, in most cases hippocampal sclerosis. In 19% of patients, the etiology was unknown. Overall, 95% of subjects had focal epilepsy, and the remainder had generalized epilepsy. All underwent serum testing for a variety of antibodies against neuronal surface antigens that have been implicated in encephalitis, seizures, and/or psychosis. Those who tested positive then underwent confirmatory testing of their cerebrospinal fluid.
The impetus for this study, the neurologist explained, is that although it’s now well established that seizures are a common clinical expression of acute- and subacute-phase autoimmune encephalitis marked by neuronal autoantibodies, little is known about the relationship between chronic epilepsy and such antibodies.
Only five Brazilian patients with chronic epilepsy, or 2.2%, tested positive for autoantibodies, all of whom had mesial temporal lobe epilepsy with hippocampal sclerosis. This suggests a possible autoimmune etiology for hippocampal sclerosis. Three of the five patients had anti-N-methyl-D-aspartate receptor antibodies (anti-NMDA) and two had antiglutamic acid decarboxylate antibodies (anti-GAD). No one was positive for anti–leucine-rich glioma-inactivated 1 antibodies (anti-LGI1), anti–contactin-associated proteinlike 2 (anti-caspr2), anti-glutamate receptor antibodies (anti-AMPAr), or anti–gamma-aminobutyric acid receptor antibodies (anti-GABAr).
The autoantibody-negative and the much smaller autoantibody-positive groups didn’t differ significantly in terms of demographics, seizure frequency, disease duration, drug resistance, cognitive impairment, comorbid autoimmune conditions, or history of status epilepticus. Indeed, only two between-group differences were found: fluctuation in seizure control was an issue in 10.6% of autoantibody-negative and 40% of autoantibody-positive patients, and cutaneous adverse reactions to antiepileptic drugs were noted in 10.6% of antibody-negative and 60% of antibody-positive patients. Psychiatric comorbidities were present in 49.5% of autoantibody-negative patients as compared with 80% – that is, four of five – who were autoantibody-positive, a trend that didn’t achieve statistical significance.
Asked if he thinks the autoantibodies found in a small subset of patients with chronic epilepsy were a cause or an effect of repeated seizures for so long, Dr. Cendes replied, “That’s a very interesting question, and I don’t have an answer, actually. But if seizures trigger development of these antibodies – and remember, this population we’re talking about had many, many seizures over the years – I would expect antibodies to be more frequent than the figure we found.”
He reported having no financial conflicts regarding his study.
SOURCE: Watanabe N et al. IEC 2019, Abstract P004.
BANGKOK – Cutaneous reactions to antiepileptic drugs in patients with chronic epilepsy suggest increased likelihood of an autoimmune element to their seizure disorder, Fernando Cendes, MD, PhD, reported at the International Epilepsy Congress.
“My recommendation based on our findings is that if you have a patient who has a history of skin reactions to AEDs [antiepileptic drugs], or who has psychosis, or who has a very strange response to antiepileptic medication – meaning that at some points they are refractory and at other points they are very well controlled – I think those patients are probably at risk for having an autoantibody,” he said at the congress sponsored by the International League Against Epilepsy.
Screening for autoantibodies in such patients is appropriate. However, there’s a caveat: “The thing is, we don’t have evidence that treating these autoantibodies with immunotherapy will have any benefit on seizure control in these patients. We don’t have that data yet, but we are looking into it,” according to Dr. Cendes, professor of neurology at the State University of Campinas (Brazil).
He presented a study of 221 consecutive adults with severe chronic refractory epilepsy as evidenced by a mean disease duration of nearly 29 years, with an average of 5.93 seizures per month. A total of 77% had a structural etiology for their epilepsy, in most cases hippocampal sclerosis. In 19% of patients, the etiology was unknown. Overall, 95% of subjects had focal epilepsy, and the remainder had generalized epilepsy. All underwent serum testing for a variety of antibodies against neuronal surface antigens that have been implicated in encephalitis, seizures, and/or psychosis. Those who tested positive then underwent confirmatory testing of their cerebrospinal fluid.
The impetus for this study, the neurologist explained, is that although it’s now well established that seizures are a common clinical expression of acute- and subacute-phase autoimmune encephalitis marked by neuronal autoantibodies, little is known about the relationship between chronic epilepsy and such antibodies.
Only five Brazilian patients with chronic epilepsy, or 2.2%, tested positive for autoantibodies, all of whom had mesial temporal lobe epilepsy with hippocampal sclerosis. This suggests a possible autoimmune etiology for hippocampal sclerosis. Three of the five patients had anti-N-methyl-D-aspartate receptor antibodies (anti-NMDA) and two had antiglutamic acid decarboxylate antibodies (anti-GAD). No one was positive for anti–leucine-rich glioma-inactivated 1 antibodies (anti-LGI1), anti–contactin-associated proteinlike 2 (anti-caspr2), anti-glutamate receptor antibodies (anti-AMPAr), or anti–gamma-aminobutyric acid receptor antibodies (anti-GABAr).
The autoantibody-negative and the much smaller autoantibody-positive groups didn’t differ significantly in terms of demographics, seizure frequency, disease duration, drug resistance, cognitive impairment, comorbid autoimmune conditions, or history of status epilepticus. Indeed, only two between-group differences were found: fluctuation in seizure control was an issue in 10.6% of autoantibody-negative and 40% of autoantibody-positive patients, and cutaneous adverse reactions to antiepileptic drugs were noted in 10.6% of antibody-negative and 60% of antibody-positive patients. Psychiatric comorbidities were present in 49.5% of autoantibody-negative patients as compared with 80% – that is, four of five – who were autoantibody-positive, a trend that didn’t achieve statistical significance.
Asked if he thinks the autoantibodies found in a small subset of patients with chronic epilepsy were a cause or an effect of repeated seizures for so long, Dr. Cendes replied, “That’s a very interesting question, and I don’t have an answer, actually. But if seizures trigger development of these antibodies – and remember, this population we’re talking about had many, many seizures over the years – I would expect antibodies to be more frequent than the figure we found.”
He reported having no financial conflicts regarding his study.
SOURCE: Watanabe N et al. IEC 2019, Abstract P004.
REPORTING FROM IEC 2019
Repeated ANA testing after negative result of little diagnostic value
MADRID – Repeated antinuclear antibody testing after a negative result has limited use for the diagnosis of ANA-associated rheumatologic conditions, according to data from a multicenter, retrospective analysis that considered a 7-year period.

Considering more than 7,875 repeated ANA tests in 4,887 patients, “the vast majority of results didn’t change,” Ai Li Yeo, MBBS, a PhD candidate, rheumatologist, and infectious disease fellow at Monash University, Melbourne, reported at the European Congress of Rheumatology.
ANA tests were repeated between 2 and as many as 45 times in individual patients, she reported, but the results of 79% of these tests remained unchanged – 45% of tests were persistently negative and 34% persistently positive using a cutoff titer of 1:160.
“Our study showed that there was a very low yield in repeating an ANA test for the diagnosis of ANA-associated rheumatological conditions unless there was evidence of evolving multisystem clinical features,” Dr. Yeo said.
Indeed, the positive predictive value was just 0.01. “So for a hundred patients staring off with a negative ANA results that on repeat testing became positive, the probability is that one patient will have a new ANA-associated rheumatological condition diagnosis,” Dr. Yeo said.
“ANA testing is frequently performed and is part of the classification criteria for autoimmune conditions such as lupus and scleroderma,” she observed. However, the test provides no information on the severity or activity of the disease, and the value of serial monitoring for such conditions is unclear.
“Minimizing unnecessary tests is a global health economic priority,” Dr. Yeo said. She noted that there are multiple initiatives in place to try to open a dialog about using health care resources most effectively, such as ‘Choosing Wisely’ set up by the American Board of Internal Medicine (ABIM) Foundation.
The aim of the present analysis was to calculate the cost of repeated ANA testing and to see if any change in the ANA result was associated with new diagnoses of ANA-associated rheumatological conditions.
The analysis considered more than 36,700 tests that were performed on samples from more than 28,800 patients within the Monash Health tertiary health network between 2011 and 2018. Of these, 22,657 (62%) had given a negative result and 14,058 (38%) had given a positive result.
“Not surprisingly, the age of those who tested positive was significantly higher than those who tested negative,” Dr. Yeo said (52.6 vs. 48.9 years; P less than .001). There was also a higher number of women than men tested, and women more often tested positive.
Around one-fifth of tests performed were repeat tests, of which 511 (6.5%) changed from being negative to positive over a median of 1.71 years.
“A small percentage of people alternated between results,” Dr. Yeo acknowledged, with 9.4% of people going from a positive to a negative result; 10.5% moving from a negative to a positive result, and 1.9% going from positive to negative to positive.
With repeated tests, just five new diagnoses of ANA-associated rheumatologic conditions were made: two cases of systemic lupus erythematosus (SLE), one case of scleroderma, and two cases of undifferentiated connective tissue disease. There was a range of ANA titers and patterns and evolving clinical features of a multisystem disease.
Based on the direct costs of ANA testing in her health care system, not performing repeated tests could yield significant savings, Dr. Yeo said, a 21.4% reduction, in fact, based on this analysis. The cost of an ANA test in Australia ranges from 15 to 46 euros, making the cost of all tests in this analysis 564,745 euros. Taking away the cost of all the single ANA tests performed (443,209 euros) gives a potential cost saving of more than 121,000 euros, she said.
“We now have an opportunity to prevent unnecessary ANA testing, Dr. Yeo said. “Ultimately, our aim is to change behavior at the start of the ordering cycle by educating medical students and doctors about inappropriate test ordering.”
The majority of repeated tests had been ordered by nonrheumatologists (82% of cases), and Dr. Yeo said that rheumatologists ordered repeat tests in 11% of cases. However, there was little information available in this retrospective analysis as to why the tests had been repeated.
The research was picked as one of the six best clinical abstracts at the meeting, out of a total of almost 5,000 submitted abstracts.
Dr. Yeo reported having no conflicts of interest.
SOURCE: Yeo AL et al. Ann Rheum Dis. Jun 2019;78(suppl 2):76-7, Abstract OP0020. doi: 10.1136/annrheumdis-2019-eular.4517.
MADRID – Repeated antinuclear antibody testing after a negative result has limited use for the diagnosis of ANA-associated rheumatologic conditions, according to data from a multicenter, retrospective analysis that considered a 7-year period.
Considering more than 7,875 repeated ANA tests in 4,887 patients, “the vast majority of results didn’t change,” Ai Li Yeo, MBBS, a PhD candidate, rheumatologist, and infectious disease fellow at Monash University, Melbourne, reported at the European Congress of Rheumatology.
ANA tests were repeated between 2 and as many as 45 times in individual patients, she reported, but the results of 79% of these tests remained unchanged – 45% of tests were persistently negative and 34% persistently positive using a cutoff titer of 1:160.
“Our study showed that there was a very low yield in repeating an ANA test for the diagnosis of ANA-associated rheumatological conditions unless there was evidence of evolving multisystem clinical features,” Dr. Yeo said.
Indeed, the positive predictive value was just 0.01. “So for a hundred patients staring off with a negative ANA results that on repeat testing became positive, the probability is that one patient will have a new ANA-associated rheumatological condition diagnosis,” Dr. Yeo said.
“ANA testing is frequently performed and is part of the classification criteria for autoimmune conditions such as lupus and scleroderma,” she observed. However, the test provides no information on the severity or activity of the disease, and the value of serial monitoring for such conditions is unclear.
“Minimizing unnecessary tests is a global health economic priority,” Dr. Yeo said. She noted that there are multiple initiatives in place to try to open a dialog about using health care resources most effectively, such as ‘Choosing Wisely’ set up by the American Board of Internal Medicine (ABIM) Foundation.
The aim of the present analysis was to calculate the cost of repeated ANA testing and to see if any change in the ANA result was associated with new diagnoses of ANA-associated rheumatological conditions.
The analysis considered more than 36,700 tests that were performed on samples from more than 28,800 patients within the Monash Health tertiary health network between 2011 and 2018. Of these, 22,657 (62%) had given a negative result and 14,058 (38%) had given a positive result.
“Not surprisingly, the age of those who tested positive was significantly higher than those who tested negative,” Dr. Yeo said (52.6 vs. 48.9 years; P less than .001). There was also a higher number of women than men tested, and women more often tested positive.
Around one-fifth of tests performed were repeat tests, of which 511 (6.5%) changed from being negative to positive over a median of 1.71 years.
“A small percentage of people alternated between results,” Dr. Yeo acknowledged, with 9.4% of people going from a positive to a negative result; 10.5% moving from a negative to a positive result, and 1.9% going from positive to negative to positive.
With repeated tests, just five new diagnoses of ANA-associated rheumatologic conditions were made: two cases of systemic lupus erythematosus (SLE), one case of scleroderma, and two cases of undifferentiated connective tissue disease. There was a range of ANA titers and patterns and evolving clinical features of a multisystem disease.
Based on the direct costs of ANA testing in her health care system, not performing repeated tests could yield significant savings, Dr. Yeo said, a 21.4% reduction, in fact, based on this analysis. The cost of an ANA test in Australia ranges from 15 to 46 euros, making the cost of all tests in this analysis 564,745 euros. Taking away the cost of all the single ANA tests performed (443,209 euros) gives a potential cost saving of more than 121,000 euros, she said.
“We now have an opportunity to prevent unnecessary ANA testing, Dr. Yeo said. “Ultimately, our aim is to change behavior at the start of the ordering cycle by educating medical students and doctors about inappropriate test ordering.”
The majority of repeated tests had been ordered by nonrheumatologists (82% of cases), and Dr. Yeo said that rheumatologists ordered repeat tests in 11% of cases. However, there was little information available in this retrospective analysis as to why the tests had been repeated.
The research was picked as one of the six best clinical abstracts at the meeting, out of a total of almost 5,000 submitted abstracts.
Dr. Yeo reported having no conflicts of interest.
SOURCE: Yeo AL et al. Ann Rheum Dis. Jun 2019;78(suppl 2):76-7, Abstract OP0020. doi: 10.1136/annrheumdis-2019-eular.4517.
MADRID – Repeated antinuclear antibody testing after a negative result has limited use for the diagnosis of ANA-associated rheumatologic conditions, according to data from a multicenter, retrospective analysis that considered a 7-year period.
Considering more than 7,875 repeated ANA tests in 4,887 patients, “the vast majority of results didn’t change,” Ai Li Yeo, MBBS, a PhD candidate, rheumatologist, and infectious disease fellow at Monash University, Melbourne, reported at the European Congress of Rheumatology.
ANA tests were repeated between 2 and as many as 45 times in individual patients, she reported, but the results of 79% of these tests remained unchanged – 45% of tests were persistently negative and 34% persistently positive using a cutoff titer of 1:160.
“Our study showed that there was a very low yield in repeating an ANA test for the diagnosis of ANA-associated rheumatological conditions unless there was evidence of evolving multisystem clinical features,” Dr. Yeo said.
Indeed, the positive predictive value was just 0.01. “So for a hundred patients staring off with a negative ANA results that on repeat testing became positive, the probability is that one patient will have a new ANA-associated rheumatological condition diagnosis,” Dr. Yeo said.
“ANA testing is frequently performed and is part of the classification criteria for autoimmune conditions such as lupus and scleroderma,” she observed. However, the test provides no information on the severity or activity of the disease, and the value of serial monitoring for such conditions is unclear.
“Minimizing unnecessary tests is a global health economic priority,” Dr. Yeo said. She noted that there are multiple initiatives in place to try to open a dialog about using health care resources most effectively, such as ‘Choosing Wisely’ set up by the American Board of Internal Medicine (ABIM) Foundation.
The aim of the present analysis was to calculate the cost of repeated ANA testing and to see if any change in the ANA result was associated with new diagnoses of ANA-associated rheumatological conditions.
The analysis considered more than 36,700 tests that were performed on samples from more than 28,800 patients within the Monash Health tertiary health network between 2011 and 2018. Of these, 22,657 (62%) had given a negative result and 14,058 (38%) had given a positive result.
“Not surprisingly, the age of those who tested positive was significantly higher than those who tested negative,” Dr. Yeo said (52.6 vs. 48.9 years; P less than .001). There was also a higher number of women than men tested, and women more often tested positive.
Around one-fifth of tests performed were repeat tests, of which 511 (6.5%) changed from being negative to positive over a median of 1.71 years.
“A small percentage of people alternated between results,” Dr. Yeo acknowledged, with 9.4% of people going from a positive to a negative result; 10.5% moving from a negative to a positive result, and 1.9% going from positive to negative to positive.
With repeated tests, just five new diagnoses of ANA-associated rheumatologic conditions were made: two cases of systemic lupus erythematosus (SLE), one case of scleroderma, and two cases of undifferentiated connective tissue disease. There was a range of ANA titers and patterns and evolving clinical features of a multisystem disease.
Based on the direct costs of ANA testing in her health care system, not performing repeated tests could yield significant savings, Dr. Yeo said, a 21.4% reduction, in fact, based on this analysis. The cost of an ANA test in Australia ranges from 15 to 46 euros, making the cost of all tests in this analysis 564,745 euros. Taking away the cost of all the single ANA tests performed (443,209 euros) gives a potential cost saving of more than 121,000 euros, she said.
“We now have an opportunity to prevent unnecessary ANA testing, Dr. Yeo said. “Ultimately, our aim is to change behavior at the start of the ordering cycle by educating medical students and doctors about inappropriate test ordering.”
The majority of repeated tests had been ordered by nonrheumatologists (82% of cases), and Dr. Yeo said that rheumatologists ordered repeat tests in 11% of cases. However, there was little information available in this retrospective analysis as to why the tests had been repeated.
The research was picked as one of the six best clinical abstracts at the meeting, out of a total of almost 5,000 submitted abstracts.
Dr. Yeo reported having no conflicts of interest.
SOURCE: Yeo AL et al. Ann Rheum Dis. Jun 2019;78(suppl 2):76-7, Abstract OP0020. doi: 10.1136/annrheumdis-2019-eular.4517.
REPORTING FROM EULAR 2019 CONGRESS
Genetic variant could dictate rituximab response in lupus
MADRID – Response to rituximab in patients with systemic lupus erythematosus (SLE) might be dictated by the presence of a genetic variant that encodes the Fc gamma receptors (FcGRs), expressed on natural killer (NK) cells, according to findings from a single-center, longitudinal cohort study.

It is well known that not everyone with SLE will respond well to rituximab, but that some will, first author Md Yuzaiful Md Yusof, MBChB, PhD, explained in an interview at the European Congress of Rheumatology.
Although data from clinical trials with rituximab in this patient setting have been essentially negative, the methodology of those trials has since been disputed, he observed. Indeed, subsequent data (Ann Rheum Dis. 2017;76:1829-36) have suggested that as many as 80% of patients could achieve a response with rituximab, particularly if there is complete B-cell depletion.
Previous researchers (Ann Rheum Dis. 2012;71:875-7) have shown that a polymorphism (158V) in the Fc gamma receptor IIIA (FCGR3A) gene is associated with the response to rituximab-based therapy in patients with rheumatoid arthritis (RA). This gene is important for antibody-dependent cellular-mediated cytotoxicity (ADCC).
The objective of the current study – an observational, prospective, longitudinal cohort study conducted in Leeds (England) – was therefore to see if the FCGR3A-158V polymorphism might influence response in patients with SLE.
“We were trying to find pretreatment biomarkers that could predict response to rituximab in SLE,” Dr. Md Yusof explained.
For the study, 85 patients who were treated with rituximab were assessed. The cohort was predominantly female (96%), with a mean age of 40 years. All of the patients had antinuclear antibodies, with just over half having anti–double-stranded DNA antibodies, and two-thirds having extractable nuclear antigens. One-third had low complement (C3/C4) levels.
Complete B-cell depletion occurred in 63% of patients with the FCGR3A-158V allele, a significantly higher rate than the 40% observed among those with 158 FF genotype (odds ratio, 2.73; P = .041). A significantly higher percentage of patients with the FCGR3A-158V allele also achieved a major BILAG (British Isles Lupus Assessment Group) response when compared against patients with the 158 FF variant (48% vs. 23%), with an odds ratio of 3.06 (P = .033).
Rituximab’s effect on NK cell-mediated B-cell killing may have played a key role in treatment response. Carrying the FCGR3A-158V allele was associated with greater degranulation activity versus the 158 FF variant.
Lastly, patients were more likely to remain on treatment with rituximab over a 10-year period if they had the FCGR3A-158V allele, compared with the 158 FF variant.
“These data suggest one mechanism by which patients with SLE might become resistant to the effects of rituximab, and could be used to guide therapy in the future,” Dr. Md Yusof suggested.
“Once this finding is validated, the clinical implication is that this genetic testing could be done prior to rituximab to identify those who will respond to therapy,” he postulated. “People with SLE who have this genetic variant with high affinity for rituximab are the ones that are better suited for rituximab therapy,” he added, otherwise a different CD20-directed antibody or alternative B-cell blockade therapies should be used.
The U.K. National Institute for Health Research funded the study. Dr. Md Yusof had no conflicts of interest to disclose; some coauthors disclosed ties to Roche, GlaxoSmithKline, and AstraZeneca, among other companies.
SOURCE: Md Yusof MY et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):1069-70. Abstract SAT0009, doi: 10.1136/annrheumdis-2019-eular.6919.
MADRID – Response to rituximab in patients with systemic lupus erythematosus (SLE) might be dictated by the presence of a genetic variant that encodes the Fc gamma receptors (FcGRs), expressed on natural killer (NK) cells, according to findings from a single-center, longitudinal cohort study.
It is well known that not everyone with SLE will respond well to rituximab, but that some will, first author Md Yuzaiful Md Yusof, MBChB, PhD, explained in an interview at the European Congress of Rheumatology.
Although data from clinical trials with rituximab in this patient setting have been essentially negative, the methodology of those trials has since been disputed, he observed. Indeed, subsequent data (Ann Rheum Dis. 2017;76:1829-36) have suggested that as many as 80% of patients could achieve a response with rituximab, particularly if there is complete B-cell depletion.
Previous researchers (Ann Rheum Dis. 2012;71:875-7) have shown that a polymorphism (158V) in the Fc gamma receptor IIIA (FCGR3A) gene is associated with the response to rituximab-based therapy in patients with rheumatoid arthritis (RA). This gene is important for antibody-dependent cellular-mediated cytotoxicity (ADCC).
The objective of the current study – an observational, prospective, longitudinal cohort study conducted in Leeds (England) – was therefore to see if the FCGR3A-158V polymorphism might influence response in patients with SLE.
“We were trying to find pretreatment biomarkers that could predict response to rituximab in SLE,” Dr. Md Yusof explained.
For the study, 85 patients who were treated with rituximab were assessed. The cohort was predominantly female (96%), with a mean age of 40 years. All of the patients had antinuclear antibodies, with just over half having anti–double-stranded DNA antibodies, and two-thirds having extractable nuclear antigens. One-third had low complement (C3/C4) levels.
Complete B-cell depletion occurred in 63% of patients with the FCGR3A-158V allele, a significantly higher rate than the 40% observed among those with 158 FF genotype (odds ratio, 2.73; P = .041). A significantly higher percentage of patients with the FCGR3A-158V allele also achieved a major BILAG (British Isles Lupus Assessment Group) response when compared against patients with the 158 FF variant (48% vs. 23%), with an odds ratio of 3.06 (P = .033).
Rituximab’s effect on NK cell-mediated B-cell killing may have played a key role in treatment response. Carrying the FCGR3A-158V allele was associated with greater degranulation activity versus the 158 FF variant.
Lastly, patients were more likely to remain on treatment with rituximab over a 10-year period if they had the FCGR3A-158V allele, compared with the 158 FF variant.
“These data suggest one mechanism by which patients with SLE might become resistant to the effects of rituximab, and could be used to guide therapy in the future,” Dr. Md Yusof suggested.
“Once this finding is validated, the clinical implication is that this genetic testing could be done prior to rituximab to identify those who will respond to therapy,” he postulated. “People with SLE who have this genetic variant with high affinity for rituximab are the ones that are better suited for rituximab therapy,” he added, otherwise a different CD20-directed antibody or alternative B-cell blockade therapies should be used.
The U.K. National Institute for Health Research funded the study. Dr. Md Yusof had no conflicts of interest to disclose; some coauthors disclosed ties to Roche, GlaxoSmithKline, and AstraZeneca, among other companies.
SOURCE: Md Yusof MY et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):1069-70. Abstract SAT0009, doi: 10.1136/annrheumdis-2019-eular.6919.
MADRID – Response to rituximab in patients with systemic lupus erythematosus (SLE) might be dictated by the presence of a genetic variant that encodes the Fc gamma receptors (FcGRs), expressed on natural killer (NK) cells, according to findings from a single-center, longitudinal cohort study.
It is well known that not everyone with SLE will respond well to rituximab, but that some will, first author Md Yuzaiful Md Yusof, MBChB, PhD, explained in an interview at the European Congress of Rheumatology.
Although data from clinical trials with rituximab in this patient setting have been essentially negative, the methodology of those trials has since been disputed, he observed. Indeed, subsequent data (Ann Rheum Dis. 2017;76:1829-36) have suggested that as many as 80% of patients could achieve a response with rituximab, particularly if there is complete B-cell depletion.
Previous researchers (Ann Rheum Dis. 2012;71:875-7) have shown that a polymorphism (158V) in the Fc gamma receptor IIIA (FCGR3A) gene is associated with the response to rituximab-based therapy in patients with rheumatoid arthritis (RA). This gene is important for antibody-dependent cellular-mediated cytotoxicity (ADCC).
The objective of the current study – an observational, prospective, longitudinal cohort study conducted in Leeds (England) – was therefore to see if the FCGR3A-158V polymorphism might influence response in patients with SLE.
“We were trying to find pretreatment biomarkers that could predict response to rituximab in SLE,” Dr. Md Yusof explained.
For the study, 85 patients who were treated with rituximab were assessed. The cohort was predominantly female (96%), with a mean age of 40 years. All of the patients had antinuclear antibodies, with just over half having anti–double-stranded DNA antibodies, and two-thirds having extractable nuclear antigens. One-third had low complement (C3/C4) levels.
Complete B-cell depletion occurred in 63% of patients with the FCGR3A-158V allele, a significantly higher rate than the 40% observed among those with 158 FF genotype (odds ratio, 2.73; P = .041). A significantly higher percentage of patients with the FCGR3A-158V allele also achieved a major BILAG (British Isles Lupus Assessment Group) response when compared against patients with the 158 FF variant (48% vs. 23%), with an odds ratio of 3.06 (P = .033).
Rituximab’s effect on NK cell-mediated B-cell killing may have played a key role in treatment response. Carrying the FCGR3A-158V allele was associated with greater degranulation activity versus the 158 FF variant.
Lastly, patients were more likely to remain on treatment with rituximab over a 10-year period if they had the FCGR3A-158V allele, compared with the 158 FF variant.
“These data suggest one mechanism by which patients with SLE might become resistant to the effects of rituximab, and could be used to guide therapy in the future,” Dr. Md Yusof suggested.
“Once this finding is validated, the clinical implication is that this genetic testing could be done prior to rituximab to identify those who will respond to therapy,” he postulated. “People with SLE who have this genetic variant with high affinity for rituximab are the ones that are better suited for rituximab therapy,” he added, otherwise a different CD20-directed antibody or alternative B-cell blockade therapies should be used.
The U.K. National Institute for Health Research funded the study. Dr. Md Yusof had no conflicts of interest to disclose; some coauthors disclosed ties to Roche, GlaxoSmithKline, and AstraZeneca, among other companies.
SOURCE: Md Yusof MY et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):1069-70. Abstract SAT0009, doi: 10.1136/annrheumdis-2019-eular.6919.
REPORTING FROM EULAR 2019 CONGRESS

Tocilizumab preserves lung function in systemic sclerosis
MADRID – , according to a secondary endpoint analysis of the phase 3, double-blind, randomized, controlled focuSSced trial.

After 48 weeks, a significantly lower proportion of patients treated with tocilizumab than placebo experienced any decline in lung function from baseline (50.5% versus 70.3% (P = .015), as defined by the percentage increase in predicted forced vital capacity (%pFVC). When only patients with interstitial lung disease (ILD) were considered, the respective percentages were 51.7% and 75.5% (P = .003).
In SSc-ILD patients, a clinically meaningful decline of 10% or more of the %pFVC in lung function was seen in 24.5% given placebo but in just 8.6% of those treated with tocilizumab.
“ILD is a major complication of scleroderma; it has high morbidity and mortality ... and it’s largely irreversible,” Dinesh Khanna, MD, said at the European Congress of Rheumatology.
“In this day and age, when we treat ILD, we wait for a patient to develop clinical ILD,” added Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor. Clinical ILD can be defined by symptoms, abnormal pulmonary function tests, and marked abnormalities on high resolution computed tomography (HRCT) scans. He indicated that if improving ILD was not possible, then the next best thing would be to stabilize the disease and ensure there was no worsening in lung function.
As yet, there are no disease-modifying treatments available to treat SSc but there are “ample data that interleukin-6 plays a very important role in the pathogenesis of scleroderma,” Dr. Khanna observed. Tocilizumab is a humanized monoclonal antibody against the interleukin-6 receptor.
Data from the phase 2 faSScinate trial showed initial promise for the drug in SSc where a numerical, but not statistically significant, improvement in skin thickening was seen, and the results had hinted at a possible benefit on lung function (Lancet. 2016 Jun 25;387:2630-40).
However, in the phase 3 focuSSced trial, there was no statistically significant difference in the change from baseline to week 48 modified Rodnan skin score (mRSS) between tocilizumab and placebo, which was the primary endpoint. The least square mean change in mRSS was –6.14 for tocilizumab and –4.41 for placebo (P = .0983).
A total of 205 patients with SSc were studied and randomized, 1:1 in a double-blind fashion, to receive either a once-weekly, subcutaneous dose of 162 mg tocilizumab or a weekly subcutaneous placebo injection for 48 weeks.
For inclusion in the study, patients had to have SSc that met American College of Rheumatology and European League Against Rheumatism (EULAR) criteria and be diagnosed less than 60 months previously. Patients had to have an mRSS of 10-35 units and active disease with one or more of the following: C-reactive protein of 6 mg/L or higher; erythrocyte sedimentation rate of 28 mm/h or higher; and platelet count of330 x 109 L.
“What was astonishing in the trial was that every patient had HRCT at baseline and at the end of the study,” Dr. Khanna reported. These scans showed that 64% of patients had evidence of ILD at baseline and that those treated with tocilizumab had less evidence of fibrosis at week 48 versus placebo, indicating a stabilization rather than worsening of disease.
A time to treatment failure analysis also favored tocilizumab over placebo, but there were no significant changes in patient-reported outcomes.
Dr. Khanna’s slides stated that “given that the primary endpoint for mRSS was not met, all other P values are presented for information purposes only and cannot be considered statistically significant despite the strength of the evidence.” During the Q&A after his presentation, he noted that it was unlikely that the study’s sponsors (Roche/Genentech) will now pursue a license for tocilizumab in SSc.
Nevertheless, Dr. Khanna concluded, “we have the opportunity, based on these data, to treat these patients early on, where you can preserve the lung function, which is a paradigm shift versus waiting for the lung function to decline, become clinically meaningful, significant, and then treat this patient population.”
Roche/Genentech sponsored the study. Dr. Khanna acts as a consultant to Roche/Genentech and eight other pharmaceutical companies. He owns stock in Eicos Sciences.
SOURCE: Khanna D et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):202-3. Abstract OP0245, doi: 10.1136/annrheumdis-2019-eular.2120
MADRID – , according to a secondary endpoint analysis of the phase 3, double-blind, randomized, controlled focuSSced trial.
After 48 weeks, a significantly lower proportion of patients treated with tocilizumab than placebo experienced any decline in lung function from baseline (50.5% versus 70.3% (P = .015), as defined by the percentage increase in predicted forced vital capacity (%pFVC). When only patients with interstitial lung disease (ILD) were considered, the respective percentages were 51.7% and 75.5% (P = .003).
In SSc-ILD patients, a clinically meaningful decline of 10% or more of the %pFVC in lung function was seen in 24.5% given placebo but in just 8.6% of those treated with tocilizumab.
“ILD is a major complication of scleroderma; it has high morbidity and mortality ... and it’s largely irreversible,” Dinesh Khanna, MD, said at the European Congress of Rheumatology.
“In this day and age, when we treat ILD, we wait for a patient to develop clinical ILD,” added Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor. Clinical ILD can be defined by symptoms, abnormal pulmonary function tests, and marked abnormalities on high resolution computed tomography (HRCT) scans. He indicated that if improving ILD was not possible, then the next best thing would be to stabilize the disease and ensure there was no worsening in lung function.
As yet, there are no disease-modifying treatments available to treat SSc but there are “ample data that interleukin-6 plays a very important role in the pathogenesis of scleroderma,” Dr. Khanna observed. Tocilizumab is a humanized monoclonal antibody against the interleukin-6 receptor.
Data from the phase 2 faSScinate trial showed initial promise for the drug in SSc where a numerical, but not statistically significant, improvement in skin thickening was seen, and the results had hinted at a possible benefit on lung function (Lancet. 2016 Jun 25;387:2630-40).
However, in the phase 3 focuSSced trial, there was no statistically significant difference in the change from baseline to week 48 modified Rodnan skin score (mRSS) between tocilizumab and placebo, which was the primary endpoint. The least square mean change in mRSS was –6.14 for tocilizumab and –4.41 for placebo (P = .0983).
A total of 205 patients with SSc were studied and randomized, 1:1 in a double-blind fashion, to receive either a once-weekly, subcutaneous dose of 162 mg tocilizumab or a weekly subcutaneous placebo injection for 48 weeks.
For inclusion in the study, patients had to have SSc that met American College of Rheumatology and European League Against Rheumatism (EULAR) criteria and be diagnosed less than 60 months previously. Patients had to have an mRSS of 10-35 units and active disease with one or more of the following: C-reactive protein of 6 mg/L or higher; erythrocyte sedimentation rate of 28 mm/h or higher; and platelet count of330 x 109 L.
“What was astonishing in the trial was that every patient had HRCT at baseline and at the end of the study,” Dr. Khanna reported. These scans showed that 64% of patients had evidence of ILD at baseline and that those treated with tocilizumab had less evidence of fibrosis at week 48 versus placebo, indicating a stabilization rather than worsening of disease.
A time to treatment failure analysis also favored tocilizumab over placebo, but there were no significant changes in patient-reported outcomes.
Dr. Khanna’s slides stated that “given that the primary endpoint for mRSS was not met, all other P values are presented for information purposes only and cannot be considered statistically significant despite the strength of the evidence.” During the Q&A after his presentation, he noted that it was unlikely that the study’s sponsors (Roche/Genentech) will now pursue a license for tocilizumab in SSc.
Nevertheless, Dr. Khanna concluded, “we have the opportunity, based on these data, to treat these patients early on, where you can preserve the lung function, which is a paradigm shift versus waiting for the lung function to decline, become clinically meaningful, significant, and then treat this patient population.”
Roche/Genentech sponsored the study. Dr. Khanna acts as a consultant to Roche/Genentech and eight other pharmaceutical companies. He owns stock in Eicos Sciences.
SOURCE: Khanna D et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):202-3. Abstract OP0245, doi: 10.1136/annrheumdis-2019-eular.2120
MADRID – , according to a secondary endpoint analysis of the phase 3, double-blind, randomized, controlled focuSSced trial.
After 48 weeks, a significantly lower proportion of patients treated with tocilizumab than placebo experienced any decline in lung function from baseline (50.5% versus 70.3% (P = .015), as defined by the percentage increase in predicted forced vital capacity (%pFVC). When only patients with interstitial lung disease (ILD) were considered, the respective percentages were 51.7% and 75.5% (P = .003).
In SSc-ILD patients, a clinically meaningful decline of 10% or more of the %pFVC in lung function was seen in 24.5% given placebo but in just 8.6% of those treated with tocilizumab.
“ILD is a major complication of scleroderma; it has high morbidity and mortality ... and it’s largely irreversible,” Dinesh Khanna, MD, said at the European Congress of Rheumatology.
“In this day and age, when we treat ILD, we wait for a patient to develop clinical ILD,” added Dr. Khanna, director of the scleroderma program at the University of Michigan, Ann Arbor. Clinical ILD can be defined by symptoms, abnormal pulmonary function tests, and marked abnormalities on high resolution computed tomography (HRCT) scans. He indicated that if improving ILD was not possible, then the next best thing would be to stabilize the disease and ensure there was no worsening in lung function.
As yet, there are no disease-modifying treatments available to treat SSc but there are “ample data that interleukin-6 plays a very important role in the pathogenesis of scleroderma,” Dr. Khanna observed. Tocilizumab is a humanized monoclonal antibody against the interleukin-6 receptor.
Data from the phase 2 faSScinate trial showed initial promise for the drug in SSc where a numerical, but not statistically significant, improvement in skin thickening was seen, and the results had hinted at a possible benefit on lung function (Lancet. 2016 Jun 25;387:2630-40).
However, in the phase 3 focuSSced trial, there was no statistically significant difference in the change from baseline to week 48 modified Rodnan skin score (mRSS) between tocilizumab and placebo, which was the primary endpoint. The least square mean change in mRSS was –6.14 for tocilizumab and –4.41 for placebo (P = .0983).
A total of 205 patients with SSc were studied and randomized, 1:1 in a double-blind fashion, to receive either a once-weekly, subcutaneous dose of 162 mg tocilizumab or a weekly subcutaneous placebo injection for 48 weeks.
For inclusion in the study, patients had to have SSc that met American College of Rheumatology and European League Against Rheumatism (EULAR) criteria and be diagnosed less than 60 months previously. Patients had to have an mRSS of 10-35 units and active disease with one or more of the following: C-reactive protein of 6 mg/L or higher; erythrocyte sedimentation rate of 28 mm/h or higher; and platelet count of330 x 109 L.
“What was astonishing in the trial was that every patient had HRCT at baseline and at the end of the study,” Dr. Khanna reported. These scans showed that 64% of patients had evidence of ILD at baseline and that those treated with tocilizumab had less evidence of fibrosis at week 48 versus placebo, indicating a stabilization rather than worsening of disease.
A time to treatment failure analysis also favored tocilizumab over placebo, but there were no significant changes in patient-reported outcomes.
Dr. Khanna’s slides stated that “given that the primary endpoint for mRSS was not met, all other P values are presented for information purposes only and cannot be considered statistically significant despite the strength of the evidence.” During the Q&A after his presentation, he noted that it was unlikely that the study’s sponsors (Roche/Genentech) will now pursue a license for tocilizumab in SSc.
Nevertheless, Dr. Khanna concluded, “we have the opportunity, based on these data, to treat these patients early on, where you can preserve the lung function, which is a paradigm shift versus waiting for the lung function to decline, become clinically meaningful, significant, and then treat this patient population.”
Roche/Genentech sponsored the study. Dr. Khanna acts as a consultant to Roche/Genentech and eight other pharmaceutical companies. He owns stock in Eicos Sciences.
SOURCE: Khanna D et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):202-3. Abstract OP0245, doi: 10.1136/annrheumdis-2019-eular.2120
REPORTING FROM THE EULAR 2019 CONGRESS

Bullous Systemic Lupus Erythematosus Successfully Treated With Rituximab
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
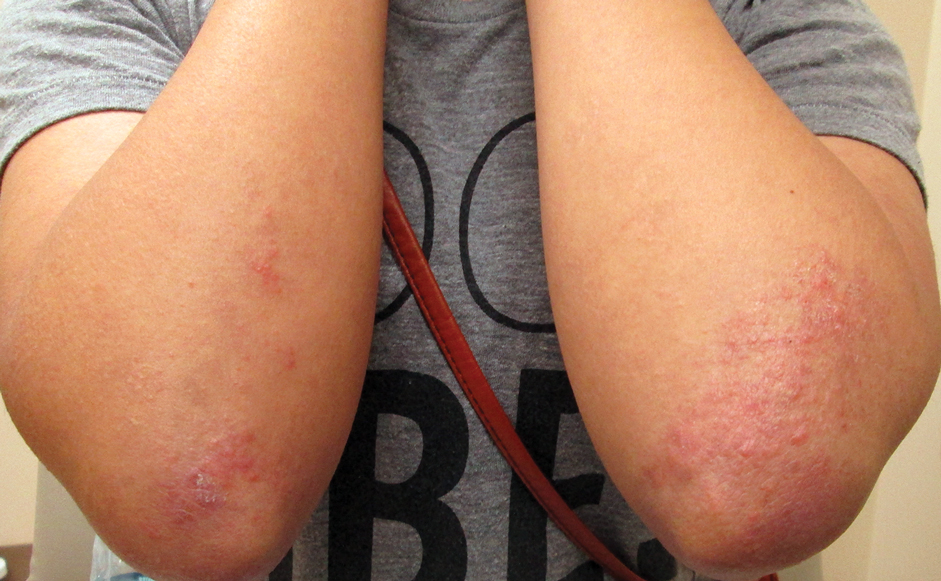
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
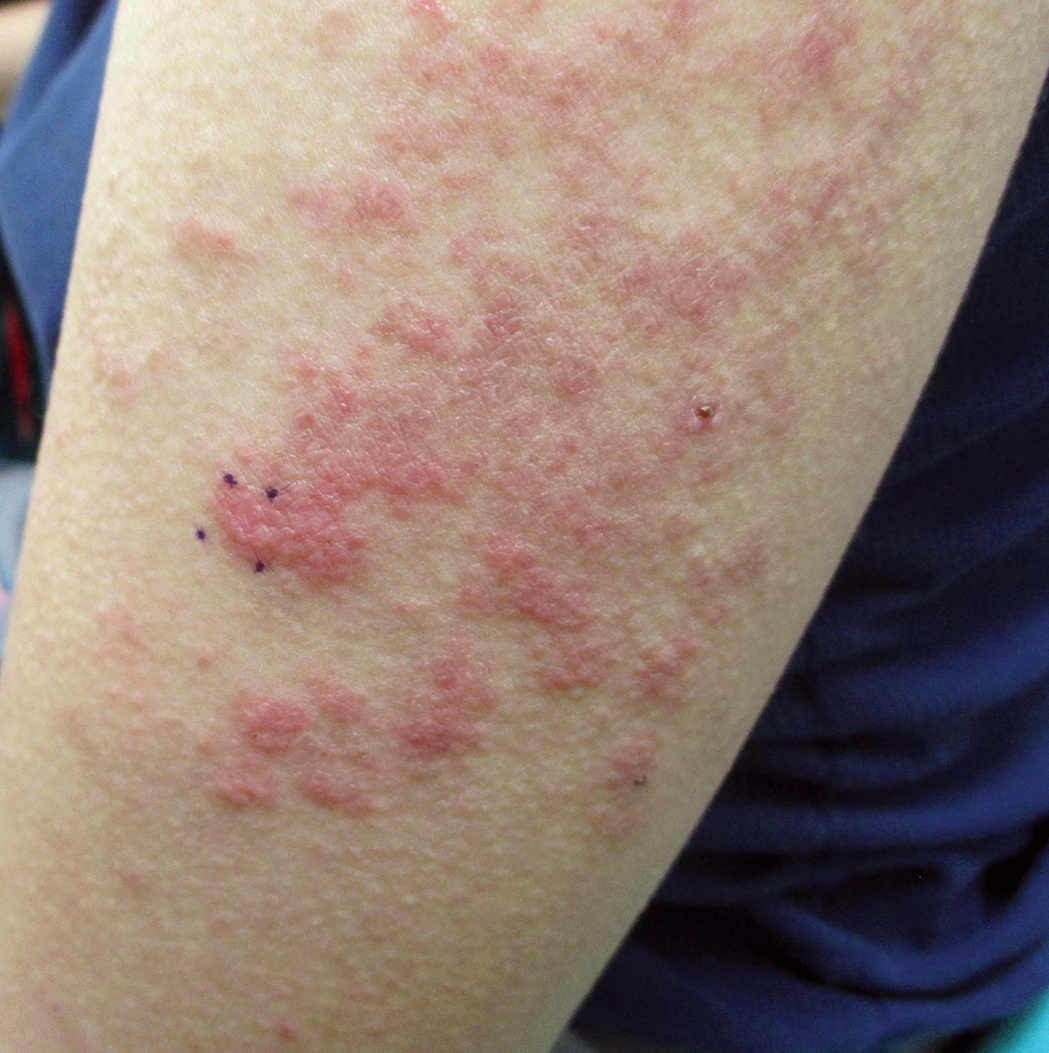
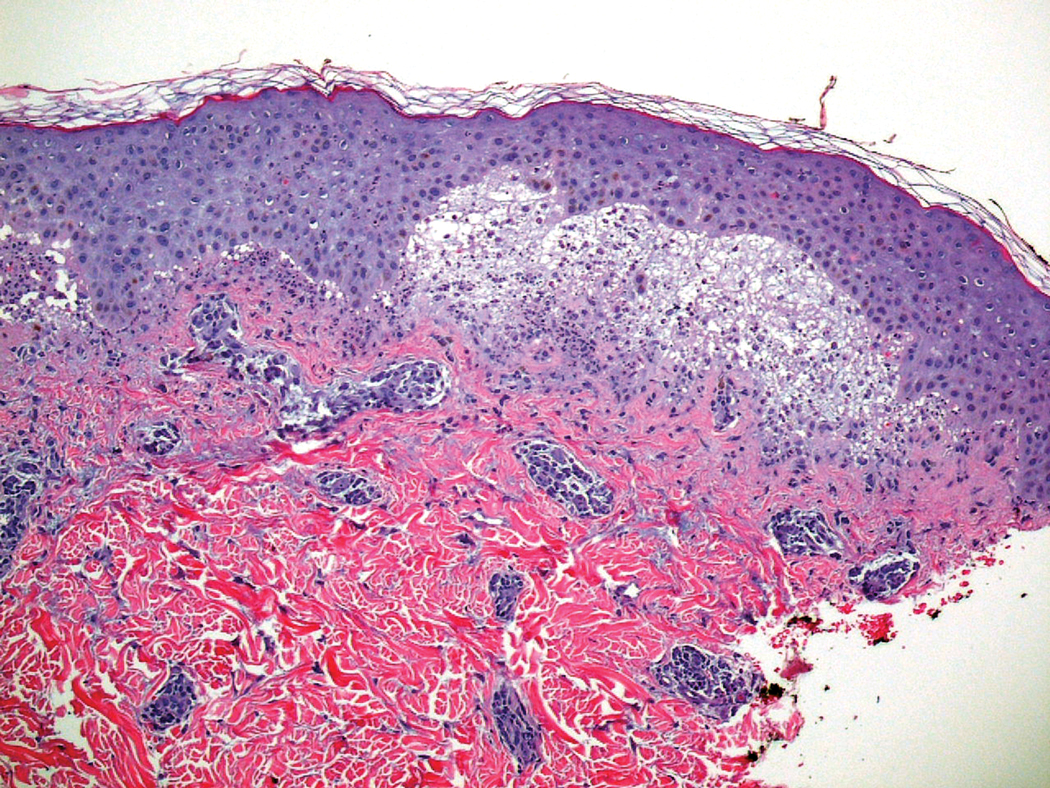
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Practice Points
- Bullous systemic lupus erythematosus (BSLE) can present with a waxing and waning course punctuated by flares.
- Different clinical presentations can occur over the disease course.
- Rituximab is a viable treatment option in BSLE.
Graham-Little-Piccardi-Lassueur Syndrome
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.

The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
Practice Points
- Graham-Little-Piccardi-Lassueur syndrome (GLPL) is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp.
- Graham-Little-Piccardi-Lassueur syndrome is considered one of the 3 clinical variants of lichen planopilaris.
- Potential therapies for GLPL include hydroxychloroquine, cyclosporine, tetracyclines, and pioglitazone.

Intradermal etanercept improves discoid lupus
BIRMINGHAM, ENGLAND – Intradermal delivery of a tumor necrosis factor inhibitor (TNFi) could offer patients with discoid lupus erythematosus (DLE) a much-needed additional treatment option, according to results of a phase 2, “proof-of-concept” study.

Overall, 14 (56%) of the 25 patients in the study achieved a 20% or greater reduction in disease activity from baseline to week 12 via intradermal injection of etanercept (Enbrel), which was assessed via the modified limited Score of Activity and Damage in DLE (ML-SADDLE). About half (48%) and one-fifth (20%) also achieved greater reductions of 50% and 70%, respectively.
“Discoid lupus is a chronic form of cutaneous lupus. Usually it occurs in visible areas like the face and scalp, causing scarring, so it’s really disabling and affects patients’ quality of life,” observed the lead study investigator Md Yuzaiful Md Yusof, MBChB, PhD, NIHR Academic Clinical Lecturer at the University of Leeds, England.
“It’s also one of the most resistant manifestations of lupus,” he said during a poster presentation at the annual conference of the British Society for Rheumatology. “Usually, when people have discoid lupus, the dermatologist gives antimalarial treatment, but only 50% of people respond to these drugs. So, what happens to the rest of them?” Basically, it is trial and error, Dr. Md Yusof said; some patients may be given disease-modifying antirheumatic drugs and in some patients this may work well, but in others there may be toxicity that contraindicates treatment.
B-cell therapy with rituximab (Rituxan) has not been successful, he said. In a previous study of 35 patients with refractory discoid lupus, none of the patients responded to rituximab and half of them actually flared after taking the drug.
There is a pathologic case for using anti-TNF therapy in DLE, but the use of TNFis is not without concern. Such treatment can increase antinuclear antibody production and make lupus worse. “In order to overcome this, as the lesion is quite small, we don’t need to use a systemic approach,” Dr. Md Yusof explained in an interview. “If you give directly, it should just be confined to the lesion and not absorbed, that’s the whole idea of thinking outside the box.” He noted that if it worked, such treatment would be for inducing remission and not for maintenance.
The study, “Targeted therapy using intradermal injection of etanercept for remission induction in discoid lupus erythematosus” (TARGET-DLE) was designed to test the validity of using intradermal rather than subcutaneous TNFi therapy in patients with discoid lupus.
Dr. Md Yusof noted that only 25 patients needed to be recruited into the single-arm, prospective trial as a “Simon’s two-stage minimized design” was used (Control Clin Trials. 1989;10[1]:1-10). This involved treating the first few patients to see if a response occurred and if it did, carrying on with treating the others, but if no response occurred in at least two patients, the trial would stop completely.
Adult patients were eligible for inclusion if they had one or more active DLE lesions and had not responded to antimalarial treatment. Stable doses of DMARDs and up to 10 mg of oral prednisolone daily was permitted if already being taken prior to entering the study.
Etanercept was injected intradermally around the most symptomatic lesion once a week for up to 12 weeks. The dosage was determined based on the radius of the selected discoid lesion. Over an 18-month period, all 25 patients were recruited, including 18 women. The median age of patients was 47 years, and six had systemic lupus erythematosus. The median number of prior DMARDs was 5 but ranged from 1 to 16, indicating a very resistant patient population.
The primary endpoint was at least 6 of the 25 patients having at least a 20% reduction in ML-SADDLE at week 12; 14 (56%) patients achieved this.
“We didn’t use CLASI [Cutaneous Lupus Area and Severity Index Activity Score] because that only includes erythema and atrophy,” Dr. Md Yusof explained. “In discoid lupus, induration is quite important as well, so that’s why we used ML-SADDLE. We called it ‘modified limited’ because the original SADDLE score is based on the whole organ score, but we only calculated the one lesion that we wanted to treat.”
In addition to meeting the primary endpoint, several secondary endpoints were met, including significant improvements in scores on visual analog scales as determined by pre- and posttreatment scoring by physicians (53.1 mm vs. 23.2 mm; P less than .001) and patients (56.9 mm vs. 29.7 mm; P = .001). Mean Dermatology Life Quality Index (DLQI) score significantly improved between pre- and post treatment, as did blood perfusion under the skin based on laser Doppler imaging and infrared thermography. However, no difference was seen with optical coherence tomography.
“There were only four grade 3/4 toxicities, and importantly, none of the SLE patients got worse, and none with DLE only converted into SLE,” Dr. Md Yusof reported. Of the four grade 3/4 adverse events, two were chest infections, one was heart failure, and one was a worsening of chilblains.
“It was a full-powered phase 2 trial, and because it was positive, now we can go to phase 3 trial,” he added.
Before conducting a phase 3 trial, however, Dr. Md Yusof wants to refine how the TNFi is delivered. Perhaps an intradermal patch with microneedles could be used. This would be left on the skin for a short amount of time to allow drug delivery and then removed. It could help ensure that all patients comply with treatment and perhaps even self-administer, he noted.
“The median compliance rate was 80%, which is not too bad, but I think when we come to run a phase 3 trial, I’m looking to improve the drug delivery,” he said. Changing the delivery method will need to be validated before a phase 3 trial can be started.
The study was not commercially funded. Dr. Md Yusof had no disclosures. Pfizer provided the study drug free of charge.
SOURCE: Md Yusof MY et al. Rheumatology. 2019;58(suppl 3): Abstract 244. doi: 10.1093/rheumatology/kez107.060.
BIRMINGHAM, ENGLAND – Intradermal delivery of a tumor necrosis factor inhibitor (TNFi) could offer patients with discoid lupus erythematosus (DLE) a much-needed additional treatment option, according to results of a phase 2, “proof-of-concept” study.
Overall, 14 (56%) of the 25 patients in the study achieved a 20% or greater reduction in disease activity from baseline to week 12 via intradermal injection of etanercept (Enbrel), which was assessed via the modified limited Score of Activity and Damage in DLE (ML-SADDLE). About half (48%) and one-fifth (20%) also achieved greater reductions of 50% and 70%, respectively.
“Discoid lupus is a chronic form of cutaneous lupus. Usually it occurs in visible areas like the face and scalp, causing scarring, so it’s really disabling and affects patients’ quality of life,” observed the lead study investigator Md Yuzaiful Md Yusof, MBChB, PhD, NIHR Academic Clinical Lecturer at the University of Leeds, England.
“It’s also one of the most resistant manifestations of lupus,” he said during a poster presentation at the annual conference of the British Society for Rheumatology. “Usually, when people have discoid lupus, the dermatologist gives antimalarial treatment, but only 50% of people respond to these drugs. So, what happens to the rest of them?” Basically, it is trial and error, Dr. Md Yusof said; some patients may be given disease-modifying antirheumatic drugs and in some patients this may work well, but in others there may be toxicity that contraindicates treatment.
B-cell therapy with rituximab (Rituxan) has not been successful, he said. In a previous study of 35 patients with refractory discoid lupus, none of the patients responded to rituximab and half of them actually flared after taking the drug.
There is a pathologic case for using anti-TNF therapy in DLE, but the use of TNFis is not without concern. Such treatment can increase antinuclear antibody production and make lupus worse. “In order to overcome this, as the lesion is quite small, we don’t need to use a systemic approach,” Dr. Md Yusof explained in an interview. “If you give directly, it should just be confined to the lesion and not absorbed, that’s the whole idea of thinking outside the box.” He noted that if it worked, such treatment would be for inducing remission and not for maintenance.
The study, “Targeted therapy using intradermal injection of etanercept for remission induction in discoid lupus erythematosus” (TARGET-DLE) was designed to test the validity of using intradermal rather than subcutaneous TNFi therapy in patients with discoid lupus.
Dr. Md Yusof noted that only 25 patients needed to be recruited into the single-arm, prospective trial as a “Simon’s two-stage minimized design” was used (Control Clin Trials. 1989;10[1]:1-10). This involved treating the first few patients to see if a response occurred and if it did, carrying on with treating the others, but if no response occurred in at least two patients, the trial would stop completely.
Adult patients were eligible for inclusion if they had one or more active DLE lesions and had not responded to antimalarial treatment. Stable doses of DMARDs and up to 10 mg of oral prednisolone daily was permitted if already being taken prior to entering the study.
Etanercept was injected intradermally around the most symptomatic lesion once a week for up to 12 weeks. The dosage was determined based on the radius of the selected discoid lesion. Over an 18-month period, all 25 patients were recruited, including 18 women. The median age of patients was 47 years, and six had systemic lupus erythematosus. The median number of prior DMARDs was 5 but ranged from 1 to 16, indicating a very resistant patient population.
The primary endpoint was at least 6 of the 25 patients having at least a 20% reduction in ML-SADDLE at week 12; 14 (56%) patients achieved this.
“We didn’t use CLASI [Cutaneous Lupus Area and Severity Index Activity Score] because that only includes erythema and atrophy,” Dr. Md Yusof explained. “In discoid lupus, induration is quite important as well, so that’s why we used ML-SADDLE. We called it ‘modified limited’ because the original SADDLE score is based on the whole organ score, but we only calculated the one lesion that we wanted to treat.”
In addition to meeting the primary endpoint, several secondary endpoints were met, including significant improvements in scores on visual analog scales as determined by pre- and posttreatment scoring by physicians (53.1 mm vs. 23.2 mm; P less than .001) and patients (56.9 mm vs. 29.7 mm; P = .001). Mean Dermatology Life Quality Index (DLQI) score significantly improved between pre- and post treatment, as did blood perfusion under the skin based on laser Doppler imaging and infrared thermography. However, no difference was seen with optical coherence tomography.
“There were only four grade 3/4 toxicities, and importantly, none of the SLE patients got worse, and none with DLE only converted into SLE,” Dr. Md Yusof reported. Of the four grade 3/4 adverse events, two were chest infections, one was heart failure, and one was a worsening of chilblains.
“It was a full-powered phase 2 trial, and because it was positive, now we can go to phase 3 trial,” he added.
Before conducting a phase 3 trial, however, Dr. Md Yusof wants to refine how the TNFi is delivered. Perhaps an intradermal patch with microneedles could be used. This would be left on the skin for a short amount of time to allow drug delivery and then removed. It could help ensure that all patients comply with treatment and perhaps even self-administer, he noted.
“The median compliance rate was 80%, which is not too bad, but I think when we come to run a phase 3 trial, I’m looking to improve the drug delivery,” he said. Changing the delivery method will need to be validated before a phase 3 trial can be started.
The study was not commercially funded. Dr. Md Yusof had no disclosures. Pfizer provided the study drug free of charge.
SOURCE: Md Yusof MY et al. Rheumatology. 2019;58(suppl 3): Abstract 244. doi: 10.1093/rheumatology/kez107.060.
BIRMINGHAM, ENGLAND – Intradermal delivery of a tumor necrosis factor inhibitor (TNFi) could offer patients with discoid lupus erythematosus (DLE) a much-needed additional treatment option, according to results of a phase 2, “proof-of-concept” study.
Overall, 14 (56%) of the 25 patients in the study achieved a 20% or greater reduction in disease activity from baseline to week 12 via intradermal injection of etanercept (Enbrel), which was assessed via the modified limited Score of Activity and Damage in DLE (ML-SADDLE). About half (48%) and one-fifth (20%) also achieved greater reductions of 50% and 70%, respectively.
“Discoid lupus is a chronic form of cutaneous lupus. Usually it occurs in visible areas like the face and scalp, causing scarring, so it’s really disabling and affects patients’ quality of life,” observed the lead study investigator Md Yuzaiful Md Yusof, MBChB, PhD, NIHR Academic Clinical Lecturer at the University of Leeds, England.
“It’s also one of the most resistant manifestations of lupus,” he said during a poster presentation at the annual conference of the British Society for Rheumatology. “Usually, when people have discoid lupus, the dermatologist gives antimalarial treatment, but only 50% of people respond to these drugs. So, what happens to the rest of them?” Basically, it is trial and error, Dr. Md Yusof said; some patients may be given disease-modifying antirheumatic drugs and in some patients this may work well, but in others there may be toxicity that contraindicates treatment.
B-cell therapy with rituximab (Rituxan) has not been successful, he said. In a previous study of 35 patients with refractory discoid lupus, none of the patients responded to rituximab and half of them actually flared after taking the drug.
There is a pathologic case for using anti-TNF therapy in DLE, but the use of TNFis is not without concern. Such treatment can increase antinuclear antibody production and make lupus worse. “In order to overcome this, as the lesion is quite small, we don’t need to use a systemic approach,” Dr. Md Yusof explained in an interview. “If you give directly, it should just be confined to the lesion and not absorbed, that’s the whole idea of thinking outside the box.” He noted that if it worked, such treatment would be for inducing remission and not for maintenance.
The study, “Targeted therapy using intradermal injection of etanercept for remission induction in discoid lupus erythematosus” (TARGET-DLE) was designed to test the validity of using intradermal rather than subcutaneous TNFi therapy in patients with discoid lupus.
Dr. Md Yusof noted that only 25 patients needed to be recruited into the single-arm, prospective trial as a “Simon’s two-stage minimized design” was used (Control Clin Trials. 1989;10[1]:1-10). This involved treating the first few patients to see if a response occurred and if it did, carrying on with treating the others, but if no response occurred in at least two patients, the trial would stop completely.
Adult patients were eligible for inclusion if they had one or more active DLE lesions and had not responded to antimalarial treatment. Stable doses of DMARDs and up to 10 mg of oral prednisolone daily was permitted if already being taken prior to entering the study.
Etanercept was injected intradermally around the most symptomatic lesion once a week for up to 12 weeks. The dosage was determined based on the radius of the selected discoid lesion. Over an 18-month period, all 25 patients were recruited, including 18 women. The median age of patients was 47 years, and six had systemic lupus erythematosus. The median number of prior DMARDs was 5 but ranged from 1 to 16, indicating a very resistant patient population.
The primary endpoint was at least 6 of the 25 patients having at least a 20% reduction in ML-SADDLE at week 12; 14 (56%) patients achieved this.
“We didn’t use CLASI [Cutaneous Lupus Area and Severity Index Activity Score] because that only includes erythema and atrophy,” Dr. Md Yusof explained. “In discoid lupus, induration is quite important as well, so that’s why we used ML-SADDLE. We called it ‘modified limited’ because the original SADDLE score is based on the whole organ score, but we only calculated the one lesion that we wanted to treat.”
In addition to meeting the primary endpoint, several secondary endpoints were met, including significant improvements in scores on visual analog scales as determined by pre- and posttreatment scoring by physicians (53.1 mm vs. 23.2 mm; P less than .001) and patients (56.9 mm vs. 29.7 mm; P = .001). Mean Dermatology Life Quality Index (DLQI) score significantly improved between pre- and post treatment, as did blood perfusion under the skin based on laser Doppler imaging and infrared thermography. However, no difference was seen with optical coherence tomography.
“There were only four grade 3/4 toxicities, and importantly, none of the SLE patients got worse, and none with DLE only converted into SLE,” Dr. Md Yusof reported. Of the four grade 3/4 adverse events, two were chest infections, one was heart failure, and one was a worsening of chilblains.
“It was a full-powered phase 2 trial, and because it was positive, now we can go to phase 3 trial,” he added.
Before conducting a phase 3 trial, however, Dr. Md Yusof wants to refine how the TNFi is delivered. Perhaps an intradermal patch with microneedles could be used. This would be left on the skin for a short amount of time to allow drug delivery and then removed. It could help ensure that all patients comply with treatment and perhaps even self-administer, he noted.
“The median compliance rate was 80%, which is not too bad, but I think when we come to run a phase 3 trial, I’m looking to improve the drug delivery,” he said. Changing the delivery method will need to be validated before a phase 3 trial can be started.
The study was not commercially funded. Dr. Md Yusof had no disclosures. Pfizer provided the study drug free of charge.
SOURCE: Md Yusof MY et al. Rheumatology. 2019;58(suppl 3): Abstract 244. doi: 10.1093/rheumatology/kez107.060.
REPORTING FROM BSR 2019
Infographic: Hyperhidrosis Survey Results
Studies begin to pinpoint ways to diagnose SLE earlier
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.

These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.
These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
SAN FRANCISCO – A host of novel clinical and serologic findings that physicians can put to good use right now in helping to distinguish early SLE from its many mimickers have been identified in a large study conducted on four continents, Marta Mosca, MD, PhD, observed at an international congress on systemic lupus erythematosus.
These useful findings aren’t incorporated into the current American College of Rheumatology (ACR) or Systemic Lupus International Collaborating Clinics (SLICC) lupus classification criteria, which have come under criticism for limited sensitivity in identifying early SLE. Some of the novel findings provide support for increased suspicion of early SLE, while others suggest a need to veer in another direction and assess a patient for a disease other than lupus. The study has served to provide input for the proposed new ACR/EULAR weighted SLE classification criteria, although that scheme is meant to be used only for research and not in clinical practice, explained Dr. Mosca of the University of Pisa (Italy).
She was lead author of the four-continent study, which included 616 patients referred to experienced academic lupus centers for possible SLE with a symptom duration of less than 1 year. During up to 3 years of follow-up, 389 patients were diagnosed as having SLE by experienced rheumatologists based upon their clinical judgment, without any requirement to meet the full ACR or SLICC classification criteria. The other 227 patients were determined to be SLE mimickers with conditions including lymphoma, Sjögren’s syndrome, systemic sclerosis, interstitial lung disease, fibromyalgia, antinuclear antibody–positive thyroiditis, and undifferentiated connective tissue disease.
Dr. Mosca also highlighted key recent work by other investigators aimed at speeding the diagnosis of SLE and shortening the duration of what she called “the gray zone” of diagnostic uncertainty, when autoantibodies and insidious symptoms are present but not yet sufficient to make the diagnosis of SLE by conventional criteria. It’s well established that 60%-70% of patients with mild undifferentiated connective tissue disease will remain stable without evolving into SLE during long years of follow-up.
The ultimate objective of all this work is to try to change the natural history of the disease through targeted early aggressive therapy aimed at minimizing the extent of active disease and preventing severe organ involvement.
Among the key takeaways from the four-continent study led by Dr. Mosca: Fever not related to infection was far more prevalent in early SLE than in mimicking conditions, by a margin of 34.5% versus 13.7%. On the other hand, Raynaud’s phenomenon was more than twice as prevalent among patients with mimicking conditions: 22.1% in early SLE, compared with 48.5% in SLE mimickers. Sicca symptoms were present in just 4.4% of early SLE patients versus 34.4% of SLE mimickers. Only 0.3% of early SLE patients complained of dysphagia; the rate was 20-fold higher in the SLE mimickers. Rashes atypical for lupus were twice as frequent in the SLE mimicking conditions (Arthritis Rheumatol. 2019 Jan;71[1]:91-8).
Turning to key differentiating serologic findings, Dr. Mosca noted that anti-double stranded DNA (anti-dsDNA) and anti-Sm antibodies were present in 71.7% and 30.2% of early SLE patients, respectively, compared with 6.9% and 2.6% of SLE mimickers. Anticardiolipin IgM, a positive Coombs test, anti-beta2 glycoprotein-I antibodies, leukopenia, autoimmune hemolytic anemia, and hypocomplementemia were all significantly more common in the early SLE cohort.
In contrast, antibodies to Ro (SS-A) and La (SS-B) were of no value in separating early SLE from its mimickers, according to Dr. Mosca.
Other tipoffs to early SLE
Two separate teams of British researchers have advanced the field in a highly practical way. One group showed in a study of 1,739 newly diagnosed SLE patients and 6,956 controls that in the 5 years prior to diagnosis, the SLE group averaged 9.2 primary care visits per year, compared with 3.8 for controls. The visits clustered around nonspecific complaints of arthritis and arthralgias, alopecia, and rash (Arthritis Care Res. 2017 Jun;69[6]:833-41).
“An accumulating number of primary care office visits and referrals over time should raise suspicion,” Dr. Mosca said.
Other investigators, working with 1,426 cases of newly diagnosed SLE in the U.K. Clinical Practice Research Database, observed that the proportion of patients with disease manifestations in three or more British Isles Lupus Activity Group (BILAG) symptom domains rose from 18.7% at 3 years prior to diagnosis to 39.7% in the year before diagnosis (Lupus Sci Med. 2017 Feb 10;4[1]:e000172. doi: 10.1136/lupus-2016-000172).
“These patients accrue clinical manifestations. It’s not just one symptom, it’s more of a state of being unwell. This is a suspicious factor for the development of lupus,” she continued.
And just as patients who will eventually be diagnosed with SLE accrue a growing number of signs and symptoms during the run up to diagnosis, they also accrue multiple autoantibodies. Moreover, as demonstrated by a multicenter group of U.S. investigators, patients also develop elevated levels of multiple soluble inflammatory markers more than 3.5 years prior to diagnosis of SLE. These include interleukins-5 and -6 and interferon-gamma. And less than 10 months prior to being classified as having SLE, patients develop significantly higher levels of B lymphocyte stimulator (BLyS) and a proliferation-inducing ligand known as APRIL. The investigators developed a predictive model incorporating IL-5, -6, and interferon-gamma levels with antinuclear antibody status that identified future SLE patients with 84% accuracy more than 3.5 years before diagnosis. This could prove useful in selecting high-risk patients for clinical prevention trials (J Autoimmun. 2016 Nov;74:182-93).
Researchers at the University of Leeds (England) have also zeroed in on interferon activity as playing a key role in progression from asymptomatic antinuclear antigen positivity, which is present in up to 25% of the general population, to symptomatic autoimmune connective tissue disease, which affects less than 1%. A multivariate logistic regression analysis identified two independent predictors of development of autoimmune connective tissue disease within the next 12 months: a family history of autoimmune rheumatic disease, which was associated with an 8.2-fold increased risk; and positivity for a pattern of interferon-stimulated gene activity they call IFN-Score-B (Ann Rheum Dis. 2018 Oct;77[10]:1432-9).
All of this work has led up to what Dr. Mosca called “a glance into the future”: the National Institutes of Health–supported Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE), which is now recruiting patients. This randomized, double-blind, placebo-controlled multicenter U.S. trial involves patients who are antinuclear antibody positive at a titer of 1:80 or more plus one or two additional criteria from the SLICC classification scheme. Participants are being randomized to 96 weeks of hydroxychloroquine or placebo. The goal is to learn whether hydroxychloroquine can slow disease progression, with the primary endpoint being the number of SLICC criteria met at the study’s end. The trial will also scrutinize potential biomarkers that could be used to guide treatment decisions (Trials. 2018 Dec 20;19[1]:694. doi: 10.1186/s13063-018-3076-7).
Dr. Mosca reported serving as an adviser to UCB and Lilly.
REPORTING FROM LUPUS 2019
Hydroxychloroquine adherence in SLE: worse than you thought
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.

First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.
First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
SAN FRANCISCO – Routine office measurement of hydroxychloroquine blood levels in systemic lupus erythematosus (SLE) patients accomplishes two major objectives, Nathalie Costedoat-Chalumeau, MD, asserted at an international congress on systemic lupus erythematosus.
First, measuring hydroxychloroquine levels identifies the surprisingly large number of individuals who are severely nonadherent to this cornerstone of lupus therapy despite its excellent benefit/risk ratio. Also, serial measurements coupled with brief counseling have been shown to boost poor adherence rates, noted Dr. Costedoat-Chalumeau, professor of rheumatology at Paris Descartes University.
Numerous studies have documented startlingly low adherence to hydroxychloroquine among SLE patients. Some of the same studies show prescribing physicians are often clueless as to their patients’ adherence or lack thereof.
Just how bad is the adherence problem? A recent study of 10,406 Medicaid patients with SLE who started on hydroxychloroquine showed that 85% of them were nonadherent as defined by pharmacy refill data, indicating insufficient medication on hand to cover a minimum of 80% of days during at least 1 year of follow-up.
In a novel finding, the investigators also broke down the Medicaid data month by month and identified four broad patterns of adherence/nonadherence. A total of 17% of patients were persistently adherent throughout the first year after the drug was dispensed. Another 36% were persistent nonadherers right from the get-go. A further 24% remained partially adherent, dropping down to a plateau of 30%-40% monthly adherence after the first couple of months and staying there. And 23% dropped steadily from roughly 50% adherence at month 3 to nearly total nonadherence from month 9 onward. Overall, adherence in the Medicaid cohort declined over the course of the first year (Semin Arthritis Rheum. 2018 Oct;48[2]:205-13).
Dr. Costedoat-Chalumeau was the lead investigator in a large French multicenter clinical trial known as the PLUS Study, which established that increasing the hydroxychloroquine daily dose to raise blood levels to a target of at least 1,000 ng/mL didn’t reduce the risk of flares (Ann Rheum Dis. 2013;72[11]:1786-92).
“So there is no reason to use blood drug measurements to adjust hydroxychloroquine daily dose or blood levels to prevent SLE flares. But drug levels teach us something regarding adherence,” she said.
Why routinely measuring hydroxychloroquine levels matters
Dr. Costedoat-Chalumeau and other investigators have shown that whole blood drug levels below 200 ng/mL indicate a patient is severely nonadherent. In various studies, that’s 7%-29% of SLE patients who are supposedly on hydroxychloroquine.
Also, investigators at Johns Hopkins University, Baltimore, have analyzed prospective data from the Hopkins Lupus Cohort and determined that at the first clinic visit after going on a maximum of 400 mg/day of hydroxychloroquine, only 44% of participants had a blood drug level above the 500-ng/mL threshold indicative of adherence.
Importantly, however, the Hopkins researchers also demonstrated that with repeated brief counseling of nonadherent patients as to why hydroxychloroquine is the most important medication they take for their SLE, adherence climbed in stepwise fashion with each visit in which the drug blood level was assessed: With no prior measurement, adherence was 56%; with one prior measurement, it jumped to 69%; with two, 77% of patients were adherent to hydroxychloroquine; and with three or more prior blood level checks, adherence rose to 80% (J Rheumatol. 2015 Nov;42[11]:2092-7).
It is well established that hydroxychloroquine prevents SLE flares, protects against thrombotic events, diabetes, dyslipidemia, and lupus-induced organ damage, and improves survival. Dr. Costedoat-Chalumeau’s final words on hydroxychloroquine adherence: ”Drugs don’t work in people who don’t take them.”
She reported having no financial conflicts regarding her presentation.
REPORTING FROM LUPUS 2019