User login
Unexpected Findings: A Rare Case of Signet Ring Cell Adenocarcinoma in the Small Intestine
Introduction
Signet ring cell carcinoma (SRCC) of the small intestine is very rare. It is characterized by the presence of malignant cells that contain mucin that push nuclei to the periphery. It is more aggressive compared to other adenocarcinomas due to early metastasis and poorer prognosis.
Case Presentation
A 59-year-old male with a history of HIV/AIDS, presented with complaints of anorexia, vomiting and weight loss. Initial abdominal CT showed a retroperitoneal mass causing gastric outlet obstruction. The patient elected to go home after supportive treatment and follow up as an outpatient, however, he presented 10 days later with worsening symptoms. Evaluation with CT abdomen and pelvis showed enlarging soft tissue density in the retrocrural space extending into the retroperitoneum around the aorta, as well as a 1.5 cm intraluminal cystic lesion in the duodenum. Endoscopic ultrasound revealed lymphadenopathy of celiac and porta hepatis regions, along with duodenal stenosis, stent placement for decompression was not feasible and biopsies were inconclusive. The decision was made to proceed with laparotomy for decompression and additional biopsies from the retroperitoneal mass and omental lymph nodes, which confirmed poorly differentiated adenocarcinoma with signet ring cells. The presence of a mass in the duodenum strongly suggested adenocarcinoma of small intestine origin. As the patient’s symptoms worsened, imaging revealed progression with lung metastases. The patient continued to deteriorate rapidly requiring dialysis and gangrenous cholecystitis. Given his complex medical history, patient decided to transition to comfort care.
Discussion
SRCC can present with any GI symptoms. Most important step in diagnosing SRCC is biopsy. Current treatment options for small intestinal malignancies include wide resection that includes the mesentery and corresponding lymph nodes. The use of adjuvant chemotherapy has been described only in small retrospective studies. Due to its scarcity, there isn’t sufficient data for optimal treatment strategies compared to gastric SRCC.
Conclusions
This case report highlights the importance of how rare and aggressive signet ring cell adenocarcinoma of the small intestine. There are only a few cases documented in the literature, which is why we lack data on how to manage the disease.
Introduction
Signet ring cell carcinoma (SRCC) of the small intestine is very rare. It is characterized by the presence of malignant cells that contain mucin that push nuclei to the periphery. It is more aggressive compared to other adenocarcinomas due to early metastasis and poorer prognosis.
Case Presentation
A 59-year-old male with a history of HIV/AIDS, presented with complaints of anorexia, vomiting and weight loss. Initial abdominal CT showed a retroperitoneal mass causing gastric outlet obstruction. The patient elected to go home after supportive treatment and follow up as an outpatient, however, he presented 10 days later with worsening symptoms. Evaluation with CT abdomen and pelvis showed enlarging soft tissue density in the retrocrural space extending into the retroperitoneum around the aorta, as well as a 1.5 cm intraluminal cystic lesion in the duodenum. Endoscopic ultrasound revealed lymphadenopathy of celiac and porta hepatis regions, along with duodenal stenosis, stent placement for decompression was not feasible and biopsies were inconclusive. The decision was made to proceed with laparotomy for decompression and additional biopsies from the retroperitoneal mass and omental lymph nodes, which confirmed poorly differentiated adenocarcinoma with signet ring cells. The presence of a mass in the duodenum strongly suggested adenocarcinoma of small intestine origin. As the patient’s symptoms worsened, imaging revealed progression with lung metastases. The patient continued to deteriorate rapidly requiring dialysis and gangrenous cholecystitis. Given his complex medical history, patient decided to transition to comfort care.
Discussion
SRCC can present with any GI symptoms. Most important step in diagnosing SRCC is biopsy. Current treatment options for small intestinal malignancies include wide resection that includes the mesentery and corresponding lymph nodes. The use of adjuvant chemotherapy has been described only in small retrospective studies. Due to its scarcity, there isn’t sufficient data for optimal treatment strategies compared to gastric SRCC.
Conclusions
This case report highlights the importance of how rare and aggressive signet ring cell adenocarcinoma of the small intestine. There are only a few cases documented in the literature, which is why we lack data on how to manage the disease.
Introduction
Signet ring cell carcinoma (SRCC) of the small intestine is very rare. It is characterized by the presence of malignant cells that contain mucin that push nuclei to the periphery. It is more aggressive compared to other adenocarcinomas due to early metastasis and poorer prognosis.
Case Presentation
A 59-year-old male with a history of HIV/AIDS, presented with complaints of anorexia, vomiting and weight loss. Initial abdominal CT showed a retroperitoneal mass causing gastric outlet obstruction. The patient elected to go home after supportive treatment and follow up as an outpatient, however, he presented 10 days later with worsening symptoms. Evaluation with CT abdomen and pelvis showed enlarging soft tissue density in the retrocrural space extending into the retroperitoneum around the aorta, as well as a 1.5 cm intraluminal cystic lesion in the duodenum. Endoscopic ultrasound revealed lymphadenopathy of celiac and porta hepatis regions, along with duodenal stenosis, stent placement for decompression was not feasible and biopsies were inconclusive. The decision was made to proceed with laparotomy for decompression and additional biopsies from the retroperitoneal mass and omental lymph nodes, which confirmed poorly differentiated adenocarcinoma with signet ring cells. The presence of a mass in the duodenum strongly suggested adenocarcinoma of small intestine origin. As the patient’s symptoms worsened, imaging revealed progression with lung metastases. The patient continued to deteriorate rapidly requiring dialysis and gangrenous cholecystitis. Given his complex medical history, patient decided to transition to comfort care.
Discussion
SRCC can present with any GI symptoms. Most important step in diagnosing SRCC is biopsy. Current treatment options for small intestinal malignancies include wide resection that includes the mesentery and corresponding lymph nodes. The use of adjuvant chemotherapy has been described only in small retrospective studies. Due to its scarcity, there isn’t sufficient data for optimal treatment strategies compared to gastric SRCC.
Conclusions
This case report highlights the importance of how rare and aggressive signet ring cell adenocarcinoma of the small intestine. There are only a few cases documented in the literature, which is why we lack data on how to manage the disease.
Acquired Factor VIII Deficiency Presenting as Compartment Syndrome
Compartment syndrome occurs when the interstitial tissue pressures within a confined space are elevated to a level at which the arterial perfusion is diminished. Multiple etiologies exist and can be extrinsic (a cast that is too tight or prolonged compression on a limb), iatrogenic (aggressive resuscitation, drug infiltration, arterial puncture, or a spontaneous bleed from anticoagulation), and traumatic (fracture, snake envenomation, circumferential burn, or electrocution). If the compartments are not released, irreversible changes happen to the cells, including nerve and muscle death.1 Definitive management of this emergency requires prompt fasciotomy to decompress the compartment(s).1-3
Case Presentation
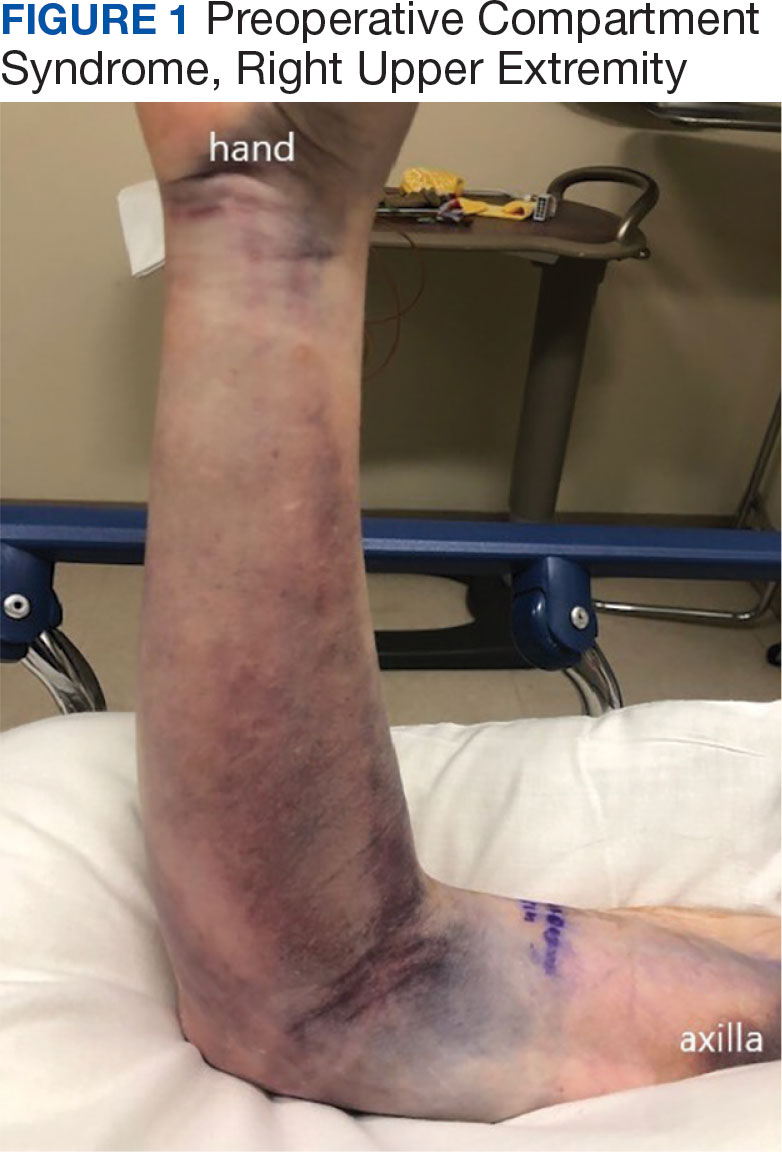
A 76-year-old right-handed woman with a history of chronic obstructive pulmonary disease, hypertension, and hyperlipidemia presented to the emergency department with 2 days of extensive right upper extremity ecchymosis and severe pain that was localized to her forearm (Figure 1). She was taking low-dose aspirin (81 mg/d) for left subclavian stenosis and over-the-counter ginkgo biloba. Leading up to the presentation, the patient was able to perform routine household chores, including yard work, cleaning, and taking care of her cats. Wrist and elbow X-rays were negative for a fracture. An upper extremity ultrasound found no venous occlusion. A computed tomography (CT) angiogram of her arm and chest found diffuse edema around the right elbow and forearm without pulmonary or right upper extremity emboli, fractures, hematoma, abscess, or air in the tissues.
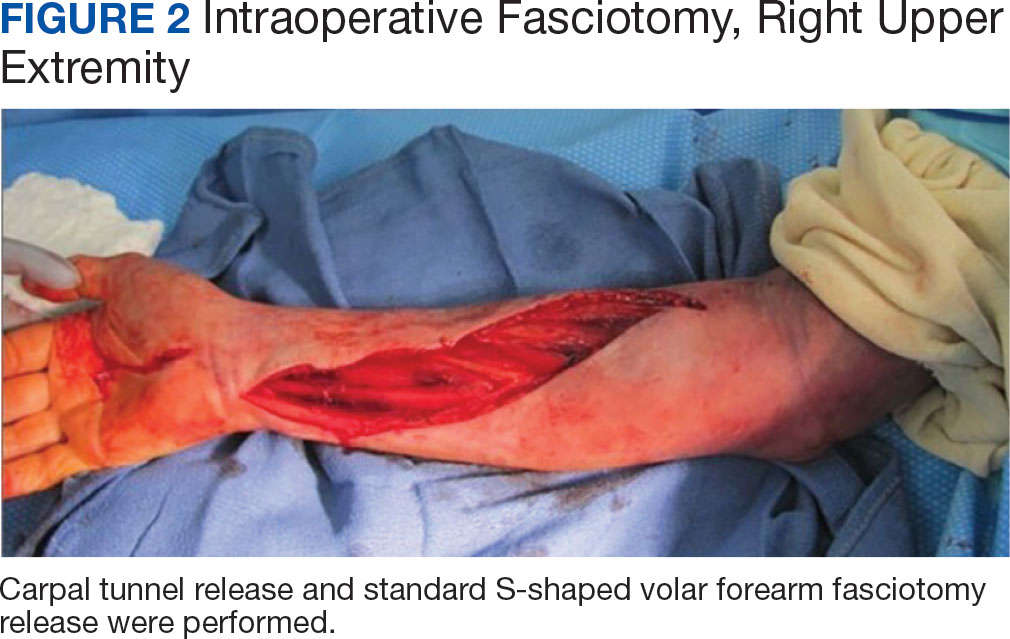
The plastic surgery service was consulted. The patient was found to have a very tense forearm and pain to passive digital extension. The 2-point discrimination and pulses were intact. The patient was diagnosed with compartment syndrome based on the examination alone and gave consent for an emergent forearm and hand fasciotomy. A carpal tunnel release and a standard S-shaped volar forearm fasciotomy release were performed, which provided immediate decompression (Figure 2). The rest of the hand and extremity were soft. Edematous, healthy flexor muscle belly was identified without a hematoma. Most of the forearm wound was left open because the skin could not be reapproximated. Oxidized regenerated cellulose (Surgicel) was placed around the wound edges and the muscle was covered with a nonadherent dressing. Hemoglobin on admission was 12.9 g/dL(reference range, 12 to 16 g/dL). Kidney function was within normal limits. The rest of the complete blood count was unremarkable. Postoperative hemoglobin was 8.6 g/dL. Over the next several days, the patient's skin edges and muscle bellies continued to slowly bleed, and her hemoglobin fell to 5.6 g/dL by postoperative Day 2. The bleeding was managed with topical oxidized regenerated cellulose, thrombin spray, a hemostatic dressing made with kaolin (QuikClot), and a transfusion of 2 units of packed red blood cells.
A hematology consultation was requested. The patient was noted to have an elevated partial thromboplastin time (PTT) since admission measuring between 39.9 to 61.7 seconds (reference range, 26.2 to 37.2 seconds) and a normal prothrombin time test with an international normalized ratio. A PTT measured 17 months prior to admission was within the normal range. She reported no personal or family history of bleeding disorders. Until recently, she had never had easy bruisability. She reported no history of heavy menses or epistaxis. The patient had no children and had never been pregnant. She had tolerated an exploratory laparotomy 40 years prior to admission without bleeding complications and had never required blood transfusions before. A PTT 1:1 mixing study revealed incomplete correction. Subsequent workup included factor VIII (FVIII) activity, factor IX activity, factor XI activity, von Willebrand factor antigen, ristocetin cofactor assay, and von Willebrand factor multimers. FVIII activity was severely reduced at 7.8% (reference, > 54%) with a positive Bethesda assay of 300 to 400 Bodansky units (BU), indicating a strong FVIII inhibitor was present and establishing a diagnosis of acquired hemophilia A. Further workup for secondary causes of acquired hemophilia A including abdominal and pelvic CT, serum protein electrophoresis, and serum free light chains, were negative. She was started on prednisone 1 mg/kg daily and rituximab 375 mg/m2. Her hemoglobin stabilized, and she required no further blood transfusions.
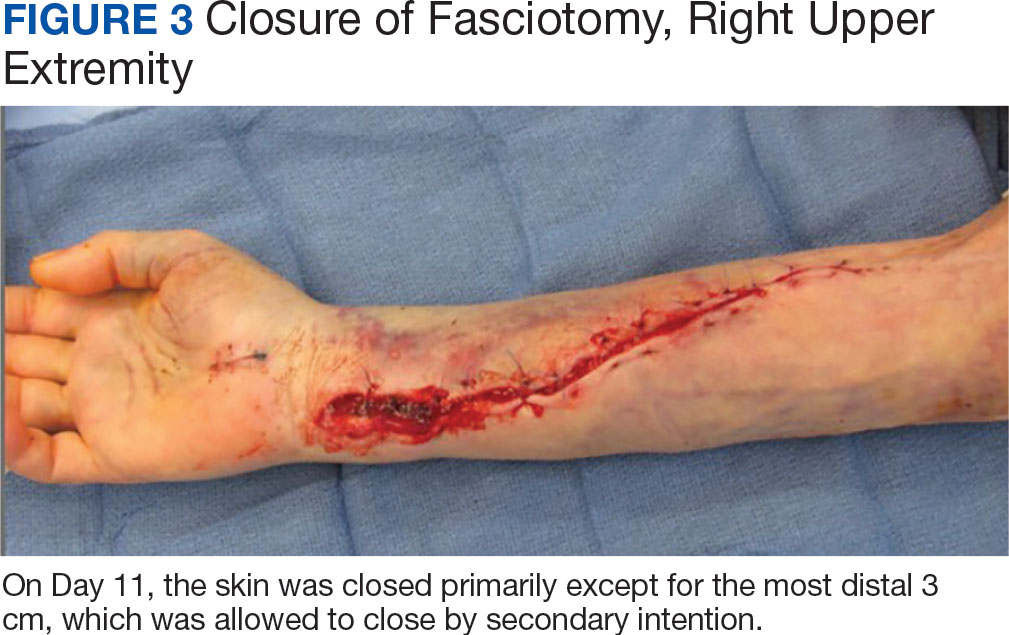
The patient underwent wound closure on postoperative Day 11. At the time of the second surgery, there was still no improvement in her FVIII levels or PTT; therefore, 70 mcg/kg of recombinant coagulation-activated FVII was given just before surgery with no bleeding complications. The skin was closed primarily except for the most distal 3 cm (Figure 3). Due to concerns regarding further bleeding with skin graft, the remaining wound was allowed to close by secondary intention. As a precaution, the wound was covered with oxidized regenerated cellulose and thrombin spray. The patient continued to progress postoperatively without bleeding complications or a need for additional transfusions. She was seen by the hand therapist before and after the second surgery to help with edema management and joint mobility. She completed 4 weekly doses of 375 mg/m² rituximab and prednisone was tapered by 10 mg weekly.
Three weeks after starting treatment, her PTT normalized, and her FVIII increased to 33.7%. The Bethesda assay remained high at 198 BU, although it was lower than at admission. She was discharged home with dressing changes and monthly follow-up appointments. The wounds were fully closed at her 3-month appointment when she proudly demonstrated full digital extension and flexion into her palm.
Discussion
Forearm compartment syndrome is most often caused by fractures—distal radius in adults and supracondylar in children.2 This case initially presented as a diagnostic puzzle to the emergency department due to the patient’s lucid review of several days of nontraumatic injury.
The clinical hallmarks of compartment syndrome are the 5 Ps: pain, pallor, paresthesia, paralysis, and pulselessness. Patients will describe the pain as out of proportion to the nature of the injury; the compartments will be tense and swollen, they will have pain to passive muscle stretch, and sensation will progressively diminish. Distal pulses are the last to go, and permanent tissue damage can still occur when pulses are present.1
Compartment Syndrome
Compartment syndrome is generally a clinical diagnosis; however, in patients who are sedated or uncooperative, or if the clinical findings are equivocal, the examination can be supplemented with intercompartmental pressures using an arterial line transducer system.2 In general, a tissue pressure of 30 mm Hg or a 20- to 30-mm Hg difference between the diastolic and compartment pressures are indications for fasciotomy.1 The hand is treated with an open carpal tunnel release, interosseous muscle release through 2 dorsal hand incisions, and thenar and hypothenar muscle release. The forearm is treated through a curved volar incision that usually decompresses the dorsal compartment, as it did in our patient. If pressures are still high in the forearm, a longitudinal dorsal incision over the mobile wad is necessary. Wounds can be closed primarily days later, left open to close by secondary intention, or reconstructed with skin grafts.2 In our patient, compartment syndrome was isolated to her forearm and the carpal tunnel release was performed prophylactically since it did not add significant time or morbidity to the surgery.
Nontraumatic upper extremity compartment syndrome is rare. A 2021 review of acute nontraumatic upper extremity compartment syndrome found a bleeding disorder as the etiology in 3 cases published in the literature between 1993 and 2016.4 One of these cases was secondary to a known diagnosis of hemophilia A in a teenager.5 Ogrodnik and colleagues described a spontaneous hand hematoma secondary to previously undiagnosed acquired hemophilia A and Waldenström macroglobulinemia.4 Ilyas and colleagues described a spontaneous hematoma in the forearm dorsal compartment in a 67-year-old woman, which presented as compartment syndrome and elevated PTT and led to a diagnosis of acquired FVIII inhibitor. The authors recommended prompt hematology consultation to coordinate treatment once this diagnosis issuspected.6 Compartment syndrome also has been found to develop slowly over weeks in patients with acquired FVIII deficiency, suggesting a high index of suspicion and frequent examinations are needed when patients with known acquired hemophilia A present with a painful extremity.7
Nontraumatic compartment syndrome in the lower extremity in patients with previously undiagnosed acquired hemophilia A has also been described in the literature.8-11 Case reports describe the delay in diagnosis as the patients were originally seen by clinicians for lower extremity pain and swelling within days of presenting to the emergency room with compartment syndrome. Persistent bleeding and abnormal laboratory results prompted further tests and examinations.8,9,11 This underscores the need to be suspicious of this unusual pathology without a history of trauma.
Acquired Hemophilia A
Acquired hemophilia A is an autoimmune disease most often found in older individuals, with a mean age of approximately 70 years.12 It is caused by the spontaneous production of neutralizing immunoglobin autoantibodies that target endogenous FVIII. Many cases are idiopathic; however, up to 50% of cases are associated with underlying autoimmunity, malignancy (especially lymphoproliferative disorders), or pregnancy. It often presents as bleeding that is subcutaneous or in the gastrointestinal system, muscle, retroperitoneal space, or genitourinary system. Unlike congenital hemophilia A, joint bleeding is rare.13
The diagnosis is suspected with an isolated elevated PTT in the absence of other coagulation abnormalities. A 1:1 mixing study will typically show incomplete correction, which suggests the presence of an inhibitor. FVIII activity is reduced, and the FVIII inhibitor is confirmed with the Bethesda assay. Clinically active bleeding is treated with bypassing agents such as recombinant coagulation-activated FVII, activated prothrombin complex concentrates such as anti-inhibitor coagulant complex (FEIBA), or recombinant porcine FVIII.12,14 Not all patients require hemostatic treatment, but close monitoring, education, recognition, and immediate treatment, if needed, are indicated.13 Immunosuppressive therapy (corticosteroids, rituximab, and/or cyclophosphamide) is prescribed to eradicate the antibodies and induce remission.12
Conclusions
An older woman without a preceding trauma was diagnosed with an unusual case of acute compartment syndrome in the forearm. No hematoma was found, but muscle and skin bleeding plus an elevated PTT prompted a hematology workup, and, ultimately, the diagnosis of FVIII inhibitor secondary to acquired hemophilia A.
While a nontraumatic cause of compartment syndrome is rare, it should be considered in differential diagnosis for clinicians who see hand and upper extremity emergencies. An isolated elevated PTT in a patient with a bleed should raise suspicions and trigger immediate further evaluation. Once suspected, multidisciplinary treatment is indicated for immediate and long-term successful outcomes.
Acknowledgments
This manuscript is the result of work supported withresources and the use of facilities at the North Florida/South Georgia Veterans Health System, Gainesville, Florida.
1. Leversedge FJ, Moore TJ, Peterson BC, Seiler JG 3rd. Compartment syndrome of the upper extremity. J Hand Surg Am. 2011;36:544-559. doi:10.1016/j.jhsa.2010.12.008
2. Kalyani BS, Fisher BE, Roberts CS, Giannoudis PV. Compartment syndrome of the forearm: a systematic review. J Hand Surg Am. 2011;36:535-543. doi:10.1016/j.jhsa.2010.12.007
3. Steadman W, Wu R, Hamilton AT, Richardson MD, Wall CJ. Review article: a comprehensive review of unusual causes of acute limb compartment syndrome. Emerg Med Australas. 2022;34:871-876. doi:10.1111/1742-6723.14098
4. Ogrodnik J, Oliver JD, Cani D, Boczar D, Huayllani MT, Restrepo DJ, et al. Clinical case of acute non-traumatic hand compartment syndrome and systematic review for the upper extremity. Hand (N Y). 2021;16:285-291. doi:10.1177/1558944719856106
5. Kim J, Zelken J, Sacks JM. Case report. Spontaneous forearm compartment syndrome in a boy with hemophilia a: a therapeutic dilemma. Eplasty. 2013:13:e16.
6. Ilyas AM, Wisbeck JM, Shaffer GW, Thoder JJ. Upper extremity compartment syndrome secondary to acquired factor VIII inhibitor. A case report. J Bone Joint Surg Am. 2005;87:1606-1608. doi:10.2106/JBJS.C.01720
7. Adeclat GJ, Hayes M, Amick M, Kahan J, Halim A. Acute forearm compartment syndrome in the setting of acquired hemophilia A. Case Reports Plast Surg Hand Surg. 2022;9:140-144. doi:10.1080/23320885.2022.2071274
8. Abudaqqa RY, Arun KP, Mas AJA, Abushaaban FA. Acute atraumatic compartment syndrome of the thigh due to acquired coagulopathy disorder: a case report in known healthy patient. J Orthop Case Rep. 2021;11:59-62. doi:10.13107/jocr.2021.v11.i08.2366
9. Alidoost M, Conte GA, Chaudry R, Nahum K, Marchesani D. A unique presentation of spontaneous compartment syndrome due to acquired hemophilia A and associated malignancy: case report and literature review. World J Oncol. 2020;11:72-75. doi:10.14740/wjon1260
10. Jentzsch T, Brand-Staufer B, Schäfer FP, Wanner GA, Simmen H-P. Illustrated operative management of spontaneous bleeding and compartment syndrome of the lower extremity in a patient with acquired hemophilia A: a case report. J Med Case Rep. 2014;8:132. doi:10.1186/1752-1947-8-132
11. Pham TV, Sorenson CA, Nable JV. Acquired factor VIII deficiency presenting with compartment syndrome. Am J Emerg Med. 2014;32:195.e1-2. doi:10.1016/j.ajem.2013.09.022
12. Tiede A, Zieger B, Lisman T. Acquired bleeding disorders. Haemophilia. 2022;28(suppl 4):68-76. doi:10.1111/hae.14548
13. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92:695-705. doi:10.1002/ajh.24777
14. Ilkhchoui Y, Koshkin E, Windsor JJ, Petersen TR, Charles M, Pack JD. Perioperative management of acquired hemophilia A: a case report and review of literature. Anesth Pain Med. 2013;4:e11906. doi:10.5812/aapm.11906
Compartment syndrome occurs when the interstitial tissue pressures within a confined space are elevated to a level at which the arterial perfusion is diminished. Multiple etiologies exist and can be extrinsic (a cast that is too tight or prolonged compression on a limb), iatrogenic (aggressive resuscitation, drug infiltration, arterial puncture, or a spontaneous bleed from anticoagulation), and traumatic (fracture, snake envenomation, circumferential burn, or electrocution). If the compartments are not released, irreversible changes happen to the cells, including nerve and muscle death.1 Definitive management of this emergency requires prompt fasciotomy to decompress the compartment(s).1-3
Case Presentation
A 76-year-old right-handed woman with a history of chronic obstructive pulmonary disease, hypertension, and hyperlipidemia presented to the emergency department with 2 days of extensive right upper extremity ecchymosis and severe pain that was localized to her forearm (Figure 1). She was taking low-dose aspirin (81 mg/d) for left subclavian stenosis and over-the-counter ginkgo biloba. Leading up to the presentation, the patient was able to perform routine household chores, including yard work, cleaning, and taking care of her cats. Wrist and elbow X-rays were negative for a fracture. An upper extremity ultrasound found no venous occlusion. A computed tomography (CT) angiogram of her arm and chest found diffuse edema around the right elbow and forearm without pulmonary or right upper extremity emboli, fractures, hematoma, abscess, or air in the tissues.
The plastic surgery service was consulted. The patient was found to have a very tense forearm and pain to passive digital extension. The 2-point discrimination and pulses were intact. The patient was diagnosed with compartment syndrome based on the examination alone and gave consent for an emergent forearm and hand fasciotomy. A carpal tunnel release and a standard S-shaped volar forearm fasciotomy release were performed, which provided immediate decompression (Figure 2). The rest of the hand and extremity were soft. Edematous, healthy flexor muscle belly was identified without a hematoma. Most of the forearm wound was left open because the skin could not be reapproximated. Oxidized regenerated cellulose (Surgicel) was placed around the wound edges and the muscle was covered with a nonadherent dressing. Hemoglobin on admission was 12.9 g/dL(reference range, 12 to 16 g/dL). Kidney function was within normal limits. The rest of the complete blood count was unremarkable. Postoperative hemoglobin was 8.6 g/dL. Over the next several days, the patient's skin edges and muscle bellies continued to slowly bleed, and her hemoglobin fell to 5.6 g/dL by postoperative Day 2. The bleeding was managed with topical oxidized regenerated cellulose, thrombin spray, a hemostatic dressing made with kaolin (QuikClot), and a transfusion of 2 units of packed red blood cells.
A hematology consultation was requested. The patient was noted to have an elevated partial thromboplastin time (PTT) since admission measuring between 39.9 to 61.7 seconds (reference range, 26.2 to 37.2 seconds) and a normal prothrombin time test with an international normalized ratio. A PTT measured 17 months prior to admission was within the normal range. She reported no personal or family history of bleeding disorders. Until recently, she had never had easy bruisability. She reported no history of heavy menses or epistaxis. The patient had no children and had never been pregnant. She had tolerated an exploratory laparotomy 40 years prior to admission without bleeding complications and had never required blood transfusions before. A PTT 1:1 mixing study revealed incomplete correction. Subsequent workup included factor VIII (FVIII) activity, factor IX activity, factor XI activity, von Willebrand factor antigen, ristocetin cofactor assay, and von Willebrand factor multimers. FVIII activity was severely reduced at 7.8% (reference, > 54%) with a positive Bethesda assay of 300 to 400 Bodansky units (BU), indicating a strong FVIII inhibitor was present and establishing a diagnosis of acquired hemophilia A. Further workup for secondary causes of acquired hemophilia A including abdominal and pelvic CT, serum protein electrophoresis, and serum free light chains, were negative. She was started on prednisone 1 mg/kg daily and rituximab 375 mg/m2. Her hemoglobin stabilized, and she required no further blood transfusions.
The patient underwent wound closure on postoperative Day 11. At the time of the second surgery, there was still no improvement in her FVIII levels or PTT; therefore, 70 mcg/kg of recombinant coagulation-activated FVII was given just before surgery with no bleeding complications. The skin was closed primarily except for the most distal 3 cm (Figure 3). Due to concerns regarding further bleeding with skin graft, the remaining wound was allowed to close by secondary intention. As a precaution, the wound was covered with oxidized regenerated cellulose and thrombin spray. The patient continued to progress postoperatively without bleeding complications or a need for additional transfusions. She was seen by the hand therapist before and after the second surgery to help with edema management and joint mobility. She completed 4 weekly doses of 375 mg/m² rituximab and prednisone was tapered by 10 mg weekly.
Three weeks after starting treatment, her PTT normalized, and her FVIII increased to 33.7%. The Bethesda assay remained high at 198 BU, although it was lower than at admission. She was discharged home with dressing changes and monthly follow-up appointments. The wounds were fully closed at her 3-month appointment when she proudly demonstrated full digital extension and flexion into her palm.
Discussion
Forearm compartment syndrome is most often caused by fractures—distal radius in adults and supracondylar in children.2 This case initially presented as a diagnostic puzzle to the emergency department due to the patient’s lucid review of several days of nontraumatic injury.
The clinical hallmarks of compartment syndrome are the 5 Ps: pain, pallor, paresthesia, paralysis, and pulselessness. Patients will describe the pain as out of proportion to the nature of the injury; the compartments will be tense and swollen, they will have pain to passive muscle stretch, and sensation will progressively diminish. Distal pulses are the last to go, and permanent tissue damage can still occur when pulses are present.1
Compartment Syndrome
Compartment syndrome is generally a clinical diagnosis; however, in patients who are sedated or uncooperative, or if the clinical findings are equivocal, the examination can be supplemented with intercompartmental pressures using an arterial line transducer system.2 In general, a tissue pressure of 30 mm Hg or a 20- to 30-mm Hg difference between the diastolic and compartment pressures are indications for fasciotomy.1 The hand is treated with an open carpal tunnel release, interosseous muscle release through 2 dorsal hand incisions, and thenar and hypothenar muscle release. The forearm is treated through a curved volar incision that usually decompresses the dorsal compartment, as it did in our patient. If pressures are still high in the forearm, a longitudinal dorsal incision over the mobile wad is necessary. Wounds can be closed primarily days later, left open to close by secondary intention, or reconstructed with skin grafts.2 In our patient, compartment syndrome was isolated to her forearm and the carpal tunnel release was performed prophylactically since it did not add significant time or morbidity to the surgery.
Nontraumatic upper extremity compartment syndrome is rare. A 2021 review of acute nontraumatic upper extremity compartment syndrome found a bleeding disorder as the etiology in 3 cases published in the literature between 1993 and 2016.4 One of these cases was secondary to a known diagnosis of hemophilia A in a teenager.5 Ogrodnik and colleagues described a spontaneous hand hematoma secondary to previously undiagnosed acquired hemophilia A and Waldenström macroglobulinemia.4 Ilyas and colleagues described a spontaneous hematoma in the forearm dorsal compartment in a 67-year-old woman, which presented as compartment syndrome and elevated PTT and led to a diagnosis of acquired FVIII inhibitor. The authors recommended prompt hematology consultation to coordinate treatment once this diagnosis issuspected.6 Compartment syndrome also has been found to develop slowly over weeks in patients with acquired FVIII deficiency, suggesting a high index of suspicion and frequent examinations are needed when patients with known acquired hemophilia A present with a painful extremity.7
Nontraumatic compartment syndrome in the lower extremity in patients with previously undiagnosed acquired hemophilia A has also been described in the literature.8-11 Case reports describe the delay in diagnosis as the patients were originally seen by clinicians for lower extremity pain and swelling within days of presenting to the emergency room with compartment syndrome. Persistent bleeding and abnormal laboratory results prompted further tests and examinations.8,9,11 This underscores the need to be suspicious of this unusual pathology without a history of trauma.
Acquired Hemophilia A
Acquired hemophilia A is an autoimmune disease most often found in older individuals, with a mean age of approximately 70 years.12 It is caused by the spontaneous production of neutralizing immunoglobin autoantibodies that target endogenous FVIII. Many cases are idiopathic; however, up to 50% of cases are associated with underlying autoimmunity, malignancy (especially lymphoproliferative disorders), or pregnancy. It often presents as bleeding that is subcutaneous or in the gastrointestinal system, muscle, retroperitoneal space, or genitourinary system. Unlike congenital hemophilia A, joint bleeding is rare.13
The diagnosis is suspected with an isolated elevated PTT in the absence of other coagulation abnormalities. A 1:1 mixing study will typically show incomplete correction, which suggests the presence of an inhibitor. FVIII activity is reduced, and the FVIII inhibitor is confirmed with the Bethesda assay. Clinically active bleeding is treated with bypassing agents such as recombinant coagulation-activated FVII, activated prothrombin complex concentrates such as anti-inhibitor coagulant complex (FEIBA), or recombinant porcine FVIII.12,14 Not all patients require hemostatic treatment, but close monitoring, education, recognition, and immediate treatment, if needed, are indicated.13 Immunosuppressive therapy (corticosteroids, rituximab, and/or cyclophosphamide) is prescribed to eradicate the antibodies and induce remission.12
Conclusions
An older woman without a preceding trauma was diagnosed with an unusual case of acute compartment syndrome in the forearm. No hematoma was found, but muscle and skin bleeding plus an elevated PTT prompted a hematology workup, and, ultimately, the diagnosis of FVIII inhibitor secondary to acquired hemophilia A.
While a nontraumatic cause of compartment syndrome is rare, it should be considered in differential diagnosis for clinicians who see hand and upper extremity emergencies. An isolated elevated PTT in a patient with a bleed should raise suspicions and trigger immediate further evaluation. Once suspected, multidisciplinary treatment is indicated for immediate and long-term successful outcomes.
Acknowledgments
This manuscript is the result of work supported withresources and the use of facilities at the North Florida/South Georgia Veterans Health System, Gainesville, Florida.
Compartment syndrome occurs when the interstitial tissue pressures within a confined space are elevated to a level at which the arterial perfusion is diminished. Multiple etiologies exist and can be extrinsic (a cast that is too tight or prolonged compression on a limb), iatrogenic (aggressive resuscitation, drug infiltration, arterial puncture, or a spontaneous bleed from anticoagulation), and traumatic (fracture, snake envenomation, circumferential burn, or electrocution). If the compartments are not released, irreversible changes happen to the cells, including nerve and muscle death.1 Definitive management of this emergency requires prompt fasciotomy to decompress the compartment(s).1-3
Case Presentation
A 76-year-old right-handed woman with a history of chronic obstructive pulmonary disease, hypertension, and hyperlipidemia presented to the emergency department with 2 days of extensive right upper extremity ecchymosis and severe pain that was localized to her forearm (Figure 1). She was taking low-dose aspirin (81 mg/d) for left subclavian stenosis and over-the-counter ginkgo biloba. Leading up to the presentation, the patient was able to perform routine household chores, including yard work, cleaning, and taking care of her cats. Wrist and elbow X-rays were negative for a fracture. An upper extremity ultrasound found no venous occlusion. A computed tomography (CT) angiogram of her arm and chest found diffuse edema around the right elbow and forearm without pulmonary or right upper extremity emboli, fractures, hematoma, abscess, or air in the tissues.
The plastic surgery service was consulted. The patient was found to have a very tense forearm and pain to passive digital extension. The 2-point discrimination and pulses were intact. The patient was diagnosed with compartment syndrome based on the examination alone and gave consent for an emergent forearm and hand fasciotomy. A carpal tunnel release and a standard S-shaped volar forearm fasciotomy release were performed, which provided immediate decompression (Figure 2). The rest of the hand and extremity were soft. Edematous, healthy flexor muscle belly was identified without a hematoma. Most of the forearm wound was left open because the skin could not be reapproximated. Oxidized regenerated cellulose (Surgicel) was placed around the wound edges and the muscle was covered with a nonadherent dressing. Hemoglobin on admission was 12.9 g/dL(reference range, 12 to 16 g/dL). Kidney function was within normal limits. The rest of the complete blood count was unremarkable. Postoperative hemoglobin was 8.6 g/dL. Over the next several days, the patient's skin edges and muscle bellies continued to slowly bleed, and her hemoglobin fell to 5.6 g/dL by postoperative Day 2. The bleeding was managed with topical oxidized regenerated cellulose, thrombin spray, a hemostatic dressing made with kaolin (QuikClot), and a transfusion of 2 units of packed red blood cells.
A hematology consultation was requested. The patient was noted to have an elevated partial thromboplastin time (PTT) since admission measuring between 39.9 to 61.7 seconds (reference range, 26.2 to 37.2 seconds) and a normal prothrombin time test with an international normalized ratio. A PTT measured 17 months prior to admission was within the normal range. She reported no personal or family history of bleeding disorders. Until recently, she had never had easy bruisability. She reported no history of heavy menses or epistaxis. The patient had no children and had never been pregnant. She had tolerated an exploratory laparotomy 40 years prior to admission without bleeding complications and had never required blood transfusions before. A PTT 1:1 mixing study revealed incomplete correction. Subsequent workup included factor VIII (FVIII) activity, factor IX activity, factor XI activity, von Willebrand factor antigen, ristocetin cofactor assay, and von Willebrand factor multimers. FVIII activity was severely reduced at 7.8% (reference, > 54%) with a positive Bethesda assay of 300 to 400 Bodansky units (BU), indicating a strong FVIII inhibitor was present and establishing a diagnosis of acquired hemophilia A. Further workup for secondary causes of acquired hemophilia A including abdominal and pelvic CT, serum protein electrophoresis, and serum free light chains, were negative. She was started on prednisone 1 mg/kg daily and rituximab 375 mg/m2. Her hemoglobin stabilized, and she required no further blood transfusions.
The patient underwent wound closure on postoperative Day 11. At the time of the second surgery, there was still no improvement in her FVIII levels or PTT; therefore, 70 mcg/kg of recombinant coagulation-activated FVII was given just before surgery with no bleeding complications. The skin was closed primarily except for the most distal 3 cm (Figure 3). Due to concerns regarding further bleeding with skin graft, the remaining wound was allowed to close by secondary intention. As a precaution, the wound was covered with oxidized regenerated cellulose and thrombin spray. The patient continued to progress postoperatively without bleeding complications or a need for additional transfusions. She was seen by the hand therapist before and after the second surgery to help with edema management and joint mobility. She completed 4 weekly doses of 375 mg/m² rituximab and prednisone was tapered by 10 mg weekly.
Three weeks after starting treatment, her PTT normalized, and her FVIII increased to 33.7%. The Bethesda assay remained high at 198 BU, although it was lower than at admission. She was discharged home with dressing changes and monthly follow-up appointments. The wounds were fully closed at her 3-month appointment when she proudly demonstrated full digital extension and flexion into her palm.
Discussion
Forearm compartment syndrome is most often caused by fractures—distal radius in adults and supracondylar in children.2 This case initially presented as a diagnostic puzzle to the emergency department due to the patient’s lucid review of several days of nontraumatic injury.
The clinical hallmarks of compartment syndrome are the 5 Ps: pain, pallor, paresthesia, paralysis, and pulselessness. Patients will describe the pain as out of proportion to the nature of the injury; the compartments will be tense and swollen, they will have pain to passive muscle stretch, and sensation will progressively diminish. Distal pulses are the last to go, and permanent tissue damage can still occur when pulses are present.1
Compartment Syndrome
Compartment syndrome is generally a clinical diagnosis; however, in patients who are sedated or uncooperative, or if the clinical findings are equivocal, the examination can be supplemented with intercompartmental pressures using an arterial line transducer system.2 In general, a tissue pressure of 30 mm Hg or a 20- to 30-mm Hg difference between the diastolic and compartment pressures are indications for fasciotomy.1 The hand is treated with an open carpal tunnel release, interosseous muscle release through 2 dorsal hand incisions, and thenar and hypothenar muscle release. The forearm is treated through a curved volar incision that usually decompresses the dorsal compartment, as it did in our patient. If pressures are still high in the forearm, a longitudinal dorsal incision over the mobile wad is necessary. Wounds can be closed primarily days later, left open to close by secondary intention, or reconstructed with skin grafts.2 In our patient, compartment syndrome was isolated to her forearm and the carpal tunnel release was performed prophylactically since it did not add significant time or morbidity to the surgery.
Nontraumatic upper extremity compartment syndrome is rare. A 2021 review of acute nontraumatic upper extremity compartment syndrome found a bleeding disorder as the etiology in 3 cases published in the literature between 1993 and 2016.4 One of these cases was secondary to a known diagnosis of hemophilia A in a teenager.5 Ogrodnik and colleagues described a spontaneous hand hematoma secondary to previously undiagnosed acquired hemophilia A and Waldenström macroglobulinemia.4 Ilyas and colleagues described a spontaneous hematoma in the forearm dorsal compartment in a 67-year-old woman, which presented as compartment syndrome and elevated PTT and led to a diagnosis of acquired FVIII inhibitor. The authors recommended prompt hematology consultation to coordinate treatment once this diagnosis issuspected.6 Compartment syndrome also has been found to develop slowly over weeks in patients with acquired FVIII deficiency, suggesting a high index of suspicion and frequent examinations are needed when patients with known acquired hemophilia A present with a painful extremity.7
Nontraumatic compartment syndrome in the lower extremity in patients with previously undiagnosed acquired hemophilia A has also been described in the literature.8-11 Case reports describe the delay in diagnosis as the patients were originally seen by clinicians for lower extremity pain and swelling within days of presenting to the emergency room with compartment syndrome. Persistent bleeding and abnormal laboratory results prompted further tests and examinations.8,9,11 This underscores the need to be suspicious of this unusual pathology without a history of trauma.
Acquired Hemophilia A
Acquired hemophilia A is an autoimmune disease most often found in older individuals, with a mean age of approximately 70 years.12 It is caused by the spontaneous production of neutralizing immunoglobin autoantibodies that target endogenous FVIII. Many cases are idiopathic; however, up to 50% of cases are associated with underlying autoimmunity, malignancy (especially lymphoproliferative disorders), or pregnancy. It often presents as bleeding that is subcutaneous or in the gastrointestinal system, muscle, retroperitoneal space, or genitourinary system. Unlike congenital hemophilia A, joint bleeding is rare.13
The diagnosis is suspected with an isolated elevated PTT in the absence of other coagulation abnormalities. A 1:1 mixing study will typically show incomplete correction, which suggests the presence of an inhibitor. FVIII activity is reduced, and the FVIII inhibitor is confirmed with the Bethesda assay. Clinically active bleeding is treated with bypassing agents such as recombinant coagulation-activated FVII, activated prothrombin complex concentrates such as anti-inhibitor coagulant complex (FEIBA), or recombinant porcine FVIII.12,14 Not all patients require hemostatic treatment, but close monitoring, education, recognition, and immediate treatment, if needed, are indicated.13 Immunosuppressive therapy (corticosteroids, rituximab, and/or cyclophosphamide) is prescribed to eradicate the antibodies and induce remission.12
Conclusions
An older woman without a preceding trauma was diagnosed with an unusual case of acute compartment syndrome in the forearm. No hematoma was found, but muscle and skin bleeding plus an elevated PTT prompted a hematology workup, and, ultimately, the diagnosis of FVIII inhibitor secondary to acquired hemophilia A.
While a nontraumatic cause of compartment syndrome is rare, it should be considered in differential diagnosis for clinicians who see hand and upper extremity emergencies. An isolated elevated PTT in a patient with a bleed should raise suspicions and trigger immediate further evaluation. Once suspected, multidisciplinary treatment is indicated for immediate and long-term successful outcomes.
Acknowledgments
This manuscript is the result of work supported withresources and the use of facilities at the North Florida/South Georgia Veterans Health System, Gainesville, Florida.
1. Leversedge FJ, Moore TJ, Peterson BC, Seiler JG 3rd. Compartment syndrome of the upper extremity. J Hand Surg Am. 2011;36:544-559. doi:10.1016/j.jhsa.2010.12.008
2. Kalyani BS, Fisher BE, Roberts CS, Giannoudis PV. Compartment syndrome of the forearm: a systematic review. J Hand Surg Am. 2011;36:535-543. doi:10.1016/j.jhsa.2010.12.007
3. Steadman W, Wu R, Hamilton AT, Richardson MD, Wall CJ. Review article: a comprehensive review of unusual causes of acute limb compartment syndrome. Emerg Med Australas. 2022;34:871-876. doi:10.1111/1742-6723.14098
4. Ogrodnik J, Oliver JD, Cani D, Boczar D, Huayllani MT, Restrepo DJ, et al. Clinical case of acute non-traumatic hand compartment syndrome and systematic review for the upper extremity. Hand (N Y). 2021;16:285-291. doi:10.1177/1558944719856106
5. Kim J, Zelken J, Sacks JM. Case report. Spontaneous forearm compartment syndrome in a boy with hemophilia a: a therapeutic dilemma. Eplasty. 2013:13:e16.
6. Ilyas AM, Wisbeck JM, Shaffer GW, Thoder JJ. Upper extremity compartment syndrome secondary to acquired factor VIII inhibitor. A case report. J Bone Joint Surg Am. 2005;87:1606-1608. doi:10.2106/JBJS.C.01720
7. Adeclat GJ, Hayes M, Amick M, Kahan J, Halim A. Acute forearm compartment syndrome in the setting of acquired hemophilia A. Case Reports Plast Surg Hand Surg. 2022;9:140-144. doi:10.1080/23320885.2022.2071274
8. Abudaqqa RY, Arun KP, Mas AJA, Abushaaban FA. Acute atraumatic compartment syndrome of the thigh due to acquired coagulopathy disorder: a case report in known healthy patient. J Orthop Case Rep. 2021;11:59-62. doi:10.13107/jocr.2021.v11.i08.2366
9. Alidoost M, Conte GA, Chaudry R, Nahum K, Marchesani D. A unique presentation of spontaneous compartment syndrome due to acquired hemophilia A and associated malignancy: case report and literature review. World J Oncol. 2020;11:72-75. doi:10.14740/wjon1260
10. Jentzsch T, Brand-Staufer B, Schäfer FP, Wanner GA, Simmen H-P. Illustrated operative management of spontaneous bleeding and compartment syndrome of the lower extremity in a patient with acquired hemophilia A: a case report. J Med Case Rep. 2014;8:132. doi:10.1186/1752-1947-8-132
11. Pham TV, Sorenson CA, Nable JV. Acquired factor VIII deficiency presenting with compartment syndrome. Am J Emerg Med. 2014;32:195.e1-2. doi:10.1016/j.ajem.2013.09.022
12. Tiede A, Zieger B, Lisman T. Acquired bleeding disorders. Haemophilia. 2022;28(suppl 4):68-76. doi:10.1111/hae.14548
13. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92:695-705. doi:10.1002/ajh.24777
14. Ilkhchoui Y, Koshkin E, Windsor JJ, Petersen TR, Charles M, Pack JD. Perioperative management of acquired hemophilia A: a case report and review of literature. Anesth Pain Med. 2013;4:e11906. doi:10.5812/aapm.11906
1. Leversedge FJ, Moore TJ, Peterson BC, Seiler JG 3rd. Compartment syndrome of the upper extremity. J Hand Surg Am. 2011;36:544-559. doi:10.1016/j.jhsa.2010.12.008
2. Kalyani BS, Fisher BE, Roberts CS, Giannoudis PV. Compartment syndrome of the forearm: a systematic review. J Hand Surg Am. 2011;36:535-543. doi:10.1016/j.jhsa.2010.12.007
3. Steadman W, Wu R, Hamilton AT, Richardson MD, Wall CJ. Review article: a comprehensive review of unusual causes of acute limb compartment syndrome. Emerg Med Australas. 2022;34:871-876. doi:10.1111/1742-6723.14098
4. Ogrodnik J, Oliver JD, Cani D, Boczar D, Huayllani MT, Restrepo DJ, et al. Clinical case of acute non-traumatic hand compartment syndrome and systematic review for the upper extremity. Hand (N Y). 2021;16:285-291. doi:10.1177/1558944719856106
5. Kim J, Zelken J, Sacks JM. Case report. Spontaneous forearm compartment syndrome in a boy with hemophilia a: a therapeutic dilemma. Eplasty. 2013:13:e16.
6. Ilyas AM, Wisbeck JM, Shaffer GW, Thoder JJ. Upper extremity compartment syndrome secondary to acquired factor VIII inhibitor. A case report. J Bone Joint Surg Am. 2005;87:1606-1608. doi:10.2106/JBJS.C.01720
7. Adeclat GJ, Hayes M, Amick M, Kahan J, Halim A. Acute forearm compartment syndrome in the setting of acquired hemophilia A. Case Reports Plast Surg Hand Surg. 2022;9:140-144. doi:10.1080/23320885.2022.2071274
8. Abudaqqa RY, Arun KP, Mas AJA, Abushaaban FA. Acute atraumatic compartment syndrome of the thigh due to acquired coagulopathy disorder: a case report in known healthy patient. J Orthop Case Rep. 2021;11:59-62. doi:10.13107/jocr.2021.v11.i08.2366
9. Alidoost M, Conte GA, Chaudry R, Nahum K, Marchesani D. A unique presentation of spontaneous compartment syndrome due to acquired hemophilia A and associated malignancy: case report and literature review. World J Oncol. 2020;11:72-75. doi:10.14740/wjon1260
10. Jentzsch T, Brand-Staufer B, Schäfer FP, Wanner GA, Simmen H-P. Illustrated operative management of spontaneous bleeding and compartment syndrome of the lower extremity in a patient with acquired hemophilia A: a case report. J Med Case Rep. 2014;8:132. doi:10.1186/1752-1947-8-132
11. Pham TV, Sorenson CA, Nable JV. Acquired factor VIII deficiency presenting with compartment syndrome. Am J Emerg Med. 2014;32:195.e1-2. doi:10.1016/j.ajem.2013.09.022
12. Tiede A, Zieger B, Lisman T. Acquired bleeding disorders. Haemophilia. 2022;28(suppl 4):68-76. doi:10.1111/hae.14548
13. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92:695-705. doi:10.1002/ajh.24777
14. Ilkhchoui Y, Koshkin E, Windsor JJ, Petersen TR, Charles M, Pack JD. Perioperative management of acquired hemophilia A: a case report and review of literature. Anesth Pain Med. 2013;4:e11906. doi:10.5812/aapm.11906
Prognostication in Hospice Care: Challenges, Opportunities, and the Importance of Functional Status
Predicting life expectancy and providing an end-of-life diagnosis in hospice and palliative care is a challenge for most clinicians. Lack of training, limited communication skills, and relationships with patients are all contributing factors. These skills can improve with the use of functional scoring tools in conjunction with the patient’s comorbidities and physical/psychological symptoms. The Palliative Performance Scale (PPS), Karnofsky Performance Scale (KPS), and Eastern Cooperative Oncology Group Performance Status Scale (ECOG) are commonly used functional scoring tools.
The PPS measures 5 functional dimensions including ambulation, activity level, ability to administer self-care, oral intake, and level of consciousness.1 It has been shown to be valid for a broad range of palliative care patients, including those with advanced cancer or life-threatening noncancer diagnoses in hospitals or hospice care.2 The scale, measured in 10% increments, runs from 100% (completely functional) to 0% (dead). A PPS ≤ 70% helps meet hospice eligibility criteria.
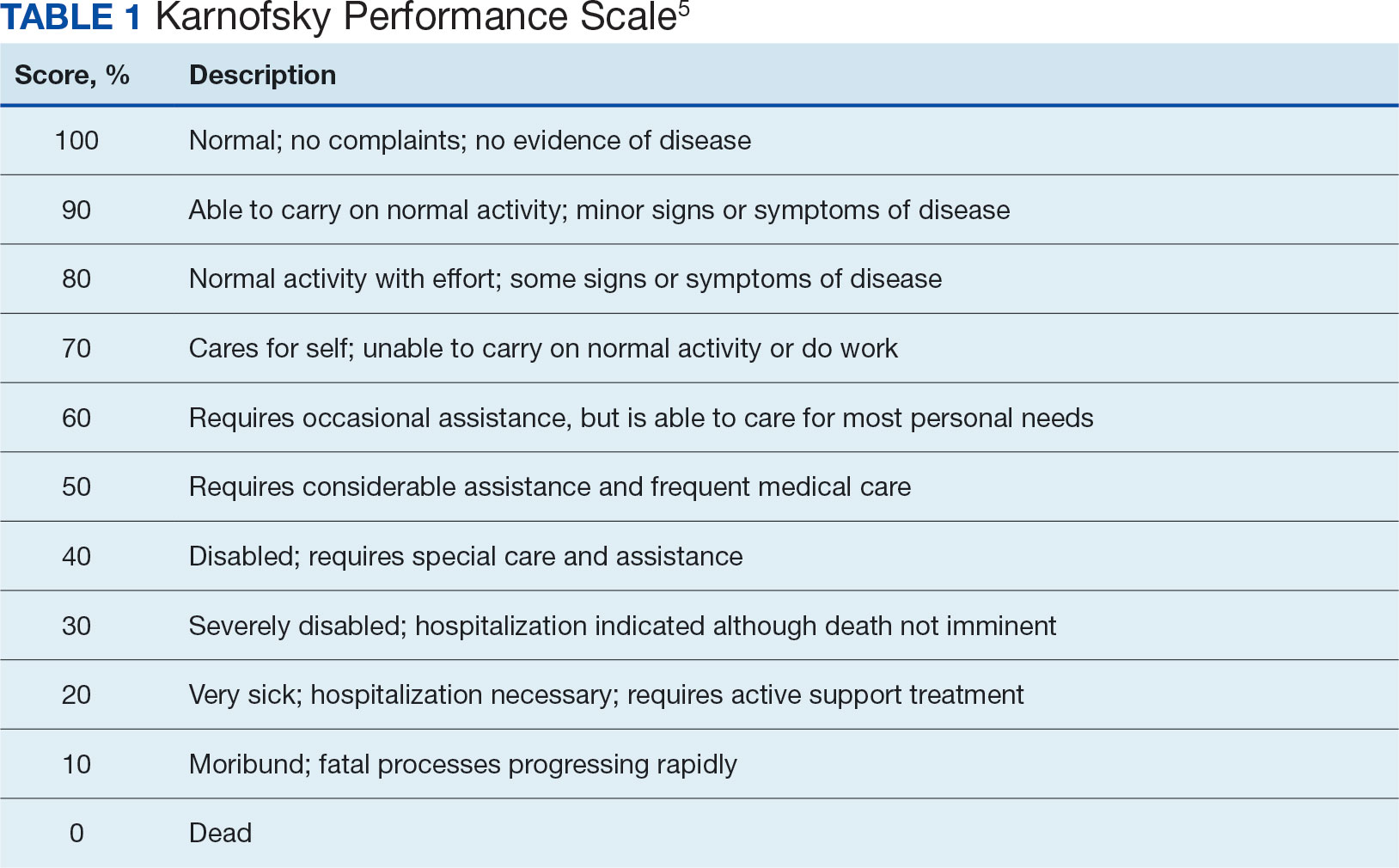
The KPS evaluates functional impairment and helps with prognostication. Developed in 1948, it evaluates a patient’s functional ability to tolerate chemotherapy, specifically in lung cancer, and has since been validated to predict mortality across older adults and in chronic disease populations.3,4 The KPS is also measured in 10% increments ranging from 100% (completely functional without assistance) to 0% (dead). A KPS ≤ 70% assists with hospice eligibility criteria (Table 1).5

Developed in 1974, the ECOG has been identified as one of the most important functional status tools in adult cancer care.6 It describes a cancer patient’s functional ability, evaluating their ability to care for oneself and participate in daily activities.7 The ECOG is a 6-point scale; patients can receive scores ranging from 0 (fully active) to 5 (dead). An ECOG score of 4 (sometimes 3) is generally supportive of meeting hospice eligibility (Table 2).6
CASE Presentation
An 80-year-old patient was admitted to the hospice service at the Veterans Affairs Puget Sound Health Care System (VAPSHCS) community living center (CLC) in Tacoma, Washington, from a community-based acute care hospital. His medical history included prostate cancer with metastasis to his pelvis and type 2 diabetes mellitus, which was stable with treatment with oral medication. Six weeks earlier the patient reported a severe frontal headache that was not responding to over-the-counter analgesics. After 2 days with these symptoms, including a ground-level fall without injuries, he presented to the VAPSHCS emergency department (ED) where a complete neurological examination, including magnetic resonance imaging, revealed a left frontoparietal brain lesion that was 4.2 cm × 3.4 cm × 4.2 cm.
The patient experienced a seizure during his ED evaluation and was admitted for treatment. He underwent a craniotomy where most, but not all the lesions were successfully removed. Postoperatively, the patient exhibited right-sided neglect, gait instability, emotional lability, and cognitive communication disorder. The patient completed 15 of 20 planned radiation treatments but declined further radiation or chemotherapy. The patient decided to halt radiation treatments after being informed by the oncology service that the treatments would likely only add 1 to 2 months to his overall survival, which was < 6 months. The patient elected to focus his goals of care on comfort, dignity, and respect at the end of life and accepted recommendations to be placed into end-of-life hospice care. He was then transferred to the VAPSHCS CLC in Tacoma, Washington, for hospice care.
Upon admission, the patient weighed 94 kg, his vital signs were within reference range, and he reported no pain or headaches. His initial laboratory results revealed a 13.2 g/dL hemoglobin, 3.6 g/dL serum albumin, and a 5.5% hemoglobin A1c, all of which fall into a normal reference range. He had a reported ECOG score of 3 and a KPS score of 50% by the transferring medical team. The patient’s medications included scheduled dexamethasone, metformin, senna, levetiracetam, and as-needed midazolam nasal spray for breakthrough seizures. He also had as-needed acetaminophen for pain. He was alert, oriented ×3, and fully ambulatory but continuously used a 4-wheeled walker for safety and gait instability.
After the patient’s first night, the hospice team met with him to discuss his understanding of his health issues. The patient appeared to have low health literacy but told the team, “I know I am dying.” He had completed written advance directives and a Portable Order for Life-Sustaining Treatment indicating that life-sustaining treatments, including cardiopulmonary resuscitation, supplemental mechanical feeding, or intubation, were not to be used to keep him alive.
At his first 90-day recertification, the patient had gained 8 kg and laboratory results revealed a 14.6 g/dL hemoglobin, 3.8 g/dL serum albumin, and a 6.1% hemoglobin A1c. His ECOG score remained at 3, but his KPS score had increased to 60%. The patient exhibited no new neurologic symptoms or seizures and reported no headaches but had 2 ground-level falls without injury. On both occasions the patient chose not to use his walker to go to the bathroom because it was “too far from my bed.” Per VA policy, after discussions with the hospice team, he was recertified for 90 more days of hospice care. At the end of 6 months in CLC, the patient’s weight remained stable, as did his complete blood count and comprehensive medical panel. He had 1 additional noninjurious ground-level fall and again reported no pain and no use of as-needed acetaminophen. His only medical complication was testing positive for COVID-19, but he remained asymptomatic. The patient was graduated from hospice care and referred to a nearby non-VA adult family home in the community after 180 days. At that time his ECOG score was 2 and his KPS score had increased to 70%.
DISCUSSION
Primary brain tumors account for about 2% of all malignant neoplasms in adults. About half of them represent gliomas. Glioblastoma multiforme derived from neuroepithelial cells is the most frequent and deadly primary malignant central nervous system tumor in adults.8 About 50% of patients with glioblastomas are aged ≥ 65 years at diagnosis.9 A retrospective study of Centers for Medicare and Medicaid Services claims data paired with the Surveillance, Epidemiology, and End Results database indicated a median survival of 4 months for patients with glioblastoma multiforme aged > 65 years, including all treatment modalities.10 Surgical resection combined with radiation and chemotherapy offers the best prognosis for the preservation of neurologic function.11 However, comorbidities, adverse drug effects, and the potential for postoperative complications pose significant risks, especially for older patients. Ultimately, goals of care conversations and advance directives play a very important role in evaluating benefits vs risks with this malignancy.
Our patient was aged 80 years and had previously been diagnosed with metastatic prostate malignancy. His goals of care focused on spending time with his friends, leaving his room to eat in the facility dining area, and continuing his daily walks. He remained clear that he did not want his care team to institute life-sustaining treatments to be kept alive and felt the information regarding the risks vs benefits of accepting chemotherapy was not aligned with his goals of care. Over the 6 months that he received hospice care, he gained weight, improved his hemoglobin and serum albumin levels, and ambulated with the use of a 4-wheeled walker. As the patient exhibited no functional decline or new comorbidities and his functional status improved, the clinical staff felt he no longer needed hospice services. The patient had an ECOG score of 2 and a KPS score of 70% at his hospice graduation.
Medical prognostication is one of the biggest challenges clinicians face. Clinicians are generally “over prognosticators,” and their thoughts tend to be based on the patient relationship, overall experiences in health care, and desire to treat and cure patients.12 In hospice we are asked to define the usual, normal, or expected course of a disease, but what does that mean? Although metastatic malignancies usually have a predictable course in comparison to diagnoses such as dementia, chronic obstructive pulmonary disease, or congestive heart failure, the challenges to improve prognostic ability andpredict disease course continue.13-15 Focusing on functional status, goals of care, and comorbidities are keys to helping with prognosis. Given the challenge, we find the PPS, KPS, and ECOG scales important tools.
When prognosticating, we attempt to define quantity and quality of life (which our patients must define independently or from the voice of their surrogate) and their ability to perform daily activities. Quality of life in patients with glioblastoma is progressively and significantly impacted due to the emergence of debilitating neurologic symptoms arising from infiltrative tumor growth into functionally intact brain tissue that restricts and disrupts normal day-to-day activities. However, functional status plays a significant role in helping the hospice team improve its overall prognosis.
Conclusions
This case study illustrates the difficulty that comes with prognostication(s) despite a patient's severely morbid disease, history of metastatic prostate cancer, and advanced age. Although a diagnosis may be concerning, documenting a patient’s status using functional scales prior to hospice admission and during the recertification process is helpful in prognostication. Doing so will allow health care professionals to have an accepted medical standard to use regardless how distinct the patient's diagnosis. The expression, “as the disease does not read the textbook,” may serve as a helpful reminder in talking with patients and their families. This is important as most patient’s clinical disease courses are different and having the opportunity to use performance status scales may help improve prognostic skills.
1. Cleary TA. The Palliative Performance Scale (PPSv2) Version 2. In: Downing GM, ed. Medical Care of the Dying. 4th ed. Victoria Hospice Society, Learning Centre for Palliative Care; 2006:120.
2. Palliative Performance Scale. ePrognosis, University of California San Francisco. Accessed June 14, 2024. https://eprognosis.ucsf.edu/pps.php
3. Karnofsky DA, Burchenal JH. The Clinical Evaluation of Chemotherapeutic Agents in Cancer. In: MacLeod CM, ed. Evaluation of Chemotherapeutic Agents. Columbia University Press; 1949:191-205.
4. Khalid MA, Achakzai IK, Ahmed Khan S, et al. The use of Karnofsky Performance Status (KPS) as a predictor of 3 month post discharge mortality in cirrhotic patients. Gastroenterol Hepatol Bed Bench. 2018;11(4):301-305.
5. Karnofsky Performance Scale. US Dept of Veterans Affairs. Accessed June 14, 2024. https://www.hiv.va.gov/provider/tools/karnofsky-performance-scale.asp
6. Mischel A-M, Rosielle DA. Eastern Cooperative Oncology Group Performance Status. Palliative Care Network of Wisconsin. December 10, 2021. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/eastern-cooperative-oncology-group-performance-status/
7. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649-655.
8. Nizamutdinov D, Stock EM, Dandashi JA, et al. Prognostication of survival outcomes in patients diagnosed with glioblastoma. World Neurosurg. 2018;109:e67-e74. doi:10.1016/j.wneu.2017.09.104
9. Kita D Ciernik IFVaccarella S Age as a predictive factor in glioblastomas: population-based study. Neuroepidemiology. 2009;33(1):17-22. doi:10.1159/000210017
10. Jordan JT, Gerstner ER, Batchelor TT, Cahill DP, Plotkin SR. Glioblastoma care in the elderly. Cancer. 2016;122(2):189-197. doi:10.1002/cnr.29742
11. Brown, NF, Ottaviani D, Tazare J, et al. Survival outcomes and prognostic factors in glioblastoma. Cancers (Basel). 2022;14(13):3161. doi:10.3390/cancers14133161
12. Christalakis NA. Death Foretold: Prophecy and Prognosis in Medical Care. University of Chicago Press; 2000.
13. Weissman DE. Determining Prognosis in Advanced Cancer. Palliative Care Network of Wisconsin. January 28, 2019. Accessed June 14, 2014. https://www.mypcnow.org/fast-fact/determining-prognosis-in-advanced-cancer/
14. Childers JW, Arnold R, Curtis JR. Prognosis in End-Stage COPD. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognosis-in-end-stage-copd/
15. Reisfield GM, Wilson GR. Prognostication in Heart Failure. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognostication-in-heart-failure/
Predicting life expectancy and providing an end-of-life diagnosis in hospice and palliative care is a challenge for most clinicians. Lack of training, limited communication skills, and relationships with patients are all contributing factors. These skills can improve with the use of functional scoring tools in conjunction with the patient’s comorbidities and physical/psychological symptoms. The Palliative Performance Scale (PPS), Karnofsky Performance Scale (KPS), and Eastern Cooperative Oncology Group Performance Status Scale (ECOG) are commonly used functional scoring tools.
The PPS measures 5 functional dimensions including ambulation, activity level, ability to administer self-care, oral intake, and level of consciousness.1 It has been shown to be valid for a broad range of palliative care patients, including those with advanced cancer or life-threatening noncancer diagnoses in hospitals or hospice care.2 The scale, measured in 10% increments, runs from 100% (completely functional) to 0% (dead). A PPS ≤ 70% helps meet hospice eligibility criteria.
The KPS evaluates functional impairment and helps with prognostication. Developed in 1948, it evaluates a patient’s functional ability to tolerate chemotherapy, specifically in lung cancer, and has since been validated to predict mortality across older adults and in chronic disease populations.3,4 The KPS is also measured in 10% increments ranging from 100% (completely functional without assistance) to 0% (dead). A KPS ≤ 70% assists with hospice eligibility criteria (Table 1).5
Developed in 1974, the ECOG has been identified as one of the most important functional status tools in adult cancer care.6 It describes a cancer patient’s functional ability, evaluating their ability to care for oneself and participate in daily activities.7 The ECOG is a 6-point scale; patients can receive scores ranging from 0 (fully active) to 5 (dead). An ECOG score of 4 (sometimes 3) is generally supportive of meeting hospice eligibility (Table 2).6
CASE Presentation
An 80-year-old patient was admitted to the hospice service at the Veterans Affairs Puget Sound Health Care System (VAPSHCS) community living center (CLC) in Tacoma, Washington, from a community-based acute care hospital. His medical history included prostate cancer with metastasis to his pelvis and type 2 diabetes mellitus, which was stable with treatment with oral medication. Six weeks earlier the patient reported a severe frontal headache that was not responding to over-the-counter analgesics. After 2 days with these symptoms, including a ground-level fall without injuries, he presented to the VAPSHCS emergency department (ED) where a complete neurological examination, including magnetic resonance imaging, revealed a left frontoparietal brain lesion that was 4.2 cm × 3.4 cm × 4.2 cm.
The patient experienced a seizure during his ED evaluation and was admitted for treatment. He underwent a craniotomy where most, but not all the lesions were successfully removed. Postoperatively, the patient exhibited right-sided neglect, gait instability, emotional lability, and cognitive communication disorder. The patient completed 15 of 20 planned radiation treatments but declined further radiation or chemotherapy. The patient decided to halt radiation treatments after being informed by the oncology service that the treatments would likely only add 1 to 2 months to his overall survival, which was < 6 months. The patient elected to focus his goals of care on comfort, dignity, and respect at the end of life and accepted recommendations to be placed into end-of-life hospice care. He was then transferred to the VAPSHCS CLC in Tacoma, Washington, for hospice care.
Upon admission, the patient weighed 94 kg, his vital signs were within reference range, and he reported no pain or headaches. His initial laboratory results revealed a 13.2 g/dL hemoglobin, 3.6 g/dL serum albumin, and a 5.5% hemoglobin A1c, all of which fall into a normal reference range. He had a reported ECOG score of 3 and a KPS score of 50% by the transferring medical team. The patient’s medications included scheduled dexamethasone, metformin, senna, levetiracetam, and as-needed midazolam nasal spray for breakthrough seizures. He also had as-needed acetaminophen for pain. He was alert, oriented ×3, and fully ambulatory but continuously used a 4-wheeled walker for safety and gait instability.
After the patient’s first night, the hospice team met with him to discuss his understanding of his health issues. The patient appeared to have low health literacy but told the team, “I know I am dying.” He had completed written advance directives and a Portable Order for Life-Sustaining Treatment indicating that life-sustaining treatments, including cardiopulmonary resuscitation, supplemental mechanical feeding, or intubation, were not to be used to keep him alive.
At his first 90-day recertification, the patient had gained 8 kg and laboratory results revealed a 14.6 g/dL hemoglobin, 3.8 g/dL serum albumin, and a 6.1% hemoglobin A1c. His ECOG score remained at 3, but his KPS score had increased to 60%. The patient exhibited no new neurologic symptoms or seizures and reported no headaches but had 2 ground-level falls without injury. On both occasions the patient chose not to use his walker to go to the bathroom because it was “too far from my bed.” Per VA policy, after discussions with the hospice team, he was recertified for 90 more days of hospice care. At the end of 6 months in CLC, the patient’s weight remained stable, as did his complete blood count and comprehensive medical panel. He had 1 additional noninjurious ground-level fall and again reported no pain and no use of as-needed acetaminophen. His only medical complication was testing positive for COVID-19, but he remained asymptomatic. The patient was graduated from hospice care and referred to a nearby non-VA adult family home in the community after 180 days. At that time his ECOG score was 2 and his KPS score had increased to 70%.
DISCUSSION
Primary brain tumors account for about 2% of all malignant neoplasms in adults. About half of them represent gliomas. Glioblastoma multiforme derived from neuroepithelial cells is the most frequent and deadly primary malignant central nervous system tumor in adults.8 About 50% of patients with glioblastomas are aged ≥ 65 years at diagnosis.9 A retrospective study of Centers for Medicare and Medicaid Services claims data paired with the Surveillance, Epidemiology, and End Results database indicated a median survival of 4 months for patients with glioblastoma multiforme aged > 65 years, including all treatment modalities.10 Surgical resection combined with radiation and chemotherapy offers the best prognosis for the preservation of neurologic function.11 However, comorbidities, adverse drug effects, and the potential for postoperative complications pose significant risks, especially for older patients. Ultimately, goals of care conversations and advance directives play a very important role in evaluating benefits vs risks with this malignancy.
Our patient was aged 80 years and had previously been diagnosed with metastatic prostate malignancy. His goals of care focused on spending time with his friends, leaving his room to eat in the facility dining area, and continuing his daily walks. He remained clear that he did not want his care team to institute life-sustaining treatments to be kept alive and felt the information regarding the risks vs benefits of accepting chemotherapy was not aligned with his goals of care. Over the 6 months that he received hospice care, he gained weight, improved his hemoglobin and serum albumin levels, and ambulated with the use of a 4-wheeled walker. As the patient exhibited no functional decline or new comorbidities and his functional status improved, the clinical staff felt he no longer needed hospice services. The patient had an ECOG score of 2 and a KPS score of 70% at his hospice graduation.
Medical prognostication is one of the biggest challenges clinicians face. Clinicians are generally “over prognosticators,” and their thoughts tend to be based on the patient relationship, overall experiences in health care, and desire to treat and cure patients.12 In hospice we are asked to define the usual, normal, or expected course of a disease, but what does that mean? Although metastatic malignancies usually have a predictable course in comparison to diagnoses such as dementia, chronic obstructive pulmonary disease, or congestive heart failure, the challenges to improve prognostic ability andpredict disease course continue.13-15 Focusing on functional status, goals of care, and comorbidities are keys to helping with prognosis. Given the challenge, we find the PPS, KPS, and ECOG scales important tools.
When prognosticating, we attempt to define quantity and quality of life (which our patients must define independently or from the voice of their surrogate) and their ability to perform daily activities. Quality of life in patients with glioblastoma is progressively and significantly impacted due to the emergence of debilitating neurologic symptoms arising from infiltrative tumor growth into functionally intact brain tissue that restricts and disrupts normal day-to-day activities. However, functional status plays a significant role in helping the hospice team improve its overall prognosis.
Conclusions
This case study illustrates the difficulty that comes with prognostication(s) despite a patient's severely morbid disease, history of metastatic prostate cancer, and advanced age. Although a diagnosis may be concerning, documenting a patient’s status using functional scales prior to hospice admission and during the recertification process is helpful in prognostication. Doing so will allow health care professionals to have an accepted medical standard to use regardless how distinct the patient's diagnosis. The expression, “as the disease does not read the textbook,” may serve as a helpful reminder in talking with patients and their families. This is important as most patient’s clinical disease courses are different and having the opportunity to use performance status scales may help improve prognostic skills.
Predicting life expectancy and providing an end-of-life diagnosis in hospice and palliative care is a challenge for most clinicians. Lack of training, limited communication skills, and relationships with patients are all contributing factors. These skills can improve with the use of functional scoring tools in conjunction with the patient’s comorbidities and physical/psychological symptoms. The Palliative Performance Scale (PPS), Karnofsky Performance Scale (KPS), and Eastern Cooperative Oncology Group Performance Status Scale (ECOG) are commonly used functional scoring tools.
The PPS measures 5 functional dimensions including ambulation, activity level, ability to administer self-care, oral intake, and level of consciousness.1 It has been shown to be valid for a broad range of palliative care patients, including those with advanced cancer or life-threatening noncancer diagnoses in hospitals or hospice care.2 The scale, measured in 10% increments, runs from 100% (completely functional) to 0% (dead). A PPS ≤ 70% helps meet hospice eligibility criteria.
The KPS evaluates functional impairment and helps with prognostication. Developed in 1948, it evaluates a patient’s functional ability to tolerate chemotherapy, specifically in lung cancer, and has since been validated to predict mortality across older adults and in chronic disease populations.3,4 The KPS is also measured in 10% increments ranging from 100% (completely functional without assistance) to 0% (dead). A KPS ≤ 70% assists with hospice eligibility criteria (Table 1).5
Developed in 1974, the ECOG has been identified as one of the most important functional status tools in adult cancer care.6 It describes a cancer patient’s functional ability, evaluating their ability to care for oneself and participate in daily activities.7 The ECOG is a 6-point scale; patients can receive scores ranging from 0 (fully active) to 5 (dead). An ECOG score of 4 (sometimes 3) is generally supportive of meeting hospice eligibility (Table 2).6
CASE Presentation
An 80-year-old patient was admitted to the hospice service at the Veterans Affairs Puget Sound Health Care System (VAPSHCS) community living center (CLC) in Tacoma, Washington, from a community-based acute care hospital. His medical history included prostate cancer with metastasis to his pelvis and type 2 diabetes mellitus, which was stable with treatment with oral medication. Six weeks earlier the patient reported a severe frontal headache that was not responding to over-the-counter analgesics. After 2 days with these symptoms, including a ground-level fall without injuries, he presented to the VAPSHCS emergency department (ED) where a complete neurological examination, including magnetic resonance imaging, revealed a left frontoparietal brain lesion that was 4.2 cm × 3.4 cm × 4.2 cm.
The patient experienced a seizure during his ED evaluation and was admitted for treatment. He underwent a craniotomy where most, but not all the lesions were successfully removed. Postoperatively, the patient exhibited right-sided neglect, gait instability, emotional lability, and cognitive communication disorder. The patient completed 15 of 20 planned radiation treatments but declined further radiation or chemotherapy. The patient decided to halt radiation treatments after being informed by the oncology service that the treatments would likely only add 1 to 2 months to his overall survival, which was < 6 months. The patient elected to focus his goals of care on comfort, dignity, and respect at the end of life and accepted recommendations to be placed into end-of-life hospice care. He was then transferred to the VAPSHCS CLC in Tacoma, Washington, for hospice care.
Upon admission, the patient weighed 94 kg, his vital signs were within reference range, and he reported no pain or headaches. His initial laboratory results revealed a 13.2 g/dL hemoglobin, 3.6 g/dL serum albumin, and a 5.5% hemoglobin A1c, all of which fall into a normal reference range. He had a reported ECOG score of 3 and a KPS score of 50% by the transferring medical team. The patient’s medications included scheduled dexamethasone, metformin, senna, levetiracetam, and as-needed midazolam nasal spray for breakthrough seizures. He also had as-needed acetaminophen for pain. He was alert, oriented ×3, and fully ambulatory but continuously used a 4-wheeled walker for safety and gait instability.
After the patient’s first night, the hospice team met with him to discuss his understanding of his health issues. The patient appeared to have low health literacy but told the team, “I know I am dying.” He had completed written advance directives and a Portable Order for Life-Sustaining Treatment indicating that life-sustaining treatments, including cardiopulmonary resuscitation, supplemental mechanical feeding, or intubation, were not to be used to keep him alive.
At his first 90-day recertification, the patient had gained 8 kg and laboratory results revealed a 14.6 g/dL hemoglobin, 3.8 g/dL serum albumin, and a 6.1% hemoglobin A1c. His ECOG score remained at 3, but his KPS score had increased to 60%. The patient exhibited no new neurologic symptoms or seizures and reported no headaches but had 2 ground-level falls without injury. On both occasions the patient chose not to use his walker to go to the bathroom because it was “too far from my bed.” Per VA policy, after discussions with the hospice team, he was recertified for 90 more days of hospice care. At the end of 6 months in CLC, the patient’s weight remained stable, as did his complete blood count and comprehensive medical panel. He had 1 additional noninjurious ground-level fall and again reported no pain and no use of as-needed acetaminophen. His only medical complication was testing positive for COVID-19, but he remained asymptomatic. The patient was graduated from hospice care and referred to a nearby non-VA adult family home in the community after 180 days. At that time his ECOG score was 2 and his KPS score had increased to 70%.
DISCUSSION
Primary brain tumors account for about 2% of all malignant neoplasms in adults. About half of them represent gliomas. Glioblastoma multiforme derived from neuroepithelial cells is the most frequent and deadly primary malignant central nervous system tumor in adults.8 About 50% of patients with glioblastomas are aged ≥ 65 years at diagnosis.9 A retrospective study of Centers for Medicare and Medicaid Services claims data paired with the Surveillance, Epidemiology, and End Results database indicated a median survival of 4 months for patients with glioblastoma multiforme aged > 65 years, including all treatment modalities.10 Surgical resection combined with radiation and chemotherapy offers the best prognosis for the preservation of neurologic function.11 However, comorbidities, adverse drug effects, and the potential for postoperative complications pose significant risks, especially for older patients. Ultimately, goals of care conversations and advance directives play a very important role in evaluating benefits vs risks with this malignancy.
Our patient was aged 80 years and had previously been diagnosed with metastatic prostate malignancy. His goals of care focused on spending time with his friends, leaving his room to eat in the facility dining area, and continuing his daily walks. He remained clear that he did not want his care team to institute life-sustaining treatments to be kept alive and felt the information regarding the risks vs benefits of accepting chemotherapy was not aligned with his goals of care. Over the 6 months that he received hospice care, he gained weight, improved his hemoglobin and serum albumin levels, and ambulated with the use of a 4-wheeled walker. As the patient exhibited no functional decline or new comorbidities and his functional status improved, the clinical staff felt he no longer needed hospice services. The patient had an ECOG score of 2 and a KPS score of 70% at his hospice graduation.
Medical prognostication is one of the biggest challenges clinicians face. Clinicians are generally “over prognosticators,” and their thoughts tend to be based on the patient relationship, overall experiences in health care, and desire to treat and cure patients.12 In hospice we are asked to define the usual, normal, or expected course of a disease, but what does that mean? Although metastatic malignancies usually have a predictable course in comparison to diagnoses such as dementia, chronic obstructive pulmonary disease, or congestive heart failure, the challenges to improve prognostic ability andpredict disease course continue.13-15 Focusing on functional status, goals of care, and comorbidities are keys to helping with prognosis. Given the challenge, we find the PPS, KPS, and ECOG scales important tools.
When prognosticating, we attempt to define quantity and quality of life (which our patients must define independently or from the voice of their surrogate) and their ability to perform daily activities. Quality of life in patients with glioblastoma is progressively and significantly impacted due to the emergence of debilitating neurologic symptoms arising from infiltrative tumor growth into functionally intact brain tissue that restricts and disrupts normal day-to-day activities. However, functional status plays a significant role in helping the hospice team improve its overall prognosis.
Conclusions
This case study illustrates the difficulty that comes with prognostication(s) despite a patient's severely morbid disease, history of metastatic prostate cancer, and advanced age. Although a diagnosis may be concerning, documenting a patient’s status using functional scales prior to hospice admission and during the recertification process is helpful in prognostication. Doing so will allow health care professionals to have an accepted medical standard to use regardless how distinct the patient's diagnosis. The expression, “as the disease does not read the textbook,” may serve as a helpful reminder in talking with patients and their families. This is important as most patient’s clinical disease courses are different and having the opportunity to use performance status scales may help improve prognostic skills.
1. Cleary TA. The Palliative Performance Scale (PPSv2) Version 2. In: Downing GM, ed. Medical Care of the Dying. 4th ed. Victoria Hospice Society, Learning Centre for Palliative Care; 2006:120.
2. Palliative Performance Scale. ePrognosis, University of California San Francisco. Accessed June 14, 2024. https://eprognosis.ucsf.edu/pps.php
3. Karnofsky DA, Burchenal JH. The Clinical Evaluation of Chemotherapeutic Agents in Cancer. In: MacLeod CM, ed. Evaluation of Chemotherapeutic Agents. Columbia University Press; 1949:191-205.
4. Khalid MA, Achakzai IK, Ahmed Khan S, et al. The use of Karnofsky Performance Status (KPS) as a predictor of 3 month post discharge mortality in cirrhotic patients. Gastroenterol Hepatol Bed Bench. 2018;11(4):301-305.
5. Karnofsky Performance Scale. US Dept of Veterans Affairs. Accessed June 14, 2024. https://www.hiv.va.gov/provider/tools/karnofsky-performance-scale.asp
6. Mischel A-M, Rosielle DA. Eastern Cooperative Oncology Group Performance Status. Palliative Care Network of Wisconsin. December 10, 2021. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/eastern-cooperative-oncology-group-performance-status/
7. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649-655.
8. Nizamutdinov D, Stock EM, Dandashi JA, et al. Prognostication of survival outcomes in patients diagnosed with glioblastoma. World Neurosurg. 2018;109:e67-e74. doi:10.1016/j.wneu.2017.09.104
9. Kita D Ciernik IFVaccarella S Age as a predictive factor in glioblastomas: population-based study. Neuroepidemiology. 2009;33(1):17-22. doi:10.1159/000210017
10. Jordan JT, Gerstner ER, Batchelor TT, Cahill DP, Plotkin SR. Glioblastoma care in the elderly. Cancer. 2016;122(2):189-197. doi:10.1002/cnr.29742
11. Brown, NF, Ottaviani D, Tazare J, et al. Survival outcomes and prognostic factors in glioblastoma. Cancers (Basel). 2022;14(13):3161. doi:10.3390/cancers14133161
12. Christalakis NA. Death Foretold: Prophecy and Prognosis in Medical Care. University of Chicago Press; 2000.
13. Weissman DE. Determining Prognosis in Advanced Cancer. Palliative Care Network of Wisconsin. January 28, 2019. Accessed June 14, 2014. https://www.mypcnow.org/fast-fact/determining-prognosis-in-advanced-cancer/
14. Childers JW, Arnold R, Curtis JR. Prognosis in End-Stage COPD. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognosis-in-end-stage-copd/
15. Reisfield GM, Wilson GR. Prognostication in Heart Failure. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognostication-in-heart-failure/
1. Cleary TA. The Palliative Performance Scale (PPSv2) Version 2. In: Downing GM, ed. Medical Care of the Dying. 4th ed. Victoria Hospice Society, Learning Centre for Palliative Care; 2006:120.
2. Palliative Performance Scale. ePrognosis, University of California San Francisco. Accessed June 14, 2024. https://eprognosis.ucsf.edu/pps.php
3. Karnofsky DA, Burchenal JH. The Clinical Evaluation of Chemotherapeutic Agents in Cancer. In: MacLeod CM, ed. Evaluation of Chemotherapeutic Agents. Columbia University Press; 1949:191-205.
4. Khalid MA, Achakzai IK, Ahmed Khan S, et al. The use of Karnofsky Performance Status (KPS) as a predictor of 3 month post discharge mortality in cirrhotic patients. Gastroenterol Hepatol Bed Bench. 2018;11(4):301-305.
5. Karnofsky Performance Scale. US Dept of Veterans Affairs. Accessed June 14, 2024. https://www.hiv.va.gov/provider/tools/karnofsky-performance-scale.asp
6. Mischel A-M, Rosielle DA. Eastern Cooperative Oncology Group Performance Status. Palliative Care Network of Wisconsin. December 10, 2021. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/eastern-cooperative-oncology-group-performance-status/
7. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5(6):649-655.
8. Nizamutdinov D, Stock EM, Dandashi JA, et al. Prognostication of survival outcomes in patients diagnosed with glioblastoma. World Neurosurg. 2018;109:e67-e74. doi:10.1016/j.wneu.2017.09.104
9. Kita D Ciernik IFVaccarella S Age as a predictive factor in glioblastomas: population-based study. Neuroepidemiology. 2009;33(1):17-22. doi:10.1159/000210017
10. Jordan JT, Gerstner ER, Batchelor TT, Cahill DP, Plotkin SR. Glioblastoma care in the elderly. Cancer. 2016;122(2):189-197. doi:10.1002/cnr.29742
11. Brown, NF, Ottaviani D, Tazare J, et al. Survival outcomes and prognostic factors in glioblastoma. Cancers (Basel). 2022;14(13):3161. doi:10.3390/cancers14133161
12. Christalakis NA. Death Foretold: Prophecy and Prognosis in Medical Care. University of Chicago Press; 2000.
13. Weissman DE. Determining Prognosis in Advanced Cancer. Palliative Care Network of Wisconsin. January 28, 2019. Accessed June 14, 2014. https://www.mypcnow.org/fast-fact/determining-prognosis-in-advanced-cancer/
14. Childers JW, Arnold R, Curtis JR. Prognosis in End-Stage COPD. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognosis-in-end-stage-copd/
15. Reisfield GM, Wilson GR. Prognostication in Heart Failure. Palliative Care Network of Wisconsin. February 11, 2019. Accessed June 14, 2024. https://www.mypcnow.org/fast-fact/prognostication-in-heart-failure/
Generalized Fixed Drug Eruptions Require Urgent Care: A Case Series
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
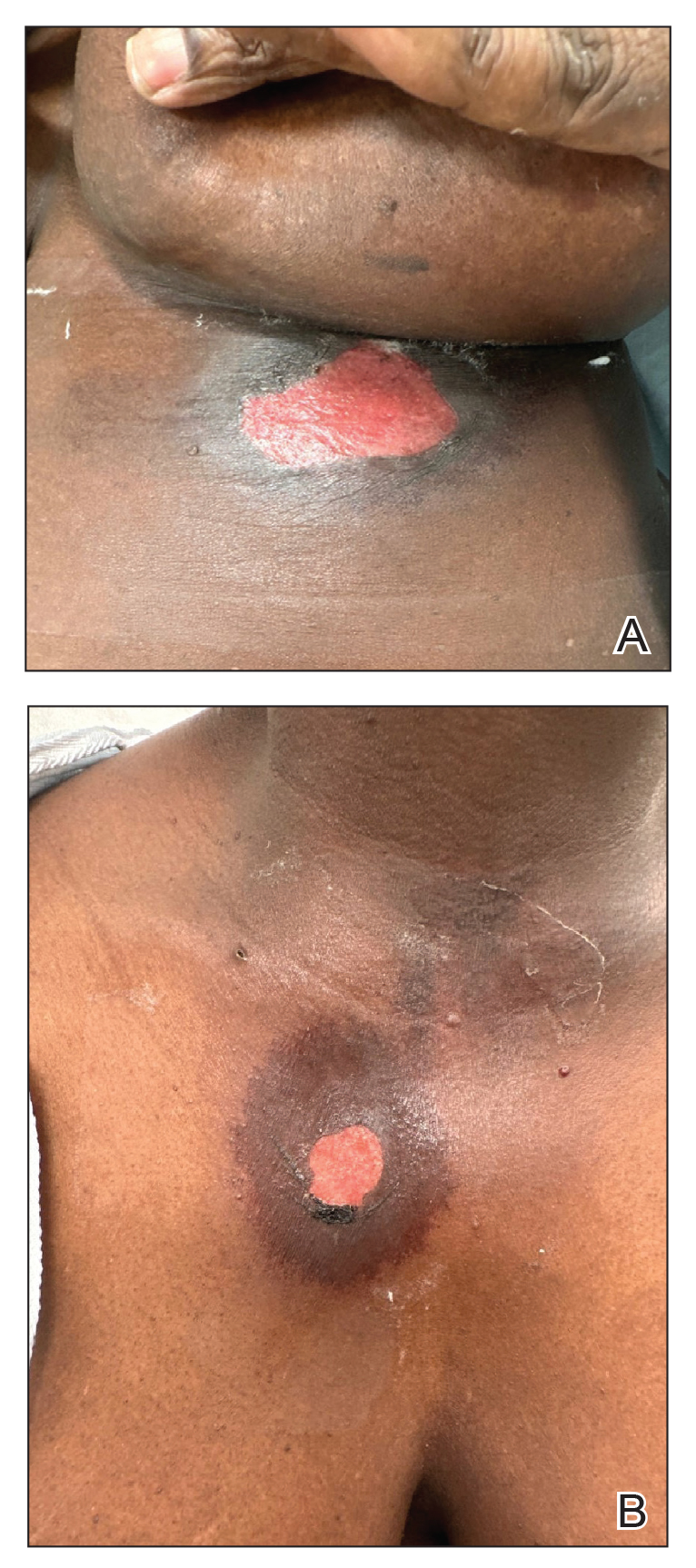
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
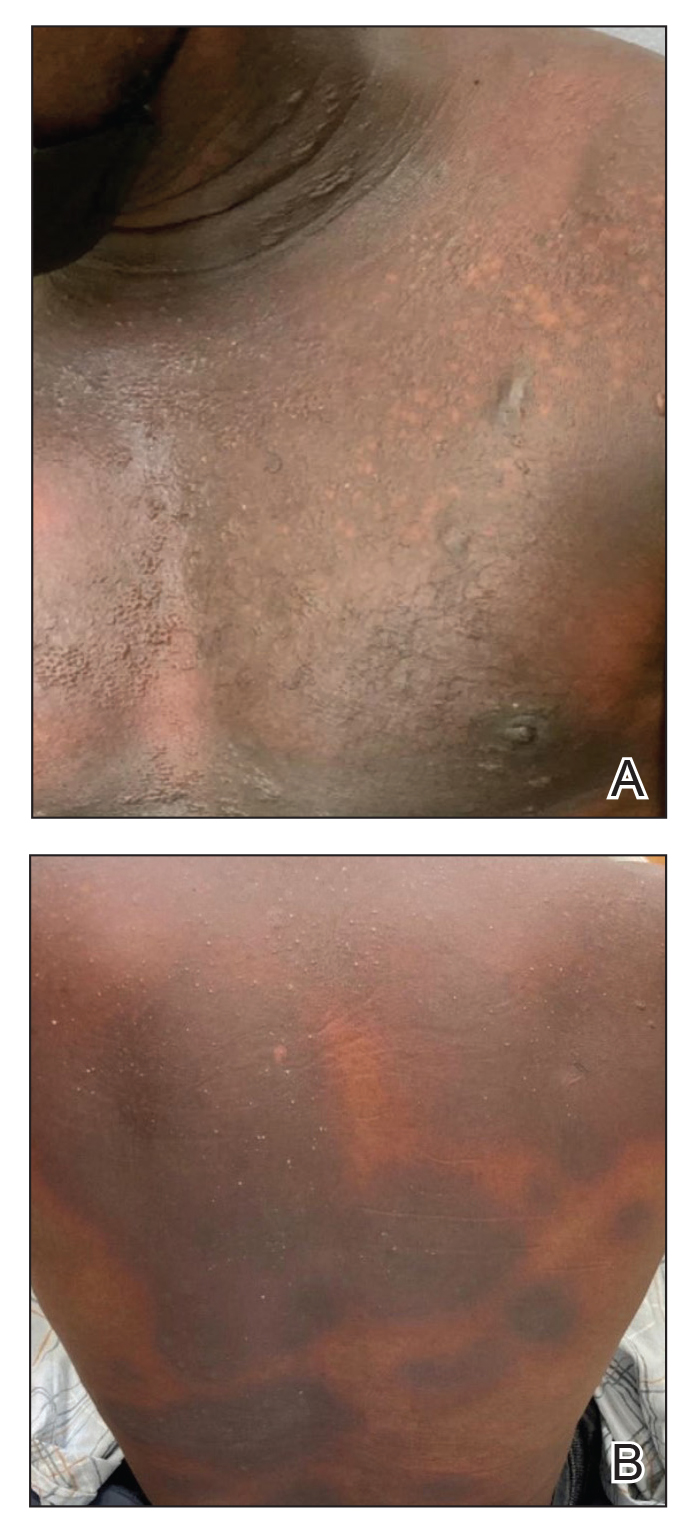
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
Recognizing cutaneous drug eruptions is important for treatment and prevention of recurrence. Fixed drug eruptions (FDEs) typically are harmless but can have major negative cosmetic consequences for patients. In its more severe forms, patients are at risk for widespread epithelial necrosis with accompanying complications. We report 1 patient with generalized FDE and 2 with generalized bullous FDE. We also discuss the recognition and treatment of the condition. Two patients previously had been diagnosed with systemic lupus erythematosus (SLE).
Case Series
Patient 1—A 60-year-old woman presented to dermatology with a rash on the trunk and groin folds of 4 days’ duration. She had a history of SLE and cutaneous lupus treated with hydroxychloroquine 200 mg twice daily and topical corticosteroids. She had started sulfamethoxazole-trimethoprim for a urinary tract infection with a rash appearing 1 day later. She reported burning skin pain with progression to blisters that “sloughed” off. She denied any known history of allergy to sulfa drugs. Prior to evaluation by dermatology, she visited an urgent care facility and was prescribed hydroxyzine and intramuscular corticosteroids. At presentation to dermatology 3 days after taking sulfamethoxazole-trimethoprim, she had annular flaccid bullae and superficial erosions with dusky borders on the right posterior thigh, right side of the chest, left inframammary fold, and right inguinal fold (Figure 1). She had no ocular, oral, or vaginal erosions. A diagnosis of generalized bullous FDE was favored over erythema multiforme or Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Shave biopsies from lesions on the right posterior thigh and right inguinal fold demonstrated interface dermatitis with epidermal necrosis, pigment incontinence, and numerous eosinophils. Direct immunofluorescence of the perilesional skin was negative for immunoprotein deposition. These findings were consistent with the clinical impression of generalized bullous FDE. Prior to receiving the histopathology report, the patient was initiated on a regimen of cyclosporine 5 mg/kg/d in the setting of normal renal function and followed until the eruption resolved completely. Cyclosporine was tapered at 2 weeks and discontinued at 3 weeks.
Patient 2—A 32-year-old woman presented for follow-up management of discoid lupus erythematosus. She had a history of systemic and cutaneous lupus, juvenile rheumatoid arthritis, and mixed connective tissue disease managed with prednisone, hydroxychloroquine, azathioprine, and belimumab. Physical examination revealed scarring alopecia with dyspigmentation and active inflammation consistent with uncontrolled cutaneous lupus. However, she also had oval-shaped hyperpigmented patches over the left breast, clavicle, and anterior chest consistent with a generalized FDE (Figure 2). The patient did not recall a history of similar lesions and could not identify a possible trigger. She was counseled on possible culprits and advised to avoid unnecessary medications. She had an unremarkable clinical course; therefore, no further intervention was necessary.
Patient 3—A 33-year-old man presented to the emergency department with a painful rash on the chest and back of 2 days’ duration that began 1 hour after taking naproxen (dosage unknown) for back pain. He had no notable medical history. The patient stated that the rash had slowly worsened and started to develop blisters. He visited an urgent care facility 1 day prior to the current presentation and was started on a 5-day course of prednisone 40 mg daily; the first 2 doses did not help. He denied any mucosal involvement apart from a tender lesion on the penis. He reported a history of an allergic reaction to penicillin. Physical examination revealed extensive dusky violaceous annular plaques with erythematous borders across the anterior and posterior trunk (Figure 3). Multiple flaccid bullae developed within these plaques, involving 15% of the body surface area. He was diagnosed with generalized bullous FDE based on the clinical history and histopathology. He was admitted to the burn intensive care unit and treated with cyclosporine 3 mg/kg/d with subsequent resolution of the eruption.
Comment
Presentation of FDEs—A fixed drug eruption manifests with 1 or more well-demarcated, red or violaceous, annular patches that resolve with postinflammatory hyperpigmentation; it occasionally may manifest with bullae. Initial eruptions may occur up to 2 weeks following medication exposure, but recurrent eruptions usually happen within minutes to hours later. They often are in the same location as prior lesions. A fixed drug eruption can be solitary, scattered, or generalized; a generalized FDE typically demonstrates multiple bilateral lesions that may itch, burn, or cause no symptoms. Patients can experience an FDE at any age, though the median age is reported as 35 to 60 years of age.1 A fixed drug eruption usually occurs after ingestion of oral medications, though there have been a few reports with iodinated contrast.2 Well-known culprits include antibiotics (eg, sulfamethoxazole-trimethoprim, tetracyclines, penicillins/cephalosporins, quinolones, dapsone), nonsteroidal anti-inflammatory drugs, acetaminophen (eg, paracetamol), barbiturates, antimalarials, and anticonvulsants. It also can occur with vaccines or with certain foods (fixed food eruption).3,4 Clinicians may try an oral drug challenge to identify the cause of an FDE, but in patients with a history of a generalized FDE, the risk for developing an increasingly severe reaction with repeated exposure to the medication is too high.5
Histopathology—Patch testing at the site of prior eruption with suspected drug culprits may be useful.6 Histopathology of FDE typically demonstrates vacuolar changes at the dermoepidermal junction with a lichenoid lymphocytic infiltrate. Early lesions often show a predominance of eosinophils. Subepidermal clefting is a feature of the bullous variant. In an active lesion, there are large numbers of CD8+ T lymphocytes expressing natural killer cell–associated molecules.7 The pathologic mechanism is not well understood, though it has been hypothesized that memory CD8+ cells are maintained in specific regions of the epidermis by IL-15 produced in the microenvironment and are activated upon rechallenge.7Considerations in Generalized Bullous FDE—Generalized FDE is defined in the literature as an FDE with involvement of 3 of 6 body areas: head, neck, trunk, upper limbs, lower limbs, and genital area. It may cover more or less than 10% of the body surface area.8-10 Although an isolated FDE frequently is asymptomatic and may not be cause for alarm, recurring drug eruptions increase the risk for development of generalized bullous FDE. Generalized bullous FDE is a rare subset. It is frequently misdiagnosed, and data on its incidence are uncertain.11 Of note, several pathologies causing bullous lesions may be in the differential diagnosis, including bullous pemphigoid; pemphigus vulgaris; bullous SLE; or bullae from cutaneous lupus, staphylococcal scalded skin syndrome, erythema multiforme, or SJS/TEN.12 When matched for body surface area involvement with SJS/TEN, generalized bullous FDE shares nearly identical mortality rates10; therefore, these patients should be treated with the same level of urgency and admitted to a critical care or burn unit, as they are at serious risk for infection and other complications.13
Clinical history and presentation along with histopathologic findings help to narrow down the differential diagnosis. Clinically, generalized bullous FDE does not affect the surrounding skin and manifests sooner after drug exposure (1–24 hours) with less mucosal involvement than SJS/TEN.9 Additionally, SJS/TEN patients frequently have generalized malaise and/or fever, while generalized bullous FDE patients do not. Finally, patients with generalized bullous FDE may report a history of a cutaneous eruption similar in morphology or in the same location.
Histopathologically, generalized bullous FDE may be similar to FDE with the addition of a subepidermal blister. Generalized bullous FDE patients have greater eosinophil infiltration and dermal melanophages than patients with SJS/TEN.9 Cellular infiltrates in generalized bullous FDE include more dermal CD41 cells, such as Foxp31 regulatory T cells; fewer intraepidermal CD561 cells; and fewer intraepidermal cells with granulysin.9 Occasionally, generalized bullous FDE causes full-thickness necrosis. In those cases, generalized bullous FDE cannot reliably be distinguished from other conditions with epidermal necrolysis on histopathology.13
FDE Diagnostics—A cytotoxin produced by
Management—Avoidance of the inciting drug often is sufficient for patients with an FDE, as demonstrated in patient 2 in our case series. Clinicians also should counsel patients on avoidance of potential cross-reacting drugs. Symptomatic treatment for itch or pain is appropriate and may include antihistamines or topical steroids. Nonsteroidal anti-inflammatory drugs may exacerbate or be causative of FDE. For generalized bullous FDE, cyclosporine is favored in the literature15,16 and was used to successfully treat both patients 1 and 3 in our case series. A short course of systemic corticosteroids or intravenous immunoglobulin also may be considered. Mild cases of generalized bullous FDE may be treated with close outpatient follow-up (patient 1), while severe cases require inpatient or even critical care monitoring with aggressive medical management to prevent the progression of skin desquamation (patient 3). Patients with severe oral lesions may require inpatient support for fluid maintenance.
Lupus History—Two patients in our case series had a history of lupus. Lupus itself can cause primary bullous lesions. Similar to FDE, bullous SLE can involve sun-exposed and nonexposed areas of the skin as well as the mucous membranes with a predilection for the lower vermilion lip.17 In bullous SLE, tense subepidermal blisters with a neutrophil-rich infiltrate form due to circulating antibodies to type VII collagen. These blisters have an erythematous or urticated base, most commonly on the face, upper trunk, and proximal extremities.18 In both SLE with skin manifestations and lupus limited to the skin, bullae may form due to extensive vacuolar degeneration. Similar to TEN, they can form rapidly in a widespread distribution.17 However, there is limited mucosal involvement, no clear drug association, and a better prognosis. Bullae caused by lupus will frequently demonstrate deposition of immunoproteins IgG, IgM, IgA, and complement component 3 at the basement membrane zone in perilesional skin on direct immunofluorescence. However, negative direct immunofluorescence does not rule out lupus.12 At the same time, patients with lupus frequently have comorbidities requiring multiple medications; the need for these medications may predispose patients to higher rates of cutaneous drug eruptions.19 To our knowledge, there is no known association between FDE and lupus.
Conclusion
Patients with acute eruptions following the initiation of a new prescription or over-the-counter medication require urgent evaluation. Generalized bullous FDE requires timely diagnosis and intervention. Patients with lupus have an increased risk for cutaneous drug eruptions due to polypharmacy. Further investigation is necessary to determine if there is a pathophysiologic mechanism responsible for the development of FDE in lupus patients.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
- Anderson HJ, Lee JB. A review of fixed drug eruption with a special focus on generalized bullous fixed drug eruption. Medicina (Kaunas). 2021;57:925.
- Gavin M, Sharp L, Walker K, et al. Contrast-induced generalized bullous fixed drug eruption resembling Stevens-Johnson syndrome. Proc (Bayl Univ Med Cent). 2019;32:601-602.
- Kabir S, Feit EJ, Heilman ER. Generalized fixed drug eruption following Pfizer-BioNtech COVID-19 vaccination. Clin Case Rep. 2022;10:E6684.
- Choi S, Kim SH, Hwang JH, et al. Rapidly progressing generalized bullous fixed drug eruption after the first dose of COVID-19 messenger RNA vaccination. J Dermatol. 2023;50:1190-1193.
- Mahboob A, Haroon TS. Drugs causing fixed eruptions: a study of 450 cases. Int J Dermatol. 1998;37:833-838.
- Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. 2009;9:316-321.
- Mizukawa Y, Yamazaki Y, Shiohara T. In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol. 2008;158:1230-1238.
- Lee CH, Chen YC, Cho YT, et al. Fixed-drug eruption: a retrospective study in a single referral center in northern Taiwan. Dermatologica Sinica. 2012;30:11-15.
- Cho YT, Lin JW, Chen YC, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539-548.
- Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726-732.
- Patel S, John AM, Handler MZ, et al. Fixed drug eruptions: an update, emphasizing the potentially lethal generalized bullous fixed drug eruption. Am J Clin Dermatol. 2020;21:393-399.
- Ranario JS, Smith JL. Bullous lesions in a patient with systemic lupus erythematosus. J Clin Aesthet Dermatol. 2014;7:44-49.
- Perron E, Viarnaud A, Marciano L, et al. Clinical and histological features of fixed drug eruption: a single-centre series of 73 cases with comparison between bullous and non-bullous forms. Eur J Dermatol. 2021;31:372-380.
- Chen CB, Kuo KL, Wang CW, et al. Detecting lesional granulysin levels for rapid diagnosis of cytotoxic T lymphocyte-mediated bullous skin disorders. J Allergy Clin Immunol Pract. 2021;9:1327-1337.e3.
- Beniwal R, Gupta LK, Khare AK, et al. Cyclosporine in generalized bullous-fixed drug eruption. Indian J Dermatol. 2018;63:432-433.
- Vargas Mora P, García S, Valenzuela F, et al. Generalized bullous fixed drug eruption successfully treated with cyclosporine. Dermatol Ther. 2020;33:E13492.
- Montagnon CM, Tolkachjov SN, Murrell DF, et al. Subepithelial autoimmune blistering dermatoses: clinical features and diagnosis. J Am Acad Dermatol. 2021;85:1-14.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Zonzits E, Aberer W, Tappeiner G. Drug eruptions from mesna. After cyclophosphamide treatment of patients with systemic lupus erythematosus and dermatomyositis. Arch Dermatol. 1992;128:80-82.
Practice Points
- Although localized fixed drug eruption (FDE) is a relatively benign diagnosis, generalized bullous FDE requires urgent management and may necessitate intensive burn care.
- Patients with lupus are at increased risk for drug eruptions due to polypharmacy, and there is a wide differential for bullous eruptions in these patients.
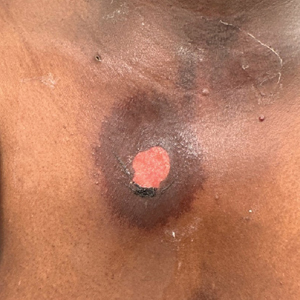
Barriers to Mohs Micrographic Surgery in Japanese Patients With Basal Cell Carcinoma
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
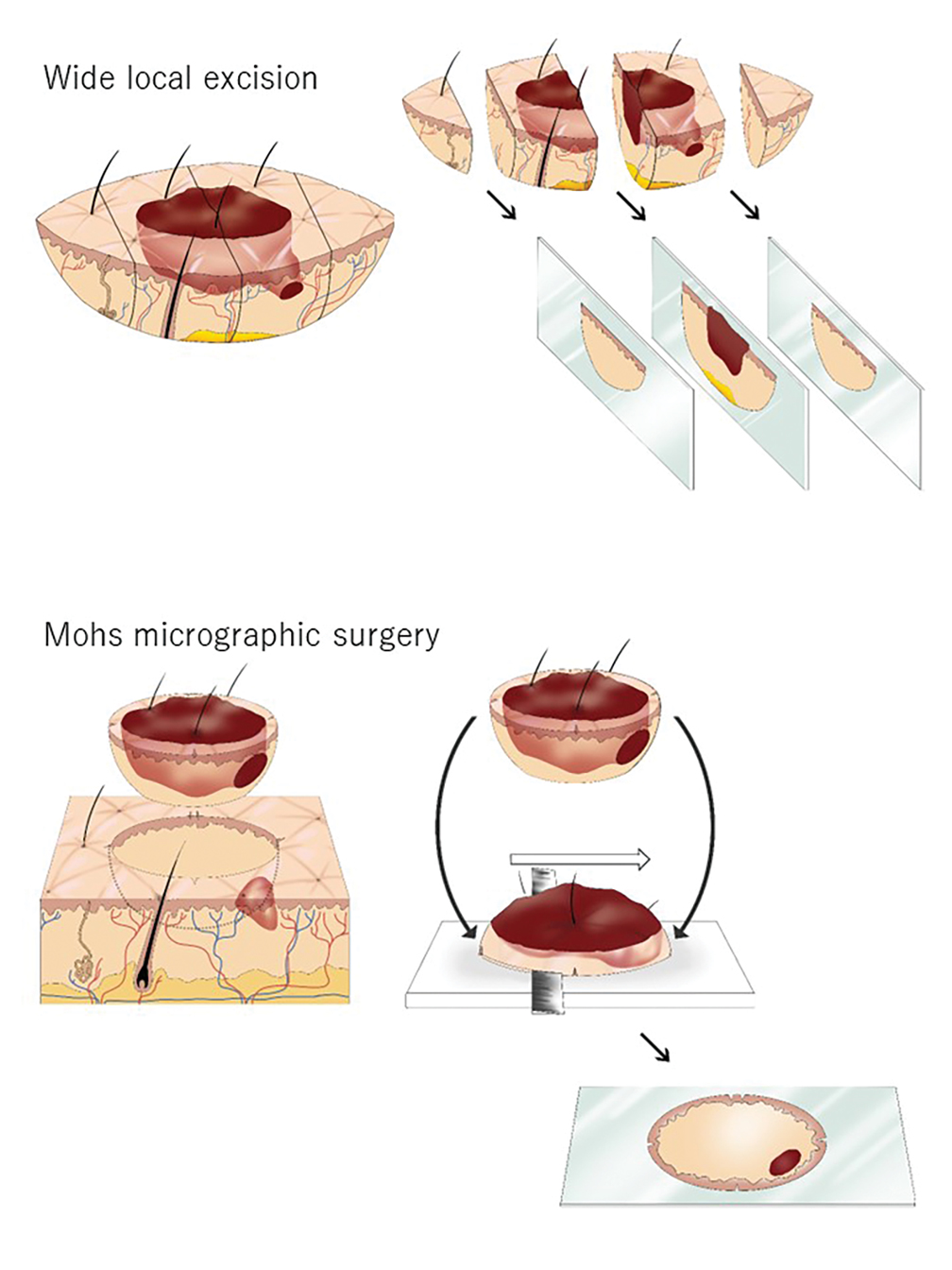
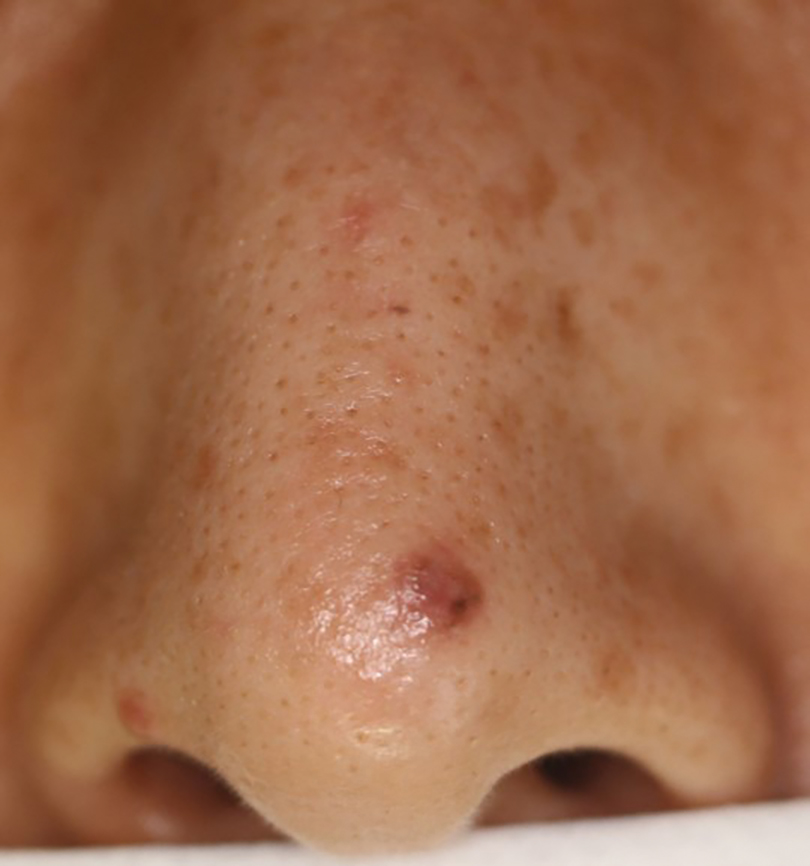
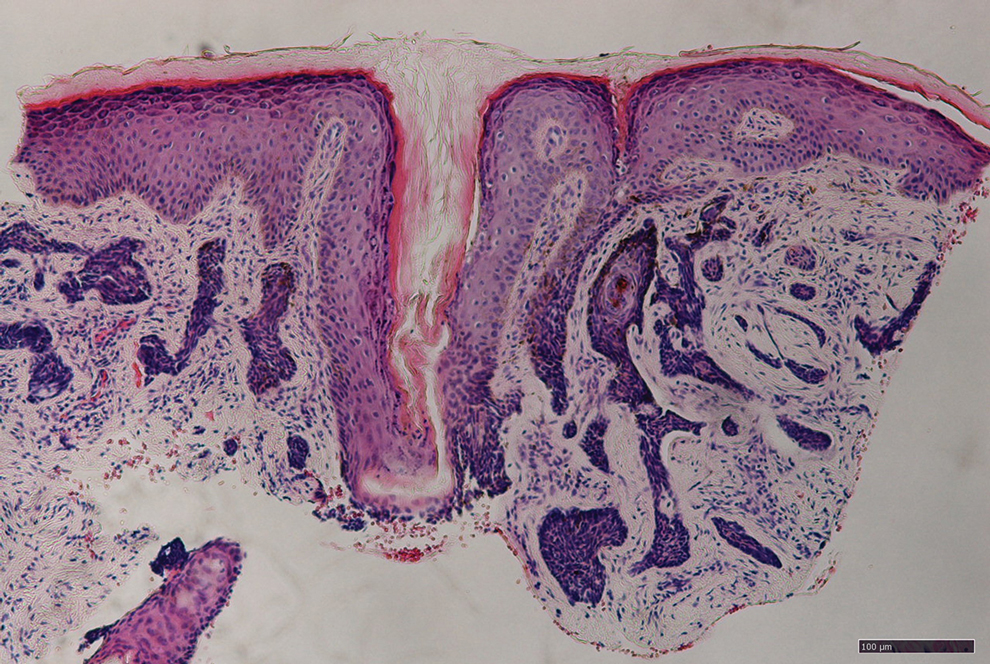
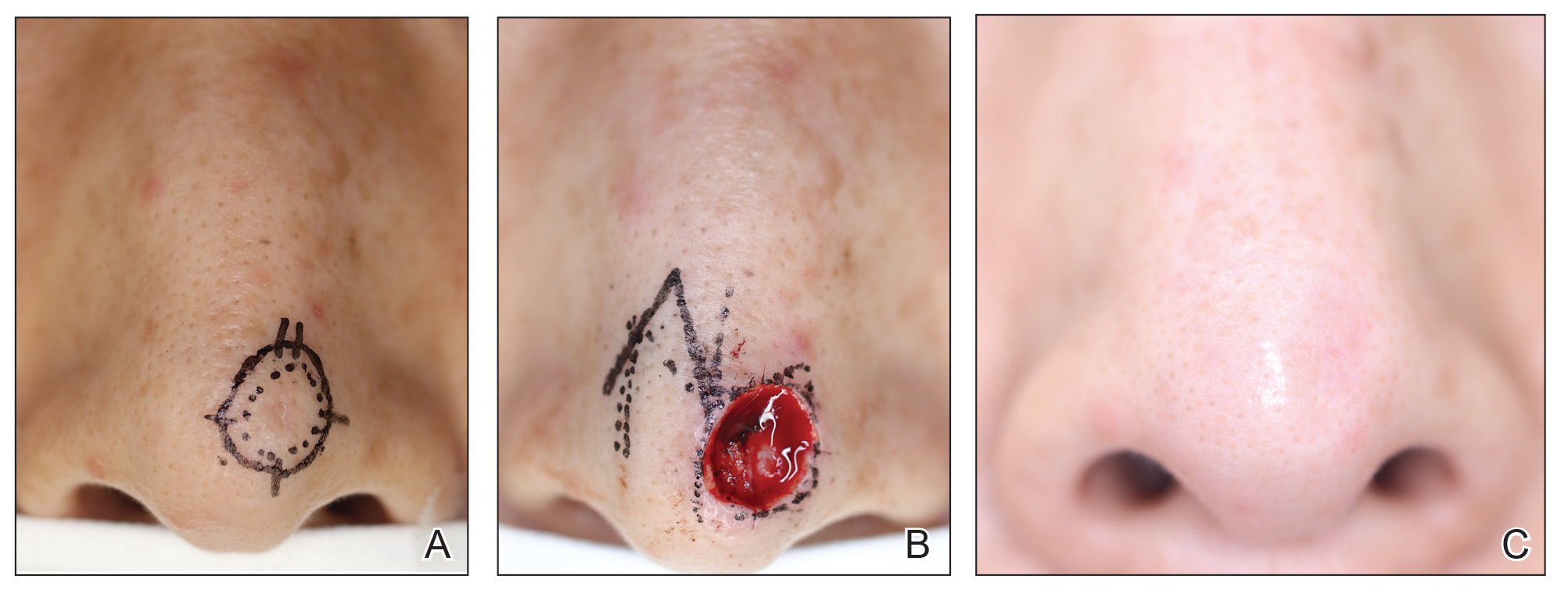
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
Margin-controlled surgery for squamous cell carcinoma (SCC) on the lower lip was first performed by Dr. Frederic Mohs on June 30, 1936. Since then, thousands of skin cancer surgeons have refined and adopted the technique. Due to the high cure rate and sparing of normal tissue, Mohs micrographic surgery (MMS) has become the gold standard treatment for facial and special-site nonmelanoma skin cancer worldwide. Mohs micrographic surgery is performed on more than 876,000 tumors annually in the United States.1 Among 3.5 million Americans diagnosed with nonmelanoma skin cancer in 2006, one-quarter were treated with MMS.2 In Japan, basal cell carcinoma (BCC) is the most common skin malignancy, with an incidence of 3.34 cases per 100,000 individuals; SCC is the second most common, with an incidence of 2.5 cases per 100,000 individuals.3
The essential element that makes MMS unique is the careful microscopic examination of the entire margin of the removed specimen. Tissue processing is done with careful en face orientation to ensure that circumferential and deep margins are entirely visible. The surgeon interprets the slides and proceeds to remove the additional tumor as necessary. Because the same physician performs both the surgery and the pathologic assessment throughout the procedure, a precise correlation between the microscopic and surgical findings can be made. The surgeon can begin with smaller margins, removing minimal healthy tissue while removing all the cancer cells, which results in the smallest-possible skin defect and the best prognosis for the malignancy (Figure 1).
At the only facility in Japan offering MMS, the lead author (S.S.) has treated 52 lesions with MMS in 46 patients (2020-2022). Of these patients, 40 were White, 5 were Japanese, and 1 was of African descent. In this case series, we present 5 Japanese patients who had BCC treated with MMS.
Case Series
Patient 1—A 50-year-old Japanese woman presented to dermatology with a brown papule on the nasal tip of 1.25 year’s duration (Figure 2). A biopsy revealed infiltrative BCC (Figure 3), and the patient was referred to the dermatology department at a nearby university hospital. Because the BCC was an aggressive variant, wide local excision (WLE) with subsequent flap reconstruction was recommended as well as radiation therapy. The patient learned about MMS through an internet search and refused both options, seeking MMS treatment at our clinic. Although Japanese health insurance does not cover MMS, the patient had supplemental private insurance that did cover the cost. She provided consent to undergo the procedure. Physical examination revealed a 7.5×6-mm, brown-red macule with ill-defined borders on the tip of the nose. We used a 1.5-mm margin for the first stage of MMS (Figure 4A). The frozen section revealed that the tumor had been entirely excised in the first stage, leaving only a 10.5×9-mm skin defect that was reconstructed with a Dufourmentel flap (Figure 4B). No signs of recurrence were noted at 3.5-year follow-up, and the cosmetic outcome was favorable (Figure 4C). National Comprehensive Cancer Network guidelines recommend a margin greater than 4 mm for infiltrative BCCs4; therefore, our technique reduced the total defect by at least 4 mm in a cosmetically sensitive area. The patient also did not need radiation therapy, which reduced morbidity. She continues to be recurrence free at 3.5-year follow-up.
Patient 2—A 63-year-old Japanese man presented to dermatology with a brown macule on the right lower eyelid of 2 years’ duration. A biopsy of the lesion was positive for nodular BCC. After being advised to undergo WLE and extensive reconstruction with plastic surgery, the patient learned of MMS through an internet search and found our clinic. Physical examination revealed a 7×5-mm brown macule on the right lower eyelid. The patient had supplemental private insurance that covered the cost of MMS, and he provided consent for the procedure. A 1.5-mm margin was taken for the first stage, resulting in a 10×8-mm defect superficial to the orbicularis oculi muscle. The frozen section revealed residual tumor exposure in the dermis at the 9- to 10-o’clock position. A second-stage excision was performed to remove an additional 1.5 mm of skin at the 9- to 12-o’clock position with a thin layer of the orbicularis oculi muscle. The subsequent histologic examination revealed no residual BCC, and the final 13×9-mm skin defect was reconstructed with a rotation flap. There were no signs of recurrence at 2.5-year follow-up with an excellent cosmetic outcome.
Patient 3—A 73-year-old Japanese man presented to a local university dermatology clinic with a new papule on the nose. The dermatologist suggested WLE with 4-mm margins and reconstruction of the skin defect 2 weeks later by a plastic surgeon. The patient was not satisfied with the proposed surgical plan, which led him to learn about MMS on the internet; he subsequently found our clinic. Physical examination revealed a 4×3.5-mm brown papule on the tip of the nose. He understood the nature of MMS and chose to pay out-of-pocket because Japanese health insurance did not cover the procedure. We used a 2-mm margin for the first stage, which created a 7.5×7-mm skin defect. The frozen section pathology revealed no residual BCC at the cut surface. The skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Patient 4—A 45-year-old man presented to a dermatology clinic with a papule on the right side of the nose of 1 year’s duration. A biopsy revealed the lesion was a nodular BCC. The dermatologist recommended WLE at a general hospital, but the patient refused after learning about MMS. He subsequently made an appointment with our clinic. Physical examination revealed a 7×4-mm white papule on the right side of the nose. The patient had private insurance that covered the cost of MMS. The first stage was performed with 1.5-mm margins and was clear of residual tumor. A Limberg rhombic flap from the adjacent cheek was used to repair the final 10×7-mm skin defect. There were no signs of recurrence at 1 year and 9 months’ follow-up with a favorable cosmetic outcome.
Patient 5—A 76-year-old Japanese woman presented to a university hospital near Tokyo with a black papule on the left cutaneous lip of 5 years’ duration. A biopsy revealed nodular BCC, and WLE with flap reconstruction was recommended. The patient’s son learned about MMS through internet research and referred her to our clinic. Physical examination revealed a 7×5-mm black papule on the left upper lip. The patient’s private insurance covered the cost of MMS, and she consented to the procedure. We used a 2-mm initial margin, and the immediate frozen section revealed no signs of BCC at the cut surface. The 11×9-mm skin defect was reconstructed with a Limberg rhombic flap. There were no signs of recurrence at 1.5-year follow-up with a favorable cosmetic outcome.
Comment
We presented 5 cases of MMS in Japanese patients with BCC. More than 7000 new cases of nonmelanoma skin cancer occur every year in Japan.3 Only 0.04% of these cases—the 5 cases presented here—were treated with MMS in Japan in 2020 and 2021, in contrast to 25% in the United States in 2006.2
MMS vs Other BCC Treatments—Mohs micrographic surgery offers 2 distinct advantages over conventional excision: an improved cure rate while achieving a smaller final defect size, generally leading to better cosmetic outcomes. Overall 5-year recurrence rates of BCC are 10% for conventional surgical excision vs 1% for MMS, while the recurrence rates for SCC are 8% and 3%, respectively.5 A study of well-demarcated BCCs smaller than 2 cm that were treated with MMS with 2-mm increments revealed that 95% of the cases were free of malignancy within a 4-mm margin of the normal-appearing skin surrounding the tumor.6 Several articles have reported a 95% cure rate or higher with conventional excision of localized BCC,7 but 4- to 5-mm excision margins are required, resulting in a greater skin defect and a lower cure rate compared to MMS.
Aggressive subtypes of BCC have a higher recurrence rate. Rowe et al8 reported the following 5-year recurrence rates: 5.6% for MMS, 17.4% for conventional surgical excision, 40.0% for curettage and electrodesiccation, and 9.8% for radiation therapy. Primary BCCs with high-risk histologic subtypes has a 10-year recurrence rate of 4.4% with MMS vs 12.2% with conventional excision.9 These findings reveal that MMS yields a better prognosis compared to traditional treatment methods for recurrent BCCs and BCCs of high-risk histologic subtypes.
The primary reason for the excellent cure rate seen in MMS is the ability to perform complete margin assessment. Peripheral and deep en face margin assessment (PDEMA) is crucial in achieving high cure rates with narrow margins. In WLE (Figure 1), vertical sectioning (also known as bread-loafing) does not achieve direct visualization of the entire surgical margin, as this technique only evaluates random sections and does not achieve PDEMA.10 The bread-loafing method is used almost exclusively in Japan and visualizes only 0.1% of the entire margin compared to 100% with MMS.11 Beyond the superior cure rate, the MMS technique often yields smaller final defects compared to WLE. All 5 of our patients achieved complete tumor removal while sparing more normal tissue compared to conventional WLE, which takes at least a 4-mm margin in all directions.
Barriers to Adopting MMS in Japan—There are many barriers to the broader adoption of MMS in Japan. A guideline of the Japanese Dermatological Association says, MMS “is complicated, requires special training for acquisition, and requires time and labor for implementation of a series of processes, and it has not gained wide acceptance in Japan because of these disadvantages.”3 There currently are no MMS training programs in Japan. We refute this statement from the Japanese Dermatological Association because, in our experience, only 1 surgeon plus a single histotechnician familiar with MMS is sufficient for a facility to offer the procedure (the lead author of this study [S.S.] acts as both the surgeon and the histotechnician). Another misconception among some physicians in Japan is that cancer on ethnically Japanese skin is uniquely suited to excision without microscopic verification of tumor clearance because the borders of the tumors are easily identified, which was based on good cure rates for the excision of well-demarcated pigmented BCCs in a Japanese cohort. This study of a Japanese cohort investigated the specimens with the conventional bread-loafing technique but not with the PDEMA.12
Eighty percent (4/5) of our patients presented with nodular BCC, and only 1 required a second stage. In comparison, we also treated 16 White patients with nodular BCC with MMS during the same period, and 31% (5/16) required more than 1 stage, with 1 patient requiring 3 stages. This cohort, however, is too small to demonstrate a statistically significant difference (S.S., unpublished data, 2020-2022).
A study in Singapore reported the postsurgical complication rate and 5-year recurrence rate for 481 tumors (92% BCC and 7.5% SCC). The median follow-up duration after MMS was 36 months, and the recurrence rate was 0.6%. The postsurgical complications included 11 (2.3%) cases with superficial tip necrosis of surgical flaps/grafts, 2 (0.4%) with mild wound dehiscence, 1 (0.2%) with minor surgical site bleeding, and 1 (0.2%) with minor wound infection.13 This study supports the notion that MMS is equally effective for Asian patients.
Awareness of MMS in Japan is lacking, and most Japanese dermatologists do not know about the technique. All 5 patients in our case series asked their dermatologists about alternative treatment options and were not offered MMS. In each case, the patients learned of the technique through internet research.
The lack of insurance reimbursement for MMS in Japan is another barrier. Because the national health insurance does not reimburse for MMS, the procedure is relatively unavailable to most Japanese citizens who cannot pay out-of-pocket for the treatment and do not have supplemental insurance. Mohs micrographic surgery may seem expensive compared to WLE followed by repair; however, in the authors’ experience, in Japan, excision without MMS may require general sedation and multiple surgeries to reconstruct larger skin defects, leading to greater morbidity and risk for the patient.
Conclusion
Mohs micrographic surgery in Japan is in its infancy, and further studies showing recurrence rates and long-term prognosis are needed. Such data should help increase awareness of MMS among Japanese physicians as an excellent treatment option for their patients. Furthermore, as Japan becomes more heterogenous as a society and the US Military increases its presence in the region, the need for MMS is likely to increase.
Acknowledgments—We appreciate the proofreading support by Mark Bivens, MBA, MSc (Tokyo, Japan), as well as the technical support from Ben Tallon, MBChB, and Robyn Mason (both in Tauranga, New Zealand) to start MMS at our clinic.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
- Asgari MM, Olson J, Alam M. Needs assessment for Mohs micrographic surgery. Dermatol Clin. 2012;30:167-175. doi:10.1016/j.det.2011.08.010
- Connolly SM, Baker DR, Baker DR, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550.
- Ansai SI, Umebayashi Y, Katsumata N, et al. Japanese Dermatological Association Guidelines: outlines of guidelines for cutaneous squamous cell carcinoma 2020. J Dermatol. 2021;48:E288-E311.
- Schmults CD, Blitzblau R, Aasi SZ, et at. Basal cell skin cancer, version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2023;21:1181-1203. doi:10.6004/jncn.2023.0056
- Snow SN, Gunkel J. Mohs surgery. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:2445-2455. doi:10.1016/b978-0-070-94171-3.00041-7
- Wolf DJ, Zitelli JA. Surgical margins for basal cell carcinoma. Arch Dermatol. 1987;123:340-344.
- Quazi SJ, Aslam N, Saleem H, et al. Surgical margin of excision in basal cell carcinoma: a systematic review of literature. Cureus. 2020;12:E9211.
- Rowe DE, Carroll RJ, Day Jus CL. Mohs surgery is the treatment of choice for recurrent (previously treated) basal cell carcinoma. J Dermatol Surg Oncol. 1989;15:424-431.
- Van Loo, Mosterd K, Krekels GA. Surgical excision versus Mohs’ micrographic surgery for basal cell carcinoma of the face. Eur J Cancer. 2014;50:3011-3020.
- Schmults CD, Blitzblau R, Aasi SZ, et al. NCCN Guidelines Insights: Squamous Cell Skin Cancer, Version 1.2022. J Natl Compr Canc Netw. 2021;19:1382-1394.
- Hui AM, Jacobson M, Markowitz O, et al. Mohs micrographic surgery for the treatment of melanoma. Dermatol Clin. 2012;30:503-515.
- Ito T, Inatomi Y, Nagae K, et al. Narrow-margin excision is a safe, reliable treatment for well-defined, primary pigmented basal cell carcinoma: an analysis of 288 lesions in Japan. J Eur Acad Dermatol Venereol. 2015;29:1828-1831.
- Ho WYB, Zhao X, Tan WPM. Mohs micrographic surgery in Singapore: a long-term follow-up review. Ann Acad Med Singap. 2021;50:922-923.
Practice Points
- Mohs micrographic surgery (MMS) is a safe and effective treatment method for nonmelanoma skin cancer. In some cases, this procedure is superior to standard wide local excision and repair.
- For the broader adaptation of this vital technique in Japan—where MMS is not well established—increased awareness of treatment outcomes among Japanese physicians is needed.
Suspected Orbital Compartment Syndrome Leading to Visual Loss After Pterional Craniotomy
Perioperative visual loss (POVL) is a well-documented yet uncommon complication of nonocular surgery. Patients undergoing cardiac and spinal surgery are at the greatest risk, though POVL may occur during other neurosurgical and vascular procedures as well. The most common causes of POVL are central retinal artery occlusion (CRAO) and ischemic optic neuropathy (ION),1-3 though cases of orbital compartment syndrome (OCS) have also been reported.4-7
We describe a case of POVL during a temporal meningioma excision using the pterional approach. Though the etiology is not fully understood, the patient’s clinical course was complicated by a third cranial nerve (CN III) palsy and CRAO, which, together with the patient’s presentation, were consistent with previously documented cases of OCS. The goals of this case report are to increase awareness of this surgical outcome, identify practices that may have contributed to its development, and delineate methods to minimize its occurrence.
Informed consent regarding this research was obtained from the patient and an institutional Health Insurance Portability and Accountability Act authorization form was completed. This manuscript adheres to the applicable Enhancing the Quality and Transparency of Health Research guideline.8
Case Presentation

A 47-year-old woman underwent a left temporal craniotomy for resection of a sphenoid wing meningioma discovered during a workup for persistent headaches. She had no medical history of diabetes, hypertension, coronary artery disease, or ophthalmic disease. Two months before her scheduled surgery, the patient reported bilateral blurry vision and underwent ophthalmologic evaluation. Her intraocular pressure (IOP) was normal, and she had no pupillary or retinal disease. She showed evidence of decreased vision in her left eye, suggesting a possible mass effect from her meningioma. Subsequent imaging of the optic nerve and retina had unremarkable physiology (Figure 1). Preoperative magnetic resonance imaging (MRI) demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus(Figure 2). There was a superior mass effect on the left middle cerebral artery, but all vessels were patent without evidence of thrombosis.

The patient underwent general anesthesia with invasive hemodynamic monitoring used throughout the procedure. She was induced with fentanyl, propofol, and rocuronium; anesthesia was maintained with isoflurane and a remifentanil infusion. Hypotension was treated with phenylephrine and intravenous fluids. Intraoperative neuromonitoring with electroencephalogram (EEG) and somatosensory evoked potentials was performed. During the surgery, the patient was positioned supine in a Mayfield 3-point head fixation system. All pressure points were padded appropriately and continually checked. A standard left pterional craniotomy was performed, and the scalp was reflected anteriorly and secured using fish hooks with rubber bands. The operation did not violate the cavernous sinus or orbital compartment. There was no evidence of active bleeding upon inspection nor with the Valsalva maneuver. No changes were noted in EEG or somatosensory evoked potentials; blood pressure remained within 20 mm Hg of the patient’s baseline. She was extubated at the end of the 10-hour case and was hemodynamically stable upon transport to the surgical intensive care unit. Postoperative imaging confirmed the successful removal of the left sphenoid wing meningioma.
The patient’s postoperative examination demonstrated a 5 mm dilated, nonresponsive left pupil, though the patient did not report visual loss at that time. Defects were noted in the inferior oblique, superior, inferior, and medial rectus muscles, consistent with CN III palsy. The surgery included manipulation of CN III, which made this a possible outcome, but an alternate causative pathology like OCS was not immediately suspected. Postoperative computed tomography (CT) showed an expected pneumocephalus and left scalp swelling without evidence of mass effect or midline shift.
On the morning of postoperative Day 1, the patient reported vision loss in her left eye, while her clinical examination revealed erythema and conjunctival chemosis with left eyelid swelling. The ophthalmologic evaluation was notable for a continued leftCN III palsy with intact lateral rectus and superior oblique function, a nonreactive and dilated left eye with 3+ afferent pupillary defect by reverse (light perception), pallor throughout, a flat cherry red macula with blurred disc margins, left upper eyelid edema, and 18 mm Hg intraocular pressure bilaterally (reference range, 8 to 21 mm Hg). Fundoscopic examination showed a clear vitreous without plaques or occlusions, no perivascular sheathing, and no retinal hemorrhages. CT angiography revealed small outpouchings at the superolateral aspect of the left and right cavernous carotid, consistent with atherosclerotic calcifications. An echocardiogram revealed a Valsalva-dependent patent foramen ovale, but a venous Doppler ultrasound yielded negative results.
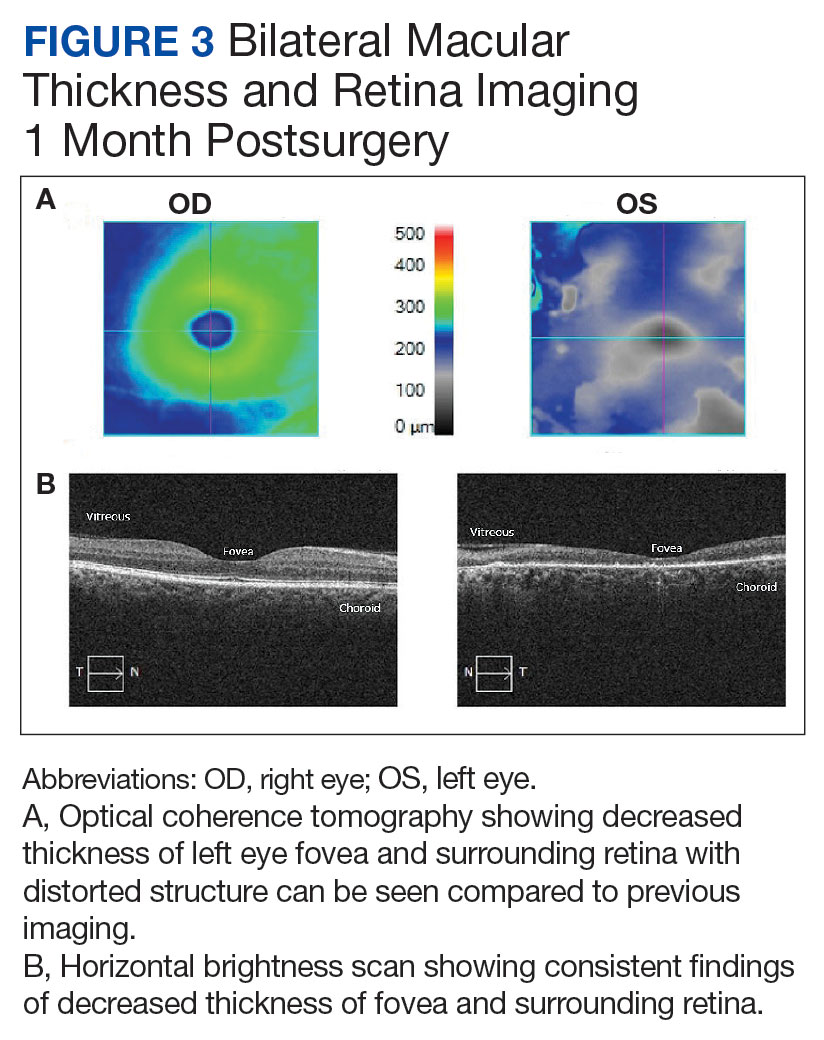
Repeat MRI showed denervation of the left medial rectus and minimal left-sided proptosis. A 3-month ophthalmologic follow-up revealed a persistent CN III palsy, including an afferent pupillary defect, absence of light perception in her left eye, and continued ophthalmoplegia. Repeat examination showed a left-sided 4+ afferent pupillary defect unreactive to light, 4+ pallor surrounding the optic nerve, macular atrophy, sclerotic vessels, and 17 mm Hg intraocular pressure bilaterally. The eye had diffuse atrophy of the inner retina and significant patchy atrophy of the outer retinal components without neovascularization of the iris. Postoperative retinal imaging can be seen in Figure 3. Her vision loss persisted at this encounter and has continued through subsequent follow-up examinations.
Discussion
Perioperative visual loss is a rare surgical complication, with an estimated incidence of once in every 60,000 to 125,000 cases.9 The mechanism of injury is variable and dependent upon the type of surgical intervention, with cardiac and spine surgeries carrying the greatest risk.10,11 The injury often results in either CRAO or ION, which may result in visual loss.1-3 POVL can also occur in the aftermath of rapid changes in intracranial pressure during decompressive craniotomies, though the pathophysiology in such cases is not well understood.5
Among the myriad ways in which POVL can occur, neurosurgical cases carry the unique risk of direct cranial nerve injury. Such an insult can lead to vision loss via optic nerve damage or ophthalmoplegia if damage occurs to CN III, IV, or VI. This can occur during manipulation or resection, especially if the surgical approach involves the orbital cavity or the cavernous sinus. Though neither space was entered in this patient, direct injury cannot be ruled out as the etiology for either her vision loss or persistent ophthalmoplegia. An alternate causative scenario for both symptoms involve an impaired blood supply, with the vision loss potentially occurring secondary to CRAO and the ophthalmoplegia to an alternate cause of decreased blood flow. It is unclear which of these 2 conditions occurred first or if they occurred due to the same insult, but OCS could lead to both. Though it is a less common etiology for POVL, this patient’s presentation was similar to those in previously reported cases, and OCS was identified as the likely diagnosis.
OCS is precipitated by an elevated orbital pressure, which leads to ischemia of the retina and damage to orbital contents. Though associated with retrobulbar hemorrhage and orbital trauma, another proposed mechanism for OCS is extrinsic orbital compression, resulting in increased IOP and subsequent CRAO.10 A cherry red spot is visible on fundoscopy, as only the macula with its thin retinal layer will permit the choroidal vessels to be visualized. In a separate process, the relative increase in orbital pressure may lead to impaired perfusion or damage of CN III. However, a causative relationship between the 2 may be difficult to establish. Such an injury to the oculomotor nerve is demonstrated by impaired function of the inferior oblique, superior rectus, inferior rectus, and medial rectus muscles, which may persist even after the compressive symptoms of OCS have resolved.12 Other reported symptoms of OCS include erythema, ophthalmoplegia, conjunctival chemosis, ptosis, corneal abrasion, and eyelid edema.12-15
Alternate Diagnoses
OCS is a diagnosis of exclusion, and several alternate mechanisms were considered before identifying it as the likely etiology. The patient’s preoperative imaging demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus, with displacement of the left middle cerebral artery, left cavernous internal carotid artery, and left optic canal. Dissection and removal of this tumor could have compromised the arterial or venous blood supply to the orbit, thus causing ischemia to the retina and other ocular structures. CN III was manipulated during surgery, and it may have been inadvertently damaged during exposure or resection of the tumor.
The patient’s Valsalva-dependent patent foramen ovale put her at risk of a paroxysmal embolus as an alternate explanation, particularly as a Valsalva maneuver was utilized to confirm hemostasis. The patient did not, however, demonstrate any evidence of venous thromboembolism (VTE) on ultrasound, nor did she have the common risk factors of hypertension, diabetes, or smoking history that would increase VTE risk.16 Her cancer diagnosis and surgical status may have put her at risk of VTE, but she did not have any clinical or laboratory values suggestive of hypercoagulability. Had an embolism occurred, it may have compromised the orbital blood supply and led to the CRAO. A similar scenario may have occurred from an atherosclerotic plaque in either of her carotid arteries, as she did have evidence of atherosclerosis on postoperative CT angiography. However, atherosclerosis as a risk factor for POVL appears to be related more to its impact upon impaired blood supply rather than as an embolic source. The patient did not have any significant intraoperative hypotensive episodes, making ION in the setting of atherosclerosis and hypotension a less likely etiology.17
This patient differed from other reported OCS cases. She was never placed in a prone or jackknife position, nor was she agitated or straining for a sustained period. These factors, along with the fact that the orbital compartment was not entered, decreased the likelihood of intraorbital hemorrhage and other intrinsic causes of elevated IOP.12 Additionally, the presentation of our patient’s vision loss was delayed compared with other cases, despite clinicians observing a dilated left pupil and CN III palsy on examination immediately after surgery.14 It is significant to note that OCS may not demonstrate a significant increase in IOP once the source of compression is removed, which may explain the absence of proptosis on her postoperative examination.
The diagnosis of OCS was primarily implicated by the positioning of the myocutaneous flap during the pterional approach to craniotomy. It was retracted anteriorly and superiorly, ultimately resting over her left orbit for most of the 10-hour surgery. Kim and colleagues found that myocutaneous flaps may increase IOP as much as 17.5 mm Hg if improperly positioned, providing an unrecognized source of compression and increasing the risk of damage to orbital contents. According to their review, elevated IOP > 40 mm Hg, particularly over several hours, can compromise blood flow to the optic nerve and increase the risk for POVL.18 The flap was secured using fish hooks and rubber bands. However, it is suspected that the orbital rim did not fully support its pressure, thereby resting to some degree directly on the globe for an extended period and compromising the orbital blood supply. There are no current methods for measuring intraoperative IOP, though surrogate markers are under investigation and may yield clinical utility.18 The myocutaneous flap was created and positioned by the surgeons, but it may be that increased vigilance and communication from the anesthesia and nursing teams could have prevented it from remaining in an improper position.
Conclusions
Despite having few reported cases, OCS must be considered in neurosurgical patients with ophthalmoplegia and CRAO on postoperative examinations. Myocutaneous flaps that are retracted across the orbit can lead to significant elevations in IOP, leading to vision loss, which likely occurred with the patient in this case. Though protecting neurovascular structures is within the purview of the surgeon, all members of the intraoperative team should assist with ensuring proper flap positioning. These measures can help ensure adequate blood flow to the ophthalmic artery, decrease the likelihood of elevated IOP due to extrinsic compression, and help prevent the development of POVL and OCS in these patients.
1. Biousse V, Nahab F, Newman NJ. Management of acute retinal ischemia: follow the guidelines! Ophthalmology. 2018;125(10):1597-1607. doi:10.1016/j.ophtha.2018.03.054
2. Biousse V, Newman NJ. Ischemic optic neuropathies. N Engl J Med. 2015;372(25):2428-2436. doi:10.1056/NEJMra1413352
3. Shah SH, Chen YF, Moss HE, Rubin DS, Joslin CE, Roth S. Predicting risk of perioperative ischemic optic neuropathy in spine fusion surgery: a cohort study using the national inpatient sample. Anesth Analg. 2020;130(4):967-974. doi:10.1213/ANE.0000000000004383
4. Habets JGV, Haeren RHL, Lie SAN, Bauer NJC, Dings JTA. Acute monocular blindness due to orbital compartment syndrome following pterional craniotomy. World Neurosurg. 2018;114:72-75. doi:10.1016/j.wneu.2018.03.013
5. Vahedi P, Meshkini A, Mohajernezhadfard Z, Tubbs RS. Post-craniotomy blindness in the supine position: Unlikely or ignored? Asian J Neurosurg. 2013;8(1):36-41. doi:10.4103/1793-5482.110278
6. Kang S, Yang Y, Kim T, Kim J. Sudden unilateral blindness after intracranial aneurysm surgery. Acta Neurochir (Wien). 1997;139(3):221-226. doi:10.1007/BF01844755
7. Zimmerman CF, Van Patten PD, Golnik KC, Kopitnik TA Jr, Anand R. Orbital infarction syndrome after surgery for intracranial aneurysms. Ophthalmology. 1995;102(4):594-598. doi:10.1016/s0161-6420(95)30979-7
8. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 23;2013:bcr2013201554. doi:10.1136/bcr-2013-201554
9. Raphael J, Moss HE, Roth S. Perioperative visual loss in cardiac surgery. J Cardiothorac Vasc Anesth. 2019;33(5):1420-429. doi:10.1053/j.jvca.2018.11.035
10. Kansakar P, Sundar G. Vision loss associated with orbital surgery - a major review. Orbit. 2020;39(3):197-208. doi:10.1080/01676830.2019.1658790
11. Dohlman JC, Yoon MK. Principles of protection of the eye and vision in orbital surgery. J Neurol Surg B Skull Base. 2020;81(4):381-384. doi:10.1055/s-0040-1714077
12. Pahl FH, de Oliveira MF, Dal Col Lúcio JE, Souza E Castro EF. Orbital compartment syndrome after frontotemporal craniotomy: case report and review of literature. World Neurosurg. 2018;109:218-221. doi:10.1016/j.wneu.2017.09.167
13. Grossman W, Ward WT. Central retinal artery occlusion after scoliosis surgery with a horseshoe headrest. Case report and literature review. Spine (Phila Pa 1976). 1993;18(9):1226-1228. doi:10.1097/00007632-199307000-00017
14. Newman NJ. Perioperative visual loss after nonocular surgeries. Am J Ophthalmol. 2008;145(4):604-610. doi:10.1016/j.ajo.2007.09.016
15. Roth S, Tung A, Ksiazek S. Visual loss in a prone-positioned spine surgery patient with the head on a foam headrest and goggles covering the eyes: an old complication with a new mechanism. Anesth Analg. 2007;104(5):1185-1187. doi:10.1213/01.ane.0000264319.57758.55
16. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85. doi:10.1097/01.brs.0000152169.48117.c7
17. Nickels TJ, Manlapaz MR, Farag E. Perioperative visual loss after spine surgery. World J Orthop. 2014;5(2):100-106. Published 2014 April 18. doi:10.5312/wjo.v5.i2.100
18. Kim TS, Hur JW, Park DH, et al. Extraocular ressure measurements to avoid orbital compartment syndrome in aneurysm surgery. World Neurosurg. 2018;118:e601-e609. doi:10.1016/j.wneu.2018.06.248
Perioperative visual loss (POVL) is a well-documented yet uncommon complication of nonocular surgery. Patients undergoing cardiac and spinal surgery are at the greatest risk, though POVL may occur during other neurosurgical and vascular procedures as well. The most common causes of POVL are central retinal artery occlusion (CRAO) and ischemic optic neuropathy (ION),1-3 though cases of orbital compartment syndrome (OCS) have also been reported.4-7
We describe a case of POVL during a temporal meningioma excision using the pterional approach. Though the etiology is not fully understood, the patient’s clinical course was complicated by a third cranial nerve (CN III) palsy and CRAO, which, together with the patient’s presentation, were consistent with previously documented cases of OCS. The goals of this case report are to increase awareness of this surgical outcome, identify practices that may have contributed to its development, and delineate methods to minimize its occurrence.
Informed consent regarding this research was obtained from the patient and an institutional Health Insurance Portability and Accountability Act authorization form was completed. This manuscript adheres to the applicable Enhancing the Quality and Transparency of Health Research guideline.8
Case Presentation
A 47-year-old woman underwent a left temporal craniotomy for resection of a sphenoid wing meningioma discovered during a workup for persistent headaches. She had no medical history of diabetes, hypertension, coronary artery disease, or ophthalmic disease. Two months before her scheduled surgery, the patient reported bilateral blurry vision and underwent ophthalmologic evaluation. Her intraocular pressure (IOP) was normal, and she had no pupillary or retinal disease. She showed evidence of decreased vision in her left eye, suggesting a possible mass effect from her meningioma. Subsequent imaging of the optic nerve and retina had unremarkable physiology (Figure 1). Preoperative magnetic resonance imaging (MRI) demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus(Figure 2). There was a superior mass effect on the left middle cerebral artery, but all vessels were patent without evidence of thrombosis.
The patient underwent general anesthesia with invasive hemodynamic monitoring used throughout the procedure. She was induced with fentanyl, propofol, and rocuronium; anesthesia was maintained with isoflurane and a remifentanil infusion. Hypotension was treated with phenylephrine and intravenous fluids. Intraoperative neuromonitoring with electroencephalogram (EEG) and somatosensory evoked potentials was performed. During the surgery, the patient was positioned supine in a Mayfield 3-point head fixation system. All pressure points were padded appropriately and continually checked. A standard left pterional craniotomy was performed, and the scalp was reflected anteriorly and secured using fish hooks with rubber bands. The operation did not violate the cavernous sinus or orbital compartment. There was no evidence of active bleeding upon inspection nor with the Valsalva maneuver. No changes were noted in EEG or somatosensory evoked potentials; blood pressure remained within 20 mm Hg of the patient’s baseline. She was extubated at the end of the 10-hour case and was hemodynamically stable upon transport to the surgical intensive care unit. Postoperative imaging confirmed the successful removal of the left sphenoid wing meningioma.
The patient’s postoperative examination demonstrated a 5 mm dilated, nonresponsive left pupil, though the patient did not report visual loss at that time. Defects were noted in the inferior oblique, superior, inferior, and medial rectus muscles, consistent with CN III palsy. The surgery included manipulation of CN III, which made this a possible outcome, but an alternate causative pathology like OCS was not immediately suspected. Postoperative computed tomography (CT) showed an expected pneumocephalus and left scalp swelling without evidence of mass effect or midline shift.
On the morning of postoperative Day 1, the patient reported vision loss in her left eye, while her clinical examination revealed erythema and conjunctival chemosis with left eyelid swelling. The ophthalmologic evaluation was notable for a continued leftCN III palsy with intact lateral rectus and superior oblique function, a nonreactive and dilated left eye with 3+ afferent pupillary defect by reverse (light perception), pallor throughout, a flat cherry red macula with blurred disc margins, left upper eyelid edema, and 18 mm Hg intraocular pressure bilaterally (reference range, 8 to 21 mm Hg). Fundoscopic examination showed a clear vitreous without plaques or occlusions, no perivascular sheathing, and no retinal hemorrhages. CT angiography revealed small outpouchings at the superolateral aspect of the left and right cavernous carotid, consistent with atherosclerotic calcifications. An echocardiogram revealed a Valsalva-dependent patent foramen ovale, but a venous Doppler ultrasound yielded negative results.
Repeat MRI showed denervation of the left medial rectus and minimal left-sided proptosis. A 3-month ophthalmologic follow-up revealed a persistent CN III palsy, including an afferent pupillary defect, absence of light perception in her left eye, and continued ophthalmoplegia. Repeat examination showed a left-sided 4+ afferent pupillary defect unreactive to light, 4+ pallor surrounding the optic nerve, macular atrophy, sclerotic vessels, and 17 mm Hg intraocular pressure bilaterally. The eye had diffuse atrophy of the inner retina and significant patchy atrophy of the outer retinal components without neovascularization of the iris. Postoperative retinal imaging can be seen in Figure 3. Her vision loss persisted at this encounter and has continued through subsequent follow-up examinations.
Discussion
Perioperative visual loss is a rare surgical complication, with an estimated incidence of once in every 60,000 to 125,000 cases.9 The mechanism of injury is variable and dependent upon the type of surgical intervention, with cardiac and spine surgeries carrying the greatest risk.10,11 The injury often results in either CRAO or ION, which may result in visual loss.1-3 POVL can also occur in the aftermath of rapid changes in intracranial pressure during decompressive craniotomies, though the pathophysiology in such cases is not well understood.5
Among the myriad ways in which POVL can occur, neurosurgical cases carry the unique risk of direct cranial nerve injury. Such an insult can lead to vision loss via optic nerve damage or ophthalmoplegia if damage occurs to CN III, IV, or VI. This can occur during manipulation or resection, especially if the surgical approach involves the orbital cavity or the cavernous sinus. Though neither space was entered in this patient, direct injury cannot be ruled out as the etiology for either her vision loss or persistent ophthalmoplegia. An alternate causative scenario for both symptoms involve an impaired blood supply, with the vision loss potentially occurring secondary to CRAO and the ophthalmoplegia to an alternate cause of decreased blood flow. It is unclear which of these 2 conditions occurred first or if they occurred due to the same insult, but OCS could lead to both. Though it is a less common etiology for POVL, this patient’s presentation was similar to those in previously reported cases, and OCS was identified as the likely diagnosis.
OCS is precipitated by an elevated orbital pressure, which leads to ischemia of the retina and damage to orbital contents. Though associated with retrobulbar hemorrhage and orbital trauma, another proposed mechanism for OCS is extrinsic orbital compression, resulting in increased IOP and subsequent CRAO.10 A cherry red spot is visible on fundoscopy, as only the macula with its thin retinal layer will permit the choroidal vessels to be visualized. In a separate process, the relative increase in orbital pressure may lead to impaired perfusion or damage of CN III. However, a causative relationship between the 2 may be difficult to establish. Such an injury to the oculomotor nerve is demonstrated by impaired function of the inferior oblique, superior rectus, inferior rectus, and medial rectus muscles, which may persist even after the compressive symptoms of OCS have resolved.12 Other reported symptoms of OCS include erythema, ophthalmoplegia, conjunctival chemosis, ptosis, corneal abrasion, and eyelid edema.12-15
Alternate Diagnoses
OCS is a diagnosis of exclusion, and several alternate mechanisms were considered before identifying it as the likely etiology. The patient’s preoperative imaging demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus, with displacement of the left middle cerebral artery, left cavernous internal carotid artery, and left optic canal. Dissection and removal of this tumor could have compromised the arterial or venous blood supply to the orbit, thus causing ischemia to the retina and other ocular structures. CN III was manipulated during surgery, and it may have been inadvertently damaged during exposure or resection of the tumor.
The patient’s Valsalva-dependent patent foramen ovale put her at risk of a paroxysmal embolus as an alternate explanation, particularly as a Valsalva maneuver was utilized to confirm hemostasis. The patient did not, however, demonstrate any evidence of venous thromboembolism (VTE) on ultrasound, nor did she have the common risk factors of hypertension, diabetes, or smoking history that would increase VTE risk.16 Her cancer diagnosis and surgical status may have put her at risk of VTE, but she did not have any clinical or laboratory values suggestive of hypercoagulability. Had an embolism occurred, it may have compromised the orbital blood supply and led to the CRAO. A similar scenario may have occurred from an atherosclerotic plaque in either of her carotid arteries, as she did have evidence of atherosclerosis on postoperative CT angiography. However, atherosclerosis as a risk factor for POVL appears to be related more to its impact upon impaired blood supply rather than as an embolic source. The patient did not have any significant intraoperative hypotensive episodes, making ION in the setting of atherosclerosis and hypotension a less likely etiology.17
This patient differed from other reported OCS cases. She was never placed in a prone or jackknife position, nor was she agitated or straining for a sustained period. These factors, along with the fact that the orbital compartment was not entered, decreased the likelihood of intraorbital hemorrhage and other intrinsic causes of elevated IOP.12 Additionally, the presentation of our patient’s vision loss was delayed compared with other cases, despite clinicians observing a dilated left pupil and CN III palsy on examination immediately after surgery.14 It is significant to note that OCS may not demonstrate a significant increase in IOP once the source of compression is removed, which may explain the absence of proptosis on her postoperative examination.
The diagnosis of OCS was primarily implicated by the positioning of the myocutaneous flap during the pterional approach to craniotomy. It was retracted anteriorly and superiorly, ultimately resting over her left orbit for most of the 10-hour surgery. Kim and colleagues found that myocutaneous flaps may increase IOP as much as 17.5 mm Hg if improperly positioned, providing an unrecognized source of compression and increasing the risk of damage to orbital contents. According to their review, elevated IOP > 40 mm Hg, particularly over several hours, can compromise blood flow to the optic nerve and increase the risk for POVL.18 The flap was secured using fish hooks and rubber bands. However, it is suspected that the orbital rim did not fully support its pressure, thereby resting to some degree directly on the globe for an extended period and compromising the orbital blood supply. There are no current methods for measuring intraoperative IOP, though surrogate markers are under investigation and may yield clinical utility.18 The myocutaneous flap was created and positioned by the surgeons, but it may be that increased vigilance and communication from the anesthesia and nursing teams could have prevented it from remaining in an improper position.
Conclusions
Despite having few reported cases, OCS must be considered in neurosurgical patients with ophthalmoplegia and CRAO on postoperative examinations. Myocutaneous flaps that are retracted across the orbit can lead to significant elevations in IOP, leading to vision loss, which likely occurred with the patient in this case. Though protecting neurovascular structures is within the purview of the surgeon, all members of the intraoperative team should assist with ensuring proper flap positioning. These measures can help ensure adequate blood flow to the ophthalmic artery, decrease the likelihood of elevated IOP due to extrinsic compression, and help prevent the development of POVL and OCS in these patients.
Perioperative visual loss (POVL) is a well-documented yet uncommon complication of nonocular surgery. Patients undergoing cardiac and spinal surgery are at the greatest risk, though POVL may occur during other neurosurgical and vascular procedures as well. The most common causes of POVL are central retinal artery occlusion (CRAO) and ischemic optic neuropathy (ION),1-3 though cases of orbital compartment syndrome (OCS) have also been reported.4-7
We describe a case of POVL during a temporal meningioma excision using the pterional approach. Though the etiology is not fully understood, the patient’s clinical course was complicated by a third cranial nerve (CN III) palsy and CRAO, which, together with the patient’s presentation, were consistent with previously documented cases of OCS. The goals of this case report are to increase awareness of this surgical outcome, identify practices that may have contributed to its development, and delineate methods to minimize its occurrence.
Informed consent regarding this research was obtained from the patient and an institutional Health Insurance Portability and Accountability Act authorization form was completed. This manuscript adheres to the applicable Enhancing the Quality and Transparency of Health Research guideline.8
Case Presentation
A 47-year-old woman underwent a left temporal craniotomy for resection of a sphenoid wing meningioma discovered during a workup for persistent headaches. She had no medical history of diabetes, hypertension, coronary artery disease, or ophthalmic disease. Two months before her scheduled surgery, the patient reported bilateral blurry vision and underwent ophthalmologic evaluation. Her intraocular pressure (IOP) was normal, and she had no pupillary or retinal disease. She showed evidence of decreased vision in her left eye, suggesting a possible mass effect from her meningioma. Subsequent imaging of the optic nerve and retina had unremarkable physiology (Figure 1). Preoperative magnetic resonance imaging (MRI) demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus(Figure 2). There was a superior mass effect on the left middle cerebral artery, but all vessels were patent without evidence of thrombosis.
The patient underwent general anesthesia with invasive hemodynamic monitoring used throughout the procedure. She was induced with fentanyl, propofol, and rocuronium; anesthesia was maintained with isoflurane and a remifentanil infusion. Hypotension was treated with phenylephrine and intravenous fluids. Intraoperative neuromonitoring with electroencephalogram (EEG) and somatosensory evoked potentials was performed. During the surgery, the patient was positioned supine in a Mayfield 3-point head fixation system. All pressure points were padded appropriately and continually checked. A standard left pterional craniotomy was performed, and the scalp was reflected anteriorly and secured using fish hooks with rubber bands. The operation did not violate the cavernous sinus or orbital compartment. There was no evidence of active bleeding upon inspection nor with the Valsalva maneuver. No changes were noted in EEG or somatosensory evoked potentials; blood pressure remained within 20 mm Hg of the patient’s baseline. She was extubated at the end of the 10-hour case and was hemodynamically stable upon transport to the surgical intensive care unit. Postoperative imaging confirmed the successful removal of the left sphenoid wing meningioma.
The patient’s postoperative examination demonstrated a 5 mm dilated, nonresponsive left pupil, though the patient did not report visual loss at that time. Defects were noted in the inferior oblique, superior, inferior, and medial rectus muscles, consistent with CN III palsy. The surgery included manipulation of CN III, which made this a possible outcome, but an alternate causative pathology like OCS was not immediately suspected. Postoperative computed tomography (CT) showed an expected pneumocephalus and left scalp swelling without evidence of mass effect or midline shift.
On the morning of postoperative Day 1, the patient reported vision loss in her left eye, while her clinical examination revealed erythema and conjunctival chemosis with left eyelid swelling. The ophthalmologic evaluation was notable for a continued leftCN III palsy with intact lateral rectus and superior oblique function, a nonreactive and dilated left eye with 3+ afferent pupillary defect by reverse (light perception), pallor throughout, a flat cherry red macula with blurred disc margins, left upper eyelid edema, and 18 mm Hg intraocular pressure bilaterally (reference range, 8 to 21 mm Hg). Fundoscopic examination showed a clear vitreous without plaques or occlusions, no perivascular sheathing, and no retinal hemorrhages. CT angiography revealed small outpouchings at the superolateral aspect of the left and right cavernous carotid, consistent with atherosclerotic calcifications. An echocardiogram revealed a Valsalva-dependent patent foramen ovale, but a venous Doppler ultrasound yielded negative results.
Repeat MRI showed denervation of the left medial rectus and minimal left-sided proptosis. A 3-month ophthalmologic follow-up revealed a persistent CN III palsy, including an afferent pupillary defect, absence of light perception in her left eye, and continued ophthalmoplegia. Repeat examination showed a left-sided 4+ afferent pupillary defect unreactive to light, 4+ pallor surrounding the optic nerve, macular atrophy, sclerotic vessels, and 17 mm Hg intraocular pressure bilaterally. The eye had diffuse atrophy of the inner retina and significant patchy atrophy of the outer retinal components without neovascularization of the iris. Postoperative retinal imaging can be seen in Figure 3. Her vision loss persisted at this encounter and has continued through subsequent follow-up examinations.
Discussion
Perioperative visual loss is a rare surgical complication, with an estimated incidence of once in every 60,000 to 125,000 cases.9 The mechanism of injury is variable and dependent upon the type of surgical intervention, with cardiac and spine surgeries carrying the greatest risk.10,11 The injury often results in either CRAO or ION, which may result in visual loss.1-3 POVL can also occur in the aftermath of rapid changes in intracranial pressure during decompressive craniotomies, though the pathophysiology in such cases is not well understood.5
Among the myriad ways in which POVL can occur, neurosurgical cases carry the unique risk of direct cranial nerve injury. Such an insult can lead to vision loss via optic nerve damage or ophthalmoplegia if damage occurs to CN III, IV, or VI. This can occur during manipulation or resection, especially if the surgical approach involves the orbital cavity or the cavernous sinus. Though neither space was entered in this patient, direct injury cannot be ruled out as the etiology for either her vision loss or persistent ophthalmoplegia. An alternate causative scenario for both symptoms involve an impaired blood supply, with the vision loss potentially occurring secondary to CRAO and the ophthalmoplegia to an alternate cause of decreased blood flow. It is unclear which of these 2 conditions occurred first or if they occurred due to the same insult, but OCS could lead to both. Though it is a less common etiology for POVL, this patient’s presentation was similar to those in previously reported cases, and OCS was identified as the likely diagnosis.
OCS is precipitated by an elevated orbital pressure, which leads to ischemia of the retina and damage to orbital contents. Though associated with retrobulbar hemorrhage and orbital trauma, another proposed mechanism for OCS is extrinsic orbital compression, resulting in increased IOP and subsequent CRAO.10 A cherry red spot is visible on fundoscopy, as only the macula with its thin retinal layer will permit the choroidal vessels to be visualized. In a separate process, the relative increase in orbital pressure may lead to impaired perfusion or damage of CN III. However, a causative relationship between the 2 may be difficult to establish. Such an injury to the oculomotor nerve is demonstrated by impaired function of the inferior oblique, superior rectus, inferior rectus, and medial rectus muscles, which may persist even after the compressive symptoms of OCS have resolved.12 Other reported symptoms of OCS include erythema, ophthalmoplegia, conjunctival chemosis, ptosis, corneal abrasion, and eyelid edema.12-15
Alternate Diagnoses
OCS is a diagnosis of exclusion, and several alternate mechanisms were considered before identifying it as the likely etiology. The patient’s preoperative imaging demonstrated a stable enhancing mass involving the left great sphenoid wing and left cavernous sinus, with displacement of the left middle cerebral artery, left cavernous internal carotid artery, and left optic canal. Dissection and removal of this tumor could have compromised the arterial or venous blood supply to the orbit, thus causing ischemia to the retina and other ocular structures. CN III was manipulated during surgery, and it may have been inadvertently damaged during exposure or resection of the tumor.
The patient’s Valsalva-dependent patent foramen ovale put her at risk of a paroxysmal embolus as an alternate explanation, particularly as a Valsalva maneuver was utilized to confirm hemostasis. The patient did not, however, demonstrate any evidence of venous thromboembolism (VTE) on ultrasound, nor did she have the common risk factors of hypertension, diabetes, or smoking history that would increase VTE risk.16 Her cancer diagnosis and surgical status may have put her at risk of VTE, but she did not have any clinical or laboratory values suggestive of hypercoagulability. Had an embolism occurred, it may have compromised the orbital blood supply and led to the CRAO. A similar scenario may have occurred from an atherosclerotic plaque in either of her carotid arteries, as she did have evidence of atherosclerosis on postoperative CT angiography. However, atherosclerosis as a risk factor for POVL appears to be related more to its impact upon impaired blood supply rather than as an embolic source. The patient did not have any significant intraoperative hypotensive episodes, making ION in the setting of atherosclerosis and hypotension a less likely etiology.17
This patient differed from other reported OCS cases. She was never placed in a prone or jackknife position, nor was she agitated or straining for a sustained period. These factors, along with the fact that the orbital compartment was not entered, decreased the likelihood of intraorbital hemorrhage and other intrinsic causes of elevated IOP.12 Additionally, the presentation of our patient’s vision loss was delayed compared with other cases, despite clinicians observing a dilated left pupil and CN III palsy on examination immediately after surgery.14 It is significant to note that OCS may not demonstrate a significant increase in IOP once the source of compression is removed, which may explain the absence of proptosis on her postoperative examination.
The diagnosis of OCS was primarily implicated by the positioning of the myocutaneous flap during the pterional approach to craniotomy. It was retracted anteriorly and superiorly, ultimately resting over her left orbit for most of the 10-hour surgery. Kim and colleagues found that myocutaneous flaps may increase IOP as much as 17.5 mm Hg if improperly positioned, providing an unrecognized source of compression and increasing the risk of damage to orbital contents. According to their review, elevated IOP > 40 mm Hg, particularly over several hours, can compromise blood flow to the optic nerve and increase the risk for POVL.18 The flap was secured using fish hooks and rubber bands. However, it is suspected that the orbital rim did not fully support its pressure, thereby resting to some degree directly on the globe for an extended period and compromising the orbital blood supply. There are no current methods for measuring intraoperative IOP, though surrogate markers are under investigation and may yield clinical utility.18 The myocutaneous flap was created and positioned by the surgeons, but it may be that increased vigilance and communication from the anesthesia and nursing teams could have prevented it from remaining in an improper position.
Conclusions
Despite having few reported cases, OCS must be considered in neurosurgical patients with ophthalmoplegia and CRAO on postoperative examinations. Myocutaneous flaps that are retracted across the orbit can lead to significant elevations in IOP, leading to vision loss, which likely occurred with the patient in this case. Though protecting neurovascular structures is within the purview of the surgeon, all members of the intraoperative team should assist with ensuring proper flap positioning. These measures can help ensure adequate blood flow to the ophthalmic artery, decrease the likelihood of elevated IOP due to extrinsic compression, and help prevent the development of POVL and OCS in these patients.
1. Biousse V, Nahab F, Newman NJ. Management of acute retinal ischemia: follow the guidelines! Ophthalmology. 2018;125(10):1597-1607. doi:10.1016/j.ophtha.2018.03.054
2. Biousse V, Newman NJ. Ischemic optic neuropathies. N Engl J Med. 2015;372(25):2428-2436. doi:10.1056/NEJMra1413352
3. Shah SH, Chen YF, Moss HE, Rubin DS, Joslin CE, Roth S. Predicting risk of perioperative ischemic optic neuropathy in spine fusion surgery: a cohort study using the national inpatient sample. Anesth Analg. 2020;130(4):967-974. doi:10.1213/ANE.0000000000004383
4. Habets JGV, Haeren RHL, Lie SAN, Bauer NJC, Dings JTA. Acute monocular blindness due to orbital compartment syndrome following pterional craniotomy. World Neurosurg. 2018;114:72-75. doi:10.1016/j.wneu.2018.03.013
5. Vahedi P, Meshkini A, Mohajernezhadfard Z, Tubbs RS. Post-craniotomy blindness in the supine position: Unlikely or ignored? Asian J Neurosurg. 2013;8(1):36-41. doi:10.4103/1793-5482.110278
6. Kang S, Yang Y, Kim T, Kim J. Sudden unilateral blindness after intracranial aneurysm surgery. Acta Neurochir (Wien). 1997;139(3):221-226. doi:10.1007/BF01844755
7. Zimmerman CF, Van Patten PD, Golnik KC, Kopitnik TA Jr, Anand R. Orbital infarction syndrome after surgery for intracranial aneurysms. Ophthalmology. 1995;102(4):594-598. doi:10.1016/s0161-6420(95)30979-7
8. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 23;2013:bcr2013201554. doi:10.1136/bcr-2013-201554
9. Raphael J, Moss HE, Roth S. Perioperative visual loss in cardiac surgery. J Cardiothorac Vasc Anesth. 2019;33(5):1420-429. doi:10.1053/j.jvca.2018.11.035
10. Kansakar P, Sundar G. Vision loss associated with orbital surgery - a major review. Orbit. 2020;39(3):197-208. doi:10.1080/01676830.2019.1658790
11. Dohlman JC, Yoon MK. Principles of protection of the eye and vision in orbital surgery. J Neurol Surg B Skull Base. 2020;81(4):381-384. doi:10.1055/s-0040-1714077
12. Pahl FH, de Oliveira MF, Dal Col Lúcio JE, Souza E Castro EF. Orbital compartment syndrome after frontotemporal craniotomy: case report and review of literature. World Neurosurg. 2018;109:218-221. doi:10.1016/j.wneu.2017.09.167
13. Grossman W, Ward WT. Central retinal artery occlusion after scoliosis surgery with a horseshoe headrest. Case report and literature review. Spine (Phila Pa 1976). 1993;18(9):1226-1228. doi:10.1097/00007632-199307000-00017
14. Newman NJ. Perioperative visual loss after nonocular surgeries. Am J Ophthalmol. 2008;145(4):604-610. doi:10.1016/j.ajo.2007.09.016
15. Roth S, Tung A, Ksiazek S. Visual loss in a prone-positioned spine surgery patient with the head on a foam headrest and goggles covering the eyes: an old complication with a new mechanism. Anesth Analg. 2007;104(5):1185-1187. doi:10.1213/01.ane.0000264319.57758.55
16. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85. doi:10.1097/01.brs.0000152169.48117.c7
17. Nickels TJ, Manlapaz MR, Farag E. Perioperative visual loss after spine surgery. World J Orthop. 2014;5(2):100-106. Published 2014 April 18. doi:10.5312/wjo.v5.i2.100
18. Kim TS, Hur JW, Park DH, et al. Extraocular ressure measurements to avoid orbital compartment syndrome in aneurysm surgery. World Neurosurg. 2018;118:e601-e609. doi:10.1016/j.wneu.2018.06.248
1. Biousse V, Nahab F, Newman NJ. Management of acute retinal ischemia: follow the guidelines! Ophthalmology. 2018;125(10):1597-1607. doi:10.1016/j.ophtha.2018.03.054
2. Biousse V, Newman NJ. Ischemic optic neuropathies. N Engl J Med. 2015;372(25):2428-2436. doi:10.1056/NEJMra1413352
3. Shah SH, Chen YF, Moss HE, Rubin DS, Joslin CE, Roth S. Predicting risk of perioperative ischemic optic neuropathy in spine fusion surgery: a cohort study using the national inpatient sample. Anesth Analg. 2020;130(4):967-974. doi:10.1213/ANE.0000000000004383
4. Habets JGV, Haeren RHL, Lie SAN, Bauer NJC, Dings JTA. Acute monocular blindness due to orbital compartment syndrome following pterional craniotomy. World Neurosurg. 2018;114:72-75. doi:10.1016/j.wneu.2018.03.013
5. Vahedi P, Meshkini A, Mohajernezhadfard Z, Tubbs RS. Post-craniotomy blindness in the supine position: Unlikely or ignored? Asian J Neurosurg. 2013;8(1):36-41. doi:10.4103/1793-5482.110278
6. Kang S, Yang Y, Kim T, Kim J. Sudden unilateral blindness after intracranial aneurysm surgery. Acta Neurochir (Wien). 1997;139(3):221-226. doi:10.1007/BF01844755
7. Zimmerman CF, Van Patten PD, Golnik KC, Kopitnik TA Jr, Anand R. Orbital infarction syndrome after surgery for intracranial aneurysms. Ophthalmology. 1995;102(4):594-598. doi:10.1016/s0161-6420(95)30979-7
8. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. BMJ Case Rep. 23;2013:bcr2013201554. doi:10.1136/bcr-2013-201554
9. Raphael J, Moss HE, Roth S. Perioperative visual loss in cardiac surgery. J Cardiothorac Vasc Anesth. 2019;33(5):1420-429. doi:10.1053/j.jvca.2018.11.035
10. Kansakar P, Sundar G. Vision loss associated with orbital surgery - a major review. Orbit. 2020;39(3):197-208. doi:10.1080/01676830.2019.1658790
11. Dohlman JC, Yoon MK. Principles of protection of the eye and vision in orbital surgery. J Neurol Surg B Skull Base. 2020;81(4):381-384. doi:10.1055/s-0040-1714077
12. Pahl FH, de Oliveira MF, Dal Col Lúcio JE, Souza E Castro EF. Orbital compartment syndrome after frontotemporal craniotomy: case report and review of literature. World Neurosurg. 2018;109:218-221. doi:10.1016/j.wneu.2017.09.167
13. Grossman W, Ward WT. Central retinal artery occlusion after scoliosis surgery with a horseshoe headrest. Case report and literature review. Spine (Phila Pa 1976). 1993;18(9):1226-1228. doi:10.1097/00007632-199307000-00017
14. Newman NJ. Perioperative visual loss after nonocular surgeries. Am J Ophthalmol. 2008;145(4):604-610. doi:10.1016/j.ajo.2007.09.016
15. Roth S, Tung A, Ksiazek S. Visual loss in a prone-positioned spine surgery patient with the head on a foam headrest and goggles covering the eyes: an old complication with a new mechanism. Anesth Analg. 2007;104(5):1185-1187. doi:10.1213/01.ane.0000264319.57758.55
16. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85. doi:10.1097/01.brs.0000152169.48117.c7
17. Nickels TJ, Manlapaz MR, Farag E. Perioperative visual loss after spine surgery. World J Orthop. 2014;5(2):100-106. Published 2014 April 18. doi:10.5312/wjo.v5.i2.100
18. Kim TS, Hur JW, Park DH, et al. Extraocular ressure measurements to avoid orbital compartment syndrome in aneurysm surgery. World Neurosurg. 2018;118:e601-e609. doi:10.1016/j.wneu.2018.06.248
Isotretinoin-Induced Skin Fragility in an Aerialist
Isotretinoin was introduced more than 3 decades ago and marked a major advancement in the treatment of severe refractory cystic acne. The most common adverse effects linked to isotretinoin usage are mucocutaneous in nature, manifesting as xerosis and cheilitis.1 Skin fragility and poor wound healing also have been reported.2-6 Current recommendations for avoiding these adverse effects include refraining from waxing, laser procedures, and other elective cutaneous procedures for at least 6 months.7 We present a case of isotretinoin-induced cutaneous fragility resulting in blistering and erosions on the palms of a competitive aerial trapeze artist.
Case Report
A 25-year-old woman presented for follow-up during week 12 of isotretinoin therapy (40 mg twice daily) prescribed for acne. She reported peeling of the skin on the palms following intense aerial acrobatic workouts. She had been a performing aerialist for many years and had never sustained a similar injury. The wounds were painful and led to decreased activity. She had no notable medical history. Physical examination of the palms revealed erosions in a distribution that corresponded to horizontal bar contact and friction (Figure). The patient was advised on proper wound care, application of emollients, and minimizing friction. She completed the course of isotretinoin and has continued aerialist activity without recurrence of skin fragility.
Comment
Skin fragility is a well-known adverse effect of isotretinoin therapy.8 Pavlis and Lieblich9 reported skin fragility in a young wrestler who experienced similar skin erosions due to isotretinoin therapy. The proposed mechanism of isotretinoin-induced skin fragility is multifactorial. It involves an apoptotic effect on sebocytes,5 which results in reduced stratum corneum hydration and an associated increase in transepidermal water loss.6,10,11 Retinoids also are known to cause thinning of the skin, likely due to the disadhesion of both the epidermis and the stratum corneum, which was demonstrated by the easy removal of cornified cells through tape stripping in hairless mice treated with isotretinoin.12 In further investigations, human patients and hairless mice treated with isotretinoin readily developed friction blisters through pencil eraser abrasion.13 Examination of the friction blisters using light and electron microscopy revealed fraying or loss of the stratum corneum and viable epidermis as well as loss of desmosomes and tonofilaments. Additionally, intracellular and intercellular deposits of an unidentified amorphous material were noted.13
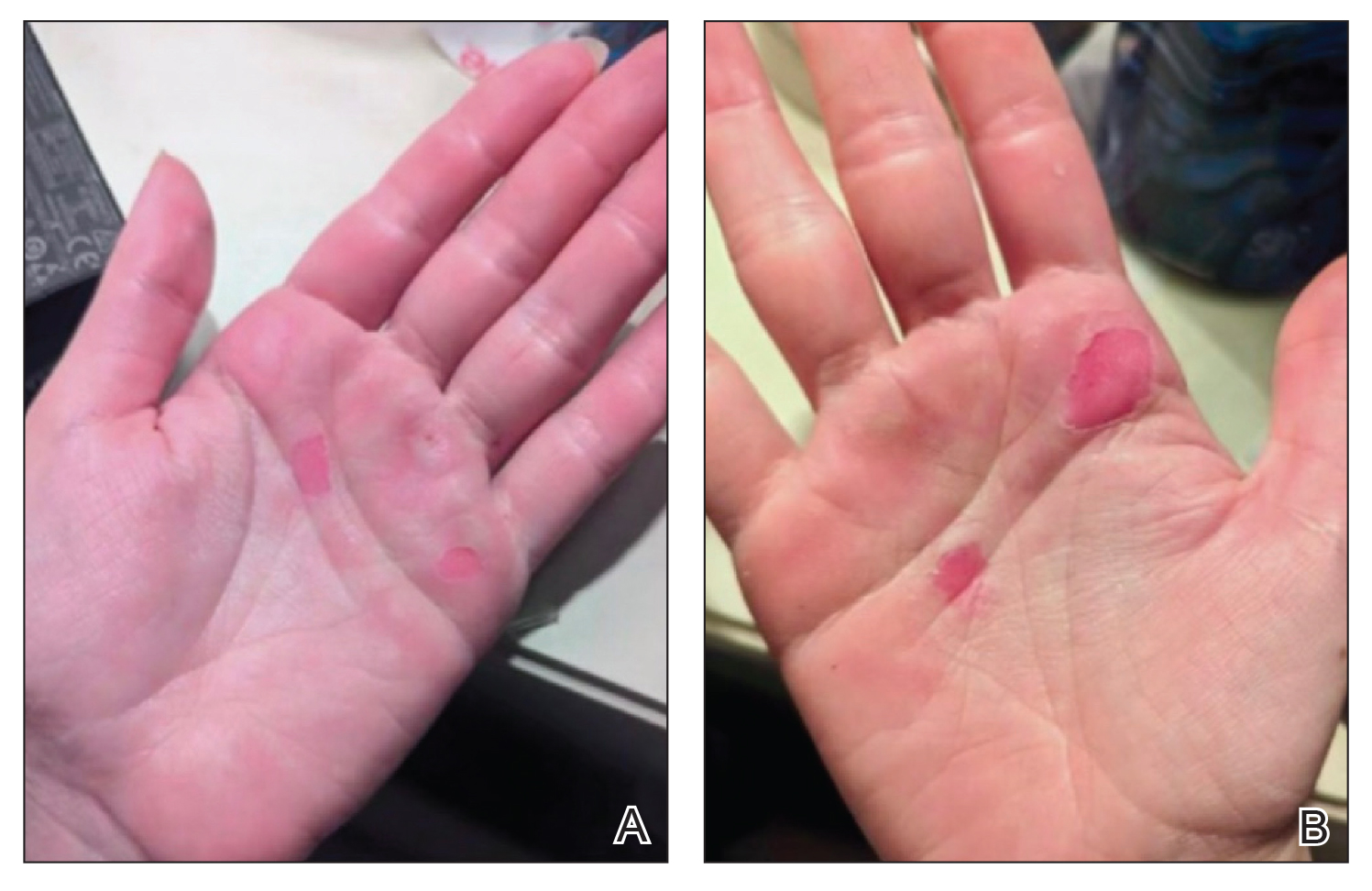
Overall, the origin of skin fragility induced by isotretinoin is supported by its effect on sebocytes, increased transepidermal water loss, and profound disruption of the integrity of the epidermis, resulting in an elevated risk for inadvertent skin damage. Patients were encouraged to avoid cosmetic procedures in prior case reports,14-16 and because our case demonstrates the risk for cutaneous injury in athletes due to isotretinoin-induced skin fragility, we propose an extension of these warnings to encompass athletes receiving isotretinoin treatment. Offering early guidance on wound prevention is of paramount importance in maintaining athletic performance and minimizing painful injuries.
- Rajput I, Anjankar VP. Side effects of treating acne vulgaris with isotretinoin: a systematic review. Cureus. 2024;16:E55946. doi:10.7759/cureus.55946
- Hatami P, Balighi K, Asl HN, et al. Isotretinoin and timing of procedural interventions: clinical implications and practical points. J Cosmet Dermatol. 2023;22:2146-2149. doi:10.1111/jocd.15874
- McDonald KA, Shelley AJ, Alavi A. A systematic review on oral isotretinoin therapy and clinically observable wound healing in acne patients. J Cutan Med Surg. 2017;21:325-333. doi:10.1177/1203475417701419
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169. doi:10.4161/derm.1.3.9364
- Zouboulis CC. Isotretinoin revisited: pluripotent effects on human sebaceous gland cells. J Invest Dermatol. 2006;126:2154-2156. doi:10.1038/sj.jid.5700418
- Kmiec´ ML, Pajor A, Broniarczyk-Dyła G. Evaluation of biophysical skin parameters and assessment of hair growth in patients with acne treated with isotretinoin. Postepy Dermatol Alergol. 2013;30:343-349. doi:10.5114/pdia.2013.39432
- Waldman A, Bolotin D, Arndt KA, et al. ASDS Guidelines Task Force: Consensus recommendations regarding the safety of lasers, dermabrasion, chemical peels, energy devices, and skin surgery during and after isotretinoin use. Dermatolog Surg. 2017;43:1249-1262. doi:10.1097/DSS.0000000000001166
- Aksoy H, Aksoy B, Calikoglu E. Systemic retinoids and scar dehiscence. Indian J Dermatol. 2019;64:68. doi:10.4103/ijd.IJD_148_18
- Pavlis MB, Lieblich L. Isotretinoin-induced skin fragility in a teenaged athlete: a case report. Cutis. 2013;92:33-34.
- Herane MI, Fuenzalida H, Zegpi E, et al. Specific gel-cream as adjuvant to oral isotretinoin improved hydration and prevented TEWL increase—a double-blind, randomized, placebo-controlled study. J Cosmet Dermatol. 2009;8:181-185. doi:10.1111/j.1473-2165.2009.00455.x
- Park KY, Ko EJ, Kim IS, et al. The effect of evening primrose oil for the prevention of xerotic cheilitis in acne patients being treated with isotretinoin: a pilot study. Ann Dermatol. 2014;26:706-712. doi:10.5021/ad.2014.26.6.706
- Elias PM, Fritsch PO, Lampe M, et al. Retinoid effects on epidermal structure, differentiation, and permeability. Lab Invest. 1981;44:531-540.
- Williams ML, Elias PM. Nature of skin fragility in patients receiving retinoids for systemic effect. Arch Dermatol. 1981;117:611-619.
- Rubenstein R, Roenigk HH, Stegman SJ, et al. Atypical keloids after dermabrasion of patients taking isotretinoin. J Am Acad Dermatol. 1986;15:280-285. doi:10.1016/S0190-9622(86)70167-9
- Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706. doi:10.1111/j.1365-2133.1988.tb02574.x
- Katz BE, Mac Farlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853. doi:10.1016/S0190-9622(94)70096-6
Isotretinoin was introduced more than 3 decades ago and marked a major advancement in the treatment of severe refractory cystic acne. The most common adverse effects linked to isotretinoin usage are mucocutaneous in nature, manifesting as xerosis and cheilitis.1 Skin fragility and poor wound healing also have been reported.2-6 Current recommendations for avoiding these adverse effects include refraining from waxing, laser procedures, and other elective cutaneous procedures for at least 6 months.7 We present a case of isotretinoin-induced cutaneous fragility resulting in blistering and erosions on the palms of a competitive aerial trapeze artist.
Case Report
A 25-year-old woman presented for follow-up during week 12 of isotretinoin therapy (40 mg twice daily) prescribed for acne. She reported peeling of the skin on the palms following intense aerial acrobatic workouts. She had been a performing aerialist for many years and had never sustained a similar injury. The wounds were painful and led to decreased activity. She had no notable medical history. Physical examination of the palms revealed erosions in a distribution that corresponded to horizontal bar contact and friction (Figure). The patient was advised on proper wound care, application of emollients, and minimizing friction. She completed the course of isotretinoin and has continued aerialist activity without recurrence of skin fragility.
Comment
Skin fragility is a well-known adverse effect of isotretinoin therapy.8 Pavlis and Lieblich9 reported skin fragility in a young wrestler who experienced similar skin erosions due to isotretinoin therapy. The proposed mechanism of isotretinoin-induced skin fragility is multifactorial. It involves an apoptotic effect on sebocytes,5 which results in reduced stratum corneum hydration and an associated increase in transepidermal water loss.6,10,11 Retinoids also are known to cause thinning of the skin, likely due to the disadhesion of both the epidermis and the stratum corneum, which was demonstrated by the easy removal of cornified cells through tape stripping in hairless mice treated with isotretinoin.12 In further investigations, human patients and hairless mice treated with isotretinoin readily developed friction blisters through pencil eraser abrasion.13 Examination of the friction blisters using light and electron microscopy revealed fraying or loss of the stratum corneum and viable epidermis as well as loss of desmosomes and tonofilaments. Additionally, intracellular and intercellular deposits of an unidentified amorphous material were noted.13
Overall, the origin of skin fragility induced by isotretinoin is supported by its effect on sebocytes, increased transepidermal water loss, and profound disruption of the integrity of the epidermis, resulting in an elevated risk for inadvertent skin damage. Patients were encouraged to avoid cosmetic procedures in prior case reports,14-16 and because our case demonstrates the risk for cutaneous injury in athletes due to isotretinoin-induced skin fragility, we propose an extension of these warnings to encompass athletes receiving isotretinoin treatment. Offering early guidance on wound prevention is of paramount importance in maintaining athletic performance and minimizing painful injuries.
Isotretinoin was introduced more than 3 decades ago and marked a major advancement in the treatment of severe refractory cystic acne. The most common adverse effects linked to isotretinoin usage are mucocutaneous in nature, manifesting as xerosis and cheilitis.1 Skin fragility and poor wound healing also have been reported.2-6 Current recommendations for avoiding these adverse effects include refraining from waxing, laser procedures, and other elective cutaneous procedures for at least 6 months.7 We present a case of isotretinoin-induced cutaneous fragility resulting in blistering and erosions on the palms of a competitive aerial trapeze artist.
Case Report
A 25-year-old woman presented for follow-up during week 12 of isotretinoin therapy (40 mg twice daily) prescribed for acne. She reported peeling of the skin on the palms following intense aerial acrobatic workouts. She had been a performing aerialist for many years and had never sustained a similar injury. The wounds were painful and led to decreased activity. She had no notable medical history. Physical examination of the palms revealed erosions in a distribution that corresponded to horizontal bar contact and friction (Figure). The patient was advised on proper wound care, application of emollients, and minimizing friction. She completed the course of isotretinoin and has continued aerialist activity without recurrence of skin fragility.
Comment
Skin fragility is a well-known adverse effect of isotretinoin therapy.8 Pavlis and Lieblich9 reported skin fragility in a young wrestler who experienced similar skin erosions due to isotretinoin therapy. The proposed mechanism of isotretinoin-induced skin fragility is multifactorial. It involves an apoptotic effect on sebocytes,5 which results in reduced stratum corneum hydration and an associated increase in transepidermal water loss.6,10,11 Retinoids also are known to cause thinning of the skin, likely due to the disadhesion of both the epidermis and the stratum corneum, which was demonstrated by the easy removal of cornified cells through tape stripping in hairless mice treated with isotretinoin.12 In further investigations, human patients and hairless mice treated with isotretinoin readily developed friction blisters through pencil eraser abrasion.13 Examination of the friction blisters using light and electron microscopy revealed fraying or loss of the stratum corneum and viable epidermis as well as loss of desmosomes and tonofilaments. Additionally, intracellular and intercellular deposits of an unidentified amorphous material were noted.13
Overall, the origin of skin fragility induced by isotretinoin is supported by its effect on sebocytes, increased transepidermal water loss, and profound disruption of the integrity of the epidermis, resulting in an elevated risk for inadvertent skin damage. Patients were encouraged to avoid cosmetic procedures in prior case reports,14-16 and because our case demonstrates the risk for cutaneous injury in athletes due to isotretinoin-induced skin fragility, we propose an extension of these warnings to encompass athletes receiving isotretinoin treatment. Offering early guidance on wound prevention is of paramount importance in maintaining athletic performance and minimizing painful injuries.
- Rajput I, Anjankar VP. Side effects of treating acne vulgaris with isotretinoin: a systematic review. Cureus. 2024;16:E55946. doi:10.7759/cureus.55946
- Hatami P, Balighi K, Asl HN, et al. Isotretinoin and timing of procedural interventions: clinical implications and practical points. J Cosmet Dermatol. 2023;22:2146-2149. doi:10.1111/jocd.15874
- McDonald KA, Shelley AJ, Alavi A. A systematic review on oral isotretinoin therapy and clinically observable wound healing in acne patients. J Cutan Med Surg. 2017;21:325-333. doi:10.1177/1203475417701419
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169. doi:10.4161/derm.1.3.9364
- Zouboulis CC. Isotretinoin revisited: pluripotent effects on human sebaceous gland cells. J Invest Dermatol. 2006;126:2154-2156. doi:10.1038/sj.jid.5700418
- Kmiec´ ML, Pajor A, Broniarczyk-Dyła G. Evaluation of biophysical skin parameters and assessment of hair growth in patients with acne treated with isotretinoin. Postepy Dermatol Alergol. 2013;30:343-349. doi:10.5114/pdia.2013.39432
- Waldman A, Bolotin D, Arndt KA, et al. ASDS Guidelines Task Force: Consensus recommendations regarding the safety of lasers, dermabrasion, chemical peels, energy devices, and skin surgery during and after isotretinoin use. Dermatolog Surg. 2017;43:1249-1262. doi:10.1097/DSS.0000000000001166
- Aksoy H, Aksoy B, Calikoglu E. Systemic retinoids and scar dehiscence. Indian J Dermatol. 2019;64:68. doi:10.4103/ijd.IJD_148_18
- Pavlis MB, Lieblich L. Isotretinoin-induced skin fragility in a teenaged athlete: a case report. Cutis. 2013;92:33-34.
- Herane MI, Fuenzalida H, Zegpi E, et al. Specific gel-cream as adjuvant to oral isotretinoin improved hydration and prevented TEWL increase—a double-blind, randomized, placebo-controlled study. J Cosmet Dermatol. 2009;8:181-185. doi:10.1111/j.1473-2165.2009.00455.x
- Park KY, Ko EJ, Kim IS, et al. The effect of evening primrose oil for the prevention of xerotic cheilitis in acne patients being treated with isotretinoin: a pilot study. Ann Dermatol. 2014;26:706-712. doi:10.5021/ad.2014.26.6.706
- Elias PM, Fritsch PO, Lampe M, et al. Retinoid effects on epidermal structure, differentiation, and permeability. Lab Invest. 1981;44:531-540.
- Williams ML, Elias PM. Nature of skin fragility in patients receiving retinoids for systemic effect. Arch Dermatol. 1981;117:611-619.
- Rubenstein R, Roenigk HH, Stegman SJ, et al. Atypical keloids after dermabrasion of patients taking isotretinoin. J Am Acad Dermatol. 1986;15:280-285. doi:10.1016/S0190-9622(86)70167-9
- Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706. doi:10.1111/j.1365-2133.1988.tb02574.x
- Katz BE, Mac Farlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853. doi:10.1016/S0190-9622(94)70096-6
- Rajput I, Anjankar VP. Side effects of treating acne vulgaris with isotretinoin: a systematic review. Cureus. 2024;16:E55946. doi:10.7759/cureus.55946
- Hatami P, Balighi K, Asl HN, et al. Isotretinoin and timing of procedural interventions: clinical implications and practical points. J Cosmet Dermatol. 2023;22:2146-2149. doi:10.1111/jocd.15874
- McDonald KA, Shelley AJ, Alavi A. A systematic review on oral isotretinoin therapy and clinically observable wound healing in acne patients. J Cutan Med Surg. 2017;21:325-333. doi:10.1177/1203475417701419
- Layton A. The use of isotretinoin in acne. Dermatoendocrinol. 2009;1:162-169. doi:10.4161/derm.1.3.9364
- Zouboulis CC. Isotretinoin revisited: pluripotent effects on human sebaceous gland cells. J Invest Dermatol. 2006;126:2154-2156. doi:10.1038/sj.jid.5700418
- Kmiec´ ML, Pajor A, Broniarczyk-Dyła G. Evaluation of biophysical skin parameters and assessment of hair growth in patients with acne treated with isotretinoin. Postepy Dermatol Alergol. 2013;30:343-349. doi:10.5114/pdia.2013.39432
- Waldman A, Bolotin D, Arndt KA, et al. ASDS Guidelines Task Force: Consensus recommendations regarding the safety of lasers, dermabrasion, chemical peels, energy devices, and skin surgery during and after isotretinoin use. Dermatolog Surg. 2017;43:1249-1262. doi:10.1097/DSS.0000000000001166
- Aksoy H, Aksoy B, Calikoglu E. Systemic retinoids and scar dehiscence. Indian J Dermatol. 2019;64:68. doi:10.4103/ijd.IJD_148_18
- Pavlis MB, Lieblich L. Isotretinoin-induced skin fragility in a teenaged athlete: a case report. Cutis. 2013;92:33-34.
- Herane MI, Fuenzalida H, Zegpi E, et al. Specific gel-cream as adjuvant to oral isotretinoin improved hydration and prevented TEWL increase—a double-blind, randomized, placebo-controlled study. J Cosmet Dermatol. 2009;8:181-185. doi:10.1111/j.1473-2165.2009.00455.x
- Park KY, Ko EJ, Kim IS, et al. The effect of evening primrose oil for the prevention of xerotic cheilitis in acne patients being treated with isotretinoin: a pilot study. Ann Dermatol. 2014;26:706-712. doi:10.5021/ad.2014.26.6.706
- Elias PM, Fritsch PO, Lampe M, et al. Retinoid effects on epidermal structure, differentiation, and permeability. Lab Invest. 1981;44:531-540.
- Williams ML, Elias PM. Nature of skin fragility in patients receiving retinoids for systemic effect. Arch Dermatol. 1981;117:611-619.
- Rubenstein R, Roenigk HH, Stegman SJ, et al. Atypical keloids after dermabrasion of patients taking isotretinoin. J Am Acad Dermatol. 1986;15:280-285. doi:10.1016/S0190-9622(86)70167-9
- Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706. doi:10.1111/j.1365-2133.1988.tb02574.x
- Katz BE, Mac Farlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853. doi:10.1016/S0190-9622(94)70096-6
Practice Points
- Isotretinoin is used to treat severe nodulocystic acne but can cause adverse effects such as skin fragility, xerosis, and poor wound healing.
- Dermatologists should inform athletes of heightened skin vulnerability while undergoing isotretinoin treatment.
- Isotretinoin-induced skin fragility involves the effects of isotretinoin on sebocytes, transepidermal water loss, and disruption of the integrity of the epidermis.
Uncommon Locations for Brain Herniations Into Arachnoid Granulations: 5 Cases and Literature Review
The circulation of cerebrospinal fluid (CSF) is crucial for maintaining homeostasis for the optimal functioning of the multiple complex activities of the brain and spinal cord, including the disposal of metabolic waste products of brain and spinal cord activity into the cerebral venous drainage. Throughout the brain, the arachnoid mater forms small outpouchings or diverticula that penetrate the dura mater and communicate with the dural venous sinuses. These outpuchings are called arachnoid granulations or arachnoid villi, and most are found within the dural sinuses, primarily in the transverse sinuses and superior sagittal sinus, but can occasionally be seen extending into the inner table of the calvarium.1,2

The amount of arachnoid granulations seen in bone, particularly around the superior sagittal sinus, may increase with age.2 Arachnoid granulations are generally small but the largest ones can be seen on gross examination during intracranial procedures or autopsy.3 Magnetic resonance imaging (MRI) can detect arachnoid granulations, which are characterized as T1 hypointense and T2 hyperintense (CSF isointense), well-circumscribed, small, nonenhancing masses within the dural sinuses or in the diploic space (Figure 1). Even small arachnoid granulations < 1 mm in length can be detected.2
Smaller arachnoid granulations have been described histologically as entirely covered by a dural membrane, thus creating a subdural space that separates the body of the arachnoid granulation from the lumen of the accompanying venous sinus.4 However, larger arachnoid granulations may not be completely covered by a dural membrane, thus creating a point of contact between the arachnoid granulation and the venous sinus.4 Larger arachnoid granulations are normally filled with CSF, and their signal characteristics are similar to CSF on imaging.5,6 Arachnoid granulations also often contain vessels draining into the adjacent venous sinus.5,6
When larger arachnoid granulations are present, they may permit the protrusion of herniated brain tissue. There has been an increasing number of reports of these brain herniations into arachnoid granulations (BHAGs) in the literature.7-10 While these herniations have been associated with nonspecific neurologic symptoms like tinnitus and idiopathic intracranial hypertension, their true clinical significance remains undetermined.10,11 This article presents 5 cases of BHAG, discusses their clinical presentations and image findings, and reviews the current literature.
Case 1
A 30-year-old male with a history of multiple traumatic brain injuries presented for evaluation of seizures. The patient described the semiology of the seizures as a bright, colorful light in his right visual field, followed by loss of vision, then loss of awareness and full body convulsion. The semiology of this patient’s seizures was consistent with left temporo-occipital lobe seizure. The only abnormality seen in the brain MRI was the herniation of brain parenchyma originating from the occipital lobe into the transverse sinus, presumably through an arachnoid granulation (Figure 1). An electroencephalogram (EEG) was unremarkable, though the semiology of the seizure historically described by the patient was localized to the area of BHAG. The patient is currently taking antiseizure medications and has experienced no additional seizures.
Case 2

A male aged 53 years with a history of peripheral artery disease presented with a 6-month history of headaches and dizziness. The patient reported the onset of visual aura to his right visual field, starting as a fingernail-sized scintillating kaleidoscope light that would gradually increase in size to a round shape with fading kaleidoscope colors. This episode would last for a few minutes and was immediately followed by a headache. There was no alteration of consciousness during visual aura, although sometimes the patient would have right-sided scalp tingling. These episodes were often unprovoked, but occasionally triggered by bright lights. A single routine EEG was unremarkable. The patient reported headaches without aura, but not aura without headaches, which made occipital lobe seizure less likely. MRI demonstrated a small herniation of brain parenchyma into the inner table of the left occipital bone (Figure 2). The patient was diagnosed with migraine with aura, and the semiology of the visual aura corresponded to the location of the herniation in the left occipital region.
Case 3

A 77-year-old male with a history of left ear diving injury presented with left-sided asymmetric hearing loss and word recognition difficulty for several years. MRI obtained as part of his work-up to evaluate for possible schwannoma of the eighth left cranial nerve instead demonstrated an incidental right cerebellar herniation within an arachnoid granulation into the diploic space of the occipital bone (Figure 3). The BHAG for this patient appeared to be an incidental finding unrelated to his asymmetric hearing loss.
Case 4

A male aged 62 years with a history of metastatic esophageal cancer, substance abuse, and a prior presumed alcohol withdrawal seizure underwent an MRI for evaluation of brain metastasis after presenting to the hospital with confusion 1 day after starting chemotherapy (Figure 4). Nine years prior, the patient had an isolated generalized tonic-clonic seizure approximately 72 hours following a period of alcohol cessation. The MRI demonstrated an incidental left parasagittal herniation of left parietal lobe tissue through an arachnoid granulation into the superior sagittal sinus, in addition to metastatic brain lesions. An EEG showed mild encephalopathy without evidence of seizures. It was determined that the patient's confusion was most likely due to toxic-metabolic encephalopathy from chemotherapy.
Case 5

A 51-year-old male presented with worsening headache severity and frequency. He had a history of chronic headaches for about 20 years that occurred annually, but were now occurring twice weekly. The headaches often started with a left eye visual aura followed by pressure in the left eye, left frontal region, and left ear, with at times a cervicogenic component. No cervical spine imaging was available. An MRI revealed 2 small adjacent areas of cerebellar herniation into arachnoid granulations in the left occipital bone (Figure 5).
Discussion
Arachnoid granulations appear very early in life, although they are uncommon before age 2 years.2 Classically, they have been understood to act as 1-way valves permitting the outflow of CSF from the subarachnoid space to the dural venous sinuses. However, increasing evidence shows they may only play a minor role in that process.12 The structure of arachnoid granulations is being reexamined. A recent microscopy study demonstrated structural heterogeneity with a fine, porous lining that permits flow.13 Additionally, associated immune components in the microenvironment suggests that arachnoid granulations may function similarly to lymph nodes as part of a central nervous system lymphatic network.13 Evidence is lacking for arachnoid granulations being the primary route of CSF outflow, and newer models include CSF exit pathways along the cranial nerves and drainage through lymphatics within the dura mater.12
New MRI systems have demonstrated that the prevalence of arachnoid granulations increases with age. One study found that all subjects in the aged 40 years cohort had detectable arachnoid granulations on images obtained with a 3T MRI system, with the main site being the superior sagittal sinus.2 The prevalence increased until age 40 years and then noticeably decreased. Not only did the prevalence increase in this pattern, but the total number of detectable arachnoid granulations followed a similar pattern.2 In addition, the detectable arachnoid granulations tend to be larger in older patients. Arachnoid granulations are very common in adults, but little is known about when and why brain tissue herniates through these structures.
This case series illustrates how a small amount of adult cerebral or cerebellar matter in large arachnoid granulations can herniate into the dural sinuses and diploic space. Although arachnoid granulations extending into the dural sinuses and diploic space are a relatively common finding on MRI, BHAGs are rare in these locations.1,2,8 Improved spatial resolution afforded by newer high-field scanners with thinner sections, such as very thin (1 mm) T1- and heavily T2-weighted 3 dimensional sequences may lead to increased detection of BHAG. Some of these herniations are small and may be easily missed or confused for normal arachnoid granulations on 3 to 5 mm thickness MRIs.
Despite increased recognition, it is still uncertain to what degree these herniations contribute to the clinical presentations. Associated neurologic symptoms may include seizures, headaches, tinnitus, syncope, and increased intracranial pressure.7-10
Three cases presented in this article demonstrated abnormal signals adjacent to the herniated brain, presumably due to dysplasia of gliotic tissue. In 1 study, parenchymal signal and structural changes occurred in about one-half of the reported BHAG, all of which were cerebellar herniations.7 In Case 1, the herniation and adjacent abnormal MRI signal corresponded to localization of the seizure semiology as obtained from patient history, strongly suggesting the BHAG played a role in the presentation. Signal abnormality accompanying an adjacent BHAG may suggest a higher likelihood that the BHAG has clinical relevance. However, the patient in Case 2 had a visual aura that corresponded to the BHAG location, so a signal abnormality may not be necessary for a patient to develop symptoms. Case 1 also included a history of documented traumatic brain injuries, suggesting that perhaps head trauma may facilitate BHAG development. Regardless, there is likely also a congenital component to their formation, as BHAG has been observed in the pediatric population.14
The patient's asymmetric left-sided hearing loss in Case 3 appeared unrelated to the BHAG as its location was in the contralateral cerebellar region and did not correspond to the patient’s clinical findings. The patient in Case 4 had a limited history regarding localization details of their prior presumed alcohol withdrawal seizure, such as head movements, eye deviation, or lateralized onset of convulsions. Given this limited data, it is unclear whether their prior seizure could have been related to BHAG or not. The patient in case 5 reported worsening headaches on the left side of his head, which corresponded to BHAG occurring on the left side. However, given that the increased T2 signal occurred in the left cerebellar hemisphere with BHAG in the left occipital bone, the occipital cortex was not involved. In this case, the BHAG would not explain the patient’s visual aura as such a lesion would have been expected in the right occipital cortex rather than its actual location in this patient’s left cerebellar hemisphere.
CONCLUSIONS
Understanding the clinical impact of brain herniations is important because they are probably more common than previously thought. Improved MRI capabilities suggest that more BHAG will be detected moving forward as radiologists interpret images with higher resolution and thinner slices. Until its significance is fully understood, BHAG will continue to complicate the diagnosis of patients with neurologic complaints whose brain MRIs and EEGs are otherwise unremarkable.

There have been no cases of surgical BHAG intervention and pathology analysis that would help determine their clinical significance. A related entity, temporal lobe encephalocele, has been linked to focal temporal lobe epilepsy, which has demonstrated significant symptom improvement following surgical correction.15 However, encephaloceles have been distinguished from BHAG in part because they do not necessarily herniate through an arachnoid granulation.8 BHAG has only begun to be characterized in detail over the last decade, so more research is needed to understand how it develops and what clinical significance it truly holds.
1. Ikushima I, Korogi Y, Makita O, et al. MRI of arachnoid granulations within the dural sinuses using a FLAIR pulse sequence. Br J Radiol. 1999;72(863):1046-1051. doi:10.1259/bjr.72.863.10700819
2. Rados M, Zivko M, Perisa A, Oreskovic D, Klarica M. No arachnoid granulations-no problems: number, size, and distribution of arachnoid granulations from birth to 80 years of age. Front Aging Neurosci. 2021;13:698865. doi:10.3389/fnagi.2021.698865
3. Grossman CB, Potts DG. Arachnoid granulations: radiology and anatomy. Radiology. 1974;113(1):95-100. doi:10.1148/113.1.95
4. Wolpow ER, Schaumburg HH. Structure of the human arachnoid granulation. J Neurosurg. 1972;37(6):724-727. doi:10.3171/jns.1972.37.6.0724
5. Leach JL, Jones BV, Tomsick TA, Stewart CA, Balko MG. Normal appearance of arachnoid granulations on contrast-enhanced CT and MR of the brain: differentiation from dural sinus disease. AJNR Am J Neuroradiol. 1996;17(8):1523-1532.
6. Roche J, Warner D. Arachnoid granulations in the transverse and sigmoid sinuses: CT, MR, and MR angiographic appearance of a normal anatomic variation. AJNR Am J Neuroradiol. 1996;17(4):677-683.
7. Malekzadehlashkariani S, Wanke I, Rufenacht DA, San Millan D. Brain herniations into arachnoid granulations: about 68 cases in 38 patients and review of the literature. Neuroradiology. 2016;58(5):443-457. doi:10.1007/s00234-016-1662-5
8. Battal B, Castillo M. Brain herniations into the dural venous sinuses or calvarium: MRI of a recently recognized entity. Neuroradiol J. 2014;27(1):55-62. doi:10.15274/NRJ-2014-10006
9. Liebo GB, Lane JJ, Van Gompel JJ, Eckel LJ, Schwartz KM, Lehman VT. Brain herniation into arachnoid granulations: clinical and neuroimaging features. J Neuroimaging. 2016;26(6):592-598. doi:10.1111/jon.12366
10. Smith ER, Caton MT, Villanueva-Meyer JE, et al. Brain herniation (encephalocele) into arachnoid granulations: Prevalence and association with pulsatile tinnitus and idiopathic intracranial hypertension. Neuroradiology. 2022;64(9):1747-1754.
11. Battal B, Hamcan S, Akgun V, et al. Brain herniations into the dural venous sinus or calvarium: MRI findings, possible causes and clinical significance. Eur Radiol. 2016;26(6):1723-1731.
12. Proulx ST. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell Mol Life Sci. 2021;78(6):2429-2457.
13. Shah T, Leurgans SE, Mehta RI, et al. Arachnoid granulations are lymphatic conduits that communicate with bone marrow and dura-arachnoid stroma. J Exp Med. 2023;220(2).
14. Sade R, Ogul H, Polat G, Pirimoglu B, Kantarci M. Brain herniation into the transverse sinuses’ arachnoid granulations in the pediatric population investigated with 3 T MRI. Acta Neurol Belg. 2019;119(2):225-231.
15. Saavalainen T, Jutila L, Mervaala E, Kalviainen R, Vanninen R, Immonen A. Temporal anteroinferior encephalocele: An underrecognized etiology of temporal lobe epilepsy? Neurology. 2015;85(17):1467-1474.
The circulation of cerebrospinal fluid (CSF) is crucial for maintaining homeostasis for the optimal functioning of the multiple complex activities of the brain and spinal cord, including the disposal of metabolic waste products of brain and spinal cord activity into the cerebral venous drainage. Throughout the brain, the arachnoid mater forms small outpouchings or diverticula that penetrate the dura mater and communicate with the dural venous sinuses. These outpuchings are called arachnoid granulations or arachnoid villi, and most are found within the dural sinuses, primarily in the transverse sinuses and superior sagittal sinus, but can occasionally be seen extending into the inner table of the calvarium.1,2
The amount of arachnoid granulations seen in bone, particularly around the superior sagittal sinus, may increase with age.2 Arachnoid granulations are generally small but the largest ones can be seen on gross examination during intracranial procedures or autopsy.3 Magnetic resonance imaging (MRI) can detect arachnoid granulations, which are characterized as T1 hypointense and T2 hyperintense (CSF isointense), well-circumscribed, small, nonenhancing masses within the dural sinuses or in the diploic space (Figure 1). Even small arachnoid granulations < 1 mm in length can be detected.2
Smaller arachnoid granulations have been described histologically as entirely covered by a dural membrane, thus creating a subdural space that separates the body of the arachnoid granulation from the lumen of the accompanying venous sinus.4 However, larger arachnoid granulations may not be completely covered by a dural membrane, thus creating a point of contact between the arachnoid granulation and the venous sinus.4 Larger arachnoid granulations are normally filled with CSF, and their signal characteristics are similar to CSF on imaging.5,6 Arachnoid granulations also often contain vessels draining into the adjacent venous sinus.5,6
When larger arachnoid granulations are present, they may permit the protrusion of herniated brain tissue. There has been an increasing number of reports of these brain herniations into arachnoid granulations (BHAGs) in the literature.7-10 While these herniations have been associated with nonspecific neurologic symptoms like tinnitus and idiopathic intracranial hypertension, their true clinical significance remains undetermined.10,11 This article presents 5 cases of BHAG, discusses their clinical presentations and image findings, and reviews the current literature.
Case 1
A 30-year-old male with a history of multiple traumatic brain injuries presented for evaluation of seizures. The patient described the semiology of the seizures as a bright, colorful light in his right visual field, followed by loss of vision, then loss of awareness and full body convulsion. The semiology of this patient’s seizures was consistent with left temporo-occipital lobe seizure. The only abnormality seen in the brain MRI was the herniation of brain parenchyma originating from the occipital lobe into the transverse sinus, presumably through an arachnoid granulation (Figure 1). An electroencephalogram (EEG) was unremarkable, though the semiology of the seizure historically described by the patient was localized to the area of BHAG. The patient is currently taking antiseizure medications and has experienced no additional seizures.
Case 2
A male aged 53 years with a history of peripheral artery disease presented with a 6-month history of headaches and dizziness. The patient reported the onset of visual aura to his right visual field, starting as a fingernail-sized scintillating kaleidoscope light that would gradually increase in size to a round shape with fading kaleidoscope colors. This episode would last for a few minutes and was immediately followed by a headache. There was no alteration of consciousness during visual aura, although sometimes the patient would have right-sided scalp tingling. These episodes were often unprovoked, but occasionally triggered by bright lights. A single routine EEG was unremarkable. The patient reported headaches without aura, but not aura without headaches, which made occipital lobe seizure less likely. MRI demonstrated a small herniation of brain parenchyma into the inner table of the left occipital bone (Figure 2). The patient was diagnosed with migraine with aura, and the semiology of the visual aura corresponded to the location of the herniation in the left occipital region.
Case 3
A 77-year-old male with a history of left ear diving injury presented with left-sided asymmetric hearing loss and word recognition difficulty for several years. MRI obtained as part of his work-up to evaluate for possible schwannoma of the eighth left cranial nerve instead demonstrated an incidental right cerebellar herniation within an arachnoid granulation into the diploic space of the occipital bone (Figure 3). The BHAG for this patient appeared to be an incidental finding unrelated to his asymmetric hearing loss.
Case 4
A male aged 62 years with a history of metastatic esophageal cancer, substance abuse, and a prior presumed alcohol withdrawal seizure underwent an MRI for evaluation of brain metastasis after presenting to the hospital with confusion 1 day after starting chemotherapy (Figure 4). Nine years prior, the patient had an isolated generalized tonic-clonic seizure approximately 72 hours following a period of alcohol cessation. The MRI demonstrated an incidental left parasagittal herniation of left parietal lobe tissue through an arachnoid granulation into the superior sagittal sinus, in addition to metastatic brain lesions. An EEG showed mild encephalopathy without evidence of seizures. It was determined that the patient's confusion was most likely due to toxic-metabolic encephalopathy from chemotherapy.
Case 5
A 51-year-old male presented with worsening headache severity and frequency. He had a history of chronic headaches for about 20 years that occurred annually, but were now occurring twice weekly. The headaches often started with a left eye visual aura followed by pressure in the left eye, left frontal region, and left ear, with at times a cervicogenic component. No cervical spine imaging was available. An MRI revealed 2 small adjacent areas of cerebellar herniation into arachnoid granulations in the left occipital bone (Figure 5).
Discussion
Arachnoid granulations appear very early in life, although they are uncommon before age 2 years.2 Classically, they have been understood to act as 1-way valves permitting the outflow of CSF from the subarachnoid space to the dural venous sinuses. However, increasing evidence shows they may only play a minor role in that process.12 The structure of arachnoid granulations is being reexamined. A recent microscopy study demonstrated structural heterogeneity with a fine, porous lining that permits flow.13 Additionally, associated immune components in the microenvironment suggests that arachnoid granulations may function similarly to lymph nodes as part of a central nervous system lymphatic network.13 Evidence is lacking for arachnoid granulations being the primary route of CSF outflow, and newer models include CSF exit pathways along the cranial nerves and drainage through lymphatics within the dura mater.12
New MRI systems have demonstrated that the prevalence of arachnoid granulations increases with age. One study found that all subjects in the aged 40 years cohort had detectable arachnoid granulations on images obtained with a 3T MRI system, with the main site being the superior sagittal sinus.2 The prevalence increased until age 40 years and then noticeably decreased. Not only did the prevalence increase in this pattern, but the total number of detectable arachnoid granulations followed a similar pattern.2 In addition, the detectable arachnoid granulations tend to be larger in older patients. Arachnoid granulations are very common in adults, but little is known about when and why brain tissue herniates through these structures.
This case series illustrates how a small amount of adult cerebral or cerebellar matter in large arachnoid granulations can herniate into the dural sinuses and diploic space. Although arachnoid granulations extending into the dural sinuses and diploic space are a relatively common finding on MRI, BHAGs are rare in these locations.1,2,8 Improved spatial resolution afforded by newer high-field scanners with thinner sections, such as very thin (1 mm) T1- and heavily T2-weighted 3 dimensional sequences may lead to increased detection of BHAG. Some of these herniations are small and may be easily missed or confused for normal arachnoid granulations on 3 to 5 mm thickness MRIs.
Despite increased recognition, it is still uncertain to what degree these herniations contribute to the clinical presentations. Associated neurologic symptoms may include seizures, headaches, tinnitus, syncope, and increased intracranial pressure.7-10
Three cases presented in this article demonstrated abnormal signals adjacent to the herniated brain, presumably due to dysplasia of gliotic tissue. In 1 study, parenchymal signal and structural changes occurred in about one-half of the reported BHAG, all of which were cerebellar herniations.7 In Case 1, the herniation and adjacent abnormal MRI signal corresponded to localization of the seizure semiology as obtained from patient history, strongly suggesting the BHAG played a role in the presentation. Signal abnormality accompanying an adjacent BHAG may suggest a higher likelihood that the BHAG has clinical relevance. However, the patient in Case 2 had a visual aura that corresponded to the BHAG location, so a signal abnormality may not be necessary for a patient to develop symptoms. Case 1 also included a history of documented traumatic brain injuries, suggesting that perhaps head trauma may facilitate BHAG development. Regardless, there is likely also a congenital component to their formation, as BHAG has been observed in the pediatric population.14
The patient's asymmetric left-sided hearing loss in Case 3 appeared unrelated to the BHAG as its location was in the contralateral cerebellar region and did not correspond to the patient’s clinical findings. The patient in Case 4 had a limited history regarding localization details of their prior presumed alcohol withdrawal seizure, such as head movements, eye deviation, or lateralized onset of convulsions. Given this limited data, it is unclear whether their prior seizure could have been related to BHAG or not. The patient in case 5 reported worsening headaches on the left side of his head, which corresponded to BHAG occurring on the left side. However, given that the increased T2 signal occurred in the left cerebellar hemisphere with BHAG in the left occipital bone, the occipital cortex was not involved. In this case, the BHAG would not explain the patient’s visual aura as such a lesion would have been expected in the right occipital cortex rather than its actual location in this patient’s left cerebellar hemisphere.
CONCLUSIONS
Understanding the clinical impact of brain herniations is important because they are probably more common than previously thought. Improved MRI capabilities suggest that more BHAG will be detected moving forward as radiologists interpret images with higher resolution and thinner slices. Until its significance is fully understood, BHAG will continue to complicate the diagnosis of patients with neurologic complaints whose brain MRIs and EEGs are otherwise unremarkable.
There have been no cases of surgical BHAG intervention and pathology analysis that would help determine their clinical significance. A related entity, temporal lobe encephalocele, has been linked to focal temporal lobe epilepsy, which has demonstrated significant symptom improvement following surgical correction.15 However, encephaloceles have been distinguished from BHAG in part because they do not necessarily herniate through an arachnoid granulation.8 BHAG has only begun to be characterized in detail over the last decade, so more research is needed to understand how it develops and what clinical significance it truly holds.
The circulation of cerebrospinal fluid (CSF) is crucial for maintaining homeostasis for the optimal functioning of the multiple complex activities of the brain and spinal cord, including the disposal of metabolic waste products of brain and spinal cord activity into the cerebral venous drainage. Throughout the brain, the arachnoid mater forms small outpouchings or diverticula that penetrate the dura mater and communicate with the dural venous sinuses. These outpuchings are called arachnoid granulations or arachnoid villi, and most are found within the dural sinuses, primarily in the transverse sinuses and superior sagittal sinus, but can occasionally be seen extending into the inner table of the calvarium.1,2
The amount of arachnoid granulations seen in bone, particularly around the superior sagittal sinus, may increase with age.2 Arachnoid granulations are generally small but the largest ones can be seen on gross examination during intracranial procedures or autopsy.3 Magnetic resonance imaging (MRI) can detect arachnoid granulations, which are characterized as T1 hypointense and T2 hyperintense (CSF isointense), well-circumscribed, small, nonenhancing masses within the dural sinuses or in the diploic space (Figure 1). Even small arachnoid granulations < 1 mm in length can be detected.2
Smaller arachnoid granulations have been described histologically as entirely covered by a dural membrane, thus creating a subdural space that separates the body of the arachnoid granulation from the lumen of the accompanying venous sinus.4 However, larger arachnoid granulations may not be completely covered by a dural membrane, thus creating a point of contact between the arachnoid granulation and the venous sinus.4 Larger arachnoid granulations are normally filled with CSF, and their signal characteristics are similar to CSF on imaging.5,6 Arachnoid granulations also often contain vessels draining into the adjacent venous sinus.5,6
When larger arachnoid granulations are present, they may permit the protrusion of herniated brain tissue. There has been an increasing number of reports of these brain herniations into arachnoid granulations (BHAGs) in the literature.7-10 While these herniations have been associated with nonspecific neurologic symptoms like tinnitus and idiopathic intracranial hypertension, their true clinical significance remains undetermined.10,11 This article presents 5 cases of BHAG, discusses their clinical presentations and image findings, and reviews the current literature.
Case 1
A 30-year-old male with a history of multiple traumatic brain injuries presented for evaluation of seizures. The patient described the semiology of the seizures as a bright, colorful light in his right visual field, followed by loss of vision, then loss of awareness and full body convulsion. The semiology of this patient’s seizures was consistent with left temporo-occipital lobe seizure. The only abnormality seen in the brain MRI was the herniation of brain parenchyma originating from the occipital lobe into the transverse sinus, presumably through an arachnoid granulation (Figure 1). An electroencephalogram (EEG) was unremarkable, though the semiology of the seizure historically described by the patient was localized to the area of BHAG. The patient is currently taking antiseizure medications and has experienced no additional seizures.
Case 2
A male aged 53 years with a history of peripheral artery disease presented with a 6-month history of headaches and dizziness. The patient reported the onset of visual aura to his right visual field, starting as a fingernail-sized scintillating kaleidoscope light that would gradually increase in size to a round shape with fading kaleidoscope colors. This episode would last for a few minutes and was immediately followed by a headache. There was no alteration of consciousness during visual aura, although sometimes the patient would have right-sided scalp tingling. These episodes were often unprovoked, but occasionally triggered by bright lights. A single routine EEG was unremarkable. The patient reported headaches without aura, but not aura without headaches, which made occipital lobe seizure less likely. MRI demonstrated a small herniation of brain parenchyma into the inner table of the left occipital bone (Figure 2). The patient was diagnosed with migraine with aura, and the semiology of the visual aura corresponded to the location of the herniation in the left occipital region.
Case 3
A 77-year-old male with a history of left ear diving injury presented with left-sided asymmetric hearing loss and word recognition difficulty for several years. MRI obtained as part of his work-up to evaluate for possible schwannoma of the eighth left cranial nerve instead demonstrated an incidental right cerebellar herniation within an arachnoid granulation into the diploic space of the occipital bone (Figure 3). The BHAG for this patient appeared to be an incidental finding unrelated to his asymmetric hearing loss.
Case 4
A male aged 62 years with a history of metastatic esophageal cancer, substance abuse, and a prior presumed alcohol withdrawal seizure underwent an MRI for evaluation of brain metastasis after presenting to the hospital with confusion 1 day after starting chemotherapy (Figure 4). Nine years prior, the patient had an isolated generalized tonic-clonic seizure approximately 72 hours following a period of alcohol cessation. The MRI demonstrated an incidental left parasagittal herniation of left parietal lobe tissue through an arachnoid granulation into the superior sagittal sinus, in addition to metastatic brain lesions. An EEG showed mild encephalopathy without evidence of seizures. It was determined that the patient's confusion was most likely due to toxic-metabolic encephalopathy from chemotherapy.
Case 5
A 51-year-old male presented with worsening headache severity and frequency. He had a history of chronic headaches for about 20 years that occurred annually, but were now occurring twice weekly. The headaches often started with a left eye visual aura followed by pressure in the left eye, left frontal region, and left ear, with at times a cervicogenic component. No cervical spine imaging was available. An MRI revealed 2 small adjacent areas of cerebellar herniation into arachnoid granulations in the left occipital bone (Figure 5).
Discussion
Arachnoid granulations appear very early in life, although they are uncommon before age 2 years.2 Classically, they have been understood to act as 1-way valves permitting the outflow of CSF from the subarachnoid space to the dural venous sinuses. However, increasing evidence shows they may only play a minor role in that process.12 The structure of arachnoid granulations is being reexamined. A recent microscopy study demonstrated structural heterogeneity with a fine, porous lining that permits flow.13 Additionally, associated immune components in the microenvironment suggests that arachnoid granulations may function similarly to lymph nodes as part of a central nervous system lymphatic network.13 Evidence is lacking for arachnoid granulations being the primary route of CSF outflow, and newer models include CSF exit pathways along the cranial nerves and drainage through lymphatics within the dura mater.12
New MRI systems have demonstrated that the prevalence of arachnoid granulations increases with age. One study found that all subjects in the aged 40 years cohort had detectable arachnoid granulations on images obtained with a 3T MRI system, with the main site being the superior sagittal sinus.2 The prevalence increased until age 40 years and then noticeably decreased. Not only did the prevalence increase in this pattern, but the total number of detectable arachnoid granulations followed a similar pattern.2 In addition, the detectable arachnoid granulations tend to be larger in older patients. Arachnoid granulations are very common in adults, but little is known about when and why brain tissue herniates through these structures.
This case series illustrates how a small amount of adult cerebral or cerebellar matter in large arachnoid granulations can herniate into the dural sinuses and diploic space. Although arachnoid granulations extending into the dural sinuses and diploic space are a relatively common finding on MRI, BHAGs are rare in these locations.1,2,8 Improved spatial resolution afforded by newer high-field scanners with thinner sections, such as very thin (1 mm) T1- and heavily T2-weighted 3 dimensional sequences may lead to increased detection of BHAG. Some of these herniations are small and may be easily missed or confused for normal arachnoid granulations on 3 to 5 mm thickness MRIs.
Despite increased recognition, it is still uncertain to what degree these herniations contribute to the clinical presentations. Associated neurologic symptoms may include seizures, headaches, tinnitus, syncope, and increased intracranial pressure.7-10
Three cases presented in this article demonstrated abnormal signals adjacent to the herniated brain, presumably due to dysplasia of gliotic tissue. In 1 study, parenchymal signal and structural changes occurred in about one-half of the reported BHAG, all of which were cerebellar herniations.7 In Case 1, the herniation and adjacent abnormal MRI signal corresponded to localization of the seizure semiology as obtained from patient history, strongly suggesting the BHAG played a role in the presentation. Signal abnormality accompanying an adjacent BHAG may suggest a higher likelihood that the BHAG has clinical relevance. However, the patient in Case 2 had a visual aura that corresponded to the BHAG location, so a signal abnormality may not be necessary for a patient to develop symptoms. Case 1 also included a history of documented traumatic brain injuries, suggesting that perhaps head trauma may facilitate BHAG development. Regardless, there is likely also a congenital component to their formation, as BHAG has been observed in the pediatric population.14
The patient's asymmetric left-sided hearing loss in Case 3 appeared unrelated to the BHAG as its location was in the contralateral cerebellar region and did not correspond to the patient’s clinical findings. The patient in Case 4 had a limited history regarding localization details of their prior presumed alcohol withdrawal seizure, such as head movements, eye deviation, or lateralized onset of convulsions. Given this limited data, it is unclear whether their prior seizure could have been related to BHAG or not. The patient in case 5 reported worsening headaches on the left side of his head, which corresponded to BHAG occurring on the left side. However, given that the increased T2 signal occurred in the left cerebellar hemisphere with BHAG in the left occipital bone, the occipital cortex was not involved. In this case, the BHAG would not explain the patient’s visual aura as such a lesion would have been expected in the right occipital cortex rather than its actual location in this patient’s left cerebellar hemisphere.
CONCLUSIONS
Understanding the clinical impact of brain herniations is important because they are probably more common than previously thought. Improved MRI capabilities suggest that more BHAG will be detected moving forward as radiologists interpret images with higher resolution and thinner slices. Until its significance is fully understood, BHAG will continue to complicate the diagnosis of patients with neurologic complaints whose brain MRIs and EEGs are otherwise unremarkable.
There have been no cases of surgical BHAG intervention and pathology analysis that would help determine their clinical significance. A related entity, temporal lobe encephalocele, has been linked to focal temporal lobe epilepsy, which has demonstrated significant symptom improvement following surgical correction.15 However, encephaloceles have been distinguished from BHAG in part because they do not necessarily herniate through an arachnoid granulation.8 BHAG has only begun to be characterized in detail over the last decade, so more research is needed to understand how it develops and what clinical significance it truly holds.
1. Ikushima I, Korogi Y, Makita O, et al. MRI of arachnoid granulations within the dural sinuses using a FLAIR pulse sequence. Br J Radiol. 1999;72(863):1046-1051. doi:10.1259/bjr.72.863.10700819
2. Rados M, Zivko M, Perisa A, Oreskovic D, Klarica M. No arachnoid granulations-no problems: number, size, and distribution of arachnoid granulations from birth to 80 years of age. Front Aging Neurosci. 2021;13:698865. doi:10.3389/fnagi.2021.698865
3. Grossman CB, Potts DG. Arachnoid granulations: radiology and anatomy. Radiology. 1974;113(1):95-100. doi:10.1148/113.1.95
4. Wolpow ER, Schaumburg HH. Structure of the human arachnoid granulation. J Neurosurg. 1972;37(6):724-727. doi:10.3171/jns.1972.37.6.0724
5. Leach JL, Jones BV, Tomsick TA, Stewart CA, Balko MG. Normal appearance of arachnoid granulations on contrast-enhanced CT and MR of the brain: differentiation from dural sinus disease. AJNR Am J Neuroradiol. 1996;17(8):1523-1532.
6. Roche J, Warner D. Arachnoid granulations in the transverse and sigmoid sinuses: CT, MR, and MR angiographic appearance of a normal anatomic variation. AJNR Am J Neuroradiol. 1996;17(4):677-683.
7. Malekzadehlashkariani S, Wanke I, Rufenacht DA, San Millan D. Brain herniations into arachnoid granulations: about 68 cases in 38 patients and review of the literature. Neuroradiology. 2016;58(5):443-457. doi:10.1007/s00234-016-1662-5
8. Battal B, Castillo M. Brain herniations into the dural venous sinuses or calvarium: MRI of a recently recognized entity. Neuroradiol J. 2014;27(1):55-62. doi:10.15274/NRJ-2014-10006
9. Liebo GB, Lane JJ, Van Gompel JJ, Eckel LJ, Schwartz KM, Lehman VT. Brain herniation into arachnoid granulations: clinical and neuroimaging features. J Neuroimaging. 2016;26(6):592-598. doi:10.1111/jon.12366
10. Smith ER, Caton MT, Villanueva-Meyer JE, et al. Brain herniation (encephalocele) into arachnoid granulations: Prevalence and association with pulsatile tinnitus and idiopathic intracranial hypertension. Neuroradiology. 2022;64(9):1747-1754.
11. Battal B, Hamcan S, Akgun V, et al. Brain herniations into the dural venous sinus or calvarium: MRI findings, possible causes and clinical significance. Eur Radiol. 2016;26(6):1723-1731.
12. Proulx ST. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell Mol Life Sci. 2021;78(6):2429-2457.
13. Shah T, Leurgans SE, Mehta RI, et al. Arachnoid granulations are lymphatic conduits that communicate with bone marrow and dura-arachnoid stroma. J Exp Med. 2023;220(2).
14. Sade R, Ogul H, Polat G, Pirimoglu B, Kantarci M. Brain herniation into the transverse sinuses’ arachnoid granulations in the pediatric population investigated with 3 T MRI. Acta Neurol Belg. 2019;119(2):225-231.
15. Saavalainen T, Jutila L, Mervaala E, Kalviainen R, Vanninen R, Immonen A. Temporal anteroinferior encephalocele: An underrecognized etiology of temporal lobe epilepsy? Neurology. 2015;85(17):1467-1474.
1. Ikushima I, Korogi Y, Makita O, et al. MRI of arachnoid granulations within the dural sinuses using a FLAIR pulse sequence. Br J Radiol. 1999;72(863):1046-1051. doi:10.1259/bjr.72.863.10700819
2. Rados M, Zivko M, Perisa A, Oreskovic D, Klarica M. No arachnoid granulations-no problems: number, size, and distribution of arachnoid granulations from birth to 80 years of age. Front Aging Neurosci. 2021;13:698865. doi:10.3389/fnagi.2021.698865
3. Grossman CB, Potts DG. Arachnoid granulations: radiology and anatomy. Radiology. 1974;113(1):95-100. doi:10.1148/113.1.95
4. Wolpow ER, Schaumburg HH. Structure of the human arachnoid granulation. J Neurosurg. 1972;37(6):724-727. doi:10.3171/jns.1972.37.6.0724
5. Leach JL, Jones BV, Tomsick TA, Stewart CA, Balko MG. Normal appearance of arachnoid granulations on contrast-enhanced CT and MR of the brain: differentiation from dural sinus disease. AJNR Am J Neuroradiol. 1996;17(8):1523-1532.
6. Roche J, Warner D. Arachnoid granulations in the transverse and sigmoid sinuses: CT, MR, and MR angiographic appearance of a normal anatomic variation. AJNR Am J Neuroradiol. 1996;17(4):677-683.
7. Malekzadehlashkariani S, Wanke I, Rufenacht DA, San Millan D. Brain herniations into arachnoid granulations: about 68 cases in 38 patients and review of the literature. Neuroradiology. 2016;58(5):443-457. doi:10.1007/s00234-016-1662-5
8. Battal B, Castillo M. Brain herniations into the dural venous sinuses or calvarium: MRI of a recently recognized entity. Neuroradiol J. 2014;27(1):55-62. doi:10.15274/NRJ-2014-10006
9. Liebo GB, Lane JJ, Van Gompel JJ, Eckel LJ, Schwartz KM, Lehman VT. Brain herniation into arachnoid granulations: clinical and neuroimaging features. J Neuroimaging. 2016;26(6):592-598. doi:10.1111/jon.12366
10. Smith ER, Caton MT, Villanueva-Meyer JE, et al. Brain herniation (encephalocele) into arachnoid granulations: Prevalence and association with pulsatile tinnitus and idiopathic intracranial hypertension. Neuroradiology. 2022;64(9):1747-1754.
11. Battal B, Hamcan S, Akgun V, et al. Brain herniations into the dural venous sinus or calvarium: MRI findings, possible causes and clinical significance. Eur Radiol. 2016;26(6):1723-1731.
12. Proulx ST. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell Mol Life Sci. 2021;78(6):2429-2457.
13. Shah T, Leurgans SE, Mehta RI, et al. Arachnoid granulations are lymphatic conduits that communicate with bone marrow and dura-arachnoid stroma. J Exp Med. 2023;220(2).
14. Sade R, Ogul H, Polat G, Pirimoglu B, Kantarci M. Brain herniation into the transverse sinuses’ arachnoid granulations in the pediatric population investigated with 3 T MRI. Acta Neurol Belg. 2019;119(2):225-231.
15. Saavalainen T, Jutila L, Mervaala E, Kalviainen R, Vanninen R, Immonen A. Temporal anteroinferior encephalocele: An underrecognized etiology of temporal lobe epilepsy? Neurology. 2015;85(17):1467-1474.
The Challenges of Delivering Allergen Immunotherapy in the Military Health System
Allergic rhinoconjunctivitis causes onerous symptoms of sneezing, rhinorrhea, postnasal drip, nasal congestion, and itchy, watery eyes. It is a common condition that affects 10% to 25% of the US population and up to 23% of military members with increased symptoms during deployments.1-3 Allergen immunotherapy (AIT), commonly known as allergy shots, is an effective treatment for allergic rhinoconjunctivitis, especially for patients whose symptoms are not controlled by allergy medications.4 Many military personnel who would like to receive AIT cannot continue with their immunotherapy because of frequent moves, deployments, and temporary duty assignments. This case report highlights the difficulty of managing AIT in the Military Health System.
Case Presentation
A 34-year-old active-duty US Air Force male surgeon with a medical history of allergic rhinoconjunctivitis was referred to the allergy clinic for evaluation and consideration of AIT. His symptoms included rhinorrhea, sneezing, nasal congestion, and itchy, watery eyes. The symptoms had been present for several years, occurring predominantly in the spring and fall, but also perennially when exposed to animals such as cats, dogs, and horses. The patient was raised on a ranch where he was exposed to these animals.
The patient had prior skin testing at the University of Nebraska Medical Center (UNMC) for aeroallergens and was positive for trees, grasses, weeds, molds, dust mites, cats, dogs, and horses. He received AIT at UNMC with great success for18 months. Regrettably, the patient discontinued AIT following a military move to Keesler Air Force Base in Mississippi. The patient’s examination was notable for injected conjunctiva, nasal mucosa edema, and a cobblestone throat. His symptoms were not alleviated with oral cetirizine and nasal fluticasone.

His skin testing was positive for trees, weeds, mold, cats, dogs, dust mites, and horsehair (Table). The risks and benefits of AIT were discussed with the patient, who elected to proceed with restarting AIT and received counseling on aeroallergen avoidance. The patient was unable to continue AIT at Keesler Medical Center because of a military deployment.
Discussion
There are several barriers to receiving AIT for active-duty patients with allergies. Due to previous skin test extracts, our patient had become desensitized to them. Though he had received aeroallergen immunotherapy with success for 18 months, the patients had to restart the build up phase of AIT due to a military-related move.
For patients to benefit from AIT, they must build up and maintain their immunotherapy injections for at least 3 to 5 years.4 The build-up period of immunotherapy lasts about 3 to 4 months. Patients typically receive weekly injections until they reach a maintenance immunotherapy dose of 0.5 mL of a 1:1 concentration ratio.4
Frequent deployments or temporary duty assignments are other barriers to AIT for active-duty patients. AIT is not usually given on deployments or temporary duty assignments unless the patient is located near a major military medical center. The US Air Force and Army operate allergy extender clinics at smaller bases and overseas locations to facilitate the maintenance of immunotherapy for military patients. Primary care physicians act as allergy extenders. These smaller allergy clinics are supervised by regional allergists at major military medical centers via telehealth and electronic/telephonic communication. These allergy clinics are not more widely available because there are not enough allergists and allergy medical technicians.
Allergen immunotherapy is not standardized, meaning civilian allergists use different aeroallergen immunotherapy formulations. While AIT is standardized in the US military through the Extract Laboratory Management System (ELMS), many active-duty patients are evaluated by civilian allergists in the TRICARE system who do not use ELMS, and when they move, AIT is not maintained.
Because up to 25% of active-duty personnel suffer from allergic rhinoconjunctivitis and AIT is not administered in many deployed settings, this issue could affect mission readiness and capabilities.3-6 These personnel may suffer from frequent and severe nasal and ocular allergy symptoms without being able to continue AIT. There is the potential for adverse effects on the military missions because of these impaired military personnel.5,6
Potential steps to improve the availability of allergen immunotherapy in the deployed setting include training deployed physicians, medical technicians, and other health care practitioners in administering and treating AIT so deployed personnel can receive therapy. Additionally, AIT should be standardized and ordered via the ELMS. Civilian allergists should be highly encouraged to use ELMS. This would create standardization of AIT for all active-duty allergy patients. The allergy extender system could be expanded to all military treatment facilities to provide easy access to allergen immunotherapy. The US Navy has the fewest allergists and allergy extenders, and would need to expand its network of allergy extenders to provide AIT at its health care facilities.
Conclusions
We present an active-duty servicemember with allergic rhinoconjunctivitis to trees, grasses, weeds, cats, dogs, dust mites, mold, and horses who had intermittent therapy that was interrupted by deployments. Our case highlights the difficulty of managing AIT in the military health system due to frequent moves, deployments, and temporary duty assignments. We also suggest steps that could help expand AIT for military personnel, including those deployed internationally.
1. Maciag MC, Phipatanakul W. Update on indoor allergens and their impact on pediatric asthma. Ann Allergy Asthma Immunol. 2022;128(6):652-658. doi:10.1016/j.anai.2022.02.009
2. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351(9111):1225-1232.
3. Roop SA, Niven AS, Calvin BE, Bader J, Zacher LL. The prevalence and impact of respiratory symptoms in asthmatics and nonasthmatics during deployment. Mil Med. 2007;172:1264–1269. doi:10.7205/milmed.172.12.1264
4. Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. [published correction appears in J Allergy Clin Immunol. 2011 Mar;127(3):840]. J Allergy Clin Immunol. 2011;127(1 Suppl):S1-S55. doi:10.1016/j.jaci.2010.09.034
5. Szema AM, Peters MC, Weissinger KM, Gagliano CA, Chen JJ. Increased allergic rhinitis rates among U.S. military personnel after deployment to the Persian Gulf. J Allergy Clin Immunol. 2008;121,S230. doi:10.1016/j.jaci.2007.12.909
6. Garshick E, Abraham JH, Baird CP, Ciminera P, et al. Respiratory ealth after military service in Southwest Asia and Afghanistan. An official American Thoracic Society Workshop report. Ann Am Thorac Soc. 2019;16(8):e1-e16. doi:10.1513/AnnalsATS.201904-344WS
Allergic rhinoconjunctivitis causes onerous symptoms of sneezing, rhinorrhea, postnasal drip, nasal congestion, and itchy, watery eyes. It is a common condition that affects 10% to 25% of the US population and up to 23% of military members with increased symptoms during deployments.1-3 Allergen immunotherapy (AIT), commonly known as allergy shots, is an effective treatment for allergic rhinoconjunctivitis, especially for patients whose symptoms are not controlled by allergy medications.4 Many military personnel who would like to receive AIT cannot continue with their immunotherapy because of frequent moves, deployments, and temporary duty assignments. This case report highlights the difficulty of managing AIT in the Military Health System.
Case Presentation
A 34-year-old active-duty US Air Force male surgeon with a medical history of allergic rhinoconjunctivitis was referred to the allergy clinic for evaluation and consideration of AIT. His symptoms included rhinorrhea, sneezing, nasal congestion, and itchy, watery eyes. The symptoms had been present for several years, occurring predominantly in the spring and fall, but also perennially when exposed to animals such as cats, dogs, and horses. The patient was raised on a ranch where he was exposed to these animals.
The patient had prior skin testing at the University of Nebraska Medical Center (UNMC) for aeroallergens and was positive for trees, grasses, weeds, molds, dust mites, cats, dogs, and horses. He received AIT at UNMC with great success for18 months. Regrettably, the patient discontinued AIT following a military move to Keesler Air Force Base in Mississippi. The patient’s examination was notable for injected conjunctiva, nasal mucosa edema, and a cobblestone throat. His symptoms were not alleviated with oral cetirizine and nasal fluticasone.
His skin testing was positive for trees, weeds, mold, cats, dogs, dust mites, and horsehair (Table). The risks and benefits of AIT were discussed with the patient, who elected to proceed with restarting AIT and received counseling on aeroallergen avoidance. The patient was unable to continue AIT at Keesler Medical Center because of a military deployment.
Discussion
There are several barriers to receiving AIT for active-duty patients with allergies. Due to previous skin test extracts, our patient had become desensitized to them. Though he had received aeroallergen immunotherapy with success for 18 months, the patients had to restart the build up phase of AIT due to a military-related move.
For patients to benefit from AIT, they must build up and maintain their immunotherapy injections for at least 3 to 5 years.4 The build-up period of immunotherapy lasts about 3 to 4 months. Patients typically receive weekly injections until they reach a maintenance immunotherapy dose of 0.5 mL of a 1:1 concentration ratio.4
Frequent deployments or temporary duty assignments are other barriers to AIT for active-duty patients. AIT is not usually given on deployments or temporary duty assignments unless the patient is located near a major military medical center. The US Air Force and Army operate allergy extender clinics at smaller bases and overseas locations to facilitate the maintenance of immunotherapy for military patients. Primary care physicians act as allergy extenders. These smaller allergy clinics are supervised by regional allergists at major military medical centers via telehealth and electronic/telephonic communication. These allergy clinics are not more widely available because there are not enough allergists and allergy medical technicians.
Allergen immunotherapy is not standardized, meaning civilian allergists use different aeroallergen immunotherapy formulations. While AIT is standardized in the US military through the Extract Laboratory Management System (ELMS), many active-duty patients are evaluated by civilian allergists in the TRICARE system who do not use ELMS, and when they move, AIT is not maintained.
Because up to 25% of active-duty personnel suffer from allergic rhinoconjunctivitis and AIT is not administered in many deployed settings, this issue could affect mission readiness and capabilities.3-6 These personnel may suffer from frequent and severe nasal and ocular allergy symptoms without being able to continue AIT. There is the potential for adverse effects on the military missions because of these impaired military personnel.5,6
Potential steps to improve the availability of allergen immunotherapy in the deployed setting include training deployed physicians, medical technicians, and other health care practitioners in administering and treating AIT so deployed personnel can receive therapy. Additionally, AIT should be standardized and ordered via the ELMS. Civilian allergists should be highly encouraged to use ELMS. This would create standardization of AIT for all active-duty allergy patients. The allergy extender system could be expanded to all military treatment facilities to provide easy access to allergen immunotherapy. The US Navy has the fewest allergists and allergy extenders, and would need to expand its network of allergy extenders to provide AIT at its health care facilities.
Conclusions
We present an active-duty servicemember with allergic rhinoconjunctivitis to trees, grasses, weeds, cats, dogs, dust mites, mold, and horses who had intermittent therapy that was interrupted by deployments. Our case highlights the difficulty of managing AIT in the military health system due to frequent moves, deployments, and temporary duty assignments. We also suggest steps that could help expand AIT for military personnel, including those deployed internationally.
Allergic rhinoconjunctivitis causes onerous symptoms of sneezing, rhinorrhea, postnasal drip, nasal congestion, and itchy, watery eyes. It is a common condition that affects 10% to 25% of the US population and up to 23% of military members with increased symptoms during deployments.1-3 Allergen immunotherapy (AIT), commonly known as allergy shots, is an effective treatment for allergic rhinoconjunctivitis, especially for patients whose symptoms are not controlled by allergy medications.4 Many military personnel who would like to receive AIT cannot continue with their immunotherapy because of frequent moves, deployments, and temporary duty assignments. This case report highlights the difficulty of managing AIT in the Military Health System.
Case Presentation
A 34-year-old active-duty US Air Force male surgeon with a medical history of allergic rhinoconjunctivitis was referred to the allergy clinic for evaluation and consideration of AIT. His symptoms included rhinorrhea, sneezing, nasal congestion, and itchy, watery eyes. The symptoms had been present for several years, occurring predominantly in the spring and fall, but also perennially when exposed to animals such as cats, dogs, and horses. The patient was raised on a ranch where he was exposed to these animals.
The patient had prior skin testing at the University of Nebraska Medical Center (UNMC) for aeroallergens and was positive for trees, grasses, weeds, molds, dust mites, cats, dogs, and horses. He received AIT at UNMC with great success for18 months. Regrettably, the patient discontinued AIT following a military move to Keesler Air Force Base in Mississippi. The patient’s examination was notable for injected conjunctiva, nasal mucosa edema, and a cobblestone throat. His symptoms were not alleviated with oral cetirizine and nasal fluticasone.
His skin testing was positive for trees, weeds, mold, cats, dogs, dust mites, and horsehair (Table). The risks and benefits of AIT were discussed with the patient, who elected to proceed with restarting AIT and received counseling on aeroallergen avoidance. The patient was unable to continue AIT at Keesler Medical Center because of a military deployment.
Discussion
There are several barriers to receiving AIT for active-duty patients with allergies. Due to previous skin test extracts, our patient had become desensitized to them. Though he had received aeroallergen immunotherapy with success for 18 months, the patients had to restart the build up phase of AIT due to a military-related move.
For patients to benefit from AIT, they must build up and maintain their immunotherapy injections for at least 3 to 5 years.4 The build-up period of immunotherapy lasts about 3 to 4 months. Patients typically receive weekly injections until they reach a maintenance immunotherapy dose of 0.5 mL of a 1:1 concentration ratio.4
Frequent deployments or temporary duty assignments are other barriers to AIT for active-duty patients. AIT is not usually given on deployments or temporary duty assignments unless the patient is located near a major military medical center. The US Air Force and Army operate allergy extender clinics at smaller bases and overseas locations to facilitate the maintenance of immunotherapy for military patients. Primary care physicians act as allergy extenders. These smaller allergy clinics are supervised by regional allergists at major military medical centers via telehealth and electronic/telephonic communication. These allergy clinics are not more widely available because there are not enough allergists and allergy medical technicians.
Allergen immunotherapy is not standardized, meaning civilian allergists use different aeroallergen immunotherapy formulations. While AIT is standardized in the US military through the Extract Laboratory Management System (ELMS), many active-duty patients are evaluated by civilian allergists in the TRICARE system who do not use ELMS, and when they move, AIT is not maintained.
Because up to 25% of active-duty personnel suffer from allergic rhinoconjunctivitis and AIT is not administered in many deployed settings, this issue could affect mission readiness and capabilities.3-6 These personnel may suffer from frequent and severe nasal and ocular allergy symptoms without being able to continue AIT. There is the potential for adverse effects on the military missions because of these impaired military personnel.5,6
Potential steps to improve the availability of allergen immunotherapy in the deployed setting include training deployed physicians, medical technicians, and other health care practitioners in administering and treating AIT so deployed personnel can receive therapy. Additionally, AIT should be standardized and ordered via the ELMS. Civilian allergists should be highly encouraged to use ELMS. This would create standardization of AIT for all active-duty allergy patients. The allergy extender system could be expanded to all military treatment facilities to provide easy access to allergen immunotherapy. The US Navy has the fewest allergists and allergy extenders, and would need to expand its network of allergy extenders to provide AIT at its health care facilities.
Conclusions
We present an active-duty servicemember with allergic rhinoconjunctivitis to trees, grasses, weeds, cats, dogs, dust mites, mold, and horses who had intermittent therapy that was interrupted by deployments. Our case highlights the difficulty of managing AIT in the military health system due to frequent moves, deployments, and temporary duty assignments. We also suggest steps that could help expand AIT for military personnel, including those deployed internationally.
1. Maciag MC, Phipatanakul W. Update on indoor allergens and their impact on pediatric asthma. Ann Allergy Asthma Immunol. 2022;128(6):652-658. doi:10.1016/j.anai.2022.02.009
2. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351(9111):1225-1232.
3. Roop SA, Niven AS, Calvin BE, Bader J, Zacher LL. The prevalence and impact of respiratory symptoms in asthmatics and nonasthmatics during deployment. Mil Med. 2007;172:1264–1269. doi:10.7205/milmed.172.12.1264
4. Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. [published correction appears in J Allergy Clin Immunol. 2011 Mar;127(3):840]. J Allergy Clin Immunol. 2011;127(1 Suppl):S1-S55. doi:10.1016/j.jaci.2010.09.034
5. Szema AM, Peters MC, Weissinger KM, Gagliano CA, Chen JJ. Increased allergic rhinitis rates among U.S. military personnel after deployment to the Persian Gulf. J Allergy Clin Immunol. 2008;121,S230. doi:10.1016/j.jaci.2007.12.909
6. Garshick E, Abraham JH, Baird CP, Ciminera P, et al. Respiratory ealth after military service in Southwest Asia and Afghanistan. An official American Thoracic Society Workshop report. Ann Am Thorac Soc. 2019;16(8):e1-e16. doi:10.1513/AnnalsATS.201904-344WS
1. Maciag MC, Phipatanakul W. Update on indoor allergens and their impact on pediatric asthma. Ann Allergy Asthma Immunol. 2022;128(6):652-658. doi:10.1016/j.anai.2022.02.009
2. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet. 1998;351(9111):1225-1232.
3. Roop SA, Niven AS, Calvin BE, Bader J, Zacher LL. The prevalence and impact of respiratory symptoms in asthmatics and nonasthmatics during deployment. Mil Med. 2007;172:1264–1269. doi:10.7205/milmed.172.12.1264
4. Cox L, Nelson H, Lockey R, et al. Allergen immunotherapy: a practice parameter third update. [published correction appears in J Allergy Clin Immunol. 2011 Mar;127(3):840]. J Allergy Clin Immunol. 2011;127(1 Suppl):S1-S55. doi:10.1016/j.jaci.2010.09.034
5. Szema AM, Peters MC, Weissinger KM, Gagliano CA, Chen JJ. Increased allergic rhinitis rates among U.S. military personnel after deployment to the Persian Gulf. J Allergy Clin Immunol. 2008;121,S230. doi:10.1016/j.jaci.2007.12.909
6. Garshick E, Abraham JH, Baird CP, Ciminera P, et al. Respiratory ealth after military service in Southwest Asia and Afghanistan. An official American Thoracic Society Workshop report. Ann Am Thorac Soc. 2019;16(8):e1-e16. doi:10.1513/AnnalsATS.201904-344WS
The Clinical Utility of Teledermatology in Triaging and Diagnosing Skin Malignancies: Case Series
With the increasing utilization of telemedicine since the COVID-19 pandemic, it is critical that clinicians have an appropriate understanding of the application of virtual care resources, including teledermatology. We present a case series of 3 patients to demonstrate the clinical utility of teledermatology in reducing the time to diagnosis of various rare and/or aggressive cutaneous malignancies, including Merkel cell carcinoma, malignant melanoma, and atypical fibroxanthoma. Cases were obtained from one large Midwestern medical center during the month of July 2021. Each case presented includes a description of the initial teledermatology presentation and reviews the clinical timeline from initial consultation submission to in-person clinic visit with lesion biopsy. This case series demonstrates real-world examples of how teledermatology can be utilized to expedite the care of specific vulnerable patient populations.
Teledermatology is a rapidly growing digital resource with specific utility in triaging patients to determine those requiring in-person evaluation for early and accurate detection of skin malignancies. Approximately one-third of teledermatology consultations result in face-to-face clinical encounters, with malignant neoplasms being the leading cause for biopsy.1,2 For specific populations, such as geriatric and immunocompromised patients, teledermatology may serve as a valuable tool, particularly in the wake of the COVID-19 pandemic. Furthermore, telemedicine may aid in addressing health disparities within the field of medicine and ultimately may improve access to care for vulnerable populations.3 Along with increasing access to specific subspecialty expertise, the use of teledermatology may reduce health care costs and improve the overall quality of care delivered to patients.4,5
We describe the clinical utility of teledermatology in triaging and diagnosing skin malignancies through a series of 3 cases obtained from digital image review at one large Midwestern medical center during the month of July 2021. Three unique cases with a final diagnosis of a rare or aggressive skin cancer were selected as examples, including a 75-year-old man with Merkle cell carcinoma, a 55-year-old man with aggressive pT3b malignant melanoma, and a 72-year-old man with an atypical fibroxanthoma. A clinical timeline of each case is presented, including the time intervals from initial image submission to image review, image submission to face-to-face clinical encounter, and image submission to final diagnosis. In all cases, the primary care provider submitted an order for teledermatology, and the teledermatology team obtained the images.
Case Series
Patient 1—Images of the right hand of a 75-year-old man with a medical history of basal cell carcinoma were submitted for teledermatology consultation utilizing store-and-forward image-capturing technology (day 1). The patient history provided with image submission indicated that the lesion had been present for 6 months and there were no associated symptoms. Clinical imaging demonstrated a pink-red pearly papule located on the proximal fourth digit of the dorsal aspect of the right hand (Figure 1). One day following the teledermatology request (day 2), the patient’s case was reviewed and triaged for an in-person visit. The patient was brought to clinic on day 34, and a biopsy was performed. On day 36, dermatopathology results indicated a diagnosis of Merkel cell carcinoma. On day 37, the patient was referred to surgical oncology, and on day 44, the patient underwent an initial surgical oncology visit with a plan for wide local excision of the right fourth digit with right axillary sentinel lymph node biopsy.
.")
Patient 2—Images of the left flank of a 55-year-old man were submitted for teledermatology consultation via store-and-forward technology (day 1). A patient history provided with the image indicated that the lesion had been present for months to years and there were no associated symptoms, but the lesion recently had changed in color and size. Teledermatology images were reviewed on day 3 and demonstrated a 2- to 3-cm brown plaque on the left flank with color variegation and a prominent red papule protruding centrally (Figure 2). The patient was scheduled for an urgent in-person visit with biopsy. On day 6, the patient presented to clinic and an excision biopsy was performed. Dermatopathology was ordered with a RUSH indication, with results on day 7 revealing a pT3b malignant melanoma. An urgent consultation to surgical oncology was placed on the same day, and the patient underwent an initial surgical oncology visit on day 24 with a plan for wide local excision with left axillary and inguinal sentinel lymph node biopsy.
.")
Patient 3—Images of the left ear of a 72-year-old man were submitted for teledermatology consultation utilizing review via store-and-forward technology (day 1). A patient history indicated that the lesion had been present for 3 months with associated bleeding. Image review demonstrated a solitary pearly pink papule located on the crura of the antihelix (Figure 3). Initial teledermatology consultation was reviewed on day 2 with notification of the need for in-person evaluation. The patient presented to clinic on day 33 for a biopsy, with dermatopathology results on day 36 consistent with an atypical fibroxanthoma. The patient was scheduled for Mohs micrographic surgery on day 37 and underwent surgical treatment on day 64.
.")
Comment
Teledermatology consultations from all patients demonstrated adequate image quality to be able to evaluate the lesion of concern and yielded a request for in-person evaluation with possible biopsy (Table). In this case series, the average time interval from teledermatology consultation placement to teledermatology image report was 2 days (range, 1–3 days). The average time from teledermatology consultation placement to face-to-face encounter with biopsy was 24.3 days for the 3 cases presented in this series (range, 6–34 days). The initial surgical oncology visits took place an average of 34 days after the initial teledermatology consultation was placed for the 2 patients requiring referral (44 days for patient 1; 24 days for patient 2). For patient 3, Mohs micrographic surgery was required for treatment, which was scheduled by day 37 and subsequently performed on day 64.

When specifically looking at the diagnosis of cutaneous malignancies, studies have found that the incidence of skin cancer detection is similar for teledermatology compared to in-person clinic visits.6,7 Creighton-Smith et al6 performed a retrospective cohort study comparing prebiopsy and postbiopsy diagnostic accuracy and detection rates of skin cancer between store-and-forward technology and face-to-face consultation. When adjusting for possible compounding factors including personal and family history of skin cancer, there was no notable difference in detection rates of any skin cancer, including melanoma and nonmelanoma skin cancers. Furthermore, the 2 cohorts of patients were found to have similar prebiopsy and postbiopsy diagnostic concordance, with similar times from consultation being placed to requested biopsy and time from biopsy to final treatment.6
Clarke et al7 similarly analyzed the accuracy of store-and-forward teledermatology and found that there was overall concordance in diagnosis when comparing clinical dermatologists to teledermatologists. Moreover, when melanocytic lesions were excluded from the study, the decision to biopsy did not differ substantially.7
Areas of further study include determining what percentage of teledermatology lesions of concern for malignancy were proven to be skin cancer after in-person evaluation and biopsy, as well as investigating the effectiveness of teledermatology for melanocytic lesions, which frequently are removed from analysis in large-scale teledermatology studies.
Although teledermatology has substantial clinical utility and may serve as a great resource for specific populations, including geriatric patients and those who are immunocompromised, it is important to recognize notable limitations. Specifically, brief history and image review should not serve as replacements for a face-to-face visit with physical examination in cases where the diagnosis remains uncertain or when high-risk skin malignancies are suspected or included in the differential. Certain aggressive cutaneous malignancies such as Merkel cell carcinoma may appear as less aggressive via teledermatology due to restrictions of technology.
Conclusion
Teledermatology has had a major impact on the way health care is delivered to patients and may increase access to care, reducing unnecessary in-person visits and decreasing the number of in-person visit no-shows. With the appropriate use of a brief clinical history and image review, teledermatology can be effective to evaluate specific lesions of concern. We report 3 unique cases identified during a 1-month period at a large Midwestern medical center. These cases serve as important examples of the application of teledermatology in reducing the time to diagnosis of aggressive skin malignancies. Further research on the clinical utility of teledermatology is warranted.
Acknowledgments—The authors thank the additional providers from the University of Wisconsin and William S. Middleton Memorial Veterans Hospital (both in Madison, Wisconsin) involved in the medical care of the patients included in this case series.
- Bianchi MG, Santos A, Cordioli E. Benefits of teledermatology for geriatric patients: population-based cross-sectional study. J Med Internet Res. 2020;22:E16700.
- Mortimer S, Rosin A. A retrospective review of incidental malignancies in veterans seen for face-to-face follow-up after teledermatology consultation. J Am Acad Dermatol. 2021;84:1130-1132.
- Costello CM, Cumsky HJL, Maly CJ, et al. Improving access to care through the establishment of a local, teledermatology network. Telemed J E Health. 2020;26:935-940. doi:10.1089/tmj.2019.0051
- Lee JJ, English JC 3rd. Teledermatology: a review and update. Am J Clin Dermatol. 2018;19:253-260. doi:10.1007/s40257-017-0317-6
- Hadeler E, Beer J, Nouri K. The influence of teledermatology on health care access and equity. J Am Acad Dermatol. 2021;84:E219-E220. doi:10.1016/j.jaad.2020.12.036
- Creighton-Smith M, Murgia RD 3rd, Konnikov N, et al. Incidence of melanoma and keratinocytic carcinomas in patients evaluated by store-and-forward teledermatology vs dermatology clinic. Int J Dermatol. 2017;56:1026-1031. doi:10.1111/ijd.13672
- Clarke EL, Reichenberg JS, Ahmed AM, et al. The utility of teledermatology in the evaluation of skin lesions. J Telemed Telecare. 2023;29:382-389. doi:10.1177/1357633X20987423
With the increasing utilization of telemedicine since the COVID-19 pandemic, it is critical that clinicians have an appropriate understanding of the application of virtual care resources, including teledermatology. We present a case series of 3 patients to demonstrate the clinical utility of teledermatology in reducing the time to diagnosis of various rare and/or aggressive cutaneous malignancies, including Merkel cell carcinoma, malignant melanoma, and atypical fibroxanthoma. Cases were obtained from one large Midwestern medical center during the month of July 2021. Each case presented includes a description of the initial teledermatology presentation and reviews the clinical timeline from initial consultation submission to in-person clinic visit with lesion biopsy. This case series demonstrates real-world examples of how teledermatology can be utilized to expedite the care of specific vulnerable patient populations.
Teledermatology is a rapidly growing digital resource with specific utility in triaging patients to determine those requiring in-person evaluation for early and accurate detection of skin malignancies. Approximately one-third of teledermatology consultations result in face-to-face clinical encounters, with malignant neoplasms being the leading cause for biopsy.1,2 For specific populations, such as geriatric and immunocompromised patients, teledermatology may serve as a valuable tool, particularly in the wake of the COVID-19 pandemic. Furthermore, telemedicine may aid in addressing health disparities within the field of medicine and ultimately may improve access to care for vulnerable populations.3 Along with increasing access to specific subspecialty expertise, the use of teledermatology may reduce health care costs and improve the overall quality of care delivered to patients.4,5
We describe the clinical utility of teledermatology in triaging and diagnosing skin malignancies through a series of 3 cases obtained from digital image review at one large Midwestern medical center during the month of July 2021. Three unique cases with a final diagnosis of a rare or aggressive skin cancer were selected as examples, including a 75-year-old man with Merkle cell carcinoma, a 55-year-old man with aggressive pT3b malignant melanoma, and a 72-year-old man with an atypical fibroxanthoma. A clinical timeline of each case is presented, including the time intervals from initial image submission to image review, image submission to face-to-face clinical encounter, and image submission to final diagnosis. In all cases, the primary care provider submitted an order for teledermatology, and the teledermatology team obtained the images.
Case Series
Patient 1—Images of the right hand of a 75-year-old man with a medical history of basal cell carcinoma were submitted for teledermatology consultation utilizing store-and-forward image-capturing technology (day 1). The patient history provided with image submission indicated that the lesion had been present for 6 months and there were no associated symptoms. Clinical imaging demonstrated a pink-red pearly papule located on the proximal fourth digit of the dorsal aspect of the right hand (Figure 1). One day following the teledermatology request (day 2), the patient’s case was reviewed and triaged for an in-person visit. The patient was brought to clinic on day 34, and a biopsy was performed. On day 36, dermatopathology results indicated a diagnosis of Merkel cell carcinoma. On day 37, the patient was referred to surgical oncology, and on day 44, the patient underwent an initial surgical oncology visit with a plan for wide local excision of the right fourth digit with right axillary sentinel lymph node biopsy.
Patient 2—Images of the left flank of a 55-year-old man were submitted for teledermatology consultation via store-and-forward technology (day 1). A patient history provided with the image indicated that the lesion had been present for months to years and there were no associated symptoms, but the lesion recently had changed in color and size. Teledermatology images were reviewed on day 3 and demonstrated a 2- to 3-cm brown plaque on the left flank with color variegation and a prominent red papule protruding centrally (Figure 2). The patient was scheduled for an urgent in-person visit with biopsy. On day 6, the patient presented to clinic and an excision biopsy was performed. Dermatopathology was ordered with a RUSH indication, with results on day 7 revealing a pT3b malignant melanoma. An urgent consultation to surgical oncology was placed on the same day, and the patient underwent an initial surgical oncology visit on day 24 with a plan for wide local excision with left axillary and inguinal sentinel lymph node biopsy.
Patient 3—Images of the left ear of a 72-year-old man were submitted for teledermatology consultation utilizing review via store-and-forward technology (day 1). A patient history indicated that the lesion had been present for 3 months with associated bleeding. Image review demonstrated a solitary pearly pink papule located on the crura of the antihelix (Figure 3). Initial teledermatology consultation was reviewed on day 2 with notification of the need for in-person evaluation. The patient presented to clinic on day 33 for a biopsy, with dermatopathology results on day 36 consistent with an atypical fibroxanthoma. The patient was scheduled for Mohs micrographic surgery on day 37 and underwent surgical treatment on day 64.
Comment
Teledermatology consultations from all patients demonstrated adequate image quality to be able to evaluate the lesion of concern and yielded a request for in-person evaluation with possible biopsy (Table). In this case series, the average time interval from teledermatology consultation placement to teledermatology image report was 2 days (range, 1–3 days). The average time from teledermatology consultation placement to face-to-face encounter with biopsy was 24.3 days for the 3 cases presented in this series (range, 6–34 days). The initial surgical oncology visits took place an average of 34 days after the initial teledermatology consultation was placed for the 2 patients requiring referral (44 days for patient 1; 24 days for patient 2). For patient 3, Mohs micrographic surgery was required for treatment, which was scheduled by day 37 and subsequently performed on day 64.
When specifically looking at the diagnosis of cutaneous malignancies, studies have found that the incidence of skin cancer detection is similar for teledermatology compared to in-person clinic visits.6,7 Creighton-Smith et al6 performed a retrospective cohort study comparing prebiopsy and postbiopsy diagnostic accuracy and detection rates of skin cancer between store-and-forward technology and face-to-face consultation. When adjusting for possible compounding factors including personal and family history of skin cancer, there was no notable difference in detection rates of any skin cancer, including melanoma and nonmelanoma skin cancers. Furthermore, the 2 cohorts of patients were found to have similar prebiopsy and postbiopsy diagnostic concordance, with similar times from consultation being placed to requested biopsy and time from biopsy to final treatment.6
Clarke et al7 similarly analyzed the accuracy of store-and-forward teledermatology and found that there was overall concordance in diagnosis when comparing clinical dermatologists to teledermatologists. Moreover, when melanocytic lesions were excluded from the study, the decision to biopsy did not differ substantially.7
Areas of further study include determining what percentage of teledermatology lesions of concern for malignancy were proven to be skin cancer after in-person evaluation and biopsy, as well as investigating the effectiveness of teledermatology for melanocytic lesions, which frequently are removed from analysis in large-scale teledermatology studies.
Although teledermatology has substantial clinical utility and may serve as a great resource for specific populations, including geriatric patients and those who are immunocompromised, it is important to recognize notable limitations. Specifically, brief history and image review should not serve as replacements for a face-to-face visit with physical examination in cases where the diagnosis remains uncertain or when high-risk skin malignancies are suspected or included in the differential. Certain aggressive cutaneous malignancies such as Merkel cell carcinoma may appear as less aggressive via teledermatology due to restrictions of technology.
Conclusion
Teledermatology has had a major impact on the way health care is delivered to patients and may increase access to care, reducing unnecessary in-person visits and decreasing the number of in-person visit no-shows. With the appropriate use of a brief clinical history and image review, teledermatology can be effective to evaluate specific lesions of concern. We report 3 unique cases identified during a 1-month period at a large Midwestern medical center. These cases serve as important examples of the application of teledermatology in reducing the time to diagnosis of aggressive skin malignancies. Further research on the clinical utility of teledermatology is warranted.
Acknowledgments—The authors thank the additional providers from the University of Wisconsin and William S. Middleton Memorial Veterans Hospital (both in Madison, Wisconsin) involved in the medical care of the patients included in this case series.
With the increasing utilization of telemedicine since the COVID-19 pandemic, it is critical that clinicians have an appropriate understanding of the application of virtual care resources, including teledermatology. We present a case series of 3 patients to demonstrate the clinical utility of teledermatology in reducing the time to diagnosis of various rare and/or aggressive cutaneous malignancies, including Merkel cell carcinoma, malignant melanoma, and atypical fibroxanthoma. Cases were obtained from one large Midwestern medical center during the month of July 2021. Each case presented includes a description of the initial teledermatology presentation and reviews the clinical timeline from initial consultation submission to in-person clinic visit with lesion biopsy. This case series demonstrates real-world examples of how teledermatology can be utilized to expedite the care of specific vulnerable patient populations.
Teledermatology is a rapidly growing digital resource with specific utility in triaging patients to determine those requiring in-person evaluation for early and accurate detection of skin malignancies. Approximately one-third of teledermatology consultations result in face-to-face clinical encounters, with malignant neoplasms being the leading cause for biopsy.1,2 For specific populations, such as geriatric and immunocompromised patients, teledermatology may serve as a valuable tool, particularly in the wake of the COVID-19 pandemic. Furthermore, telemedicine may aid in addressing health disparities within the field of medicine and ultimately may improve access to care for vulnerable populations.3 Along with increasing access to specific subspecialty expertise, the use of teledermatology may reduce health care costs and improve the overall quality of care delivered to patients.4,5
We describe the clinical utility of teledermatology in triaging and diagnosing skin malignancies through a series of 3 cases obtained from digital image review at one large Midwestern medical center during the month of July 2021. Three unique cases with a final diagnosis of a rare or aggressive skin cancer were selected as examples, including a 75-year-old man with Merkle cell carcinoma, a 55-year-old man with aggressive pT3b malignant melanoma, and a 72-year-old man with an atypical fibroxanthoma. A clinical timeline of each case is presented, including the time intervals from initial image submission to image review, image submission to face-to-face clinical encounter, and image submission to final diagnosis. In all cases, the primary care provider submitted an order for teledermatology, and the teledermatology team obtained the images.
Case Series
Patient 1—Images of the right hand of a 75-year-old man with a medical history of basal cell carcinoma were submitted for teledermatology consultation utilizing store-and-forward image-capturing technology (day 1). The patient history provided with image submission indicated that the lesion had been present for 6 months and there were no associated symptoms. Clinical imaging demonstrated a pink-red pearly papule located on the proximal fourth digit of the dorsal aspect of the right hand (Figure 1). One day following the teledermatology request (day 2), the patient’s case was reviewed and triaged for an in-person visit. The patient was brought to clinic on day 34, and a biopsy was performed. On day 36, dermatopathology results indicated a diagnosis of Merkel cell carcinoma. On day 37, the patient was referred to surgical oncology, and on day 44, the patient underwent an initial surgical oncology visit with a plan for wide local excision of the right fourth digit with right axillary sentinel lymph node biopsy.
Patient 2—Images of the left flank of a 55-year-old man were submitted for teledermatology consultation via store-and-forward technology (day 1). A patient history provided with the image indicated that the lesion had been present for months to years and there were no associated symptoms, but the lesion recently had changed in color and size. Teledermatology images were reviewed on day 3 and demonstrated a 2- to 3-cm brown plaque on the left flank with color variegation and a prominent red papule protruding centrally (Figure 2). The patient was scheduled for an urgent in-person visit with biopsy. On day 6, the patient presented to clinic and an excision biopsy was performed. Dermatopathology was ordered with a RUSH indication, with results on day 7 revealing a pT3b malignant melanoma. An urgent consultation to surgical oncology was placed on the same day, and the patient underwent an initial surgical oncology visit on day 24 with a plan for wide local excision with left axillary and inguinal sentinel lymph node biopsy.
Patient 3—Images of the left ear of a 72-year-old man were submitted for teledermatology consultation utilizing review via store-and-forward technology (day 1). A patient history indicated that the lesion had been present for 3 months with associated bleeding. Image review demonstrated a solitary pearly pink papule located on the crura of the antihelix (Figure 3). Initial teledermatology consultation was reviewed on day 2 with notification of the need for in-person evaluation. The patient presented to clinic on day 33 for a biopsy, with dermatopathology results on day 36 consistent with an atypical fibroxanthoma. The patient was scheduled for Mohs micrographic surgery on day 37 and underwent surgical treatment on day 64.
Comment
Teledermatology consultations from all patients demonstrated adequate image quality to be able to evaluate the lesion of concern and yielded a request for in-person evaluation with possible biopsy (Table). In this case series, the average time interval from teledermatology consultation placement to teledermatology image report was 2 days (range, 1–3 days). The average time from teledermatology consultation placement to face-to-face encounter with biopsy was 24.3 days for the 3 cases presented in this series (range, 6–34 days). The initial surgical oncology visits took place an average of 34 days after the initial teledermatology consultation was placed for the 2 patients requiring referral (44 days for patient 1; 24 days for patient 2). For patient 3, Mohs micrographic surgery was required for treatment, which was scheduled by day 37 and subsequently performed on day 64.
When specifically looking at the diagnosis of cutaneous malignancies, studies have found that the incidence of skin cancer detection is similar for teledermatology compared to in-person clinic visits.6,7 Creighton-Smith et al6 performed a retrospective cohort study comparing prebiopsy and postbiopsy diagnostic accuracy and detection rates of skin cancer between store-and-forward technology and face-to-face consultation. When adjusting for possible compounding factors including personal and family history of skin cancer, there was no notable difference in detection rates of any skin cancer, including melanoma and nonmelanoma skin cancers. Furthermore, the 2 cohorts of patients were found to have similar prebiopsy and postbiopsy diagnostic concordance, with similar times from consultation being placed to requested biopsy and time from biopsy to final treatment.6
Clarke et al7 similarly analyzed the accuracy of store-and-forward teledermatology and found that there was overall concordance in diagnosis when comparing clinical dermatologists to teledermatologists. Moreover, when melanocytic lesions were excluded from the study, the decision to biopsy did not differ substantially.7
Areas of further study include determining what percentage of teledermatology lesions of concern for malignancy were proven to be skin cancer after in-person evaluation and biopsy, as well as investigating the effectiveness of teledermatology for melanocytic lesions, which frequently are removed from analysis in large-scale teledermatology studies.
Although teledermatology has substantial clinical utility and may serve as a great resource for specific populations, including geriatric patients and those who are immunocompromised, it is important to recognize notable limitations. Specifically, brief history and image review should not serve as replacements for a face-to-face visit with physical examination in cases where the diagnosis remains uncertain or when high-risk skin malignancies are suspected or included in the differential. Certain aggressive cutaneous malignancies such as Merkel cell carcinoma may appear as less aggressive via teledermatology due to restrictions of technology.
Conclusion
Teledermatology has had a major impact on the way health care is delivered to patients and may increase access to care, reducing unnecessary in-person visits and decreasing the number of in-person visit no-shows. With the appropriate use of a brief clinical history and image review, teledermatology can be effective to evaluate specific lesions of concern. We report 3 unique cases identified during a 1-month period at a large Midwestern medical center. These cases serve as important examples of the application of teledermatology in reducing the time to diagnosis of aggressive skin malignancies. Further research on the clinical utility of teledermatology is warranted.
Acknowledgments—The authors thank the additional providers from the University of Wisconsin and William S. Middleton Memorial Veterans Hospital (both in Madison, Wisconsin) involved in the medical care of the patients included in this case series.
- Bianchi MG, Santos A, Cordioli E. Benefits of teledermatology for geriatric patients: population-based cross-sectional study. J Med Internet Res. 2020;22:E16700.
- Mortimer S, Rosin A. A retrospective review of incidental malignancies in veterans seen for face-to-face follow-up after teledermatology consultation. J Am Acad Dermatol. 2021;84:1130-1132.
- Costello CM, Cumsky HJL, Maly CJ, et al. Improving access to care through the establishment of a local, teledermatology network. Telemed J E Health. 2020;26:935-940. doi:10.1089/tmj.2019.0051
- Lee JJ, English JC 3rd. Teledermatology: a review and update. Am J Clin Dermatol. 2018;19:253-260. doi:10.1007/s40257-017-0317-6
- Hadeler E, Beer J, Nouri K. The influence of teledermatology on health care access and equity. J Am Acad Dermatol. 2021;84:E219-E220. doi:10.1016/j.jaad.2020.12.036
- Creighton-Smith M, Murgia RD 3rd, Konnikov N, et al. Incidence of melanoma and keratinocytic carcinomas in patients evaluated by store-and-forward teledermatology vs dermatology clinic. Int J Dermatol. 2017;56:1026-1031. doi:10.1111/ijd.13672
- Clarke EL, Reichenberg JS, Ahmed AM, et al. The utility of teledermatology in the evaluation of skin lesions. J Telemed Telecare. 2023;29:382-389. doi:10.1177/1357633X20987423
- Bianchi MG, Santos A, Cordioli E. Benefits of teledermatology for geriatric patients: population-based cross-sectional study. J Med Internet Res. 2020;22:E16700.
- Mortimer S, Rosin A. A retrospective review of incidental malignancies in veterans seen for face-to-face follow-up after teledermatology consultation. J Am Acad Dermatol. 2021;84:1130-1132.
- Costello CM, Cumsky HJL, Maly CJ, et al. Improving access to care through the establishment of a local, teledermatology network. Telemed J E Health. 2020;26:935-940. doi:10.1089/tmj.2019.0051
- Lee JJ, English JC 3rd. Teledermatology: a review and update. Am J Clin Dermatol. 2018;19:253-260. doi:10.1007/s40257-017-0317-6
- Hadeler E, Beer J, Nouri K. The influence of teledermatology on health care access and equity. J Am Acad Dermatol. 2021;84:E219-E220. doi:10.1016/j.jaad.2020.12.036
- Creighton-Smith M, Murgia RD 3rd, Konnikov N, et al. Incidence of melanoma and keratinocytic carcinomas in patients evaluated by store-and-forward teledermatology vs dermatology clinic. Int J Dermatol. 2017;56:1026-1031. doi:10.1111/ijd.13672
- Clarke EL, Reichenberg JS, Ahmed AM, et al. The utility of teledermatology in the evaluation of skin lesions. J Telemed Telecare. 2023;29:382-389. doi:10.1177/1357633X20987423
Practice Points
- Teledermatology via store-and-forward technology has been demonstrated to be effective in assessing and triaging various cutaneous malignancies.
- The use of teledermatology has increased because of the COVID-19 pandemic and may be useful for specific vulnerable populations.
- When used appropriately, teledermatology may function as a useful resource to triage patients requiring in-person evaluation for the diagnosis of aggressive skin malignancies and may aid in reducing the time to diagnosis of various skin cancers.