User login
Bringing you the latest case reports, original research, clinical trial reviews, perspectives, patient resources, and more.
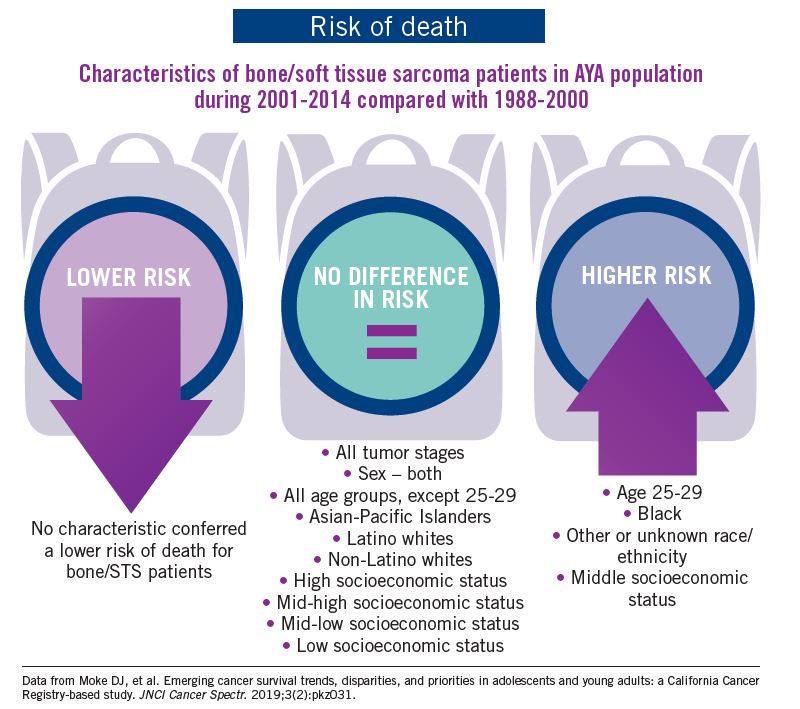
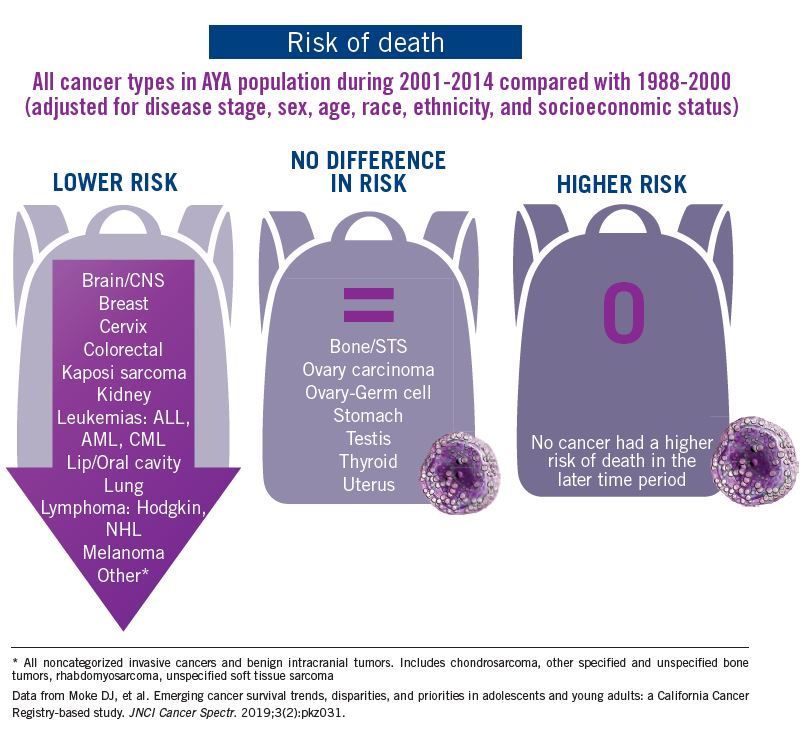
Adolescent and young adult (AYA) survival trends
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
Conference Coverage: ASCO 2019
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
SFA awards grants to 15 researchers
Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant

Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant
Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant
Significant clinical response induced by vismodegib in advanced sarcoma: Hedgehog pathway inhibition
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
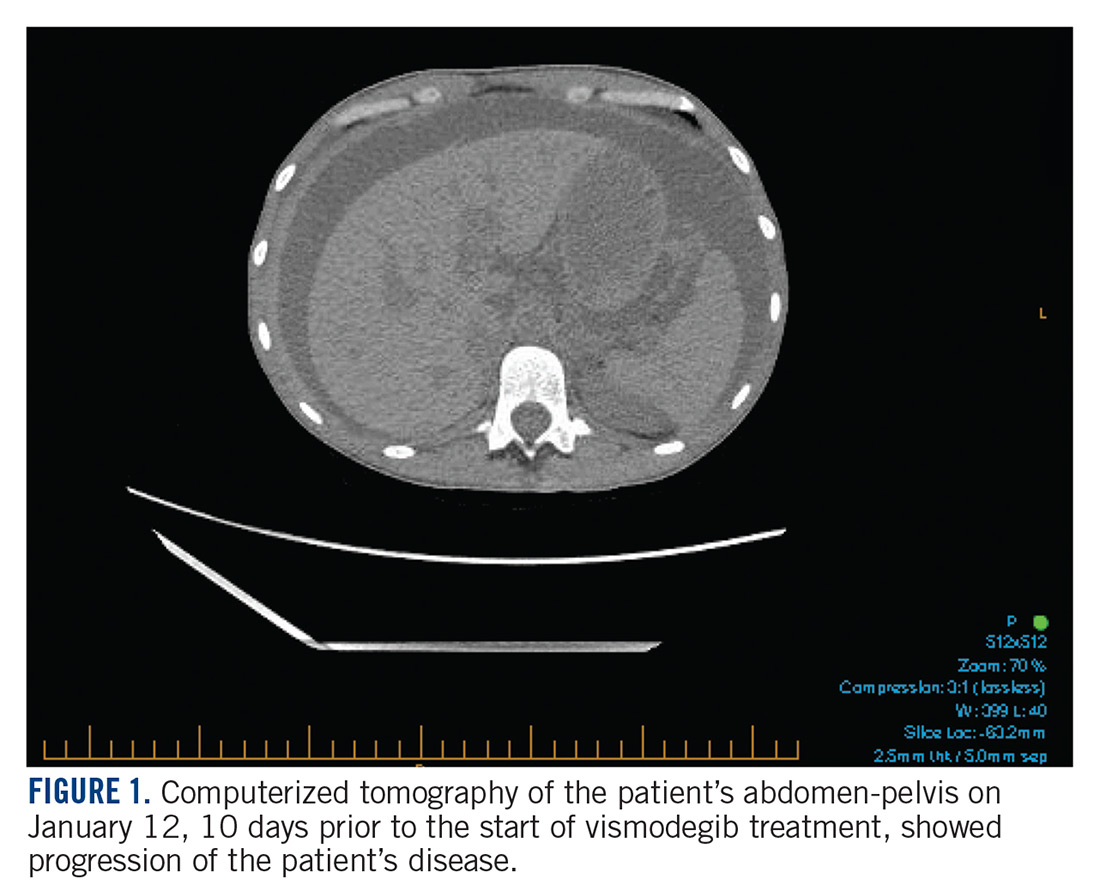
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
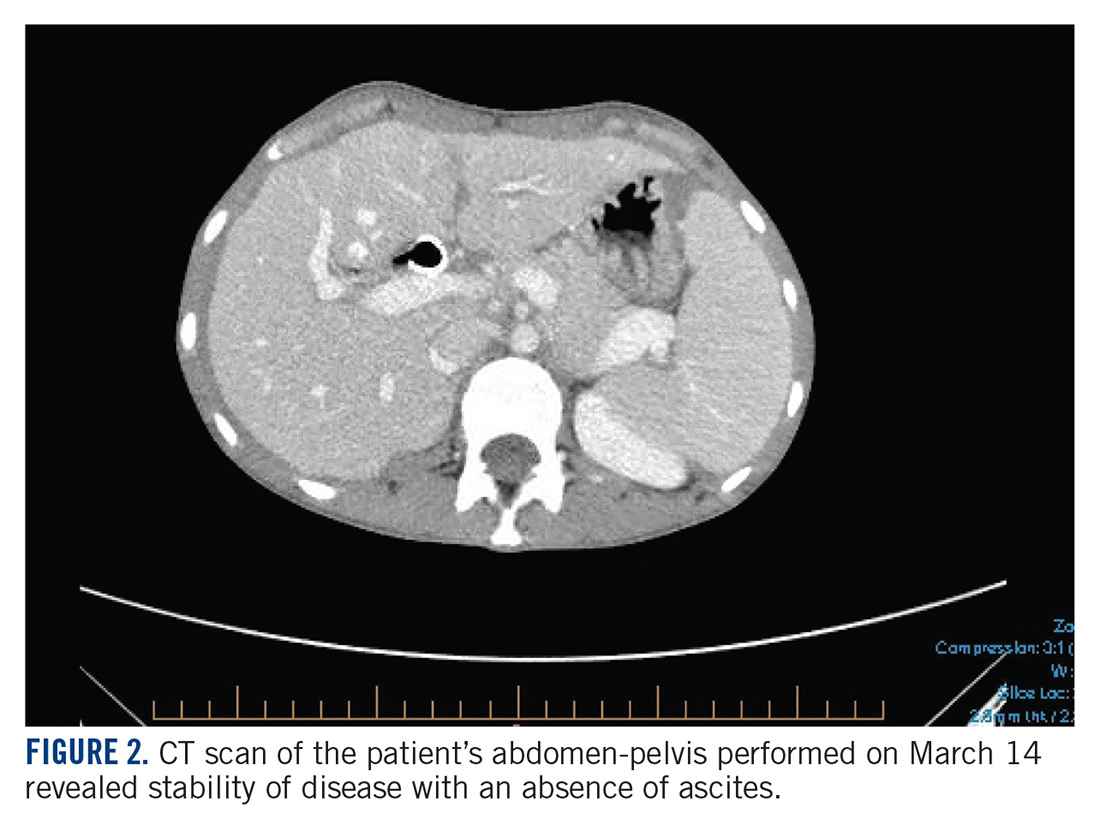
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Clinical trials in sarcoma bring hope and promise
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
The Sarcoma Journal Author Guidelines
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to eriley@mdedge.com. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact fiorio@mdedge.com to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to eriley@mdedge.com
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to eriley@mdedge.com. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact fiorio@mdedge.com to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to eriley@mdedge.com
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to eriley@mdedge.com. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact fiorio@mdedge.com to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to eriley@mdedge.com
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Rozlytrek approved for ROS1-positive metastatic NSCLC, cancers with NTRK gene fusion defects
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.

Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.
Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.
Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
FDA approves Turalio for symptomatic tenosynovial giant cell tumor
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.