User login
Neurology Reviews covers innovative and emerging news in neurology and neuroscience every month, with a focus on practical approaches to treating Parkinson's disease, epilepsy, headache, stroke, multiple sclerosis, Alzheimer's disease, and other neurologic disorders.
PML
Progressive multifocal leukoencephalopathy
Rituxan
The leading independent newspaper covering neurology news and commentary.
COVID-19 claims more than 675,000 U.S. lives, surpassing the 1918 flu
, according to data collected by Johns Hopkins University.
Although the raw numbers match, epidemiologists point out that 675,000 deaths in 1918 was a much greater proportion of the population. In 1918, the U.S. population was 105 million, less than one third of what it is today.
The AIDS pandemic of the 1980s remains the deadliest of the 20th Century, claiming the lives of 700,000 Americans. But at our current pace of 2,000 COVID deaths a day, we could quickly eclipse that death toll, too.
Even though the 1918 epidemic is often called the “Spanish Flu,” there is no universal consensus regarding where the virus originated, according to the Centers for Disease Control and Prevention.
Still, the almost incomprehensible loss harkens back to a time when medicine and technology were far less advanced than they are today.
In 1918, the United States didn’t have access to a vaccine, or near real-time tools to trace the spread and communicate the threat.
In some ways, the United States has failed to learn from the mistakes of the past.
There are many similarities between the two pandemics. In the spring of 1918, when the first wave of influenza hit, the United States and its allies were nearing victory in Europe in World War I. Just this summer the United States has ended its longest war, the conflict in Afghanistan, as COVID cases surge.
In both pandemics, hospitals and funeral homes were overrun and makeshift clinics were opened where space was available. Mask mandates were installed; schools, churches, and theaters closed; and social distancing was encouraged.
As is the case today, different jurisdictions took different steps to fight the pandemic and some were more successful than others.
According to History.com, in 1918, Philadelphia’s mayor said a popular annual parade could be held, and an estimated 200,000 people attended. In less than 2 weeks, more than 1,000 local residents were dead. But in St. Louis, public gatherings were banned, schools and theaters closed, and the death toll there was one eighth of Philadelphia’s.
Just as in 1918, America has at times continued to fan the flames of the epidemic by relaxing restrictions too quickly and relying on unproven treatments. Poor communication allowed younger people to feel that they wouldn’t necessarily face the worst consequences of the virus, contributing to a false sense of security in the age group that was fueling the spread.
“A lot of the mistakes that we definitely fell into in 1918, we hoped we wouldn’t fall into in 2020,” epidemiologist Stephen Kissler, PhD, of the Harvard T.H. Chan School of Public Health, told CNN. “We did.”
A version of this article first appeared on Medscape.com.
, according to data collected by Johns Hopkins University.
Although the raw numbers match, epidemiologists point out that 675,000 deaths in 1918 was a much greater proportion of the population. In 1918, the U.S. population was 105 million, less than one third of what it is today.
The AIDS pandemic of the 1980s remains the deadliest of the 20th Century, claiming the lives of 700,000 Americans. But at our current pace of 2,000 COVID deaths a day, we could quickly eclipse that death toll, too.
Even though the 1918 epidemic is often called the “Spanish Flu,” there is no universal consensus regarding where the virus originated, according to the Centers for Disease Control and Prevention.
Still, the almost incomprehensible loss harkens back to a time when medicine and technology were far less advanced than they are today.
In 1918, the United States didn’t have access to a vaccine, or near real-time tools to trace the spread and communicate the threat.
In some ways, the United States has failed to learn from the mistakes of the past.
There are many similarities between the two pandemics. In the spring of 1918, when the first wave of influenza hit, the United States and its allies were nearing victory in Europe in World War I. Just this summer the United States has ended its longest war, the conflict in Afghanistan, as COVID cases surge.
In both pandemics, hospitals and funeral homes were overrun and makeshift clinics were opened where space was available. Mask mandates were installed; schools, churches, and theaters closed; and social distancing was encouraged.
As is the case today, different jurisdictions took different steps to fight the pandemic and some were more successful than others.
According to History.com, in 1918, Philadelphia’s mayor said a popular annual parade could be held, and an estimated 200,000 people attended. In less than 2 weeks, more than 1,000 local residents were dead. But in St. Louis, public gatherings were banned, schools and theaters closed, and the death toll there was one eighth of Philadelphia’s.
Just as in 1918, America has at times continued to fan the flames of the epidemic by relaxing restrictions too quickly and relying on unproven treatments. Poor communication allowed younger people to feel that they wouldn’t necessarily face the worst consequences of the virus, contributing to a false sense of security in the age group that was fueling the spread.
“A lot of the mistakes that we definitely fell into in 1918, we hoped we wouldn’t fall into in 2020,” epidemiologist Stephen Kissler, PhD, of the Harvard T.H. Chan School of Public Health, told CNN. “We did.”
A version of this article first appeared on Medscape.com.
, according to data collected by Johns Hopkins University.
Although the raw numbers match, epidemiologists point out that 675,000 deaths in 1918 was a much greater proportion of the population. In 1918, the U.S. population was 105 million, less than one third of what it is today.
The AIDS pandemic of the 1980s remains the deadliest of the 20th Century, claiming the lives of 700,000 Americans. But at our current pace of 2,000 COVID deaths a day, we could quickly eclipse that death toll, too.
Even though the 1918 epidemic is often called the “Spanish Flu,” there is no universal consensus regarding where the virus originated, according to the Centers for Disease Control and Prevention.
Still, the almost incomprehensible loss harkens back to a time when medicine and technology were far less advanced than they are today.
In 1918, the United States didn’t have access to a vaccine, or near real-time tools to trace the spread and communicate the threat.
In some ways, the United States has failed to learn from the mistakes of the past.
There are many similarities between the two pandemics. In the spring of 1918, when the first wave of influenza hit, the United States and its allies were nearing victory in Europe in World War I. Just this summer the United States has ended its longest war, the conflict in Afghanistan, as COVID cases surge.
In both pandemics, hospitals and funeral homes were overrun and makeshift clinics were opened where space was available. Mask mandates were installed; schools, churches, and theaters closed; and social distancing was encouraged.
As is the case today, different jurisdictions took different steps to fight the pandemic and some were more successful than others.
According to History.com, in 1918, Philadelphia’s mayor said a popular annual parade could be held, and an estimated 200,000 people attended. In less than 2 weeks, more than 1,000 local residents were dead. But in St. Louis, public gatherings were banned, schools and theaters closed, and the death toll there was one eighth of Philadelphia’s.
Just as in 1918, America has at times continued to fan the flames of the epidemic by relaxing restrictions too quickly and relying on unproven treatments. Poor communication allowed younger people to feel that they wouldn’t necessarily face the worst consequences of the virus, contributing to a false sense of security in the age group that was fueling the spread.
“A lot of the mistakes that we definitely fell into in 1918, we hoped we wouldn’t fall into in 2020,” epidemiologist Stephen Kissler, PhD, of the Harvard T.H. Chan School of Public Health, told CNN. “We did.”
A version of this article first appeared on Medscape.com.
Sublingual film well tolerated for Parkinson ‘off’ episodes
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.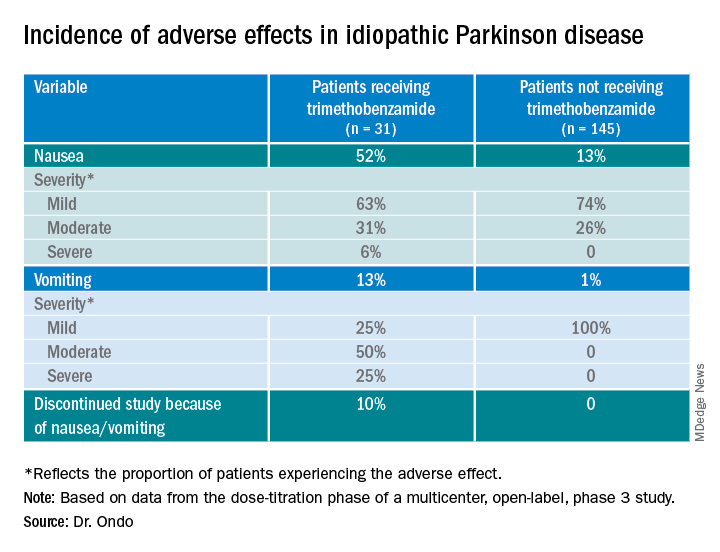
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
new research shows.
“The bottom line was that the majority of patients did not have dose-limiting nausea or vomiting,” said coinvestigator William Ondo, MD, from Houston Methodist Neurological Institute. “And although it really did not compare in a prospective, placebo-controlled manner use of [trimethobenzamide antiemetic] ... versus not using [it], anecdotally and based on historic data, nausea really seemed to be about the same even without the antinausea medication.”
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
This study was the dose-titration phase to determine the effective and tolerable dose of the drug as part of a longer study looking at safety and efficacy.
Only 13% of patients experienced nausea and/or vomiting, and of those, 74% cases were of mild severity and 26% were of moderate severity. These rates of nausea/vomiting were lower than those seen when trimethobenzamide (Tigan, Pfizer) was needed to be administered during the titration period, at the discretion of the investigator.
This multicenter, ongoing, open-label, phase 3 study enrolled 176 patients (mean age, 64.4 years) who had idiopathic Parkinson’s disease for a mean of 8.0 years and had no prior exposure to SL-apo, with modified Hoehn and Yahr stage 1-3 disease (83% stage 2 or 2.5 during “on” time).
Study participants had Mini-Mental State Examination scores greater than 25, were receiving stable doses of levodopa/carbidopa, and had 1 or more (mean, 4.2) “off” episodes per day with a total daily “off” time of 2 hours or more. Patients with mouth cankers or sores within 30 days of screening were excluded.
Open-label dose titration occurred during sequential office visits while patients were “off,” with escalating doses of 10-35 mg in 5-mg increments to determine a tolerable dose leading to a full “on” period within 45 minutes. Patients self-administered this achieved dose of SL-apo for up to five “off” episodes per day with a minimum of 2 hours between doses for the full 48-week study period.
The study protocol prohibited antiemetic use except when clinically warranted at the investigator’s discretion. Of the 176 patients, 31 (18%) received the antiemetic trimethobenzamide and 145 (82%) did not.
Of the 176 patients, 76% received their effective and tolerated dose within the first three doses. Just over half (55%) received 10 mg or 15 mg. Only 24% received the highest doses of 25 mg or 30 mg.
About 52%of patients who received trimethobenzamide experienced treatment-related nausea and 13% experienced vomiting; in comparison, 13% not receiving trimethobenzamide had nausea and 1% had vomiting. About 10%of patients in the former group and none in the latter discontinued the study because of nausea and/or vomiting.
The apomorphine sublingual film has “the advantage of ease of use compared to the injectable form,” Dr. Ondo said. “I think the injectable form, purely based on anecdotal experience, might start to work a minute or 2 faster than the sublingual form, but overall I would say efficacy as far as potency of turning ‘on’ and consistency of turning ‘on’ is comparable.”
In addition to the known adverse effects of nausea, vomiting, and hypotension with the use of any apomorphine, he said that long-term use of the sublingual form can lead to gingival irritation. Two recommendations are to place the film in a different site and to use a more basic toothpaste, such as one containing baking powder, because irritation may result from the acidity of the apomorphine.
Good news
Commenting on the study, Ludy Shih, MD, MMSc, from Boston University, noted that the drug label reports that “13%-15% had oropharyngeal soft tissue swelling or pain ... and 7% had oral ulcers and stomatitis.”
In addition, oral trimethobenzamide has been discontinued, although an injectable form is still available. This situation may present a problem, she said. “Most antinausea drugs block dopamine, so ... I would say they’re contraindicated for treating people with Parkinson’s disease. But trimethobenzamide in particular is one that we often reach for. ... But that appears to be constrained and may, in fact, be expensive for patients.”
Turning to the study findings, she said they suggest that “not everyone needs prophylactic use of trimethobenzamide before they take the apomorphine sublingual film, which is good news that helps doctors try to decide whether or not it’s reasonable to recommend people trying it without the trimethobenzamide.”
Although some patients did experience mild nausea, she said the fact that no needle is involved may attract some patients. Moreover, taking this medication may be easier than administering an injection during an “off” episode.
Dr. Ondo is a consultant for Sunovion Pharmaceuticals, which sponsored the study. Dr. Shih had no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM MDS VIRTUAL CONGRESS 2021
Friedreich’s ataxia treatment shows extended benefit
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.

The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
according to results of a clinical trial presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders.
The study, labeled the Delayed-Start Study, is an extension study of the two-part MOXIE phase 2 trial of omaveloxolone.
“This study shows two things,” said David Lynch, MD, PhD, of Children’s Hospital of Philadelphia. “It doesn’t matter when you started omaveloxolone for you to see a benefit; and that the benefit that the active group saw in the first part of the study was maintained as they went into the delayed-start part. So in fact omaveloxolone does modify the long-term behavior of the disease.”
Friedreich’s ataxia only affects about 22,000 people worldwide, and children typically present between the ages of 5 and 15, Dr. Lynch said.
The extension study included 73 patients who completed either of the first two parts of the MOXIe trial. The MOXIe trial randomized patients on a 3:1 basis to either omaveloxolone 2.5-300 mg or placebo for 12 weeks in the first part. The second part was a double-blind trial of 103 patients randomized on a 1:1 basis to 150 mg omaveloxolone or placebo for 48 weeks. Participants had a baseline modified Friedreich’s ataxia scale (mFARS) of 20-80 and were aged 16-40 years.
Patients in the extension study did not have severe pes cavus. The extension study was a 72-week evaluation of patients who were in either the treatment or placebo groups in the first two parts. There was a 4-week off-treatment period between the end of MOXIe part 2 and the beginning of the extension study, in which all patients received omaveloxolone.
At the end of the placebo-controlled study, patients taking omaveloxolone showed a –2.18-point (±0.96) difference in improvement in mFARS score (P = .027), compared with the placebo group, which was preserved at the end of the delayed-start period, with a –2.92-point (±2.13) improvement (P = .179), Dr. Lynch said.
In the extension study, former placebo patients who went on omaveloxolone had annualized mFARS slopes similar to the previously treated patients – 0.29 (±0.68) and 0.17 (±0.61), respectively (P = .85) – from weeks 48 to 144, Dr. Lynch said.
“This study showed that, when analyzed in a delayed-start fashion, it does not matter when you start omaveloxolone to see a benefit: Each cohort benefited almost equally once they started the drug,” Dr. Lynch said in an interview. “Also, in both groups, once they started omaveloxolone, they changed slower than people in natural history studies.”
A clinically meaningful difference?
Reached for comment, Massimo Pandolfo, MD, a neurologist at McGill University, Montreal, noted that the Delayed-Start Study included only patients without pes cavus, an indication that the patients had less severe disease. “It would be important to see how overall patients with Friedreich’s ataxia would have responded to the medication without this kind of selection,” Dr. Pandolfo said in an interview.
He also noted that the seemingly modest improvement in mFARS score could be an issue. “It’s a very difficult question: What is a clinically meaningful difference in this kind of rating scale? I would argue that probably 2 points is not a huge difference by itself, but it may be meaningful and one indicator of that is that if it was accompanied by also a significant difference in activities of daily living scale.”
In any event, Dr. Pandolfo said this is the first medication for Friedreich’s ataxia that has “survived” a randomized clinical trial.
Dr. Lynch said the study sponsor, Reata, may prepare a new drug application for omaveloxolone in patients ages 16 and older. “That would leave a need for investigation in younger FA patients.”
Dr. Lynch disclosed that his institution receives a grant from trial sponsor Reata to conduct the MOXIe trial. Dr. Pandolfo reports financial relationships with Design Therapeutics, Exicure and Voyager Therapeutics.
FROM MDS VIRTUAL CONGRESS 2021
Survey identifies clinicians’ unease with genetic testing
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
Before getting to work on developing guidelines for genetic testing in Parkinson’s disease, a task force of the Movement Disorders Society surveyed members worldwide to identify concerns they have about using genetic testing in practice. In results presented as a late-breaking abstract at the International Congress of Parkinson’s Disease and Movement Disorders,
“Some of the major outstanding issues are the clinical actionability of genetic testing – and this was highlighted by some survey participants,” senior study author Rachel Saunders-Pullman, MD, MPH, professor of neurology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview. The issue is “dynamic,” and will change even more radically when genetic therapies for Parkinson’s disease become available. “It is planned that, in the development of the MDS Task Force guidelines, scenarios which outline the changes in consideration of testing will depend on the availability of clinically actionable data,” she said.
Barriers to genetic testing
The MDS Task Force for Genetic Testing in Parkinson Disease conducted the survey, completed online by 568 MDS members. Respondents were from the four regions from which the MDS draws members: Africa, Europe, Asia/Oceania, and Pan-America. Half of the respondents considered themselves movement disorder specialists and 31% as general neurologists, said Maggie Markgraf, research coordinator at Mount Sinai Beth Israel in New York, who presented the survey findings.
Barriers to genetic testing that the clinicians cited included cost (57%), lack of availability of genetic counseling (37%), time for testing (20%) or time for counseling (17%). About 14%also cited a lack of knowledge, and only 8.5 % said they saw no barriers for genetic testing. Other concerns included a lack of therapeutic options if tests are positive and low overall positivity rates.
“Perceived barriers for general neurologists differed slightly, with limited knowledge being the most widely reported barrier, followed closely by cost and access to testing and genetic counseling,” Ms. Markgraf said.
Respondents were also asked to identify what they thought their patients perceived as barriers to genetic testing. The major one was cost (65%), followed by limited knowledge about genetics (43%), lack of access to genetic counseling (34%), and lack of access to testing separate from cost (30%). “Across all MDS regions, the perceived level of a patient’s knowledge about genetic testing is considered to be exceedingly low,” Ms. Markgraf said.
Europe had the highest availability to genetic tests, with 41.8% saying they’re accessible to general neurologists, followed by Asia/Oceania (31%) and Pan-America (30%).
“The area of most unmet need when it comes to PD genetic testing was cost for each MDS region, although the intertwined issue of access was also high, and over 50% reported that knowledge was an unmet need in their region,” Dr. Saunders-Pullman said.
Insurance coverage was another issue the survey respondents identified. In Europe, 53.6% said insurance or government programs cover genetic testing for PD, while only 14% in Pan-America and 10.3% in Asia/Oceania (and 0% in Africa) said such coverage was available.
“While there are limitations to this study, greater awareness of availability and barriers to genetic testing and counseling across different regions, as well as disparities among regions, will help inform development of the MDS Task Force guidelines,” Dr. Saunders-Pullman said.
Unmet needs
Connie Marras, MD, PhD, a professor of neurology at the University of Toronto, noted the survey suggested neurologists exhibit a “lack of comfort or lack of time” with genetic testing and counseling for Parkinson’s disease. “Even if we make genetic testing more widely available, we need health care providers that are comfortable and available to counsel patients before and after the testing, and clearly these are unmet needs,” Dr. Marras said in an interview.
“To date, pharmacologic treatment of Parkinson’s disease did not depend on genetics,” Dr. Marras said. “This may well change in the near future with treatments specifically targeting mechanisms related to two of the most common genetic risk factors for PD: LRRK2 and GBA gene variants being in clinical trials.” These developments may soon raise the urgency to reduce barriers to genetic testing.
Dr. Saunders-Pullman and Dr. Marras have no relevant relationships to disclose.
FROM MDS VIRTUAL CONGRESS 2021
Guideline gives weak support to trying oral medical cannabis for chronic pain
“Evidence alone is not sufficient for clinical decision-making, particularly in chronic pain,” said Jason Busse, DC, PhD, director of Michael G. DeGroote Centre for Medicinal Cannabis Research at McMaster University, Hamilton, Ont., and lead author of a newly released rapid guideline on medical cannabis or cannabinoids for chronic pain.

The recommendations, published online Sept. 9, 2021 in the British Medical Journal, suggest that providers offer patients with chronic pain a trial of noninhaled medical cannabis or cannabinoids if standard care or management is ineffective. However, the “weak” rating attached to the recommendation may compel some clinicians to automatically write off the panel’s recommendations.
“Because of the close balance between benefits and harms and wide variability in patient attitudes, the panel came to the conclusion that [some] patients presented with the current best evidence would likely choose to engage in a trial of medicinal cannabis, if their current care was felt to be suboptimal,” Dr. Busse explained in an interview.
But more importantly, “the recommendation allows for shared decision making to occur, and for different patients to make different decisions based on individual preferences and circumstances,” he said.
Evidence supports improved pain and sleep quality, physical functioning
Evidence supporting the use of medical cannabis in chronic pain is derived from a rigorous systematic review and meta-analysis of 32 studies enrolling 5,174 patients randomized to oral (capsule, spray, sublingual drops) or topical (transdermal cream) medical cannabis or placebo. Of note, three types of cannabinoids were represented: phytocannabinoids, synthetic, and endocannabinoids.
The studies included both patients with chronic noncancer pain (28 studies, n = 3,812) and chronic cancer pain not receiving palliative care (4 studies, n = 1,362). On average, baseline pain scores were a median 6.28 cm on a 10-cm visual analog scale (VAS), and median participant age was 53 years. 60% of trials reporting sex differences enrolled female participants. Overall, patients were followed for roughly 2 months (median, 50 days).
Findings (27 studies, n = 3,939) showed that, compared with placebo, medical cannabis resulted in a small, albeit important, improvement in the proportion of patients experiencing pain relief at or above the minimally important difference (MID) (moderate-certainty evidence, 10% modeled risk difference [RD; 95% confidence interval, 5%-15%] for achieving at least the MID of 1 cm).
Medical cannabis (15 studies, n = 2,425) also provided a small increase in the proportion of patients experiencing improvements in physical functioning at or above the MID (high certainty evidence, 4% modeled RD [95% CI, 0.1%-8%] for achieving at least a MID of 10 points).
Additionally, participants experienced significant improvements in sleep quality, compared with placebo (16 studies, 3,124 participants, high-quality evidence), demonstrating a weighted mean difference of –0.53 cm on a 10-cm VAS (95% CI, –0.75 to –0.30 cm). A total of nine larger trials (n = 2,652, high-certainty evidence) saw a small increase in the proportion of patients experiencing improved sleep quality at or above the MID: 6% modeled RD (95% CI, 2%-9%).
On the other hand, benefits did not extend to emotional, role, or social functioning (high-certainty evidence).
First do no harm: Start low, go slow
While these findings provide a rationale for medical cannabis in chronic pain, exploring options with patients can be challenging. Studies on medical cannabis consistently note that patients want information, but data also show that many providers express a lack of knowledge to provide adequate counseling.
There are also legal hurdles. Despite the authorization of medicinal cannabis across a majority of states and territories, cannabis is still a schedule I substance under the Federal Controlled Substances Act. In addition, the absence of standards around formulations, potency, and dosing has also been cited as a major barrier to recommending medical cannabis, as have concerns about adverse events (AEs), especially with inhaled and tetrahydrocannabinol (THC)-predominant formulations.
Like most medications, medical cannabis dosing should be individualized depending on product, patient, and ability to titrate the dose, but the guidelines provide a general rule of thumb. Providers considering therapeutic noninhaled medical cannabis trials are encouraged to start with a low-dose cannabidiol (CBD) oral tablet, spray, or sublingual oil drops 5 mg twice daily, increasing it by 10 mg every 2-3 days depending on the clinical response (to a maximum daily dose of 40 mg/day). If patient response is unsatisfactory, they should consider adding 1-2.5 mg THC/daily, titrated every 2-7 days to a maximum of 40 mg/day.
Still, an important caveat is whether or not adjunctive CBD alone is effective for chronic pain.

“While we know that one out of seven U.S. adults are using cannabidiol, we know very little about its therapeutic effects when given by itself for pain,” Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, and an associate professor at-large of psychology and behavioral science, said in an interview. (Dr. Cooper was not involved in the guideline development.)
“But patients tend to self-report that CBD is helpful, and at low doses, we know that it is unlikely to have adverse effects of any significant concern,” Dr. Cooper noted.
Depending on its components, medical cannabis is associated with a wide range of AEs. Studies comprising the evidence base for the guideline reported transient cognitive impairment (relative risk, 2.39; 95% CI, 1.06-5.38), vomiting (RR, 1.46; 95% CI, 1.07-1.99), and drowsiness (RR, 2.14; 95% CI, 1.55-2.95), attention impairment (RR, 4.04; 95% CI, 1.67-9.74), and nausea (RR, 1.59; 95% CI, 1.28-1.99). Of note, findings of a subgroup analysis showed that the risk of dizziness increased with treatment duration, starting at 3 months (test of interaction P = .002).
However, Dr. Cooper explained that, because the included studies were inconsistent in terms of cannabis type (e.g., some looked at synthetic THC or THC-like substances where others looked at a THC/CBD combination) and formulation (capsules, oral mucosal sprays), it’s difficult to tease out component-specific AEs.
“These are really important things to note, especially when you think about different populations that might be using these types of medicines moving forward,” she said.
Toward that end, the guideline specifically states that there is “no reason why the expected benefits would be systematically different among adolescents and emerging adults.”
Among children with cancer, prior study findings reinforce the conclusion that benefits are similar to adults, but studies in this area are limited to end-of-life treatment, childhood cancer with primarily palliative intent, or progressive or relapsed cancer. Because THC’s safety profile is less certain in children, it’s also important to consider adverse neurocognitive effects before initiating a medical cannabis trial in this population.
Navigating the landscape
Although promising, the medical cannabis landscape is undoubtedly difficult to navigate, with land mines ranging from a limited inability to simply pick up a prescribing pad to quality control.
With the exception of three Food and Drug Administration–approved products – dronabinol, cannabidiol Rx, and nabilone – U.S. providers are only able to ‘certify,’ not prescribe, medical cannabis for chronic pain, and only if it is included within the state cannabis board’s list of eligible conditions. (A state-by-state guide is available.)

Quality control also varies by product but is critical. “You want to look for certificates of quality assurance,” Jenny Wilkerson, PhD, a research assistant professor of pharmacodynamics at the University of Florida, Gainesville, said in an interview. (Dr. Wilkerson was not involved in the guideline development.)
“A good dispensary should have that information or at least be willing to get that information, but generally speaking, that is something that patients need to ask for,” she emphasized, noting that “most available mass readouts are not divided by lots.”
Initial counseling and AE monitoring and regular follow-up is important, especially among patients who’ve never tried medical cannabis (or older patients whose prior experience may be limited to weaker recreational marijuana).
Notably, the reliance on medical dispensaries to deliver the right information at the right time may prove to be faulty. While recent data show that frontline dispensary workers regularly provide information to customers on their medical conditions and available products, they rarely, if ever, base recommendations on provider input, and never or rarely discuss potential AEs and other risks.
Per the new guideline, inexperienced patients should be seen monthly until a stable dose is achieved; longer times between visits can be considered in those who are more experienced. Still, patients should be advised to contact their provider when pain relief or other goals are insufficient, or when response or problematic AEs occur. This facilitates down-titration to a previously tolerated dose, up-titration in CBD and/or THC, or a different route of administration/formulation altogether.
Dr. Wilkerson pointed out that follow-up visits also provide an opportunity to do a blood draw and ask the lab to conduct pharmacokinetic analysis.
If possible, “ask patients to [ensure that they] take a standard dose before the visit so that the lab can assess the blood percentage of primary compounds and metabolites in the product that they are using,” she explained, noting that the information is helping to determine how “the different ratios may be affecting therapeutic response in individual patients.”
Granted, the guideline is only a start. But it is a good one.
“A lot of physicians want to be able to hang their hat on evidence of the safety and efficacy of these products, and the analysis that was leveraged for this guideline was very rigorous,” Dr. Cooper said.
Not only do they reinforce that “oral cannabinoids can produce small improvements in pain and provide a dosing structure that minimizes risk to the patient, [but they] should be able to help educate physicians who [are looking] for a sense of what the literature tells us at this time,” she added.
“With chronic pain, we often find that different treatments will show small potential benefits and they have a certain risk profile,” Dr. Busse said.
“It’s almost impossible to know what patients think about this option unless you present them with the evidence and ask them to make a decision based on their values and preferences,” he said.
The Michael G. DeGroote Centre for Medicinal Cannabis Research funded the MAGIC Evidence Ecosystem Foundation to support the creation of the guideline. The center receives no funding from industry Dr. Busse, Dr. Cooper, and Dr. Wilkerson reported having no relevant financial relationships.
“Evidence alone is not sufficient for clinical decision-making, particularly in chronic pain,” said Jason Busse, DC, PhD, director of Michael G. DeGroote Centre for Medicinal Cannabis Research at McMaster University, Hamilton, Ont., and lead author of a newly released rapid guideline on medical cannabis or cannabinoids for chronic pain.
The recommendations, published online Sept. 9, 2021 in the British Medical Journal, suggest that providers offer patients with chronic pain a trial of noninhaled medical cannabis or cannabinoids if standard care or management is ineffective. However, the “weak” rating attached to the recommendation may compel some clinicians to automatically write off the panel’s recommendations.
“Because of the close balance between benefits and harms and wide variability in patient attitudes, the panel came to the conclusion that [some] patients presented with the current best evidence would likely choose to engage in a trial of medicinal cannabis, if their current care was felt to be suboptimal,” Dr. Busse explained in an interview.
But more importantly, “the recommendation allows for shared decision making to occur, and for different patients to make different decisions based on individual preferences and circumstances,” he said.
Evidence supports improved pain and sleep quality, physical functioning
Evidence supporting the use of medical cannabis in chronic pain is derived from a rigorous systematic review and meta-analysis of 32 studies enrolling 5,174 patients randomized to oral (capsule, spray, sublingual drops) or topical (transdermal cream) medical cannabis or placebo. Of note, three types of cannabinoids were represented: phytocannabinoids, synthetic, and endocannabinoids.
The studies included both patients with chronic noncancer pain (28 studies, n = 3,812) and chronic cancer pain not receiving palliative care (4 studies, n = 1,362). On average, baseline pain scores were a median 6.28 cm on a 10-cm visual analog scale (VAS), and median participant age was 53 years. 60% of trials reporting sex differences enrolled female participants. Overall, patients were followed for roughly 2 months (median, 50 days).
Findings (27 studies, n = 3,939) showed that, compared with placebo, medical cannabis resulted in a small, albeit important, improvement in the proportion of patients experiencing pain relief at or above the minimally important difference (MID) (moderate-certainty evidence, 10% modeled risk difference [RD; 95% confidence interval, 5%-15%] for achieving at least the MID of 1 cm).
Medical cannabis (15 studies, n = 2,425) also provided a small increase in the proportion of patients experiencing improvements in physical functioning at or above the MID (high certainty evidence, 4% modeled RD [95% CI, 0.1%-8%] for achieving at least a MID of 10 points).
Additionally, participants experienced significant improvements in sleep quality, compared with placebo (16 studies, 3,124 participants, high-quality evidence), demonstrating a weighted mean difference of –0.53 cm on a 10-cm VAS (95% CI, –0.75 to –0.30 cm). A total of nine larger trials (n = 2,652, high-certainty evidence) saw a small increase in the proportion of patients experiencing improved sleep quality at or above the MID: 6% modeled RD (95% CI, 2%-9%).
On the other hand, benefits did not extend to emotional, role, or social functioning (high-certainty evidence).
First do no harm: Start low, go slow
While these findings provide a rationale for medical cannabis in chronic pain, exploring options with patients can be challenging. Studies on medical cannabis consistently note that patients want information, but data also show that many providers express a lack of knowledge to provide adequate counseling.
There are also legal hurdles. Despite the authorization of medicinal cannabis across a majority of states and territories, cannabis is still a schedule I substance under the Federal Controlled Substances Act. In addition, the absence of standards around formulations, potency, and dosing has also been cited as a major barrier to recommending medical cannabis, as have concerns about adverse events (AEs), especially with inhaled and tetrahydrocannabinol (THC)-predominant formulations.
Like most medications, medical cannabis dosing should be individualized depending on product, patient, and ability to titrate the dose, but the guidelines provide a general rule of thumb. Providers considering therapeutic noninhaled medical cannabis trials are encouraged to start with a low-dose cannabidiol (CBD) oral tablet, spray, or sublingual oil drops 5 mg twice daily, increasing it by 10 mg every 2-3 days depending on the clinical response (to a maximum daily dose of 40 mg/day). If patient response is unsatisfactory, they should consider adding 1-2.5 mg THC/daily, titrated every 2-7 days to a maximum of 40 mg/day.
Still, an important caveat is whether or not adjunctive CBD alone is effective for chronic pain.
“While we know that one out of seven U.S. adults are using cannabidiol, we know very little about its therapeutic effects when given by itself for pain,” Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, and an associate professor at-large of psychology and behavioral science, said in an interview. (Dr. Cooper was not involved in the guideline development.)
“But patients tend to self-report that CBD is helpful, and at low doses, we know that it is unlikely to have adverse effects of any significant concern,” Dr. Cooper noted.
Depending on its components, medical cannabis is associated with a wide range of AEs. Studies comprising the evidence base for the guideline reported transient cognitive impairment (relative risk, 2.39; 95% CI, 1.06-5.38), vomiting (RR, 1.46; 95% CI, 1.07-1.99), and drowsiness (RR, 2.14; 95% CI, 1.55-2.95), attention impairment (RR, 4.04; 95% CI, 1.67-9.74), and nausea (RR, 1.59; 95% CI, 1.28-1.99). Of note, findings of a subgroup analysis showed that the risk of dizziness increased with treatment duration, starting at 3 months (test of interaction P = .002).
However, Dr. Cooper explained that, because the included studies were inconsistent in terms of cannabis type (e.g., some looked at synthetic THC or THC-like substances where others looked at a THC/CBD combination) and formulation (capsules, oral mucosal sprays), it’s difficult to tease out component-specific AEs.
“These are really important things to note, especially when you think about different populations that might be using these types of medicines moving forward,” she said.
Toward that end, the guideline specifically states that there is “no reason why the expected benefits would be systematically different among adolescents and emerging adults.”
Among children with cancer, prior study findings reinforce the conclusion that benefits are similar to adults, but studies in this area are limited to end-of-life treatment, childhood cancer with primarily palliative intent, or progressive or relapsed cancer. Because THC’s safety profile is less certain in children, it’s also important to consider adverse neurocognitive effects before initiating a medical cannabis trial in this population.
Navigating the landscape
Although promising, the medical cannabis landscape is undoubtedly difficult to navigate, with land mines ranging from a limited inability to simply pick up a prescribing pad to quality control.
With the exception of three Food and Drug Administration–approved products – dronabinol, cannabidiol Rx, and nabilone – U.S. providers are only able to ‘certify,’ not prescribe, medical cannabis for chronic pain, and only if it is included within the state cannabis board’s list of eligible conditions. (A state-by-state guide is available.)
Quality control also varies by product but is critical. “You want to look for certificates of quality assurance,” Jenny Wilkerson, PhD, a research assistant professor of pharmacodynamics at the University of Florida, Gainesville, said in an interview. (Dr. Wilkerson was not involved in the guideline development.)
“A good dispensary should have that information or at least be willing to get that information, but generally speaking, that is something that patients need to ask for,” she emphasized, noting that “most available mass readouts are not divided by lots.”
Initial counseling and AE monitoring and regular follow-up is important, especially among patients who’ve never tried medical cannabis (or older patients whose prior experience may be limited to weaker recreational marijuana).
Notably, the reliance on medical dispensaries to deliver the right information at the right time may prove to be faulty. While recent data show that frontline dispensary workers regularly provide information to customers on their medical conditions and available products, they rarely, if ever, base recommendations on provider input, and never or rarely discuss potential AEs and other risks.
Per the new guideline, inexperienced patients should be seen monthly until a stable dose is achieved; longer times between visits can be considered in those who are more experienced. Still, patients should be advised to contact their provider when pain relief or other goals are insufficient, or when response or problematic AEs occur. This facilitates down-titration to a previously tolerated dose, up-titration in CBD and/or THC, or a different route of administration/formulation altogether.
Dr. Wilkerson pointed out that follow-up visits also provide an opportunity to do a blood draw and ask the lab to conduct pharmacokinetic analysis.
If possible, “ask patients to [ensure that they] take a standard dose before the visit so that the lab can assess the blood percentage of primary compounds and metabolites in the product that they are using,” she explained, noting that the information is helping to determine how “the different ratios may be affecting therapeutic response in individual patients.”
Granted, the guideline is only a start. But it is a good one.
“A lot of physicians want to be able to hang their hat on evidence of the safety and efficacy of these products, and the analysis that was leveraged for this guideline was very rigorous,” Dr. Cooper said.
Not only do they reinforce that “oral cannabinoids can produce small improvements in pain and provide a dosing structure that minimizes risk to the patient, [but they] should be able to help educate physicians who [are looking] for a sense of what the literature tells us at this time,” she added.
“With chronic pain, we often find that different treatments will show small potential benefits and they have a certain risk profile,” Dr. Busse said.
“It’s almost impossible to know what patients think about this option unless you present them with the evidence and ask them to make a decision based on their values and preferences,” he said.
The Michael G. DeGroote Centre for Medicinal Cannabis Research funded the MAGIC Evidence Ecosystem Foundation to support the creation of the guideline. The center receives no funding from industry Dr. Busse, Dr. Cooper, and Dr. Wilkerson reported having no relevant financial relationships.
“Evidence alone is not sufficient for clinical decision-making, particularly in chronic pain,” said Jason Busse, DC, PhD, director of Michael G. DeGroote Centre for Medicinal Cannabis Research at McMaster University, Hamilton, Ont., and lead author of a newly released rapid guideline on medical cannabis or cannabinoids for chronic pain.
The recommendations, published online Sept. 9, 2021 in the British Medical Journal, suggest that providers offer patients with chronic pain a trial of noninhaled medical cannabis or cannabinoids if standard care or management is ineffective. However, the “weak” rating attached to the recommendation may compel some clinicians to automatically write off the panel’s recommendations.
“Because of the close balance between benefits and harms and wide variability in patient attitudes, the panel came to the conclusion that [some] patients presented with the current best evidence would likely choose to engage in a trial of medicinal cannabis, if their current care was felt to be suboptimal,” Dr. Busse explained in an interview.
But more importantly, “the recommendation allows for shared decision making to occur, and for different patients to make different decisions based on individual preferences and circumstances,” he said.
Evidence supports improved pain and sleep quality, physical functioning
Evidence supporting the use of medical cannabis in chronic pain is derived from a rigorous systematic review and meta-analysis of 32 studies enrolling 5,174 patients randomized to oral (capsule, spray, sublingual drops) or topical (transdermal cream) medical cannabis or placebo. Of note, three types of cannabinoids were represented: phytocannabinoids, synthetic, and endocannabinoids.
The studies included both patients with chronic noncancer pain (28 studies, n = 3,812) and chronic cancer pain not receiving palliative care (4 studies, n = 1,362). On average, baseline pain scores were a median 6.28 cm on a 10-cm visual analog scale (VAS), and median participant age was 53 years. 60% of trials reporting sex differences enrolled female participants. Overall, patients were followed for roughly 2 months (median, 50 days).
Findings (27 studies, n = 3,939) showed that, compared with placebo, medical cannabis resulted in a small, albeit important, improvement in the proportion of patients experiencing pain relief at or above the minimally important difference (MID) (moderate-certainty evidence, 10% modeled risk difference [RD; 95% confidence interval, 5%-15%] for achieving at least the MID of 1 cm).
Medical cannabis (15 studies, n = 2,425) also provided a small increase in the proportion of patients experiencing improvements in physical functioning at or above the MID (high certainty evidence, 4% modeled RD [95% CI, 0.1%-8%] for achieving at least a MID of 10 points).
Additionally, participants experienced significant improvements in sleep quality, compared with placebo (16 studies, 3,124 participants, high-quality evidence), demonstrating a weighted mean difference of –0.53 cm on a 10-cm VAS (95% CI, –0.75 to –0.30 cm). A total of nine larger trials (n = 2,652, high-certainty evidence) saw a small increase in the proportion of patients experiencing improved sleep quality at or above the MID: 6% modeled RD (95% CI, 2%-9%).
On the other hand, benefits did not extend to emotional, role, or social functioning (high-certainty evidence).
First do no harm: Start low, go slow
While these findings provide a rationale for medical cannabis in chronic pain, exploring options with patients can be challenging. Studies on medical cannabis consistently note that patients want information, but data also show that many providers express a lack of knowledge to provide adequate counseling.
There are also legal hurdles. Despite the authorization of medicinal cannabis across a majority of states and territories, cannabis is still a schedule I substance under the Federal Controlled Substances Act. In addition, the absence of standards around formulations, potency, and dosing has also been cited as a major barrier to recommending medical cannabis, as have concerns about adverse events (AEs), especially with inhaled and tetrahydrocannabinol (THC)-predominant formulations.
Like most medications, medical cannabis dosing should be individualized depending on product, patient, and ability to titrate the dose, but the guidelines provide a general rule of thumb. Providers considering therapeutic noninhaled medical cannabis trials are encouraged to start with a low-dose cannabidiol (CBD) oral tablet, spray, or sublingual oil drops 5 mg twice daily, increasing it by 10 mg every 2-3 days depending on the clinical response (to a maximum daily dose of 40 mg/day). If patient response is unsatisfactory, they should consider adding 1-2.5 mg THC/daily, titrated every 2-7 days to a maximum of 40 mg/day.
Still, an important caveat is whether or not adjunctive CBD alone is effective for chronic pain.
“While we know that one out of seven U.S. adults are using cannabidiol, we know very little about its therapeutic effects when given by itself for pain,” Ziva Cooper, PhD, director of the Cannabis Research Initiative at the University of California, Los Angeles, and an associate professor at-large of psychology and behavioral science, said in an interview. (Dr. Cooper was not involved in the guideline development.)
“But patients tend to self-report that CBD is helpful, and at low doses, we know that it is unlikely to have adverse effects of any significant concern,” Dr. Cooper noted.
Depending on its components, medical cannabis is associated with a wide range of AEs. Studies comprising the evidence base for the guideline reported transient cognitive impairment (relative risk, 2.39; 95% CI, 1.06-5.38), vomiting (RR, 1.46; 95% CI, 1.07-1.99), and drowsiness (RR, 2.14; 95% CI, 1.55-2.95), attention impairment (RR, 4.04; 95% CI, 1.67-9.74), and nausea (RR, 1.59; 95% CI, 1.28-1.99). Of note, findings of a subgroup analysis showed that the risk of dizziness increased with treatment duration, starting at 3 months (test of interaction P = .002).
However, Dr. Cooper explained that, because the included studies were inconsistent in terms of cannabis type (e.g., some looked at synthetic THC or THC-like substances where others looked at a THC/CBD combination) and formulation (capsules, oral mucosal sprays), it’s difficult to tease out component-specific AEs.
“These are really important things to note, especially when you think about different populations that might be using these types of medicines moving forward,” she said.
Toward that end, the guideline specifically states that there is “no reason why the expected benefits would be systematically different among adolescents and emerging adults.”
Among children with cancer, prior study findings reinforce the conclusion that benefits are similar to adults, but studies in this area are limited to end-of-life treatment, childhood cancer with primarily palliative intent, or progressive or relapsed cancer. Because THC’s safety profile is less certain in children, it’s also important to consider adverse neurocognitive effects before initiating a medical cannabis trial in this population.
Navigating the landscape
Although promising, the medical cannabis landscape is undoubtedly difficult to navigate, with land mines ranging from a limited inability to simply pick up a prescribing pad to quality control.
With the exception of three Food and Drug Administration–approved products – dronabinol, cannabidiol Rx, and nabilone – U.S. providers are only able to ‘certify,’ not prescribe, medical cannabis for chronic pain, and only if it is included within the state cannabis board’s list of eligible conditions. (A state-by-state guide is available.)
Quality control also varies by product but is critical. “You want to look for certificates of quality assurance,” Jenny Wilkerson, PhD, a research assistant professor of pharmacodynamics at the University of Florida, Gainesville, said in an interview. (Dr. Wilkerson was not involved in the guideline development.)
“A good dispensary should have that information or at least be willing to get that information, but generally speaking, that is something that patients need to ask for,” she emphasized, noting that “most available mass readouts are not divided by lots.”
Initial counseling and AE monitoring and regular follow-up is important, especially among patients who’ve never tried medical cannabis (or older patients whose prior experience may be limited to weaker recreational marijuana).
Notably, the reliance on medical dispensaries to deliver the right information at the right time may prove to be faulty. While recent data show that frontline dispensary workers regularly provide information to customers on their medical conditions and available products, they rarely, if ever, base recommendations on provider input, and never or rarely discuss potential AEs and other risks.
Per the new guideline, inexperienced patients should be seen monthly until a stable dose is achieved; longer times between visits can be considered in those who are more experienced. Still, patients should be advised to contact their provider when pain relief or other goals are insufficient, or when response or problematic AEs occur. This facilitates down-titration to a previously tolerated dose, up-titration in CBD and/or THC, or a different route of administration/formulation altogether.
Dr. Wilkerson pointed out that follow-up visits also provide an opportunity to do a blood draw and ask the lab to conduct pharmacokinetic analysis.
If possible, “ask patients to [ensure that they] take a standard dose before the visit so that the lab can assess the blood percentage of primary compounds and metabolites in the product that they are using,” she explained, noting that the information is helping to determine how “the different ratios may be affecting therapeutic response in individual patients.”
Granted, the guideline is only a start. But it is a good one.
“A lot of physicians want to be able to hang their hat on evidence of the safety and efficacy of these products, and the analysis that was leveraged for this guideline was very rigorous,” Dr. Cooper said.
Not only do they reinforce that “oral cannabinoids can produce small improvements in pain and provide a dosing structure that minimizes risk to the patient, [but they] should be able to help educate physicians who [are looking] for a sense of what the literature tells us at this time,” she added.
“With chronic pain, we often find that different treatments will show small potential benefits and they have a certain risk profile,” Dr. Busse said.
“It’s almost impossible to know what patients think about this option unless you present them with the evidence and ask them to make a decision based on their values and preferences,” he said.
The Michael G. DeGroote Centre for Medicinal Cannabis Research funded the MAGIC Evidence Ecosystem Foundation to support the creation of the guideline. The center receives no funding from industry Dr. Busse, Dr. Cooper, and Dr. Wilkerson reported having no relevant financial relationships.
FROM THE BMJ
Moderna vaccine more effective than Pfizer and J&J
the Centers for Disease Control and Protection has said.
“Among U.S. adults without immunocompromising conditions, vaccine effectiveness against COVID-19 hospitalization during March 11–Aug. 15, 2021, was higher for the Moderna vaccine (93%) than the Pfizer-BioNTech vaccine (88%) and the Janssen vaccine (71%),” the agency’s Morbidity and Mortality Weekly Report said. Janssen refers to the Johnson & Johnson vaccine.
The CDC said the data could help people make informed decisions.
“Understanding differences in VE [vaccine effectiveness] by vaccine product can guide individual choices and policy recommendations regarding vaccine boosters. All Food and Drug Administration–approved or authorized COVID-19 vaccines provide substantial protection against COVID-19 hospitalization,” the report said.
The study also broke down effectiveness for longer periods. Moderna came out on top again.
After 120 days, the Moderna vaccine provided 92% effectiveness against hospitalization, whereas the Pfizer vaccine’s effectiveness dropped to 77%, the CDC said. There was no similar calculation for the Johnson & Johnson vaccine.
The CDC studied 3,689 adults at 21 hospitals in 18 states who got the two-shot Pfizer or Moderna vaccine or the one-shot Johnson & Johnson vaccine between March and August.
The agency noted some factors that could have come into play.
“Differences in vaccine effectiveness between the Moderna and Pfizer-BioNTech vaccine might be due to higher mRNA content in the Moderna vaccine, differences in timing between doses (3 weeks for Pfizer-BioNTech vs. 4 weeks for Moderna), or possible differences between groups that received each vaccine that were not accounted for in the analysis,” the report said.
The CDC noted limitations in the findings. Children, immunocompromised adults, and vaccine effectiveness against COVID-19 that did not result in hospitalization were not studied.
Other studies have shown all three U.S. vaccines provide a high rate of protection against coronavirus.
A version of this article first appeared on WebMD.com.
the Centers for Disease Control and Protection has said.
“Among U.S. adults without immunocompromising conditions, vaccine effectiveness against COVID-19 hospitalization during March 11–Aug. 15, 2021, was higher for the Moderna vaccine (93%) than the Pfizer-BioNTech vaccine (88%) and the Janssen vaccine (71%),” the agency’s Morbidity and Mortality Weekly Report said. Janssen refers to the Johnson & Johnson vaccine.
The CDC said the data could help people make informed decisions.
“Understanding differences in VE [vaccine effectiveness] by vaccine product can guide individual choices and policy recommendations regarding vaccine boosters. All Food and Drug Administration–approved or authorized COVID-19 vaccines provide substantial protection against COVID-19 hospitalization,” the report said.
The study also broke down effectiveness for longer periods. Moderna came out on top again.
After 120 days, the Moderna vaccine provided 92% effectiveness against hospitalization, whereas the Pfizer vaccine’s effectiveness dropped to 77%, the CDC said. There was no similar calculation for the Johnson & Johnson vaccine.
The CDC studied 3,689 adults at 21 hospitals in 18 states who got the two-shot Pfizer or Moderna vaccine or the one-shot Johnson & Johnson vaccine between March and August.
The agency noted some factors that could have come into play.
“Differences in vaccine effectiveness between the Moderna and Pfizer-BioNTech vaccine might be due to higher mRNA content in the Moderna vaccine, differences in timing between doses (3 weeks for Pfizer-BioNTech vs. 4 weeks for Moderna), or possible differences between groups that received each vaccine that were not accounted for in the analysis,” the report said.
The CDC noted limitations in the findings. Children, immunocompromised adults, and vaccine effectiveness against COVID-19 that did not result in hospitalization were not studied.
Other studies have shown all three U.S. vaccines provide a high rate of protection against coronavirus.
A version of this article first appeared on WebMD.com.
the Centers for Disease Control and Protection has said.
“Among U.S. adults without immunocompromising conditions, vaccine effectiveness against COVID-19 hospitalization during March 11–Aug. 15, 2021, was higher for the Moderna vaccine (93%) than the Pfizer-BioNTech vaccine (88%) and the Janssen vaccine (71%),” the agency’s Morbidity and Mortality Weekly Report said. Janssen refers to the Johnson & Johnson vaccine.
The CDC said the data could help people make informed decisions.
“Understanding differences in VE [vaccine effectiveness] by vaccine product can guide individual choices and policy recommendations regarding vaccine boosters. All Food and Drug Administration–approved or authorized COVID-19 vaccines provide substantial protection against COVID-19 hospitalization,” the report said.
The study also broke down effectiveness for longer periods. Moderna came out on top again.
After 120 days, the Moderna vaccine provided 92% effectiveness against hospitalization, whereas the Pfizer vaccine’s effectiveness dropped to 77%, the CDC said. There was no similar calculation for the Johnson & Johnson vaccine.
The CDC studied 3,689 adults at 21 hospitals in 18 states who got the two-shot Pfizer or Moderna vaccine or the one-shot Johnson & Johnson vaccine between March and August.
The agency noted some factors that could have come into play.
“Differences in vaccine effectiveness between the Moderna and Pfizer-BioNTech vaccine might be due to higher mRNA content in the Moderna vaccine, differences in timing between doses (3 weeks for Pfizer-BioNTech vs. 4 weeks for Moderna), or possible differences between groups that received each vaccine that were not accounted for in the analysis,” the report said.
The CDC noted limitations in the findings. Children, immunocompromised adults, and vaccine effectiveness against COVID-19 that did not result in hospitalization were not studied.
Other studies have shown all three U.S. vaccines provide a high rate of protection against coronavirus.
A version of this article first appeared on WebMD.com.
Parent-led intervention linked with decreased autism symptoms in at-risk infants
These findings, which were published in JAMA Pediatrics, were the first evidence worldwide that a preemptive intervention during infancy could lead to such a significant improvement in children’s social development, resulting in “three times fewer diagnoses of autism at age 3,” said lead author Andrew Whitehouse, PhD, in a statement.
“No trial of a preemptive infant intervention, applied prior to diagnosis, has to date shown such an effect to impact diagnostic outcomes – until now,” he said.
Study intervention is a nontraditonal approach
Dr. Whitehouse, who is professor of Autism Research at Telethon Kids and University of Western Australia, and Director of CliniKids in Perth, said the intervention is a departure from traditional approaches. “Traditionally, therapy seeks to train children to learn ‘typical’ behaviors,” he said in an email. “The difference of this therapy is that we help parents understand the unique abilities of their baby, and to use these strengths as a foundation for future development.”
Dr. Whitehouse’s study included 103 children (aged approximately 12 months), who displayed at least three of five behaviors indicating a high likelihood of ASD as defined by the Social Attention and Communication Surveillance–Revised (SACS-R) 12-month checklist. The infants were randomized to receive either usual care or the intervention, which is called the iBASIS–Video Interaction to Promote Positive Parenting (iBASIS-VIPP). Usual care was delivered by community physicians, whereas the intervention involved 10 sessions delivered at home by a trained therapist.
“The iBASIS-VIPP uses video-feedback as a means of helping parents recognize their baby’s communication cues so they can respond in a way that builds their social communication development,” Dr. Whitehouse explained in an interview. “The therapist then provides guidance to the parent as to how their baby is communicating with them, and they can communicate back to have back-and-forth conversations.”
“We know these back-and-forth conversations are crucial to support early social communication development, and are a precursor to more complex skills, such as verbal language,” he added.
Reassessment of the children at age 3 years showed a “small but enduring” benefit of the intervention, noted the authors. Children in the intervention group had a reduction in ASD symptom severity (P = .04), and reduced odds of ASD classification, compared with children receiving usual care (6.7% vs. 20.5%; odds ratio, 0.18; P = .02).
The findings provide “initial evidence of efficacy for a new clinical model that uses a specific developmentally focused intervention,” noted the authors. “The children falling below the diagnostic threshold still had developmental difficulties, but by working with each child’s unique differences, rather than trying to counter them, the therapy has effectively supported their development through the early childhood years,” noted Dr. Whitehouse in a statement.
Other research has shown benefits of new study approach
This is a “solid” study, “but, as acknowledged by the authors, the main effects are small in magnitude, and longer-term outcomes will be important to capture,” said Jessica Brian, PhD, C Psych, associate professor in the department of pediatrics at the University of Toronto, colead at the Autism Research Centre, and psychologist and clinician-investigator at Holland Bloorview Kids Rehab Hospital in Toronto.
Dr. Brian said she and her coauthors of a paper published in Autism Research and others have shown that the kind of approach used in the new study can be helpful for enhancing different areas of toddler development, but “the specific finding of reduced likelihood of a clinical ASD diagnosis is a bit different.”
The goal of reducing the likelihood of an ASD diagnosis “needs to be considered carefully, from the perspective of autism acceptance,” she added. “From an acceptance lens, the primary objective of early intervention in ASD might be better positioned as aiming to enhance or support a young child’s development, help them make developmental progress, build on their strengths, optimize outcomes, or reduce impairment. … I think the authors do a good job of balancing this perspective.”
New study shows value of parent-mediated interventions
Overall, Dr. Brian, who coauthored the Canadian Paediatric Society’s position statement on ASD diagnosis, lauded the findings as good news.
“It shows the value of using parent-mediated interventions, which are far less costly and are more resource-efficient than most therapist-delivered models.”
“In cases where parent-mediated approaches are made available to families prior to diagnosis, there is potential for strong effects, when the brain is most amenable to learning. Such models may also be an ideal fit before diagnosis, since they are less resource-intensive than therapist-delivered models, which may only be funded by governments once a diagnosis is confirmed,” she said.
“Finally, parent-mediated programs have the potential to support parents during what, for many families, is a particularly challenging time as they identify their child’s developmental differences or receive a diagnosis. Such programs have potential to increase parents’ confidence in parenting a young child with unique learning needs.”
What Dr. Brian thought was missing from the paper was acknowledgment that, “despite early developmental gains from parent-mediated interventions, it is likely that most children with ASD will need additional supports throughout development.”
This study was sponsored by the Telethon Kids Institute. Dr. Whitehouse reported no conflicts of interest. Dr. Brian codeveloped a parent-mediated intervention for toddlers with probable or confirmed ASD (the Social ABCs), for which she does not receive any royalties.
These findings, which were published in JAMA Pediatrics, were the first evidence worldwide that a preemptive intervention during infancy could lead to such a significant improvement in children’s social development, resulting in “three times fewer diagnoses of autism at age 3,” said lead author Andrew Whitehouse, PhD, in a statement.
“No trial of a preemptive infant intervention, applied prior to diagnosis, has to date shown such an effect to impact diagnostic outcomes – until now,” he said.
Study intervention is a nontraditonal approach
Dr. Whitehouse, who is professor of Autism Research at Telethon Kids and University of Western Australia, and Director of CliniKids in Perth, said the intervention is a departure from traditional approaches. “Traditionally, therapy seeks to train children to learn ‘typical’ behaviors,” he said in an email. “The difference of this therapy is that we help parents understand the unique abilities of their baby, and to use these strengths as a foundation for future development.”
Dr. Whitehouse’s study included 103 children (aged approximately 12 months), who displayed at least three of five behaviors indicating a high likelihood of ASD as defined by the Social Attention and Communication Surveillance–Revised (SACS-R) 12-month checklist. The infants were randomized to receive either usual care or the intervention, which is called the iBASIS–Video Interaction to Promote Positive Parenting (iBASIS-VIPP). Usual care was delivered by community physicians, whereas the intervention involved 10 sessions delivered at home by a trained therapist.
“The iBASIS-VIPP uses video-feedback as a means of helping parents recognize their baby’s communication cues so they can respond in a way that builds their social communication development,” Dr. Whitehouse explained in an interview. “The therapist then provides guidance to the parent as to how their baby is communicating with them, and they can communicate back to have back-and-forth conversations.”
“We know these back-and-forth conversations are crucial to support early social communication development, and are a precursor to more complex skills, such as verbal language,” he added.
Reassessment of the children at age 3 years showed a “small but enduring” benefit of the intervention, noted the authors. Children in the intervention group had a reduction in ASD symptom severity (P = .04), and reduced odds of ASD classification, compared with children receiving usual care (6.7% vs. 20.5%; odds ratio, 0.18; P = .02).
The findings provide “initial evidence of efficacy for a new clinical model that uses a specific developmentally focused intervention,” noted the authors. “The children falling below the diagnostic threshold still had developmental difficulties, but by working with each child’s unique differences, rather than trying to counter them, the therapy has effectively supported their development through the early childhood years,” noted Dr. Whitehouse in a statement.
Other research has shown benefits of new study approach
This is a “solid” study, “but, as acknowledged by the authors, the main effects are small in magnitude, and longer-term outcomes will be important to capture,” said Jessica Brian, PhD, C Psych, associate professor in the department of pediatrics at the University of Toronto, colead at the Autism Research Centre, and psychologist and clinician-investigator at Holland Bloorview Kids Rehab Hospital in Toronto.
Dr. Brian said she and her coauthors of a paper published in Autism Research and others have shown that the kind of approach used in the new study can be helpful for enhancing different areas of toddler development, but “the specific finding of reduced likelihood of a clinical ASD diagnosis is a bit different.”
The goal of reducing the likelihood of an ASD diagnosis “needs to be considered carefully, from the perspective of autism acceptance,” she added. “From an acceptance lens, the primary objective of early intervention in ASD might be better positioned as aiming to enhance or support a young child’s development, help them make developmental progress, build on their strengths, optimize outcomes, or reduce impairment. … I think the authors do a good job of balancing this perspective.”
New study shows value of parent-mediated interventions
Overall, Dr. Brian, who coauthored the Canadian Paediatric Society’s position statement on ASD diagnosis, lauded the findings as good news.
“It shows the value of using parent-mediated interventions, which are far less costly and are more resource-efficient than most therapist-delivered models.”
“In cases where parent-mediated approaches are made available to families prior to diagnosis, there is potential for strong effects, when the brain is most amenable to learning. Such models may also be an ideal fit before diagnosis, since they are less resource-intensive than therapist-delivered models, which may only be funded by governments once a diagnosis is confirmed,” she said.
“Finally, parent-mediated programs have the potential to support parents during what, for many families, is a particularly challenging time as they identify their child’s developmental differences or receive a diagnosis. Such programs have potential to increase parents’ confidence in parenting a young child with unique learning needs.”
What Dr. Brian thought was missing from the paper was acknowledgment that, “despite early developmental gains from parent-mediated interventions, it is likely that most children with ASD will need additional supports throughout development.”
This study was sponsored by the Telethon Kids Institute. Dr. Whitehouse reported no conflicts of interest. Dr. Brian codeveloped a parent-mediated intervention for toddlers with probable or confirmed ASD (the Social ABCs), for which she does not receive any royalties.
These findings, which were published in JAMA Pediatrics, were the first evidence worldwide that a preemptive intervention during infancy could lead to such a significant improvement in children’s social development, resulting in “three times fewer diagnoses of autism at age 3,” said lead author Andrew Whitehouse, PhD, in a statement.
“No trial of a preemptive infant intervention, applied prior to diagnosis, has to date shown such an effect to impact diagnostic outcomes – until now,” he said.
Study intervention is a nontraditonal approach
Dr. Whitehouse, who is professor of Autism Research at Telethon Kids and University of Western Australia, and Director of CliniKids in Perth, said the intervention is a departure from traditional approaches. “Traditionally, therapy seeks to train children to learn ‘typical’ behaviors,” he said in an email. “The difference of this therapy is that we help parents understand the unique abilities of their baby, and to use these strengths as a foundation for future development.”
Dr. Whitehouse’s study included 103 children (aged approximately 12 months), who displayed at least three of five behaviors indicating a high likelihood of ASD as defined by the Social Attention and Communication Surveillance–Revised (SACS-R) 12-month checklist. The infants were randomized to receive either usual care or the intervention, which is called the iBASIS–Video Interaction to Promote Positive Parenting (iBASIS-VIPP). Usual care was delivered by community physicians, whereas the intervention involved 10 sessions delivered at home by a trained therapist.
“The iBASIS-VIPP uses video-feedback as a means of helping parents recognize their baby’s communication cues so they can respond in a way that builds their social communication development,” Dr. Whitehouse explained in an interview. “The therapist then provides guidance to the parent as to how their baby is communicating with them, and they can communicate back to have back-and-forth conversations.”
“We know these back-and-forth conversations are crucial to support early social communication development, and are a precursor to more complex skills, such as verbal language,” he added.
Reassessment of the children at age 3 years showed a “small but enduring” benefit of the intervention, noted the authors. Children in the intervention group had a reduction in ASD symptom severity (P = .04), and reduced odds of ASD classification, compared with children receiving usual care (6.7% vs. 20.5%; odds ratio, 0.18; P = .02).
The findings provide “initial evidence of efficacy for a new clinical model that uses a specific developmentally focused intervention,” noted the authors. “The children falling below the diagnostic threshold still had developmental difficulties, but by working with each child’s unique differences, rather than trying to counter them, the therapy has effectively supported their development through the early childhood years,” noted Dr. Whitehouse in a statement.
Other research has shown benefits of new study approach
This is a “solid” study, “but, as acknowledged by the authors, the main effects are small in magnitude, and longer-term outcomes will be important to capture,” said Jessica Brian, PhD, C Psych, associate professor in the department of pediatrics at the University of Toronto, colead at the Autism Research Centre, and psychologist and clinician-investigator at Holland Bloorview Kids Rehab Hospital in Toronto.
Dr. Brian said she and her coauthors of a paper published in Autism Research and others have shown that the kind of approach used in the new study can be helpful for enhancing different areas of toddler development, but “the specific finding of reduced likelihood of a clinical ASD diagnosis is a bit different.”
The goal of reducing the likelihood of an ASD diagnosis “needs to be considered carefully, from the perspective of autism acceptance,” she added. “From an acceptance lens, the primary objective of early intervention in ASD might be better positioned as aiming to enhance or support a young child’s development, help them make developmental progress, build on their strengths, optimize outcomes, or reduce impairment. … I think the authors do a good job of balancing this perspective.”
New study shows value of parent-mediated interventions
Overall, Dr. Brian, who coauthored the Canadian Paediatric Society’s position statement on ASD diagnosis, lauded the findings as good news.
“It shows the value of using parent-mediated interventions, which are far less costly and are more resource-efficient than most therapist-delivered models.”
“In cases where parent-mediated approaches are made available to families prior to diagnosis, there is potential for strong effects, when the brain is most amenable to learning. Such models may also be an ideal fit before diagnosis, since they are less resource-intensive than therapist-delivered models, which may only be funded by governments once a diagnosis is confirmed,” she said.
“Finally, parent-mediated programs have the potential to support parents during what, for many families, is a particularly challenging time as they identify their child’s developmental differences or receive a diagnosis. Such programs have potential to increase parents’ confidence in parenting a young child with unique learning needs.”
What Dr. Brian thought was missing from the paper was acknowledgment that, “despite early developmental gains from parent-mediated interventions, it is likely that most children with ASD will need additional supports throughout development.”
This study was sponsored by the Telethon Kids Institute. Dr. Whitehouse reported no conflicts of interest. Dr. Brian codeveloped a parent-mediated intervention for toddlers with probable or confirmed ASD (the Social ABCs), for which she does not receive any royalties.
FROM JAMA PEDIATRICS
Nonopioid med promising for neuropathic pain
Top-line results from a phase 2 study suggest vixotrigine (BIIB074, Biogen), a nonopioid investigational oral pain medication, reduces chronic neuropathic pain caused by small fiber neuropathy (SFN) and is generally well tolerated.
“We are encouraged by the overall results of the CONVEY study, especially given the significant unmet medical need for additional agents to treat chronic painful neuropathy,” Katherine Dawson, MD, senior vice president and head of the therapeutics development unit at Biogen, said in a news release.
Vixotrigine (BIIB074) is a peripherally and centrally acting, orally administered, voltage- and use-dependent voltage-gated sodium channel blocker.
CONVEY was a phase 2, placebo-controlled, double-blind, randomized withdrawal study of 265 patients experiencing pain from confirmed idiopathic or diabetes-associated SFN.
Following a 4-week open-label run-in period, 123 responders to vixotrigine were randomly allocated to 200 mg or 350 mg vixotrigine or placebo twice daily for 12 weeks in the double-blind portion of the study.
At week 12, vixotrigine 200 mg twice daily met the primary endpoint of a statistically significant reduction from baseline in the mean average daily pain (ADP) score versus placebo (P = .0501).
A subgroup analysis showed a treatment effect in patients with diabetes-associated SFN but not in the smaller subgroup of patients with idiopathic SFN.
The 200-mg dose also led to a significant improvement over placebo in mean worst daily pain score at 12 weeks (P = .0455).
A numeric advantage of 200 mg vixotrigine over placebo was observed in additional secondary endpoints, including the proportion of patients with at least a 2-point improvement in ADP score and the proportion with at least a 30% reduction in ADP at week 12, but these failed to reach statistical significance.
Vixotrigine 350 mg twice daily did not meet the primary endpoint of mean change in ADP at 12 weeks.
However, treatment at the higher dose led to a significant increase in the proportion of patients who reported being “very much improved” or “much improved” over baseline (P = .0580), Biogen reported.
In addition, a numeric advantage of 350 mg over placebo was observed in the proportion of patients with a 2-point or greater improvement in ADP score and the proportion with at least a 30% reduction in ADP at 12 weeks, but these also did not reach statistical significance.
Both doses of vixotrigine were “generally well tolerated and the safety profile was consistent with previous studies of vixotrigine with no evidence of abuse potential,” the company said.
In the open-label period, common adverse events seen in at least 2.5% of patients were dizziness, headache, vertigo, and nausea; adverse events led 5.3% of patients to discontinue the open-label portion of the study. Across the entire study, most adverse events were mild or moderate in severity.
“The totality of data from the vixotrigine program will inform potential doses for study in future phase 3 clinical trials,” the company said.
A version of this article first appeared on Medscape.com.
Top-line results from a phase 2 study suggest vixotrigine (BIIB074, Biogen), a nonopioid investigational oral pain medication, reduces chronic neuropathic pain caused by small fiber neuropathy (SFN) and is generally well tolerated.
“We are encouraged by the overall results of the CONVEY study, especially given the significant unmet medical need for additional agents to treat chronic painful neuropathy,” Katherine Dawson, MD, senior vice president and head of the therapeutics development unit at Biogen, said in a news release.
Vixotrigine (BIIB074) is a peripherally and centrally acting, orally administered, voltage- and use-dependent voltage-gated sodium channel blocker.
CONVEY was a phase 2, placebo-controlled, double-blind, randomized withdrawal study of 265 patients experiencing pain from confirmed idiopathic or diabetes-associated SFN.
Following a 4-week open-label run-in period, 123 responders to vixotrigine were randomly allocated to 200 mg or 350 mg vixotrigine or placebo twice daily for 12 weeks in the double-blind portion of the study.
At week 12, vixotrigine 200 mg twice daily met the primary endpoint of a statistically significant reduction from baseline in the mean average daily pain (ADP) score versus placebo (P = .0501).
A subgroup analysis showed a treatment effect in patients with diabetes-associated SFN but not in the smaller subgroup of patients with idiopathic SFN.
The 200-mg dose also led to a significant improvement over placebo in mean worst daily pain score at 12 weeks (P = .0455).
A numeric advantage of 200 mg vixotrigine over placebo was observed in additional secondary endpoints, including the proportion of patients with at least a 2-point improvement in ADP score and the proportion with at least a 30% reduction in ADP at week 12, but these failed to reach statistical significance.
Vixotrigine 350 mg twice daily did not meet the primary endpoint of mean change in ADP at 12 weeks.
However, treatment at the higher dose led to a significant increase in the proportion of patients who reported being “very much improved” or “much improved” over baseline (P = .0580), Biogen reported.
In addition, a numeric advantage of 350 mg over placebo was observed in the proportion of patients with a 2-point or greater improvement in ADP score and the proportion with at least a 30% reduction in ADP at 12 weeks, but these also did not reach statistical significance.
Both doses of vixotrigine were “generally well tolerated and the safety profile was consistent with previous studies of vixotrigine with no evidence of abuse potential,” the company said.
In the open-label period, common adverse events seen in at least 2.5% of patients were dizziness, headache, vertigo, and nausea; adverse events led 5.3% of patients to discontinue the open-label portion of the study. Across the entire study, most adverse events were mild or moderate in severity.
“The totality of data from the vixotrigine program will inform potential doses for study in future phase 3 clinical trials,” the company said.
A version of this article first appeared on Medscape.com.
Top-line results from a phase 2 study suggest vixotrigine (BIIB074, Biogen), a nonopioid investigational oral pain medication, reduces chronic neuropathic pain caused by small fiber neuropathy (SFN) and is generally well tolerated.
“We are encouraged by the overall results of the CONVEY study, especially given the significant unmet medical need for additional agents to treat chronic painful neuropathy,” Katherine Dawson, MD, senior vice president and head of the therapeutics development unit at Biogen, said in a news release.
Vixotrigine (BIIB074) is a peripherally and centrally acting, orally administered, voltage- and use-dependent voltage-gated sodium channel blocker.
CONVEY was a phase 2, placebo-controlled, double-blind, randomized withdrawal study of 265 patients experiencing pain from confirmed idiopathic or diabetes-associated SFN.
Following a 4-week open-label run-in period, 123 responders to vixotrigine were randomly allocated to 200 mg or 350 mg vixotrigine or placebo twice daily for 12 weeks in the double-blind portion of the study.
At week 12, vixotrigine 200 mg twice daily met the primary endpoint of a statistically significant reduction from baseline in the mean average daily pain (ADP) score versus placebo (P = .0501).
A subgroup analysis showed a treatment effect in patients with diabetes-associated SFN but not in the smaller subgroup of patients with idiopathic SFN.
The 200-mg dose also led to a significant improvement over placebo in mean worst daily pain score at 12 weeks (P = .0455).
A numeric advantage of 200 mg vixotrigine over placebo was observed in additional secondary endpoints, including the proportion of patients with at least a 2-point improvement in ADP score and the proportion with at least a 30% reduction in ADP at week 12, but these failed to reach statistical significance.
Vixotrigine 350 mg twice daily did not meet the primary endpoint of mean change in ADP at 12 weeks.
However, treatment at the higher dose led to a significant increase in the proportion of patients who reported being “very much improved” or “much improved” over baseline (P = .0580), Biogen reported.
In addition, a numeric advantage of 350 mg over placebo was observed in the proportion of patients with a 2-point or greater improvement in ADP score and the proportion with at least a 30% reduction in ADP at 12 weeks, but these also did not reach statistical significance.
Both doses of vixotrigine were “generally well tolerated and the safety profile was consistent with previous studies of vixotrigine with no evidence of abuse potential,” the company said.
In the open-label period, common adverse events seen in at least 2.5% of patients were dizziness, headache, vertigo, and nausea; adverse events led 5.3% of patients to discontinue the open-label portion of the study. Across the entire study, most adverse events were mild or moderate in severity.
“The totality of data from the vixotrigine program will inform potential doses for study in future phase 3 clinical trials,” the company said.
A version of this article first appeared on Medscape.com.
FDA panel backs Pfizer's COVID booster for 65 and older, those at high risk
An expert panel that advises the Food and Drug Administration on its regulatory decisions voted Sept. 17 against recommending third doses of Pfizer’s COVID-19 vaccine for younger Americans.
But they didn’t kill the idea of booster shots completely.
In a dramatic, last-minute pivot, the 18 members of the FDA’s Vaccines and Related Biological Products Advisory Committee unanimously voted to recommend the FDA make boosters available for seniors and others at high risk of severe outcomes from COVID-19, including health care workers.
The 16-2 vote was a rebuttal to Pfizer’s initial request. The company had asked the FDA to allow it to offer third doses to all Americans over the age of 16 at least six months after their second shot.
The company requested an amendment to the full approval the FDA granted in August. That is the typical way boosters are authorized in the U.S., but it requires a higher bar of evidence and more regulatory scrutiny than the agency had been able to give since Pfizer filed for the change just days after its vaccine was granted full approval.
The committee’s actions were also a rebuff to the Biden administration, which announced before the FDA approved them that boosters would be rolled out to the general public Sept. 20. The announcement triggered the resignations of two of the agency’s top vaccine reviewers, who both participated in the Sept. 17 meeting.
After initially voting against Pfizer’s request to amend its license, the committee then worked on the fly with FDA officials to craft a strategy that would allow third doses to be offered under an emergency use authorization (EUA).
An EUA requires a lower standard of evidence and is more specific. It will restrict third doses to a more defined population than a change to the license would. It will also require Pfizer to continue to monitor the safety of third doses as they begin to be administered.
“This should demonstrate to the public that the members of this committee are independent of the FDA and that we do, in fact, bring our voices to the table when we are asked to serve on this committee,” said Archana Chattergee, MD, a pediatric infectious disease specialist who is dean of the Chicago Medical School at Rosalind Franklin University in Illinois.
The FDA doesn’t have to follow the committee’s recommendation, but almost certainly will, though regulators said they may still make some changes.
“We are not bound at FDA by your vote, we can tweak this,” said Peter Marks, MD, director of the Center for Biologics Evaluation and Research at the FDA. Dr. Marks participated in the meeting and helped to draft the revised proposal.
If the FDA issues the recommended EUA, a council of independent advisors to the CDC will make specific recommendations about how the third doses should be given. After the CDC director weighs in, boosters will begin rolling out to the public.
Moderna submitted data to the FDA on Sept. 1 in support of adding a booster dose to its regimen. The agency has not yet scheduled a public review of that data.
The Biden administration is prepared to administer shots as soon as they get the green light, Surgeon General Vivek Murthy, MD, said at a White House briefing earlier Sept. 17.
"This process is consistent with what we outlined in August where our goals were to stay ahead of the virus," Dr. Murthy said. "Our goal then and now is to protect the health and well-being of the public. As soon as the FDA and CDC complete their evaluations, we will be ready to move forward accordingly."
He added, "We've used this time since our August announcement to communicate and coordinate with pharmacy partners, nursing homes, states, and localities."
White House COVID-19 Response Coordinator Jeff Zients said vaccine supply is "in good shape for all Americans to get boosters as recommended."
Taking cues from Israel
In considering Pfizer’s original request, the committee overwhelmingly felt that they didn’t have enough information to say that the benefits of an additional dose of vaccine in 16- and 17-year-olds would outweigh its risk. Teens have the highest risk of rare heart inflammation after vaccination, a side effect known as myocarditis. It is not known how the vaccines are causing these cases of heart swelling. Most who have been diagnosed with the condition have recovered, though some have needed hospital care.
Pfizer didn’t include 16- and 17-year-olds in its studies of boosters, which included about 300 people between the ages of 18 and 55. The company acknowledged that gap in its data but pointed to FDA guidance that said evidence from adults could be extrapolated to teens.
“We don’t know that much about risks,” said committee member Eric Rubin, MD, who is editor-in-chief of the New England Journal of Medicine.
Much of the data on the potential benefits and harms of third Pfizer doses comes from Israel, which first began rolling out boosters to older adults in July.
In a highly anticipated presentation, Sharon Alroy-Preis, Israel’s director of public health services, joined the meeting to describe Israel’s experience with boosters.
Israel began to see a third surge of COVID-19 cases in December.
“This was after having two waves and two lockdowns,” Ms. Alroy-Preis said. By the third surge, she said, Israelis were tired.
“We decided on a lockdown, but the compliance of the public wasn’t as it was in the previous two waves,” she said.
Then the vaccine arrived. Israel started vaccinations as soon as the FDA approved it, and they quickly vaccinated a high percentage of their population, about 3 months faster than the rest of the world.
All vaccinations are reported and tracked by the Ministry of Health, so the country is able to keep close tabs on how well the shots are working.
As vaccines rolled out, cases fell dramatically. The pandemic seemed to be behind them. Delta arrived in March. By June, their cases were doubling every 10 days, despite about 80% of their most vulnerable adults being fully vaccinated, she said.
Most concerning was that about 60% of severe cases were breakthrough cases in fully vaccinated individuals.
“We had to stop and figure out, was this a Delta issue,” she said. “Or was this a waning immunity issue.”
“We had some clue that it might not be the Delta variant, at least not alone,” she said.
People who had originally been first in line for the vaccines, seniors and health care workers, were having the highest rates of breakthrough infections. People further away from their second dose were more likely to get a breakthrough infection.
Ms. Alroy-Preis said that if they had not started booster doses in July, their hospitals would have been overwhelmed. They had projected that they would have 2,000 cases in the hospital each day.
Boosters have helped to flatten the curve, though they are still seeing a significant number of infections.
Data from Israel presented at the meeting show boosters are largely safe and effective at reducing severe outcomes in seniors. Israeli experience also showed that third doses, which generate very high levels of neutralizing antibodies—the first and fastest line of the body’s immune defense - -may also slow transmission of the virus.
Key differences in the U.S.
The benefit of slowing down the explosive spread of a highly contagious virus was tantalizing, but many members noted that circumstances in Israel are very different than in the United States. Israel went into its current Delta surge already having high levels of vaccination in its population. They also relied on the Pfizer vaccine almost exclusively for their campaign.
The United States used a different mix of vaccines – Pfizer, Moderna, and Johnson & Johnson -- and doesn’t have the same high level of vaccination coverage of its population.
In the United States, transmission is mainly being driven by unvaccinated people, Dr. Rubin noted.
“That really means the primary benefit is going to be in reducing disease,” he said, “And we know the people who are going to benefit from that … and those are the kinds of people the FDA has already approved a third dose for,” he said, referring to those with underlying health conditions.
But Israel only began vaccinating younger people a few weeks ago. Most are still within a window where rare risks like myocarditis could appear, Rubin noted.
He and other members of the committee said they wished they had more information about the safety of third doses in younger adults.
“We don’t have that right now, and I don’t think I would be comfortable giving it to a 16-year-old,” he said.
At the same time, the primary benefit for third doses would be in preventing severe disease, and overall, data from the United States and other countries show that two doses of the vaccines remain highly effective at preventing hospitalization and death.
Asked why Israel began to see more severe cases in fully vaccinated people, the CDC’s Sara Oliver, MD, a disease detective with the CDC, said it was probably due to a mix of factors including the fact that Israel defines severe cases a little differently.
In the United States, a severe case is generally a person who has to be hospitalized or who has died from the infection. In Israel, a person with a severe case is someone who has an elevated respiratory rate and someone who has a blood oxygen level less than 94%. In the United States, that kind of patient wouldn’t necessarily be hospitalized.
In the end, one of the two committee members who wanted full approval for Pfizer’s third doses said he was satisfied with the outcome.
Mark Sawyer, MD, a professor of pediatrics and infectious disease at the University of California at San Diego, said he voted yes on the first question because he thought full approval was the best way to give doctors the flexibility to prescribe the shots to vulnerable individuals.
“I’m really glad we authorized a vaccine for a third dose, and I plan to go out and get my vaccine this afternoon,” Dr. Sawyer said, noting that he was at high risk as a health care provider.
This article was updated 9/19/21.
A version of this article first appeared on Medscape.com.
An expert panel that advises the Food and Drug Administration on its regulatory decisions voted Sept. 17 against recommending third doses of Pfizer’s COVID-19 vaccine for younger Americans.
But they didn’t kill the idea of booster shots completely.
In a dramatic, last-minute pivot, the 18 members of the FDA’s Vaccines and Related Biological Products Advisory Committee unanimously voted to recommend the FDA make boosters available for seniors and others at high risk of severe outcomes from COVID-19, including health care workers.
The 16-2 vote was a rebuttal to Pfizer’s initial request. The company had asked the FDA to allow it to offer third doses to all Americans over the age of 16 at least six months after their second shot.
The company requested an amendment to the full approval the FDA granted in August. That is the typical way boosters are authorized in the U.S., but it requires a higher bar of evidence and more regulatory scrutiny than the agency had been able to give since Pfizer filed for the change just days after its vaccine was granted full approval.
The committee’s actions were also a rebuff to the Biden administration, which announced before the FDA approved them that boosters would be rolled out to the general public Sept. 20. The announcement triggered the resignations of two of the agency’s top vaccine reviewers, who both participated in the Sept. 17 meeting.
After initially voting against Pfizer’s request to amend its license, the committee then worked on the fly with FDA officials to craft a strategy that would allow third doses to be offered under an emergency use authorization (EUA).
An EUA requires a lower standard of evidence and is more specific. It will restrict third doses to a more defined population than a change to the license would. It will also require Pfizer to continue to monitor the safety of third doses as they begin to be administered.
“This should demonstrate to the public that the members of this committee are independent of the FDA and that we do, in fact, bring our voices to the table when we are asked to serve on this committee,” said Archana Chattergee, MD, a pediatric infectious disease specialist who is dean of the Chicago Medical School at Rosalind Franklin University in Illinois.
The FDA doesn’t have to follow the committee’s recommendation, but almost certainly will, though regulators said they may still make some changes.
“We are not bound at FDA by your vote, we can tweak this,” said Peter Marks, MD, director of the Center for Biologics Evaluation and Research at the FDA. Dr. Marks participated in the meeting and helped to draft the revised proposal.
If the FDA issues the recommended EUA, a council of independent advisors to the CDC will make specific recommendations about how the third doses should be given. After the CDC director weighs in, boosters will begin rolling out to the public.
Moderna submitted data to the FDA on Sept. 1 in support of adding a booster dose to its regimen. The agency has not yet scheduled a public review of that data.
The Biden administration is prepared to administer shots as soon as they get the green light, Surgeon General Vivek Murthy, MD, said at a White House briefing earlier Sept. 17.
"This process is consistent with what we outlined in August where our goals were to stay ahead of the virus," Dr. Murthy said. "Our goal then and now is to protect the health and well-being of the public. As soon as the FDA and CDC complete their evaluations, we will be ready to move forward accordingly."
He added, "We've used this time since our August announcement to communicate and coordinate with pharmacy partners, nursing homes, states, and localities."
White House COVID-19 Response Coordinator Jeff Zients said vaccine supply is "in good shape for all Americans to get boosters as recommended."
Taking cues from Israel
In considering Pfizer’s original request, the committee overwhelmingly felt that they didn’t have enough information to say that the benefits of an additional dose of vaccine in 16- and 17-year-olds would outweigh its risk. Teens have the highest risk of rare heart inflammation after vaccination, a side effect known as myocarditis. It is not known how the vaccines are causing these cases of heart swelling. Most who have been diagnosed with the condition have recovered, though some have needed hospital care.
Pfizer didn’t include 16- and 17-year-olds in its studies of boosters, which included about 300 people between the ages of 18 and 55. The company acknowledged that gap in its data but pointed to FDA guidance that said evidence from adults could be extrapolated to teens.
“We don’t know that much about risks,” said committee member Eric Rubin, MD, who is editor-in-chief of the New England Journal of Medicine.
Much of the data on the potential benefits and harms of third Pfizer doses comes from Israel, which first began rolling out boosters to older adults in July.
In a highly anticipated presentation, Sharon Alroy-Preis, Israel’s director of public health services, joined the meeting to describe Israel’s experience with boosters.
Israel began to see a third surge of COVID-19 cases in December.
“This was after having two waves and two lockdowns,” Ms. Alroy-Preis said. By the third surge, she said, Israelis were tired.
“We decided on a lockdown, but the compliance of the public wasn’t as it was in the previous two waves,” she said.
Then the vaccine arrived. Israel started vaccinations as soon as the FDA approved it, and they quickly vaccinated a high percentage of their population, about 3 months faster than the rest of the world.
All vaccinations are reported and tracked by the Ministry of Health, so the country is able to keep close tabs on how well the shots are working.
As vaccines rolled out, cases fell dramatically. The pandemic seemed to be behind them. Delta arrived in March. By June, their cases were doubling every 10 days, despite about 80% of their most vulnerable adults being fully vaccinated, she said.
Most concerning was that about 60% of severe cases were breakthrough cases in fully vaccinated individuals.
“We had to stop and figure out, was this a Delta issue,” she said. “Or was this a waning immunity issue.”
“We had some clue that it might not be the Delta variant, at least not alone,” she said.
People who had originally been first in line for the vaccines, seniors and health care workers, were having the highest rates of breakthrough infections. People further away from their second dose were more likely to get a breakthrough infection.
Ms. Alroy-Preis said that if they had not started booster doses in July, their hospitals would have been overwhelmed. They had projected that they would have 2,000 cases in the hospital each day.
Boosters have helped to flatten the curve, though they are still seeing a significant number of infections.
Data from Israel presented at the meeting show boosters are largely safe and effective at reducing severe outcomes in seniors. Israeli experience also showed that third doses, which generate very high levels of neutralizing antibodies—the first and fastest line of the body’s immune defense - -may also slow transmission of the virus.
Key differences in the U.S.
The benefit of slowing down the explosive spread of a highly contagious virus was tantalizing, but many members noted that circumstances in Israel are very different than in the United States. Israel went into its current Delta surge already having high levels of vaccination in its population. They also relied on the Pfizer vaccine almost exclusively for their campaign.
The United States used a different mix of vaccines – Pfizer, Moderna, and Johnson & Johnson -- and doesn’t have the same high level of vaccination coverage of its population.
In the United States, transmission is mainly being driven by unvaccinated people, Dr. Rubin noted.
“That really means the primary benefit is going to be in reducing disease,” he said, “And we know the people who are going to benefit from that … and those are the kinds of people the FDA has already approved a third dose for,” he said, referring to those with underlying health conditions.
But Israel only began vaccinating younger people a few weeks ago. Most are still within a window where rare risks like myocarditis could appear, Rubin noted.
He and other members of the committee said they wished they had more information about the safety of third doses in younger adults.
“We don’t have that right now, and I don’t think I would be comfortable giving it to a 16-year-old,” he said.
At the same time, the primary benefit for third doses would be in preventing severe disease, and overall, data from the United States and other countries show that two doses of the vaccines remain highly effective at preventing hospitalization and death.
Asked why Israel began to see more severe cases in fully vaccinated people, the CDC’s Sara Oliver, MD, a disease detective with the CDC, said it was probably due to a mix of factors including the fact that Israel defines severe cases a little differently.
In the United States, a severe case is generally a person who has to be hospitalized or who has died from the infection. In Israel, a person with a severe case is someone who has an elevated respiratory rate and someone who has a blood oxygen level less than 94%. In the United States, that kind of patient wouldn’t necessarily be hospitalized.
In the end, one of the two committee members who wanted full approval for Pfizer’s third doses said he was satisfied with the outcome.
Mark Sawyer, MD, a professor of pediatrics and infectious disease at the University of California at San Diego, said he voted yes on the first question because he thought full approval was the best way to give doctors the flexibility to prescribe the shots to vulnerable individuals.
“I’m really glad we authorized a vaccine for a third dose, and I plan to go out and get my vaccine this afternoon,” Dr. Sawyer said, noting that he was at high risk as a health care provider.
This article was updated 9/19/21.
A version of this article first appeared on Medscape.com.
An expert panel that advises the Food and Drug Administration on its regulatory decisions voted Sept. 17 against recommending third doses of Pfizer’s COVID-19 vaccine for younger Americans.
But they didn’t kill the idea of booster shots completely.
In a dramatic, last-minute pivot, the 18 members of the FDA’s Vaccines and Related Biological Products Advisory Committee unanimously voted to recommend the FDA make boosters available for seniors and others at high risk of severe outcomes from COVID-19, including health care workers.
The 16-2 vote was a rebuttal to Pfizer’s initial request. The company had asked the FDA to allow it to offer third doses to all Americans over the age of 16 at least six months after their second shot.
The company requested an amendment to the full approval the FDA granted in August. That is the typical way boosters are authorized in the U.S., but it requires a higher bar of evidence and more regulatory scrutiny than the agency had been able to give since Pfizer filed for the change just days after its vaccine was granted full approval.
The committee’s actions were also a rebuff to the Biden administration, which announced before the FDA approved them that boosters would be rolled out to the general public Sept. 20. The announcement triggered the resignations of two of the agency’s top vaccine reviewers, who both participated in the Sept. 17 meeting.
After initially voting against Pfizer’s request to amend its license, the committee then worked on the fly with FDA officials to craft a strategy that would allow third doses to be offered under an emergency use authorization (EUA).
An EUA requires a lower standard of evidence and is more specific. It will restrict third doses to a more defined population than a change to the license would. It will also require Pfizer to continue to monitor the safety of third doses as they begin to be administered.
“This should demonstrate to the public that the members of this committee are independent of the FDA and that we do, in fact, bring our voices to the table when we are asked to serve on this committee,” said Archana Chattergee, MD, a pediatric infectious disease specialist who is dean of the Chicago Medical School at Rosalind Franklin University in Illinois.
The FDA doesn’t have to follow the committee’s recommendation, but almost certainly will, though regulators said they may still make some changes.
“We are not bound at FDA by your vote, we can tweak this,” said Peter Marks, MD, director of the Center for Biologics Evaluation and Research at the FDA. Dr. Marks participated in the meeting and helped to draft the revised proposal.
If the FDA issues the recommended EUA, a council of independent advisors to the CDC will make specific recommendations about how the third doses should be given. After the CDC director weighs in, boosters will begin rolling out to the public.
Moderna submitted data to the FDA on Sept. 1 in support of adding a booster dose to its regimen. The agency has not yet scheduled a public review of that data.
The Biden administration is prepared to administer shots as soon as they get the green light, Surgeon General Vivek Murthy, MD, said at a White House briefing earlier Sept. 17.
"This process is consistent with what we outlined in August where our goals were to stay ahead of the virus," Dr. Murthy said. "Our goal then and now is to protect the health and well-being of the public. As soon as the FDA and CDC complete their evaluations, we will be ready to move forward accordingly."
He added, "We've used this time since our August announcement to communicate and coordinate with pharmacy partners, nursing homes, states, and localities."
White House COVID-19 Response Coordinator Jeff Zients said vaccine supply is "in good shape for all Americans to get boosters as recommended."
Taking cues from Israel
In considering Pfizer’s original request, the committee overwhelmingly felt that they didn’t have enough information to say that the benefits of an additional dose of vaccine in 16- and 17-year-olds would outweigh its risk. Teens have the highest risk of rare heart inflammation after vaccination, a side effect known as myocarditis. It is not known how the vaccines are causing these cases of heart swelling. Most who have been diagnosed with the condition have recovered, though some have needed hospital care.
Pfizer didn’t include 16- and 17-year-olds in its studies of boosters, which included about 300 people between the ages of 18 and 55. The company acknowledged that gap in its data but pointed to FDA guidance that said evidence from adults could be extrapolated to teens.
“We don’t know that much about risks,” said committee member Eric Rubin, MD, who is editor-in-chief of the New England Journal of Medicine.
Much of the data on the potential benefits and harms of third Pfizer doses comes from Israel, which first began rolling out boosters to older adults in July.
In a highly anticipated presentation, Sharon Alroy-Preis, Israel’s director of public health services, joined the meeting to describe Israel’s experience with boosters.
Israel began to see a third surge of COVID-19 cases in December.
“This was after having two waves and two lockdowns,” Ms. Alroy-Preis said. By the third surge, she said, Israelis were tired.
“We decided on a lockdown, but the compliance of the public wasn’t as it was in the previous two waves,” she said.
Then the vaccine arrived. Israel started vaccinations as soon as the FDA approved it, and they quickly vaccinated a high percentage of their population, about 3 months faster than the rest of the world.
All vaccinations are reported and tracked by the Ministry of Health, so the country is able to keep close tabs on how well the shots are working.
As vaccines rolled out, cases fell dramatically. The pandemic seemed to be behind them. Delta arrived in March. By June, their cases were doubling every 10 days, despite about 80% of their most vulnerable adults being fully vaccinated, she said.
Most concerning was that about 60% of severe cases were breakthrough cases in fully vaccinated individuals.
“We had to stop and figure out, was this a Delta issue,” she said. “Or was this a waning immunity issue.”
“We had some clue that it might not be the Delta variant, at least not alone,” she said.
People who had originally been first in line for the vaccines, seniors and health care workers, were having the highest rates of breakthrough infections. People further away from their second dose were more likely to get a breakthrough infection.
Ms. Alroy-Preis said that if they had not started booster doses in July, their hospitals would have been overwhelmed. They had projected that they would have 2,000 cases in the hospital each day.
Boosters have helped to flatten the curve, though they are still seeing a significant number of infections.
Data from Israel presented at the meeting show boosters are largely safe and effective at reducing severe outcomes in seniors. Israeli experience also showed that third doses, which generate very high levels of neutralizing antibodies—the first and fastest line of the body’s immune defense - -may also slow transmission of the virus.
Key differences in the U.S.
The benefit of slowing down the explosive spread of a highly contagious virus was tantalizing, but many members noted that circumstances in Israel are very different than in the United States. Israel went into its current Delta surge already having high levels of vaccination in its population. They also relied on the Pfizer vaccine almost exclusively for their campaign.
The United States used a different mix of vaccines – Pfizer, Moderna, and Johnson & Johnson -- and doesn’t have the same high level of vaccination coverage of its population.
In the United States, transmission is mainly being driven by unvaccinated people, Dr. Rubin noted.
“That really means the primary benefit is going to be in reducing disease,” he said, “And we know the people who are going to benefit from that … and those are the kinds of people the FDA has already approved a third dose for,” he said, referring to those with underlying health conditions.
But Israel only began vaccinating younger people a few weeks ago. Most are still within a window where rare risks like myocarditis could appear, Rubin noted.
He and other members of the committee said they wished they had more information about the safety of third doses in younger adults.
“We don’t have that right now, and I don’t think I would be comfortable giving it to a 16-year-old,” he said.
At the same time, the primary benefit for third doses would be in preventing severe disease, and overall, data from the United States and other countries show that two doses of the vaccines remain highly effective at preventing hospitalization and death.
Asked why Israel began to see more severe cases in fully vaccinated people, the CDC’s Sara Oliver, MD, a disease detective with the CDC, said it was probably due to a mix of factors including the fact that Israel defines severe cases a little differently.
In the United States, a severe case is generally a person who has to be hospitalized or who has died from the infection. In Israel, a person with a severe case is someone who has an elevated respiratory rate and someone who has a blood oxygen level less than 94%. In the United States, that kind of patient wouldn’t necessarily be hospitalized.
In the end, one of the two committee members who wanted full approval for Pfizer’s third doses said he was satisfied with the outcome.
Mark Sawyer, MD, a professor of pediatrics and infectious disease at the University of California at San Diego, said he voted yes on the first question because he thought full approval was the best way to give doctors the flexibility to prescribe the shots to vulnerable individuals.
“I’m really glad we authorized a vaccine for a third dose, and I plan to go out and get my vaccine this afternoon,” Dr. Sawyer said, noting that he was at high risk as a health care provider.
This article was updated 9/19/21.
A version of this article first appeared on Medscape.com.
As opioid deaths climb, human trials begin for vaccine
Opioid-related drug overdose deaths in the United States exploded to an estimated record high of 69,031 people in 2020, topping the 49,860 deaths logged in 2019, according to a new report from the Centers for Disease Control and Prevention. Most of the deaths involved synthetic opioids such as fentanyl.
President Joe Biden has pledged more than $10 billion to expand access to prevention, treatment, and recovery services. The money is important as people receiving treatment for opioid use disorder have a high risk for relapse, and that means a high risk for opioid overdose.
Now, researchers are studying a possible bridge to successful recovery: A vaccine that could blunt the drugs’ ability to cause harm.
The first such vaccines are now entering clinical trials, raising hopes of adding another tool to the antiaddiction armamentarium. But even if the vaccines prove safe and effective, their success could generate some new problems to solve.
An advantage of vaccines is that their effects can last for several months, said trial investigator Sandra Comer, PhD, professor of neurobiology and psychiatry at Columbia University Irving Medical Center, New York. Dropout rates for existing medical therapies for opioid use disorder are as high as 50% at 6 months, and a vaccine could protect people from overdose and give them time to re-enter treatment.
“It serves as a bit of a safety net,” she said.
The first vaccine to enter a trial targets oxycodone. Volunteers are being recruited who have a diagnosis of opioid use disorder but are not being medically treated and are still using opioids. A third of them will receive a placebo vaccine, a third will receive a low-dose injection of vaccine, and the other third will receive a high-dose vaccine.
A shot against oxycodone
Researchers are primarily tracking the safety of the shot, but they’re also looking at whether vaccination prevents the euphoria that opioids usually produce. They expect to enroll 24 people initially but expand to 45 if results look promising.
In response to the shot, the body produces antibodies, proteins that tag oxycodone and keep it from reaching the brain. If the drug can’t reach brain cells, it can’t produce euphoria. And more important for lifesaving effects, it can’t block the brain’s signals to the body to breathe. The vaccine has already performed well in animal studies.
Previous trials of vaccines for cocaine and nicotine failed. Those vaccines made it to the last clinical trial stage, but didn’t prove effective overall. So this time, investigators plan to track antibody levels in participants, examining blood samples for signs of a good immune response to the vaccine.
But even though earlier cocaine and nicotine vaccines didn’t work for everybody, there were some people they seemed to help. This is why investigators involved in opioid vaccine trials want to track immune responses, said Marco Pravetoni, PhD, associate professor of pharmacology and medicine at the University of Minnesota, Minneapolis, whose team will be assessing the blood samples. Ultimately, a doctor might even be able to use this information to tailor vaccine selection to a specific person.
Dr. Pravetoni also said that oxycodone is one of three vaccine targets – the other two are heroin and fentanyl – that researchers hope to combine into a single shot. Recipients might need to have one shot a month for the first 3 to 4 months and then receive annual boosters.
Stopping the pain
The vaccines also raise some issues that need attention, said Cody Wenthur, PharmD, PhD, assistant professor of pharmacy at the University of Wisconsin–Madison, who is not involved in the vaccine trials.
“If you’re vaccinated against oxycodone, you might not have access to adequate pain control if you get into a car accident, for example,” he said.
Clinicians could use other opioids for pain management, but limiting the opioids that the vaccine targets is a “double-edged sword,” said Dr. Wenthur, because vaccinated people could just switch their opioid of choice to one that a vaccine does not inhibit.
Although these issues need to be addressed, vaccines, if successful, will have an important role. Dr. Wenthur noted a survey of pharmacists and pharmacy students that he and his group conducted showing that respondents “overwhelmingly” viewed a potential vaccine as helpful.
said Dr. Pravetoni. He mentioned the 2002 incident when terrorists took over a theater in Moscow and Russian special forces are thought to have used an aerosolized form of fentanyl to incapacitate everyone in the room. More than 100 of the hostages died, and the episode raised the specter of opioids being used in chemical attacks.
Dr. Pravetoni said vaccination could offer protection for first responders, law enforcement or other people whose professions place them at risk for inhalation, either accidentally or through such attacks.
These or other real-world applications for people at risk for exposure are several years away. Dr. Pravetoni said it took 10 years to get to this phase and estimates that, in about 5 years, a vaccine that targets multiple opioid drugs might enter the first clinical trial.
A version of this article first appeared on WebMD.com.
Opioid-related drug overdose deaths in the United States exploded to an estimated record high of 69,031 people in 2020, topping the 49,860 deaths logged in 2019, according to a new report from the Centers for Disease Control and Prevention. Most of the deaths involved synthetic opioids such as fentanyl.
President Joe Biden has pledged more than $10 billion to expand access to prevention, treatment, and recovery services. The money is important as people receiving treatment for opioid use disorder have a high risk for relapse, and that means a high risk for opioid overdose.
Now, researchers are studying a possible bridge to successful recovery: A vaccine that could blunt the drugs’ ability to cause harm.
The first such vaccines are now entering clinical trials, raising hopes of adding another tool to the antiaddiction armamentarium. But even if the vaccines prove safe and effective, their success could generate some new problems to solve.
An advantage of vaccines is that their effects can last for several months, said trial investigator Sandra Comer, PhD, professor of neurobiology and psychiatry at Columbia University Irving Medical Center, New York. Dropout rates for existing medical therapies for opioid use disorder are as high as 50% at 6 months, and a vaccine could protect people from overdose and give them time to re-enter treatment.
“It serves as a bit of a safety net,” she said.
The first vaccine to enter a trial targets oxycodone. Volunteers are being recruited who have a diagnosis of opioid use disorder but are not being medically treated and are still using opioids. A third of them will receive a placebo vaccine, a third will receive a low-dose injection of vaccine, and the other third will receive a high-dose vaccine.
A shot against oxycodone
Researchers are primarily tracking the safety of the shot, but they’re also looking at whether vaccination prevents the euphoria that opioids usually produce. They expect to enroll 24 people initially but expand to 45 if results look promising.
In response to the shot, the body produces antibodies, proteins that tag oxycodone and keep it from reaching the brain. If the drug can’t reach brain cells, it can’t produce euphoria. And more important for lifesaving effects, it can’t block the brain’s signals to the body to breathe. The vaccine has already performed well in animal studies.
Previous trials of vaccines for cocaine and nicotine failed. Those vaccines made it to the last clinical trial stage, but didn’t prove effective overall. So this time, investigators plan to track antibody levels in participants, examining blood samples for signs of a good immune response to the vaccine.
But even though earlier cocaine and nicotine vaccines didn’t work for everybody, there were some people they seemed to help. This is why investigators involved in opioid vaccine trials want to track immune responses, said Marco Pravetoni, PhD, associate professor of pharmacology and medicine at the University of Minnesota, Minneapolis, whose team will be assessing the blood samples. Ultimately, a doctor might even be able to use this information to tailor vaccine selection to a specific person.
Dr. Pravetoni also said that oxycodone is one of three vaccine targets – the other two are heroin and fentanyl – that researchers hope to combine into a single shot. Recipients might need to have one shot a month for the first 3 to 4 months and then receive annual boosters.
Stopping the pain
The vaccines also raise some issues that need attention, said Cody Wenthur, PharmD, PhD, assistant professor of pharmacy at the University of Wisconsin–Madison, who is not involved in the vaccine trials.
“If you’re vaccinated against oxycodone, you might not have access to adequate pain control if you get into a car accident, for example,” he said.
Clinicians could use other opioids for pain management, but limiting the opioids that the vaccine targets is a “double-edged sword,” said Dr. Wenthur, because vaccinated people could just switch their opioid of choice to one that a vaccine does not inhibit.
Although these issues need to be addressed, vaccines, if successful, will have an important role. Dr. Wenthur noted a survey of pharmacists and pharmacy students that he and his group conducted showing that respondents “overwhelmingly” viewed a potential vaccine as helpful.
said Dr. Pravetoni. He mentioned the 2002 incident when terrorists took over a theater in Moscow and Russian special forces are thought to have used an aerosolized form of fentanyl to incapacitate everyone in the room. More than 100 of the hostages died, and the episode raised the specter of opioids being used in chemical attacks.
Dr. Pravetoni said vaccination could offer protection for first responders, law enforcement or other people whose professions place them at risk for inhalation, either accidentally or through such attacks.
These or other real-world applications for people at risk for exposure are several years away. Dr. Pravetoni said it took 10 years to get to this phase and estimates that, in about 5 years, a vaccine that targets multiple opioid drugs might enter the first clinical trial.
A version of this article first appeared on WebMD.com.
Opioid-related drug overdose deaths in the United States exploded to an estimated record high of 69,031 people in 2020, topping the 49,860 deaths logged in 2019, according to a new report from the Centers for Disease Control and Prevention. Most of the deaths involved synthetic opioids such as fentanyl.
President Joe Biden has pledged more than $10 billion to expand access to prevention, treatment, and recovery services. The money is important as people receiving treatment for opioid use disorder have a high risk for relapse, and that means a high risk for opioid overdose.
Now, researchers are studying a possible bridge to successful recovery: A vaccine that could blunt the drugs’ ability to cause harm.
The first such vaccines are now entering clinical trials, raising hopes of adding another tool to the antiaddiction armamentarium. But even if the vaccines prove safe and effective, their success could generate some new problems to solve.
An advantage of vaccines is that their effects can last for several months, said trial investigator Sandra Comer, PhD, professor of neurobiology and psychiatry at Columbia University Irving Medical Center, New York. Dropout rates for existing medical therapies for opioid use disorder are as high as 50% at 6 months, and a vaccine could protect people from overdose and give them time to re-enter treatment.
“It serves as a bit of a safety net,” she said.
The first vaccine to enter a trial targets oxycodone. Volunteers are being recruited who have a diagnosis of opioid use disorder but are not being medically treated and are still using opioids. A third of them will receive a placebo vaccine, a third will receive a low-dose injection of vaccine, and the other third will receive a high-dose vaccine.
A shot against oxycodone
Researchers are primarily tracking the safety of the shot, but they’re also looking at whether vaccination prevents the euphoria that opioids usually produce. They expect to enroll 24 people initially but expand to 45 if results look promising.
In response to the shot, the body produces antibodies, proteins that tag oxycodone and keep it from reaching the brain. If the drug can’t reach brain cells, it can’t produce euphoria. And more important for lifesaving effects, it can’t block the brain’s signals to the body to breathe. The vaccine has already performed well in animal studies.
Previous trials of vaccines for cocaine and nicotine failed. Those vaccines made it to the last clinical trial stage, but didn’t prove effective overall. So this time, investigators plan to track antibody levels in participants, examining blood samples for signs of a good immune response to the vaccine.
But even though earlier cocaine and nicotine vaccines didn’t work for everybody, there were some people they seemed to help. This is why investigators involved in opioid vaccine trials want to track immune responses, said Marco Pravetoni, PhD, associate professor of pharmacology and medicine at the University of Minnesota, Minneapolis, whose team will be assessing the blood samples. Ultimately, a doctor might even be able to use this information to tailor vaccine selection to a specific person.
Dr. Pravetoni also said that oxycodone is one of three vaccine targets – the other two are heroin and fentanyl – that researchers hope to combine into a single shot. Recipients might need to have one shot a month for the first 3 to 4 months and then receive annual boosters.
Stopping the pain
The vaccines also raise some issues that need attention, said Cody Wenthur, PharmD, PhD, assistant professor of pharmacy at the University of Wisconsin–Madison, who is not involved in the vaccine trials.
“If you’re vaccinated against oxycodone, you might not have access to adequate pain control if you get into a car accident, for example,” he said.
Clinicians could use other opioids for pain management, but limiting the opioids that the vaccine targets is a “double-edged sword,” said Dr. Wenthur, because vaccinated people could just switch their opioid of choice to one that a vaccine does not inhibit.
Although these issues need to be addressed, vaccines, if successful, will have an important role. Dr. Wenthur noted a survey of pharmacists and pharmacy students that he and his group conducted showing that respondents “overwhelmingly” viewed a potential vaccine as helpful.
said Dr. Pravetoni. He mentioned the 2002 incident when terrorists took over a theater in Moscow and Russian special forces are thought to have used an aerosolized form of fentanyl to incapacitate everyone in the room. More than 100 of the hostages died, and the episode raised the specter of opioids being used in chemical attacks.
Dr. Pravetoni said vaccination could offer protection for first responders, law enforcement or other people whose professions place them at risk for inhalation, either accidentally or through such attacks.
These or other real-world applications for people at risk for exposure are several years away. Dr. Pravetoni said it took 10 years to get to this phase and estimates that, in about 5 years, a vaccine that targets multiple opioid drugs might enter the first clinical trial.
A version of this article first appeared on WebMD.com.