User login
Transapical valve replacement relieves mitral regurgitation
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
FROM TCT 2023
Short aspirin therapy noninferior to DAPT for 1 year after PCI for ACS
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.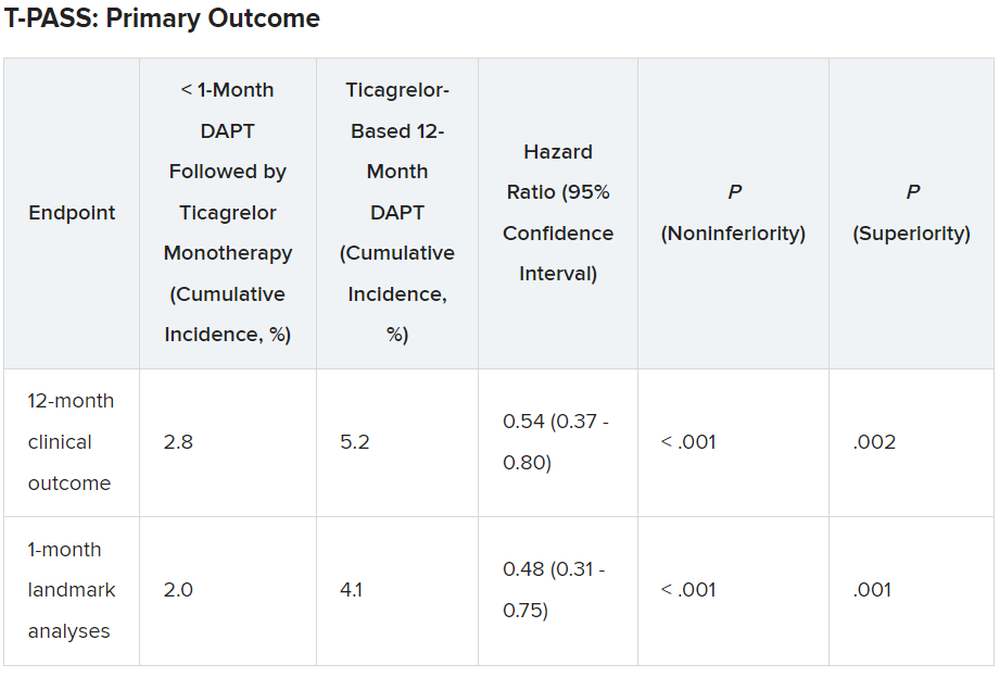
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
AT TCT 2023
Drug-coated balloon beats conventional angioplasty for high-risk patients with in-stent restenosis
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.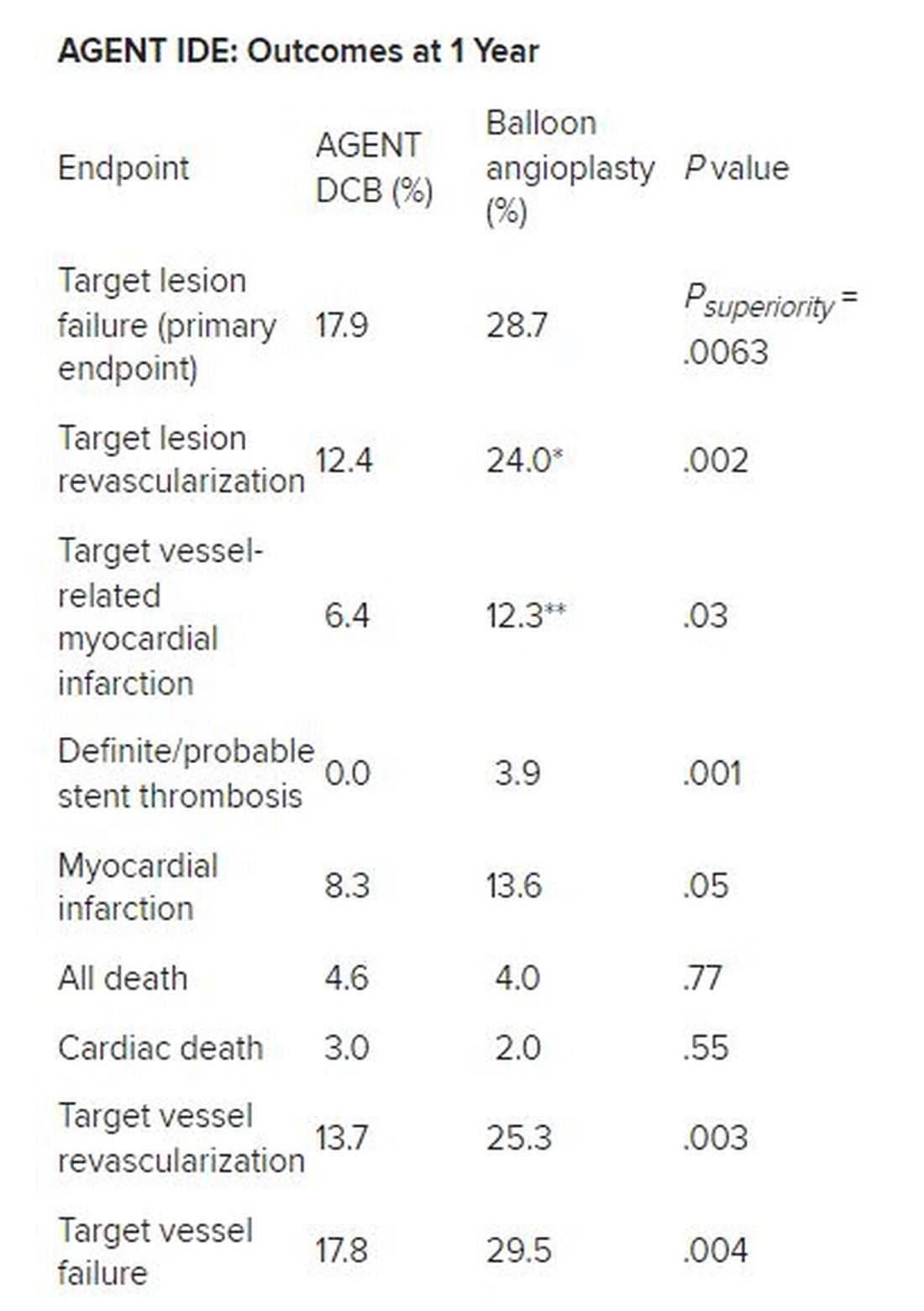
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
AT TCT 2023
Drug-eluting resorbable scaffold beats angioplasty for infrapopliteal artery disease
SAN FRANCISCO – An everolimus-eluting stent with a resorbable scaffold showed superior efficacy in a randomized multicenter trial when compared with angioplasty for the treatment of patients with chronic limb threatening ischemia (CLTI) resulting from infrapopliteal artery disease.
The stent (Esprit BTK, Abbott Vascular) was also noninferior to angioplasty in terms of safety.
Presenting results of the LIFE-BTK trial at the Transcatheter Cardiovascular Therapeutics annual meeting, Ramon Varcoe, MBBS, MS, PhD, MMed, of Prince of Wales Hospital, Sydney, said that peripheral artery disease is a global epidemic, the most serious manifestation of which is CTLI.
“If not treated expeditiously, this could lead to high rates of amputation, which, as we all know, has a severe impact on patients’ quality of life and even worse impact on their life prognosis, with prognosis rates worse than most cancers.”
He said that for infrapopliteal or below-the-knee (BTK) arterial disease, treatment with angioplasty has proven superior to bypass graft surgery, but some limitations of angioplasty are elastic recoil, dissection, and restenosis, thus limiting its durability. Coronary drug-eluting stents showed promise in BTK procedures but can interfere with reintervention. Thus, LIFE-BTK compared a drug-eluting stent with resorbable scaffolding to surgery.
Esprit BTK is a drug-eluting resorbable scaffold consisting of a temporary scaffold backbone of poly(L-lactide) and a strut thickness of 99 μm. It is coated with everolimus and bioresorbable poly(D,L-lactide). Two platinum markers at each end provide radiopacity.
LIFE-BTK enrolled patients aged 18 years or older with CLTI associated with ischemic rest pain or minor tissue loss and who had infrapopliteal artery stenosis or occlusion. The trial was prospective, international multicenter, and single-blind and randomly assigned 261 patients aged 18 years or older in the ratio of 2:1 to Esprit BTK (n = 173) or to angioplasty (n = 88). Treatment of up to two target lesions was allowed with a total scaffold length less than 170 mm.
The primary efficacy endpoint was superiority of Esprit BTK over angioplasty in terms of freedom of above-ankle amputation in the index limb, binary restenosis of the target lesion, and clinically driven target lesion revascularization evaluated at 1 year.
The primary safety endpoint, evaluated at 6 months, consisted of freedom from above-ankle amputation, major reintervention at 6 months, and perioperative mortality at 30 days.
An independent committee adjudicated clinical events, and core laboratories with assessors blinded to trial group assignment adjudicated imaging results and wound assessments.
Superior efficacy, noninferior safety
Participants were about two-thirds men, largely White, with about 15% of participants being Black/African American, and more than 90% of patients in each arm had hypertension. Lesion lengths were approximately 44 mm in each group with reference vessel diameters averaging 2.82-2.94 mm before intervention. Less than 4% in each group had severe lesion calcification.
Clinical follow-up rate at 1 year in the Esprit BTK arm was 90.2% and in the angioplasty arm 90.9%. Six patients in the former arm died versus five in the latter.
At the meeting, sponsored by the Cardiovascular Research Foundation, Dr. Varcoe showed a graph of the primary efficacy results at 453 days, the extra time being allowed to achieve a diagnostic ultrasound. (P < .0001).
“As you can see, those bars start to diverge in about 5 months and continue to separate over time, showing clear superiority of the scaffold over angioplasty, absolute risk difference of 30.8% and a highly statistically significant P value,” he said. Very similar primary efficacy outcomes were seen at 393 days.
At 1 year, the secondary efficacy endpoint of binary restenosis of the target lesion occurred in 24.2% of scaffold patients versus 46.5% of the angioplasty group (P < .0001). Another secondary endpoint, freedom from above-ankle amputation, 100% total occlusion of the target vessel, or clinically driven target lesion revascularization, occurred in 82.5% of the scaffold group versus 70.4% in the angioplasty group (P = .0081).
The primary safety endpoint of freedom from a major adverse limb event plus perioperative death was 100% for angioplasty and 96.9% for Esprit BTK (P = .0019)
All subgroup analyses assessing interaction by sex, race, geographic region, or age showed Esprit BTK was superior to angioplasty, with relative risks ranging from 0.27 to 0.61.
“If this technology is approved by the FDA, it will provide a new option for our patients with very difficult-to-treat disease, which will provide them additional durability and fewer reinterventions,” Dr. Varcoe concluded. “And I think we all know deep down that’s going to translate to improved clinical outcomes and few amputations.”
David Kandzari, MD, of Peidmont Heart Institute, Atlanta, was asked to comment on the study. He said that as with other vascular interventions, the ideal technology “would be first to provide a safe and effective antiproliferative therapy that would mitigate against restenosis and for scaffolding to prevent elastic recoil and reocclusion ... and ultimately fulfill these two promises without the requisite of a permanent implant.
“Despite their common use in femoral popliteal disease, drug-coated balloons had at best demonstrated inconsistent results below the knee, and drug-coated balloons, therefore, are not approved for such indications.”
He said that drug-eluting stents have demonstrated efficacy in this indication but that these studies have been limited to fairly discrete proximal disease.
“LIFE-BTK therefore represents one of the most rigorous trials in the space of endovascular interventions as a single line and randomized trial,” Dr. Kandzari said, “showing a primary composite endpoint of both safety and effectiveness relative to conventional angioplasty.”
Dr. Kandzari pointed to strengths of the study in that it used standardization of technique with independent adjudication of both imaging and wound healing assessments. “And the study population, too, was relevant to clinical practice, representing oftentimes underrepresented groups, including those with extensive disease burden [and] clinical severity,” he said. “Importantly, in this study, nearly one-third of the population will be women.”
Panelist Jennifer Rymer, MD, of Duke University Medical Center, Durham, N.C., commented that she treats a lot of African American patients with CTLI and applauded the researchers for including those patients. “I think that this will be a groundbreaking new change in our practice,” she said.
The trial results were published simultaneously with the presentation at TCT 2023 in the New England Journal of Medicine.
Dr. Varcoe reported receiving consulting fees/honoraria from Boston Scientific Corporation, Medtronic, Abbott, BD, Intervene, Surmodics, Philips, Nectero, Alucent, W.L. Gore, Vesteck, Bard Medical, Cook Medical, and R3 Vascular. He has equity, stock, or stock options in EBR Systems and has an executive role or ownership interest in Provisio Medical and Vesteck. Dr. Kandzari received grant support/research contract from Medtronic, Teleflex, Biotronik, and CSI; consultant fees/honoraria from and is on the speaker’s bureau of CSI and Medtronic; and has equity, stock, or options in Biostar Ventures. Dr. Rymer receives grant support/research contract from Chiesi, Abbott Vascular, Abiomed, and Pfizer. The study was funded by Abbott.
A version of this article appeared on Medscape.com.
SAN FRANCISCO – An everolimus-eluting stent with a resorbable scaffold showed superior efficacy in a randomized multicenter trial when compared with angioplasty for the treatment of patients with chronic limb threatening ischemia (CLTI) resulting from infrapopliteal artery disease.
The stent (Esprit BTK, Abbott Vascular) was also noninferior to angioplasty in terms of safety.
Presenting results of the LIFE-BTK trial at the Transcatheter Cardiovascular Therapeutics annual meeting, Ramon Varcoe, MBBS, MS, PhD, MMed, of Prince of Wales Hospital, Sydney, said that peripheral artery disease is a global epidemic, the most serious manifestation of which is CTLI.
“If not treated expeditiously, this could lead to high rates of amputation, which, as we all know, has a severe impact on patients’ quality of life and even worse impact on their life prognosis, with prognosis rates worse than most cancers.”
He said that for infrapopliteal or below-the-knee (BTK) arterial disease, treatment with angioplasty has proven superior to bypass graft surgery, but some limitations of angioplasty are elastic recoil, dissection, and restenosis, thus limiting its durability. Coronary drug-eluting stents showed promise in BTK procedures but can interfere with reintervention. Thus, LIFE-BTK compared a drug-eluting stent with resorbable scaffolding to surgery.
Esprit BTK is a drug-eluting resorbable scaffold consisting of a temporary scaffold backbone of poly(L-lactide) and a strut thickness of 99 μm. It is coated with everolimus and bioresorbable poly(D,L-lactide). Two platinum markers at each end provide radiopacity.
LIFE-BTK enrolled patients aged 18 years or older with CLTI associated with ischemic rest pain or minor tissue loss and who had infrapopliteal artery stenosis or occlusion. The trial was prospective, international multicenter, and single-blind and randomly assigned 261 patients aged 18 years or older in the ratio of 2:1 to Esprit BTK (n = 173) or to angioplasty (n = 88). Treatment of up to two target lesions was allowed with a total scaffold length less than 170 mm.
The primary efficacy endpoint was superiority of Esprit BTK over angioplasty in terms of freedom of above-ankle amputation in the index limb, binary restenosis of the target lesion, and clinically driven target lesion revascularization evaluated at 1 year.
The primary safety endpoint, evaluated at 6 months, consisted of freedom from above-ankle amputation, major reintervention at 6 months, and perioperative mortality at 30 days.
An independent committee adjudicated clinical events, and core laboratories with assessors blinded to trial group assignment adjudicated imaging results and wound assessments.
Superior efficacy, noninferior safety
Participants were about two-thirds men, largely White, with about 15% of participants being Black/African American, and more than 90% of patients in each arm had hypertension. Lesion lengths were approximately 44 mm in each group with reference vessel diameters averaging 2.82-2.94 mm before intervention. Less than 4% in each group had severe lesion calcification.
Clinical follow-up rate at 1 year in the Esprit BTK arm was 90.2% and in the angioplasty arm 90.9%. Six patients in the former arm died versus five in the latter.
At the meeting, sponsored by the Cardiovascular Research Foundation, Dr. Varcoe showed a graph of the primary efficacy results at 453 days, the extra time being allowed to achieve a diagnostic ultrasound. (P < .0001).
“As you can see, those bars start to diverge in about 5 months and continue to separate over time, showing clear superiority of the scaffold over angioplasty, absolute risk difference of 30.8% and a highly statistically significant P value,” he said. Very similar primary efficacy outcomes were seen at 393 days.
At 1 year, the secondary efficacy endpoint of binary restenosis of the target lesion occurred in 24.2% of scaffold patients versus 46.5% of the angioplasty group (P < .0001). Another secondary endpoint, freedom from above-ankle amputation, 100% total occlusion of the target vessel, or clinically driven target lesion revascularization, occurred in 82.5% of the scaffold group versus 70.4% in the angioplasty group (P = .0081).
The primary safety endpoint of freedom from a major adverse limb event plus perioperative death was 100% for angioplasty and 96.9% for Esprit BTK (P = .0019)
All subgroup analyses assessing interaction by sex, race, geographic region, or age showed Esprit BTK was superior to angioplasty, with relative risks ranging from 0.27 to 0.61.
“If this technology is approved by the FDA, it will provide a new option for our patients with very difficult-to-treat disease, which will provide them additional durability and fewer reinterventions,” Dr. Varcoe concluded. “And I think we all know deep down that’s going to translate to improved clinical outcomes and few amputations.”
David Kandzari, MD, of Peidmont Heart Institute, Atlanta, was asked to comment on the study. He said that as with other vascular interventions, the ideal technology “would be first to provide a safe and effective antiproliferative therapy that would mitigate against restenosis and for scaffolding to prevent elastic recoil and reocclusion ... and ultimately fulfill these two promises without the requisite of a permanent implant.
“Despite their common use in femoral popliteal disease, drug-coated balloons had at best demonstrated inconsistent results below the knee, and drug-coated balloons, therefore, are not approved for such indications.”
He said that drug-eluting stents have demonstrated efficacy in this indication but that these studies have been limited to fairly discrete proximal disease.
“LIFE-BTK therefore represents one of the most rigorous trials in the space of endovascular interventions as a single line and randomized trial,” Dr. Kandzari said, “showing a primary composite endpoint of both safety and effectiveness relative to conventional angioplasty.”
Dr. Kandzari pointed to strengths of the study in that it used standardization of technique with independent adjudication of both imaging and wound healing assessments. “And the study population, too, was relevant to clinical practice, representing oftentimes underrepresented groups, including those with extensive disease burden [and] clinical severity,” he said. “Importantly, in this study, nearly one-third of the population will be women.”
Panelist Jennifer Rymer, MD, of Duke University Medical Center, Durham, N.C., commented that she treats a lot of African American patients with CTLI and applauded the researchers for including those patients. “I think that this will be a groundbreaking new change in our practice,” she said.
The trial results were published simultaneously with the presentation at TCT 2023 in the New England Journal of Medicine.
Dr. Varcoe reported receiving consulting fees/honoraria from Boston Scientific Corporation, Medtronic, Abbott, BD, Intervene, Surmodics, Philips, Nectero, Alucent, W.L. Gore, Vesteck, Bard Medical, Cook Medical, and R3 Vascular. He has equity, stock, or stock options in EBR Systems and has an executive role or ownership interest in Provisio Medical and Vesteck. Dr. Kandzari received grant support/research contract from Medtronic, Teleflex, Biotronik, and CSI; consultant fees/honoraria from and is on the speaker’s bureau of CSI and Medtronic; and has equity, stock, or options in Biostar Ventures. Dr. Rymer receives grant support/research contract from Chiesi, Abbott Vascular, Abiomed, and Pfizer. The study was funded by Abbott.
A version of this article appeared on Medscape.com.
SAN FRANCISCO – An everolimus-eluting stent with a resorbable scaffold showed superior efficacy in a randomized multicenter trial when compared with angioplasty for the treatment of patients with chronic limb threatening ischemia (CLTI) resulting from infrapopliteal artery disease.
The stent (Esprit BTK, Abbott Vascular) was also noninferior to angioplasty in terms of safety.
Presenting results of the LIFE-BTK trial at the Transcatheter Cardiovascular Therapeutics annual meeting, Ramon Varcoe, MBBS, MS, PhD, MMed, of Prince of Wales Hospital, Sydney, said that peripheral artery disease is a global epidemic, the most serious manifestation of which is CTLI.
“If not treated expeditiously, this could lead to high rates of amputation, which, as we all know, has a severe impact on patients’ quality of life and even worse impact on their life prognosis, with prognosis rates worse than most cancers.”
He said that for infrapopliteal or below-the-knee (BTK) arterial disease, treatment with angioplasty has proven superior to bypass graft surgery, but some limitations of angioplasty are elastic recoil, dissection, and restenosis, thus limiting its durability. Coronary drug-eluting stents showed promise in BTK procedures but can interfere with reintervention. Thus, LIFE-BTK compared a drug-eluting stent with resorbable scaffolding to surgery.
Esprit BTK is a drug-eluting resorbable scaffold consisting of a temporary scaffold backbone of poly(L-lactide) and a strut thickness of 99 μm. It is coated with everolimus and bioresorbable poly(D,L-lactide). Two platinum markers at each end provide radiopacity.
LIFE-BTK enrolled patients aged 18 years or older with CLTI associated with ischemic rest pain or minor tissue loss and who had infrapopliteal artery stenosis or occlusion. The trial was prospective, international multicenter, and single-blind and randomly assigned 261 patients aged 18 years or older in the ratio of 2:1 to Esprit BTK (n = 173) or to angioplasty (n = 88). Treatment of up to two target lesions was allowed with a total scaffold length less than 170 mm.
The primary efficacy endpoint was superiority of Esprit BTK over angioplasty in terms of freedom of above-ankle amputation in the index limb, binary restenosis of the target lesion, and clinically driven target lesion revascularization evaluated at 1 year.
The primary safety endpoint, evaluated at 6 months, consisted of freedom from above-ankle amputation, major reintervention at 6 months, and perioperative mortality at 30 days.
An independent committee adjudicated clinical events, and core laboratories with assessors blinded to trial group assignment adjudicated imaging results and wound assessments.
Superior efficacy, noninferior safety
Participants were about two-thirds men, largely White, with about 15% of participants being Black/African American, and more than 90% of patients in each arm had hypertension. Lesion lengths were approximately 44 mm in each group with reference vessel diameters averaging 2.82-2.94 mm before intervention. Less than 4% in each group had severe lesion calcification.
Clinical follow-up rate at 1 year in the Esprit BTK arm was 90.2% and in the angioplasty arm 90.9%. Six patients in the former arm died versus five in the latter.
At the meeting, sponsored by the Cardiovascular Research Foundation, Dr. Varcoe showed a graph of the primary efficacy results at 453 days, the extra time being allowed to achieve a diagnostic ultrasound. (P < .0001).
“As you can see, those bars start to diverge in about 5 months and continue to separate over time, showing clear superiority of the scaffold over angioplasty, absolute risk difference of 30.8% and a highly statistically significant P value,” he said. Very similar primary efficacy outcomes were seen at 393 days.
At 1 year, the secondary efficacy endpoint of binary restenosis of the target lesion occurred in 24.2% of scaffold patients versus 46.5% of the angioplasty group (P < .0001). Another secondary endpoint, freedom from above-ankle amputation, 100% total occlusion of the target vessel, or clinically driven target lesion revascularization, occurred in 82.5% of the scaffold group versus 70.4% in the angioplasty group (P = .0081).
The primary safety endpoint of freedom from a major adverse limb event plus perioperative death was 100% for angioplasty and 96.9% for Esprit BTK (P = .0019)
All subgroup analyses assessing interaction by sex, race, geographic region, or age showed Esprit BTK was superior to angioplasty, with relative risks ranging from 0.27 to 0.61.
“If this technology is approved by the FDA, it will provide a new option for our patients with very difficult-to-treat disease, which will provide them additional durability and fewer reinterventions,” Dr. Varcoe concluded. “And I think we all know deep down that’s going to translate to improved clinical outcomes and few amputations.”
David Kandzari, MD, of Peidmont Heart Institute, Atlanta, was asked to comment on the study. He said that as with other vascular interventions, the ideal technology “would be first to provide a safe and effective antiproliferative therapy that would mitigate against restenosis and for scaffolding to prevent elastic recoil and reocclusion ... and ultimately fulfill these two promises without the requisite of a permanent implant.
“Despite their common use in femoral popliteal disease, drug-coated balloons had at best demonstrated inconsistent results below the knee, and drug-coated balloons, therefore, are not approved for such indications.”
He said that drug-eluting stents have demonstrated efficacy in this indication but that these studies have been limited to fairly discrete proximal disease.
“LIFE-BTK therefore represents one of the most rigorous trials in the space of endovascular interventions as a single line and randomized trial,” Dr. Kandzari said, “showing a primary composite endpoint of both safety and effectiveness relative to conventional angioplasty.”
Dr. Kandzari pointed to strengths of the study in that it used standardization of technique with independent adjudication of both imaging and wound healing assessments. “And the study population, too, was relevant to clinical practice, representing oftentimes underrepresented groups, including those with extensive disease burden [and] clinical severity,” he said. “Importantly, in this study, nearly one-third of the population will be women.”
Panelist Jennifer Rymer, MD, of Duke University Medical Center, Durham, N.C., commented that she treats a lot of African American patients with CTLI and applauded the researchers for including those patients. “I think that this will be a groundbreaking new change in our practice,” she said.
The trial results were published simultaneously with the presentation at TCT 2023 in the New England Journal of Medicine.
Dr. Varcoe reported receiving consulting fees/honoraria from Boston Scientific Corporation, Medtronic, Abbott, BD, Intervene, Surmodics, Philips, Nectero, Alucent, W.L. Gore, Vesteck, Bard Medical, Cook Medical, and R3 Vascular. He has equity, stock, or stock options in EBR Systems and has an executive role or ownership interest in Provisio Medical and Vesteck. Dr. Kandzari received grant support/research contract from Medtronic, Teleflex, Biotronik, and CSI; consultant fees/honoraria from and is on the speaker’s bureau of CSI and Medtronic; and has equity, stock, or options in Biostar Ventures. Dr. Rymer receives grant support/research contract from Chiesi, Abbott Vascular, Abiomed, and Pfizer. The study was funded by Abbott.
A version of this article appeared on Medscape.com.
AT TCT 2023
Low-risk TAVR studies: Divergent long-term results
The PARTNER-3 and Evolut trials were heralded as a landmark advance in medicine when the 1-year results from the two studies were presented back in 2019. Both trials suggested benefits of the less-invasive TAVR approach over surgery.
But because these low-surgical-risk patients are younger and will likely have a longer lifespan than will higher risk patients for whom the TAVR technique was first established, patient outcomes and information on how the TAVR devices hold up over the long-term are critical to inform clinical decision-making.
Latest results from the two trials show that the initial benefits of TAVR over surgery seen in PARTNER-3 seem to have attenuated over the longer-term, with main outcomes looking very similar in both groups after 5 years.
However, in the Evolut trial, the early benefit in all-cause mortality or disabling stroke seen in the TAVR group is continuing to increase, with current results showing a 26% relative reduction in this endpoint with TAVR vs. surgery at 4 years.
The 5-year results of the PARTNER-3 trial and the 4-year results of the Evolut study were presented Transcatheter Cardiovascular Therapeutics annual meeting sponsored by the Cardiovascular Research Foundation. Both sets of results were simultaneously published online: PARTNER-3 in The New England Journal of Medicine and Evolut in JACC.
Marty Leon, MD, of NewYork-Presbyterian Columbia University Irving Medical Center, who presented the PARTNER-3 results, said in an interview that both trials are good news for TAVR:
“Both trials have clearly reaffirmed clinical and echocardiographic benefits of TAVR as a meaningful alternative therapy to surgery in low-risk severe symptomatic aortic stenosis patients.” Michael Reardon, MD, Houston Methodist Debakey Heart & Vascular Center, who presented the Evolut results, agreed that both trials were positive for TAVR “as TAVR just has to be as good as surgery to be a winner because clearly it is a lot less invasive.”
But Dr. Dr. Reardon added that, “In making that decision for younger lower-risk patients, then the Evolut valve is the only TAVR valve that has shown superior hemodynamics and durability at all time points with excellent outcomes and widening benefits compared with surgery over the first 4 years.”
PARTNER-3
The PARTNER-3 trial randomly assigned 1,000 patients with severe symptomatic aortic stenosis and low surgical risk to undergo either TAVR with the SAPIEN 3 transcatheter heart valve or surgery.
The results at 5 years show no difference in the two primary composite outcomes between TAVR and surgery patients.
The incidence of the composite end point of death, stroke, or rehospitalization related to the valve, the procedure, or heart failure was similar in the TAVR group and the surgery group, occurring in 22.8% of patients in the TAVR group and 27.2% in the surgery group, which is a nonsignificant difference (P = .07).
The incidence of the individual components of the composite end point were also similar in the two groups. Death occurred in 10.0% in the TAVR group and 8.2% in the surgery group; stroke in 5.8% of the TAVR group and 6.4% of the surgery group; and rehospitalization in 13.7% and 17.4%, respectively.
Aortic-valve durability also looked similar in the two groups. The hemodynamic performance of the valve, assessed according to the mean valve gradient, was 12.8 mm Hg in the TAVR group and 11.7 mm Hg in the surgery group. Bioprosthetic-valve failure occurred in 3.3% of the patients in the TAVR group and in 3.8% of those in the surgery group.
Among the secondary end points, atrial fibrillation and bleeding appeared to be less frequent in the TAVR group than in the surgery group, whereas paravalvular aortic regurgitation, valve thrombosis, and pacemaker implantation appeared to be less frequent in the surgery group.
Functional and health-status outcomes assessed according to New York Heart Association class, quality of life scores, and the percentage of patients who were alive and well at 5 years appeared to be similar in the two groups.
“These data are reassuring,” Dr. Leon said. “Cardiovascular mortality occurred at a rate of about 1% per year with both therapies, strokes at the rate of 1% per year with both therapies, and hospitalization for cardiovascular reasons at about 3% per year with both therapies. For patients in their 70s, these are very good numbers.”
Along with showing similar outcomes for TAVR and surgery at 5 years, he added, “the need for re-intervention was particularly low (2%-3%) and equivalent for both approaches. And structural valve deterioration was also very low and equivalent in both groups.”
Evolut low-risk trial
The Evolut trial enrolled 1,414 patients with low surgical risk who were randomly assigned to TAVR, a self-expanding supra-annular CoreValve Evolut R PRO, or surgery.
By 4 years, the primary endpoint of all-cause mortality or disabling stroke had occurred in 10.7% of the TAVR group and 14.1% in the surgery group (hazard ratio, 0.74; P = .05), representing a 26% relative reduction with TAVR.
The absolute difference between treatment arms for the primary endpoint continued to increase over time: 1.8% at 1 year, 2.0% at 2 years, 2.9% at 3 years, and 3.4% at 4 years.
Rates of the primary endpoint components were all-cause mortality 9.0% with TAVR vs. 12.1% with surgery (P = .07); and disabling stroke was 2.9% with TAVR) vs. 3.8% for surgery (P = .32). Aortic valve rehospitalization was 10.3% with TAVR vs. 12.1% with surgery (P = .27).
The composite of all-cause mortality, disabling stroke, or aortic valve rehospitalization was significantly lower with TAVR, compared with surgery (18.0% vs. 22.4%; HR, 0.78; P = .04).
New permanent pacemaker implantation was significantly higher in the TAVR group (24.6% vs. 9.9%).
Indicators of valve performance including aortic valve reintervention (1.3% TAVR vs. 1.7% surgery); clinical or subclinical valve thrombosis (0.7% TAVR vs. 0.6% surgery); and valve endocarditis (0.9% TAVR vs. 2.2% surgery) were similarly low between groups, the authors report.
TAVR patients had sustained improvement in hemodynamics as measured by echocardiography, with significantly lower aortic valve mean gradients (9.8 mm Hg TAVR vs. 12.1 mm Hg surgery) and greater effective orifice area (2.1 cm2 TAVR vs. 2.0 cm2 surgery).
At 4 years, 84.7% of TAVR patients and 98.4% of surgery patients had no or trace paravalvular regurgitation, and there was no difference between groups in moderate or greater paravalvular regurgitation (0.4% TAVR vs. 0.0% surgery).
“The Evolut valve has shown a superior performance to surgery,” Dr. Reardon concluded. “It has less structural valve deterioration, less severe patient prosthetic mismatch, and superior hemodynamics, compared to surgery. All these factors are translating into a widening difference in clinical event curves year on year with the Evolut valve vs. surgery.”
Why the difference between trials?
The big question is why the early benefit seen with TAVR vs. surgery in both trials was attenuated by 5 years in PARTNER-3 but seemed to become greater each year in the Evolut trial. There were no definite explanations for these observations, but several possibilities were suggested.
Dr. Leon noted that with trials of intervention vs. surgery, it is common for the intervention group to do better in the beginning and for surgery to catch up a bit in later years. “So, it is not that much of a surprise to see outcomes plateauing in PARTNER-3.”
But he also suggested some other factors that may have played a role, one of which was the COVID pandemic.
“During the 2-year COVID period more than 75% of the deaths and strokes in the trial occurred in the TAVR patients,” he said. “Surgery patients were getting more anticoagulation because they had more paroxysmal [atrial fibrillation]. We know that COVID is a stimulus of thrombogenic events so in an odd way we think there may have been some cardioprotective effects from anticoagulation therapy in the surgery group.”
He also pointed out that though hospitalization and strokes were slightly lower with TAVR vs. surgery in the PARTNER-3 trial, mortality was slightly greater in the TAVR group.
“There was a 2:1 ratio in the TAVR and surgery groups in noncardiovascular deaths which influenced the all-cause mortality numbers,” he noted.
Mortality rates in the surgery groups
Dr. Leon also pointed out differences in the mortality rates in the surgery groups in the two trials, which he suggested may contribute to the explanation for the different longer-term results.
“The baseline characteristics for patients in these two trials were almost identical, and results at 1 year were very similar, but for whatever reason, over the course of a few years, the outcomes in the Evolut trial were different to those in PARTNER-3, and in particular the difference was in the surgical arms, with a higher event rate in the surgical arm in Evolut than in the surgery arm in PARTNER-3,” he said. “When the control does not perform well it is a lot easier to show that the experimental arm is better.”
“When you look at the TAVR arms in both studies at each time point they are either similar or PARTNER-3 is actually lower,” he added. “That is why it is so difficult to compare these two trials.”
But Dr. Reardon dismissed this argument.
“What determines long-term survival after a procedure is the intrinsic risk level of the patients,” he said. “Overall mortality rates differ between the two trials because the PARTNER-3 trial enrolled a lower end of a low-risk population while Evolut enrolled an upper end of a low-risk population. You cannot look at absolute numbers between trials. That is intellectually and scientifically invalid.”
“It is the relative difference between surgery and TAVR that we are interested in, and we see in Evolut that the relative difference between the two procedures in terms of benefit with TAVR is widening every year,” he added. “That is because the superior valve performance and hemodynamics of the Evolut valve compared to surgery has translated into excellent clinical outcomes.
“In the PARTNER-3 trial – their curves are coming together. I think that is worrisome, but I don’t want to draw conclusions about their trial,” Dr. Reardon said. “All I know is that in our trial, we have excellent outcomes that are getting better year after year.”
Competition between valves
The different results of the two trials is inevitably producing some competition between the two products.
Dr. Reardon said: “In terms of which valve to use, clinicians will want to choose a valve that has the best durability and shows the best survival vs. surgery and that is clearly the Evolut valve. I think the writing is on the wall. Some clinicians are going to wait for longer term data, but the question is do we have enough long-term data now.”
But Dr. Leon countered: “There’s never been a head-to-head device to suggest that the self- expanding device performs better than the balloon expanding device. We always think about them as being similar in terms of performance. There is an aggressive effort to suggest that by virtue of the current trial results there was a superior outcome with the Medtronic device, but it’s hard to explain why that would be the case, and we should not compare between the two trials.”
Both Dr. Leon and Dr. Reardon stressed that longer-term follow-up is critical because some surgical valves are known to fail between 5-10 years, and it is not known how the TAVR valves will perform over that period.
Both the PARTNER-3 and Evolut trials are planning to keep following patients out to 10 years.
For the time being though, both Dr. Leon and Dr. Reardon agreed that these current results will probably accelerate the already rapid transition from surgery to TAVR in low-risk patients.
“TAVR will be the default therapy,” Dr. Leon commented. “It will be the first choice for patients. Whether TAVR is superior to surgery in terms of outcomes or just the same, there are sufficient benefits from a logistic and patient perspective that most people would prefer to have the less invasive therapy. TAVR is a one-day procedure, there is no need for general anesthetic, a lot of the secondary outcomes that are so problematic with surgery don’t exist, and the ability to be in a symptom-free state is dramatically accelerated.”
“This was the first serious foray into the low-risk population with TAVR,” he added. “We had an age cut of 65 years, but the vast majority of patients in both trials were over 70. We could now start looking at younger patient populations.”
But Dr. Reardon said that these younger patients are already being given TAVR, and future trials randomizing between TAVR and surgery may not be possible.
“Even though US guidelines still recommend surgery for patients under 65 years, patients want TAVR, and they get TAVR,” he said. “Recent data shows that in 2021, use of TAVR rose to 47.5% in patients under 65 needing isolated aortic valve replacement. That doesn’t meet the guidelines but there’s clearly a big shift going on. These results will just keep that momentum going.”
The PARTNER-3 trial was funded by Edwards Lifesciences. The Evolut study was funded by Medtronic. Dr. Leon reports grant support from Edwards Lifesciences and Medtronic. Dr. Reardon receives research grants from Medtronic.
A version of this article first appeared on Medscape.com.
The PARTNER-3 and Evolut trials were heralded as a landmark advance in medicine when the 1-year results from the two studies were presented back in 2019. Both trials suggested benefits of the less-invasive TAVR approach over surgery.
But because these low-surgical-risk patients are younger and will likely have a longer lifespan than will higher risk patients for whom the TAVR technique was first established, patient outcomes and information on how the TAVR devices hold up over the long-term are critical to inform clinical decision-making.
Latest results from the two trials show that the initial benefits of TAVR over surgery seen in PARTNER-3 seem to have attenuated over the longer-term, with main outcomes looking very similar in both groups after 5 years.
However, in the Evolut trial, the early benefit in all-cause mortality or disabling stroke seen in the TAVR group is continuing to increase, with current results showing a 26% relative reduction in this endpoint with TAVR vs. surgery at 4 years.
The 5-year results of the PARTNER-3 trial and the 4-year results of the Evolut study were presented Transcatheter Cardiovascular Therapeutics annual meeting sponsored by the Cardiovascular Research Foundation. Both sets of results were simultaneously published online: PARTNER-3 in The New England Journal of Medicine and Evolut in JACC.
Marty Leon, MD, of NewYork-Presbyterian Columbia University Irving Medical Center, who presented the PARTNER-3 results, said in an interview that both trials are good news for TAVR:
“Both trials have clearly reaffirmed clinical and echocardiographic benefits of TAVR as a meaningful alternative therapy to surgery in low-risk severe symptomatic aortic stenosis patients.” Michael Reardon, MD, Houston Methodist Debakey Heart & Vascular Center, who presented the Evolut results, agreed that both trials were positive for TAVR “as TAVR just has to be as good as surgery to be a winner because clearly it is a lot less invasive.”
But Dr. Dr. Reardon added that, “In making that decision for younger lower-risk patients, then the Evolut valve is the only TAVR valve that has shown superior hemodynamics and durability at all time points with excellent outcomes and widening benefits compared with surgery over the first 4 years.”
PARTNER-3
The PARTNER-3 trial randomly assigned 1,000 patients with severe symptomatic aortic stenosis and low surgical risk to undergo either TAVR with the SAPIEN 3 transcatheter heart valve or surgery.
The results at 5 years show no difference in the two primary composite outcomes between TAVR and surgery patients.
The incidence of the composite end point of death, stroke, or rehospitalization related to the valve, the procedure, or heart failure was similar in the TAVR group and the surgery group, occurring in 22.8% of patients in the TAVR group and 27.2% in the surgery group, which is a nonsignificant difference (P = .07).
The incidence of the individual components of the composite end point were also similar in the two groups. Death occurred in 10.0% in the TAVR group and 8.2% in the surgery group; stroke in 5.8% of the TAVR group and 6.4% of the surgery group; and rehospitalization in 13.7% and 17.4%, respectively.
Aortic-valve durability also looked similar in the two groups. The hemodynamic performance of the valve, assessed according to the mean valve gradient, was 12.8 mm Hg in the TAVR group and 11.7 mm Hg in the surgery group. Bioprosthetic-valve failure occurred in 3.3% of the patients in the TAVR group and in 3.8% of those in the surgery group.
Among the secondary end points, atrial fibrillation and bleeding appeared to be less frequent in the TAVR group than in the surgery group, whereas paravalvular aortic regurgitation, valve thrombosis, and pacemaker implantation appeared to be less frequent in the surgery group.
Functional and health-status outcomes assessed according to New York Heart Association class, quality of life scores, and the percentage of patients who were alive and well at 5 years appeared to be similar in the two groups.
“These data are reassuring,” Dr. Leon said. “Cardiovascular mortality occurred at a rate of about 1% per year with both therapies, strokes at the rate of 1% per year with both therapies, and hospitalization for cardiovascular reasons at about 3% per year with both therapies. For patients in their 70s, these are very good numbers.”
Along with showing similar outcomes for TAVR and surgery at 5 years, he added, “the need for re-intervention was particularly low (2%-3%) and equivalent for both approaches. And structural valve deterioration was also very low and equivalent in both groups.”
Evolut low-risk trial
The Evolut trial enrolled 1,414 patients with low surgical risk who were randomly assigned to TAVR, a self-expanding supra-annular CoreValve Evolut R PRO, or surgery.
By 4 years, the primary endpoint of all-cause mortality or disabling stroke had occurred in 10.7% of the TAVR group and 14.1% in the surgery group (hazard ratio, 0.74; P = .05), representing a 26% relative reduction with TAVR.
The absolute difference between treatment arms for the primary endpoint continued to increase over time: 1.8% at 1 year, 2.0% at 2 years, 2.9% at 3 years, and 3.4% at 4 years.
Rates of the primary endpoint components were all-cause mortality 9.0% with TAVR vs. 12.1% with surgery (P = .07); and disabling stroke was 2.9% with TAVR) vs. 3.8% for surgery (P = .32). Aortic valve rehospitalization was 10.3% with TAVR vs. 12.1% with surgery (P = .27).
The composite of all-cause mortality, disabling stroke, or aortic valve rehospitalization was significantly lower with TAVR, compared with surgery (18.0% vs. 22.4%; HR, 0.78; P = .04).
New permanent pacemaker implantation was significantly higher in the TAVR group (24.6% vs. 9.9%).
Indicators of valve performance including aortic valve reintervention (1.3% TAVR vs. 1.7% surgery); clinical or subclinical valve thrombosis (0.7% TAVR vs. 0.6% surgery); and valve endocarditis (0.9% TAVR vs. 2.2% surgery) were similarly low between groups, the authors report.
TAVR patients had sustained improvement in hemodynamics as measured by echocardiography, with significantly lower aortic valve mean gradients (9.8 mm Hg TAVR vs. 12.1 mm Hg surgery) and greater effective orifice area (2.1 cm2 TAVR vs. 2.0 cm2 surgery).
At 4 years, 84.7% of TAVR patients and 98.4% of surgery patients had no or trace paravalvular regurgitation, and there was no difference between groups in moderate or greater paravalvular regurgitation (0.4% TAVR vs. 0.0% surgery).
“The Evolut valve has shown a superior performance to surgery,” Dr. Reardon concluded. “It has less structural valve deterioration, less severe patient prosthetic mismatch, and superior hemodynamics, compared to surgery. All these factors are translating into a widening difference in clinical event curves year on year with the Evolut valve vs. surgery.”
Why the difference between trials?
The big question is why the early benefit seen with TAVR vs. surgery in both trials was attenuated by 5 years in PARTNER-3 but seemed to become greater each year in the Evolut trial. There were no definite explanations for these observations, but several possibilities were suggested.
Dr. Leon noted that with trials of intervention vs. surgery, it is common for the intervention group to do better in the beginning and for surgery to catch up a bit in later years. “So, it is not that much of a surprise to see outcomes plateauing in PARTNER-3.”
But he also suggested some other factors that may have played a role, one of which was the COVID pandemic.
“During the 2-year COVID period more than 75% of the deaths and strokes in the trial occurred in the TAVR patients,” he said. “Surgery patients were getting more anticoagulation because they had more paroxysmal [atrial fibrillation]. We know that COVID is a stimulus of thrombogenic events so in an odd way we think there may have been some cardioprotective effects from anticoagulation therapy in the surgery group.”
He also pointed out that though hospitalization and strokes were slightly lower with TAVR vs. surgery in the PARTNER-3 trial, mortality was slightly greater in the TAVR group.
“There was a 2:1 ratio in the TAVR and surgery groups in noncardiovascular deaths which influenced the all-cause mortality numbers,” he noted.
Mortality rates in the surgery groups
Dr. Leon also pointed out differences in the mortality rates in the surgery groups in the two trials, which he suggested may contribute to the explanation for the different longer-term results.
“The baseline characteristics for patients in these two trials were almost identical, and results at 1 year were very similar, but for whatever reason, over the course of a few years, the outcomes in the Evolut trial were different to those in PARTNER-3, and in particular the difference was in the surgical arms, with a higher event rate in the surgical arm in Evolut than in the surgery arm in PARTNER-3,” he said. “When the control does not perform well it is a lot easier to show that the experimental arm is better.”
“When you look at the TAVR arms in both studies at each time point they are either similar or PARTNER-3 is actually lower,” he added. “That is why it is so difficult to compare these two trials.”
But Dr. Reardon dismissed this argument.
“What determines long-term survival after a procedure is the intrinsic risk level of the patients,” he said. “Overall mortality rates differ between the two trials because the PARTNER-3 trial enrolled a lower end of a low-risk population while Evolut enrolled an upper end of a low-risk population. You cannot look at absolute numbers between trials. That is intellectually and scientifically invalid.”
“It is the relative difference between surgery and TAVR that we are interested in, and we see in Evolut that the relative difference between the two procedures in terms of benefit with TAVR is widening every year,” he added. “That is because the superior valve performance and hemodynamics of the Evolut valve compared to surgery has translated into excellent clinical outcomes.
“In the PARTNER-3 trial – their curves are coming together. I think that is worrisome, but I don’t want to draw conclusions about their trial,” Dr. Reardon said. “All I know is that in our trial, we have excellent outcomes that are getting better year after year.”
Competition between valves
The different results of the two trials is inevitably producing some competition between the two products.
Dr. Reardon said: “In terms of which valve to use, clinicians will want to choose a valve that has the best durability and shows the best survival vs. surgery and that is clearly the Evolut valve. I think the writing is on the wall. Some clinicians are going to wait for longer term data, but the question is do we have enough long-term data now.”
But Dr. Leon countered: “There’s never been a head-to-head device to suggest that the self- expanding device performs better than the balloon expanding device. We always think about them as being similar in terms of performance. There is an aggressive effort to suggest that by virtue of the current trial results there was a superior outcome with the Medtronic device, but it’s hard to explain why that would be the case, and we should not compare between the two trials.”
Both Dr. Leon and Dr. Reardon stressed that longer-term follow-up is critical because some surgical valves are known to fail between 5-10 years, and it is not known how the TAVR valves will perform over that period.
Both the PARTNER-3 and Evolut trials are planning to keep following patients out to 10 years.
For the time being though, both Dr. Leon and Dr. Reardon agreed that these current results will probably accelerate the already rapid transition from surgery to TAVR in low-risk patients.
“TAVR will be the default therapy,” Dr. Leon commented. “It will be the first choice for patients. Whether TAVR is superior to surgery in terms of outcomes or just the same, there are sufficient benefits from a logistic and patient perspective that most people would prefer to have the less invasive therapy. TAVR is a one-day procedure, there is no need for general anesthetic, a lot of the secondary outcomes that are so problematic with surgery don’t exist, and the ability to be in a symptom-free state is dramatically accelerated.”
“This was the first serious foray into the low-risk population with TAVR,” he added. “We had an age cut of 65 years, but the vast majority of patients in both trials were over 70. We could now start looking at younger patient populations.”
But Dr. Reardon said that these younger patients are already being given TAVR, and future trials randomizing between TAVR and surgery may not be possible.
“Even though US guidelines still recommend surgery for patients under 65 years, patients want TAVR, and they get TAVR,” he said. “Recent data shows that in 2021, use of TAVR rose to 47.5% in patients under 65 needing isolated aortic valve replacement. That doesn’t meet the guidelines but there’s clearly a big shift going on. These results will just keep that momentum going.”
The PARTNER-3 trial was funded by Edwards Lifesciences. The Evolut study was funded by Medtronic. Dr. Leon reports grant support from Edwards Lifesciences and Medtronic. Dr. Reardon receives research grants from Medtronic.
A version of this article first appeared on Medscape.com.
The PARTNER-3 and Evolut trials were heralded as a landmark advance in medicine when the 1-year results from the two studies were presented back in 2019. Both trials suggested benefits of the less-invasive TAVR approach over surgery.
But because these low-surgical-risk patients are younger and will likely have a longer lifespan than will higher risk patients for whom the TAVR technique was first established, patient outcomes and information on how the TAVR devices hold up over the long-term are critical to inform clinical decision-making.
Latest results from the two trials show that the initial benefits of TAVR over surgery seen in PARTNER-3 seem to have attenuated over the longer-term, with main outcomes looking very similar in both groups after 5 years.
However, in the Evolut trial, the early benefit in all-cause mortality or disabling stroke seen in the TAVR group is continuing to increase, with current results showing a 26% relative reduction in this endpoint with TAVR vs. surgery at 4 years.
The 5-year results of the PARTNER-3 trial and the 4-year results of the Evolut study were presented Transcatheter Cardiovascular Therapeutics annual meeting sponsored by the Cardiovascular Research Foundation. Both sets of results were simultaneously published online: PARTNER-3 in The New England Journal of Medicine and Evolut in JACC.
Marty Leon, MD, of NewYork-Presbyterian Columbia University Irving Medical Center, who presented the PARTNER-3 results, said in an interview that both trials are good news for TAVR:
“Both trials have clearly reaffirmed clinical and echocardiographic benefits of TAVR as a meaningful alternative therapy to surgery in low-risk severe symptomatic aortic stenosis patients.” Michael Reardon, MD, Houston Methodist Debakey Heart & Vascular Center, who presented the Evolut results, agreed that both trials were positive for TAVR “as TAVR just has to be as good as surgery to be a winner because clearly it is a lot less invasive.”
But Dr. Dr. Reardon added that, “In making that decision for younger lower-risk patients, then the Evolut valve is the only TAVR valve that has shown superior hemodynamics and durability at all time points with excellent outcomes and widening benefits compared with surgery over the first 4 years.”
PARTNER-3
The PARTNER-3 trial randomly assigned 1,000 patients with severe symptomatic aortic stenosis and low surgical risk to undergo either TAVR with the SAPIEN 3 transcatheter heart valve or surgery.
The results at 5 years show no difference in the two primary composite outcomes between TAVR and surgery patients.
The incidence of the composite end point of death, stroke, or rehospitalization related to the valve, the procedure, or heart failure was similar in the TAVR group and the surgery group, occurring in 22.8% of patients in the TAVR group and 27.2% in the surgery group, which is a nonsignificant difference (P = .07).
The incidence of the individual components of the composite end point were also similar in the two groups. Death occurred in 10.0% in the TAVR group and 8.2% in the surgery group; stroke in 5.8% of the TAVR group and 6.4% of the surgery group; and rehospitalization in 13.7% and 17.4%, respectively.
Aortic-valve durability also looked similar in the two groups. The hemodynamic performance of the valve, assessed according to the mean valve gradient, was 12.8 mm Hg in the TAVR group and 11.7 mm Hg in the surgery group. Bioprosthetic-valve failure occurred in 3.3% of the patients in the TAVR group and in 3.8% of those in the surgery group.
Among the secondary end points, atrial fibrillation and bleeding appeared to be less frequent in the TAVR group than in the surgery group, whereas paravalvular aortic regurgitation, valve thrombosis, and pacemaker implantation appeared to be less frequent in the surgery group.
Functional and health-status outcomes assessed according to New York Heart Association class, quality of life scores, and the percentage of patients who were alive and well at 5 years appeared to be similar in the two groups.
“These data are reassuring,” Dr. Leon said. “Cardiovascular mortality occurred at a rate of about 1% per year with both therapies, strokes at the rate of 1% per year with both therapies, and hospitalization for cardiovascular reasons at about 3% per year with both therapies. For patients in their 70s, these are very good numbers.”
Along with showing similar outcomes for TAVR and surgery at 5 years, he added, “the need for re-intervention was particularly low (2%-3%) and equivalent for both approaches. And structural valve deterioration was also very low and equivalent in both groups.”
Evolut low-risk trial
The Evolut trial enrolled 1,414 patients with low surgical risk who were randomly assigned to TAVR, a self-expanding supra-annular CoreValve Evolut R PRO, or surgery.
By 4 years, the primary endpoint of all-cause mortality or disabling stroke had occurred in 10.7% of the TAVR group and 14.1% in the surgery group (hazard ratio, 0.74; P = .05), representing a 26% relative reduction with TAVR.
The absolute difference between treatment arms for the primary endpoint continued to increase over time: 1.8% at 1 year, 2.0% at 2 years, 2.9% at 3 years, and 3.4% at 4 years.
Rates of the primary endpoint components were all-cause mortality 9.0% with TAVR vs. 12.1% with surgery (P = .07); and disabling stroke was 2.9% with TAVR) vs. 3.8% for surgery (P = .32). Aortic valve rehospitalization was 10.3% with TAVR vs. 12.1% with surgery (P = .27).
The composite of all-cause mortality, disabling stroke, or aortic valve rehospitalization was significantly lower with TAVR, compared with surgery (18.0% vs. 22.4%; HR, 0.78; P = .04).
New permanent pacemaker implantation was significantly higher in the TAVR group (24.6% vs. 9.9%).
Indicators of valve performance including aortic valve reintervention (1.3% TAVR vs. 1.7% surgery); clinical or subclinical valve thrombosis (0.7% TAVR vs. 0.6% surgery); and valve endocarditis (0.9% TAVR vs. 2.2% surgery) were similarly low between groups, the authors report.
TAVR patients had sustained improvement in hemodynamics as measured by echocardiography, with significantly lower aortic valve mean gradients (9.8 mm Hg TAVR vs. 12.1 mm Hg surgery) and greater effective orifice area (2.1 cm2 TAVR vs. 2.0 cm2 surgery).
At 4 years, 84.7% of TAVR patients and 98.4% of surgery patients had no or trace paravalvular regurgitation, and there was no difference between groups in moderate or greater paravalvular regurgitation (0.4% TAVR vs. 0.0% surgery).
“The Evolut valve has shown a superior performance to surgery,” Dr. Reardon concluded. “It has less structural valve deterioration, less severe patient prosthetic mismatch, and superior hemodynamics, compared to surgery. All these factors are translating into a widening difference in clinical event curves year on year with the Evolut valve vs. surgery.”
Why the difference between trials?
The big question is why the early benefit seen with TAVR vs. surgery in both trials was attenuated by 5 years in PARTNER-3 but seemed to become greater each year in the Evolut trial. There were no definite explanations for these observations, but several possibilities were suggested.
Dr. Leon noted that with trials of intervention vs. surgery, it is common for the intervention group to do better in the beginning and for surgery to catch up a bit in later years. “So, it is not that much of a surprise to see outcomes plateauing in PARTNER-3.”
But he also suggested some other factors that may have played a role, one of which was the COVID pandemic.
“During the 2-year COVID period more than 75% of the deaths and strokes in the trial occurred in the TAVR patients,” he said. “Surgery patients were getting more anticoagulation because they had more paroxysmal [atrial fibrillation]. We know that COVID is a stimulus of thrombogenic events so in an odd way we think there may have been some cardioprotective effects from anticoagulation therapy in the surgery group.”
He also pointed out that though hospitalization and strokes were slightly lower with TAVR vs. surgery in the PARTNER-3 trial, mortality was slightly greater in the TAVR group.
“There was a 2:1 ratio in the TAVR and surgery groups in noncardiovascular deaths which influenced the all-cause mortality numbers,” he noted.
Mortality rates in the surgery groups
Dr. Leon also pointed out differences in the mortality rates in the surgery groups in the two trials, which he suggested may contribute to the explanation for the different longer-term results.
“The baseline characteristics for patients in these two trials were almost identical, and results at 1 year were very similar, but for whatever reason, over the course of a few years, the outcomes in the Evolut trial were different to those in PARTNER-3, and in particular the difference was in the surgical arms, with a higher event rate in the surgical arm in Evolut than in the surgery arm in PARTNER-3,” he said. “When the control does not perform well it is a lot easier to show that the experimental arm is better.”
“When you look at the TAVR arms in both studies at each time point they are either similar or PARTNER-3 is actually lower,” he added. “That is why it is so difficult to compare these two trials.”
But Dr. Reardon dismissed this argument.
“What determines long-term survival after a procedure is the intrinsic risk level of the patients,” he said. “Overall mortality rates differ between the two trials because the PARTNER-3 trial enrolled a lower end of a low-risk population while Evolut enrolled an upper end of a low-risk population. You cannot look at absolute numbers between trials. That is intellectually and scientifically invalid.”
“It is the relative difference between surgery and TAVR that we are interested in, and we see in Evolut that the relative difference between the two procedures in terms of benefit with TAVR is widening every year,” he added. “That is because the superior valve performance and hemodynamics of the Evolut valve compared to surgery has translated into excellent clinical outcomes.
“In the PARTNER-3 trial – their curves are coming together. I think that is worrisome, but I don’t want to draw conclusions about their trial,” Dr. Reardon said. “All I know is that in our trial, we have excellent outcomes that are getting better year after year.”
Competition between valves
The different results of the two trials is inevitably producing some competition between the two products.
Dr. Reardon said: “In terms of which valve to use, clinicians will want to choose a valve that has the best durability and shows the best survival vs. surgery and that is clearly the Evolut valve. I think the writing is on the wall. Some clinicians are going to wait for longer term data, but the question is do we have enough long-term data now.”
But Dr. Leon countered: “There’s never been a head-to-head device to suggest that the self- expanding device performs better than the balloon expanding device. We always think about them as being similar in terms of performance. There is an aggressive effort to suggest that by virtue of the current trial results there was a superior outcome with the Medtronic device, but it’s hard to explain why that would be the case, and we should not compare between the two trials.”
Both Dr. Leon and Dr. Reardon stressed that longer-term follow-up is critical because some surgical valves are known to fail between 5-10 years, and it is not known how the TAVR valves will perform over that period.
Both the PARTNER-3 and Evolut trials are planning to keep following patients out to 10 years.
For the time being though, both Dr. Leon and Dr. Reardon agreed that these current results will probably accelerate the already rapid transition from surgery to TAVR in low-risk patients.
“TAVR will be the default therapy,” Dr. Leon commented. “It will be the first choice for patients. Whether TAVR is superior to surgery in terms of outcomes or just the same, there are sufficient benefits from a logistic and patient perspective that most people would prefer to have the less invasive therapy. TAVR is a one-day procedure, there is no need for general anesthetic, a lot of the secondary outcomes that are so problematic with surgery don’t exist, and the ability to be in a symptom-free state is dramatically accelerated.”
“This was the first serious foray into the low-risk population with TAVR,” he added. “We had an age cut of 65 years, but the vast majority of patients in both trials were over 70. We could now start looking at younger patient populations.”
But Dr. Reardon said that these younger patients are already being given TAVR, and future trials randomizing between TAVR and surgery may not be possible.
“Even though US guidelines still recommend surgery for patients under 65 years, patients want TAVR, and they get TAVR,” he said. “Recent data shows that in 2021, use of TAVR rose to 47.5% in patients under 65 needing isolated aortic valve replacement. That doesn’t meet the guidelines but there’s clearly a big shift going on. These results will just keep that momentum going.”
The PARTNER-3 trial was funded by Edwards Lifesciences. The Evolut study was funded by Medtronic. Dr. Leon reports grant support from Edwards Lifesciences and Medtronic. Dr. Reardon receives research grants from Medtronic.
A version of this article first appeared on Medscape.com.
FROM TCT 2023
Trilogy TAVR safe, effective in aortic regurgitation
SAN FRANCISCO – , achieving a 1-year all-cause mortality rate of 7.8%.
New pacemaker implantation was 24%, similar to previously reported outcomes.
Vinod Thourani, MD, Piedmont Heart Institute, Atlanta, presented initial outcome results of the ALIGN-AR trial at the Transcatheter Cardiovascular Therapeutics annual meeting.
Dr. Thourani concluded that the Trilogy system provides the first dedicated transcatheter aortic valve replacement options “for symptomatic patients with moderate to severe or severe aortic regurgitation or at high risk for surgery and is well positioned to become the preferred therapy upon approval for this population.”
Currently, Trilogy is not approved by the U.S. Food and Drug Administration in the United States and is for investigational use only.
Untreated, severe symptomatic aortic regurgitation (AR) is associated with high mortality, especially for those with NYHA class 3 or 4 symptoms, Dr. Thourani explained. “While surgery remains the only recommended intervention for patients with native severe AR, there are a multitude of high-risk patients who are not offered therapy.”
Off-label use of transcatheter valves for AR has been associated with “higher rates of complications, including paravalvular regurgitation and embolization,” he noted.
Dr. Thourani described the unique features of the JenaValve Trilogy valve. The system has a set of three “locators” in its own sheath that allows it to be rotated to align with the three cusps of the native aortic valve, falling into the sinuses and securely anchored to the native valve leaflets – then the valve is deployed. Inside a self-expanding nitinol frame is porcine pericardial tissue. A sealing ring provides sufficient anchoring while conforming to the annulus.
ALIGN-AR was a multicenter, single arm, non-blinded trial with follow-up out to 5 years involving patients with 3-plus or greater AR at high risk for surgical aortic valve replacement. Exclusion criteria included an aortic root diameter greater than 5 cm, a previous prosthetic aortic valve, mitral regurgitation greater than moderate, or coronary artery disease requiring revascularization.
After Trilogy valve implantation, patients were followed for 1, 6, and 12 months, as well as annually out to 5 years. Safety and efficacy endpoints were compared with prespecified performance goals. Of 180 patients enrolled, 177 were successfully implanted with the Trilogy device.
Patients had an average age of 75.5 years, 47.2% were women, 67.2% were in NYHA class III/IV, 82.8% were hypertensive, and one-third were frail. Severe AR was present in 62.4%, and 31.7% had moderate to severe AR.
The primary composite safety endpoint included all-cause mortality, any stroke, major vascular complication, major bleeding, a new pacemaker, acute kidney injury, valve dysfunction, or any intervention related to the device. The primary efficacy endpoint was all-cause mortality at 12 months.
The performance goal for primary efficacy was a weighted average of 25%, derived mainly from 1-year mortality figures for NYHA class I/II and class II/IV with conservative management.
Non-inferiority margin met
With a 25% prespecified non-inferiority margin for the primary efficacy endpoint, “We have observed a rate of 7.8%,” Dr. Thourani reported during a late-breaking clinical trials session. “The non-inferiority margin was met for the primary efficacy endpoint with a P value of less than .0001.”
“With a 40.5% prespecified non-inferiority margin of our primary safety endpoint, with a Trilogy [heart valve] we have observed a rate of 26.7%,” he said. “At 30 days there was a 2.2% mortality and a 2.2% stroke rate. There was a 26.7% primary safety endpoint, mainly driven by the 24% new pacemaker implantation rate. Without pacemaker implantation, the rate of safety events was less than 8%,” (P noninferiority < .0001).
Procedure technical success was 95%, device success 96.7%, and procedure success 92.8%. There was one ascending aortic dissection (0.6%). Moderate or greater paravalvular regurgitation also occurred in one patient. There were four cases of valve embolization.
Pacemaker implants occurred in 30% of patients in the first tercile enrolled and decreased to 14% for the third tercile enrolled. “Lower rates are most likely due to the change in the insertion technique, placing locators above the nadir of the native cusps, reduction in oversizing, and also evolution in the management of periprocedural conduction abnormalities,” Dr. Thourani proposed.
The hemodynamics of the valve improved from a gradient of 8.7 mm Hg at baseline to 3.9 mm Hg at 30 days and remained fairly stable out to 1 year. Paravalvular regurgitation was absent in 80.8% of patients at 30 days and mild in 18%. It improved over time, being absent in 93.5% at 6 months and in 92.2% at 1 year.
Left ventricular (LV) remodeling occurred over one year, with LV end systolic diameter, LV end systolic volume, LV mass, and LV mass index all decreasing significantly from baseline to one year (all P < .0001). Importantly to patients, NYHA class improved, from 32% class II, 63% class III, and 5% class IV at baseline to 54% class I, 37% class II, and 10% class III at 30 days and improving slightly out to 1 year.
These improvements resulted in better quality of life, as reflected in a 21.8-point improvement in the self-reported Kansas City Cardiomyopathy Questionnaire Overall Summary Score, with a score of 77.6 at 1 year, indicating self-perceived good health.
Encouraging data
During the session, Robert Bonow, MD, of Northwestern University Feinberg School of Medicine, Chicago, commented that Dr. Thourani presented very encouraging data from the ALIGN-AR trial of high-risk surgical patients with significant aortic regurgitation. However, he had a couple of questions for Dr. Thourani.
One related to the efficacy data being compared with historical survival data. “So, are you planning to do a randomized study of these patients? You could argue, unlike aortic stenosis, where there’s no medical therapy, there could be medical therapies for the patients.” He noted that one-third of the patients are only in NYHA functional class II, so those patients “might do well over the long haul, with medical therapy as an agent.”
Dr. Thourani said it was an excellent question. “Doing a randomized trial with a high-risk patient [is] probably less likely,” he said. “I think there is a lot of interest among physicians on the cardiology side and on the surgery side of looking at lower-risk patients, and this could include those that are intermediate and or low risk.”
He said he believes investigators and leadership of the ALIGN-AR trial have conceived of such a trial involving all comers. “I think that’s warranted if we go into younger patients,” he said.
Dr. Bonow then asked if there was significant aortic valve calcification in the study population, because it is common with regurgitation, “which is why standard approaches are not effective ... And how does this device behave in calcified valves?” But Dr. Thourani said calcification was an exclusion criterion for this trial.
He said, “deep dives are going to come,” looking at the ventricular outcomes and also looking at a lot of the echocardiographic parameters.
Dr. Bonow related this study’s findings on ventricular remodeling to what is seen with surgical aortic valve replacement, where the ventricle decompresses within days. “And that’s predictive of good outcome if you have early data and these patients show how the ventricle remodels quickly,” he said.
The trial was supported by JenaValve. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, Atricure, Boston Scientific, CroiValve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, Atricure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Bonow had no disclosures.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – , achieving a 1-year all-cause mortality rate of 7.8%.
New pacemaker implantation was 24%, similar to previously reported outcomes.
Vinod Thourani, MD, Piedmont Heart Institute, Atlanta, presented initial outcome results of the ALIGN-AR trial at the Transcatheter Cardiovascular Therapeutics annual meeting.
Dr. Thourani concluded that the Trilogy system provides the first dedicated transcatheter aortic valve replacement options “for symptomatic patients with moderate to severe or severe aortic regurgitation or at high risk for surgery and is well positioned to become the preferred therapy upon approval for this population.”
Currently, Trilogy is not approved by the U.S. Food and Drug Administration in the United States and is for investigational use only.
Untreated, severe symptomatic aortic regurgitation (AR) is associated with high mortality, especially for those with NYHA class 3 or 4 symptoms, Dr. Thourani explained. “While surgery remains the only recommended intervention for patients with native severe AR, there are a multitude of high-risk patients who are not offered therapy.”
Off-label use of transcatheter valves for AR has been associated with “higher rates of complications, including paravalvular regurgitation and embolization,” he noted.
Dr. Thourani described the unique features of the JenaValve Trilogy valve. The system has a set of three “locators” in its own sheath that allows it to be rotated to align with the three cusps of the native aortic valve, falling into the sinuses and securely anchored to the native valve leaflets – then the valve is deployed. Inside a self-expanding nitinol frame is porcine pericardial tissue. A sealing ring provides sufficient anchoring while conforming to the annulus.
ALIGN-AR was a multicenter, single arm, non-blinded trial with follow-up out to 5 years involving patients with 3-plus or greater AR at high risk for surgical aortic valve replacement. Exclusion criteria included an aortic root diameter greater than 5 cm, a previous prosthetic aortic valve, mitral regurgitation greater than moderate, or coronary artery disease requiring revascularization.
After Trilogy valve implantation, patients were followed for 1, 6, and 12 months, as well as annually out to 5 years. Safety and efficacy endpoints were compared with prespecified performance goals. Of 180 patients enrolled, 177 were successfully implanted with the Trilogy device.
Patients had an average age of 75.5 years, 47.2% were women, 67.2% were in NYHA class III/IV, 82.8% were hypertensive, and one-third were frail. Severe AR was present in 62.4%, and 31.7% had moderate to severe AR.
The primary composite safety endpoint included all-cause mortality, any stroke, major vascular complication, major bleeding, a new pacemaker, acute kidney injury, valve dysfunction, or any intervention related to the device. The primary efficacy endpoint was all-cause mortality at 12 months.
The performance goal for primary efficacy was a weighted average of 25%, derived mainly from 1-year mortality figures for NYHA class I/II and class II/IV with conservative management.
Non-inferiority margin met
With a 25% prespecified non-inferiority margin for the primary efficacy endpoint, “We have observed a rate of 7.8%,” Dr. Thourani reported during a late-breaking clinical trials session. “The non-inferiority margin was met for the primary efficacy endpoint with a P value of less than .0001.”
“With a 40.5% prespecified non-inferiority margin of our primary safety endpoint, with a Trilogy [heart valve] we have observed a rate of 26.7%,” he said. “At 30 days there was a 2.2% mortality and a 2.2% stroke rate. There was a 26.7% primary safety endpoint, mainly driven by the 24% new pacemaker implantation rate. Without pacemaker implantation, the rate of safety events was less than 8%,” (P noninferiority < .0001).
Procedure technical success was 95%, device success 96.7%, and procedure success 92.8%. There was one ascending aortic dissection (0.6%). Moderate or greater paravalvular regurgitation also occurred in one patient. There were four cases of valve embolization.
Pacemaker implants occurred in 30% of patients in the first tercile enrolled and decreased to 14% for the third tercile enrolled. “Lower rates are most likely due to the change in the insertion technique, placing locators above the nadir of the native cusps, reduction in oversizing, and also evolution in the management of periprocedural conduction abnormalities,” Dr. Thourani proposed.
The hemodynamics of the valve improved from a gradient of 8.7 mm Hg at baseline to 3.9 mm Hg at 30 days and remained fairly stable out to 1 year. Paravalvular regurgitation was absent in 80.8% of patients at 30 days and mild in 18%. It improved over time, being absent in 93.5% at 6 months and in 92.2% at 1 year.
Left ventricular (LV) remodeling occurred over one year, with LV end systolic diameter, LV end systolic volume, LV mass, and LV mass index all decreasing significantly from baseline to one year (all P < .0001). Importantly to patients, NYHA class improved, from 32% class II, 63% class III, and 5% class IV at baseline to 54% class I, 37% class II, and 10% class III at 30 days and improving slightly out to 1 year.
These improvements resulted in better quality of life, as reflected in a 21.8-point improvement in the self-reported Kansas City Cardiomyopathy Questionnaire Overall Summary Score, with a score of 77.6 at 1 year, indicating self-perceived good health.
Encouraging data
During the session, Robert Bonow, MD, of Northwestern University Feinberg School of Medicine, Chicago, commented that Dr. Thourani presented very encouraging data from the ALIGN-AR trial of high-risk surgical patients with significant aortic regurgitation. However, he had a couple of questions for Dr. Thourani.
One related to the efficacy data being compared with historical survival data. “So, are you planning to do a randomized study of these patients? You could argue, unlike aortic stenosis, where there’s no medical therapy, there could be medical therapies for the patients.” He noted that one-third of the patients are only in NYHA functional class II, so those patients “might do well over the long haul, with medical therapy as an agent.”
Dr. Thourani said it was an excellent question. “Doing a randomized trial with a high-risk patient [is] probably less likely,” he said. “I think there is a lot of interest among physicians on the cardiology side and on the surgery side of looking at lower-risk patients, and this could include those that are intermediate and or low risk.”
He said he believes investigators and leadership of the ALIGN-AR trial have conceived of such a trial involving all comers. “I think that’s warranted if we go into younger patients,” he said.
Dr. Bonow then asked if there was significant aortic valve calcification in the study population, because it is common with regurgitation, “which is why standard approaches are not effective ... And how does this device behave in calcified valves?” But Dr. Thourani said calcification was an exclusion criterion for this trial.
He said, “deep dives are going to come,” looking at the ventricular outcomes and also looking at a lot of the echocardiographic parameters.
Dr. Bonow related this study’s findings on ventricular remodeling to what is seen with surgical aortic valve replacement, where the ventricle decompresses within days. “And that’s predictive of good outcome if you have early data and these patients show how the ventricle remodels quickly,” he said.
The trial was supported by JenaValve. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, Atricure, Boston Scientific, CroiValve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, Atricure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Bonow had no disclosures.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – , achieving a 1-year all-cause mortality rate of 7.8%.
New pacemaker implantation was 24%, similar to previously reported outcomes.
Vinod Thourani, MD, Piedmont Heart Institute, Atlanta, presented initial outcome results of the ALIGN-AR trial at the Transcatheter Cardiovascular Therapeutics annual meeting.
Dr. Thourani concluded that the Trilogy system provides the first dedicated transcatheter aortic valve replacement options “for symptomatic patients with moderate to severe or severe aortic regurgitation or at high risk for surgery and is well positioned to become the preferred therapy upon approval for this population.”
Currently, Trilogy is not approved by the U.S. Food and Drug Administration in the United States and is for investigational use only.
Untreated, severe symptomatic aortic regurgitation (AR) is associated with high mortality, especially for those with NYHA class 3 or 4 symptoms, Dr. Thourani explained. “While surgery remains the only recommended intervention for patients with native severe AR, there are a multitude of high-risk patients who are not offered therapy.”
Off-label use of transcatheter valves for AR has been associated with “higher rates of complications, including paravalvular regurgitation and embolization,” he noted.
Dr. Thourani described the unique features of the JenaValve Trilogy valve. The system has a set of three “locators” in its own sheath that allows it to be rotated to align with the three cusps of the native aortic valve, falling into the sinuses and securely anchored to the native valve leaflets – then the valve is deployed. Inside a self-expanding nitinol frame is porcine pericardial tissue. A sealing ring provides sufficient anchoring while conforming to the annulus.
ALIGN-AR was a multicenter, single arm, non-blinded trial with follow-up out to 5 years involving patients with 3-plus or greater AR at high risk for surgical aortic valve replacement. Exclusion criteria included an aortic root diameter greater than 5 cm, a previous prosthetic aortic valve, mitral regurgitation greater than moderate, or coronary artery disease requiring revascularization.
After Trilogy valve implantation, patients were followed for 1, 6, and 12 months, as well as annually out to 5 years. Safety and efficacy endpoints were compared with prespecified performance goals. Of 180 patients enrolled, 177 were successfully implanted with the Trilogy device.
Patients had an average age of 75.5 years, 47.2% were women, 67.2% were in NYHA class III/IV, 82.8% were hypertensive, and one-third were frail. Severe AR was present in 62.4%, and 31.7% had moderate to severe AR.
The primary composite safety endpoint included all-cause mortality, any stroke, major vascular complication, major bleeding, a new pacemaker, acute kidney injury, valve dysfunction, or any intervention related to the device. The primary efficacy endpoint was all-cause mortality at 12 months.
The performance goal for primary efficacy was a weighted average of 25%, derived mainly from 1-year mortality figures for NYHA class I/II and class II/IV with conservative management.
Non-inferiority margin met
With a 25% prespecified non-inferiority margin for the primary efficacy endpoint, “We have observed a rate of 7.8%,” Dr. Thourani reported during a late-breaking clinical trials session. “The non-inferiority margin was met for the primary efficacy endpoint with a P value of less than .0001.”
“With a 40.5% prespecified non-inferiority margin of our primary safety endpoint, with a Trilogy [heart valve] we have observed a rate of 26.7%,” he said. “At 30 days there was a 2.2% mortality and a 2.2% stroke rate. There was a 26.7% primary safety endpoint, mainly driven by the 24% new pacemaker implantation rate. Without pacemaker implantation, the rate of safety events was less than 8%,” (P noninferiority < .0001).
Procedure technical success was 95%, device success 96.7%, and procedure success 92.8%. There was one ascending aortic dissection (0.6%). Moderate or greater paravalvular regurgitation also occurred in one patient. There were four cases of valve embolization.
Pacemaker implants occurred in 30% of patients in the first tercile enrolled and decreased to 14% for the third tercile enrolled. “Lower rates are most likely due to the change in the insertion technique, placing locators above the nadir of the native cusps, reduction in oversizing, and also evolution in the management of periprocedural conduction abnormalities,” Dr. Thourani proposed.
The hemodynamics of the valve improved from a gradient of 8.7 mm Hg at baseline to 3.9 mm Hg at 30 days and remained fairly stable out to 1 year. Paravalvular regurgitation was absent in 80.8% of patients at 30 days and mild in 18%. It improved over time, being absent in 93.5% at 6 months and in 92.2% at 1 year.
Left ventricular (LV) remodeling occurred over one year, with LV end systolic diameter, LV end systolic volume, LV mass, and LV mass index all decreasing significantly from baseline to one year (all P < .0001). Importantly to patients, NYHA class improved, from 32% class II, 63% class III, and 5% class IV at baseline to 54% class I, 37% class II, and 10% class III at 30 days and improving slightly out to 1 year.
These improvements resulted in better quality of life, as reflected in a 21.8-point improvement in the self-reported Kansas City Cardiomyopathy Questionnaire Overall Summary Score, with a score of 77.6 at 1 year, indicating self-perceived good health.
Encouraging data
During the session, Robert Bonow, MD, of Northwestern University Feinberg School of Medicine, Chicago, commented that Dr. Thourani presented very encouraging data from the ALIGN-AR trial of high-risk surgical patients with significant aortic regurgitation. However, he had a couple of questions for Dr. Thourani.
One related to the efficacy data being compared with historical survival data. “So, are you planning to do a randomized study of these patients? You could argue, unlike aortic stenosis, where there’s no medical therapy, there could be medical therapies for the patients.” He noted that one-third of the patients are only in NYHA functional class II, so those patients “might do well over the long haul, with medical therapy as an agent.”
Dr. Thourani said it was an excellent question. “Doing a randomized trial with a high-risk patient [is] probably less likely,” he said. “I think there is a lot of interest among physicians on the cardiology side and on the surgery side of looking at lower-risk patients, and this could include those that are intermediate and or low risk.”
He said he believes investigators and leadership of the ALIGN-AR trial have conceived of such a trial involving all comers. “I think that’s warranted if we go into younger patients,” he said.
Dr. Bonow then asked if there was significant aortic valve calcification in the study population, because it is common with regurgitation, “which is why standard approaches are not effective ... And how does this device behave in calcified valves?” But Dr. Thourani said calcification was an exclusion criterion for this trial.
He said, “deep dives are going to come,” looking at the ventricular outcomes and also looking at a lot of the echocardiographic parameters.
Dr. Bonow related this study’s findings on ventricular remodeling to what is seen with surgical aortic valve replacement, where the ventricle decompresses within days. “And that’s predictive of good outcome if you have early data and these patients show how the ventricle remodels quickly,” he said.
The trial was supported by JenaValve. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, Atricure, Boston Scientific, CroiValve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, Atricure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Bonow had no disclosures.
A version of this article first appeared on Medscape.com.
AT TCT 2023