User login
Methotrexate does not cause rheumatoid interstitial lung disease
BIRMINGHAM, ENGLAND – Data from two early RA inception cohorts provide reassurance that methotrexate does not cause interstitial lung disease and suggest that treatment with methotrexate might even be protective.

In the Early RA Study (ERAS) and Early RA Network (ERAN), which together include 2,701 patients with RA, 101 (3.7%) had interstitial lung disease (ILD). There were 92 patients with RA-ILD who had information available on exposure to any conventional synthetic disease-modifying antirheumatic drug (csDMARD); of these, 39 (2.5%) had been exposed to methotrexate (n = 1,578) and 53 (4.8%) to other csDMARDs (n = 1,114).
Multivariate analysis showed that methotrexate exposure was associated with a reduced risk of developing ILD, with an odds ratio of 0.48 (P = .004). In a separate analysis that excluded 25 patients who had ILD before they received any csDMARD therapy (n = 67), there was no association between methotrexate use and ILD (OR, 0.85; P = .578). In fact, there was a nonsignificant trend for a delayed onset of ILD in patients who had been treated with methotrexate (OR, 0.54; P = .072).
Methotrexate use is associated with an acute hypersensitivity pneumonitis in patients with RA, explained Patrick Kiely, MBBS, PhD, of St. George’s University Hospitals NHS Foundation Trust in London at the British Society for Rheumatology annual conference. “This is well recognized, it’s very rare [0.43%-1.00%], it’s easy to spot, and usually goes away if you stop methotrexate,” said Dr. Kiely, adding that “it’s not benign, and severe cases can be life threatening.”
Because of the association between methotrexate and pneumonitis, there has been concern that methotrexate may exacerbate or even cause ILD in RA but there are sparse data available to confirm this. The bottom line is that you should not start someone on methotrexate if you think their existing lung capacity is not up to treatment with methotrexate, Dr. Kiely said.
ILD is not always symptomatic in RA, but when it is, it is associated with very poor survival. The lung disease can be present before joint symptoms, Dr. Kiely said. Although less than 10% of cases may be symptomatic, this “is a big deal, because it has a high mortality, with death within 5 years. It’s the second-commonest cause of excess mortality in RA after cardiovascular disease.”
To look at the association between incident RA-ILD and the use of methotrexate, Dr. Kiely and associates analyzed data from ERAS (1986-2001) and ERAN (2002-2013), that together have more than 25 years of follow-up data on patients who were recruited at the first sign of RA symptoms. Patients within these cohorts have been treated according to best practice, and a range of outcomes – including RA-ILD – have been assessed at annual intervals.
In the patients who developed ILD after any csDMARD exposure, older age at RA onset (OR, 1.04; P less than .001) and having ever smoked (OR, 1.91; P = .016) were associated with the development of the lung disease. Incident ILD was also associated with being positive for rheumatoid factor (OR, 2.02; P = .029) at baseline. Being male was also associated with a higher risk for developing ILD, Dr. Kiely reported, as was a longer duration of time between the onset of first RA symptoms and the first secondary care visit. Conversely, the presence of nonrespiratory, major comorbidities at baseline appeared to be protective (OR, 0.62; P = .027).
“We found no association between methotrexate treatment and incident RA-ILD and a possibility that it may be protective,” Dr. Kiely concluded, noting that these data were now published in BMJ Open (2019;9:e028466. doi: 10.1136/bmjopen-2018-028466).
Following Dr. Kiely’s presentation, an audience member asked if the protective effect seen with methotrexate could have been caused by better disease control overall.
Dr. Kiely answered that, up until 2001, the time when ERAS was ongoing, standard practice in the United Kingdom was to use sulfasalazine, but then methotrexate started to be used in higher and higher doses, as seen in ERAN.
The interesting thing is that in ERAN more methotrexate was used in higher doses, but less RA-ILD was seen, Dr. Kiely observed. The overall prevalence of RA-ILD in the later early RA cohort was 3.2% and the median dose of methotrexate used was 20 mg. In ERAS, the prevalence was 4.2% and the median dose of methotrexate used was 10 mg.
There was a suggestion that disease control was slightly better in ERAN than ERAS, but that wasn’t statistically significant, Dr. Kiely said.
So, should a patient with RA and ILD be given methotrexate? There’s no reason not to, Dr. Kiely suggested, based on the evidence shown. Part of the challenge will now be convincing chest physician colleagues that methotrexate is not problematic in terms of causing ILD.
These findings are completely on board with the ILD group’s findings that methotrexate doesn’t cause pulmonary fibrosis in patients with RA, commented Julie Dawson, MD, of St. Helens and Knowsley Teaching Hospitals NHS Trust, St. Helens, England. Her own research, which includes a 10-year follow-up of patients with inflammatory arthritis, has shown that methotrexate does not appear to increase the risk of pulmonary fibrosis.
The study had no specific outside funding. Dr. Kiely reported having no conflicts of interest.
SOURCE: Kiely P et al. Rheumatology. 2019;58(suppl 3), Abstract 009.
BIRMINGHAM, ENGLAND – Data from two early RA inception cohorts provide reassurance that methotrexate does not cause interstitial lung disease and suggest that treatment with methotrexate might even be protective.
In the Early RA Study (ERAS) and Early RA Network (ERAN), which together include 2,701 patients with RA, 101 (3.7%) had interstitial lung disease (ILD). There were 92 patients with RA-ILD who had information available on exposure to any conventional synthetic disease-modifying antirheumatic drug (csDMARD); of these, 39 (2.5%) had been exposed to methotrexate (n = 1,578) and 53 (4.8%) to other csDMARDs (n = 1,114).
Multivariate analysis showed that methotrexate exposure was associated with a reduced risk of developing ILD, with an odds ratio of 0.48 (P = .004). In a separate analysis that excluded 25 patients who had ILD before they received any csDMARD therapy (n = 67), there was no association between methotrexate use and ILD (OR, 0.85; P = .578). In fact, there was a nonsignificant trend for a delayed onset of ILD in patients who had been treated with methotrexate (OR, 0.54; P = .072).
Methotrexate use is associated with an acute hypersensitivity pneumonitis in patients with RA, explained Patrick Kiely, MBBS, PhD, of St. George’s University Hospitals NHS Foundation Trust in London at the British Society for Rheumatology annual conference. “This is well recognized, it’s very rare [0.43%-1.00%], it’s easy to spot, and usually goes away if you stop methotrexate,” said Dr. Kiely, adding that “it’s not benign, and severe cases can be life threatening.”
Because of the association between methotrexate and pneumonitis, there has been concern that methotrexate may exacerbate or even cause ILD in RA but there are sparse data available to confirm this. The bottom line is that you should not start someone on methotrexate if you think their existing lung capacity is not up to treatment with methotrexate, Dr. Kiely said.
ILD is not always symptomatic in RA, but when it is, it is associated with very poor survival. The lung disease can be present before joint symptoms, Dr. Kiely said. Although less than 10% of cases may be symptomatic, this “is a big deal, because it has a high mortality, with death within 5 years. It’s the second-commonest cause of excess mortality in RA after cardiovascular disease.”
To look at the association between incident RA-ILD and the use of methotrexate, Dr. Kiely and associates analyzed data from ERAS (1986-2001) and ERAN (2002-2013), that together have more than 25 years of follow-up data on patients who were recruited at the first sign of RA symptoms. Patients within these cohorts have been treated according to best practice, and a range of outcomes – including RA-ILD – have been assessed at annual intervals.
In the patients who developed ILD after any csDMARD exposure, older age at RA onset (OR, 1.04; P less than .001) and having ever smoked (OR, 1.91; P = .016) were associated with the development of the lung disease. Incident ILD was also associated with being positive for rheumatoid factor (OR, 2.02; P = .029) at baseline. Being male was also associated with a higher risk for developing ILD, Dr. Kiely reported, as was a longer duration of time between the onset of first RA symptoms and the first secondary care visit. Conversely, the presence of nonrespiratory, major comorbidities at baseline appeared to be protective (OR, 0.62; P = .027).
“We found no association between methotrexate treatment and incident RA-ILD and a possibility that it may be protective,” Dr. Kiely concluded, noting that these data were now published in BMJ Open (2019;9:e028466. doi: 10.1136/bmjopen-2018-028466).
Following Dr. Kiely’s presentation, an audience member asked if the protective effect seen with methotrexate could have been caused by better disease control overall.
Dr. Kiely answered that, up until 2001, the time when ERAS was ongoing, standard practice in the United Kingdom was to use sulfasalazine, but then methotrexate started to be used in higher and higher doses, as seen in ERAN.
The interesting thing is that in ERAN more methotrexate was used in higher doses, but less RA-ILD was seen, Dr. Kiely observed. The overall prevalence of RA-ILD in the later early RA cohort was 3.2% and the median dose of methotrexate used was 20 mg. In ERAS, the prevalence was 4.2% and the median dose of methotrexate used was 10 mg.
There was a suggestion that disease control was slightly better in ERAN than ERAS, but that wasn’t statistically significant, Dr. Kiely said.
So, should a patient with RA and ILD be given methotrexate? There’s no reason not to, Dr. Kiely suggested, based on the evidence shown. Part of the challenge will now be convincing chest physician colleagues that methotrexate is not problematic in terms of causing ILD.
These findings are completely on board with the ILD group’s findings that methotrexate doesn’t cause pulmonary fibrosis in patients with RA, commented Julie Dawson, MD, of St. Helens and Knowsley Teaching Hospitals NHS Trust, St. Helens, England. Her own research, which includes a 10-year follow-up of patients with inflammatory arthritis, has shown that methotrexate does not appear to increase the risk of pulmonary fibrosis.
The study had no specific outside funding. Dr. Kiely reported having no conflicts of interest.
SOURCE: Kiely P et al. Rheumatology. 2019;58(suppl 3), Abstract 009.
BIRMINGHAM, ENGLAND – Data from two early RA inception cohorts provide reassurance that methotrexate does not cause interstitial lung disease and suggest that treatment with methotrexate might even be protective.
In the Early RA Study (ERAS) and Early RA Network (ERAN), which together include 2,701 patients with RA, 101 (3.7%) had interstitial lung disease (ILD). There were 92 patients with RA-ILD who had information available on exposure to any conventional synthetic disease-modifying antirheumatic drug (csDMARD); of these, 39 (2.5%) had been exposed to methotrexate (n = 1,578) and 53 (4.8%) to other csDMARDs (n = 1,114).
Multivariate analysis showed that methotrexate exposure was associated with a reduced risk of developing ILD, with an odds ratio of 0.48 (P = .004). In a separate analysis that excluded 25 patients who had ILD before they received any csDMARD therapy (n = 67), there was no association between methotrexate use and ILD (OR, 0.85; P = .578). In fact, there was a nonsignificant trend for a delayed onset of ILD in patients who had been treated with methotrexate (OR, 0.54; P = .072).
Methotrexate use is associated with an acute hypersensitivity pneumonitis in patients with RA, explained Patrick Kiely, MBBS, PhD, of St. George’s University Hospitals NHS Foundation Trust in London at the British Society for Rheumatology annual conference. “This is well recognized, it’s very rare [0.43%-1.00%], it’s easy to spot, and usually goes away if you stop methotrexate,” said Dr. Kiely, adding that “it’s not benign, and severe cases can be life threatening.”
Because of the association between methotrexate and pneumonitis, there has been concern that methotrexate may exacerbate or even cause ILD in RA but there are sparse data available to confirm this. The bottom line is that you should not start someone on methotrexate if you think their existing lung capacity is not up to treatment with methotrexate, Dr. Kiely said.
ILD is not always symptomatic in RA, but when it is, it is associated with very poor survival. The lung disease can be present before joint symptoms, Dr. Kiely said. Although less than 10% of cases may be symptomatic, this “is a big deal, because it has a high mortality, with death within 5 years. It’s the second-commonest cause of excess mortality in RA after cardiovascular disease.”
To look at the association between incident RA-ILD and the use of methotrexate, Dr. Kiely and associates analyzed data from ERAS (1986-2001) and ERAN (2002-2013), that together have more than 25 years of follow-up data on patients who were recruited at the first sign of RA symptoms. Patients within these cohorts have been treated according to best practice, and a range of outcomes – including RA-ILD – have been assessed at annual intervals.
In the patients who developed ILD after any csDMARD exposure, older age at RA onset (OR, 1.04; P less than .001) and having ever smoked (OR, 1.91; P = .016) were associated with the development of the lung disease. Incident ILD was also associated with being positive for rheumatoid factor (OR, 2.02; P = .029) at baseline. Being male was also associated with a higher risk for developing ILD, Dr. Kiely reported, as was a longer duration of time between the onset of first RA symptoms and the first secondary care visit. Conversely, the presence of nonrespiratory, major comorbidities at baseline appeared to be protective (OR, 0.62; P = .027).
“We found no association between methotrexate treatment and incident RA-ILD and a possibility that it may be protective,” Dr. Kiely concluded, noting that these data were now published in BMJ Open (2019;9:e028466. doi: 10.1136/bmjopen-2018-028466).
Following Dr. Kiely’s presentation, an audience member asked if the protective effect seen with methotrexate could have been caused by better disease control overall.
Dr. Kiely answered that, up until 2001, the time when ERAS was ongoing, standard practice in the United Kingdom was to use sulfasalazine, but then methotrexate started to be used in higher and higher doses, as seen in ERAN.
The interesting thing is that in ERAN more methotrexate was used in higher doses, but less RA-ILD was seen, Dr. Kiely observed. The overall prevalence of RA-ILD in the later early RA cohort was 3.2% and the median dose of methotrexate used was 20 mg. In ERAS, the prevalence was 4.2% and the median dose of methotrexate used was 10 mg.
There was a suggestion that disease control was slightly better in ERAN than ERAS, but that wasn’t statistically significant, Dr. Kiely said.
So, should a patient with RA and ILD be given methotrexate? There’s no reason not to, Dr. Kiely suggested, based on the evidence shown. Part of the challenge will now be convincing chest physician colleagues that methotrexate is not problematic in terms of causing ILD.
These findings are completely on board with the ILD group’s findings that methotrexate doesn’t cause pulmonary fibrosis in patients with RA, commented Julie Dawson, MD, of St. Helens and Knowsley Teaching Hospitals NHS Trust, St. Helens, England. Her own research, which includes a 10-year follow-up of patients with inflammatory arthritis, has shown that methotrexate does not appear to increase the risk of pulmonary fibrosis.
The study had no specific outside funding. Dr. Kiely reported having no conflicts of interest.
SOURCE: Kiely P et al. Rheumatology. 2019;58(suppl 3), Abstract 009.
REPORTING FROM BSR 2019
New recommendations on TB screening for health care workers
U.S. health care personnel no longer need to undergo routine tuberculosis testing in the absence of known exposure, according to new screening guidelines from the National Tuberculosis Controllers Association and CDC.
![]()
The revised guidelines on tuberculosis screening, testing, and treatment of U.S. health care personnel, published in Morbidity and Mortality Weekly Report, are the first update since 2005. The new recommendations reflect a reduction in concern about U.S. health care personnel’s risk of occupational exposure to latent and active tuberculosis infection.
Lynn E. Sosa, MD, from the Connecticut Department of Public Health and National Tuberculosis Controllers Association, and coauthors wrote that rates of tuberculosis infection in the United States have declined by 73% since 1991, from 10.4/100,000 population in 1991 to 2.8/100,000 in 2017. This has been matched by similar declines among health care workers, which the authors said raised questions about the cost-effectiveness of the previously recommended routine serial occupational testing.
“In addition, a recent retrospective cohort study of approximately 40,000 health care personnel at a tertiary U.S. medical center in a low TB-incidence state found an extremely low rate of TST conversion (0.3%) during 1998-2014, with a limited proportion attributable to occupational exposure,” they wrote.
The new guidelines recommend health care personnel undergo baseline or preplacement tuberculosis testing with an interferon-gamma release assay (IGRA) or a tuberculin skin test (TST), as well as individual risk assessment and symptom evaluation.
The individual risk assessment considers whether the person has lived in a country with a high tuberculosis rate, whether they are immunosuppressed, or whether they have had close contact with someone with infectious tuberculosis.
This risk assessment can help decide how to interpret an initial positive test result, the authors said.
“For example, health care personnel with a positive test who are asymptomatic, unlikely to be infected with M. [Mycobacterium] tuberculosis, and at low risk for progression on the basis of their risk assessment should have a second test (either an IGRA or a TST) as recommended in the 2017 TB diagnostic guidelines of the American Thoracic Society, Infectious Diseases Society of America, and CDC,” they wrote. “In this example, the health care personnel should be considered infected with M. tuberculosis only if both the first and second tests are positive.”
After that baseline testing, personnel do not need to undergo routine serial testing except in the case of known exposure or ongoing transmission. The guideline authors suggested serial screening might be considered for health care workers whose work puts them at greater risk – for example, pulmonologists or respiratory therapists – or for those working in settings in which transmission has happened in the past.
For personnel with latent tuberculosis infection, the guidelines recommend “encouragement of treatment” unless it is contraindicated, and annual symptom screening in those not undergoing treatment.
The guideline committee also advocated for annual tuberculosis education for all health care workers.
The new recommendations were based on a systematic review of 36 studies of tuberculosis screening and testing among health care personnel, 16 of which were performed in the United States, and all but two of which were conducted in a hospital setting.
The authors stressed that recommendations from the 2005 CDC guidelines – which do not pertain to health care personnel screening, testing, treatment and education – remain unchanged.
One author declared personal fees from the National Tuberculosis Controllers Association during the conduct of the study. Two others reported unrelated grants and personal fees from private industry. No other conflicts of interest were disclosed.
SOURCE: Sosa L et al. MMWR. 2019;68:439-43.
U.S. health care personnel no longer need to undergo routine tuberculosis testing in the absence of known exposure, according to new screening guidelines from the National Tuberculosis Controllers Association and CDC.
![]()
The revised guidelines on tuberculosis screening, testing, and treatment of U.S. health care personnel, published in Morbidity and Mortality Weekly Report, are the first update since 2005. The new recommendations reflect a reduction in concern about U.S. health care personnel’s risk of occupational exposure to latent and active tuberculosis infection.
Lynn E. Sosa, MD, from the Connecticut Department of Public Health and National Tuberculosis Controllers Association, and coauthors wrote that rates of tuberculosis infection in the United States have declined by 73% since 1991, from 10.4/100,000 population in 1991 to 2.8/100,000 in 2017. This has been matched by similar declines among health care workers, which the authors said raised questions about the cost-effectiveness of the previously recommended routine serial occupational testing.
“In addition, a recent retrospective cohort study of approximately 40,000 health care personnel at a tertiary U.S. medical center in a low TB-incidence state found an extremely low rate of TST conversion (0.3%) during 1998-2014, with a limited proportion attributable to occupational exposure,” they wrote.
The new guidelines recommend health care personnel undergo baseline or preplacement tuberculosis testing with an interferon-gamma release assay (IGRA) or a tuberculin skin test (TST), as well as individual risk assessment and symptom evaluation.
The individual risk assessment considers whether the person has lived in a country with a high tuberculosis rate, whether they are immunosuppressed, or whether they have had close contact with someone with infectious tuberculosis.
This risk assessment can help decide how to interpret an initial positive test result, the authors said.
“For example, health care personnel with a positive test who are asymptomatic, unlikely to be infected with M. [Mycobacterium] tuberculosis, and at low risk for progression on the basis of their risk assessment should have a second test (either an IGRA or a TST) as recommended in the 2017 TB diagnostic guidelines of the American Thoracic Society, Infectious Diseases Society of America, and CDC,” they wrote. “In this example, the health care personnel should be considered infected with M. tuberculosis only if both the first and second tests are positive.”
After that baseline testing, personnel do not need to undergo routine serial testing except in the case of known exposure or ongoing transmission. The guideline authors suggested serial screening might be considered for health care workers whose work puts them at greater risk – for example, pulmonologists or respiratory therapists – or for those working in settings in which transmission has happened in the past.
For personnel with latent tuberculosis infection, the guidelines recommend “encouragement of treatment” unless it is contraindicated, and annual symptom screening in those not undergoing treatment.
The guideline committee also advocated for annual tuberculosis education for all health care workers.
The new recommendations were based on a systematic review of 36 studies of tuberculosis screening and testing among health care personnel, 16 of which were performed in the United States, and all but two of which were conducted in a hospital setting.
The authors stressed that recommendations from the 2005 CDC guidelines – which do not pertain to health care personnel screening, testing, treatment and education – remain unchanged.
One author declared personal fees from the National Tuberculosis Controllers Association during the conduct of the study. Two others reported unrelated grants and personal fees from private industry. No other conflicts of interest were disclosed.
SOURCE: Sosa L et al. MMWR. 2019;68:439-43.
U.S. health care personnel no longer need to undergo routine tuberculosis testing in the absence of known exposure, according to new screening guidelines from the National Tuberculosis Controllers Association and CDC.
![]()
The revised guidelines on tuberculosis screening, testing, and treatment of U.S. health care personnel, published in Morbidity and Mortality Weekly Report, are the first update since 2005. The new recommendations reflect a reduction in concern about U.S. health care personnel’s risk of occupational exposure to latent and active tuberculosis infection.
Lynn E. Sosa, MD, from the Connecticut Department of Public Health and National Tuberculosis Controllers Association, and coauthors wrote that rates of tuberculosis infection in the United States have declined by 73% since 1991, from 10.4/100,000 population in 1991 to 2.8/100,000 in 2017. This has been matched by similar declines among health care workers, which the authors said raised questions about the cost-effectiveness of the previously recommended routine serial occupational testing.
“In addition, a recent retrospective cohort study of approximately 40,000 health care personnel at a tertiary U.S. medical center in a low TB-incidence state found an extremely low rate of TST conversion (0.3%) during 1998-2014, with a limited proportion attributable to occupational exposure,” they wrote.
The new guidelines recommend health care personnel undergo baseline or preplacement tuberculosis testing with an interferon-gamma release assay (IGRA) or a tuberculin skin test (TST), as well as individual risk assessment and symptom evaluation.
The individual risk assessment considers whether the person has lived in a country with a high tuberculosis rate, whether they are immunosuppressed, or whether they have had close contact with someone with infectious tuberculosis.
This risk assessment can help decide how to interpret an initial positive test result, the authors said.
“For example, health care personnel with a positive test who are asymptomatic, unlikely to be infected with M. [Mycobacterium] tuberculosis, and at low risk for progression on the basis of their risk assessment should have a second test (either an IGRA or a TST) as recommended in the 2017 TB diagnostic guidelines of the American Thoracic Society, Infectious Diseases Society of America, and CDC,” they wrote. “In this example, the health care personnel should be considered infected with M. tuberculosis only if both the first and second tests are positive.”
After that baseline testing, personnel do not need to undergo routine serial testing except in the case of known exposure or ongoing transmission. The guideline authors suggested serial screening might be considered for health care workers whose work puts them at greater risk – for example, pulmonologists or respiratory therapists – or for those working in settings in which transmission has happened in the past.
For personnel with latent tuberculosis infection, the guidelines recommend “encouragement of treatment” unless it is contraindicated, and annual symptom screening in those not undergoing treatment.
The guideline committee also advocated for annual tuberculosis education for all health care workers.
The new recommendations were based on a systematic review of 36 studies of tuberculosis screening and testing among health care personnel, 16 of which were performed in the United States, and all but two of which were conducted in a hospital setting.
The authors stressed that recommendations from the 2005 CDC guidelines – which do not pertain to health care personnel screening, testing, treatment and education – remain unchanged.
One author declared personal fees from the National Tuberculosis Controllers Association during the conduct of the study. Two others reported unrelated grants and personal fees from private industry. No other conflicts of interest were disclosed.
SOURCE: Sosa L et al. MMWR. 2019;68:439-43.
FROM MMWR
Survey: Physicians predict increase in measles deaths
by real-time market insights technology firm InCrowd.
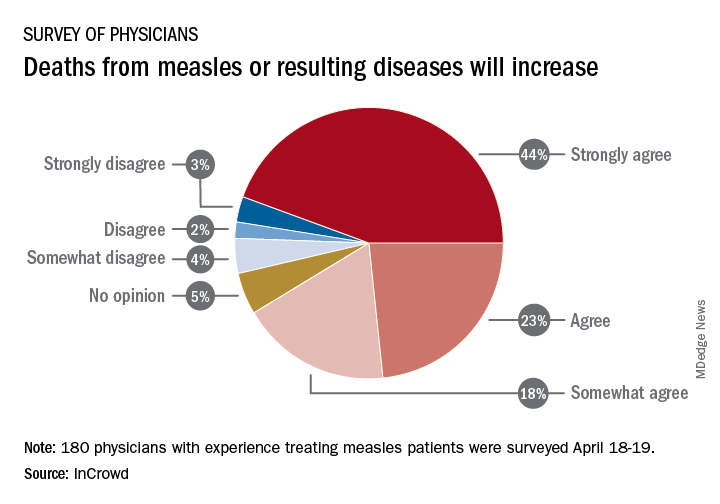
Among the 180 physicians with experience treating measles, 23% agreed and 44% said that they strongly agreed with the statement that measles deaths would increase, and another 18% said that they somewhat agreed. Only 9% expressed some level of disagreement, InCrowd said.
Most of those respondents also believe that summer travel will increase measles outbreaks (29% agreed and 30% strongly agreed) and that more communities will adopt requirements for measles vaccinations (26% and 36%). A majority also said that education about vaccinations will improve (26% agreed and 29% strongly agreed), but almost half of the physicians surveyed also expect vaccination misinformation to get worse (29% and 19%), InCrowd reported.
“With 44% of respondents predicting a high likelihood that deaths caused by measles will increase, the data show the imperative for physicians and patients to keep up the dialogue. … We have a long way to go before declaring victory,” said Diane Hayes, PhD, president and cofounder of InCrowd.
The InCrowd 5-minute microsurvey was conducted on April 18-19, 2019, and included 455 primary care physicians, of whom 40% said that they have treated or knew of colleagues in their facility or community who have treated patients with measles. Of those 180 respondents, 89 were pediatricians and 91 were in other primary care specialties.
by real-time market insights technology firm InCrowd.
Among the 180 physicians with experience treating measles, 23% agreed and 44% said that they strongly agreed with the statement that measles deaths would increase, and another 18% said that they somewhat agreed. Only 9% expressed some level of disagreement, InCrowd said.
Most of those respondents also believe that summer travel will increase measles outbreaks (29% agreed and 30% strongly agreed) and that more communities will adopt requirements for measles vaccinations (26% and 36%). A majority also said that education about vaccinations will improve (26% agreed and 29% strongly agreed), but almost half of the physicians surveyed also expect vaccination misinformation to get worse (29% and 19%), InCrowd reported.
“With 44% of respondents predicting a high likelihood that deaths caused by measles will increase, the data show the imperative for physicians and patients to keep up the dialogue. … We have a long way to go before declaring victory,” said Diane Hayes, PhD, president and cofounder of InCrowd.
The InCrowd 5-minute microsurvey was conducted on April 18-19, 2019, and included 455 primary care physicians, of whom 40% said that they have treated or knew of colleagues in their facility or community who have treated patients with measles. Of those 180 respondents, 89 were pediatricians and 91 were in other primary care specialties.
by real-time market insights technology firm InCrowd.
Among the 180 physicians with experience treating measles, 23% agreed and 44% said that they strongly agreed with the statement that measles deaths would increase, and another 18% said that they somewhat agreed. Only 9% expressed some level of disagreement, InCrowd said.
Most of those respondents also believe that summer travel will increase measles outbreaks (29% agreed and 30% strongly agreed) and that more communities will adopt requirements for measles vaccinations (26% and 36%). A majority also said that education about vaccinations will improve (26% agreed and 29% strongly agreed), but almost half of the physicians surveyed also expect vaccination misinformation to get worse (29% and 19%), InCrowd reported.
“With 44% of respondents predicting a high likelihood that deaths caused by measles will increase, the data show the imperative for physicians and patients to keep up the dialogue. … We have a long way to go before declaring victory,” said Diane Hayes, PhD, president and cofounder of InCrowd.
The InCrowd 5-minute microsurvey was conducted on April 18-19, 2019, and included 455 primary care physicians, of whom 40% said that they have treated or knew of colleagues in their facility or community who have treated patients with measles. Of those 180 respondents, 89 were pediatricians and 91 were in other primary care specialties.
Young children with neuromuscular disease are vulnerable to respiratory viruses
This highlights the need for new vaccines
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.

Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
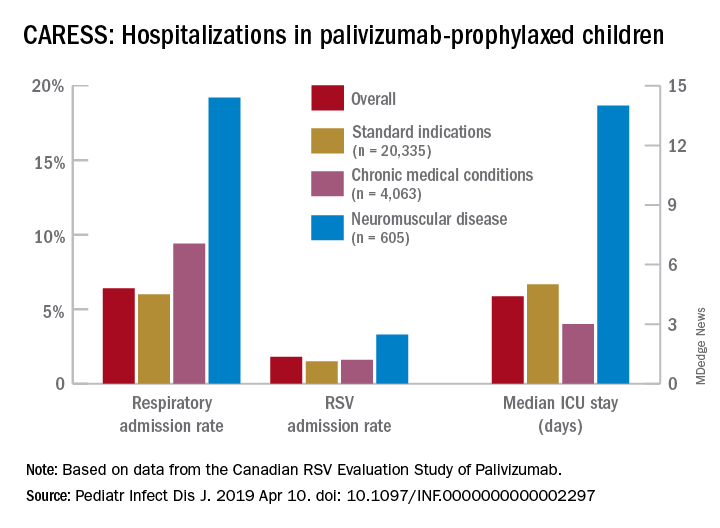
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
This highlights the need for new vaccines
This highlights the need for new vaccines
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.
Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
Influenza gets a lot of attention each winter, but respiratory syncytial virus (RSV) and other respiratory viruses have as much or more impact on pediatric populations, particularly certain high-risk groups. But currently there are no vaccines for noninfluenza respiratory viruses. That said, several are under development, for RSV and parainfluenza.
Which groups are likely to get the most benefit from these newer vaccines?
We all are aware of the extra vulnerability to respiratory viruses (RSV being the most frequent) in premature infants, those with chronic lung disease, or those with congenital heart syndromes; such vulnerable patients are not infrequently seen in routine practice. A recent report shined a brighter light on such a group.
Real-world data from a nationwide Canadian surveillance system (CARESS) was used to analyze relative risks of categories of young children who are thought to be vulnerable to respiratory viruses, with a particular focus on those with neuromuscular disease. The CARESS investigators analyzed 12 years’ data on respiratory hospitalizations from among palivizumab-prophylaxed patients (including specific data on RSV when patients were tested for RSV per standard of care).1 Unfortunately, RSV testing was not universal despite hospitalization, so the true incidence of RSV-specific hospitalizations was likely underestimated.
Nevertheless, more than 25,000 children from 2005 through 2017 were grouped into three categories of palivizumab-prophylaxed high-risk children: standard indications (SI), n = 20,335; chronic medical conditions (CMD), n = 4,063; and neuromuscular disease (NMD), n = 605. This study is notable for having a relatively large number of neuromuscular disease subjects. Two-thirds of each group were fully palivizumab adherent.
The SI group included the standard American Academy of Pediatrics–recommended groups, such as premature infants, congenital heart disease, etc.
The CMD group included conditions that lead clinicians to use palivizumab off label, such as cystic fibrosis, congenital airway anomalies, immunodeficiency, and pulmonary disorders.
The NMD participants were subdivided into two groups. Group 1 comprised general hypotonic neuromuscular diseases such as hypoxic-ischemic encephalopathy, Prader-Willi syndrome, chromosomal disorders, and migration/demyelinating diseases. Group 2 included more severe infantile neuromuscular disorders, such as spinal muscular atrophy, myotonic dystrophy, centronuclear and nemaline myopathy, mitochondrial and glycogen storage myopathies, or arthrogryposis.
Overall, 6.9% of CARESS RSV-prophylaxed subjects were hospitalized. About one in five hospitalized patients from each group was hospitalized more than once. Specific respiratory hospitalization rates for each group were 6% (n = 1,228) for SI subjects and 9.4% (n = 380) for CMD, compared with 19.2% (n = 116) for NMD subjects.
It is unclear what proportion underwent RSV testing, but a total of 334 were confirmed RSV positive: 261 were SI, 54 were CMD and 19 were NMD. The RSV-test-positive rate was 1.5% for SI, 1.6% for CMD and 3.3% for NMD; so while a higher number of SI children were RSV positive, the rate of RSV positivity was actually highest with NMD.
RSV-positive subjects needing ICU care among NMD patients also had longer ICU stays (median 14 days), compared with RSV-positive CMD or SI subjects (median 3 and 5 days, respectively). Further, hospitalized RSV-positive NMD subjects presented more frequently with pneumonia (42% vs. 30% for CMD and 20% for SI) while hospitalized RSV-positive SI subjects more often had apnea (17% vs. 10% for NMD and 5% for CMD, P less than .05).
These differences in the courses of NMD patients raise the question as to whether the NMD group was somehow different from the SI and CMD groups, other than muscular weakness that likely leads to less ability to clear secretions and a less efficient cough. It turns out that NMD children were older and had worse neonatal medical courses (longer hospital stays, more often ventilated, and used oxygen longer). It could be argued that these differences may have been in part due to the muscular weakness inherent in their underlying disease, but they appear to be predictors of worse respiratory infectious disease than other vulnerable populations as the NMD children get older.
Indeed, the overall risk of any respiratory admission among NMD subjects was nearly twice as high, compared with SI (hazard ratio, 1.90, P less than .0005); but the somewhat higher risk for NMD vs. CMD was not significant (HR, 1.33, P = .090). However, when looking specifically at RSV confirmed admissions, NMD had more than twice the hospitalization risk than either other group (HR, 2.26, P = .001 vs. SI; and HR, 2.74, P = .001 vs. CMD).
Further, an NMD subgroup analysis showed 1.69 times the overall respiratory hospitalization risk among the more severe vs. less severe NMD group, but a similar risk of RSV admission. The authors point out that one reason for this discrepancy may be a higher probability of aspiration causing hospitalization because of more dramatic acute events during respiratory infections in patients with more severe NMD. It also may be that palivizumab evened the playing field for RSV but not for other viruses such as parainfluenza, adenovirus, or even rhinovirus.
Nevertheless, these data tell us that risk of respiratory disease severe enough to need hospitalization continues to an older age in NMD than SI or CMD patients, well past 2 years of age. And the risk is not only from RSV. That said, RSV remains a player in some patients (particularly NMD patients) despite palivizumab prophylaxis, highlighting the need for RSV as well as parainfluenza vaccines. While these vaccines should help all young children, they seem likely to be even more beneficial for high-risk children including those with NMD, and particularly those with more severe NMD.
Eleven among 60 total candidate RSV vaccines (live attenuated, particle based, or vector based) are currently in clinical trials.2 Fewer parainfluenza vaccines are in the pipeline, but clinical trials also are underway.3-5 Approval of such vaccines is not expected until the mid-2020s, so at present we are left with providing palivizumab to our vulnerable patients while emphasizing nonmedical strategies that may help prevent respiratory viruses. These only partially successful preventive interventions include breastfeeding, avoiding secondhand smoke, and avoiding known high-risk exposures, such as large day care centers.
My hope is for quicker than projected progress on the vaccine front so that winter admissions for respiratory viruses might decrease in numbers similar to the decrease we have noted with another vaccine successful against a seasonally active pathogen – rotavirus.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital–Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines. The hospital also receives CDC funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus. Email Dr. Harrison at pdnews@mdedge.com.
References
1. Pediatr Infect Dis J. 2019 Apr 10. doi: 10.1097/INF.0000000000002297.
2. “Advances in RSV Vaccine Research and Development – A Global Agenda.”
3. J Pediatric Infect Dis Soc. 2015 Dec;4(4): e143-6.
4. J Virol. 2015 Oct;89(20):10319-32.
5. Vaccine. 2017 Dec 18;35(51):7139-46.
Is it measles? – Diagnosis and management for the pediatric provider
The mother of an 8-month-old calls your office and is hysterical. Her daughter has had cough for a few days with high fevers and now has developed a full body rash. She is worried about measles and is on her way to your office.
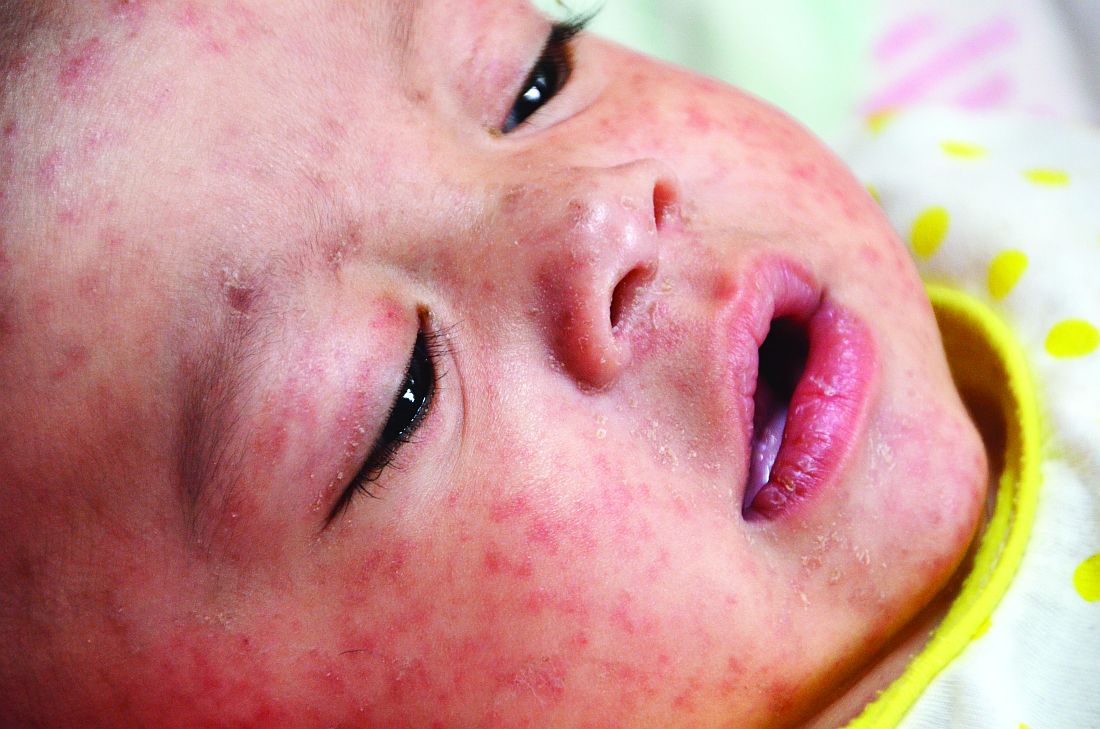
We are in the middle of a measles epidemic, there’s no denying it. Measles was declared eliminated in 2000, but reported cases in the United States have been on the rise, and are now at the highest number since 2014. Five months into 2019, there have been 839 reported cases as of May 13). Measles outbreaks (defined by the Centers for Disease Control and Prevention as three or more cases) have been reported in California, Georgia, Maryland, Michigan, New Jersey, New York, and Pennsylvania. When vaccination rates fall, it is easy for measles to spread. The virus is highly contagious in nonimmune people, because of its airborne spread and its persistence in the environment for hours.
First – is it really measles?
It can be difficult to distinguish the maculopapular rash of measles from similar rashes that occur with more benign viral illnesses. Adding to the challenge, the last major measles outbreak in the United States was over 2 decades ago, and many practicing pediatricians have never seen a single case. So, what clinical features can help distinguish measles from other febrile illnesses?
The prodromal phase of measles lasts approximately 2-4 days and children have high fevers (103°-105° F), anorexia, and malaise. Conjunctivitis, coryza, and cough develop during this phase, and precede any rash. Koplik spots appear during the prodromal phase, but are not seen in all cases. These spots are 1- to 3-mm blue-white lesions on an erythematous base on the buccal mucosa, classically opposite the first molar. The spots often slough once the rash appears. The rash appears 2-4 days after the onset of fever, and is initially maculopapular and blanching. The first lesions appear on the face and neck, and the rash spreads cranial to caudal, typically sparing palms and soles. After days 3-4, the rash will no longer blanch. High fevers persist for 2-4 more days with rash, ongoing respiratory symptoms, conjunctivitis, and pharyngitis. Note that the fever will persist even with development of the rash, unlike in roseola.
It is not only important to diagnosis measles from a public health standpoint, but also because measles can have severe complications, especially in infants and children under 5 years. During the 1989-1991 outbreak, the mortality rate was 2.2 deaths per 1,000 cases (J Infect Dis. 2004 May 1. doi: 10.1086/377694).

Six percent of patients develop pneumonia, which in infants and toddlers can lead to respiratory distress or failure requiring hospitalization. Pneumonia is responsible for 60% of measles deaths, according to the CDC “Pink Book,” Epidemiology and Prevention of Vaccine-Preventable Diseases, chapter 13 on measles, 13th Ed., 2015. Ocular complications include keratitis and corneal ulceration. Measles also can cause serious neurologic complications. Encephalitis, seen in 1 per 1,000 cases, usually arises several days after the rash and may present with seizure or encephalopathy. Acute disseminated encephalomyelitis (ADEM), an inflammatory demyelinating disease of the central nervous system, occurs in approximately 1 per 1,000 cases, typically presents during the recovery phase (1-2 weeks after rash), and can have long-term sequelae. Subacute sclerosing panencephalitis (SSPE) is a progressive and fatal neurodegenerative disorder, and presents 7-10 years after measles infection.
Should you transfer the patient to a hospital?
Unless there is a medical need for the child to be admitted, sending a patient with potential measles to the hospital is not necessary, and can cause exposure to a large group of medical personnel, and patients who cannot be vaccinated (such as infants, immunocompromised patients, and pregnant women). However, if there is concern for complications such as seizures, encephalitis, or pneumonia, then transfer is indicated. Call the accepting hospital in advance so the staff can prepare for the patient. During transfer, place a standard face mask on the patient and instruct the patient not to remove it.

For hospitals accepting a suspected measles case, meet the patient outside of the facility and ensure that the patient is wearing a standard face mask. All staff interacting with the patient should practice contact and airborne precautions (N95 respirator mask). Take the patient directly to an isolation room with negative airflow. Caution pregnant staff that they should not have contact with the patient.
Which diagnostic tests should you use?
Diagnosis can be made based on serum antibody tests (measles IgM and IgG), throat or urine viral cultures, and nasopharyngeal and throat specimen polymerase chain reaction (PCR) testing. The CDC recommends obtaining a serum sample for measles IgM testing and a throat swab for PCR in all suspected cases, but local health departments vary in their specific testing recommendations. Familiarize yourself with the tests recommended by your local department of health, and where they prefer testing on outpatients to be done. Confirmed measles should be reported to your department of health.
What are considerations for community pediatric offices?
Update families in emails to call ahead if they suspect measles. This way the office can prepare a room for the family, and have the family immediately brought back without exposing staff and other families in the waiting area. It may be more prudent to examine these children at the end of the clinic day as the virus can persist for up to 2 hours on fomites and in the air. Therefore, all waiting areas and shared air spaces (including those with shared air ducts) should be cleared for 2 hours after the patient leaves.
When should you provide prophylaxis after exposure?
A patient with suspected measles does not require immediate vaccination. If it is measles, it is already too late to vaccinate. If measles is ruled out, the child should follow the standard measles vaccination guidelines.
Individuals are contagious from 4 days before to 4 days after the rash appears.
If measles is confirmed, all people who are unvaccinated or undervaccinated and were exposed to the confirmed case during the contagious period should be vaccinated within 72 hours of exposure. Infants 6 months or older may safely receive the MMR vaccine. However, infants vaccinated with MMR before their first birthday must be vaccinated again at age 12-15 months (greater than 28 days after prior vaccine) and at 4-6 years. Immunoglobulin prophylaxis should be given intramuscularly in exposed infants ages birth to less than 6 months, and in those ages 6-12 months who present beyond the 72-hour window. Unvaccinated or undervaccinated, exposed individuals at high risk for complications from measles (immunocompromised, pregnant) also should receive immunoglobulin.
What should you tell traveling families?
Several countries have large, ongoing measles outbreaks, including Israel, Ukraine, and the Philippines. Before international travel, infants 6-11 months should receive one dose of MMR vaccine, and children 12 months and older need two doses separated by at least 28 days. For unvaccinated or undervaccinated children, consider advising families to hold off travel to high-risk countries, or understand the indications to vaccinate a child upon return.
Dr. Angelica DesPain is a pediatric emergency medicine fellow at Children’s National Medical Center in Washington. She said she has no relevant financial disclosures. Dr. Emily Willner is a pediatric emergency medicine attending at Children’s National Medical Center, and an assistant professor of pediatrics and emergency medicine at George Washington University, Washington. She has no relevant financial disclosures.
The mother of an 8-month-old calls your office and is hysterical. Her daughter has had cough for a few days with high fevers and now has developed a full body rash. She is worried about measles and is on her way to your office.
We are in the middle of a measles epidemic, there’s no denying it. Measles was declared eliminated in 2000, but reported cases in the United States have been on the rise, and are now at the highest number since 2014. Five months into 2019, there have been 839 reported cases as of May 13). Measles outbreaks (defined by the Centers for Disease Control and Prevention as three or more cases) have been reported in California, Georgia, Maryland, Michigan, New Jersey, New York, and Pennsylvania. When vaccination rates fall, it is easy for measles to spread. The virus is highly contagious in nonimmune people, because of its airborne spread and its persistence in the environment for hours.
First – is it really measles?
It can be difficult to distinguish the maculopapular rash of measles from similar rashes that occur with more benign viral illnesses. Adding to the challenge, the last major measles outbreak in the United States was over 2 decades ago, and many practicing pediatricians have never seen a single case. So, what clinical features can help distinguish measles from other febrile illnesses?
The prodromal phase of measles lasts approximately 2-4 days and children have high fevers (103°-105° F), anorexia, and malaise. Conjunctivitis, coryza, and cough develop during this phase, and precede any rash. Koplik spots appear during the prodromal phase, but are not seen in all cases. These spots are 1- to 3-mm blue-white lesions on an erythematous base on the buccal mucosa, classically opposite the first molar. The spots often slough once the rash appears. The rash appears 2-4 days after the onset of fever, and is initially maculopapular and blanching. The first lesions appear on the face and neck, and the rash spreads cranial to caudal, typically sparing palms and soles. After days 3-4, the rash will no longer blanch. High fevers persist for 2-4 more days with rash, ongoing respiratory symptoms, conjunctivitis, and pharyngitis. Note that the fever will persist even with development of the rash, unlike in roseola.
It is not only important to diagnosis measles from a public health standpoint, but also because measles can have severe complications, especially in infants and children under 5 years. During the 1989-1991 outbreak, the mortality rate was 2.2 deaths per 1,000 cases (J Infect Dis. 2004 May 1. doi: 10.1086/377694).
Six percent of patients develop pneumonia, which in infants and toddlers can lead to respiratory distress or failure requiring hospitalization. Pneumonia is responsible for 60% of measles deaths, according to the CDC “Pink Book,” Epidemiology and Prevention of Vaccine-Preventable Diseases, chapter 13 on measles, 13th Ed., 2015. Ocular complications include keratitis and corneal ulceration. Measles also can cause serious neurologic complications. Encephalitis, seen in 1 per 1,000 cases, usually arises several days after the rash and may present with seizure or encephalopathy. Acute disseminated encephalomyelitis (ADEM), an inflammatory demyelinating disease of the central nervous system, occurs in approximately 1 per 1,000 cases, typically presents during the recovery phase (1-2 weeks after rash), and can have long-term sequelae. Subacute sclerosing panencephalitis (SSPE) is a progressive and fatal neurodegenerative disorder, and presents 7-10 years after measles infection.
Should you transfer the patient to a hospital?
Unless there is a medical need for the child to be admitted, sending a patient with potential measles to the hospital is not necessary, and can cause exposure to a large group of medical personnel, and patients who cannot be vaccinated (such as infants, immunocompromised patients, and pregnant women). However, if there is concern for complications such as seizures, encephalitis, or pneumonia, then transfer is indicated. Call the accepting hospital in advance so the staff can prepare for the patient. During transfer, place a standard face mask on the patient and instruct the patient not to remove it.
For hospitals accepting a suspected measles case, meet the patient outside of the facility and ensure that the patient is wearing a standard face mask. All staff interacting with the patient should practice contact and airborne precautions (N95 respirator mask). Take the patient directly to an isolation room with negative airflow. Caution pregnant staff that they should not have contact with the patient.
Which diagnostic tests should you use?
Diagnosis can be made based on serum antibody tests (measles IgM and IgG), throat or urine viral cultures, and nasopharyngeal and throat specimen polymerase chain reaction (PCR) testing. The CDC recommends obtaining a serum sample for measles IgM testing and a throat swab for PCR in all suspected cases, but local health departments vary in their specific testing recommendations. Familiarize yourself with the tests recommended by your local department of health, and where they prefer testing on outpatients to be done. Confirmed measles should be reported to your department of health.
What are considerations for community pediatric offices?
Update families in emails to call ahead if they suspect measles. This way the office can prepare a room for the family, and have the family immediately brought back without exposing staff and other families in the waiting area. It may be more prudent to examine these children at the end of the clinic day as the virus can persist for up to 2 hours on fomites and in the air. Therefore, all waiting areas and shared air spaces (including those with shared air ducts) should be cleared for 2 hours after the patient leaves.
When should you provide prophylaxis after exposure?
A patient with suspected measles does not require immediate vaccination. If it is measles, it is already too late to vaccinate. If measles is ruled out, the child should follow the standard measles vaccination guidelines.
Individuals are contagious from 4 days before to 4 days after the rash appears.
If measles is confirmed, all people who are unvaccinated or undervaccinated and were exposed to the confirmed case during the contagious period should be vaccinated within 72 hours of exposure. Infants 6 months or older may safely receive the MMR vaccine. However, infants vaccinated with MMR before their first birthday must be vaccinated again at age 12-15 months (greater than 28 days after prior vaccine) and at 4-6 years. Immunoglobulin prophylaxis should be given intramuscularly in exposed infants ages birth to less than 6 months, and in those ages 6-12 months who present beyond the 72-hour window. Unvaccinated or undervaccinated, exposed individuals at high risk for complications from measles (immunocompromised, pregnant) also should receive immunoglobulin.
What should you tell traveling families?
Several countries have large, ongoing measles outbreaks, including Israel, Ukraine, and the Philippines. Before international travel, infants 6-11 months should receive one dose of MMR vaccine, and children 12 months and older need two doses separated by at least 28 days. For unvaccinated or undervaccinated children, consider advising families to hold off travel to high-risk countries, or understand the indications to vaccinate a child upon return.
Dr. Angelica DesPain is a pediatric emergency medicine fellow at Children’s National Medical Center in Washington. She said she has no relevant financial disclosures. Dr. Emily Willner is a pediatric emergency medicine attending at Children’s National Medical Center, and an assistant professor of pediatrics and emergency medicine at George Washington University, Washington. She has no relevant financial disclosures.
The mother of an 8-month-old calls your office and is hysterical. Her daughter has had cough for a few days with high fevers and now has developed a full body rash. She is worried about measles and is on her way to your office.
We are in the middle of a measles epidemic, there’s no denying it. Measles was declared eliminated in 2000, but reported cases in the United States have been on the rise, and are now at the highest number since 2014. Five months into 2019, there have been 839 reported cases as of May 13). Measles outbreaks (defined by the Centers for Disease Control and Prevention as three or more cases) have been reported in California, Georgia, Maryland, Michigan, New Jersey, New York, and Pennsylvania. When vaccination rates fall, it is easy for measles to spread. The virus is highly contagious in nonimmune people, because of its airborne spread and its persistence in the environment for hours.
First – is it really measles?
It can be difficult to distinguish the maculopapular rash of measles from similar rashes that occur with more benign viral illnesses. Adding to the challenge, the last major measles outbreak in the United States was over 2 decades ago, and many practicing pediatricians have never seen a single case. So, what clinical features can help distinguish measles from other febrile illnesses?
The prodromal phase of measles lasts approximately 2-4 days and children have high fevers (103°-105° F), anorexia, and malaise. Conjunctivitis, coryza, and cough develop during this phase, and precede any rash. Koplik spots appear during the prodromal phase, but are not seen in all cases. These spots are 1- to 3-mm blue-white lesions on an erythematous base on the buccal mucosa, classically opposite the first molar. The spots often slough once the rash appears. The rash appears 2-4 days after the onset of fever, and is initially maculopapular and blanching. The first lesions appear on the face and neck, and the rash spreads cranial to caudal, typically sparing palms and soles. After days 3-4, the rash will no longer blanch. High fevers persist for 2-4 more days with rash, ongoing respiratory symptoms, conjunctivitis, and pharyngitis. Note that the fever will persist even with development of the rash, unlike in roseola.
It is not only important to diagnosis measles from a public health standpoint, but also because measles can have severe complications, especially in infants and children under 5 years. During the 1989-1991 outbreak, the mortality rate was 2.2 deaths per 1,000 cases (J Infect Dis. 2004 May 1. doi: 10.1086/377694).
Six percent of patients develop pneumonia, which in infants and toddlers can lead to respiratory distress or failure requiring hospitalization. Pneumonia is responsible for 60% of measles deaths, according to the CDC “Pink Book,” Epidemiology and Prevention of Vaccine-Preventable Diseases, chapter 13 on measles, 13th Ed., 2015. Ocular complications include keratitis and corneal ulceration. Measles also can cause serious neurologic complications. Encephalitis, seen in 1 per 1,000 cases, usually arises several days after the rash and may present with seizure or encephalopathy. Acute disseminated encephalomyelitis (ADEM), an inflammatory demyelinating disease of the central nervous system, occurs in approximately 1 per 1,000 cases, typically presents during the recovery phase (1-2 weeks after rash), and can have long-term sequelae. Subacute sclerosing panencephalitis (SSPE) is a progressive and fatal neurodegenerative disorder, and presents 7-10 years after measles infection.
Should you transfer the patient to a hospital?
Unless there is a medical need for the child to be admitted, sending a patient with potential measles to the hospital is not necessary, and can cause exposure to a large group of medical personnel, and patients who cannot be vaccinated (such as infants, immunocompromised patients, and pregnant women). However, if there is concern for complications such as seizures, encephalitis, or pneumonia, then transfer is indicated. Call the accepting hospital in advance so the staff can prepare for the patient. During transfer, place a standard face mask on the patient and instruct the patient not to remove it.
For hospitals accepting a suspected measles case, meet the patient outside of the facility and ensure that the patient is wearing a standard face mask. All staff interacting with the patient should practice contact and airborne precautions (N95 respirator mask). Take the patient directly to an isolation room with negative airflow. Caution pregnant staff that they should not have contact with the patient.
Which diagnostic tests should you use?
Diagnosis can be made based on serum antibody tests (measles IgM and IgG), throat or urine viral cultures, and nasopharyngeal and throat specimen polymerase chain reaction (PCR) testing. The CDC recommends obtaining a serum sample for measles IgM testing and a throat swab for PCR in all suspected cases, but local health departments vary in their specific testing recommendations. Familiarize yourself with the tests recommended by your local department of health, and where they prefer testing on outpatients to be done. Confirmed measles should be reported to your department of health.
What are considerations for community pediatric offices?
Update families in emails to call ahead if they suspect measles. This way the office can prepare a room for the family, and have the family immediately brought back without exposing staff and other families in the waiting area. It may be more prudent to examine these children at the end of the clinic day as the virus can persist for up to 2 hours on fomites and in the air. Therefore, all waiting areas and shared air spaces (including those with shared air ducts) should be cleared for 2 hours after the patient leaves.
When should you provide prophylaxis after exposure?
A patient with suspected measles does not require immediate vaccination. If it is measles, it is already too late to vaccinate. If measles is ruled out, the child should follow the standard measles vaccination guidelines.
Individuals are contagious from 4 days before to 4 days after the rash appears.
If measles is confirmed, all people who are unvaccinated or undervaccinated and were exposed to the confirmed case during the contagious period should be vaccinated within 72 hours of exposure. Infants 6 months or older may safely receive the MMR vaccine. However, infants vaccinated with MMR before their first birthday must be vaccinated again at age 12-15 months (greater than 28 days after prior vaccine) and at 4-6 years. Immunoglobulin prophylaxis should be given intramuscularly in exposed infants ages birth to less than 6 months, and in those ages 6-12 months who present beyond the 72-hour window. Unvaccinated or undervaccinated, exposed individuals at high risk for complications from measles (immunocompromised, pregnant) also should receive immunoglobulin.
What should you tell traveling families?
Several countries have large, ongoing measles outbreaks, including Israel, Ukraine, and the Philippines. Before international travel, infants 6-11 months should receive one dose of MMR vaccine, and children 12 months and older need two doses separated by at least 28 days. For unvaccinated or undervaccinated children, consider advising families to hold off travel to high-risk countries, or understand the indications to vaccinate a child upon return.
Dr. Angelica DesPain is a pediatric emergency medicine fellow at Children’s National Medical Center in Washington. She said she has no relevant financial disclosures. Dr. Emily Willner is a pediatric emergency medicine attending at Children’s National Medical Center, and an assistant professor of pediatrics and emergency medicine at George Washington University, Washington. She has no relevant financial disclosures.
Younger patients with NSCLC tend to live longer
Younger patients with non–small cell lung cancer (NSCLC) may have better survival, despite higher rates of brain metastasis and driver mutations, according to results from a retrospective analysis.
“We carried out a comprehensive analysis of patient clinicopathologic features and clinical outcomes in both young (age ≤ 50 years) and older (age > 60 years) patients with NSCLC,” wrote Anna May Suidan of Tel Aviv University, and colleagues. The findings were published in the Journal of Global Oncology.
The researchers reviewed medical records of patients who were diagnosed with lung cancer at a large cancer treatment facility in Israel from 2010 to 2015. Patients were categorized into two groups according to age at cancer diagnosis, which was established based on tumor pathology.
Various clinical data were collected, including demographic information, history of malignancy, smoking history, histologic subtype, and survival data.
In all, 62 patients were included in the younger cohort (median age, 44.5 years) and 124 patients in the older cohort (median age, 68.0 years).
After analysis, the researchers found that younger patients had a higher incidence of brain metastasis (39% vs. 25%, respectively; P = .04), and increased rates of EGFR mutations (23% vs. 18%, respectively; P = .4) and ALK translocations (13% vs. 2%, respectively; P = .002) versus older patients.
“Our cohort, which was [composed] of white patients, demonstrated that younger patients harbored more targetable driver mutations compared with older patients (34% vs. 18%; P = .01),” the researchers wrote.
In addition, among those with a driver mutation, younger patients showed a trend toward better survival (median survival, 33 vs. 25 months, respectively; P = .4).
Two key limitations of the study were the small sample size and retrospective design.
“[These results] highlight the importance of genetic background assessments and considering lung cancer as a possible diagnosis in young symptomatic patients in clinical settings,” the researchers concluded.
No funding sources were reported. The authors reported financial affiliations with Astra Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Novartis, Roche, Teva Pharmaceuticals, and several others.
SOURCE: Suidan AM et al. J Glob Oncol. 2019 May 8. doi: 10.1200/JGO.18.00216.
Younger patients with non–small cell lung cancer (NSCLC) may have better survival, despite higher rates of brain metastasis and driver mutations, according to results from a retrospective analysis.
“We carried out a comprehensive analysis of patient clinicopathologic features and clinical outcomes in both young (age ≤ 50 years) and older (age > 60 years) patients with NSCLC,” wrote Anna May Suidan of Tel Aviv University, and colleagues. The findings were published in the Journal of Global Oncology.
The researchers reviewed medical records of patients who were diagnosed with lung cancer at a large cancer treatment facility in Israel from 2010 to 2015. Patients were categorized into two groups according to age at cancer diagnosis, which was established based on tumor pathology.
Various clinical data were collected, including demographic information, history of malignancy, smoking history, histologic subtype, and survival data.
In all, 62 patients were included in the younger cohort (median age, 44.5 years) and 124 patients in the older cohort (median age, 68.0 years).
After analysis, the researchers found that younger patients had a higher incidence of brain metastasis (39% vs. 25%, respectively; P = .04), and increased rates of EGFR mutations (23% vs. 18%, respectively; P = .4) and ALK translocations (13% vs. 2%, respectively; P = .002) versus older patients.
“Our cohort, which was [composed] of white patients, demonstrated that younger patients harbored more targetable driver mutations compared with older patients (34% vs. 18%; P = .01),” the researchers wrote.
In addition, among those with a driver mutation, younger patients showed a trend toward better survival (median survival, 33 vs. 25 months, respectively; P = .4).
Two key limitations of the study were the small sample size and retrospective design.
“[These results] highlight the importance of genetic background assessments and considering lung cancer as a possible diagnosis in young symptomatic patients in clinical settings,” the researchers concluded.
No funding sources were reported. The authors reported financial affiliations with Astra Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Novartis, Roche, Teva Pharmaceuticals, and several others.
SOURCE: Suidan AM et al. J Glob Oncol. 2019 May 8. doi: 10.1200/JGO.18.00216.
Younger patients with non–small cell lung cancer (NSCLC) may have better survival, despite higher rates of brain metastasis and driver mutations, according to results from a retrospective analysis.
“We carried out a comprehensive analysis of patient clinicopathologic features and clinical outcomes in both young (age ≤ 50 years) and older (age > 60 years) patients with NSCLC,” wrote Anna May Suidan of Tel Aviv University, and colleagues. The findings were published in the Journal of Global Oncology.
The researchers reviewed medical records of patients who were diagnosed with lung cancer at a large cancer treatment facility in Israel from 2010 to 2015. Patients were categorized into two groups according to age at cancer diagnosis, which was established based on tumor pathology.
Various clinical data were collected, including demographic information, history of malignancy, smoking history, histologic subtype, and survival data.
In all, 62 patients were included in the younger cohort (median age, 44.5 years) and 124 patients in the older cohort (median age, 68.0 years).
After analysis, the researchers found that younger patients had a higher incidence of brain metastasis (39% vs. 25%, respectively; P = .04), and increased rates of EGFR mutations (23% vs. 18%, respectively; P = .4) and ALK translocations (13% vs. 2%, respectively; P = .002) versus older patients.
“Our cohort, which was [composed] of white patients, demonstrated that younger patients harbored more targetable driver mutations compared with older patients (34% vs. 18%; P = .01),” the researchers wrote.
In addition, among those with a driver mutation, younger patients showed a trend toward better survival (median survival, 33 vs. 25 months, respectively; P = .4).
Two key limitations of the study were the small sample size and retrospective design.
“[These results] highlight the importance of genetic background assessments and considering lung cancer as a possible diagnosis in young symptomatic patients in clinical settings,” the researchers concluded.
No funding sources were reported. The authors reported financial affiliations with Astra Zeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Eli Lilly, Novartis, Roche, Teva Pharmaceuticals, and several others.
SOURCE: Suidan AM et al. J Glob Oncol. 2019 May 8. doi: 10.1200/JGO.18.00216.
FROM THE JOURNAL OF GLOBAL ONCOLOGY
Management of Late Pulmonary Complications After Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1. 
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2). 
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).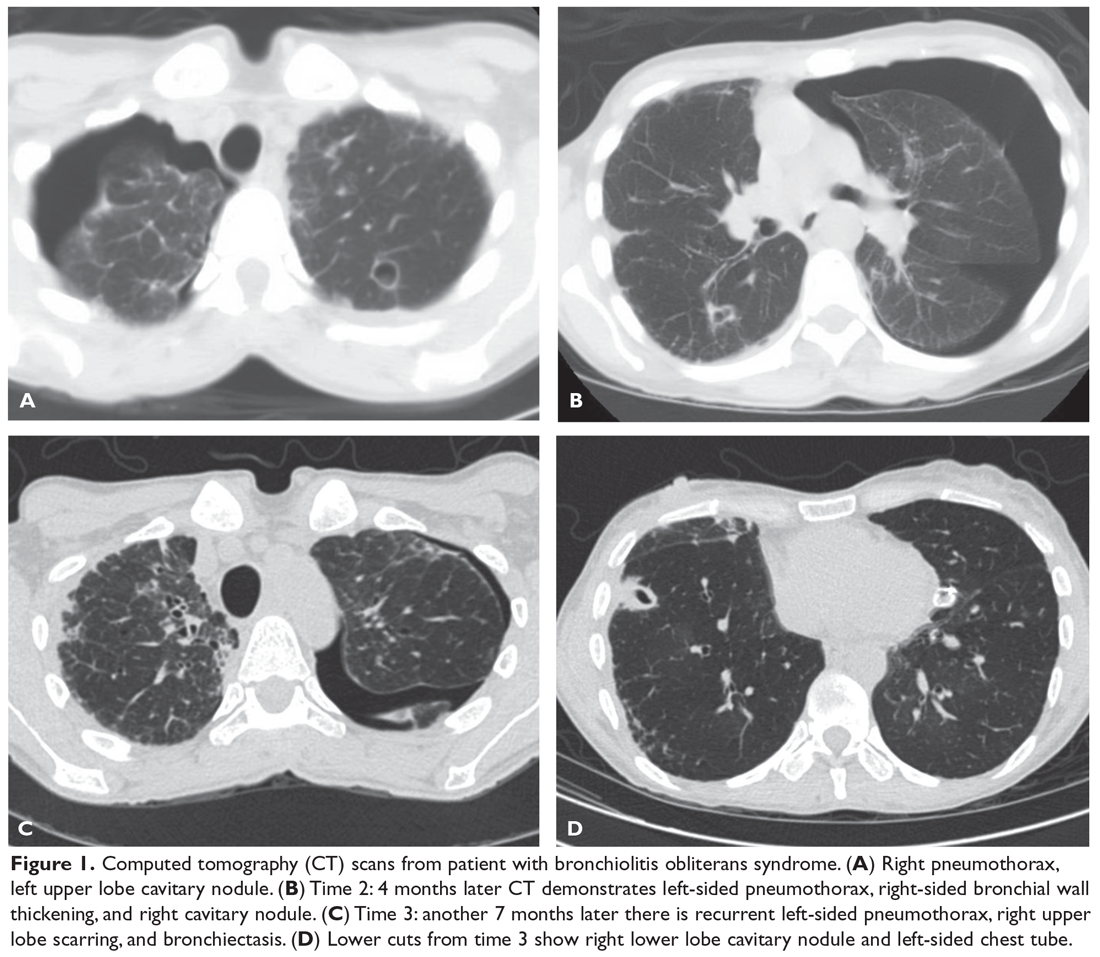
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.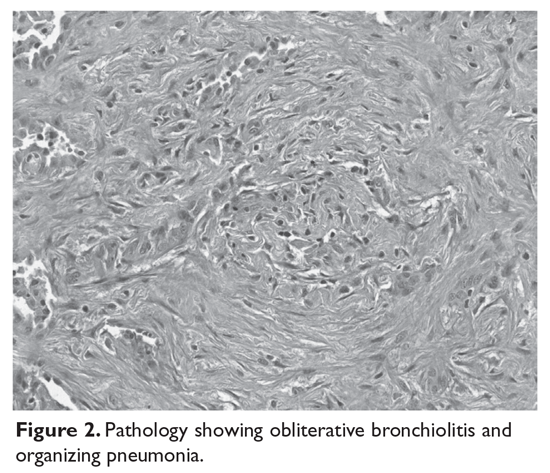
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
Hematopoietic stem cell transplantation (HSCT) is increasingly being used to treat hematologic malignancies as well as nonmalignant diseases and solid tumors. Over the past 2 decades overall survival following transplant and transplant-related mortality have improved.1 With this increased survival, there is a need to focus on late complications after transplantation. Pulmonary complications are a common but sometimes underrecognized cause of late morbidity and mortality in HSCT patients. This article, the second of 2 articles on post-HSCT pulmonary complications, reviews late-onset complications, with a focus on the evaluation and treatment of bronchiolitis obliterans syndrome (BOS), one of the most common and serious late pulmonary complications in HSCT patients. The first article reviewed the management of early-onset pulmonary complications and included a basic overview of stem cell transplantation, discussion of factors associated with pulmonary complications, and a review of methods for assessing pretransplant risk for pulmonary complications in patients undergoing HSCT.2
Case Presentation
A 40-year-old white woman with a history of acute myeloid leukemia status post peripheral blood stem cell transplant presents with dyspnea on exertion, which she states started about 1 month ago and now is limiting her with even 1 flight of stairs. She also complains of mild dry cough and a 4- to 5-lb weight loss over the past 1 to 2 months. She has an occasional runny nose, but denies gastroesophageal reflux, fevers, chills, or night sweats. She has a history of matched related sibling donor transplant with busulfan and cyclophosphamide conditioning 1 year prior to presentation. She has had significant graft-versus-host disease (GVHD), affecting the liver, gastrointestinal tract, skin, and eyes.
On physical examination, heart rate is 110 beats/min, respiratory rate is 16 breaths/min, blood pressure is 92/58 mm Hg, and the patient is afebrile. Eye exam reveals scleral injection, mouth shows dry mucous membranes with a few white plaques, and the skin has chronic changes with a rash over both arms. Cardiac exam reveals tachycardia but regular rhythm and there are no murmurs, rubs, or gallops. Lungs are clear bilaterally and abdomen shows no organomegaly.
Laboratory exam shows a white blood cell count of 7800 cells/μL, hemoglobin level of 12.4 g/dL, and platelet count of 186 × 103/μL. Liver enzymes are mildly elevated. Chest radiograph shows clear lung fields bilaterally.
- What is the differential in this patient with dyspnea 1 year after transplantation?
Late pulmonary complications are generally accepted as those occurring more than 100 days post transplant. This period of time is characterized by chronic GVHD and impaired cellular and humoral immunity. Results of longitudinal studies of infections in adult HSCT patients suggest that special attention should be paid to allogeneic HSCT recipients for post-engraftment infectious pulmonary complications.3 Encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the most frequent bacterial organisms causing late infectious pulmonary complications. Nontuberculous mycobacteria and Nocardia should also be considered. Depending upon geographic location, social and occupational risk factors, and prevalence, tuberculosis should also enter the differential.
There are many noninfectious late-onset pulmonary complications after HSCT. Unfortunately, the literature has divided pulmonary complications after HSCT using a range of criteria and classifications based upon timing, predominant pulmonary function test (PFT) findings, and etiology. These include early versus late, obstructive versus restrictive, and infectious versus noninfectious, which makes a comprehensive literature review of late pulmonary complications difficult. The most common noninfectious late-onset complications are bronchiolitis obliterans, cryptogenic organizing pneumonia (previously referred to as bronchiolitis obliterans organizing pneumonia, or BOOP), and interstitial pneumonia. Other rarely reported complications include eosinophilic pneumonia, pulmonary alveolar proteinosis, air leak syndrome, and pulmonary hypertension.
Case Continued
Because the patient does not have symptoms of infection, PFTs are obtained. Pretransplant PFTs and current PFTs are shown in Table 1.
- What is the diagnosis in this case?
Bronchiolitis Obliterans
BOS is one of the most common and most serious late-onset pulmonary diseases after allogeneic transplantation. It is considered the pulmonary form of chronic GVHD. BOS was first described in 1982 in patients with chronic GVHD after bone marrow transplantation.4 Many differing definitions of bronchiolitis obliterans have been described in the literature. A recent review of the topic cites 10 different published sets of criteria for the diagnosis of bronchiolitis obliterans.5 Traditionally, bronchiolitis obliterans was thought to occur in 2% to 8% of patients undergoing allogeneic HSCT, but these findings were from older studies that used a diagnosis based on very specific pathology findings. When more liberal diagnostic criteria are used, the incidence may be as high as 26% of allogeneic HSCT patients.6
Bronchiolitis obliterans is a progressive lung disease characterized by narrowing of the terminal airways and obliteration of the terminal bronchi. Pathology may show constrictive bronchiolitis but can also show lymphocytic bronchiolitis, which may be associated with a better outcome.7 As noted, bronchiolitis obliterans has traditionally been considered a pathologic diagnosis. Current diagnostic criteria have evolved based upon the difficulty in obtaining this diagnosis through transbronchial biopsy given the patchy nature of the disease.8 The gold standard of open lung biopsy is seldom pursued in the post-HSCT population as the procedure continues to carry a worrisome risk-benefit profile.
The 2005 National Institutes of Health (NIH) consensus development project on criteria for clinical trials in chronic GVHD developed a clinical strategy for diagnosing BOS using the following criteria: absence of active infection, decreased forced expiratory volume in 1 second (FEV1) < 75%, FEV1/forced vital capacity (FVC) ratio of < 70%, and evidence of air trapping on high-resolution computed tomography (HRCT) or PFTs (residual volume > 120%). These diagnostic criteria were applied to a small series of patients with clinically identified bronchiolitis obliterans or biopsy-proven bronchiolitis obliterans. Only 18% of these patients met the requirements for the NIH consensus definition.5 A 2011 study that applied the NIH criteria found an overall prevalence of 5.5% among all transplant recipients but a prevalence of 14% in patients with GVHD.9 In 2014, the NIH consensus development group updated their recommendations. The new criteria for diagnosis of BOS require the presence of airflow obstruction (FEV1/FVC < 70% or 5th percentile of predicted), FEV1 < 75% predicted with a ≥ 10% decline in fewer than 2 years, absence of infection, and presence of air trapping (by expiratory computed tomography [CT] scan or PFT with residual volume >120% predicted) (Table 2).
Some issues must be considered when determining airflow obstruction. The 2005 NIH working group recommends using Crapo as the reference set,11 but the National Health and Nutrition Examination Survey (NHANES) III reference values are the preferred reference set at this time12 and should be used in the United States. A recent article showed that the NHANES values were superior to older reference sets (however, they did not use Crapo as the comparison), although this study used the lower limit of normal as compared with the fixed 70% ratio.13 The 2014 NIH consensus group does not recommend a specific reference set and recognizes an FEV1/FVC ratio of 70% or less than the lower limit of normal as the cutoff value for airflow obstruction.10
Another issue in PFT interpretation is the finding of a decrease in FEV1 and FVC and normal total lung capacity, which is termed a nonspecific pattern. This pattern has been reported to occur in 9% of all PFTs and usually is associated with obstructive lung disease or obesity.14 A 2013 study described the nonspecific pattern as a BOS subgroup occurring in up to 31% of bronchiolitis obliterans patients.15
- What are the radiographic findings of BOS?
Chest radiograph is often normal in BOS. As discussed, air trapping can be documented using HRCT, according to the NIH clinical definition of bronchiolitis obliterans.16 A study that explored findings and trends seen on HRCT in HSCT patients with BOS found that the syndrome in these patients is characterized by central airway dilatation.17 Expiratory airway trapping on HRCT is the main finding, and this is best demonstrated on HRCT during inspiratory and expiratory phases.18 Other findings are bronchial wall thickening, parenchymal hypoattenuation, bronchiectasis, and centrilobular nodules.19
Galbán and colleagues developed a new technique called parametric response mapping that uses CT scanners to quantify normal parenchyma, functional small airway disease, emphysema, and parenchymal disease as relative lung volumes.20 This technique can detect airflow obstruction and small airway disease and was found to be a good method for detecting BOS after HSCT. In their study of parametric response mapping, the authors found that functional small airway disease affecting 28% or more of the total lung was highly indicative of bronchiolitis obliterans.20
- What therapies are used to treat BOS?
Traditionally, BOS has been treated with systemic immunosuppression. The recommended treatment had been systemic steroids at approximately 1 mg/ kg. However, it is increasingly recognized that BOS responds poorly to systemic steroids, and systemic steroids may actually be harmful and associated with increased mortality.15,21 The chronic GVHD recommendations from 2005 recommend ancillary therapy with inhaled corticosteroids and pulmonary rehabilitation.11 The updated 2011 German consensus statement lays out a clear management strategy for mild and moderate-severe disease with monitoring recommendations.22 The 2014 NIH chronic GVHD working group recommends fluticasone, azithromycin, and montelukast (ie, the FAM protocol) for treating BOS.23 FAM therapy in BOS may help lower the systemic steroid dose.24,25 Montelukast is not considered a treatment mainstay for BOS after lung transplant, but there is a study showing possible benefit in chronic GVHD.26 An evaluation of the natural history of a cohort of BOS patients treated with FAM therapy showed a rapid decline of FEV1 in the 6 months prior to diagnosis and treatment of BOS and subsequent stabilization following diagnosis and treatment.27 The benefit of high-dose inhaled corticosteroids or the combination of inhaled corticosteroids and long-acting beta-agonists has been demonstrated in small studies, which showed that these agents stabilized FEV1 and avoided the untoward side effects of systemic corticosteroids.28–30
Macrolide antibiotics have been explored as a treatment for BOS post HSCT because pilot studies suggested that azithromycin improved or stabilized FEV1 in patients with BOS after lung transplant or HSCT.31–33 Other studies of azithromycin have not shown benefit in the HSCT population after 3 months of therapy.34 A recent meta-analysis could neither support or refute the benefit of azithromycin for BOS after HSCT.35 In the lung transplant population, a study showed that patients who were started on azithromycin after transplant and continued on it 3 times a week had improved FEV1; these patients also had a reduced rate of BOS and improved overall and BOS-free survival 2 years after transplant.36 However, these benefits of azithromycin have not been observed in patients after HSCT. In fact, the ALLOZITHRO trial was stopped early because prophylactic azithromycin started at the time of the conditioning regimen with HSCT was associated with increased hematologic disease relapse, a decrease in airflow-decline-free survival, and reduced 2-year survival.30
Azithromycin is believed to exert an effect by its anti-inflammatory properties and perhaps by decreasing lung neutrophilia (it may be most beneficial in the subset of patients with high neutrophilia on bronchoalveolar lavage [BAL]).30 Adverse effects of chronic azithromycin include QT prolongation, cardiac arrhythmia, hearing loss, and antibiotic-resistant organism colonization.37,38
Other therapies include pulmonary rehabilitation, which may improve health-related quality of life and 6-minute walk distance,39 extracorporeal photopheresis,40 immunosuppression with calcineurin inhibitors or mycophenolate mofetil,21,41 and lung transplantation.42–44 A study with imatinib for the treatment of lung disease in steroid-refractory GVHD has shown promising results, but further validation with larger clinical trials is required.45
Case Continued
The patient is diagnosed with BOS and is treated for several months with prednisone 40 mg/day weaned over 3 months. She is started on inhaled corticosteroids, a proton pump inhibitor, and azithromycin 3 times per week, but she has a progressive decline in FEV1. She starts pulmonary rehabilitation but continues to functionally decline. Over the next year she develops bilateral pneumothoraces and bilateral cavitary nodules (Figure 1).
- What is causing this decline and the radiographic abnormalities?
Spontaneous air leak syndrome has been described in a little more than 1% of patients undergoing HSCT and has included pneumothorax and mediastinal and subcutaneous emphysema.46 It appears that air leak syndrome is more likely to occur in patients with chronic GVHD.47 The association between chronic GVHD and air leak syndrome could explain this patient’s recurrent pneumothoraces. The recurrent cavitary nodules are suspicious for infectious etiologies such as nontuberculous mycobacteria, tuberculosis, and fungal infections.
Case Continued
During an episode of pneumothorax, the patient undergoes chest tube placement, pleurodesis, and lung biopsy. Pathology reveals bronchiolitis obliterans as well as organizing pneumonia (Figure 2). No organisms are seen on acid-fast bacilli or GMS stains.
- What are the other late-onset noninfectious pulmonary complications?
Definitions of other late noninfectious pulmonary complications following HSCT are shown in Table 3.
Interstitial pneumonias may represent COP or may be idiopathic pneumonia syndrome with a later onset or a nonspecific interstitial pneumonia. This syndrome is poorly defined, with a number of differing definitions of the syndrome published in the literature.50–55
A rare pulmonary complication after HSCT is pulmonary veno-occlusive disease (PVOD). Pulmonary hypertension has been reported after HSCT,56 but PVOD is a subset of pulmonary hypertension. It is associated with pleural effusions and volume overload on chest radiography.57,58 It may present early or late after transplant and is poorly understood.
Besides obstructive and restrictive PFT abnormalities, changes in small airway function59 after transplant and loss in diffusing capacity of the lungs for carbon monoxide (D
Case Conclusion
The patient’s lung function continues to worsen, but no infectious etiologies are discovered. Ultimately, she dies of respiratory failure caused by progressive bronchiolitis obliterans.
Conclusion
Late pulmonary complications occur frequently in patients who have undergone HSCT. These complications can be classified as infectious versus noninfectious etiologies. Late-onset complications are more common in allogeneic transplantations because they are associated with chronic GVHD. These complications can be manifestations of pulmonary GHVD or can be infectious complications associated with prolonged immunosuppression. Appropriate monitoring for the development of BOS is essential. Early and aggressive treatment of respiratory infections and diagnostic bronchoscopy with BAL can help elucidate most infectious causes. Still, diagnostic challenges remain and multiple causes of respiratory deterioration can be present concurrently in the post-HSCT patient. Steroid therapy remains the mainstay treatment for most noninfectious pulmonary complications and should be strongly considered once infection is effectively ruled out.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
1. Remberger M, Ackefors M, Berglund S, et al. Improved survival after allogeneic hematopoietic stem cell transplantation in recent years. A single-center study. Biol Blood Marrow Transplant 2011;17:1688–97.
2. Wood KL, Esguerra VG. Management of late pulmonary complications after hematopoietic stem cell transplantation. Hosp Phys Hematology-Oncology Board Review Manual 2018;13(1):36–48.
3. Ninin E, Milpied N, Moreau P, et al. Longitudinal study of bacterial, viral, and fungal infections in adult recipients of bone marrow transplants. Clin Infect Dis 2001;33:41–7.
4. Roca J, Granena A, Rodriguez-Roisin R, et al. Fatal airway disease in an adult with chronic graft-versus-host disease. Thorax 1982;37:77–8.
5. Williams KM, Chien JW, Gladwin MT, Pavletic SZ. Bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. JAMA 2009;302:306–14.
6. Chien JW, Martin PJ, Gooley TA, et al. Airflow obstruction after myeloablative allogeneic hematopoietic stem cell transplantation. Am J Respir Crit Care Med 2003;168:208–14.
7. Holbro A, Lehmann T, Girsberger S, et al. Lung histology predicts outcome of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:973–80.
8. Chamberlain D, Maurer J, Chaparro C, Idolor L. Evaluation of transbronchial lung biopsy specimens in the diagnosis of bronchiolitis obliterans after lung transplantation. J Heart Lung Transplant 1994;13:963–71.
9. Au BK, Au MA, Chien JW. Bronchiolitis obliterans syndrome epidemiology after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 2011;17:1072–8.
10. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant 2015;21:389–401.
11. Couriel D, Carpenter PA, Cutler C, et al. Ancillary therapy and supportive care of chronic graft-versus-host disease: national institutes of health consensus development project on criteria for clinical trials in chronic Graft-versus-host disease: V. Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2006;12:375–96.
12. Pellegrino R, Viegi G, Brusasco V, et al. Interpretative strategies for lung function tests. Eur Respir J 2005;26:948–68.
13. Williams KM, Hnatiuk O, Mitchell SA, et al. NHANES III equations enhance early detection and mortality prediction of bronchiolitis obliterans syndrome after hematopoietic SCT. Bone Marrow Transplant 2014;49:561–6.
14. Hyatt RE, Cowl CT, Bjoraker JA, Scanlon PD. Conditions associated with an abnormal nonspecific pattern of pulmonary function tests. Chest 2009;135:419–24.
15. Bergeron A, Godet C, Chevret S, et al. Bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: phenotypes and prognosis. Bone Marrow Transplant 2013;48:819–24.
16. Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant 2005;11:945–56.
17. Gazourian L, Coronata AM, Rogers AJ, et al. Airway dilation in bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation. Respir Med 2013;107:276–83.
18. Gunn ML, Godwin JD, Kanne JP, et al. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging 2008;23:244–50.
19. Sargent MA, Cairns RA, Murdoch MJ, et al. Obstructive lung disease in children after allogeneic bone marrow transplantation: evaluation with high-resolution CT. AJR Am J Roentgenol 1995;164:693–6.
20. Galban CJ, Boes JL, Bule M, et al. Parametric response mapping as an indicator of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1592–8.
21. Meyer KC, Raghu G, Verleden GM, et al. An international ISHLT/ATS/ERS clinical practice guideline: diagnosis and management of bronchiolitis obliterans syndrome. Eur Respir J 2014;44:1479–1503.
22. Hildebrandt GC, Fazekas T, Lawitschka A, et al. Diagnosis and treatment of pulmonary chronic GVHD: report from the consensus conference on clinical practice in chronic GVHD. Bone Marrow Transplant 2011;46:1283–95.
23. Carpenter PA, Kitko CL, Elad S, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: V. The 2014 Ancillary Therapy and Supportive Care Working Group Report. Biol Blood Marrow Transplant 2015;21:1167–87.
24. Norman BC, Jacobsohn DA, Williams KM, et al. Fluticasone, azithromycin and montelukast therapy in reducing corticosteroid exposure in bronchiolitis obliterans syndrome after allogeneic hematopoietic SCT: a case series of eight patients. Bone Marrow Transplant 2011;46:1369–73.
25. Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016;22:710–6.
26. Or R, Gesundheit B, Resnick I, et al. Sparing effect by montelukast treatment for chronic graft versus host disease: a pilot study. Transplantation 2007;83:577–81.
27. Cheng GS, Storer B, Chien JW, et al. Lung function trajectory in bronchiolitis obliterans syndrome after allogeneic hematopoietic cell transplant. Ann Am Thorac Soc 2016;13:1932–9.
28. Bergeron A, Belle A, Chevret S, et al. Combined inhaled steroids and bronchodilatators in obstructive airway disease after allogeneic stem cell transplantation. Bone Marrow Transplant 2007;39:547–53.
29. Bashoura L, Gupta S, Jain A, et al. Inhaled corticosteroids stabilize constrictive bronchiolitis after hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:63–7.
30. Bergeron A, Chevret S, Granata A, et al. Effect of azithromycin on airflow decline-free survival after allogeneic hematopoietic stem cell transplant: the ALLOZITHRO randomized clinical trial. JAMA 2017;318:557–66.
31. Gerhardt SG, McDyer JF, Girgis RE, et al. Maintenance azithromycin therapy for bronchiolitis obliterans syndrome: results of a pilot study. Am J Respir Crit Care Med 2003;168:121–5.
32. Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005;25:490–3.
33. Maimon N, Lipton JH, Chan CK, Marras TK. Macrolides in the treatment of bronchiolitis obliterans in allograft recipients. Bone Marrow Transplant 2009;44:69–73.
34. Lam DC, Lam B, Wong MK, et al. Effects of azithromycin in bronchiolitis obliterans syndrome after hematopoietic SCT--a randomized double-blinded placebo-controlled study. Bone Marrow Transplant 2011;46:1551–6.
35. Yadav H, Peters SG, Keogh KA, et al. Azithromycin for the treatment of obliterative bronchiolitis after hematopoietic stem cell transplantation: a systematic review and meta-analysis. Biol Blood Marrow Transplant 2016;22:2264–9.
36. Vos R, Vanaudenaerde BM, Verleden SE, et al. A randomised controlled trial of azithromycin to prevent chronic rejection after lung transplantation. Eur Respir J 2011;37:164–72.
37. Svanstrom H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013;368:1704–12.
38. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
39. Tran J, Norder EE, Diaz PT, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2012;18:1250–4.
40. Lucid CE, Savani BN, Engelhardt BG, et al. Extracorporeal photopheresis in patients with refractory bronchiolitis obliterans developing after allo-SCT. Bone Marrow Transplant 2011;46:426–9.
41. Hostettler KE, Halter JP, Gerull S, et al. Calcineurin inhibitors in bronchiolitis obliterans syndrome following stem cell transplantation. Eur Respir J 2014;43:221–32.
42. Holm AM, Riise GC, Brinch L, et al. Lung transplantation for bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: unresolved questions. Transplantation 2013;96:e21–22.
43. Cheng GS, Edelman JD, Madtes DK, et al. Outcomes of lung transplantation after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2014;20:1169–75.
44. Okumura H, Ohtake S, Ontachi Y, et al. Living-donor lobar lung transplantation for broncho-bronchiolitis obliterans after allogeneic hematopoietic stem cell transplantation: does bronchiolitis obliterans recur in transplanted lungs? Int J Hematol 2007;86:369–73.
45. Olivieri A, Cimminiello M, Corradini P, et al. Long-term outcome and prospective validation of NIH response criteria in 39 patients receiving imatinib for steroid-refractory chronic GVHD. Blood 2013;122:4111–8.
46. Rahmanian S, Wood KL. Bronchiolitis obliterans and the risk of pneumothorax after transbronchial biopsy. Respiratory Medicine CME 2010;3:87–9.
47. Sakai R, Kanamori H, Nakaseko C, et al. Air-leak syndrome following allo-SCT in adult patients: report from the Kanto Study Group for Cell Therapy in Japan. Bone Marrow Transplant 2011;46:379–84.
48. Visscher DW, Myers JL. Histologic spectrum of idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006;3:322–9.
49. Cordier JF. Cryptogenic organising pneumonia. Eur Respir J 2006;28:422–46.
50. Nishio N, Yagasaki H, Takahashi Y, et al. Late-onset non-infectious pulmonary complications following allogeneic hematopoietic stem cell transplantation in children. Bone Marrow Transplant 2009;44:303–8.
51. Ueda K, Watadani T, Maeda E, et al. Outcome and treatment of late-onset noninfectious pulmonary complications after allogeneic haematopoietic SCT. Bone Marrow Transplant 2010;45:1719–27.
52. Schlemmer F, Chevret S, Lorillon G, et al. Late-onset noninfectious interstitial lung disease after allogeneic hematopoietic stem cell transplantation. Respir Med 2014;108:1525–33.
53. Palmas A, Tefferi A, Myers JL, et al. Late-onset noninfectious pulmonary complications after allogeneic bone marrow transplantation. Br J Haematol 1998;100:680–7.
54. Sakaida E, Nakaseko C, Harima A, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood 2003;102:4236–42.
55. Solh M, Arat M, Cao Q, et al. Late-onset noninfectious pulmonary complications in adult allogeneic hematopoietic cell transplant recipients. Transplantation 2011;91:798–803.
56. Dandoy CE, Hirsch R, Chima R, et al. Pulmonary hypertension after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013;19:1546–56.
57. Bunte MC, Patnaik MM, Pritzker MR, Burns LJ. Pulmonary veno-occlusive disease following hematopoietic stem cell transplantation: a rare model of endothelial dysfunction. Bone Marrow Transplant 2008;41:677–86.
58. Troussard X, Bernaudin JF, Cordonnier C, et al. Pulmonary veno-occlusive disease after bone marrow transplantation. Thorax 1984;39:956–7.
59. Lahzami S, Schoeffel RE, Pechey V, et al. Small airways function declines after allogeneic haematopoietic stem cell transplantation. Eur Respir J 2011;38:1180–8.
60. Jain NA, Pophali PA, Klotz JK, et al. Repair of impaired pulmonary function is possible in very-long-term allogeneic stem cell transplantation survivors. Biol Blood Marrow Transplant 2014;20:209–13.
61. Barisione G, Bacigalupo A, Crimi E, et al. Changes in lung volumes and airway responsiveness following haematopoietic stem cell transplantation. Eur Respir J 2008;32:1576–82.
62. Kovalszki A, Schumaker GL, Klein A, et al. Reduced respiratory and skeletal muscle strength in survivors of sibling or unrelated donor hematopoietic stem cell transplantation. Bone Marrow Transplant 2008;41:965–9.
63. Mathiesen S, Uhlving HH, Buchvald F, et al. Aerobic exercise capacity at long-term follow-up after paediatric allogeneic haematopoietic SCT. Bone Marrow Transplant 2014;49:1393–9.
Higher plasma cell-free DNA tracks with worse PAH survival
NEW ORLEANS – Cell-free (cf) DNA looked like an informative biomarker for both the severity of pulmonary artery hypertension and the survival prognosis for patients with this disease, based on results from two preliminary studies involving a total of 173 people.

“Plasma levels of cell-free DNA are elevated in patients with pulmonary artery hypertension, compared with healthy controls, and may predict disease severity and mortality,” Samuel B. Brusca, MD, said at the at the annual meeting of the American College of Cardiology.
A growing biomedical literature has documented a role for cfDNA in tracking the course of cancer, septic shock, and transplanted organs (Transplantation. 2019 Feb;103[2]:273-83) (Cell-Free DNA: Applications in Different Diseases, in “Cell-free DNA as Diagnostic Markers.” [New York: Humana Press, 2018, pp. 3-12]). Based on this background Dr. Brusca and his associates decided to examine whether plasma levels of cfDNA linked with pulmonary artery hypertension (PAH) severity and survival.
Their first study included seven patients with mild PAH (defined as patients with a tricuspid annular plane systolic excursion [TAPSE] of more than 18 mm and a maximum oxygen uptake [VO2] of at least 75% of predicted), eight with severe PAH (a TAPSE of 18 mm or less and a VO2 of less than 75%), and seven healthy adult controls. Measurement of plasma cfDNA showed an average level of 19.4 ng/mL among the healthy controls (prior reports had indicated that 10-20 ng/mL were normal levels), 22.0 ng/mL among patients with mild PAH, and 36.2 ng/mL in those with severe PAH. The level among the severe PAH patients was significantly higher than the level in controls by two different statistical tests, said Dr. Brusca, a critical care medicine physician at the National Institutes of Health Clinical Center in Bethesda, Md.
The second analysis by Dr. Brusca and his associates included 151 PAH patients followed by physicians at the Clinical Center for an average of 40 months. Their analysis tracked survival of these patients relative to their baseline levels of cfDNA and divided into tertiles. Patients in the lowest tertile had a starting cfDNA level of up to 39 ng/mL, those in the middle tertile had levels of 39.1-64.0 ng/mL, and those in the top tertile had levels of at least 64.1 ng/mL. A Kaplan-Meier analysis showed statistically significant differences in survival rates between each of the tertiles. Patients in the lowest tertile had a 5-year actuarial survival rate of about 65%, those in the middle tertile had a survival rate of about 48%, and those in the tertile with the highest level of cfDNA had a survival rate of about 28%.
Additional studies of cfDNA are needed in larger numbers of PAH patients, and cfDNA levels should be compared with levels of other, more established biomarkers, such as inflammatory cytokines, Dr. Brusca said in an interview.
Dr. Brusca had no disclosures. The study received no commercial funding.
SOURCE: Brusca SB et al. J Am Coll Cardiol. 2019 March 12;73(9 Suppl 1):1897.
I was very excited to hear Dr. Brusca’s report on using cell-free (cf) DNA to track the severity of pulmonary artery hypertension and survival of these patients. I’m now using cfDNA frequently to monitor heart transplant patients, and the information it provides has been very valuable. But cfDNA may be even better suited to assessing patients with pulmonary artery hypertension (PAH) because it’s a vascular disease, and increases in cfDNA appears to reflect damage to the vascular endothelium. It’s a brilliant application of this technology. Brain natriuretic peptide and troponin are markers of right heart damage, but cfDNA appears to be able to track the progression of the vascular component of PAH. It appears to be the first disease-specific biomarker we have for PAH. It’s time to start routinely measuring levels of cfDNA in trials so we can gather more data on the clinical correlates of changing levels of this biomarker.

I was very excited to hear Dr. Brusca’s report on using cell-free (cf) DNA to track the severity of pulmonary artery hypertension and survival of these patients. I’m now using cfDNA frequently to monitor heart transplant patients, and the information it provides has been very valuable. But cfDNA may be even better suited to assessing patients with pulmonary artery hypertension (PAH) because it’s a vascular disease, and increases in cfDNA appears to reflect damage to the vascular endothelium. It’s a brilliant application of this technology. Brain natriuretic peptide and troponin are markers of right heart damage, but cfDNA appears to be able to track the progression of the vascular component of PAH. It appears to be the first disease-specific biomarker we have for PAH. It’s time to start routinely measuring levels of cfDNA in trials so we can gather more data on the clinical correlates of changing levels of this biomarker.
I was very excited to hear Dr. Brusca’s report on using cell-free (cf) DNA to track the severity of pulmonary artery hypertension and survival of these patients. I’m now using cfDNA frequently to monitor heart transplant patients, and the information it provides has been very valuable. But cfDNA may be even better suited to assessing patients with pulmonary artery hypertension (PAH) because it’s a vascular disease, and increases in cfDNA appears to reflect damage to the vascular endothelium. It’s a brilliant application of this technology. Brain natriuretic peptide and troponin are markers of right heart damage, but cfDNA appears to be able to track the progression of the vascular component of PAH. It appears to be the first disease-specific biomarker we have for PAH. It’s time to start routinely measuring levels of cfDNA in trials so we can gather more data on the clinical correlates of changing levels of this biomarker.
NEW ORLEANS – Cell-free (cf) DNA looked like an informative biomarker for both the severity of pulmonary artery hypertension and the survival prognosis for patients with this disease, based on results from two preliminary studies involving a total of 173 people.
“Plasma levels of cell-free DNA are elevated in patients with pulmonary artery hypertension, compared with healthy controls, and may predict disease severity and mortality,” Samuel B. Brusca, MD, said at the at the annual meeting of the American College of Cardiology.
A growing biomedical literature has documented a role for cfDNA in tracking the course of cancer, septic shock, and transplanted organs (Transplantation. 2019 Feb;103[2]:273-83) (Cell-Free DNA: Applications in Different Diseases, in “Cell-free DNA as Diagnostic Markers.” [New York: Humana Press, 2018, pp. 3-12]). Based on this background Dr. Brusca and his associates decided to examine whether plasma levels of cfDNA linked with pulmonary artery hypertension (PAH) severity and survival.
Their first study included seven patients with mild PAH (defined as patients with a tricuspid annular plane systolic excursion [TAPSE] of more than 18 mm and a maximum oxygen uptake [VO2] of at least 75% of predicted), eight with severe PAH (a TAPSE of 18 mm or less and a VO2 of less than 75%), and seven healthy adult controls. Measurement of plasma cfDNA showed an average level of 19.4 ng/mL among the healthy controls (prior reports had indicated that 10-20 ng/mL were normal levels), 22.0 ng/mL among patients with mild PAH, and 36.2 ng/mL in those with severe PAH. The level among the severe PAH patients was significantly higher than the level in controls by two different statistical tests, said Dr. Brusca, a critical care medicine physician at the National Institutes of Health Clinical Center in Bethesda, Md.
The second analysis by Dr. Brusca and his associates included 151 PAH patients followed by physicians at the Clinical Center for an average of 40 months. Their analysis tracked survival of these patients relative to their baseline levels of cfDNA and divided into tertiles. Patients in the lowest tertile had a starting cfDNA level of up to 39 ng/mL, those in the middle tertile had levels of 39.1-64.0 ng/mL, and those in the top tertile had levels of at least 64.1 ng/mL. A Kaplan-Meier analysis showed statistically significant differences in survival rates between each of the tertiles. Patients in the lowest tertile had a 5-year actuarial survival rate of about 65%, those in the middle tertile had a survival rate of about 48%, and those in the tertile with the highest level of cfDNA had a survival rate of about 28%.
Additional studies of cfDNA are needed in larger numbers of PAH patients, and cfDNA levels should be compared with levels of other, more established biomarkers, such as inflammatory cytokines, Dr. Brusca said in an interview.
Dr. Brusca had no disclosures. The study received no commercial funding.
SOURCE: Brusca SB et al. J Am Coll Cardiol. 2019 March 12;73(9 Suppl 1):1897.
NEW ORLEANS – Cell-free (cf) DNA looked like an informative biomarker for both the severity of pulmonary artery hypertension and the survival prognosis for patients with this disease, based on results from two preliminary studies involving a total of 173 people.
“Plasma levels of cell-free DNA are elevated in patients with pulmonary artery hypertension, compared with healthy controls, and may predict disease severity and mortality,” Samuel B. Brusca, MD, said at the at the annual meeting of the American College of Cardiology.
A growing biomedical literature has documented a role for cfDNA in tracking the course of cancer, septic shock, and transplanted organs (Transplantation. 2019 Feb;103[2]:273-83) (Cell-Free DNA: Applications in Different Diseases, in “Cell-free DNA as Diagnostic Markers.” [New York: Humana Press, 2018, pp. 3-12]). Based on this background Dr. Brusca and his associates decided to examine whether plasma levels of cfDNA linked with pulmonary artery hypertension (PAH) severity and survival.
Their first study included seven patients with mild PAH (defined as patients with a tricuspid annular plane systolic excursion [TAPSE] of more than 18 mm and a maximum oxygen uptake [VO2] of at least 75% of predicted), eight with severe PAH (a TAPSE of 18 mm or less and a VO2 of less than 75%), and seven healthy adult controls. Measurement of plasma cfDNA showed an average level of 19.4 ng/mL among the healthy controls (prior reports had indicated that 10-20 ng/mL were normal levels), 22.0 ng/mL among patients with mild PAH, and 36.2 ng/mL in those with severe PAH. The level among the severe PAH patients was significantly higher than the level in controls by two different statistical tests, said Dr. Brusca, a critical care medicine physician at the National Institutes of Health Clinical Center in Bethesda, Md.
The second analysis by Dr. Brusca and his associates included 151 PAH patients followed by physicians at the Clinical Center for an average of 40 months. Their analysis tracked survival of these patients relative to their baseline levels of cfDNA and divided into tertiles. Patients in the lowest tertile had a starting cfDNA level of up to 39 ng/mL, those in the middle tertile had levels of 39.1-64.0 ng/mL, and those in the top tertile had levels of at least 64.1 ng/mL. A Kaplan-Meier analysis showed statistically significant differences in survival rates between each of the tertiles. Patients in the lowest tertile had a 5-year actuarial survival rate of about 65%, those in the middle tertile had a survival rate of about 48%, and those in the tertile with the highest level of cfDNA had a survival rate of about 28%.
Additional studies of cfDNA are needed in larger numbers of PAH patients, and cfDNA levels should be compared with levels of other, more established biomarkers, such as inflammatory cytokines, Dr. Brusca said in an interview.
Dr. Brusca had no disclosures. The study received no commercial funding.
SOURCE: Brusca SB et al. J Am Coll Cardiol. 2019 March 12;73(9 Suppl 1):1897.
REPORTING FROM ACC 2019
U.S. measles cases climb to over 800 for the year
according to the Centers for Disease Control and Prevention.
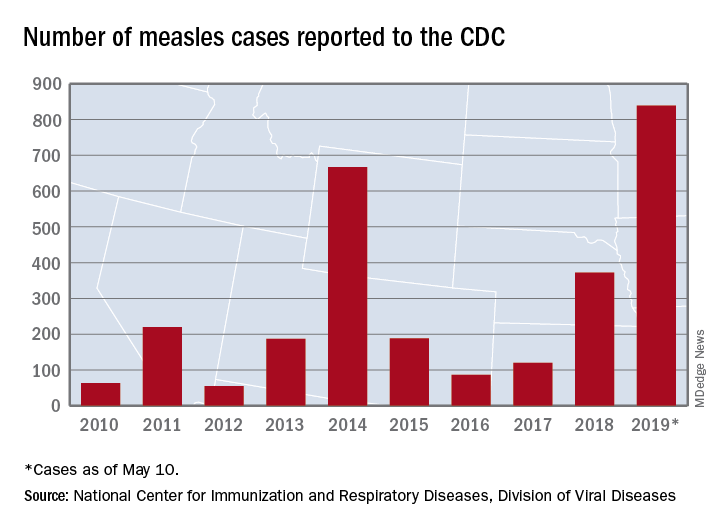
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
according to the Centers for Disease Control and Prevention.
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
according to the Centers for Disease Control and Prevention.
There are 10 states dealing with ongoing outbreaks now that Pennsylvania has been added to the list, the CDC reported May 13. The state has had five cases so far, all in Allegheny County. New York City continued to have the most active outbreak, adding 43 more cases in Brooklyn last week for a total of 410 in the city since the beginning of 2019, NYC Health said.
Several of this year’s outbreaks were predicted in an analysis published in the Lancet Infectious Diseases (2019 May 9. doi: 10.1016/S1473-3099(19)30231-2). Investigators identified the 25 counties most likely to experience a measles outbreak in 2019 – a list that includes Queens, N.Y. (adjacent to Brooklyn), Multnomah, Ore. (adjacent to Clark County, Wash., where 71 people were infected earlier this year), and San Mateo, Calif., where 4 cases have been reported.
“We recommend that public health officials and policymakers prioritize monitoring the counties we identify to be at high risk that have not yet reported cases, especially those that lie adjacent to counties with ongoing outbreaks and those that house large international airports,” senior author Lauren Gardner of Johns Hopkins University, Baltimore, said in a written statement.
The outbreak in Clark County was declared over in late April, but Gov. Jay Inslee signed a bill on May 10 that removes the personal/philosophical exemption for the MMR vaccine from the state’s school and child care immunization requirements. “We must step up our leadership to educate the public about the critical role vaccines have in keeping us healthy and safe, and continue working with communities to improve vaccination rates,” Washington State Secretary of Health John Wiesman said in a written statement.
In Oregon, a bill that would eliminate religious and philosophical exemptions to child vaccination requirements passed the state house of representatives by a 35-25 vote and is moving to the senate. Gov. Kate Brown has said that she plans to sign the bill, according to OregonLive.com.
In a tight vote, FDA panel backs mannitol for CF
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).

Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).
Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
A Food and Drug Administration Advisory Committee voted that the benefit-risk profile of an inhaled treatment for cystic fibrosis merits approval of the drug – dry powder mannitol (DPM).
Mannitol is a naturally occurring sugar alcohol that is used as a low-calorie sweetener; it is generally recognized as safe when taken enterically. Inhaled DPM, marketed as Aridol, is currently approved as a bronchoprovocation agent. For the current indication, DPM is given as 10x40-mg capsules twice daily.
In a 9-7 vote, the FDA’s Pulmonary-Allergy Drugs Advisory Committee (PADAC) decided that DPM’s modest potential to improve pulmonary function in adults with cystic fibrosis (CF) outweighed a potential signal for increased exacerbations seen in clinical trials.
Chiesi USA Inc. is seeking approval of DPM for the management of cystic fibrosis to improve pulmonary function in patients 18 years of age and older in conjunction with standard therapies. It plans to market DPM as Bronchitol.
Some committee members who voted against approval, including PADAC chair David H. Au, MD, worried that DPM’s ease of use might prompt patients and caregivers to substitute it for inhaled hypertonic saline, a medication that’s more burdensome to use but has a longer track record for efficacy and safety. While hypertonic saline requires cumbersome equipment and cleaning regimens and takes 20-30 minutes to administer, DPM is administered over about 5 minutes via a series of capsules inserted into a small inhaler device.
“I was very impressed by conversations that we heard from the community that this will be viewed as a substitute drug [for hypertonic saline],” said Dr. Au, professor of medicine at the University of Washington, Seattle. “Before we make that leap of faith ... we have to better understand how it has to be used.” He also acknowledged that making the call for DPM was “challenging.”
Other committee members were reassured by the fact that DPM is approved for adult use in 35 countries; it’s been in use since 2011 in Australia for adults and children.
Some members also noted an unmet need in CF therapies and placed confidence in those treating CF patients to find ways to use DPM safely and effectively. “I’m really counting on the cystic fibrosis clinicians who do this for a living to figure out where to use this in their armamentarium,” said John M. Kelso, MD, an allergist at Scripps Clinic, San Diego.
In 2012, the initial new drug application submitted by Pharmaxis, which then held marketing rights to DPM, resulted in a “no” vote for approval from PADAC, and eventual FDA denial of approval. The initial submission was supported by two phase 3 clinical trials, 301 and 302, that included pediatric patients. In the pediatric population, there was concern for increased hemoptysis with DPM, so the FDA advised the drug’s marketers to consider seeking approval for an adult population only in its reapplication. The current submission followed a new double-blind, randomized, placebo-controlled trial, study 303, that included adults with CF aged 18 or over.
All three studies had similar designs, tracking change from baseline in forced expiratory volume in one second (FEV1) from baseline to the end of the 26-week study period. In addition to this primary endpoint, secondary endpoints included other pulmonary function measures, as well as the number of protocol-defined pulmonary exacerbations (PDPEs). Participants also reported quality of life and symptom measures on the Cystic Fibrosis Questionnaire–Revised (CFQ-R).
In study 301, the dropout rate approached one in three participants with higher discontinuation in the intervention than the control arm, causing significant statistical problems in dealing with missing data. Thus, said the FDA’s Robert Lim, MD, though this study had positive results for FEV1, it was not “statistically robust.”
The second study, 302, did not meet its primary endpoint, and there was “no support from secondary endpoints” for efficacy, said Dr. Lim, a clinical team leader in the FDA’s Division of Pulmonary, Allergy, and Rheumatology Products.
The current submission was also supported by a new post hoc subgroup analysis of adults in studies 301 and 302. A total of 414 patients receiving DPM and 347 receiving placebo (DPM at a nontherapeutic level) were included in the integrated analysis of patients from all three studies. Studies 301 and 302 both had open-label extension arms, allowing more patients to be included in safety data.
The problems caused by the missing data from study 301 were addressed in the design of study 303 by encouraging patients who discontinued the study drug to continue data collection efforts for the study. Dropout rates were lower overall in study 303 and balanced between arms.
Over the 26-week duration of study 303, investigators saw a statistically significant improvement in FEV1 of about 50 mL, according to the FDA’s analysis. Post hoc analyses of studies 301 and 302 showed point estimate increases of approximately 80 mL, according to Dr. Lim.
In its presentations, Chiesi USA presented its integrated analysis of adult data from the three clinical trials. The analysis showed an increase in FEV1 from baseline of 73 mL for the DPM group, compared with an increase of 7 mL for the control group, using an intention-to-treat population (P less than .001). The committee heard evidence that in adults with CF, pulmonary function typically decreases by 1%-3% annually.
The PDPE rate was slightly higher in the DPM group than in the control group in studies 302 and 303, but the differences were not statistically significant. These findings have a backdrop of an overall low rate of PDPEs ranging from 0.221 to 0.995 per year, according to Chiesi presenter Scott Donaldson, MD, a pulmonologist who directs the adult cystic fibrosis center at the University of North Carolina at Chapel Hill.
When looking at the subgroup of United States study participants, the DPM integrated cohort included more patients with a history of prior pulmonary exacerbations. In the DPM group, 45% of U.S. participants had at least one exacerbation in the prior year, and 20% had two or more exacerbations, compared with 38% and 14%, respectively, in the control group. Chiesi argued that this imbalance was likely responsible for the increased exacerbation rate.
The sponsor and the FDA used different imputation methods to account for missing data from the earlier studies, complicating interpretation of the potential signal for increased exacerbations.
Quality of life data were similar between groups across the studies.
In the end, the view of the “yes” voters was encapsulated by James M. Tracy, DO, an allergist in private practice in Omaha, Neb. “This is not a drug for everybody; but absolutely, it’s a drug for somebody. Ultimately we have to make that decision – I do think that we study populations, but we really take care of people.”
The FDA usually follows the recommendations of its advisory panels.
FROM AN FDA ADVISORY COMMITTEE HEARING