User login
Peanut desensitization comes at cost of anaphylaxis
based on a meta-analysis from more than 1,000 patients published in the Lancet.

In the Peanut Allergen immunotherapy, Clarifying the Evidence (PACE) systematic review and meta-analysis, Derek K. Chu, MD, of McMaster University, Hamilton, Ont., and colleagues reviewed 12 trials conducted between 2011 and 2018 with a total of 1,041 patients (median age, 9 years).
Overall, the risk of anaphylaxis was significantly higher among children who received oral immunotherapy, compared with no therapy (risk ratio, 3.12) as was anaphylaxis frequency (incidence rate ratio, 2.72) and use of epinephrine (RR, 2.21).
In addition, oral immunotherapy increased serious adverse events, compared with no therapy (RR, 1.92). Nonanaphylactic reactions also went up among oral immunotherapy patients, with increased risk for vomiting (RR, 1.79), angioedema (RR, 2.25), upper respiratory tract reactions (RR, 1.36), and lower respiratory tract infections (RR, 1.55).
Quality of life scores were not significantly different between patients who did and did not receive oral immunotherapy, the researchers noted.
The oral immunotherapy consisted of defatted, lightly roasted peanut flour in 10 studies, and a combination of peanut paste, peanut extract, or ground and defatted peanut in the other studies.
The oral immunotherapy did induce desensitization to peanuts in support of earlier studies including the subcutaneous immunotherapy trial, but “this outcome does not translate into achieving the clinical and patient-desired aim of less allergic reactions and anaphylaxis,” Dr. Chu and associates wrote.
However, “rather than take the view that these data denounce current research in oral immunotherapy as not successful, we instead suggest that this research has reached an important milestone in mechanistic but not clinical efficacy. From a clinical or biological perspective, the apparently paradoxical desensitization versus longitudinal clinical findings show the lability and unreliability of allergen thresholds identified during oral food challenges because patients often unpredictably reacted to previously tolerated doses outside of clinic,” they emphasized.
The findings were limited by several factors including the small sample size, compared with similar studies for asthma or cardiovascular conditions, and by incomplete or inconsistent data reporting, the researchers noted. However, the results are the most comprehensive to date, and support the need for food allergy treatments with better safety profiles, using peanut allergy immunotherapy as a model for other food allergies.
Dr. Chu and two other authors reported being investigators on a federally funded ongoing peanut oral immunotherapy trial. Two authors reported receiving a variety of grants from organizations such as the National Institutes of Health; the American Academy of Allergy, Asthma, & Immunology; or pharmaceutical companies.
SOURCE: Chu DK et al. Lancet. 2019 June 1;393:2222-32.
“The key criticism of this systematic review is inherent in its method because studies with different designs were grouped together,” Graham Roberts, MD, and Elizabeth Angier, MD, wrote in an accompanying editorial. In addition, the studies chosen did not account for the development of long-term peanut tolerance after the therapy was discontinued.
Also, the researchers did not factor in the variation in patterns of anaphylactic events, with patients in the treatment groups having events at home in conjunction with daily peanut doses, while the control patients would have had events mainly away from home.
“Unfortunately, the trials have not provided information about which participants benefited most from the intervention,” they wrote.
“Trading treatment-related side effects at home for allergic reactions to accidental exposures out of the house [i.e., in social situations] might beneficial for some patients,” they added. However, more research is needed to determine which patients would benefit from different treatment options at home and outside the home. The less effective but safer option of epicutaneous immunotherapy might be preferred by some patients. And early introduction of peanut products during infancy may prevent many cases of peanut allergy.
Dr. Roberts and Dr. Angier are at the University of Southampton (England). Both are members of the European Academy of Allergy and Clinical Immunology Allergen Immunotherapy Guidelines Group, which has recently published guidelines on immunotherapy. They wrote an editorial to accompany the article by Chu et al (Lancet. 2019 June 1;393:2180-1). They had no financial conflicts to disclose.
“The key criticism of this systematic review is inherent in its method because studies with different designs were grouped together,” Graham Roberts, MD, and Elizabeth Angier, MD, wrote in an accompanying editorial. In addition, the studies chosen did not account for the development of long-term peanut tolerance after the therapy was discontinued.
Also, the researchers did not factor in the variation in patterns of anaphylactic events, with patients in the treatment groups having events at home in conjunction with daily peanut doses, while the control patients would have had events mainly away from home.
“Unfortunately, the trials have not provided information about which participants benefited most from the intervention,” they wrote.
“Trading treatment-related side effects at home for allergic reactions to accidental exposures out of the house [i.e., in social situations] might beneficial for some patients,” they added. However, more research is needed to determine which patients would benefit from different treatment options at home and outside the home. The less effective but safer option of epicutaneous immunotherapy might be preferred by some patients. And early introduction of peanut products during infancy may prevent many cases of peanut allergy.
Dr. Roberts and Dr. Angier are at the University of Southampton (England). Both are members of the European Academy of Allergy and Clinical Immunology Allergen Immunotherapy Guidelines Group, which has recently published guidelines on immunotherapy. They wrote an editorial to accompany the article by Chu et al (Lancet. 2019 June 1;393:2180-1). They had no financial conflicts to disclose.
“The key criticism of this systematic review is inherent in its method because studies with different designs were grouped together,” Graham Roberts, MD, and Elizabeth Angier, MD, wrote in an accompanying editorial. In addition, the studies chosen did not account for the development of long-term peanut tolerance after the therapy was discontinued.
Also, the researchers did not factor in the variation in patterns of anaphylactic events, with patients in the treatment groups having events at home in conjunction with daily peanut doses, while the control patients would have had events mainly away from home.
“Unfortunately, the trials have not provided information about which participants benefited most from the intervention,” they wrote.
“Trading treatment-related side effects at home for allergic reactions to accidental exposures out of the house [i.e., in social situations] might beneficial for some patients,” they added. However, more research is needed to determine which patients would benefit from different treatment options at home and outside the home. The less effective but safer option of epicutaneous immunotherapy might be preferred by some patients. And early introduction of peanut products during infancy may prevent many cases of peanut allergy.
Dr. Roberts and Dr. Angier are at the University of Southampton (England). Both are members of the European Academy of Allergy and Clinical Immunology Allergen Immunotherapy Guidelines Group, which has recently published guidelines on immunotherapy. They wrote an editorial to accompany the article by Chu et al (Lancet. 2019 June 1;393:2180-1). They had no financial conflicts to disclose.
based on a meta-analysis from more than 1,000 patients published in the Lancet.
In the Peanut Allergen immunotherapy, Clarifying the Evidence (PACE) systematic review and meta-analysis, Derek K. Chu, MD, of McMaster University, Hamilton, Ont., and colleagues reviewed 12 trials conducted between 2011 and 2018 with a total of 1,041 patients (median age, 9 years).
Overall, the risk of anaphylaxis was significantly higher among children who received oral immunotherapy, compared with no therapy (risk ratio, 3.12) as was anaphylaxis frequency (incidence rate ratio, 2.72) and use of epinephrine (RR, 2.21).
In addition, oral immunotherapy increased serious adverse events, compared with no therapy (RR, 1.92). Nonanaphylactic reactions also went up among oral immunotherapy patients, with increased risk for vomiting (RR, 1.79), angioedema (RR, 2.25), upper respiratory tract reactions (RR, 1.36), and lower respiratory tract infections (RR, 1.55).
Quality of life scores were not significantly different between patients who did and did not receive oral immunotherapy, the researchers noted.
The oral immunotherapy consisted of defatted, lightly roasted peanut flour in 10 studies, and a combination of peanut paste, peanut extract, or ground and defatted peanut in the other studies.
The oral immunotherapy did induce desensitization to peanuts in support of earlier studies including the subcutaneous immunotherapy trial, but “this outcome does not translate into achieving the clinical and patient-desired aim of less allergic reactions and anaphylaxis,” Dr. Chu and associates wrote.
However, “rather than take the view that these data denounce current research in oral immunotherapy as not successful, we instead suggest that this research has reached an important milestone in mechanistic but not clinical efficacy. From a clinical or biological perspective, the apparently paradoxical desensitization versus longitudinal clinical findings show the lability and unreliability of allergen thresholds identified during oral food challenges because patients often unpredictably reacted to previously tolerated doses outside of clinic,” they emphasized.
The findings were limited by several factors including the small sample size, compared with similar studies for asthma or cardiovascular conditions, and by incomplete or inconsistent data reporting, the researchers noted. However, the results are the most comprehensive to date, and support the need for food allergy treatments with better safety profiles, using peanut allergy immunotherapy as a model for other food allergies.
Dr. Chu and two other authors reported being investigators on a federally funded ongoing peanut oral immunotherapy trial. Two authors reported receiving a variety of grants from organizations such as the National Institutes of Health; the American Academy of Allergy, Asthma, & Immunology; or pharmaceutical companies.
SOURCE: Chu DK et al. Lancet. 2019 June 1;393:2222-32.
based on a meta-analysis from more than 1,000 patients published in the Lancet.
In the Peanut Allergen immunotherapy, Clarifying the Evidence (PACE) systematic review and meta-analysis, Derek K. Chu, MD, of McMaster University, Hamilton, Ont., and colleagues reviewed 12 trials conducted between 2011 and 2018 with a total of 1,041 patients (median age, 9 years).
Overall, the risk of anaphylaxis was significantly higher among children who received oral immunotherapy, compared with no therapy (risk ratio, 3.12) as was anaphylaxis frequency (incidence rate ratio, 2.72) and use of epinephrine (RR, 2.21).
In addition, oral immunotherapy increased serious adverse events, compared with no therapy (RR, 1.92). Nonanaphylactic reactions also went up among oral immunotherapy patients, with increased risk for vomiting (RR, 1.79), angioedema (RR, 2.25), upper respiratory tract reactions (RR, 1.36), and lower respiratory tract infections (RR, 1.55).
Quality of life scores were not significantly different between patients who did and did not receive oral immunotherapy, the researchers noted.
The oral immunotherapy consisted of defatted, lightly roasted peanut flour in 10 studies, and a combination of peanut paste, peanut extract, or ground and defatted peanut in the other studies.
The oral immunotherapy did induce desensitization to peanuts in support of earlier studies including the subcutaneous immunotherapy trial, but “this outcome does not translate into achieving the clinical and patient-desired aim of less allergic reactions and anaphylaxis,” Dr. Chu and associates wrote.
However, “rather than take the view that these data denounce current research in oral immunotherapy as not successful, we instead suggest that this research has reached an important milestone in mechanistic but not clinical efficacy. From a clinical or biological perspective, the apparently paradoxical desensitization versus longitudinal clinical findings show the lability and unreliability of allergen thresholds identified during oral food challenges because patients often unpredictably reacted to previously tolerated doses outside of clinic,” they emphasized.
The findings were limited by several factors including the small sample size, compared with similar studies for asthma or cardiovascular conditions, and by incomplete or inconsistent data reporting, the researchers noted. However, the results are the most comprehensive to date, and support the need for food allergy treatments with better safety profiles, using peanut allergy immunotherapy as a model for other food allergies.
Dr. Chu and two other authors reported being investigators on a federally funded ongoing peanut oral immunotherapy trial. Two authors reported receiving a variety of grants from organizations such as the National Institutes of Health; the American Academy of Allergy, Asthma, & Immunology; or pharmaceutical companies.
SOURCE: Chu DK et al. Lancet. 2019 June 1;393:2222-32.
FROM THE LANCET
A sleeping beast: Obstructive sleep apnea and stroke
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.

Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.

A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
Obstructive sleep apnea (OSA) is an independent risk factor for ischemic stroke and may also, infrequently, be a consequence of stroke. It is significantly underdiagnosed in the general population and is highly prevalent in patients who have had a stroke. Many patients likely had their stroke because of this chronic untreated condition.
This review focuses on OSA and its prevalence, consequences, and treatment in patients after a stroke.
DEFINING AND QUANTIFYING OSA
OSA is the most common type of sleep-disordered breathing.1,2 It involves repeated narrowing or complete collapse of the upper airway despite ongoing respiratory effort.3,4 Apneic episodes are terminated by arousals from hypoxemia or efforts to breathe.5 In contrast, central sleep apnea is characterized by a patent airway but lack of airflow due to absent respiratory effort.5
In OSA, the number of episodes of apnea (absent airflow) and hypopnea (reduced airflow) are added together and divided by hours of sleep to calculate the apnea-hypopnea index (AHI). OSA is diagnosed by either of the following3,4:
- AHI of 5 or higher, with clinical symptoms related to OSA (described below)
- AHI of 15 or higher, regardless of symptoms.
The AHI also defines OSA severity, as follows3:
- Mild: AHI 5 to 15
- Moderate: AHI 15 to 30
- Severe: AHI greater than 30.
Diagnostic criteria (eg, definition of hypopnea, testing methods, and AHI thresholds) have varied over time, an important consideration when reviewing the literature.
OSA IS MORE COMMON THAN EXPECTED AFTER STROKE
In the most methodologically sound and generalizable study of this topic to date, the Wisconsin Sleep Cohort Study6 reported in 2013 that about 14% of men and 5% of women ages 30 to 70 have an AHI greater than 5 (using 4% desaturation to score hypopneic episodes) with daytime sleepiness. Other studies suggest that 80% to 90% of people with OSA are undiagnosed and untreated.1,7
The prevalence of OSA in patients who have had a stroke is much higher, ranging from 30% to 96% depending on the study methods and population.1,8–12 A 2010 meta-analysis11 of 29 studies reported that 72% of patients who had a stroke had an AHI greater than 5, and 29% had severe OSA. In this analysis, 7% of those with sleep-disordered breathing had central sleep apnea; still, these data indicate that the prevalence of OSA in these patients is about 5 times higher than in the general population.
RISK FACTORS MAY DIFFER IN STROKE POPULATION
Several risk factors for OSA have been identified.
Obesity is one of the strongest risk factors, with increasing body mass index (BMI) associated with increased OSA prevalence.4,6,13 However, obesity appears to be a less significant risk factor in patients who have had a stroke than in the general population. In the 2010 meta-analysis11 of OSA after stroke, the average BMI was only 26.4 kg/m2 (with obesity defined as a BMI > 30.0 kg/m2), and increasing BMI was not associated with increasing AHI.
Male sex and advanced age are also OSA risk factors.4,5 They remain significant in patients after a stroke; about 65% of poststroke patients who have OSA are men, and the older the patient, the more likely the AHI is greater than 10.11
Ethnicity and genetics may also play important roles in OSA risk, with roughly 25% of OSA prevalence estimated to have a genetic basis.14,15 Some risk factors for OSA such as craniofacial shape, upper airway anatomy, upper airway muscle dysfunction, increased respiratory chemosensitivity, and poor arousal threshold during sleep are likely determined by genetics and ethnicity.14,15 Compared with people of European origin, Asians have a similar prevalence of OSA, but at a much lower average BMI, suggesting that other factors are significant.14 Possible genetically determined anatomic risk factors have not been specifically studied in the poststroke population, but it can be assumed they remain relevant.
Several studies have tried to find an association between OSA and type, location, etiology, or pattern of stroke.10,11,16–19 Although some suggest links between cardioembolic stroke and OSA,16,20 or thrombolysis and OSA,10 most have found no association between OSA and stroke features.11,12,21,22
HOW DOES OSA INCREASE STROKE RISK?
Untreated severe OSA is associated with increased cardiovascular mortality,21,22 and OSA is an independent risk factor for incident stroke.23 A number of mechanisms may explain these relationships.
Intermittent hypoxemia and recurrent sympathetic arousals resulting from OSA are thought to lead to many of the comorbid conditions with which it is associated: hypertension, coronary artery disease, heart failure, arrhythmias, pulmonary hypertension, and stroke. Repetitive decreases in ventilation lead to oxygen desaturations that result in cycles of increased sympathetic outflow and eventual sustained nocturnal hypertension and daytime chronic hypertension.1,5,9,13 Also implicated are various changes in vasodilator and vasoconstrictor substances due to endothelial dysfunction and inflammation, which are thought to play a role in the atherogenic and prothrombotic states induced by OSA.1,5,13
Cerebral circulation is altered primarily by the changes in partial pressure of carbon dioxide (Pco2). During apnea, the Pco2 rises, causing vasodilation and increased blood flow. After the apnea resolves, there is hyperpnea with resultant decreased Pco2, and vasoconstriction. In a patient who already has vascular disease, the enhanced vasoconstriction could lead to ischemia.1,5
Changes in intrathoracic pressure result in distortion of cardiac architecture. When the patient tries to breathe against an occluded airway, the intrathoracic pressure becomes more and more negative, increasing preload and afterload. When this happens repeatedly every night for years, it leads to remodeling of the heart such as left and right ventricular hypertrophy, with reduced stroke volume, myocardial ischemia, and increased risk of arrhythmia.1,5,13
Untreated OSA is believed to predispose patients to develop atrial fibrillation through sympathetic overactivity, vascular inflammation, heart rate variability, and cardiac remodeling.24 As atrial fibrillation is a major risk factor for stroke, particularly cardioembolic stroke, it may be another pathway of increased stroke risk in OSA.16,20,25
CLINICAL MANIFESTATIONS OF OSA NOT OBVIOUS AFTER STROKE
OSA typically causes both daytime symptoms (excessive sleepiness, poor concentration, morning headache, depressive symptoms) and nighttime signs and symptoms (snoring, choking, gasping, night sweats, insomnia, nocturia, witnessed episodes of apnea).3,4,26 Unfortunately, because these are nonspecific, OSA is often underdiagnosed.4,26
Identifying OSA after a stroke may be a particular challenge, as patients often do not report classic symptoms, and the typical picture of OSA may have less predictive validity in these patients.1,27,28 Within the first 24 hours after a stroke, hypersomnia, snoring history, and age are not predictive of OSA.1 Patients found to have OSA after a stroke frequently do not have the traditional symptoms (sleepiness, snoring) seen in usual OSA patients. And they have higher rates of OSA at a younger age than the usual OSA patients, so age is not a predictive risk factor. In addition, daytime sleepiness and obesity are often absent or less prominent.1,9,27,28 Finally, typical OSA signs and symptoms may be attributed to the stroke itself or to comorbidities affecting the patient, lowering suspicion for OSA.
OSA MAY HINDER STROKE RECOVERY, WORSEN OUTCOMES
OSA, particularly when moderate to severe, is linked to pathophysiologic changes that can hinder recovery from a stroke.
Intermittent hypoxemia during sleep can worsen vascular damage of at-risk tissue: nocturnal hypoxemia correlates with white matter hyperintensities on magnetic resonance imaging, a marker of ischemic demyelination.29 Oxidative stress and release of inflammatory mediators associated with intermittent hypoxemia may impair vascular blood flow to brain tissue attempting to repair itself.30 In addition, sympathetic overactivity and Pco2 fluctuations associated with OSA may impede cerebral circulation.
Taken together, such ongoing nocturnal insults can lead to clinical consequences during this vulnerable period.
A 1996 study31 of patients recovering from a stroke found that an oxygen desaturation index (number of times that the blood oxygen level drops below a certain threshold, as measured by overnight oximetry) of more than 10 per hour was associated with worse functional recovery at discharge and at 3 and 12 months after discharge. This study also noted an association between time spent with oxygen saturations below 90% and the rate of death at 1 year.
A 2003 study32 reported that patients with an AHI greater than 10 by polysomnography spent an average of 13 days longer on the rehabilitation service and had worse functional and cognitive status on discharge, even after controlling for multiple confounders. Several subsequent studies have confirmed these and similar findings.8,33,34
OSA has also been linked to depression,35 which is common after stroke and may worsen outcomes.36 The interaction between OSA, depression, and poststroke outcomes warrants further study.
In the general population, OSA has been independently associated with increased risk of stroke or death from any cause.21,22,37 These associations have also been reported in the poststroke population: a 2014 meta-analysis found that OSA increased the risk of a repeat stroke (relative risk [RR] 1.8, 95% confidence interval [CI] 1.2–2.6) and all-cause mortality (RR 1.69, 95% CI 1.4–2.1).38
TESTING FOR OSA AFTER STROKE
Because of the high prevalence of OSA in patients who have had a stroke and the potential for worse outcomes associated with untreated OSA, there should be a low threshold for evaluating for OSA soon after stroke. Objective testing is required to qualify for therapy, and the gold standard for diagnosis of OSA is formal polysomnography conducted in a sleep laboratory.2–4 Unfortunately, polysomnography may be unacceptable to some patients, is costly, and is resource-intensive, particularly in an inpatient or rehabilitation setting.28 Ideally, to optimize testing efficiency, patients should be screened for the likelihood of OSA before polysomnography is ordered.
Questionnaires can help determine the need for further testing
Questionnaires developed to assess OSA risk39 include the following:
The Berlin questionnaire, developed in 1999, has 10 questions assessing daytime and nighttime signs and symptoms and presence of hypertension.
The STOP questionnaire, developed in 2008, assesses snoring, tiredness, observed apneic episodes, and elevated blood pressure.
The STOP-BANG questionnaire, published in 2010, includes the STOP questions plus BMI over 35 kg/m2, age over 50, neck circumference over 41 cm, and male gender.
A 2017 meta-analysis39 of 108 studies with nearly 50,000 people found that the STOP-BANG questionnaire performed best with regard to sensitivity and diagnostic odds ratio, but with poor specificity.
These screening tools and modified versions of them have also been evaluated in patients who have had a stroke.
In 2015, Boulos et al28 found that the STOP-BAG (a version of STOP-BANG that excludes neck circumference) and the 4-variable (4V) questionnaire (sex, BMI, blood pressure, snoring) had moderate predictive value for OSA within 6 months after sroke.
In 2016, Katzan et al40 found that the STOP-BAG2 (STOP-BAG criteria plus continuous variables for BMI and age) had a high sensitivity for polysomnographically diagnosed OSA within the first year after a stroke. The specificity was significantly better than the STOP-BANG or the STOP-BAG questionnaire, although it remained suboptimal at 60.5%.
In 2017, Sico et al41 developed and assessed the SLEEP Inventory (sex, left heart failure, Epworth Sleepiness Scale, enlarged neck, weight in pounds, insulin resistance or diabetes, and National Institutes of Health Stroke Scale) and found that it outperformed the Berlin and STOP-BANG questionnaires in the poststroke setting. The SLEEP Inventory had the best specificity and negative predictive value, and a slightly better ability to correctly classify patients as having OSA or not, classifying 80% of patients correctly.
These newer screening tools (eg, STOP-BAG, STOP-BAG2, SLEEP) can be used to identify with reasonable accuracy which patients need definitive testing after stroke.
Pulse oximetry is another possible screening tool
Overnight pulse oximetry may also help screen for sleep apnea and stratify risk after a stroke. A 2012 study42 of overnight oximetry to screen patients before surgery found that the oxygen desaturation index was significantly associated with the AHI measured by polysomnography. However, oximetry testing cannot distinguish between OSA and central sleep apnea, so it is insufficient to diagnose OSA or qualify patients for therapy. Further study is needed to examine the ability of overnight pulse oximetry to screen or to stratify risk for OSA after stroke.
Polysomnography vs home testing
Polysomnography is the gold standard for diagnosing OSA. Benefits include technical support and trouble-shooting, determining relationships between OSA, body position, and sleep stage, and the ability to intervene with treatment.2 However, polysomnography can be cumbersome, costly, and resource-intensive.
A home sleep apnea test, ie, an unattended, limited-channel sleep study, may be an acceptable alternative.2–4,43,44 Home testing does not require a sleep technologist to be present during testing, uses fewer sensors, and is less expensive than overnight polysomnography, but its utility can be limited: it fails to accurately discriminate between episodes of OSA and central sleep apnea, there is potential for false-negative results, and it can underestimate sleep apnea burden because it does not measure sleep.2
Institutional resources and logistics may influence the choice of diagnostic modality. No data exist on outcomes from different diagnostic testing methods in poststroke patients. Further research is needed.
POSITIVE AIRWAY PRESSURE THERAPY: BENEFITS, CHALLENGES, ALTERNATIVES
The first-line treatment for OSA is positive airway pressure (PAP).3 For most patients, this is continuous PAP (CPAP) or autoadjusting PAP (APAP). In some instances, particularly for those who cannot tolerate CPAP or who have comorbid hypoventilation, bilevel PAP (BPAP) may be indicated. More advanced PAP therapies are unlikely to be used after stroke.
PAP therapy is associated with reduced daytime sleepiness, improved mood, normalization of sleep architecture, improved systemic and pulmonary artery blood pressure, reduced rates of atrial fibrillation after ablation, and improved insulin sensitivity.45–49 Whether it reduces the risk of cardiovascular events, including stroke, remains controversial; most data suggest that it does not.50,51 However, when adherence to PAP therapy is considered rather than intention to treat, treatment has been found to lead to improved cardiovascular outcomes.52
Mixed evidence of benefits after stroke
Observational studies provide evidence that CPAP may help patients with OSA after stroke, although results are mixed.53–58 The studies ranged in size from 14 to 105 patients, enrolled patients with mostly moderate to severe OSA, and followed patients from 10 days to 7 years. Adherence to therapy was generally good in the short term (50%–70%), but only 15% to 30% of patients remained adherent at 5 to 7 years. Variable outcomes were reported, with some studies finding improved symptoms in the near term and mixed evidence of cardiovascular benefit in the longer ones. However, as these studies lacked randomization, drawing definitive conclusions on CPAP efficacy is difficult.
Patients were enrolled in the index admission or when starting a rehabilitation service—generally 2 to 3 weeks after their stroke. No clear association was found between the timing of initiating PAP therapy and outcomes. All patients had ischemic strokes, but few details were provided regarding stroke location, size, and severity. Exclusion criteria included severe underlying cardiopulmonary disease, confusion, severe stroke with marked impairment, and inability to cooperate. Almost all patients had moderate to severe OSA, and patients with central sleep apnea were excluded.
The major outcomes examined were drop-out rates, PAP adherence, and neurologic improvement based on neurologic functional scales (National Institutes of Health Stroke Scale and Canadian Neurologic Scale). As expected, dropout rates were higher in patients randomized to CPAP (OR 1.83, 95% CI 1.05–3.21, P = .03), although overall adherence was better than anticipated, with mean CPAP use across trials of 4.5 hours per night (95% CI 3.97–5.08) and with about 50% to 60% of patients adhering to therapy for at least 4 hours nightly.
Improvement in neurologic outcomes favored CPAP (standard mean difference 0.54, 95% CI 0.026–1.05), although considerable heterogeneity was seen. Improved sleepiness outcomes were inconsistent. Major cardiovascular outcomes were reported in only 2 studies (using the same data set) and showed delayed time to the next cardiovascular event for those treated with CPAP but no difference in cardiovascular event-free survival.
PAP poses more challenges after stroke
The primary limitation to PAP therapy is poor acceptance and adherence to therapy.59 High rates of refusal of therapy and difficulty complying with treatment have been noted in the poststroke population, although recent studies have reported better adherence rates. How rates of adherence play out in real-world settings, outside of the controlled environment of a research study, has yet to be determined.
In general, CPAP adherence is affected by claustrophobia, difficulty tolerating a mask, problems with pressure intolerance, irritating air leaks, nasal congestion, and naso-oral dryness. Many such barriers can be overcome with use of a properly fitted mask, an appropriate pressure setting, heated humidification, nasal sprays (eg, saline, inhaled steroids), and education, encouragement, and reassurance.
After a stroke, additional obstacles may impede the ability to use PAP therapy.68 Facial paresis (hemi- or bifacial) may make fitting of the mask problematic. Paralysis or weakness of the extremities may limit the ability to adjust or remove a mask. Aphasia can impair communication and understanding of the need to use PAP therapy, and upper-airway problems related to stroke, including dysphagia, may lead to pressure intolerance or risk of aspiration. Finally, a lack of perceived benefit, particularly if the patient does not have daytime sleepiness, may limit motivation.
Consider alternatives
For patients unlikely to succeed with PAP therapy, there are alternatives. Surgery and oral appliances are not usually realistic options in the setting of recent stroke, but positional therapy, including the use of body positioners to prevent supine sleep, as well as elevating the head of the bed, may be of some benefit.69,70 A nasopharyngeal airway stenting device (nasal trumpet) may also be tolerated by some patients.
A proposed algorithm for screening, diagnosing, and treating OSA in patients after stroke is presented in Figure 1.
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
- Selim B, Roux FJ. Stroke and sleep disorders. Sleep Med Clin 2012; 7(4):597–607. doi:10.1016/j.jsmc.2012.08.007
- Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine Clinical Practice Guideline. J Clin Sleep Med 2017; 13(3):479–504. doi:10.5664/jcsm.6506
- Epstein LJ, Kristo D, Strollo PJ Jr, et al; Adult Obstructive Sleep Apnea Task Force of the American Academy of Sleep Medicine. Clinical guideline for the evaluation, management, and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5(3):263–276. pmid:19960649
- Patil SP, Schneider H, Schwartz AR, Smith PL. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest 2007; 132(1):325–337. doi:10.1378/chest.07-0040
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev 2010; 90(1):47–112. doi:10.1152/physrev.00043.2008
- Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177(9):1006–1014. doi:10.1093/aje/kws342
- Redline S, Sotres-Alvarez D, Loredo J, et al. Sleep-disordered breathing in Hispanic/Latino individuals of diverse backgrounds. The Hispanic Community Health Study/Study of Latinos. Am J Resp Crit Care Med 2014; 189(3):335–344. doi:10.1164/rccm.201309-1735OC
- Aaronson JA, van Bennekom CA, Hofman WF, et al. Obstructive sleep apnea is related to impaired cognitive and functional status after stroke. Sleep 2015; 38(9):1431–1437. doi:10.5665/sleep.4984
- Sharma S, Culebras A. Sleep apnoea and stroke. Stroke Vasc Neurol 2016; 1(4):185–191. doi:10.1136/svn-2016-000038
- Huhtakangas JK, Huhtakangas J, Bloigu R, Saaresranta T. Prevalence of sleep apnea at the acute phase of ischemic stroke with or without thrombolysis. Sleep Med 2017; 40:40–46. doi:10.1016/j.sleep.2017.08.018
- Johnson KG, Johnson DC. Frequency of sleep apnea in stroke and TIA patients: a meta-analysis. J Clin Sleep Med 2010; 6(2):131–137. pmid:20411688
- Iranzo A, Santamaria J, Berenguer J, Sanchez M, Chamorro A. Prevalence and clinical importance of sleep apnea in the first night after cerebral infarction. Neurology 2002; 58:911–916. pmid:11914407
- Javaheri S, Barbe F, Campos-Rodriguez F, et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol 2017; 69(7):841–858. doi:10.1016/j.jacc.2016.11.069
- Dudley KA, Patel SR. Disparities and genetic risk factors in obstructive sleep apnea. Sleep Med 2016; 18:96–102. doi:10.1016/j.sleep.2015.01.015
- Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev 2000; 4(6):583–602. doi:10.1053/smrv.2000.0120
- Lipford MC, Flemming KD, Calvin AD, et al. Associations between cardioembolic stroke and obstructive sleep apnea. Sleep 2015; 38(11):1699–1705. doi:10.5665/sleep.5146
- Wang Y, Wang Y, Chen J, Yi X, Dong S, Cao L. Stroke patterns, topography, and etiology in patients with obstructive sleep apnea hypopnea syndrome. Int J Clin Exp Med 2017; 10(4):7137–7143.
- Fisse AL, Kemmling A, Teuber A, et al. The association of lesion location and sleep related breathing disorder in patients with acute ischemic stroke. PLoS One 2017; 12(1):e0171243. doi:10.1371/journal.pone.0171243
- Brown DL, Mowla A, McDermott M, et al. Ischemic stroke subtype and presence of sleep-disordered breathing: the BASIC sleep apnea study. J Stroke Cerebrovasc Dis 2015; 24(2):388–393. doi:10.1016/j.jstrokecerebrovasdis.2014.09.007
- Poli M, Philip P, Taillard J, et al. Atrial fibrillation as a major cause of stroke in apneic patients: a prospective study. Sleep Med 2017; 30:251–254. doi:10.1016/j.sleep.2015.07.031
- Young T, Finn L, Peppard PE, et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008; 31(8):1071–1078. pmid:18714778
- Molnar MZ, Mucsi I, Novak M, et al. Association of incident obstructive sleep apnoea with outcomes in a large cohort of US veterans. Thorax 2015; 70(9):888–895. doi:10.1136/thoraxjnl-2015-206970
- Redline S, Yenokyan G, Gottlieb DJ, et al. Obstructive sleep apnea-hypopnea and incident stroke: the sleep heart health study. Am J Respir Crit Care Med 2010; 182(2):269–277. doi:10.1164/rccm.200911-1746OC
- Marulanda-Londono E, Chaturvedi S. The interplay between obstructive sleep apnea and atrial fibrillation. Fron Neurol 2017; 8:668. doi:10.3389/fneur.2017.00668
- Szymanski FM, Filipiak KJ, Platek AE, Hrynkiewicz-Szymanska A, Karpinski G, Opolski G. Assessment of CHADS2 and CHA 2DS 2-VASc scores in obstructive sleep apnea patients with atrial fibrillation. Sleep Breath 2015; 19(2):531–537. doi:10.1007/s11325-014-1042-5
- Stansbury RC, Strollo PJ. Clinical manifestations of sleep apnea. J Thoracic Dis 2015; 7(9):E298–E310. doi:10.3978/j.issn.2072-1439.2015.09.13
- Chan W, Coutts SB, Hanly P. Sleep apnea in patients with transient ischemic attack and minor stroke: opportunity for risk reduction of recurrent stroke? Stroke 2010; 41(12):2973–2975. doi:10.1161/STROKEAHA.110.596759
- Boulos MI, Wan A, Im J, et al. Identifying obstructive sleep apnea after stroke/TIA: evaluating four simple screening tools. Sleep Med 2016; 21:133–139. doi:10.1016/j.sleep.2015.12.013
- Patel SK, Hanly PJ, Smith EE, Chan W, Coutts SB. Nocturnal hypoxemia is associated with white matter hyperintensities in patients with a minor stroke or transient ischemic attack. J Clin Sleep Med 2015; 11(12):1417–1424. doi:10.5664/jcsm.5278
- McCarty MF, DiNicolantonio JJ, O’Keefe JH. NADPH oxidase, uncoupled endothelial nitric oxide synthase, and NF-KappaB are key mediators of the pathogenic impact of obstructive sleep apnea—therapeutic implications. J Integr Cardiol 2016; 2(5):367–374. doi:10.15761/JIC.1000177
- Good DC, Henkle JQ, Gelber D, Welsh J, Verhulst S. Sleep-disordered breathing and poor functional outcome after stroke. Stroke 1996; 27(2):252–259. pmid:8571419
- Kaneko Y, Hajek VE, Zivanovic V, Raboud J, Bradley TD. Relationship of sleep apnea to functional capacity and length of hospitalization following stroke. Sleep 2003; 26(3):293–297. pmid:12749548
- Yan-fang S, Yu-ping W. Sleep-disordered breathing: impact on functional outcome of ischemic stroke patients. Sleep Med 2009; 10(7):717–719. doi:10.1016/j.sleep.2008.08.006
- Kumar R, Suri JC, Manocha R. Study of association of severity of sleep disordered breathing and functional outcome in stroke patients. Sleep Med 2017; 34:50–56. doi:10.1016/j.sleep.2017.02.025
- Kerner NA, Roose SP. Obstructive sleep apnea is linked to depression and cognitive impairment: evidence and potential mechanisms. Am J Geriatr Psychiatry 2016; 24(6):496–508. doi:10.1016/j.jagp.2016.01.134
- Bartoli F, Lillia N, Lax A, et al. Depression after stroke and risk of mortality: a systematic review and meta-analysis. Stroke Res Treat 2013; 2013:862978. doi:10.1155/2013/862978
- Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005; 353(19):2034–2041. doi:10.1056/NEJMoa043104
- Xie W, Zheng F, Song X. Obstructive sleep apnea and serious adverse outcomes in patients with cardiovascular or cerebrovascular disease: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2014; 93(29):e336. doi:10.1097/MD.0000000000000336
- Chiu HY, Chen PY, Chuang LP, et al. Diagnostic accuracy of the Berlin questionnaire, STOP-BANG, STOP, and Epworth sleepiness scale in detecting obstructive sleep apnea: a bivariate meta-analysis. Sleep Med Rev 2017; 36:57–70. doi:10.1016/j.smrv.2016.10.004
- Katzan IL, Thompson NR, Uchino K, Foldvary-Schaefer N. A screening tool for obstructive sleep apnea in cerebrovascular patients. Sleep Med 2016; 21:70–76. doi:10.1016/j.sleep.2016.02.001
- Sico JJ, Yaggi HK, Ofner S, et al. Development, validation, and assessment of an ischemic stroke or transient ischemic attack-specific prediction tool for obstructive sleep apnea. J Stroke Cerebrovasc Dis 2017; 26(8):1745–1754. doi:10.1016/j.jstrokecerebrovasdis.2017.03.042
- Chung F, Liao P, Elsaid H, Islam S, Shapiro CM, Sun Y. Oxygen desaturation index from nocturnal oximetry: a sensitive and specific tool to detect sleep-disordered breathing in surgical patients. Anesth Analg 2012; 114(5):993–1000. doi:10.1213/ANE.0b013e318248f4f5
- Boulos MI, Elias S, Wan A, et al. Unattended hospital and home sleep apnea testing following cerebrovascular events. J Stroke Cerebrovasc Dis 2017; 26(1):143–149. doi:10.1016/j.jstrokecerebrovasdis.2016.09.001
- Saletu MT, Kotzian ST, Schwarzinger A, Haider S, Spatt J, Saletu B. Home sleep apnea testing is a feasible and accurate method to diagnose obstructive sleep apnea in stroke patients during in-hospital rehabilitation. J Clin Sleep Med 2018; 14(9):1495–1501. doi:10.5664/jcsm.7322
- Giles TL, Lasserson TJ, Smith BH, White J, Wright J, Cates CJ. Continuous positive airways pressure for obstructive sleep apnoea in adults. Cochrane Database Syst Rev 2006; (3):CD001106. doi:10.1002/14651858.CD001106.pub3
- Fatureto-Borges F, Lorenzi-Filho G, Drager LF. Effectiveness of continuous positive airway pressure in lowering blood pressure in patients with obstructive sleep apnea: a critical review of the literature. Integr Blood Press Control 2016; 9:43–47. doi:10.2147/IBPC.S70402
- Imran TF, Gharzipura M, Liu S, et al. Effect of continuous positive airway pressure treatment on pulmonary artery pressure in patients with isolated obstructive sleep apnea: a meta-analysis. Heart Fail Rev 2016; 21(5):591–598. doi:10.1007/s10741-016-9548-5
- Deng F, Raza A, Guo J. Treating obstructive sleep apnea with continuous positive airway pressure reduces risk of recurrent atrial fibrillation after catheter ablation: a meta-analysis. Sleep Med 2018; 46:5–11. doi:10.1016/j.sleep.2018.02.013
- Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes, and the metabolic syndrome. Chronic Resp Dis 2014; 11(4):257–275. doi:10.1177/1479972314552806
- Kim Y, Koo YS, Lee HY, Lee SY. Can continuous positive airway pressure reduce the risk of stroke in obstructive sleep apnea patients? A systematic review and meta-analysis. PloS ONE 2016; 11(1):e0146317. doi:10.1371/journal.pone.0146317
- Yu J, Zhou Z, McEvoy RD, et al. Association of positive airway pressure with cardiovascular events and death in adults with sleep apnea: a systematic review and meta-analysis. JAMA 2017; 318(2):156–166. doi:10.1001/jama.2017.7967
- Peker Y, Glantz H, Eulenburg C, Wegscheider K, Herlitz J, Thunström E. Effect of positive airway pressure on cardiovascular outcomes in coronary artery disease patients with nonsleepy obstructive sleep apnea. The RICCADSA randomized controlled trial. Am J Respir Crit Care Med 2016; 194(5):613–620. doi:10.1164/rccm.201601-0088OC
- Martinez-Garcia MA, Soler-Cataluna JJ, Ejarque-Martinez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med 2009; 180(1):36–41. doi:10.1164/rccm.200808-1341OC
- Broadley SA, Jorgensen L, Cheek A, et al. Early investigation and treatment of obstructive sleep apnoea after acute stroke. J Clin Neurosci 2007; 14(4):328–333. doi:10.1016/j.jocn.2006.01.017
- Wessendorf TE, Wang YM, Thilmann AF, Sorgenfrei U, Konietzko N, Teschler H. Treatment of obstructive sleep apnoea with nasal continuous positive airway pressure in stroke. Eur Respir J 2001; 18(4):623–629. pmid:11716165
- Bassetti CL, Milanova M, Gugger M. Sleep-disordered breathing and acute ischemic stroke: diagnosis, risk factors, treatment, evolution, and long-term clinical outcome. Stroke 2006; 37(4):967–972. doi:10.1161/01.STR.0000208215.49243.c3
- Palombini L, Guilleminault C. Stroke and treatment with nasal CPAP. Eur J Neurol 2006; 13(2):198–200. doi:10.1111/j.1468-1331.2006.01169.x
- Martínez-García MA, Campos-Rodríguez F, Soler-Cataluña JJ, Catalán-Serra P, Román-Sánchez P, Montserrat JM. Increased incidence of nonfatal cardiovascular events in stroke patients with sleep apnoea: effect of CPAP treatment. Eur Respir J 2012; 39(4):906–912. doi:10.1183/09031936.00011311
- Brill AK, Horvath T, Seiler A, et al. CPAP as treatment of sleep apnea after stroke: a meta-analysis of randomized trials. Neurology 2018; 90(14):e1222–e1230. doi:10.1212/WNL.0000000000005262
- Hsu C, Vennelle M, Li H, Engleman HM, Dennis MS, Douglas NJ. Sleep-disordered breathing after stroke: a randomised controlled trial of continuous positive airway pressure. J Neurol Neurosurg Psychiatry 2006; 77(10):1143–1149. doi:10.1136/jnnp.2005.086686
- Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Resp J 2011; 37(5):1128–1136. doi:10.1183/09031936.00034410
- Ryan CM, Bayley M, Green R, Murray BJ, Bradley TD. Influence of continuous positive airway pressure on outcomes of rehabilitation in stroke patients with obstructive sleep apnea. Stroke 2011; 42(4):1062–1067. doi:10.1161/STROKEAHA.110.597468
- Bravata DM, Concato J, Fried T, et al. Continuous positive airway pressure: evaluation of a novel therapy for patients with acute ischemic stroke. Sleep 2011; 34(9):1271–1277. doi:10.5665/SLEEP.1254
- Parra O, Sanchez-Armengol A, Capote F, et al. Efficacy of continuous positive airway pressure treatment on 5-year survival in patients with ischaemic stroke and obstructive sleep apnea: a randomized controlled trial. J Sleep Res 2015; 24(1):47–53. doi:10.1111/jsr.12181
- Khot SP, Davis AP, Crane DA, et al. Effect of continuous positive airway pressure on stroke rehabilitation: a pilot randomized sham-controlled trial. J Clin Sleep Med 2016; 12(7):1019–1026. doi:10.5664/jcsm.5940
- Aaronson JA, Hofman WF, van Bennekom CA, et al. Effects of continuous positive airway pressure on cognitive and functional outcome of stroke patients with obstructive sleep apnea: a randomized controlled trial. J Clin Sleep Med 2016; 12(4):533–541. doi:10.5664/jcsm.5684
- Gupta A, Shukla G, Afsar M, et al. Role of positive airway pressure therapy for obstructive sleep apnea in patients with stroke: a randomized controlled trial. J Clin Sleep Med 2018; 14(4):511–521. doi:10.5664/jcsm.7034
- Mello-Fujita L, Kim LJ, Palombini Lde O, et al. Treatment of obstructive sleep apnea syndrome associated with stroke. Sleep Med 2015; 16(6):691–696. doi:10.1016/j.sleep.2014.12.017
- Svatikova A, Chervin RD, Wing JJ, Sanchez BN, Migda EM, Brown DL. Positional therapy in ischemic stroke patients with obstructive sleep apnea. Sleep Med 2011; 12(3):262–266. doi:10.1016/j.sleep.2010.12.008
- Souza FJ, Genta PR, de Souza Filho AJ, Wellman A, Lorenzi-Filho G. The influence of head-of-bed elevation in patients with obstructive sleep apnea. Sleep Breath 2017; 21(4):815–820. doi:10.1007/s11325-017-1524-3
KEY POINTS
- A low threshold for evaluating for OSA after a stroke is warranted: the prevalence is high in this population, and risk factors for OSA and its typical clinical picture may not be present.
- Questionnaires can help screen for the likelihood of OSA and the need for more definitive assessment with polysomnography or home sleep apnea testing, tests that pose additional challenges after stroke.
- Positive airway pressure (PAP) therapy remains the first-line treatment for OSA after stroke; it may improve recovery and reduce long-term sequelae of untreated OSA.
- Acceptance of and adherence to PAP therapy can be especially problematic in this population, and alternatives should be considered if needed.
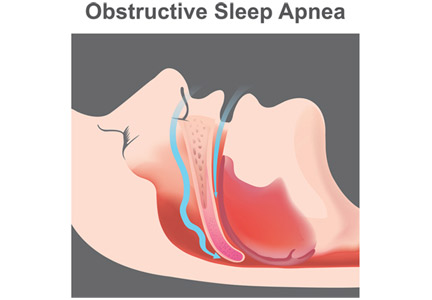
Anti-Xa assays: What is their role today in antithrombotic therapy?
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.

Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.

The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.
Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.
The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
Should clinicians abandon the activated partial thromboplastin time (aPTT) for monitoring heparin therapy in favor of tests that measure the activity of the patient’s plasma against activated factor X (anti-Xa assays)?
Although other anticoagulants are now available for preventing and treating arterial and venous thromboembolism, unfractionated heparin—which requires laboratory monitoring of therapy—is still widely used. And this monitoring can be challenging. Despite its wide use, the aPTT lacks standardization, and the role of alternative monitoring assays such as the anti-Xa assay is not well defined.
This article reviews the advantages, limitations, and clinical applicability of anti-Xa assays for monitoring therapy with unfractionated heparin and other anticoagulants.
UNFRACTIONATED HEPARIN AND WARFARIN ARE STILL WIDELY USED
Until the mid-1990s, unfractionated heparin and oral vitamin K antagonists (eg, warfarin) were the only anticoagulants widely available for clinical use. These agents have complex pharmacokinetic and pharmacodynamic properties, resulting in highly variable dosing requirements (both between patients and in individual patients) and narrow therapeutic windows, making frequent laboratory monitoring and dose adjustments mandatory.
Over the past 3 decades, other anticoagulants have been approved, including low-molecular-weight heparins, fondaparinux, parenteral direct thrombin inhibitors, and direct oral anticoagulants. While these agents have expanded the options for preventing and treating thromboembolism, unfractionated heparin and warfarin are still the most appropriate choices for many patients, eg, those with stage 4 chronic kidney disease and end-stage renal disease on dialysis, and those with mechanical heart valves.
In addition, unfractionated heparin remains the anticoagulant of choice during procedures such as hemodialysis, percutaneous transluminal angioplasty, and cardiopulmonary bypass, as well as in hospitalized and critically ill patients, who often have acute kidney injury or require frequent interruptions of therapy for invasive procedures. In these scenarios, unfractionated heparin is typically preferred because of its short plasma half-life, complete reversibility by protamine, safety regardless of renal function, and low cost compared with parenteral direct thrombin inhibitors.
As long as unfractionated heparin and warfarin remain important therapies, the need for their laboratory monitoring continues. For warfarin monitoring, the prothrombin time and international normalized ratio are validated and widely reproducible methods. But monitoring unfractionated heparin therapy remains a challenge.
UNFRACTIONATED HEPARIN’S EFFECT IS UNPREDICTABLE
Unfractionated heparin, a negatively charged mucopolysaccharide, inhibits coagulation by binding to antithrombin through the high-affinity pentasaccharide sequence.1–6 Such binding induces a conformational change in the antithrombin molecule, converting it to a rapid inhibitor of several coagulation proteins, especially factors IIa and Xa.2–4
Unfractionated heparin inhibits factors IIa and Xa in a 1:1 ratio, but low-molecular-weight heparins inhibit factor Xa more than factor IIa, with IIa-Xa inhibition ratios ranging from 1:2 to 1:4, owing to their smaller molecular size.7
One of the most important reasons for the unpredictable and highly variable individual responses to unfractionated heparin is that, infused into the blood, the large and negatively charged unfractionated heparin molecules bind nonspecifically to positively charged plasma proteins.7 In patients who are critically ill, have acute infections or inflammatory states, or have undergone major surgery, unfractionated heparin binds to acute-phase proteins that are elevated, particularly factor VIII. This results in fewer free heparin molecules and a variable anticoagulant effect.8
In contrast, low-molecular-weight heparins have longer half-lives and bind less to plasma proteins, resulting in more predictable plasma levels following subcutaneous injection.9
MONITORING UNFRACTIONATED HEPARIN IMPROVES OUTCOMES
In 1960, Barritt and Jordan10 conducted a small but landmark trial that established the clinical importance of unfractionated heparin for treating venous thromboembolism. None of the patients who received unfractionated heparin for acute pulmonary embolism developed a recurrence during the subsequent 2 weeks, while 50% of those who did not receive it had recurrent pulmonary embolism, fatal in half of the cases.
The importance of achieving a specific aPTT therapeutic target was not demonstrated until a 1972 study by Basu et al,11 in which 162 patients with venous thromboembolism were treated with heparin with a target aPTT of 1.5 to 2.5 times the control value. Patients who suffered recurrent events had subtherapeutic aPTT values on 71% of treatment days, while the rest of the patients, with no recurrences, had subtherapeutic aPTT values only 28% of treatment days. The different outcomes could not be explained by the average daily dose of unfractionated heparin, which was similar in the patients regardless of recurrence.
Subsequent studies showed that the best outcomes occur when unfractionated heparin is given in doses high enough to rapidly achieve a therapeutic prolongation of the aPTT,12–14 and that the total daily dose is also important in preventing recurrences.15,16 Failure to achieve a target aPTT within 24 hours of starting unfractionated heparin is associated with increased risk of recurrent venous thromboembolism.13,17
Raschke et al17 found that patients prospectively randomized to weight-based doses of intravenous unfractionated heparin (bolus plus infusion) achieved significantly higher rates of therapeutic aPTT within 6 hours and 24 hours after starting the infusion, and had significantly lower rates of recurrent venous thromboembolism than those randomized to a fixed unfractionated heparin protocol, without an increase in major bleeding.
Smith et al,18 in a study of 400 consecutive patients with acute pulmonary embolism treated with unfractionated heparin, found that patients who achieved a therapeutic aPTT within 24 hours had lower in-hospital and 30-day mortality rates than those who did not achieve the first therapeutic aPTT until more than 24 hours after starting unfractionated heparin infusion.
Such data lend support to the widely accepted practice and current guideline recommendation8 of using laboratory assays to adjust the dose of unfractionated heparin to achieve and maintain a therapeutic target. The use of dosing nomograms significantly reduces the time to achieve a therapeutic aPTT while minimizing subtherapeutic and supratherapeutic unfractionated heparin levels.19,20
THE aPTT REFLECTS THROMBIN INHIBITION
The aPTT has a log-linear relationship with plasma concentrations of unfractionated heparin,21 but it was not developed specifically for monitoring unfractionated heparin therapy. Originally described in 1953 as a screening tool for hemophilia,22–24 the aPTT is prolonged in the setting of factor deficiencies (typically with levels < 45%, except for factors VII and XIII), as well as lupus anticoagulants and therapy with parenteral direct thrombin inhibitors.8,25,26
Because thrombin (factor IIa) is 10 times more sensitive than factor Xa to inhibition by the heparin-antithrombin complex,4,7 thrombin inhibition appears to be the most likely mechanism by which unfractionated heparin prolongs the aPTT. In contrast, aPTT is minimally or not at all prolonged by low-molecular-weight heparins, which are predominantly factor Xa inhibitors.7
HEPARIN ASSAYS MEASURE UNFRACTIONATED HEPARIN ACTIVITY
While the aPTT is a surrogate marker of unfractionated heparin activity in plasma, unfractionated heparin activity can be measured more precisely by so-called heparin assays, which are typically not direct measures of the plasma concentration of heparins, but rather functional assays that provide indirect estimates. They include protamine sulfate titration assays and anti-Xa assays.
Protamine sulfate titration assays measure the amount of protamine sulfate required to neutralize heparin: the more protamine required, the greater the estimated concentration of unfractionated heparin in plasma.8,27–29 Protamine titration assays are technically demanding, so they are rarely used clinically.
Anti-Xa assays provide a measure of the functional level of heparins in plasma.29–33 Chromogenic anti-Xa assays are available on automated analyzers with standardized kits29,33,34 and may be faster to perform than the aPTT.35
Experiments in rabbits show that unfractionated heparin inhibits thrombus formation and extension at concentrations of 0.2 to 0.4 U/mL as measured by the protamine titration assay,27 which correlated with an anti-Xa activity of 0.35 to 0.67 U/mL in a randomized controlled trial.32
Assays that directly measure the plasma concentration of heparin exist but are not clinically relevant because they also measure heparin molecules lacking the pentasaccharide sequence, which have no anticoagulant activity.36
ANTI-Xa ASSAY VS THE aPTT
Anti-Xa assays are more expensive than the aPTT and are not available in all hospitals. For these reasons, the aPTT remains the most commonly used laboratory assay for monitoring unfractionated heparin therapy.
However, the aPTT correlates poorly with the activity level of unfractionated heparin in plasma. In one study, an anti-Xa level of 0.3 U/mL corresponded to aPTT results ranging from 47 to 108 seconds.31 Furthermore, in studies that used a heparin therapeutic target based on an aPTT ratio 1.5 to 2.5 times the control aPTT value, the lower end of that target range was often associated with subtherapeutic plasma unfractionated heparin activity measured by anti-Xa and protamine titration assays.28,31
Because of these limitations, individual laboratories should determine their own aPTT therapeutic target ranges for unfractionated heparin based on the response curves obtained with the reagent and coagulometer used. The optimal therapeutic aPTT range for treating acute venous thromboembolism should be defined as the aPTT range (in seconds) that correlates with a plasma activity level of unfractionated heparin of 0.3 to 0.7 U/mL based on a chromogenic anti-Xa assay, or 0.2 to 0.4 U/mL based on a protamine titration assay.32,34–36
Nevertheless, the anticoagulant effect of unfractionated heparin as measured by the aPTT can be unpredictable and can vary widely among individuals and in the same patient.7 This wide variability can be explained by a number of technical and biologic variables. Different commercial aPTT reagents, different lots of the same reagent, and different reagent and instrument combinations have different sensitivities to unfractionated heparin, which can lead to variable aPTT results.37 Moreover, high plasma levels of acute-phase proteins, low plasma antithrombin levels, consumptive coagulopathies, liver failure, and lupus anticoagulants may also affect the aPTT.7,25,32,36–41 These variables account for the poor correlation—ranging from 25% to 66%—reported between aPTT and anti-Xa assays.32,42–48
Such discrepancies may have serious clinical implications: if a patient’s aPTT is low (subtherapeutic) or high (supratherapeutic) but the anti-Xa assay result is within the therapeutic range (0.3–0.7 units/mL), changing the dose of unfractionated heparin (guided by an aPTT nomogram) may increase the risk of bleeding or of recurrent thromboembolism.
CLINICAL APPLICABILITY OF THE ANTI-Xa ASSAY
Neither anti-Xa nor protamine titration assays are standardized across reference laboratories, but chromogenic anti-Xa assays have better interlaboratory correlation than the aPTT49,50 and can be calibrated specifically for unfractionated or low-molecular-weight heparins.29,33
Although reagent costs are higher for chromogenic anti-Xa assays than for the aPTT, some technical variables (described below) may partially offset the cost difference.29,33,41 In addition, unlike the aPTT, anti-Xa assays do not need local calibration; the therapeutic range for unfractionated heparin is the same (0.3–0.7 U/mL) regardless of instrument or reagent.33,41
Most important, studies have found that patients monitored by anti-Xa assay achieve significantly higher rates of therapeutic anticoagulation within 24 and 48 hours after starting unfractionated heparin infusion than those monitored by the aPTT. Fewer dose adjustments and repeat tests are required, which may also result in lower cost.32,51–55
While these studies found chromogenic anti-Xa assays better for achieving laboratory end points, data regarding relevant clinical outcomes are more limited. In a retrospective, observational cohort study,51 the rate of venous thromboembolism or bleeding-related death was 2% in patients receiving unfractionated heparin therapy monitored by anti-Xa assay and 6% in patients monitored by aPTT (P = .62). Rates of major hemorrhage were also not significantly different.
In a randomized controlled trial32 in 131 patients with acute venous thromboembolism and heparin resistance, rates of recurrent venous thromboembolism were 4.6% and 6.1% in the groups randomized to anti-Xa and aPTT monitoring, respectively, whereas overall bleeding rates were 1.5% and 6.1%, respectively. Again, the differences were not statistically significant.
Heparin resistance. Some patients require unusually high doses of unfractionated heparin to achieve a therapeutic aPTT: typically, more than 35,000 U over 24 hours,7,8,32 or total daily doses that exceed their estimated weight-based requirements. Heparin resistance has been observed in various clinical settings.7,8,32,37–40,59–61 Patients with heparin resistance monitored by anti-Xa had similar rates of recurrent venous thromboembolism while receiving significantly lower doses of unfractionated heparin than those monitored by the aPTT.32
Lupus anticoagulant. Patients with the specific antiphospholipid antibody known as lupus anticoagulant frequently have a prolonged baseline aPTT,25 making it an unreliable marker of anticoagulant effect for intravenous unfractionated heparin therapy.
Critically ill infants and children. Arachchillage et al35 found that infants (< 1 year old) treated with intravenous unfractionated heparin in an intensive care department had only a 32.4% correlation between aPTT and anti-Xa levels, which was lower than that found in children ages 1 to 15 (66%) and adults (52%). In two-thirds of cases of discordant aPTT and anti-Xa levels, the aPTT was elevated (supratherapeutic) while the anti-Xa assay was within the therapeutic range (0.3–0.7 U/mL). Despite the lack of data on clinical outcomes (eg, rates of thrombosis and bleeding) with the use of an anti-Xa assay, it has been considered the method of choice for unfractionated heparin monitoring in critically ill children, and especially in those under age 1.41,44,62–64
While anti-Xa assays may also be better for unfractionated heparin monitoring in critically ill adults, the lack of clinical outcome data from large-scale randomized trials has precluded evidence-based recommendations favoring them over the aPTT.8,34
LIMITATIONS OF ANTI-Xa ASSAYS
Anti-Xa assays are hampered by some technical limitations:
Samples must be processed within 1 hour to avoid heparin neutralization.34
Samples must be clear. Hemolyzed or opaque samples (eg, due to bilirubin levels > 6.6 mg/dL or triglyceride levels > 360 mg/dL) cannot be processed, as they can cause falsely low levels.
Exposure to other anticoagulants can interfere with the results. The anti-Xa assay may be unreliable for unfractionated heparin monitoring in patients who are transitioned from low-molecular-weight heparins, fondaparinux, or an oral factor Xa inhibitor (apixaban, betrixaban, edoxaban, rivaroxaban) to intravenous unfractionated heparin, eg, due to hospitalization or acute kidney injury.65,66 Different reports have found that anti-Xa assays may be elevated for as long as 63 to 96 hours after the last dose of oral Xa inhibitors,67–69 potentially resulting in underdosing of unfractionated heparin. In such settings, unfractionated heparin therapy should be monitored by the aPTT.
ANTI-Xa ASSAYS AND LOW-MOLECULAR-WEIGHT HEPARINS
Most patients receiving low-molecular-weight heparins do not need laboratory monitoring.8 Alhenc-Gelas et al70 randomized patients to receive dalteparin in doses either based on weight or guided by anti-Xa assay results, and found that dose adjustments were rare and lacked clinical benefit.
The suggested therapeutic anti-Xa levels for low-molecular-weight heparins are:
- 0.5–1.2 U/mL for twice-daily enoxaparin
- 1.0–2.0 U/mL for once-daily enoxaparin or dalteparin.
Levels should be measured at peak plasma level (ie, 3–4 hours after subcutaneous injection, except during pregnancy, when it is 4–6 hours), and only after at least 3 doses of low-molecular-weight heparin.8,71 Unlike the anti-Xa therapeutic range recommended for unfractionated heparin therapy, these ranges are not based on prospective data, and if the assay result is outside the suggested therapeutic target range, current guidelines offer no advice on safely adjusting the dose.8,71
Measuring anti-Xa activity is particularly important for pregnant women with a mechanical prosthetic heart valve who are treated with low-molecular-weight heparins. In this setting, valve thrombosis and cardioembolic events have been reported in patients with peak low-molecular-weight heparin anti-Xa assay levels below or even at the lower end of the therapeutic range, and increased bleeding risk has been reported with elevated anti-Xa levels.71–74 Measuring trough low-molecular-weight heparin anti-Xa levels has been suggested to guide dose adjustments during pregnancy.75
Clearance of low-molecular-weight heparins as measured by the anti-Xa assay is highly correlated with creatinine clearance.76,77 A strong linear correlation has been demonstrated between creatine clearance and anti-Xa levels of enoxaparin after multiple therapeutic doses, and low-molecular-weight heparins accumulate in the plasma, especially in patients with creatine clearance less than 30 mL/min.78 The risk of major bleeding is significantly increased in patients with severe renal insufficiency (creatinine clearance < 30 mL/min) not on dialysis who are treated with either prophylactic or therapeutic doses of low-molecular-weight heparin.79–81 In a meta-analysis, the risk of bleeding with therapeutic-intensity doses of enoxaparin was 4 times higher than with prophylactic-intensity doses.79 Although bleeding risk appears to be reduced when the enoxaparin dose is reduced by 50%,8 the efficacy and safety of this strategy has not been determined by prospective trials.
ANTI-Xa ASSAYS IN PATIENTS RECEIVING DIRECT ORAL ANTICOAGULANTS
Direct oral factor Xa inhibitors cannot be measured accurately by heparin anti-Xa assays. Nevertheless, such assays may be useful to assess whether clinically relevant plasma levels are present in cases of major bleeding, suspected anticoagulant failure, or patient noncompliance.82
Intense research has focused on developing drug-specific chromogenic anti-Xa assays using calibrators and standards for apixaban, edoxaban, and rivaroxaban,82,83 and good linear correlation has been shown with some assays.82,84 In patients treated with oral factor Xa inhibitors who need to undergo an urgent invasive procedure associated with high bleeding risk, use of a specific reversal agent may be considered with drug concentrations more than 30 ng/mL measured by a drug-specific anti-Xa assay. A similar suggestion has been made for drug concentrations more than 50 ng/mL in the setting of major bleeding.85 Unfortunately, such assays are not widely available at this time.82,86
While drug-specific anti-Xa assays could become clinically important to guide reversal strategies, their relevance for drug monitoring remains uncertain. This is because no therapeutic target ranges have been established for any of the direct oral anticoagulants, which were approved on the basis of favorable clinical trial outcomes that neither measured nor were correlated with specific drug levels in plasma. Therefore, a specific anti-Xa level cannot yet be used as a marker of clinical efficacy for any specific oral direct Xa inhibitor.
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
- Abildgaard U. Highly purified antithrombin 3 with heparin cofactor activity prepared by disc electrophoresis. Scand J Clin Lab Invest 1968; 21(1):89–91. pmid:5637480
- Rosenberg RD, Lam L. Correlation between structure and function of heparin. Proc Natl Acad Sci USA 1979; 76(3):1218–1222. pmid:286307
- Lindahl U, Bäckström G, Höök M, Thunberg L, Fransson LA, Linker A. Structure of the antithrombin-binding site of heparin. Proc Natl Acad Sci USA 1979; 76(7):3198–3202. pmid:226960
- Rosenberg RD, Rosenberg JS. Natural anticoagulant mechanisms. J Clin Invest 1984; 74(1):1–6. doi:10.1172/JCI111389
- Casu B, Oreste P, Torri G, et al. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence. Chemical and 13C nuclear-magnetic-resonance studies. Biochem J 1981; 197(3):599–609. pmid:7325974
- Choay J, Lormeau JC, Petitou M, Sinaÿ P, Fareed J. Structural studies on a biologically active hexasaccharide obtained from heparin. Ann NY Acad Sci 1981; 370: 644–649. pmid:6943974
- Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy, and safety. Chest 2001; 119(suppl 1):64S–94S. pmid:11157643
- Garcia DA, Baglin TP, Weitz JI, Samama MM. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e24S–e43S. doi:10.1378/chest.11-2291
- Hirsh J, Levine M. Low-molecular weight heparin. Blood 1992; 79(1):1–17. pmid:1309422
- Barritt DW, Jordan SC. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960; 1(7138):1309–1312. pmid:13797091
- Basu D, Gallus A, Hirsh J, Cade J. A prospective study of the value of monitoring heparin treatment with the activated partial thromboplastin time. N Engl J Med 1972; 287(7):324–327. doi:10.1056/NEJM197208172870703
- Hull RD, Raskob GE, Hirsh J, et al. Continuous intravenous heparin compared with intermittent subcutaneous heparin in the initial treatment of proximal-vein thrombosis. N Engl J Med 1986; 315(18):1109–1114. doi:10.1056/NEJM198610303151801
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. Relation between the time to achieve the lower limit of the APTT therapeutic range and recurrent venous thromboembolism during heparin treatment for deep vein thrombosis. Arch Intern Med 1997; 157(22):2562–2568. pmid:9531224
- Hull RD, Raskob GE, Brant RF, Pineo GF, Valentine KA. The importance of initial heparin treatment on long-term clinical outcomes of antithrombotic therapy. The emerging theme of delayed recurrence. Arch Intern Med 1997; 157(20):2317–2321. pmid:9361572
- Anand S, Ginsberg JS, Kearon C, Gent M, Hirsh J. The relation between the activated partial thromboplastin time response and recurrence in patients with venous thrombosis treated with continuous intravenous heparin. Arch Intern Med 1996; 156(15):1677–1681. pmid:8694666
- Anand SS, Bates S, Ginsberg JS, et al. Recurrent venous thrombosis and heparin therapy: an evaluation of the importance of early activated partial thromboplastin times. Arch Intern Med 1999; 159(17):2029–2032. pmid:10510988
- Raschke RA, Reilly BM, Guidry JR, Fontana JR, Srinivas S. The weight-based heparin dosing nomogram compared with a “standard care” nomogram. A randomized controlled trial. Ann Intern Med 1993; 119(9):874–881. pmid:8214998
- Smith SB, Geske JB, Maguire JM, Zane NA, Carter RE, Morgenthaler TI. Early anticoagulation is associated with reduced mortality for acute pulmonary embolism. Chest 2010; 137(6):1382–1390. doi:10.1378/chest.09-0959
- Cruickshank MK, Levine MN, Hirsh J, Roberts R, Siguenza M. A standard heparin nomogram for the management of heparin therapy. Arch Intern Med 1991; 151(2):333–337. pmid:1789820
- Raschke RA, Gollihare B, Peirce J. The effectiveness of implementing the weight-based heparin nomogram as a practice guideline. Arch Intern Med 1996; 156(15):1645–1649. pmid:8694662
- Simko RJ, Tsung FF, Stanek EJ. Activated clotting time versus activated partial thromboplastin time for therapeutic monitoring of heparin. Ann Pharmacother 1995; 29(10):1015–1021. doi:10.1177/106002809502901012
- Langdell RD, Wagner RH, Brinkhous KM. Effect of antihemophilic factor on one-stage clotting tests; a presumptive test for hemophilia and a simple one-stage antihemophilic factor assy procedure. J Lab Clin Med 1953; 41(4):637–647.
- White GC 2nd. The partial thromboplastin time: defining an era in coagulation. J Thromb Haemost 2003; 1(11):2267–2270. pmid:14629454
- Proctor RR, Rapaport SI. The partial thromboplastin time with kaolin. A simple screening test for first stage plasma clotting factor deficiencies. Am J Clin Pathol 1961; 36:212–219. pmid:13738153
- Brandt JT, Triplett DA, Rock WA, Bovill EG, Arkin CF. Effect of lupus anticoagulants on the activated partial thromboplastin time. Results of the College of American Pathologists survey program. Arch Pathol Lab Med 1991; 115(2):109–114. pmid:1899555
- Tripodi A, Mannucci PM. Activated partial thromboplastin time (APTT). New indications for an old test? J Thromb Haemost 2006; 4(4):750–751. doi:10.1111/j.1538-7836.2006.01857.x
- Chiu HM, Hirsh J, Yung WL, Regoeczi E, Gent M. Relationship between the anticoagulant and antithrombotic effects of heparin in experimental venous thrombosis. Blood 1977; 49(2):171–184. pmid:831872
- Brill-Edwards P, Ginsberg JS, Johnston M, Hirsh J. Establishing a therapeutic range for heparin therapy. Ann Intern Med 1993; 119(2):104–109. pmid:8512158
- Vandiver JW, Vondracek TG. Antifactor Xa levels versus activated partial thromboplastin time for monitoring unfractionated heparin. Pharmacotherapy 2012; 32(6):546–558. doi:10.1002/j.1875-9114.2011.01049.x
- Newall F. Anti-factor Xa (anti-Xa). In: Monagle P, ed. Haemostasis: Methods and Protocols. New York, NY: Springer-Verlag; 2013.
- Bates SM, Weitz JI, Johnston M, Hirsh J, Ginsberg JS. Use of a fixed activated partial thromboplastin time ratio to establish a therapeutic range for unfractionated heparin. Arch Intern Med 2001; 161(3):385–391. pmid:11176764
- Levine MN, Hirsh J, Gent M, et al. A randomized trial comparing activated thromboplastin time with heparin assay in patients with acute venous thromboembolism requiring large doses of heparin. Arch Intern Med 1994; 154(1):49–56. pmid:8267489
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Lehman CM, Frank EL. Laboratory monitoring of heparin therapy: partial thromboplastin time or anti-Xa assay? Lab Med 2009; 40(1):47–51. doi:10.1309/LM9NJGW2ZIOLPHY6
- Arachchillage DR, Kamani F, Deplano S, Banya W, Laffan M. Should we abandon the aPTT for monitoring unfractionated heparin? Thromb Res 2017; 157:157–161. doi:10.1016/j.thromres.2017.07.006
- Olson JD, Arkin CA, Brandt JT, et al. College of American Pathologists Conference XXXI on Laboratory Monitoring of Anticoagulant Therapy: laboratory monitoring of unfractionated heparin therapy. Arch Pathol Lab Med 1998; 122(9):782–798. pmid:9740136
- Eikelboom JW, Hirsh J. Monitoring unfractionated heparin with the aPTT: time for a fresh look. Thromb Haemost 2006; 96(5):547–552. pmid:17080209
- Young E, Prins M, Levine MN, Hirsh J. Heparin binding to plasma proteins, an important mechanism of heparin resistance. Thromb Haemost 1992; 67(6):639–643. pmid:1509402
- Edson JR, Krivit W, White JG. Kaolin partial thromboplastin time: high levels of procoagulants producing short clotting times or masking deficiencies of other procoagulants or low concentrations of anticoagulants. J Lab Clin Med 1967; 70(3):463–470. pmid:6072020
- Whitfield LR, Lele AS, Levy G. Effect of pregnancy on the relationship between concentration and anticoagulant action of heparin. Clin Pharmacol Ther 1983; 34(1):23–28. pmid:6861435
- Marci CD, Prager D. A review of the clinical indications for the plasma heparin assay. Am J Clin Pathol 1993; 99(5):546–550.
- Takemoto CM, Streiff MB, Shermock KM, et al. Activated partial thromboplastin time and anti-Xa measurements in heparin monitoring: biochemical basis of discordance. Am J Clin Pathol 2013; 139(4):450–456. doi:10.1309/AJCPS6OW6DYNOGNH
- Adatya S, Uriel N, Yarmohammadi H, et al. Anti-factor Xa and activated partial thromboplastin time measurements for heparin monitoring in mechanical circulatory support. JACC Heart Fail 2015; 3(4):314–322. doi:10.1016/j.jchf.2014.11.009
- Kuhle S, Eulmesekian P, Kavanagh B, et al. Lack of correlation between heparin dose and standard clinical monitoring tests in treatment with unfractionated heparin in critically ill children. Haematologica 2007; 92(4):554–557. pmid:17488668
- Price EA, Jin J, Nguyen HM, Krishnan G, Bowen R, Zehnder JL. Discordant aPTT and anti-Xa values and outcomes in hospitalized patients treated with intravenous unfractionated heparin. Ann Pharmacother 2013; 47(2):151–158. doi:10.1345/aph.1R635
- Baker BA, Adelman MD, Smith PA, Osborn JC. Inability of the activated partial thromboplastin time to predict heparin levels. Arch Intern Med 1997; 157(21):2475–2479. pmid:9385299
- Koerber JM, Smythe MA, Begle RL, Mattson JC, Kershaw BP, Westley SJ. Correlation of activated clotting time and activated partial thromboplastin time to plasma heparin concentration. Pharmacotherapy 1999; 19(8):922–931. pmid:10453963
- Smythe MA, Mattson JC, Koerber JM. The heparin anti-Xa therapeutic range: are we there yet? Chest 2002; 121(1):303–304. pmid:11796474
- Cuker A, Ptashkin B, Konkle A, et al. Interlaboratory agreement in the monitoring of unfractionated heparin using the anti-factor Xa-correlated activated partial thromboplastin time. J Thromb Haemost 2009; 7(1):80–86. doi:10.1111/j.1538-7836.2008.03224.x
- Taylor CT, Petros WP, Ortel TL. Two instruments to determine activated partial thromboplastin time: implications for heparin monitoring. Pharmacotherapy 1999; 19(4):383–387. pmid:10212007
- Guervil DJ, Rosenberg AF, Winterstein AG, Harris NS, Johns TE, Zumberg MS. Activated partial thromboplastin time versus antifactor Xa heparin assay in monitoring unfractionated heparin by continuous intravenous infusion. Ann Pharmacother 2011; 45(7–8):861–868. doi:10.1345/aph.1Q161
- Fruge KS, Lee YR. Comparison of unfractionated heparin protocols using antifactor Xa monitoring or activated partial thrombin time monitoring. Am J Health Syst Pharm 2015; 72(17 suppl 2):S90–S97. doi:10.2146/sp150016
- Rosborough TK. Monitoring unfractionated heparin therapy with antifactor Xa activity results in fewer monitoring tests and dosage changes than monitoring with activated partial thromboplastin time. Pharmacotherapy 1999; 19(6):760–766. pmid:10391423
- Rosborough TK, Shepherd MF. Achieving target antifactor Xa activity with a heparin protocol based on sex, age, height, and weight. Pharmacotherapy 2004; 24(6):713–719. doi:10.1592/phco.24.8.713.36067
- Smith ML, Wheeler KE. Weight-based heparin protocol using antifactor Xa monitoring. Am J Health Syst Pharm 2010; 67(5):371–374. doi:10.2146/ajhp090123
- Bartholomew JR, Kottke-Marchant K. Monitoring anticoagulation therapy in patients with the lupus anticoagulant. J Clin Rheumatol 1998; 4(6):307–312. pmid:19078327
- Wool GD, Lu CM; Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on anticoagulation monitoring: factor X-related assays. Am J Clin Pathol 2013; 140(5):623–634. doi:10.1309/AJCPR3JTOK7NKDBJ
- Mehta TP, Smythe MA, Mattson JC. Strategies for managing heparin therapy in patients with antiphospholipid antibody syndrome. Pharmacotherapy 2011; 31(12):1221–1231. doi:10.1592/phco.31.12.1221
- Levine SP, Sorenson RR, Harris MA, Knieriem LK. The effect of platelet factor 4 (PF4) on assays of plasma heparin. Br J Haematol 1984; 57(4):585–596. pmid:6743573
- Fisher AR, Bailey CR, Shannon CN, Wielogorski AK. Heparin resistance after aprotinin. Lancet 1992; 340(8829):1230–1231. pmid:1279335
- Becker RC, Corrao JM, Bovill EG, et al. Intravenous nitroglycerin-induced heparin resistance: a qualitative antithrombin III abnormality. Am Heart J 1990; 119(6):1254–1261. pmid:2112878
- Monagle P, Chan AK, Goldenberg NA, et al. Antithrombotic therapy in neonates and children: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e737S–e801S. doi:10.1378/chest.11-2308
- Long E, Pitfield AF, Kissoon N. Anticoagulation therapy: indications, monitoring, and complications. Pediatr Emerg Care 2011; 27(1):55–61. doi:10.1097/PEC.0b013e31820461b1
- Andrew M, Schmidt B. Use of heparin in newborn infants. Semin Thromb Hemost 1988; 14(1):28–32. doi:10.1055/s-2007-1002752
- Teien AN, Lie M, Abildgaard U. Assay of heparin in plasma using a chromogenic substrate for activated factor X. Thromb Res 1976; 8(3):413–416. pmid:1265712
- Vera-Aguillera J, Yousef H, Beltran-Melgarejo D, et al. Clinical scenarios for discordant anti-Xa. Adv Hematol 2016; 2016:4054806. doi:10.1155/2016/4054806
- Macedo KA, Tatarian P, Eugenio KR. Influence of direct oral anticoagulants on anti-factor Xa measurements utilized for monitoring heparin. Ann Pharmacother 2018; 52(2):154–159. doi:10.1177/1060028017729481
- Wendte J, Voss G, Van Overschelde B. Influence of apixaban on antifactor Xa levels in a patient with acute kidney injury. Am J Health Syst Pharm 2016; 73(8):563–567. doi:10.2146/ajhp150360
- Faust AC, Kanyer D, Wittkowsky AK. Managing transitions from oral factor Xa inhibitors to unfractionated heparin infusions. Am J Health Syst Pharm 2016; 73(24):2037–2041. doi:10.2146/ajhp150596
- Alhenc-Gelas M, Jestin-Le Guernic C, Vitoux JF, Kher A, Aiach M, Fiessinger JN. Adjusted versus fixed doses of the low-molecular-weight heparin fragmin in the treatment of deep vein thrombosis. Fragmin-Study Group. Thromb Haemost 1994; 71(6):698–702. pmid:7974334
- Bates SM, Greer IA, Middeldorp S, Veenstra DL, Prabulos AM, Vandvik PO. VTE, thrombophilia, antithrombotic therapy, and pregnancy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e691S–e736S. doi:10.1378/chest.11-2300
- Bara L, Leizorovicz A, Picolet H, Samama M. Correlation between anti-Xa and occurrence of thrombosis and haemorrhage in post-surgical patients treated with either Logiparin (LMWH) or unfractionated heparin. Post-surgery Logiparin Study Group. Thromb Res 1992; 65(4–5):641–650. pmid:1319619
- Prandoni P, Lensing AW, Büller HR, et al. Comparison of subcutaneous low-molecular-weight heparin with intravenous standard heparin in proximal deep-vein thrombosis. Lancet 1992; 339(8791):441–445. pmid:1346817
- Walenga JM, Hoppensteadt D, Fareed J. Laboratory monitoring of the clinical effects of low molecular weight heparins. Thromb Res Suppl 1991;14:49–62. pmid:1658970
- Elkayam U. Anticoagulation therapy for pregnant women with mechanical prosthetic heart valves: how to improve safety? J Am Coll Cardiol 2017; 69(22):2692–2695. doi:10.1016/j.jacc.2017.04.034
- Brophy DF, Wazny LD, Gehr TW, Comstock TJ, Venitz J. The pharmacokinetics of subcutaneous enoxaparin in end-stage renal disease. Pharmacotherapy 2001; 21(2):169–174. pmid:11213853
- Becker RC, Spencer FA, Gibson M, et al; TIMI 11A Investigators. Influence of patient characteristics and renal function on factor Xa inhibition pharmacokinetics and pharmacodynamics after enoxaparin administration in non-ST-segment elevation acute coronary syndromes. Am Heart J 2002; 143(5):753–759. pmid:12040334
- Chow SL, Zammit K, West K, Dannenhoffer M, Lopez-Candales A. Correlation of antifactor Xa concentrations with renal function in patients on enoxaparin. J Clin Pharmacol 2003; 43(6):586–590. pmid:12817521
- Lim W, Dentali F, Eikelboom JW, Crowther MA. Meta-analysis: low-molecular-weight heparin and bleeding in patients with severe renal insufficiency. Ann Intern Med 2006; 144(9):673–684. pmid:16670137
- Spinler SA, Inverso SM, Cohen M, Goodman SG, Stringer KA, Antman EM; ESSENCE and TIMI 11B Investigators. Safety and efficacy of unfractionated heparin versus enoxaparin in patients who are obese and patients with severe renal impairment: analysis from the ESSENCE and TIMI 11B studies. Am Heart J 2003; 146(1):33–41. doi:10.1016/S0002-8703(03)00121-2
- Cestac P, Bagheri H, Lapeyre-Mestre M, et al. Utilisation and safety of low molecular weight heparins: prospective observational study in medical inpatients. Drug Saf 2003; 26(3):197–207. doi:10.2165/00002018-200326030-00005
- Douxfils J, Ageno W, Samama CM, et al. Laboratory testing in patients treated with direct oral anticoagulants: a practical guide for clinicians. J Thromb Haemost 2018; 16(2):209–219. doi:10.1111/jth.13912
- Samuelson BT, Cuker A, Siegal DM, Crowther M, Garcia DA. Laboratory assessment of the anticoagulant activity of direct oral anticoagulants: a systematic review. Chest 2017; 151(1):127–138. doi:10.1016/j.chest.2016.08.1462
- Gosselin RC, Francart SJ, Hawes EM, Moll S, Dager WE, Adcock DM. Heparin-calibrated chromogenic anti-Xa activity measurements in patients receiving rivaroxaban: can this test be used to quantify drug level? Ann Pharmacother 2015; 49(7):777–783. doi:10.1177/1060028015578451
- Levy JH, Ageno W, Chan NC, Crowther M, Verhamme P, Weitz JI; Subcommittee on Control of Anticoagulation. When and how to use antidotes for the reversal of direct oral anticoagulants: guidance from the SSC of the ISTH. J Thromb Haemost 2016; 14(3):623–627. doi:10.1111/jth.13227
- Cuker A, Siegal D. Monitoring and reversal of direct oral anticoagulants. Hematology Am Soc Hematol Educ Program 2015; 2015:117–124. doi:10.1182/asheducation-2015.1.117
KEY POINTS
- Intravenous unfractionated heparin treatment is typically monitored by the activated partial thromboplastin time (aPTT), with a therapeutic target defined as the range that corresponds to an anti-Xa level of 0.3 to 0.7 U/mL.
- Monitoring unfractionated heparin is important to achieve a therapeutic target within the first 24 hours and to maintain therapeutic levels thereafter.
- The heparin anti-Xa assay is unreliable for unfractionated heparin monitoring when switching from oral factor Xa inhibitor therapy to intravenous unfractionated heparin. In such cases, the aPTT is preferred.
- Most patients receiving low-molecular-weight heparin do not need monitoring, but monitoring should be considered for pregnant women with prosthetic heart valves, using an anti-Xa assay specific for low-molecular-weight heparin.
Disseminated invasive aspergillosis in an immunocompetent patient
A 57-year-old woman was admitted to our hospital for progressive hypoxic respiratory failure that developed after 10 days of empiric treatment at another hospital for an exacerbation of chronic obstructive pulmonary disease (COPD).
, with diffuse ground-glass opacities throughout.")
Computed tomography (CT) showed a lesion in the upper lobe of the left lung, with new ground-glass opacities with cystic and cavitary changes raising concern for an inflammatory or infectious cause (Figure 1). Respiratory culture of expectorated secretions grew Aspergillus. Assays for beta-d-glucan and serum Aspergillus immunoglobulin G (IgG) antibodies were positive, although given the improvement in her oxygenation requirements and overall clinical status, these were thought to be trivial. Tests for immunoglobulin deficiencies and human immunodeficiency virus were negative, ruling out primary immunodeficiency. However, within the next 48 hours, her respiratory status declined, and voriconazole was started out of concern for invasive pulmonary aspergillosis based on results of serum IgG testing.
Despite 2 days of treatment with voriconazole, the patient developed respiratory failure. Repeat CT showed that the ground-glass opacities were more dense, especially in the lower lobes, and new patchy infiltrates were noted in the left lung. The patient developed a right tension pneumothorax requiring emergency intubation and chest tube insertion.1 She subsequently developed acute abdominal pain with worsening abdominal distention, diagnosed as pneumoperitoneum. Emergency exploratory laparotomy revealed perforations in the cecum with fecal spillage, requiring ileocecectomy and ileostomy.
 of fungal hyphae (hematoxylin and eosin, × 600).")
Pathologic study of bowel specimens confirmed fungal hyphae with “tree-branch” structures consistent with fungal infection in the bowel (Figure 2).
Oral voriconazole was continued. The patient’s respiratory status improved, and she no longer required supplemental oxygen. She was discharged on a regimen of oral voriconazole 200 mg twice daily. However, over the next 12 months, she had additional hospitalizations for severe sepsis from abdominal wound infections, pneumonia, and Clostridium difficile infection. She will require lifelong antifungal treatment.
INVASIVE PULMONARY ASPERGILLOSIS
Invasive pulmonary aspergillosis is the most severe form of aspergillosis and is most often seen in immunocompromised patients. The death rate is as high as 50% in neutropenic patients regardless of the time to diagnosis or effective treatment.2 It becomes life-threatening as the infection enters the blood stream, leading to formation of thrombi and precipitating embolism and necrosis in the lungs.3
In immunocompetent patients, COPD, tuberculosis, bronchiectasis, liver disease, severe sepsis, and diabetes mellitus predispose to invasive pulmonary aspergillosis.2 Other risk factors include long-term steroid therapy at doses equivalent to prednisone 20 mg/day for at least 13 weeks4 and viral infection such as influenza.5 Chronic use of inhaled corticosteroids has been hypothesized to increase risk.4
Histopathologic confirmation of fungal elements is the gold standard for diagnosis.3 New biomarkers such as beta-d-glucan have shown promise in enabling earlier diagnosis to allow effective treatment of disseminated aspergillosis, as in our patient.6
TAKE-HOME MESSAGE
Although not common, invasive aspergillosis can occur in immunocompetent and near-immunocompetent patients, particularly those with COPD or other underlying lung disease.
Acknowledgment: The authors thank Kimberley Woodward, MD, Inova Fairfax Hospital, Falls Church, VA, for her study of the bowel specimen and for providing the histology slide.
- Vukicevic TA, Dudvarski-Ilic A, Zugic V, Stevanovic G, Rubino S, Barac A. Subacute invasive pulmonary aspergillosis as a rare cause of pneumothorax in immunocompetent patient: brief report. Infection 2017; 45(3):377–380. doi:10.1007/s15010-017-0994-3
- Moreno-González G, Ricart de Mesones A, Tazi-Mezalek R, Marron-Moya MT, Rosell A, Mañez R. Invasive pulmonary aspergillosis with disseminated infection in immunocompetent patient. Can Respir J 2016; 2016:7984032. doi:10.1155/2016/7984032
- Chen L, Liu Y, Wang W, Liu K. Adrenal and hepatic aspergillosis in an immunocompetent patient. Infect Dis (Lond) 2015; 47(6):428–432. doi:10.3109/00365548.2014.995697
- Taccone FS, Van den Abeele AM, Bulpa P, et al; AspICU Study Investigators. Epidemiology of invasive aspergillosis in critically ill patients: clinical presentation, underlying conditions, and outcomes. Crit Care 2015; 19:7. doi:10.1186/s13054-014-0722-7
- Crum-Cianflone NF. Invasive aspergillosis associated with severe influenza infections. Open Forum Infect Dis 2016; 3(3):ofw171. doi:10.1093/ofid/ofw171
- Ergene U, Akcali Z, Ozbalci D, Nese N, Senol S. Disseminated aspergillosis due to Aspergillus niger in immunocompetent patient: a case report. Case Rep Infect Dis 2013; 2013:385190. doi:10.1155/2013/385190
A 57-year-old woman was admitted to our hospital for progressive hypoxic respiratory failure that developed after 10 days of empiric treatment at another hospital for an exacerbation of chronic obstructive pulmonary disease (COPD).
Computed tomography (CT) showed a lesion in the upper lobe of the left lung, with new ground-glass opacities with cystic and cavitary changes raising concern for an inflammatory or infectious cause (Figure 1). Respiratory culture of expectorated secretions grew Aspergillus. Assays for beta-d-glucan and serum Aspergillus immunoglobulin G (IgG) antibodies were positive, although given the improvement in her oxygenation requirements and overall clinical status, these were thought to be trivial. Tests for immunoglobulin deficiencies and human immunodeficiency virus were negative, ruling out primary immunodeficiency. However, within the next 48 hours, her respiratory status declined, and voriconazole was started out of concern for invasive pulmonary aspergillosis based on results of serum IgG testing.
Despite 2 days of treatment with voriconazole, the patient developed respiratory failure. Repeat CT showed that the ground-glass opacities were more dense, especially in the lower lobes, and new patchy infiltrates were noted in the left lung. The patient developed a right tension pneumothorax requiring emergency intubation and chest tube insertion.1 She subsequently developed acute abdominal pain with worsening abdominal distention, diagnosed as pneumoperitoneum. Emergency exploratory laparotomy revealed perforations in the cecum with fecal spillage, requiring ileocecectomy and ileostomy.
Pathologic study of bowel specimens confirmed fungal hyphae with “tree-branch” structures consistent with fungal infection in the bowel (Figure 2).
Oral voriconazole was continued. The patient’s respiratory status improved, and she no longer required supplemental oxygen. She was discharged on a regimen of oral voriconazole 200 mg twice daily. However, over the next 12 months, she had additional hospitalizations for severe sepsis from abdominal wound infections, pneumonia, and Clostridium difficile infection. She will require lifelong antifungal treatment.
INVASIVE PULMONARY ASPERGILLOSIS
Invasive pulmonary aspergillosis is the most severe form of aspergillosis and is most often seen in immunocompromised patients. The death rate is as high as 50% in neutropenic patients regardless of the time to diagnosis or effective treatment.2 It becomes life-threatening as the infection enters the blood stream, leading to formation of thrombi and precipitating embolism and necrosis in the lungs.3
In immunocompetent patients, COPD, tuberculosis, bronchiectasis, liver disease, severe sepsis, and diabetes mellitus predispose to invasive pulmonary aspergillosis.2 Other risk factors include long-term steroid therapy at doses equivalent to prednisone 20 mg/day for at least 13 weeks4 and viral infection such as influenza.5 Chronic use of inhaled corticosteroids has been hypothesized to increase risk.4
Histopathologic confirmation of fungal elements is the gold standard for diagnosis.3 New biomarkers such as beta-d-glucan have shown promise in enabling earlier diagnosis to allow effective treatment of disseminated aspergillosis, as in our patient.6
TAKE-HOME MESSAGE
Although not common, invasive aspergillosis can occur in immunocompetent and near-immunocompetent patients, particularly those with COPD or other underlying lung disease.
Acknowledgment: The authors thank Kimberley Woodward, MD, Inova Fairfax Hospital, Falls Church, VA, for her study of the bowel specimen and for providing the histology slide.
A 57-year-old woman was admitted to our hospital for progressive hypoxic respiratory failure that developed after 10 days of empiric treatment at another hospital for an exacerbation of chronic obstructive pulmonary disease (COPD).
Computed tomography (CT) showed a lesion in the upper lobe of the left lung, with new ground-glass opacities with cystic and cavitary changes raising concern for an inflammatory or infectious cause (Figure 1). Respiratory culture of expectorated secretions grew Aspergillus. Assays for beta-d-glucan and serum Aspergillus immunoglobulin G (IgG) antibodies were positive, although given the improvement in her oxygenation requirements and overall clinical status, these were thought to be trivial. Tests for immunoglobulin deficiencies and human immunodeficiency virus were negative, ruling out primary immunodeficiency. However, within the next 48 hours, her respiratory status declined, and voriconazole was started out of concern for invasive pulmonary aspergillosis based on results of serum IgG testing.
Despite 2 days of treatment with voriconazole, the patient developed respiratory failure. Repeat CT showed that the ground-glass opacities were more dense, especially in the lower lobes, and new patchy infiltrates were noted in the left lung. The patient developed a right tension pneumothorax requiring emergency intubation and chest tube insertion.1 She subsequently developed acute abdominal pain with worsening abdominal distention, diagnosed as pneumoperitoneum. Emergency exploratory laparotomy revealed perforations in the cecum with fecal spillage, requiring ileocecectomy and ileostomy.
Pathologic study of bowel specimens confirmed fungal hyphae with “tree-branch” structures consistent with fungal infection in the bowel (Figure 2).
Oral voriconazole was continued. The patient’s respiratory status improved, and she no longer required supplemental oxygen. She was discharged on a regimen of oral voriconazole 200 mg twice daily. However, over the next 12 months, she had additional hospitalizations for severe sepsis from abdominal wound infections, pneumonia, and Clostridium difficile infection. She will require lifelong antifungal treatment.
INVASIVE PULMONARY ASPERGILLOSIS
Invasive pulmonary aspergillosis is the most severe form of aspergillosis and is most often seen in immunocompromised patients. The death rate is as high as 50% in neutropenic patients regardless of the time to diagnosis or effective treatment.2 It becomes life-threatening as the infection enters the blood stream, leading to formation of thrombi and precipitating embolism and necrosis in the lungs.3
In immunocompetent patients, COPD, tuberculosis, bronchiectasis, liver disease, severe sepsis, and diabetes mellitus predispose to invasive pulmonary aspergillosis.2 Other risk factors include long-term steroid therapy at doses equivalent to prednisone 20 mg/day for at least 13 weeks4 and viral infection such as influenza.5 Chronic use of inhaled corticosteroids has been hypothesized to increase risk.4
Histopathologic confirmation of fungal elements is the gold standard for diagnosis.3 New biomarkers such as beta-d-glucan have shown promise in enabling earlier diagnosis to allow effective treatment of disseminated aspergillosis, as in our patient.6
TAKE-HOME MESSAGE
Although not common, invasive aspergillosis can occur in immunocompetent and near-immunocompetent patients, particularly those with COPD or other underlying lung disease.
Acknowledgment: The authors thank Kimberley Woodward, MD, Inova Fairfax Hospital, Falls Church, VA, for her study of the bowel specimen and for providing the histology slide.
- Vukicevic TA, Dudvarski-Ilic A, Zugic V, Stevanovic G, Rubino S, Barac A. Subacute invasive pulmonary aspergillosis as a rare cause of pneumothorax in immunocompetent patient: brief report. Infection 2017; 45(3):377–380. doi:10.1007/s15010-017-0994-3
- Moreno-González G, Ricart de Mesones A, Tazi-Mezalek R, Marron-Moya MT, Rosell A, Mañez R. Invasive pulmonary aspergillosis with disseminated infection in immunocompetent patient. Can Respir J 2016; 2016:7984032. doi:10.1155/2016/7984032
- Chen L, Liu Y, Wang W, Liu K. Adrenal and hepatic aspergillosis in an immunocompetent patient. Infect Dis (Lond) 2015; 47(6):428–432. doi:10.3109/00365548.2014.995697
- Taccone FS, Van den Abeele AM, Bulpa P, et al; AspICU Study Investigators. Epidemiology of invasive aspergillosis in critically ill patients: clinical presentation, underlying conditions, and outcomes. Crit Care 2015; 19:7. doi:10.1186/s13054-014-0722-7
- Crum-Cianflone NF. Invasive aspergillosis associated with severe influenza infections. Open Forum Infect Dis 2016; 3(3):ofw171. doi:10.1093/ofid/ofw171
- Ergene U, Akcali Z, Ozbalci D, Nese N, Senol S. Disseminated aspergillosis due to Aspergillus niger in immunocompetent patient: a case report. Case Rep Infect Dis 2013; 2013:385190. doi:10.1155/2013/385190
- Vukicevic TA, Dudvarski-Ilic A, Zugic V, Stevanovic G, Rubino S, Barac A. Subacute invasive pulmonary aspergillosis as a rare cause of pneumothorax in immunocompetent patient: brief report. Infection 2017; 45(3):377–380. doi:10.1007/s15010-017-0994-3
- Moreno-González G, Ricart de Mesones A, Tazi-Mezalek R, Marron-Moya MT, Rosell A, Mañez R. Invasive pulmonary aspergillosis with disseminated infection in immunocompetent patient. Can Respir J 2016; 2016:7984032. doi:10.1155/2016/7984032
- Chen L, Liu Y, Wang W, Liu K. Adrenal and hepatic aspergillosis in an immunocompetent patient. Infect Dis (Lond) 2015; 47(6):428–432. doi:10.3109/00365548.2014.995697
- Taccone FS, Van den Abeele AM, Bulpa P, et al; AspICU Study Investigators. Epidemiology of invasive aspergillosis in critically ill patients: clinical presentation, underlying conditions, and outcomes. Crit Care 2015; 19:7. doi:10.1186/s13054-014-0722-7
- Crum-Cianflone NF. Invasive aspergillosis associated with severe influenza infections. Open Forum Infect Dis 2016; 3(3):ofw171. doi:10.1093/ofid/ofw171
- Ergene U, Akcali Z, Ozbalci D, Nese N, Senol S. Disseminated aspergillosis due to Aspergillus niger in immunocompetent patient: a case report. Case Rep Infect Dis 2013; 2013:385190. doi:10.1155/2013/385190
Is chest radiography routinely needed after thoracentesis?
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
No. After thoracentesis, chest radiography or another lung imaging study should be done only if pneumothorax is suspected, if thoracentesis requires more than 1 attempt, if the patient is on mechanical ventilation or has pre-existing lung disease, or if a large volume (> 1,500 mL) of fluid is removed. Radiography is also usually not necessary after diagnostic thoracentesis in a patient breathing spontaneously. In most cases, pneumothorax found incidentally after thoracentesis does not require decompression and can be managed supportively.
WHAT ARE THE RISKS OF THORACENTESIS?
Thoracentesis is a minimally invasive procedure usually performed at the bedside that involves insertion of a needle into the pleural cavity for drainage of fluid.1 Diagnostic thoracentesis should be done in most cases of a new pleural effusion unless the effusion is small and with a clear diagnosis, or in cases of typical heart failure.
Therapeutic thoracentesis, often called large-volume thoracentesis, aims to improve symptoms such as dyspnea attributed to the pleural effusion by removing at least 1 L of pleural fluid. The presence of active respiratory symptoms and suspicion of infected pleural effusion should lead to thoracentesis as soon as possible.
Complications of thoracentesis may be benign, such as pain and anxiety associated with the procedure and external bleeding at the site of needle insertion. Pneumothorax is the most common serious procedural complication and the principal reason to order postprocedural chest radiography.1 Less common complications include hemothorax, re-expansion pulmonary edema, infection, subdiaphragmatic organ puncture, and procedure-related death. Bleeding complications and hemothorax are rare even in patients with underlying coagulopathy.2
Point-of-care pleural ultrasonography is now considered the standard of care to guide optimal needle location for the procedure and to exclude other conditions that can mimic pleural effusion on chest radiography, such as lung consolidation and atelectasis.3 High proficiency in the use of preprocedural point-of-care ultrasonography reduces the rate of procedural complications, though it does not eliminate the risk entirely.3,4
Factors associated with higher rates of complications include lack of operator proficiency, poor understanding of the anatomy, poor patient positioning, poor patient cooperation with the procedure, lack of availability of bedside ultrasonography, and drainage of more than 1,500 mL of fluid. Addressing these factors has been shown to decrease the risk of pneumothorax and infection.1–5
HOW OFTEN DOES PNEUMOTHORAX OCCUR AFTER THORACENTESIS?
Several early studies have examined the incidence of pneumothorax after thoracentesis. Lack of ultrasonography use likely explains a higher incidence of complications in early studies: rates of pneumothorax after thoracentesis without ultrasonographic guidance ranged from 5.2% to 26%.6,7
Gervais et al8 analyzed thoracentesis with ultrasonographic guidance in 434 patients, 92 of whom were intubated, and reported that pneumothorax occurred in 10 patients, of whom 6 were intubated. Two of the intubated patients required chest tubes. Other studies have confirmed the low incidence of pneumothorax in patients undergoing thoracentesis, with rates such as 0.61%,1 5%,9 and 4%.10
The major predictor of postprocedural pneumothorax was the presence of symptoms such as chest pain and dyspnea. No intervention was necessary for most cases of pneumothorax in asymptomatic patients. The more widespread use of procedural ultrasonography may explain some discrepancies between the early5,6 and more recent studies.1,8–10
Several studies have demonstrated that postprocedural radiography is unnecessary unless a complication is suspected based on the patient’s symptoms or the need to demonstrate lung re-expansion.1,4,9,10 Clinical suspicion and the patient’s symptoms are the major predictors of procedure-related pneumothorax requiring treatment with a chest tube. Otherwise, incidentally discovered pneumothorax can usually be observed and managed supportively.
WHAT MECHANISMS UNDERLIE POSTPROCEDURAL PNEUMOTHORAX?
Major causes of pneumothorax in patients undergoing thoracentesis are direct puncture during needle or catheter insertion, the introduction of air through the needle or catheter into the pleural cavity, and the inability of the ipsilateral lung to fully expand after drainage of a large volume of fluid, known as pneumothorax ex vacuo.5
Pneumothorax ex vacuo may be seen in patients with medical conditions such as endobronchial obstruction, pleural scarring from long-standing pleural effusion, and lung malignancy, all of which can impair the lung’s ability to expand after removal of a large volume of pleural fluid. It is believed that transient parenchymal pleural fistulae form if the lung cannot expand, causing air leakage into the pleural cavity.5,8,9 Pleural manometry to monitor changes in pleural pressure and elastance can decrease the rates of pneumothorax ex vacuo in patients with the above risk factors.5
WHEN IS RADIOGRAPHY INDICATED AFTER THORACENTESIS?
Current literature suggests that imaging to evaluate for postprocedural complications should be done if there is suspicion of a complication, if thoracentesis required multiple attempts, if the procedure caused aspiration of air, if the patient has advanced lung disease, if the patient is scheduled to undergo thoracic radiation, if the patient is on mechanical ventilation, and after therapeutic thoracentesis if a large volume of fluid is removed.1–10 Routine chest radiography after thoracentesis is not supported in the literature in the absence of these risk factors.
Some practitioners order chest imaging after therapeutic thoracentesis to assess for residual pleural fluid and for visualization of other abnormalities previously hidden by pleural effusion, rather than simply to exclude postprocedural pneumothorax. Alternatively, postprocedural bedside pleural ultrasonography with recording of images can be done to assess for complications and residual pleural fluid volume without exposing the patient to radiation.11
Needle decompression and chest tube insertion should be considered in patients with tension pneumothorax, large pneumothorax (distance from the chest wall to the visceral pleural line of at least 2 cm), mechanical ventilation, progressing pneumothorax, and symptoms.
KEY POINTS
- Pneumothorax is a rare complication of thoracentesis when performed by a skilled operator using ultrasonographic guidance.
- Mechanisms behind the occurrence of pneumothorax are direct lung puncture, introduction of air into the pleural cavity, and pneumothorax ex vacuo.
- In asymptomatic patients, pneumothorax after thoracentesis rarely requires intervention beyond supportive care and close observation.
- Factors such as multiple thoracentesis attempts, symptoms, clinical suspicion, air aspiration during thoracentesis, presence of previous lung disease, and removal of a large volume of fluid may require postprocedural lung imaging (eg, bedside ultrasonography, radiography).
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
- Ault MJ, Rosen BT, Scher J, Feinglass J, Barsuk JH. Thoracentesis outcomes: a 12-year experience. Thorax 2015; 70(2):127–132. doi:10.1136/thoraxjnl-2014-206114
- Hibbert RM, Atwell TD, Lekah A, et al. Safety of ultrasound-guided thoracentesis in patients with abnormal preprocedural coagulation parameters. Chest 2013; 144(2):456–463. doi:10.1378/chest.12-2374
- Barnes TW, Morgenthaler TI, Olson EJ, Hesley GK, Decker PA, Ryu JH. Sonographically guided thoracentesis and rate of pneumothorax. J Clin Ultrasound 2005; 33(9):442–446. doi:10.1002/jcu.20163
- Gordon CE, Feller-Kopman D, Balk EM, Smetana GW. Pneumothorax following thoracentesis: a systematic review and meta-analysis. Arch Intern Med 2010; 170(4):332–339. doi:10.1001/archinternmed.2009.548
- Heidecker J, Huggins JT, Sahn SA, Doelken P. Pathophysiology of pneumothorax following ultrasound-guided thoracentesis. Chest 2006; 130(4):1173–1184. doi:10.1016/S0012-3692(15)51155-0
- Brandstetter RD, Karetzky M, Rastogi R, Lolis JD. Pneumothorax after thoracentesis in chronic obstructive pulmonary disease. Heart Lung 1994; 23(1):67–70. pmid:8150647
- Doyle JJ, Hnatiuk OW, Torrington KG, Slade AR, Howard RS. Necessity of routine chest roentgenography after thoracentesis. Ann Intern Med 1996; 124(9):816–820. pmid:8610950
- Gervais DA, Petersein A, Lee MJ, Hahn PF, Saini S, Mueller PR. US-guided thoracentesis: requirement for postprocedure chest radiography in patients who receive mechanical ventilation versus patients who breathe spontaneously. Radiology 1997; 204(2):503–506. doi:10.1148/radiology.204.2.9240544
- Capizzi SA, Prakash UB. Chest roentgenography after outpatient thoracentesis. Mayo Clin Proc 1998; 73(10):948–950. doi:10.4065/73.10.948
- Alemán C, Alegre J, Armadans L, et al. The value of chest roentgenography in the diagnosis of pneumothorax after thoracentesis. Am J Med 1999; 107(4):340–343. pmid:10527035
- Lichtenstein D. Lung ultrasound in the critically ill. Curr Opin Crit Care 2014; 20(3):315–322. doi:10.1097/MCC.0000000000000096
Complete blood cell count
To the Editor: The review by May et al1 of 3 neglected numbers in the complete blood cell count (CBC) was a good reminder to look more closely at the results of the CBCs we often order in primary care. I was surprised to see no mention of the red cell distribution width in relation to another cardiovascular disorder—obstructive sleep apnea.2,3 I wonder if the authors would comment on this association?
- May JE, Marques MB, Reddy VVB, Gangaraju R. Three neglected numbers in the CBC: The RDW, MPV, and NRBC count. Cleve Clin J Med 2019; 86(3):167–172. doi:10.3949/ccjm.86a.18072
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192
To the Editor: The review by May et al1 of 3 neglected numbers in the complete blood cell count (CBC) was a good reminder to look more closely at the results of the CBCs we often order in primary care. I was surprised to see no mention of the red cell distribution width in relation to another cardiovascular disorder—obstructive sleep apnea.2,3 I wonder if the authors would comment on this association?
To the Editor: The review by May et al1 of 3 neglected numbers in the complete blood cell count (CBC) was a good reminder to look more closely at the results of the CBCs we often order in primary care. I was surprised to see no mention of the red cell distribution width in relation to another cardiovascular disorder—obstructive sleep apnea.2,3 I wonder if the authors would comment on this association?
- May JE, Marques MB, Reddy VVB, Gangaraju R. Three neglected numbers in the CBC: The RDW, MPV, and NRBC count. Cleve Clin J Med 2019; 86(3):167–172. doi:10.3949/ccjm.86a.18072
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192
- May JE, Marques MB, Reddy VVB, Gangaraju R. Three neglected numbers in the CBC: The RDW, MPV, and NRBC count. Cleve Clin J Med 2019; 86(3):167–172. doi:10.3949/ccjm.86a.18072
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192

In reply: Complete blood cell count
In Reply: We thank Dr. Homler for his question and for highlighting another important disease state, obstructive sleep apnea, in which a high red cell distribution width (RDW) has correlated with disease severity.1,2 The 2 retrospective studies he mentioned indicated that RDW is negatively correlated with metrics such as oxygen saturation, sleep time, and sleep quality. Interestingly, another retrospective study showed that RDW was significantly higher in patients with concurrent obstructive sleep apnea and cardiovascular disease than in patients with obstructive sleep apnea alone, suggesting that the presence of anisocytosis in obstructive sleep apnea may be due to its link to cardiovascular disease.3
Although we focused on cardiovascular disease in our review, RDW has also shown prognostic significance in many other disorders including ischemic stroke,4 pneumonia,5,6 chronic kidney disease,7 and gastrointestinal disorders.8 Collectively, these studies indicate that RDW may serve as a red flag for clinicians, raising concern for increased disease severity and potential adverse outcomes. However, further research is needed to determine if and how RDW monitoring should be used to prompt interventions to improve patient outcomes.
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192
- Sunnetcioglu A, Gunbatar H, Yildiz H. Red cell distribution width and uric acid in patients with obstructive sleep apnea. Clin Respir J 2018; 12(3):1046–1052. doi:10.1111/crj.12626
- Feng G-H, Li H-P, Li Q-L, Fu Y, Huang R-B. Red blood cell distribution width and ischaemic stroke. Stroke Vasc Neurol 2017; 2(3):172-175. doi:10.1136/svn-2017-000071
- Lee JH, Chung HJ, Kim K, et al. Red cell distribution width as a prognostic marker in patients with community-acquired pneumonia. Am J Emerg Med 2013; 31:72–79. doi:10.1016/j.ajem.2012.06.004
- Miranda SJ. Validity of red cell distribution width as a predictor of clinical outcomes in pediatric patients diagnosed with pneumonia [abstract]. Chest 2017; 152(4 suppl):A843. doi:10.1016/j.chest.2017.08.877
- Kor CT, Hsieh YP, Chang CC, Chiu PF. The prognostic value of interaction between mean corpuscular volume and red cell distribution width in mortality in chronic kidney disease. Sci Rep 2018; 8(1):11870. doi:10.1038/s41598-018-19881-2
- Goyal H, Lippi G, Gjymishka A, et al. Prognostic significance of red blood cell distribution width in gastrointestinal disorders. World J Gastroenterol 2017; 23(27):4879–4891. doi:10.3748/wjg.v23.i27.4879
In Reply: We thank Dr. Homler for his question and for highlighting another important disease state, obstructive sleep apnea, in which a high red cell distribution width (RDW) has correlated with disease severity.1,2 The 2 retrospective studies he mentioned indicated that RDW is negatively correlated with metrics such as oxygen saturation, sleep time, and sleep quality. Interestingly, another retrospective study showed that RDW was significantly higher in patients with concurrent obstructive sleep apnea and cardiovascular disease than in patients with obstructive sleep apnea alone, suggesting that the presence of anisocytosis in obstructive sleep apnea may be due to its link to cardiovascular disease.3
Although we focused on cardiovascular disease in our review, RDW has also shown prognostic significance in many other disorders including ischemic stroke,4 pneumonia,5,6 chronic kidney disease,7 and gastrointestinal disorders.8 Collectively, these studies indicate that RDW may serve as a red flag for clinicians, raising concern for increased disease severity and potential adverse outcomes. However, further research is needed to determine if and how RDW monitoring should be used to prompt interventions to improve patient outcomes.
In Reply: We thank Dr. Homler for his question and for highlighting another important disease state, obstructive sleep apnea, in which a high red cell distribution width (RDW) has correlated with disease severity.1,2 The 2 retrospective studies he mentioned indicated that RDW is negatively correlated with metrics such as oxygen saturation, sleep time, and sleep quality. Interestingly, another retrospective study showed that RDW was significantly higher in patients with concurrent obstructive sleep apnea and cardiovascular disease than in patients with obstructive sleep apnea alone, suggesting that the presence of anisocytosis in obstructive sleep apnea may be due to its link to cardiovascular disease.3
Although we focused on cardiovascular disease in our review, RDW has also shown prognostic significance in many other disorders including ischemic stroke,4 pneumonia,5,6 chronic kidney disease,7 and gastrointestinal disorders.8 Collectively, these studies indicate that RDW may serve as a red flag for clinicians, raising concern for increased disease severity and potential adverse outcomes. However, further research is needed to determine if and how RDW monitoring should be used to prompt interventions to improve patient outcomes.
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192
- Sunnetcioglu A, Gunbatar H, Yildiz H. Red cell distribution width and uric acid in patients with obstructive sleep apnea. Clin Respir J 2018; 12(3):1046–1052. doi:10.1111/crj.12626
- Feng G-H, Li H-P, Li Q-L, Fu Y, Huang R-B. Red blood cell distribution width and ischaemic stroke. Stroke Vasc Neurol 2017; 2(3):172-175. doi:10.1136/svn-2017-000071
- Lee JH, Chung HJ, Kim K, et al. Red cell distribution width as a prognostic marker in patients with community-acquired pneumonia. Am J Emerg Med 2013; 31:72–79. doi:10.1016/j.ajem.2012.06.004
- Miranda SJ. Validity of red cell distribution width as a predictor of clinical outcomes in pediatric patients diagnosed with pneumonia [abstract]. Chest 2017; 152(4 suppl):A843. doi:10.1016/j.chest.2017.08.877
- Kor CT, Hsieh YP, Chang CC, Chiu PF. The prognostic value of interaction between mean corpuscular volume and red cell distribution width in mortality in chronic kidney disease. Sci Rep 2018; 8(1):11870. doi:10.1038/s41598-018-19881-2
- Goyal H, Lippi G, Gjymishka A, et al. Prognostic significance of red blood cell distribution width in gastrointestinal disorders. World J Gastroenterol 2017; 23(27):4879–4891. doi:10.3748/wjg.v23.i27.4879
- Sökücü SN, Karasulu L, Dalar L, Seyhan EC, Altın S. Can red blood cell distribution width predict severity of obstructive sleep apnea syndrome? J Clin Sleep Med 2012; 8(5):521–525. doi:10.5664/jcsm.2146
- Yousef AM, Alkhiary W. The severity of obstructive sleep apnea syndrome is related to red cell distribution width and hematocrit values. J Sleep Disord Ther 2015; 4(2):1000192. doi:10.4172/2167-0277.1000192
- Sunnetcioglu A, Gunbatar H, Yildiz H. Red cell distribution width and uric acid in patients with obstructive sleep apnea. Clin Respir J 2018; 12(3):1046–1052. doi:10.1111/crj.12626
- Feng G-H, Li H-P, Li Q-L, Fu Y, Huang R-B. Red blood cell distribution width and ischaemic stroke. Stroke Vasc Neurol 2017; 2(3):172-175. doi:10.1136/svn-2017-000071
- Lee JH, Chung HJ, Kim K, et al. Red cell distribution width as a prognostic marker in patients with community-acquired pneumonia. Am J Emerg Med 2013; 31:72–79. doi:10.1016/j.ajem.2012.06.004
- Miranda SJ. Validity of red cell distribution width as a predictor of clinical outcomes in pediatric patients diagnosed with pneumonia [abstract]. Chest 2017; 152(4 suppl):A843. doi:10.1016/j.chest.2017.08.877
- Kor CT, Hsieh YP, Chang CC, Chiu PF. The prognostic value of interaction between mean corpuscular volume and red cell distribution width in mortality in chronic kidney disease. Sci Rep 2018; 8(1):11870. doi:10.1038/s41598-018-19881-2
- Goyal H, Lippi G, Gjymishka A, et al. Prognostic significance of red blood cell distribution width in gastrointestinal disorders. World J Gastroenterol 2017; 23(27):4879–4891. doi:10.3748/wjg.v23.i27.4879

Obesity doesn’t hamper flu vaccine response in pregnancy
LJUBLJANA, SLOVENIA – ; indeed, it might actually improve their seroconversion rate, Michelle Clarke reported at the annual meeting of the European Society for Paediatric Infectious Diseases.

She presented a prospective cohort study of 90 women vaccinated against influenza during pregnancy, 24 of whom had a BMI of 30 kg/m2 or more. The impetus for the study was the investigators’ understanding that influenza in pregnancy carries an increased risk of severe complications, obesity is a known risk factor for more severe episodes of influenza, and vaccine responses could potentially be adversely affected by obesity, either because of the associated inflammatory state and altered cytokine profile or inadequate vaccine delivery via the intramuscular route. Yet the impact of obesity on vaccine responses in pregnancy has been unclear.
Blood samples obtained before and 1 month after vaccination showed similarly high-titer postvaccination seropositivity rates against influenza B, H3N2, and H1N1 regardless of the women’s weight status. Indeed, the seropositivity rate against all three influenza viruses was higher in the obese subgroup, by a margin of 92%-74%. Also, postvaccination geometric mean antibody titers were significantly higher in the obese group. Particularly impressive was the difference in H1N1 seroconversion, defined as a fourfold increase in titer 28 days after vaccination: 79% versus 55%, noted Ms. Clarke of the University of Adelaide.
Of note, influenza vaccination in the first trimester resulted in a significantly lower seropositive antibody rate than vaccination in the second or third trimesters. The implication is that gestational age at vaccination, regardless of BMI, may be an important determinant of optimal vaccine protection for mothers and their newborns. However, this tentative conclusion requires confirmation in an independent larger sample, because the patient numbers in the study were small: Seropositive antibodies to all three vaccine antigens were documented in just 7 of 12 women (58%) vaccinated in the first trimester, compared with 47 of 53 (89%) vaccinated in the second trimester and 18 of 25 (72%) in the third.
Ms. Clarke reported having no financial conflicts regarding the study, which was supported by the Women’s and Children’s Hospital Research Foundation.
LJUBLJANA, SLOVENIA – ; indeed, it might actually improve their seroconversion rate, Michelle Clarke reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
She presented a prospective cohort study of 90 women vaccinated against influenza during pregnancy, 24 of whom had a BMI of 30 kg/m2 or more. The impetus for the study was the investigators’ understanding that influenza in pregnancy carries an increased risk of severe complications, obesity is a known risk factor for more severe episodes of influenza, and vaccine responses could potentially be adversely affected by obesity, either because of the associated inflammatory state and altered cytokine profile or inadequate vaccine delivery via the intramuscular route. Yet the impact of obesity on vaccine responses in pregnancy has been unclear.
Blood samples obtained before and 1 month after vaccination showed similarly high-titer postvaccination seropositivity rates against influenza B, H3N2, and H1N1 regardless of the women’s weight status. Indeed, the seropositivity rate against all three influenza viruses was higher in the obese subgroup, by a margin of 92%-74%. Also, postvaccination geometric mean antibody titers were significantly higher in the obese group. Particularly impressive was the difference in H1N1 seroconversion, defined as a fourfold increase in titer 28 days after vaccination: 79% versus 55%, noted Ms. Clarke of the University of Adelaide.
Of note, influenza vaccination in the first trimester resulted in a significantly lower seropositive antibody rate than vaccination in the second or third trimesters. The implication is that gestational age at vaccination, regardless of BMI, may be an important determinant of optimal vaccine protection for mothers and their newborns. However, this tentative conclusion requires confirmation in an independent larger sample, because the patient numbers in the study were small: Seropositive antibodies to all three vaccine antigens were documented in just 7 of 12 women (58%) vaccinated in the first trimester, compared with 47 of 53 (89%) vaccinated in the second trimester and 18 of 25 (72%) in the third.
Ms. Clarke reported having no financial conflicts regarding the study, which was supported by the Women’s and Children’s Hospital Research Foundation.
LJUBLJANA, SLOVENIA – ; indeed, it might actually improve their seroconversion rate, Michelle Clarke reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
She presented a prospective cohort study of 90 women vaccinated against influenza during pregnancy, 24 of whom had a BMI of 30 kg/m2 or more. The impetus for the study was the investigators’ understanding that influenza in pregnancy carries an increased risk of severe complications, obesity is a known risk factor for more severe episodes of influenza, and vaccine responses could potentially be adversely affected by obesity, either because of the associated inflammatory state and altered cytokine profile or inadequate vaccine delivery via the intramuscular route. Yet the impact of obesity on vaccine responses in pregnancy has been unclear.
Blood samples obtained before and 1 month after vaccination showed similarly high-titer postvaccination seropositivity rates against influenza B, H3N2, and H1N1 regardless of the women’s weight status. Indeed, the seropositivity rate against all three influenza viruses was higher in the obese subgroup, by a margin of 92%-74%. Also, postvaccination geometric mean antibody titers were significantly higher in the obese group. Particularly impressive was the difference in H1N1 seroconversion, defined as a fourfold increase in titer 28 days after vaccination: 79% versus 55%, noted Ms. Clarke of the University of Adelaide.
Of note, influenza vaccination in the first trimester resulted in a significantly lower seropositive antibody rate than vaccination in the second or third trimesters. The implication is that gestational age at vaccination, regardless of BMI, may be an important determinant of optimal vaccine protection for mothers and their newborns. However, this tentative conclusion requires confirmation in an independent larger sample, because the patient numbers in the study were small: Seropositive antibodies to all three vaccine antigens were documented in just 7 of 12 women (58%) vaccinated in the first trimester, compared with 47 of 53 (89%) vaccinated in the second trimester and 18 of 25 (72%) in the third.
Ms. Clarke reported having no financial conflicts regarding the study, which was supported by the Women’s and Children’s Hospital Research Foundation.
REPORTING FROM ESPID 2019
Key clinical point: High BMI doesn’t impair influenza vaccine responses in pregnant women.
Major finding: Protective antibody levels against all three vaccine antigens were documented 1 month post vaccination in 92% of the obese and 74% of the nonobese mothers.
Study details: This was a prospective observational study of 90 women vaccinated against influenza during pregnancy, 24 of whom were obese.
Disclosures: The study was supported by the University of Adelaide Women’s and Children’s Hospital Research Foundation.
Warfarin found to increase adverse outcomes among patients with IPF
DALLAS – Warfarin appears to increase the risk of lung transplant or death for patients with fibrotic lung disease who need anticoagulation therapy, Christopher King, MD, said at the American Thoracic Society’s international conference.

Compared with direct oral anticoagulation (DOAC), warfarin doubled the risk of those outcomes, even after the researchers controlled for multiple morbidities that accompany the need for anticoagulation, said Dr. King, medical director of the transplant and advanced lung disease critical care program at Inova Fairfax (Va.) Hospital.
“The need for anticoagulation in patients with interstitial lung disease is already associated with an increased risk of death or transplant,” he said. Warfarin – but not oral anticoagulation – seems to increase that risk even more “no matter how you analyze it,” he said.
“We know now that fibrosis and coagulation are entwined, and there’s background epidemiologic data showing an increased incidence of venous thromboembolism and acute coronary syndrome in patients with pulmonary fibrosis. This suggests that a dysregulated coagulation cascade may play a role in the pathogenesis of fibrosis.”
The relationship has been explored for the last decade or so. Two recent meta-analyses came to similar conclusions.
In 2013, a 125-patient retrospective cohort study compared clinical characteristics and survival among patients with idiopathic pulmonary fibrosis (IPF) who received anticoagulant therapy with those who did not (Sarcoidosis Vasc Diffuse Lung Dis. 2013 Aug 1;30[2]:121-7). Those who got the treatment had worse survival outcomes at 1 and 3 years than did those who received no therapy (84% vs. 53% and 89% vs. 64%, respectively).
In 2016, a post hoc analysis of three placebo-controlled studies determined that any anticoagulant use independently increased the risk of death among patients with IPF, compared with nonuse: 15.6% vs 6.3% all-cause mortality (Eur Respir J. 2016. doi: 10.1183/13993003.02087-2015).
But these investigations didn’t parse out the types of anticoagulation. Direct oral anticoagulation (DOAC) is much more common now, however, and Dr. King and colleagues wanted to find out how warfarin and DOAC compared.
They retrospectively analyzed data from the Pulmonary Fibrosis Foundation’s database and compared the risk of lung transplant and death for patients on anticoagulation or no anticoagulation and for those receiving DOACs versus warfarin versus no anticoagulation.
The study comprised 1,918 patients, 91% of whom were not on anticoagulation therapy. The remaining 164 were either taking DOAC (n = 83) or warfarin (n = 81). Both of these groups were significantly older than those not on anticoagulation (70 vs. 67 years). As expected , they were significantly more likely to have cardiac arrhythmias, heart failure, or pulmonary embolism or deep vein thrombosis and significantly more likely to be on immunosuppressant therapy or steroids. Their diffusing capacity of lung for carbon dioxide was also significantly lower.
There were no significant lung disease–related differences in anticoagulation therapy, other than a trend toward more use among those with connective tissue disease–associated interstitial lung disease.
Over 2 years, the entire cohort experienced 110 deaths (5.7%), 52 transplants (2.7%), and 29 withdrawals (1.5%). Among patients with IPF, there were 80 deaths (6.7%), 43 transplants (3.6%) and 20 withdrawals (1.7%).
In an unadjusted analysis, anticoagulation more than doubled the risk of an event, compared with no anticoagulation (hazard ratio, 2.4). This was slightly attenuated, but still significant, in a multivariate model that controlled for age, gender, oxygen use, gastroesophageal reflux disease, obstructive sleep apnea, arrhythmia, cancer, heart failure, obesity, venous thromboembolism, and antifibrotics (HR, 1.88).
A second whole-cohort analysis looked at the survival ratios for both warfarin and DOAC, compared with no treatment. In the fully adjusted model, warfarin was associated with a significantly increased risk HR (2.28) but DOAC was not.
The investigators then examined risk in only patients with lung disease. Among those with IPF, the fully adjusted model showed that warfarin nearly tripled the risk of transplant or death (HR, 2.8), while DOAC had no significant effect.
The reason for this association remains unclear, Dr. King said. “Renal failure may be a big reason patients get warfarin instead of DOAC. It’s difficult to say whether these patients were frail or prone to bleeding. Even something like the care team not being as up to date with treatment could be affecting the numbers. And is it the direct effect of warfarin on fibrotic lung disease? Or maybe DOAC has some beneficial effect on pulmonary fibrosis? We don’t know.
“But what we can take away from this is that warfarin is associated with worse outcomes than DOAC in patients with IPF. It seems reasonable to use DOAC over warfarin if there’s no specific contraindication to DOAC. If you have a patient with pulmonary thrombosis who has indications for anticoagulation I would use DOAC, based on the evidence that we now have available.”
Dr. King had no disclosures.
DALLAS – Warfarin appears to increase the risk of lung transplant or death for patients with fibrotic lung disease who need anticoagulation therapy, Christopher King, MD, said at the American Thoracic Society’s international conference.
Compared with direct oral anticoagulation (DOAC), warfarin doubled the risk of those outcomes, even after the researchers controlled for multiple morbidities that accompany the need for anticoagulation, said Dr. King, medical director of the transplant and advanced lung disease critical care program at Inova Fairfax (Va.) Hospital.
“The need for anticoagulation in patients with interstitial lung disease is already associated with an increased risk of death or transplant,” he said. Warfarin – but not oral anticoagulation – seems to increase that risk even more “no matter how you analyze it,” he said.
“We know now that fibrosis and coagulation are entwined, and there’s background epidemiologic data showing an increased incidence of venous thromboembolism and acute coronary syndrome in patients with pulmonary fibrosis. This suggests that a dysregulated coagulation cascade may play a role in the pathogenesis of fibrosis.”
The relationship has been explored for the last decade or so. Two recent meta-analyses came to similar conclusions.
In 2013, a 125-patient retrospective cohort study compared clinical characteristics and survival among patients with idiopathic pulmonary fibrosis (IPF) who received anticoagulant therapy with those who did not (Sarcoidosis Vasc Diffuse Lung Dis. 2013 Aug 1;30[2]:121-7). Those who got the treatment had worse survival outcomes at 1 and 3 years than did those who received no therapy (84% vs. 53% and 89% vs. 64%, respectively).
In 2016, a post hoc analysis of three placebo-controlled studies determined that any anticoagulant use independently increased the risk of death among patients with IPF, compared with nonuse: 15.6% vs 6.3% all-cause mortality (Eur Respir J. 2016. doi: 10.1183/13993003.02087-2015).
But these investigations didn’t parse out the types of anticoagulation. Direct oral anticoagulation (DOAC) is much more common now, however, and Dr. King and colleagues wanted to find out how warfarin and DOAC compared.
They retrospectively analyzed data from the Pulmonary Fibrosis Foundation’s database and compared the risk of lung transplant and death for patients on anticoagulation or no anticoagulation and for those receiving DOACs versus warfarin versus no anticoagulation.
The study comprised 1,918 patients, 91% of whom were not on anticoagulation therapy. The remaining 164 were either taking DOAC (n = 83) or warfarin (n = 81). Both of these groups were significantly older than those not on anticoagulation (70 vs. 67 years). As expected , they were significantly more likely to have cardiac arrhythmias, heart failure, or pulmonary embolism or deep vein thrombosis and significantly more likely to be on immunosuppressant therapy or steroids. Their diffusing capacity of lung for carbon dioxide was also significantly lower.
There were no significant lung disease–related differences in anticoagulation therapy, other than a trend toward more use among those with connective tissue disease–associated interstitial lung disease.
Over 2 years, the entire cohort experienced 110 deaths (5.7%), 52 transplants (2.7%), and 29 withdrawals (1.5%). Among patients with IPF, there were 80 deaths (6.7%), 43 transplants (3.6%) and 20 withdrawals (1.7%).
In an unadjusted analysis, anticoagulation more than doubled the risk of an event, compared with no anticoagulation (hazard ratio, 2.4). This was slightly attenuated, but still significant, in a multivariate model that controlled for age, gender, oxygen use, gastroesophageal reflux disease, obstructive sleep apnea, arrhythmia, cancer, heart failure, obesity, venous thromboembolism, and antifibrotics (HR, 1.88).
A second whole-cohort analysis looked at the survival ratios for both warfarin and DOAC, compared with no treatment. In the fully adjusted model, warfarin was associated with a significantly increased risk HR (2.28) but DOAC was not.
The investigators then examined risk in only patients with lung disease. Among those with IPF, the fully adjusted model showed that warfarin nearly tripled the risk of transplant or death (HR, 2.8), while DOAC had no significant effect.
The reason for this association remains unclear, Dr. King said. “Renal failure may be a big reason patients get warfarin instead of DOAC. It’s difficult to say whether these patients were frail or prone to bleeding. Even something like the care team not being as up to date with treatment could be affecting the numbers. And is it the direct effect of warfarin on fibrotic lung disease? Or maybe DOAC has some beneficial effect on pulmonary fibrosis? We don’t know.
“But what we can take away from this is that warfarin is associated with worse outcomes than DOAC in patients with IPF. It seems reasonable to use DOAC over warfarin if there’s no specific contraindication to DOAC. If you have a patient with pulmonary thrombosis who has indications for anticoagulation I would use DOAC, based on the evidence that we now have available.”
Dr. King had no disclosures.
DALLAS – Warfarin appears to increase the risk of lung transplant or death for patients with fibrotic lung disease who need anticoagulation therapy, Christopher King, MD, said at the American Thoracic Society’s international conference.
Compared with direct oral anticoagulation (DOAC), warfarin doubled the risk of those outcomes, even after the researchers controlled for multiple morbidities that accompany the need for anticoagulation, said Dr. King, medical director of the transplant and advanced lung disease critical care program at Inova Fairfax (Va.) Hospital.
“The need for anticoagulation in patients with interstitial lung disease is already associated with an increased risk of death or transplant,” he said. Warfarin – but not oral anticoagulation – seems to increase that risk even more “no matter how you analyze it,” he said.
“We know now that fibrosis and coagulation are entwined, and there’s background epidemiologic data showing an increased incidence of venous thromboembolism and acute coronary syndrome in patients with pulmonary fibrosis. This suggests that a dysregulated coagulation cascade may play a role in the pathogenesis of fibrosis.”
The relationship has been explored for the last decade or so. Two recent meta-analyses came to similar conclusions.
In 2013, a 125-patient retrospective cohort study compared clinical characteristics and survival among patients with idiopathic pulmonary fibrosis (IPF) who received anticoagulant therapy with those who did not (Sarcoidosis Vasc Diffuse Lung Dis. 2013 Aug 1;30[2]:121-7). Those who got the treatment had worse survival outcomes at 1 and 3 years than did those who received no therapy (84% vs. 53% and 89% vs. 64%, respectively).
In 2016, a post hoc analysis of three placebo-controlled studies determined that any anticoagulant use independently increased the risk of death among patients with IPF, compared with nonuse: 15.6% vs 6.3% all-cause mortality (Eur Respir J. 2016. doi: 10.1183/13993003.02087-2015).
But these investigations didn’t parse out the types of anticoagulation. Direct oral anticoagulation (DOAC) is much more common now, however, and Dr. King and colleagues wanted to find out how warfarin and DOAC compared.
They retrospectively analyzed data from the Pulmonary Fibrosis Foundation’s database and compared the risk of lung transplant and death for patients on anticoagulation or no anticoagulation and for those receiving DOACs versus warfarin versus no anticoagulation.
The study comprised 1,918 patients, 91% of whom were not on anticoagulation therapy. The remaining 164 were either taking DOAC (n = 83) or warfarin (n = 81). Both of these groups were significantly older than those not on anticoagulation (70 vs. 67 years). As expected , they were significantly more likely to have cardiac arrhythmias, heart failure, or pulmonary embolism or deep vein thrombosis and significantly more likely to be on immunosuppressant therapy or steroids. Their diffusing capacity of lung for carbon dioxide was also significantly lower.
There were no significant lung disease–related differences in anticoagulation therapy, other than a trend toward more use among those with connective tissue disease–associated interstitial lung disease.
Over 2 years, the entire cohort experienced 110 deaths (5.7%), 52 transplants (2.7%), and 29 withdrawals (1.5%). Among patients with IPF, there were 80 deaths (6.7%), 43 transplants (3.6%) and 20 withdrawals (1.7%).
In an unadjusted analysis, anticoagulation more than doubled the risk of an event, compared with no anticoagulation (hazard ratio, 2.4). This was slightly attenuated, but still significant, in a multivariate model that controlled for age, gender, oxygen use, gastroesophageal reflux disease, obstructive sleep apnea, arrhythmia, cancer, heart failure, obesity, venous thromboembolism, and antifibrotics (HR, 1.88).
A second whole-cohort analysis looked at the survival ratios for both warfarin and DOAC, compared with no treatment. In the fully adjusted model, warfarin was associated with a significantly increased risk HR (2.28) but DOAC was not.
The investigators then examined risk in only patients with lung disease. Among those with IPF, the fully adjusted model showed that warfarin nearly tripled the risk of transplant or death (HR, 2.8), while DOAC had no significant effect.
The reason for this association remains unclear, Dr. King said. “Renal failure may be a big reason patients get warfarin instead of DOAC. It’s difficult to say whether these patients were frail or prone to bleeding. Even something like the care team not being as up to date with treatment could be affecting the numbers. And is it the direct effect of warfarin on fibrotic lung disease? Or maybe DOAC has some beneficial effect on pulmonary fibrosis? We don’t know.
“But what we can take away from this is that warfarin is associated with worse outcomes than DOAC in patients with IPF. It seems reasonable to use DOAC over warfarin if there’s no specific contraindication to DOAC. If you have a patient with pulmonary thrombosis who has indications for anticoagulation I would use DOAC, based on the evidence that we now have available.”
Dr. King had no disclosures.
REPORTING FROM ATS 2019
Measles cases now at highest level since 1992
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.
With 971 cases of measles reported after just 5 months of 2019, the United States has hit another dubious milestone by surpassing the 963 cases reported in the preelimination year of 1994, according to the Centers for Disease Control and Prevention.
That leaves 1992, when there were 2,237 cases reported, as the next big obstacle on measles’ current path of distinction, the CDC data show. Only 312 cases were reported in 1993.
“Outbreaks in New York City and Rockland County, New York have continued for nearly 8 months. That loss would be a huge blow for the nation and erase the hard work done by all levels of public health,” the CDC said May 30.
The CDC defines measles elimination as “the absence of continuous disease transmission for 12 months or more in a specific geographic area” and notes that “measles is no longer endemic [constantly present] in the United States.”
“Measles is preventable and the way to end this outbreak is to ensure that all children and adults who can get vaccinated, do get vaccinated. Again, I want to reassure parents that vaccines are safe, they do not cause autism. The greater danger is the disease that vaccination prevents,” CDC director Robert Redfield, MD, said in a statement.