User login
Orodental issues often associated with facial port-wine stains
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
LAKE TAHOE, CALIF. – Several years ago, David H. Darrow, MD, DDS, began to notice a pattern in the conversation threads on websites dedicated to support for parents of children with facial port-wine stains (PWS).
, and that the alveolar ridge was lower on the side of the face that harbored the lesion. “Most importantly, parents were concerned that dentists were not touching their children because they were concerned about bleeding,” Dr. Darrow said at the annual meeting of the Society for Pediatric Dermatology. A search in the medical literature for port-wine stains and oral cavity changes, did not turn up much except for a few articles on bleeding. “One said that port-wine stains or capillary malformations rarely present major problems for the oral and maxillofacial surgeon. The other said that periodontal probing should not be done, as even gentle probing can result in uncontrolled bleeding,” he noted.
This prompted Dr. Darrow, who directs the Center for Hemangiomas and Vascular Birthmarks at the Eastern Virginia Medical School, Norfolk, and coinvestigators, Megan B. Dowling, MD, and Yueqin Zhao, PhD, to characterize manifestations of PWS in the oral cavity via an anonymous paired survey of volunteers with facial PWS and their dentists who were recruited from birthmarks.com and 10 collaborating physician practices (J Am Acad Dermatol 2012;67:687-93). Volunteers ranged in age from 1 to 62 years; mean age was 29 years. A total of 30 patients participated, and most (67%) were female.
The five most common oral manifestations reported by the patients were lip hyperplasia (53%), stained gingiva (47%), malocclusion (30%), bleeding gingiva (27%), and spacing between teeth (23%). Only 3% reported bleeding from dental procedures. When the researchers evaluated the orodental findings in the paired patient-physician responses, “most of the time there was good agreement between the patient and the dentist,” Dr. Darrow said. “The only one that fell out of agreement was lip hyperplasia. That’s probably because most dentists look right past the lips and into the oral cavity.”
When the researchers examined patients who had stained gingiva versus those who did not, they found that early-stage lesions demonstrated a reddish blush of the oral mucosa and gingiva, while late-stage lesions demonstrated increased blush of the oral tissues, as well as hyperplasia of the soft tissue or bone in the affected area. “Based on our review of the literature, bleeding of gums is rarely prolonged and dental procedures are safe,” Dr. Darrow said.
The findings are important, he continued, because capillary malformations of the oral cavity may result in periodontal disease associated with gingival overgrowth. The depth of the gingival pocket should be no more than 2-3 mm. “When you have areas of inflammation and deep-pocket formation, plaque and bacteria slowly erode the connection between the tooth and the soft tissue,” he explained. “At some point, that pocket becomes so deep that it reaches down into the bone in which the tooth is anchored. As that bone is eroded, the teeth loosen and begin to fall out. The goals of therapy are prevention of periodontal disease with meticulous oral hygiene.”
Soft tissue hyperplasia may be exacerbated by medications such as calcium channel blockers, cyclosporine, and phenytoin and phenobarbital, which are sometimes used by patients with Sturge-Weber syndrome, he said.
Dr. Darrow reported having no financial disclosures.
REPORTING FROM SPD 2018
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
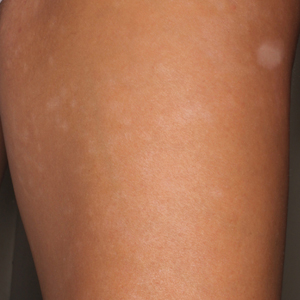
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.
Melasma Treatment With Oral Tranexamic Acid and a Novel Adjuvant Topical Therapy
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
To the Editor:
I read with interest the informative article by Sheu1 published online in Cutis in February 2018, which succinctly described the pharmacologic characteristics of tranexamic acid, a synthetic lysine derivative, and its mechanism of action in the management of melasma by mitigating UV radiation-induced melanogenesis and neovascularization by inhibiting plasminogen activation. Additionally, the author summarized a study in which oral tranexamic acid was used to successfully treat melasma patients. After 4 months of treatment, 90% of 561 patients treated at a single center in Singapore demonstrated improvement in melasma severity.2 Sheu1 also discussed daily oral doses of tranexamic acid (500-1500 mg) that demonstrated improvement in melasma patients and reviewed potential adverse events (eg, abdominal pain and bloating, deep venous thrombosis, pulmonary embolism) for which patients should be screened and counseled prior to initiating treatment.
Recently, another study showed oral tranexamic acid to be an effective treatment in women with moderate to severe melasma. An important observation by the investigators was that once the initial phase of their study--250 mg of oral tranexamic acid twice daily and sunscreen applied to the face each morning and every 2 hours during daylight hours for 3 months--concluded and a second phase during which all participants only applied sunscreen for an additional 3 months, those with severe melasma lost most of their improvement.3 An adjuvant topical treatment, such as tranexamic acid or an inhibitor of tyrosinase (hydroquinone), might improve the results; however, initiating therapy with a topical agent whose mode of action is directed toward other melasma etiologic factors, such as the increased expression of estrogen receptors and vascular endothelial growth factor in affected skin, might be more beneficial.4,5
I recently proposed a novel approach for melasma management that would be appropriate as an adjuvant topical therapy for patients concurrently being treated with oral tranexamic acid.6 The therapeutic intervention utilizes active agents that specifically affect etiologic factors in the pathogenesis of melasma--estrogen and angiogenesis--that previously have not been targeted topically. Indeed, the topical agent contains an antiestrogen--either a selective estrogen receptor modulator (eg, tamoxifen, raloxifene), aromatase inhibitor (eg, anastrozole, letrozole, exemestane), or a selective estrogen receptor degrader (eg, fulvestrant)--and a vascular endothelial growth factor inhibitor (eg, bevacizumab).6
In conclusion, the therapeutic armamentarium for managing patients with melasma includes topical agents, oral therapies, and physical modalities. Optimizing the approach to treating melasma patients should incorporate therapies that are specifically directed toward various etiologic factors of the condition. The concurrent use of a topical agent that contains an antiestrogen and an inhibitor of vascular endothelial growth factor in women with melasma who are being treated with oral tranexamic acid warrants further investigation to assess not only for enhanced but also sustained reduction in facial skin pigmentation.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.
- Sheu SL. Treatment of melasma using tranexamic acid: what's known and what's next. Cutis. 2018;101:E7-E8.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma: a retrospective study. J Am Acad Dermatol. 2016;75:385-392.
- Del Rosaria E, Forez-Pollack S, Zapata L Jr, et al. Randomized, placebo-controlled, double-blind study of oral tranexamic acid in the treatment of moderate-to-severe melasma. J Am Acad Dermatol. 2018;78:363-369.
- Jang YH, Lee JY, Kang HY, et al. Oestrogen and progesterone receptor expression in melasma: an immunohistochemical analysis. J Eur Acad Dermatol Venereol. 2010;24:1312-1316.
- Kim EH, Kim YC, Lee ES, et al. The vascular characteristics of melasma. J Dermatol Sci. 2007;46:111-116.
- Cohen PR. Melasma treatment: a novel approach using a topical agent that contains an anti-estrogen and a vascular endothelial growth factor inhibitor [published online February 3, 2017]. Med Hypotheses. 2017;101:1-5.
Pediatric vitiligo primarily affects those aged 10-17
LAKE TAHOE, CALIF. – Among children and adolescents, vitiligo appears to predominately affect nonwhite boys and girls between the ages of 10 and 17 years, results from a large cross-sectional analysis demonstrated.

During an interview at the annual meeting of the Society for Pediatric Dermatology, lead study author Jessica Haber, MD, said that, while it’s known vitiligo can have its onset in childhood, there have been no population-based analyses in the United States specific to children and adolescents with the condition.
“We wanted to examine disease burden in the U.S. specifically, because we have such a diverse population,” said Dr. Haber, a second-year resident in the department of dermatology at Northwell Health, New York.
For the study, she and her associates used IBM’s Explorys research analytics platform to conduct a cross-sectional analysis of more than 55 million unique patients across all census regions of the United States. There were 1,630 vitiligo cases identified from a total of 4,242,400 pediatric patients, for an overall standard prevalence of 0.04%, or 40.1 per 100,000 children and adolescents. The proportion of female and male patients with vitiligo was similar (49.1% and 50.9%, respectively), and nearly three-fourths (72.3%) were 10 years of age or older.
The researchers observed no significant difference in the prevalence of vitiligo between males and females (40.2 per 100,000 vs. 40 per 100,000, respectively). The standardized prevalence of vitiligo was greatest in pediatric patients who were of “other” races and ethnicities (including Asian, Hispanic, multiracial, and other; 69.1 per 100,000), followed by African Americans (51.5 per 100,000) and whites (37.9 per 100,000). There were too few vitiligo cases among biracial patients to determine standardized estimates, but the crude prevalence was greatest in this group (68.7 per 100,000).
Two factors could contribute to the increased prevalence of vitiligo observed in nonwhite children and adolescents, Dr. Haber said. One is selection bias.
“It has been reported that both children and adults with higher Fitzpatrick skin types tend to have increased morbidity of their vitiligo, so it may be a selection bias that these patients are seeking out treatment for their disease,” she said. (J Am Acad Dermatol. 2017;77[1]:1-13). That might explain some of our findings, as well.”
While the study findings “don’t necessarily change clinical practice, it is good for us to have a sense of the burden of disease in the pediatric patient population of vitiligo, and to be aware that this is a disease that predominately affects non-Caucasian children and adolescents,” Dr. Haber concluded.
She reported having no financial disclosures.
LAKE TAHOE, CALIF. – Among children and adolescents, vitiligo appears to predominately affect nonwhite boys and girls between the ages of 10 and 17 years, results from a large cross-sectional analysis demonstrated.
During an interview at the annual meeting of the Society for Pediatric Dermatology, lead study author Jessica Haber, MD, said that, while it’s known vitiligo can have its onset in childhood, there have been no population-based analyses in the United States specific to children and adolescents with the condition.
“We wanted to examine disease burden in the U.S. specifically, because we have such a diverse population,” said Dr. Haber, a second-year resident in the department of dermatology at Northwell Health, New York.
For the study, she and her associates used IBM’s Explorys research analytics platform to conduct a cross-sectional analysis of more than 55 million unique patients across all census regions of the United States. There were 1,630 vitiligo cases identified from a total of 4,242,400 pediatric patients, for an overall standard prevalence of 0.04%, or 40.1 per 100,000 children and adolescents. The proportion of female and male patients with vitiligo was similar (49.1% and 50.9%, respectively), and nearly three-fourths (72.3%) were 10 years of age or older.
The researchers observed no significant difference in the prevalence of vitiligo between males and females (40.2 per 100,000 vs. 40 per 100,000, respectively). The standardized prevalence of vitiligo was greatest in pediatric patients who were of “other” races and ethnicities (including Asian, Hispanic, multiracial, and other; 69.1 per 100,000), followed by African Americans (51.5 per 100,000) and whites (37.9 per 100,000). There were too few vitiligo cases among biracial patients to determine standardized estimates, but the crude prevalence was greatest in this group (68.7 per 100,000).
Two factors could contribute to the increased prevalence of vitiligo observed in nonwhite children and adolescents, Dr. Haber said. One is selection bias.
“It has been reported that both children and adults with higher Fitzpatrick skin types tend to have increased morbidity of their vitiligo, so it may be a selection bias that these patients are seeking out treatment for their disease,” she said. (J Am Acad Dermatol. 2017;77[1]:1-13). That might explain some of our findings, as well.”
While the study findings “don’t necessarily change clinical practice, it is good for us to have a sense of the burden of disease in the pediatric patient population of vitiligo, and to be aware that this is a disease that predominately affects non-Caucasian children and adolescents,” Dr. Haber concluded.
She reported having no financial disclosures.
LAKE TAHOE, CALIF. – Among children and adolescents, vitiligo appears to predominately affect nonwhite boys and girls between the ages of 10 and 17 years, results from a large cross-sectional analysis demonstrated.
During an interview at the annual meeting of the Society for Pediatric Dermatology, lead study author Jessica Haber, MD, said that, while it’s known vitiligo can have its onset in childhood, there have been no population-based analyses in the United States specific to children and adolescents with the condition.
“We wanted to examine disease burden in the U.S. specifically, because we have such a diverse population,” said Dr. Haber, a second-year resident in the department of dermatology at Northwell Health, New York.
For the study, she and her associates used IBM’s Explorys research analytics platform to conduct a cross-sectional analysis of more than 55 million unique patients across all census regions of the United States. There were 1,630 vitiligo cases identified from a total of 4,242,400 pediatric patients, for an overall standard prevalence of 0.04%, or 40.1 per 100,000 children and adolescents. The proportion of female and male patients with vitiligo was similar (49.1% and 50.9%, respectively), and nearly three-fourths (72.3%) were 10 years of age or older.
The researchers observed no significant difference in the prevalence of vitiligo between males and females (40.2 per 100,000 vs. 40 per 100,000, respectively). The standardized prevalence of vitiligo was greatest in pediatric patients who were of “other” races and ethnicities (including Asian, Hispanic, multiracial, and other; 69.1 per 100,000), followed by African Americans (51.5 per 100,000) and whites (37.9 per 100,000). There were too few vitiligo cases among biracial patients to determine standardized estimates, but the crude prevalence was greatest in this group (68.7 per 100,000).
Two factors could contribute to the increased prevalence of vitiligo observed in nonwhite children and adolescents, Dr. Haber said. One is selection bias.
“It has been reported that both children and adults with higher Fitzpatrick skin types tend to have increased morbidity of their vitiligo, so it may be a selection bias that these patients are seeking out treatment for their disease,” she said. (J Am Acad Dermatol. 2017;77[1]:1-13). That might explain some of our findings, as well.”
While the study findings “don’t necessarily change clinical practice, it is good for us to have a sense of the burden of disease in the pediatric patient population of vitiligo, and to be aware that this is a disease that predominately affects non-Caucasian children and adolescents,” Dr. Haber concluded.
She reported having no financial disclosures.
REPORTING FROM SPD 2018
Key clinical point: Vitiligo appears to predominately affect nonwhite boys and girls 10 years of age and older in the pediatric population.
Major finding: Of pediatric patients with vitiligo, 72.3% were 10 years of age or older.
Study details: A cross-sectional analysis of 1,630 vitiligo cases identified from a total of 4,242,400 pediatric patients.
Disclosures: Dr. Haber reported having no relevant financial disclosures.
Hypopigmentation on the Ear
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
The Diagnosis: Corticosteroid-Induced Hypopigmentation
This patient received several intralesional injections of triamcinolone acetonide once monthly for treatment of the keloid scar on the left ear at an outside institution. There was improvement in the size of the keloid over time. On physical examination during the most recent visit there was a prominent streak of hypopigmentation and atrophy near the corticosteroid injection site with extension to the postauricular region. There also was telangiectasia noted within the area of hypopigmentation. Intralesional triamcinolone injections were discontinued and the patient was advised to return for monitoring.
Intra-articular and intralesional corticosteroid injections frequently are used by clinicians. Cutaneous complications associated with these injections include atrophy, pigmentary changes, hypersensitivity reactions, flushing, cellulitis, and necrotizing fasciitis. Tendon rupture also has been reported.1
There are several case reports in the literature describing hypopigmentation and/or subcutaneous atrophy after intralesional or intra-articular corticosteroid injections. A variety of underlying conditions were treated including alopecia areata, keloids, rheumatoid arthritis, de Quervain tendonitis, and psoriasis.2-6 The lesions typically are described as linear rays of atrophy and hypopigmentation at or near the injection site, with some cases noting extension along lymph channels and proximal veins.4,6 There usually is no associated pruritus or pain.3 This phenomenon can be seen after single or multiple injections.4,6
Extension of hypopigmentation from the site of injection has been postulated to be due to venous or lymphatic uptake.2,4-6 The mechanism of hypopigmentation is not known. Biopsy of a previously described case showed intact melanocytes along the dermoepidermal junction.2 Biopsy from another case revealed a decrease in melanin staining, which suggests a decrease in number or activity of melanocytes.4 It was proposed that hypopigmentation was secondary to loss of melanocyte function instead of loss of melanocytes.2 Spontaneous improvement or resolution of the hypopigmentation were noted in some cases ranging from 1 month to 1 year after initial presentation, but the hypopigmentation also can be persistent.3-6
Hypopigmented sarcoidosis and hypopigmented mycosis fungoides, both often present on dark-skinned individuals, are included in the differential diagnosis. Hypopigmented sarcoidosis presents with hypopigmented macules or patches, some with central papules, and hypopigmented mycosis fungoides presents with hypopigmented patches or plaques with fine scale and onset often in childhood or adolescence.7,8 Morphea can present with an initial inflammatory stage that develops into a sclerotic firm plaque or nodule with hyperpigmentation or hypopigmentation.9 Vitiligo usually presents with depigmented macules or patches and depigmented hair within the lesion.10
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
- Brinks A, Koes BW, Volkers AC, et al. Adverse effects of extra-articular corticosteroid injections: a systematic review. BMC Musculoskelet Disord. 2010;11:206.
- Venkatesan P, Fangman WL. Linear hypopigmentation and cutaneous atrophy following intra-articular steroid injections for de Quervain's tendonitis. J Drugs Dermatol. 2009;8:492-493.
- Evans AV, McGibbon DH. Symmetrical hypopigmentation following triamcinolone injection for de Quervain's tenosynovitis. Clin Exp Dermatol. 2002;27:247-251.
- Friedman SJ, Butler DF, Pittelkow MR. Perilesional linear atrophy and hypopigmentation after intralesional corticosteroid therapy. report of two cases and review of the literature. J Am Acad Dermatol. 1988;19:537-541.
- van Vendeloo SN, Ettema HB. Skin depigmentation along lymph vessels of the lower leg following local corticosteroid injection for interdigital neuroma. Foot Ankle Surg. 2016;22:139-141.
- Kumar P, Adolph S. Hypopigmentation along subcutaneous veins following intrakeloid triamcinolone injection: a case report and review of literature. Burns. 1998;24:487-488.
- Elgart ML. Cutaneous sarcoidosis: definitions and types of lesions. Clin Dermatol. 1986;4:35-45.
- El-Shabrawi-Caelen L, Cerroni L, Medeiros LJ, et al. Hypopigmented mycosis fungoides: frequent expression of a CD8+ T-cell phenotype. Am J Surg Pathol. 2002;26:450-457.
- Marzano AV, Menni S, Parodi A, et al. Localized scleroderma in adults and children. clinical and laboratory investigations on 239 cases. Eur J Dermatol. 2003;13:171-176.
- Yaghoobi R, Omidian M, Bagherani N. Vitiligo: a review of the published work. J Dermatol. 2011;38:419-431.
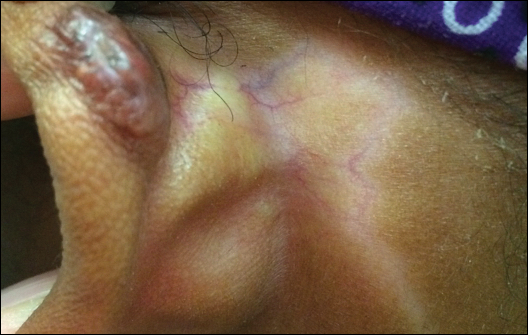
A 20-year-old black woman underwent multiple intralesional corticosteroid injections for treatment of a keloid on the superior aspect of the left helix and subsequently presented with a streak of atrophy and hypopigmentation in the postauricular region of unknown duration due to the lesion location.
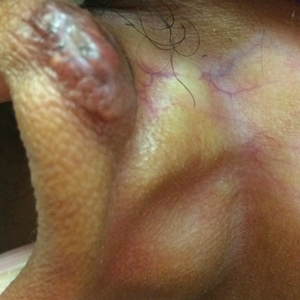
New analysis improves understanding of PHACE syndrome
LAKE TAHOE, CALIF. –
In addition, children with isolated S2 or parotid hemangiomas should be recognized as having lower risk for PHACE, and specifics of evaluation should be discussed with parents on a case-by-case basis.

Those are key findings from a retrospective cohort study presented by Colleen Cotton, MD, at the annual meeting of the Society for Pediatric Dermatology.
An association between large facial hemangiomas and multiple abnormalities was described as early as 1978, but it wasn’t until 1996 that researchers first proposed the term PHACE to describe the association (Arch Dermatol. 1996;132[3]:307-11). As the National Institutes of Health explain, “PHACE is an acronym for a neurocutaneous syndrome encompassing the following features: posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities.” Official diagnostic criteria for PHACE were not established until 2009 (Pediatrics. 2009;124[5]:1447-56) and were updated in 2016 (J Pediatr. 2016;178:24-33.e2).
“A multicenter, prospective, cohort study published in 2010 estimated the incidence of PHACE to be 31% in patients with large facial hemangiomas, while a retrospective study published in 2017 estimated the incidence to be as high as 58%,” Dr. Cotton, chief dermatology resident at the University of Arizona, Tucson, said in an interview in advance of the meeting. “With the current understanding of risk for PHACE, any child with a facial hemangioma of greater than or equal to 5 cm in diameter receives a full work-up for the syndrome. However, there has been anecdotal evidence that patients with certain subtypes of hemangiomas (such as parotid hemangiomas) may not carry this same risk.”
In what is believed to be the largest study of its kind, Dr. Cotton and her associates retrospectively analyzed data from 244 patients from 13 pediatric dermatology centers who were fully evaluated for PHACE between August 2009 and December 2014. The investigators also performed subgroup analyses on different hemangioma characteristics, including parotid hemangiomas and specific facial segments of involvement. All patients underwent magnetic resonance imaging/magnetic resonance angiography of the head and neck, and the researchers collected data on age at diagnosis; gender; patterns of hemangioma presentation, including location, size, and depth; diagnostic procedures and results; and type and number of associated anomalies. An expert reviewed photographs or diagrams to confirm facial segment locations.
Of the 244 patients, 34.7% met criteria for PHACE syndrome. On multivariate analysis, the following factors were found to be independently and significantly associated with a risk for PHACE: bilateral location (positive predictive value, 54.9%), S1 involvement (PPV, 49.5%), S3 involvement (PPV, 39.5%), and area greater than 25cm2 (PPV, 44.8%), with a P value less than .05 for all associations.
Risk of PHACE also increased with the number of locations involved, with a sharp increase observed at three or more locations (PPV, 65.5%; P less than .001). In patients with one unilateral segment involved, S2 and S3 carried a significantly lower risk (P less than .03). Parotid hemangiomas had a negative predictive value of 80.4% (P = .035).
“While we found that patients with parotid hemangiomas had a lower risk of PHACE, 10 patients with parotid hemangiomas did have PHACE, and 90% of those patients had cerebral arterial anomalies,” Dr. Cotton said. “However, only one of these patients had an isolated unilateral parotid hemangioma without other facial segment involvement. Additionally, two patients with isolated involvement of the midcheek below the eye [the S2 location, which was another low risk segment] also had PHACE, both of whom would have been missed without MRI/MRA [magnetic resonance angiography].”
She acknowledged certain limitations of the study, including its retrospective design. “Additionally, many of the very large hemangiomas were not measured in size, and so, estimated sizes needed to be used in calculating relationship of hemangioma size with risk of PHACE,” she said.
The study was funded in part by a grant from the Pediatric Dermatology Research Alliance.* Dr. Cotton reported having no relevant financial disclosures.
Correction, 7/20/18: An earlier version of this article misstated the name of the Pediatric Dermatology Research Alliance.
LAKE TAHOE, CALIF. –
In addition, children with isolated S2 or parotid hemangiomas should be recognized as having lower risk for PHACE, and specifics of evaluation should be discussed with parents on a case-by-case basis.
Those are key findings from a retrospective cohort study presented by Colleen Cotton, MD, at the annual meeting of the Society for Pediatric Dermatology.
An association between large facial hemangiomas and multiple abnormalities was described as early as 1978, but it wasn’t until 1996 that researchers first proposed the term PHACE to describe the association (Arch Dermatol. 1996;132[3]:307-11). As the National Institutes of Health explain, “PHACE is an acronym for a neurocutaneous syndrome encompassing the following features: posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities.” Official diagnostic criteria for PHACE were not established until 2009 (Pediatrics. 2009;124[5]:1447-56) and were updated in 2016 (J Pediatr. 2016;178:24-33.e2).
“A multicenter, prospective, cohort study published in 2010 estimated the incidence of PHACE to be 31% in patients with large facial hemangiomas, while a retrospective study published in 2017 estimated the incidence to be as high as 58%,” Dr. Cotton, chief dermatology resident at the University of Arizona, Tucson, said in an interview in advance of the meeting. “With the current understanding of risk for PHACE, any child with a facial hemangioma of greater than or equal to 5 cm in diameter receives a full work-up for the syndrome. However, there has been anecdotal evidence that patients with certain subtypes of hemangiomas (such as parotid hemangiomas) may not carry this same risk.”
In what is believed to be the largest study of its kind, Dr. Cotton and her associates retrospectively analyzed data from 244 patients from 13 pediatric dermatology centers who were fully evaluated for PHACE between August 2009 and December 2014. The investigators also performed subgroup analyses on different hemangioma characteristics, including parotid hemangiomas and specific facial segments of involvement. All patients underwent magnetic resonance imaging/magnetic resonance angiography of the head and neck, and the researchers collected data on age at diagnosis; gender; patterns of hemangioma presentation, including location, size, and depth; diagnostic procedures and results; and type and number of associated anomalies. An expert reviewed photographs or diagrams to confirm facial segment locations.
Of the 244 patients, 34.7% met criteria for PHACE syndrome. On multivariate analysis, the following factors were found to be independently and significantly associated with a risk for PHACE: bilateral location (positive predictive value, 54.9%), S1 involvement (PPV, 49.5%), S3 involvement (PPV, 39.5%), and area greater than 25cm2 (PPV, 44.8%), with a P value less than .05 for all associations.
Risk of PHACE also increased with the number of locations involved, with a sharp increase observed at three or more locations (PPV, 65.5%; P less than .001). In patients with one unilateral segment involved, S2 and S3 carried a significantly lower risk (P less than .03). Parotid hemangiomas had a negative predictive value of 80.4% (P = .035).
“While we found that patients with parotid hemangiomas had a lower risk of PHACE, 10 patients with parotid hemangiomas did have PHACE, and 90% of those patients had cerebral arterial anomalies,” Dr. Cotton said. “However, only one of these patients had an isolated unilateral parotid hemangioma without other facial segment involvement. Additionally, two patients with isolated involvement of the midcheek below the eye [the S2 location, which was another low risk segment] also had PHACE, both of whom would have been missed without MRI/MRA [magnetic resonance angiography].”
She acknowledged certain limitations of the study, including its retrospective design. “Additionally, many of the very large hemangiomas were not measured in size, and so, estimated sizes needed to be used in calculating relationship of hemangioma size with risk of PHACE,” she said.
The study was funded in part by a grant from the Pediatric Dermatology Research Alliance.* Dr. Cotton reported having no relevant financial disclosures.
Correction, 7/20/18: An earlier version of this article misstated the name of the Pediatric Dermatology Research Alliance.
LAKE TAHOE, CALIF. –
In addition, children with isolated S2 or parotid hemangiomas should be recognized as having lower risk for PHACE, and specifics of evaluation should be discussed with parents on a case-by-case basis.
Those are key findings from a retrospective cohort study presented by Colleen Cotton, MD, at the annual meeting of the Society for Pediatric Dermatology.
An association between large facial hemangiomas and multiple abnormalities was described as early as 1978, but it wasn’t until 1996 that researchers first proposed the term PHACE to describe the association (Arch Dermatol. 1996;132[3]:307-11). As the National Institutes of Health explain, “PHACE is an acronym for a neurocutaneous syndrome encompassing the following features: posterior fossa brain malformations, hemangiomas of the face, arterial anomalies, cardiac anomalies, and eye abnormalities.” Official diagnostic criteria for PHACE were not established until 2009 (Pediatrics. 2009;124[5]:1447-56) and were updated in 2016 (J Pediatr. 2016;178:24-33.e2).
“A multicenter, prospective, cohort study published in 2010 estimated the incidence of PHACE to be 31% in patients with large facial hemangiomas, while a retrospective study published in 2017 estimated the incidence to be as high as 58%,” Dr. Cotton, chief dermatology resident at the University of Arizona, Tucson, said in an interview in advance of the meeting. “With the current understanding of risk for PHACE, any child with a facial hemangioma of greater than or equal to 5 cm in diameter receives a full work-up for the syndrome. However, there has been anecdotal evidence that patients with certain subtypes of hemangiomas (such as parotid hemangiomas) may not carry this same risk.”
In what is believed to be the largest study of its kind, Dr. Cotton and her associates retrospectively analyzed data from 244 patients from 13 pediatric dermatology centers who were fully evaluated for PHACE between August 2009 and December 2014. The investigators also performed subgroup analyses on different hemangioma characteristics, including parotid hemangiomas and specific facial segments of involvement. All patients underwent magnetic resonance imaging/magnetic resonance angiography of the head and neck, and the researchers collected data on age at diagnosis; gender; patterns of hemangioma presentation, including location, size, and depth; diagnostic procedures and results; and type and number of associated anomalies. An expert reviewed photographs or diagrams to confirm facial segment locations.
Of the 244 patients, 34.7% met criteria for PHACE syndrome. On multivariate analysis, the following factors were found to be independently and significantly associated with a risk for PHACE: bilateral location (positive predictive value, 54.9%), S1 involvement (PPV, 49.5%), S3 involvement (PPV, 39.5%), and area greater than 25cm2 (PPV, 44.8%), with a P value less than .05 for all associations.
Risk of PHACE also increased with the number of locations involved, with a sharp increase observed at three or more locations (PPV, 65.5%; P less than .001). In patients with one unilateral segment involved, S2 and S3 carried a significantly lower risk (P less than .03). Parotid hemangiomas had a negative predictive value of 80.4% (P = .035).
“While we found that patients with parotid hemangiomas had a lower risk of PHACE, 10 patients with parotid hemangiomas did have PHACE, and 90% of those patients had cerebral arterial anomalies,” Dr. Cotton said. “However, only one of these patients had an isolated unilateral parotid hemangioma without other facial segment involvement. Additionally, two patients with isolated involvement of the midcheek below the eye [the S2 location, which was another low risk segment] also had PHACE, both of whom would have been missed without MRI/MRA [magnetic resonance angiography].”
She acknowledged certain limitations of the study, including its retrospective design. “Additionally, many of the very large hemangiomas were not measured in size, and so, estimated sizes needed to be used in calculating relationship of hemangioma size with risk of PHACE,” she said.
The study was funded in part by a grant from the Pediatric Dermatology Research Alliance.* Dr. Cotton reported having no relevant financial disclosures.
Correction, 7/20/18: An earlier version of this article misstated the name of the Pediatric Dermatology Research Alliance.
FROM SPD 2018
Key clinical point: Children with large, high-risk facial hemangiomas should be prioritized for PHACE syndrome work-up.
Major finding: On multivariate analysis, the following factors were found to be independently and significantly associated with a risk for PHACE: bilateral location (positive predictive value, 54.9%), S1 involvement (PPV, 49.5%), S3 involvement (PPV, 39.5%), and area greater than 25 cm2 (PPV, 44.8%; P less than .05 for all associations).
Study details: A retrospective evaluation of 244 patients from 13 pediatric dermatology who were fully evaluated for PHACE between August 2009 and December 2014.
Disclosures: The study was funded in part by a grant from the Pediatric Dermatology Research Association. Dr. Cotton reported having no financial disclosures.
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.
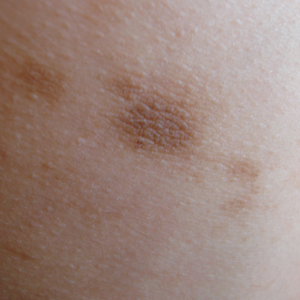
Imiquimod-Induced Hypopigmentation Following Treatment of Periungual Verruca Vulgaris
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.

 and the right elbow (B).")
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Practice Points
- Imiquimod commonly is used off label to treat viral and neoplastic processes.
- Clinicians should be aware of the potential for dyspigmentation or depigmentation as a side effect from treatment.
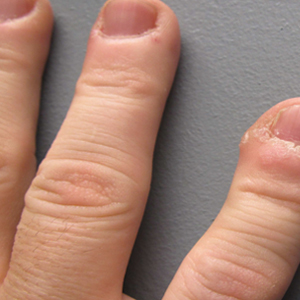
Beltlike Lichen Planus Pigmentosus Complicated With Focal Amyloidosis
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.

Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.

.")
.")
.")
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
Practice Points
- Lichen planus pigmentosus can present in a unique beltlike distribution pattern.
- Focal amyloidosis due to chronic friction and scratching cannot be excluded from the differential diagnosis.
Practice management pearls: Advice from seasoned doctors for residents looking to start a practice
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.

In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.
In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.
The notion that residency training falls short when it comes to preparing residents and doctors for starting their own practice is a common thread across the board, whether you’re just getting started or have been managing your own practice for years. I did a survey on LinkedIn and over 50 dermatology and plastic surgery colleagues generously provided their own personal insights and words of wisdom to help young doctors avoid common practice management problems.
I could not quote everyone, but here are some of the best tips that I received:
Choose your staff carefully – and invest in the right candidates
One of the biggest pieces of practice management advice that doctors had to offer was to hire the right employees from the beginning, even if that means spending a little more time in the hiring process. This will eliminate headaches and frustration later.
In his own practice in Palm Beach Gardens, Fla., Dr. Lickstein has chosen a stable group of staff members who are, “first and foremost, nice, compassionate, and mature,” he said. “They need to be able to relate to cosmetic and medical patients of all ages. My office manager screens them, and then we have potential candidates shadow us for at least a half-day in the office. Afterward, we seek feedback from the current staff. I also try and talk with the candidate for a while, because I’ve found that once you get them to loosen up, you can get an actual sense of how they really are.”
Along the same lines, Cincinnati plastic surgeon Alex Donath, MD, suggests incentivizing employees and giving them an active role in the hiring process. “Give everyone in the office a chance to meet new employee candidates,” he said, “as that will both give the employees a sense of involvement in the process and allow more opportunities to catch glimpses of poor interpersonal skills that could hurt your reputation.”
Many doctors stressed the importance of the interview process, detailed job descriptions, a 60-day trial period, and background checks prior to hiring. This advice goes along with the famous quote “Be slow to hire and quick to fire (in the first 60 days)” that I have seen in many business books.
Foster teamwork
Another important aspect of managing your practice is building a sense of teamwork and camaraderie among employees and other doctors. Sean Weiss, MD, a facial plastic surgeon in New Orleans, has a great team-building tip that he uses daily.
“I plan a daily morning huddle with my staff. During the huddle, we review the prior day’s performance, those patients that need following up on, and whether or not the prior day’s goals were met. We then review the patient list for the current day to identify patient needs. We look specifically for ways to improve efficiency and avoid slowing down the work flow. We also try to identify opportunities to cross-promote our offerings to increase awareness of our services. In about 10 minutes, the entire team becomes focused and ready for a productive day.”
For Lacey Elwyn, DO, making staff feel appreciated can be as simple as telling them thank you on a regular basis. “The success of a dermatology practice encompasses every staff member of the team,” Dr. Elwyn, a medical and cosmetic dermatologist in South Florida, said. “The physician should respect and value all staff members. Tell them when they are doing a great job and tell them that you appreciate them every day, but also let them know right away when something is wrong.”
Don’t forget about patient education
Janet Trowbridge, MD, PhD, who practices in Edmond, Wash., expressed a great point that not only do patients need to be educated about their medical or cosmetic concerns, but they also need to be educated about the way that health care works in general. “I would say that 50% of my time as a physician is spent educating patients not about their disease, but about how medicine works – or doesn’t work,” she said. “I am constantly amazed by how little the average person understands about how health care is delivered. I talk about copays, coinsurance, annual deductibles, and why their prescriptions are not being covered. Patients feel that the system has let them down.”
Play the dual role of doctor and businessperson
At the end of the day, if you are managing your own practice, you must be able to split your time and skill set between being a physician and being a businessperson. Having realized the importance of the business aspect of running a practice, Justin Bryant, DO, a plastic and reconstructive surgeon in Walled Lake, Mich., enrolled in a dual-degree program during medical school in order to obtain his MBA.
“That investment already has proven priceless, as I’ve helped attendings and colleagues with their practice in marketing, finance, technology, and simply in translating business terms and contracts with physicians,” he said. “Although I don’t think it’s necessary for all physicians to pursue an MBA, and it’s not the answer to every business problem in the field of medicine, when applied, it can be very powerful!”
Build and protect your online reputation
Now more than ever, it is imperative to build and protect your online reputation, as online reviews can make or break your business. For plastic surgeon Nirmal Nathan, MD, in Plantation, Fla., managing your reputation is one of the most important considerations when starting a practice. “I would tell residents to start early on reputation management,” he said. “Reviews are so important, even with patients referred by word of mouth. Good reputation management also allows you to quickly ramp up if you decide to move your practice location.”
A large portion of building your online reputation now as to do with what you post (and don’t post) on social media. For Haena Kim, MD, a facial plastic and reconstructive surgeon practicing in Walnut Creek, Calif., figuring out how you would like others to perceive you is the first step.
“In this day and age of social media,” she said, “it’s so hard not to feel the pressure to follow the crowd and be the loudest person out there, and it’s incredibly hard to be patient with your practice growth. It’s important to figure out how you want to present yourself and what you want patients to come away with.”
Sweat the small stuff
Seemingly small administrative and business-related tasks can quickly add up and create much larger problems if not addressed early on. Tito Vasquez, MD, who practices in Southport, Conn., summed this up with an excellent piece of advice to remember: “Sweat the small stuff now, so you don’t have to sweat over the big stuff later.”
In terms of the “small stuff” you’ll need to manage, Dr. Vasquez points to items such as learning local economics and politics, daily finances, office regulations, and documentation, investment and planning, internal and external marketing, and human resources. “While most of us would view this as mundane or at least secondary to the craft we learn,” he said, “it will actually take far greater importance to taking care of patients if you really want your business to succeed and thrive.”
Another essential aspect of business planning that may seem daunting or mundane to many doctors when first starting out is putting together the necessary training manuals to effectively run your practice. Robert Bader, MD, stressed the importance of creating manuals for the front office, back office, Material Safety Data Sheets, and Occupational Safety and Health Administration.
“This is the time, while you have some extra time, to take an active role in forming the foundation of your practice,” Dr. Bader of Deerfield Beach, Fla., said. “Set aside time every year to go over and make necessary changes to these manuals.”
Make decisions now that reflect long-term goals
When you start your practice, deciding on a location might seem like a secondary detail, but the fact of the matter is that location will ultimately play a large role in the future of your business and your life. Beverly Hills, Calif., plastic surgeon John Layke, DO, suggested “choosing where you would like to live, and then building a practice around that location. Being happy in the area you live will make a big difference,” he says. “No one will ultimately be happy making $1 million-plus per year if they are miserable living in the area. In the beginning, share office space with reputable people where you become ‘visible,’ then build the office of your dreams when you are ready.”
Summary
I was amazed at the number of responses that I received in response to this survey. It is my goal to help doctors mentor each other on these important issues so that we do not all have to recreate the wheel. Connect with me on LinkedIn if you want to participate in these surveys or if you want to see the results of them. I want to wish the residents who are graduating and going into their own practice the best of luck. My final advice is to reach out for help – it’s obvious that many people are willing to provide advice.
Dr. Baumann is a private practice dermatologist, researcher, author, and entrepreneur who practices in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote two textbooks: “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and “Cosmeceuticals and Cosmetic Ingredients,” (New York: McGraw-Hill, 2014), and a New York Times Best Sellers book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). Dr. Baumann has received funding for advisory boards and/or clinical research trials from Allergan, Evolus, Galderma, and Revance. She is the founder and CEO of Skin Type Solutions Franchise Systems. She is the author of the monthly “Cosmeceutical Critique” column in Dermatology News.