User login
Debate: Is MRD ready for prime time in multiple myeloma?
NEW YORK – Evidence of minimal residual disease (MRD) has been shown to be an important prognostic factor in several different hematologic malignancies, including acute and chronic myeloid leukemias, but its clinical utility in day-to-day practice in multiple myeloma is still uncertain.
At the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies, C. Ola Landgren, MD, PhD, chief of the myeloma service at Memorial Sloan Kettering Cancer Center in New York, and Paul G. Richardson, MD, clinical program leader and director of clinical research at the Jerome Lipper Myeloma Center at the Dana-Farber Cancer Institute in Boston, debated the question: “Is MRD ready for prime time?”
Yes: Dr. Landgren
“As we all know, with older drugs for myeloma, only a small proportion of patients reached a complete response, so there was really no reason to talk about MRD. But this belongs to the past: using the modern combination therapies, about 100% of our patients have a response, an overall response, and up to 80% of patients are reaching a complete response. So it’s really necessary, a logical step to go forward, to look at MRD,” Dr. Landgren said.
He cited evidence from two meta-analyses showing that MRD negativity is a strong predictor of clinical outcomes, including long-term survival.

“Our results show that MRD negativity is a strong predictor of clinical outcomes, supportive of MRD becoming a regulatory end point for drug approval in newly diagnosed multiple myeloma,” they wrote.
In a second meta-analysis, Nikhil Munshi, MD, from the Dana-Farber Cancer Institute, and his colleagues, reviewed PFS data from 14 studies with a total of 1,273 patients, and OS data from 12 studies with a total of 1,100 patients.
This second meta-analysis found that MRD negativity was associated with significantly better PFS (HR, 0.41; P less than .001), including among patients in studies looking specifically at complete response (CR) (HR, 0.44; P less than .001).
Munshi et al. also saw a significant benefit for MRD negativity among all patients in trials with OS as the endpoint (HR, 0.57; P less than .001) and in those focusing on patients with a CR (HR, 0.47; P less than .001).
They concluded that MRD-negative status after treatment for new newly diagnosed multiple myeloma is associated with long-term survival, and like Landgren et al. contended that their findings “provide quantitative evidence to support the integration of MRD assessment as an end point in clinical trials of multiple myeloma.”
Dr. Landgren noted that 2016 International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma, coauthored by both Dr. Landgren and Dr. Richardson, now incorporate MRD.
In addition, in the IFM/DFCI 2009 trial comparing induction therapy for patients with newly diagnosed multiple myeloma with or without autologous stem cell transplant after three cycles of lenalidomide, bortezomib, and dexamethasone, patients in each trial arm who were MRD negative had significantly better PFS than patients who were MRD positive after consolidation, regardless of assigned treatment, Dr. Landgren noted.
In the relapsed/refractory multiple myeloma setting, MRD negativity was associated with better PFS for patients in the POLLUX trial, whether subjects were assigned to receive daratumumab plus lenalidomide and dexamethasone, or len-dex alone.
“This raises an important question: Is MRD more important than treatment modality?” Dr. Landgren said.
“The debate is: Is MRD ready for prime time? And as I have shown you with all the data, the answer is ‘Yes’,” he concluded.
No: Dr. Richardson
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena. The question for me as a clinician in my clinic is: ‘Do I apply it to everyday practice?’ And I would simply suggest to you at this point we’re not ready for that, and we’re not ready for that for a variety of complex reasons,” Dr. Richardson said in his rebuttal.
He cited a definition of MRD offered by Simone Ferrero, MD, and his colleagues from the University of Turin (Italy) in 2011 in Hematological Oncology: “Any approach aimed at detecting and possibly quantifying residual tumor cells beyond the sensitivity level of routine imaging and laboratory techniques.”

“We recognize in hematologic malignancies in particular, and increasingly in myeloma, that achievement of complete clinical remission and assessing this needs a variety of different scenarios,” he said.
These scenarios may include establishing the full eradication of the neoplastic clone, determining the long-term persistence of quiescent or nonclonogenic or immunologically regulated tumor cells, or persistence of clonogenic cells capable of giving rise to a full clinical relapse within a number of years.
Myeloma specialists recognize that MRD is a real phenomenon, made more challenging by the “extraordinary” heterogeneity of myeloma, he said.
Determination of MRD using sensitive molecular techniques may allow analysis of treatments that induce a greater depth of response than others, and may also identify patients who are experiencing early relapse and will need further treatment, Dr. Richardson acknowledged.
“The question is, should it dictate what you and I do every afternoon in the clinic with a particular patient, for example, outside of a clinical trial?”
He noted that MRD is still a secondary endpoint in trials for acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, and chronic lymphocytic leukemia, although it has been accepted by the FDA as a primary endpoint to assess molecular responses to second-generation tyrosine kinase inhibitors.
MRD is also still a secondary endpoint in trials for therapies against follicular and mantle cell lymphomas as well.
“So my hypothesis, or suggestion to you this morning, is that whilst MRD clearly is a vital area of research – and I especially applaud Ola for being on the forefront of this, and I fully support all the points he made – I would just simply suggest to you that it’s less advanced than in leukemia and lymphoma, and we are currently at the point where MRD assessments are clearly secondary endpoints and an important research tool,” he said,
MRD remains a research tool in multiple myeloma because, despite the wealth of new therapies and combinations approved in just the past few years, “we’re not able to eradicate it completely, and cure remains in myeloma, frankly, evasive,” he added.
Immunotherapy, for example, is not the “mutationally agnostic” approach that clinicians had hoped for, with recent evidence suggesting that it cannot overcome every genetic variation that may give rise to multiple myeloma.
For MRD to become a useful clinical tool rather than a research/regulatory tool, standardization of testing methods will be needed. Flow cytometry until recently has been the mainstay for detecting MRD, but molecular strategies are currently being investigated, and rapid next-generation sequencing has the potential to become a gold standard, with its ability to identify and quantify all clonotypes in a sample.
“What’s critical is, therapeutic adjustment for what? What is the difference? For example, [if] one arm of a trial has 20% MRD positivity vs. 40% in another, what does that really mean for overall survival? These are enormous challenges that we still face,” Dr. Richardson said.
“I do think the lack of standardization broadly across the country is a challenge, and so with that in mind, I would simply suggest that it is not yet a standard of care in clinical practice, but may be,” he concluded.
Dr. Landgren disclosed serving as a consultant to AbbVie, Amgen, Bristol-Myers Squibb, Celgene and Janssen. Dr. Richardson disclosed consulting for Celgene and Takeda.
NEW YORK – Evidence of minimal residual disease (MRD) has been shown to be an important prognostic factor in several different hematologic malignancies, including acute and chronic myeloid leukemias, but its clinical utility in day-to-day practice in multiple myeloma is still uncertain.
At the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies, C. Ola Landgren, MD, PhD, chief of the myeloma service at Memorial Sloan Kettering Cancer Center in New York, and Paul G. Richardson, MD, clinical program leader and director of clinical research at the Jerome Lipper Myeloma Center at the Dana-Farber Cancer Institute in Boston, debated the question: “Is MRD ready for prime time?”
Yes: Dr. Landgren
“As we all know, with older drugs for myeloma, only a small proportion of patients reached a complete response, so there was really no reason to talk about MRD. But this belongs to the past: using the modern combination therapies, about 100% of our patients have a response, an overall response, and up to 80% of patients are reaching a complete response. So it’s really necessary, a logical step to go forward, to look at MRD,” Dr. Landgren said.
He cited evidence from two meta-analyses showing that MRD negativity is a strong predictor of clinical outcomes, including long-term survival.
“Our results show that MRD negativity is a strong predictor of clinical outcomes, supportive of MRD becoming a regulatory end point for drug approval in newly diagnosed multiple myeloma,” they wrote.
In a second meta-analysis, Nikhil Munshi, MD, from the Dana-Farber Cancer Institute, and his colleagues, reviewed PFS data from 14 studies with a total of 1,273 patients, and OS data from 12 studies with a total of 1,100 patients.
This second meta-analysis found that MRD negativity was associated with significantly better PFS (HR, 0.41; P less than .001), including among patients in studies looking specifically at complete response (CR) (HR, 0.44; P less than .001).
Munshi et al. also saw a significant benefit for MRD negativity among all patients in trials with OS as the endpoint (HR, 0.57; P less than .001) and in those focusing on patients with a CR (HR, 0.47; P less than .001).
They concluded that MRD-negative status after treatment for new newly diagnosed multiple myeloma is associated with long-term survival, and like Landgren et al. contended that their findings “provide quantitative evidence to support the integration of MRD assessment as an end point in clinical trials of multiple myeloma.”
Dr. Landgren noted that 2016 International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma, coauthored by both Dr. Landgren and Dr. Richardson, now incorporate MRD.
In addition, in the IFM/DFCI 2009 trial comparing induction therapy for patients with newly diagnosed multiple myeloma with or without autologous stem cell transplant after three cycles of lenalidomide, bortezomib, and dexamethasone, patients in each trial arm who were MRD negative had significantly better PFS than patients who were MRD positive after consolidation, regardless of assigned treatment, Dr. Landgren noted.
In the relapsed/refractory multiple myeloma setting, MRD negativity was associated with better PFS for patients in the POLLUX trial, whether subjects were assigned to receive daratumumab plus lenalidomide and dexamethasone, or len-dex alone.
“This raises an important question: Is MRD more important than treatment modality?” Dr. Landgren said.
“The debate is: Is MRD ready for prime time? And as I have shown you with all the data, the answer is ‘Yes’,” he concluded.
No: Dr. Richardson
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena. The question for me as a clinician in my clinic is: ‘Do I apply it to everyday practice?’ And I would simply suggest to you at this point we’re not ready for that, and we’re not ready for that for a variety of complex reasons,” Dr. Richardson said in his rebuttal.
He cited a definition of MRD offered by Simone Ferrero, MD, and his colleagues from the University of Turin (Italy) in 2011 in Hematological Oncology: “Any approach aimed at detecting and possibly quantifying residual tumor cells beyond the sensitivity level of routine imaging and laboratory techniques.”
“We recognize in hematologic malignancies in particular, and increasingly in myeloma, that achievement of complete clinical remission and assessing this needs a variety of different scenarios,” he said.
These scenarios may include establishing the full eradication of the neoplastic clone, determining the long-term persistence of quiescent or nonclonogenic or immunologically regulated tumor cells, or persistence of clonogenic cells capable of giving rise to a full clinical relapse within a number of years.
Myeloma specialists recognize that MRD is a real phenomenon, made more challenging by the “extraordinary” heterogeneity of myeloma, he said.
Determination of MRD using sensitive molecular techniques may allow analysis of treatments that induce a greater depth of response than others, and may also identify patients who are experiencing early relapse and will need further treatment, Dr. Richardson acknowledged.
“The question is, should it dictate what you and I do every afternoon in the clinic with a particular patient, for example, outside of a clinical trial?”
He noted that MRD is still a secondary endpoint in trials for acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, and chronic lymphocytic leukemia, although it has been accepted by the FDA as a primary endpoint to assess molecular responses to second-generation tyrosine kinase inhibitors.
MRD is also still a secondary endpoint in trials for therapies against follicular and mantle cell lymphomas as well.
“So my hypothesis, or suggestion to you this morning, is that whilst MRD clearly is a vital area of research – and I especially applaud Ola for being on the forefront of this, and I fully support all the points he made – I would just simply suggest to you that it’s less advanced than in leukemia and lymphoma, and we are currently at the point where MRD assessments are clearly secondary endpoints and an important research tool,” he said,
MRD remains a research tool in multiple myeloma because, despite the wealth of new therapies and combinations approved in just the past few years, “we’re not able to eradicate it completely, and cure remains in myeloma, frankly, evasive,” he added.
Immunotherapy, for example, is not the “mutationally agnostic” approach that clinicians had hoped for, with recent evidence suggesting that it cannot overcome every genetic variation that may give rise to multiple myeloma.
For MRD to become a useful clinical tool rather than a research/regulatory tool, standardization of testing methods will be needed. Flow cytometry until recently has been the mainstay for detecting MRD, but molecular strategies are currently being investigated, and rapid next-generation sequencing has the potential to become a gold standard, with its ability to identify and quantify all clonotypes in a sample.
“What’s critical is, therapeutic adjustment for what? What is the difference? For example, [if] one arm of a trial has 20% MRD positivity vs. 40% in another, what does that really mean for overall survival? These are enormous challenges that we still face,” Dr. Richardson said.
“I do think the lack of standardization broadly across the country is a challenge, and so with that in mind, I would simply suggest that it is not yet a standard of care in clinical practice, but may be,” he concluded.
Dr. Landgren disclosed serving as a consultant to AbbVie, Amgen, Bristol-Myers Squibb, Celgene and Janssen. Dr. Richardson disclosed consulting for Celgene and Takeda.
NEW YORK – Evidence of minimal residual disease (MRD) has been shown to be an important prognostic factor in several different hematologic malignancies, including acute and chronic myeloid leukemias, but its clinical utility in day-to-day practice in multiple myeloma is still uncertain.
At the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies, C. Ola Landgren, MD, PhD, chief of the myeloma service at Memorial Sloan Kettering Cancer Center in New York, and Paul G. Richardson, MD, clinical program leader and director of clinical research at the Jerome Lipper Myeloma Center at the Dana-Farber Cancer Institute in Boston, debated the question: “Is MRD ready for prime time?”
Yes: Dr. Landgren
“As we all know, with older drugs for myeloma, only a small proportion of patients reached a complete response, so there was really no reason to talk about MRD. But this belongs to the past: using the modern combination therapies, about 100% of our patients have a response, an overall response, and up to 80% of patients are reaching a complete response. So it’s really necessary, a logical step to go forward, to look at MRD,” Dr. Landgren said.
He cited evidence from two meta-analyses showing that MRD negativity is a strong predictor of clinical outcomes, including long-term survival.
“Our results show that MRD negativity is a strong predictor of clinical outcomes, supportive of MRD becoming a regulatory end point for drug approval in newly diagnosed multiple myeloma,” they wrote.
In a second meta-analysis, Nikhil Munshi, MD, from the Dana-Farber Cancer Institute, and his colleagues, reviewed PFS data from 14 studies with a total of 1,273 patients, and OS data from 12 studies with a total of 1,100 patients.
This second meta-analysis found that MRD negativity was associated with significantly better PFS (HR, 0.41; P less than .001), including among patients in studies looking specifically at complete response (CR) (HR, 0.44; P less than .001).
Munshi et al. also saw a significant benefit for MRD negativity among all patients in trials with OS as the endpoint (HR, 0.57; P less than .001) and in those focusing on patients with a CR (HR, 0.47; P less than .001).
They concluded that MRD-negative status after treatment for new newly diagnosed multiple myeloma is associated with long-term survival, and like Landgren et al. contended that their findings “provide quantitative evidence to support the integration of MRD assessment as an end point in clinical trials of multiple myeloma.”
Dr. Landgren noted that 2016 International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma, coauthored by both Dr. Landgren and Dr. Richardson, now incorporate MRD.
In addition, in the IFM/DFCI 2009 trial comparing induction therapy for patients with newly diagnosed multiple myeloma with or without autologous stem cell transplant after three cycles of lenalidomide, bortezomib, and dexamethasone, patients in each trial arm who were MRD negative had significantly better PFS than patients who were MRD positive after consolidation, regardless of assigned treatment, Dr. Landgren noted.
In the relapsed/refractory multiple myeloma setting, MRD negativity was associated with better PFS for patients in the POLLUX trial, whether subjects were assigned to receive daratumumab plus lenalidomide and dexamethasone, or len-dex alone.
“This raises an important question: Is MRD more important than treatment modality?” Dr. Landgren said.
“The debate is: Is MRD ready for prime time? And as I have shown you with all the data, the answer is ‘Yes’,” he concluded.
No: Dr. Richardson
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena. The question for me as a clinician in my clinic is: ‘Do I apply it to everyday practice?’ And I would simply suggest to you at this point we’re not ready for that, and we’re not ready for that for a variety of complex reasons,” Dr. Richardson said in his rebuttal.
He cited a definition of MRD offered by Simone Ferrero, MD, and his colleagues from the University of Turin (Italy) in 2011 in Hematological Oncology: “Any approach aimed at detecting and possibly quantifying residual tumor cells beyond the sensitivity level of routine imaging and laboratory techniques.”
“We recognize in hematologic malignancies in particular, and increasingly in myeloma, that achievement of complete clinical remission and assessing this needs a variety of different scenarios,” he said.
These scenarios may include establishing the full eradication of the neoplastic clone, determining the long-term persistence of quiescent or nonclonogenic or immunologically regulated tumor cells, or persistence of clonogenic cells capable of giving rise to a full clinical relapse within a number of years.
Myeloma specialists recognize that MRD is a real phenomenon, made more challenging by the “extraordinary” heterogeneity of myeloma, he said.
Determination of MRD using sensitive molecular techniques may allow analysis of treatments that induce a greater depth of response than others, and may also identify patients who are experiencing early relapse and will need further treatment, Dr. Richardson acknowledged.
“The question is, should it dictate what you and I do every afternoon in the clinic with a particular patient, for example, outside of a clinical trial?”
He noted that MRD is still a secondary endpoint in trials for acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, and chronic lymphocytic leukemia, although it has been accepted by the FDA as a primary endpoint to assess molecular responses to second-generation tyrosine kinase inhibitors.
MRD is also still a secondary endpoint in trials for therapies against follicular and mantle cell lymphomas as well.
“So my hypothesis, or suggestion to you this morning, is that whilst MRD clearly is a vital area of research – and I especially applaud Ola for being on the forefront of this, and I fully support all the points he made – I would just simply suggest to you that it’s less advanced than in leukemia and lymphoma, and we are currently at the point where MRD assessments are clearly secondary endpoints and an important research tool,” he said,
MRD remains a research tool in multiple myeloma because, despite the wealth of new therapies and combinations approved in just the past few years, “we’re not able to eradicate it completely, and cure remains in myeloma, frankly, evasive,” he added.
Immunotherapy, for example, is not the “mutationally agnostic” approach that clinicians had hoped for, with recent evidence suggesting that it cannot overcome every genetic variation that may give rise to multiple myeloma.
For MRD to become a useful clinical tool rather than a research/regulatory tool, standardization of testing methods will be needed. Flow cytometry until recently has been the mainstay for detecting MRD, but molecular strategies are currently being investigated, and rapid next-generation sequencing has the potential to become a gold standard, with its ability to identify and quantify all clonotypes in a sample.
“What’s critical is, therapeutic adjustment for what? What is the difference? For example, [if] one arm of a trial has 20% MRD positivity vs. 40% in another, what does that really mean for overall survival? These are enormous challenges that we still face,” Dr. Richardson said.
“I do think the lack of standardization broadly across the country is a challenge, and so with that in mind, I would simply suggest that it is not yet a standard of care in clinical practice, but may be,” he concluded.
Dr. Landgren disclosed serving as a consultant to AbbVie, Amgen, Bristol-Myers Squibb, Celgene and Janssen. Dr. Richardson disclosed consulting for Celgene and Takeda.
AT LYMPHOMA & MYELOMA 2017
Parity laws don’t lower oral cancer drug costs for everyone

US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
US state laws intended to ensure fair prices for oral cancer drugs have had a mixed impact on patients’ pocketbooks, according to a study published in JAMA Oncology.
A total of 43 states and Washington, DC, have enacted parity laws, which require that patients pay no more for an oral cancer treatment than they would for an infusion of the same treatment.
Researchers analyzed the impact of these laws and observed modest improvements in costs for some patients.
However, patients who were already paying the most for their medications saw their monthly costs go up.
“Although parity laws appear to help reduce out-of-pocket spending for some patients, they may not fully address affordability for patients needing cancer drugs,” said study author Stacie B. Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
“We need to consider ways to address drug pricing directly and to improve benefit design to make sure that all patients can access prescribed drugs.”
To gauge the impact of parity laws on treatment costs, Dr Dusetzina and her colleagues analyzed health claims data for 63,780 adults from 3 large, nationwide insurance companies before and after the laws were enacted, from 2008 to 2012.
The team compared the cost of filling an oral cancer drug prescription for patients with health insurance plans that were covered by the state laws (fully insured) and patients whose plans were not (self-funded). All patients lived in 1 of 16 states that had passed parity laws at the time of the study.
About half of patients (51.4%) had fully insured plans, and the other half (48.6%) had self-funded plans.
For the entire cohort, the use of oral cancer drugs increased from 18% in the months before parity laws were passed to 22% in the months after (adjusted difference-in-differences risk ratio [aDDRR], 1.04; 95% confidence interval [CI], 0.96-1.13; P=0.34).
The proportion of prescription fills for oral drugs without copayment increased from 15.0% to 53.0% for patients with fully insured plans and from 12.3% to 18.0% in patients with self-funded plans (aDDRR, 2.36; 95% CI, 2.00-2.79; P<0.001).
“From our results, it looks like many plans decided they would just set the co-pay for oral drugs to $0,” Dr Dusetzina said. “Instead of $30 per month, those fills were now $0.”
The proportion of prescription fills with out-of-pocket cost of more than $100 per month increased from 8.4% to 11.1% for patients with fully insured plans but decreased from 12.0% to 11.7% for those with self-funded plans (aDDRR, 1.36; 95% CI, 1.11-1.68; P=0.004).
Patients paying the most for their oral cancer drug prescriptions experienced increases in their monthly out-of-pocket costs after parity laws were passed.
For patients whose costs were more expensive than 95% of other patients, their out-of-pocket costs increased an estimated $143.25 per month. Those paying more than 90% of what other patients paid saw their costs increase by $37.19 per month.
“One of the biggest problems with parity laws as they are written is that they don’t address the prices of these medications, which can be very high,” Dr Dusetzina noted.
“Parity can be reached as long as the coverage is the same for both oral and infused cancer therapies. Because we’re now seeing more people insured by plans with high deductibles or plans that require them to pay a percentage of their drug costs, parity may not reduce spending for some patients.”
However, Dr Dusetzina and her colleagues did find that patients who paid the least for their oral cancer treatments saw their estimated monthly out-of-pocket spending decrease.
Patients in the 25th percentile saw an estimated decrease of $19.44 per month, those in the 50th percentile saw a $32.13 decrease, and patients in the 75th percentile saw a decrease of $10.83.
On the other hand, the researchers also found that average 6-month healthcare costs—including what was paid by insurance companies and patients—did not change significantly as a result of parity laws.
The aDDRR was 0.96 (95% CI, 0.90-1.02; P=0.09) for all cancer treatments and 1.06 (95% CI, 0.93-1.20; P=0.40) for oral cancer drugs.
“One of the key arguments against passing parity, both for states that haven’t passed it and for legislation at the federal level, has been that it may increase costs to health plans,” Dr Dusetzina said. “But we didn’t find evidence of that.” ![]()
CHMP wants to expand use of BV to include CTCL

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for brentuximab vedotin (BV, Adcetris).
The CHMP is recommending authorization of BV to treat adults with CD30+ cutaneous T-cell lymphoma (CTCL) who have received at least 1 prior systemic therapy.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The EC previously approved BV to treat:
- Adults with relapsed or refractory CD30+ Hodgkin lymphoma (HL) following autologous stem cell transplant (ASCT) or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- Adults with CD30+ HL at increased risk of relapse or progression following ASCT
- Adults with relapsed or refractory systemic anaplastic large-cell lymphoma.
The CHMP’s recommendation to approve BV for CTCL is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in July 2015 and August 2015.
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June. ![]()
TP53 mutations could help stratify MCL patients
TP53 mutations identified a phenotypically distinct and aggressive form of mantle cell lymphoma (MCL) that did not respond to standard-of-care treatments, according to results from 183 patients younger than 66 years from the Nordic MCL2 and MCL3 trials.
, and that patients with the mutations should be considered for experimental trials of novel agents, wrote Christian W. Eskelund of the department of hematology at Rigshospitalet, Copenhagen, and colleagues.
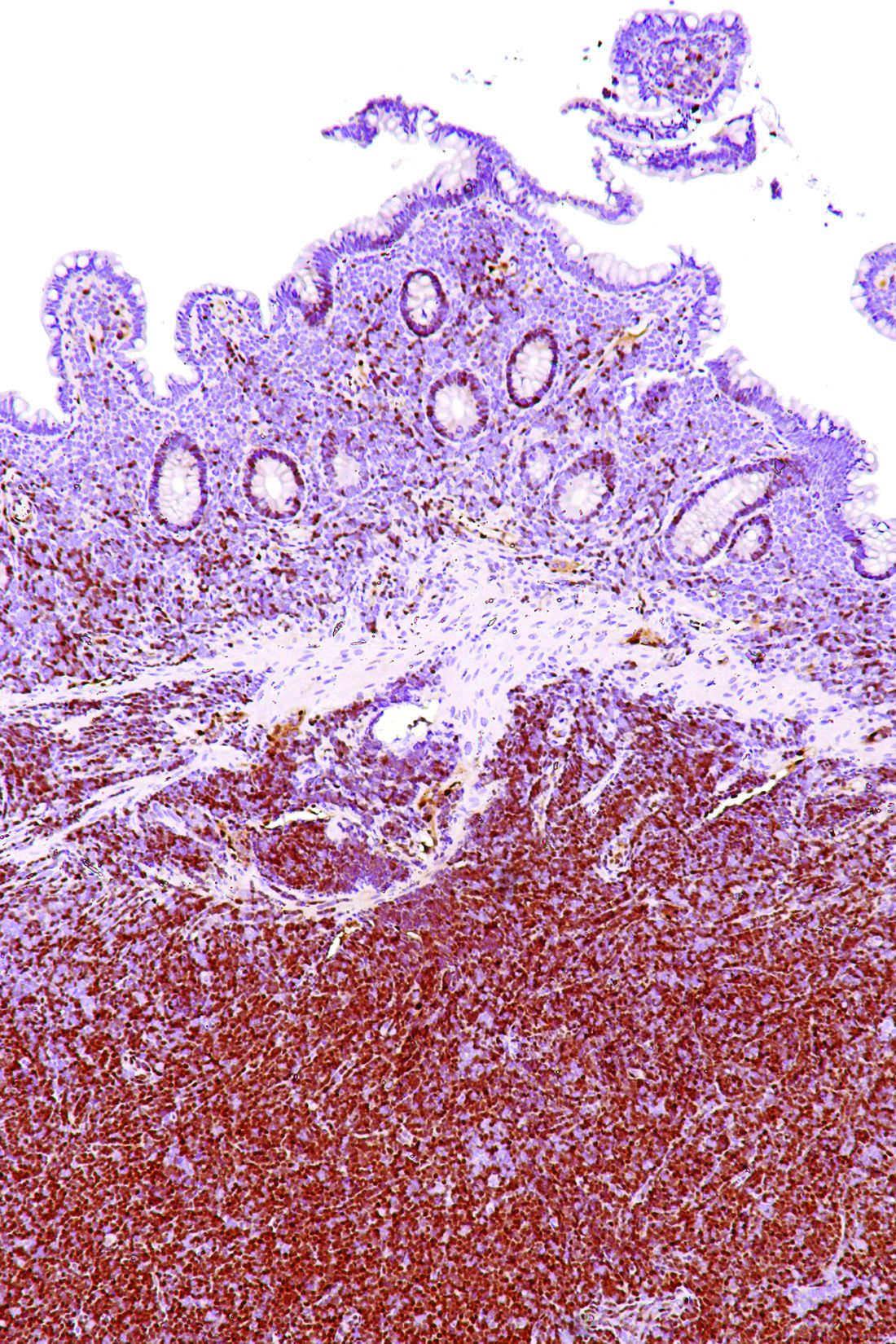
The researchers collected DNA from the Nordic MCL2 and MCL3 trials and 183 samples were of sufficient quality for genetic analyses. They examined the prognostic value of eight recurrently mutated and two recurrently deleted genes. Only TP53 mutations showed an independent prognostic effect for overall survival (hazard ratio 6.2; P less than .0001) in multivariate Cox regression analyses.
“Our data show that TP53 mutations identify a unique MCL subtype associated with high-risk baseline characteristics, dismal response to standard treatment, and poor clinical outcome,” the researchers wrote.
Read the full study in Blood (2017 Oct 26;130[17]:1903-10).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
TP53 mutations identified a phenotypically distinct and aggressive form of mantle cell lymphoma (MCL) that did not respond to standard-of-care treatments, according to results from 183 patients younger than 66 years from the Nordic MCL2 and MCL3 trials.
, and that patients with the mutations should be considered for experimental trials of novel agents, wrote Christian W. Eskelund of the department of hematology at Rigshospitalet, Copenhagen, and colleagues.
The researchers collected DNA from the Nordic MCL2 and MCL3 trials and 183 samples were of sufficient quality for genetic analyses. They examined the prognostic value of eight recurrently mutated and two recurrently deleted genes. Only TP53 mutations showed an independent prognostic effect for overall survival (hazard ratio 6.2; P less than .0001) in multivariate Cox regression analyses.
“Our data show that TP53 mutations identify a unique MCL subtype associated with high-risk baseline characteristics, dismal response to standard treatment, and poor clinical outcome,” the researchers wrote.
Read the full study in Blood (2017 Oct 26;130[17]:1903-10).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
TP53 mutations identified a phenotypically distinct and aggressive form of mantle cell lymphoma (MCL) that did not respond to standard-of-care treatments, according to results from 183 patients younger than 66 years from the Nordic MCL2 and MCL3 trials.
, and that patients with the mutations should be considered for experimental trials of novel agents, wrote Christian W. Eskelund of the department of hematology at Rigshospitalet, Copenhagen, and colleagues.
The researchers collected DNA from the Nordic MCL2 and MCL3 trials and 183 samples were of sufficient quality for genetic analyses. They examined the prognostic value of eight recurrently mutated and two recurrently deleted genes. Only TP53 mutations showed an independent prognostic effect for overall survival (hazard ratio 6.2; P less than .0001) in multivariate Cox regression analyses.
“Our data show that TP53 mutations identify a unique MCL subtype associated with high-risk baseline characteristics, dismal response to standard treatment, and poor clinical outcome,” the researchers wrote.
Read the full study in Blood (2017 Oct 26;130[17]:1903-10).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
FROM BLOOD
Lenalidomide shows clinical activity in relapsed/refractory MCL
Lenalidomide alone and in combination showed “clinically significant activity” and no new safety signals in patients with mantle cell lymphoma (MCL) who had previously failed on ibrutinib, according to findings from a retrospective, observational study.
Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues enrolled 58 MCL patients across 11 study sites. The patients had a median age of 71 years and 88% of patients had received three or more prior therapies. Most had received ibrutinib as monotherapy and used a lenalidomide-containing therapy next.
The overall response rate was 29% (95% confidence interval, 18%-43%). The rate was similar between patients with MCL refractory to ibrutinib and patients who relapsed/progressed on or following ibrutinib use (32% versus 30%, respectively). There was a 14% complete response, though it varied by subgroup with 8% among MCL patients refractory to ibrutinib and 22% among relapsed/progressed patients. There was a 20-week median duration of response, but 82% of responders were censored so the researchers urged caution in interpreting that finding.
Among the 58 patients, more than 80% reported one or more treatment-emergent adverse events during lenalidomide treatment and 20 patients (34%) had serious events. Nine patients (16%) discontinued the drug because of adverse events.
“Lenalidomide addresses an unmet medical need and widens the therapeutic options in a difficult-to-treat patient population,” the researchers wrote.
Read the full study in the Journal of Hematology Oncology (2017 Nov 2;10[1]:171).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Lenalidomide alone and in combination showed “clinically significant activity” and no new safety signals in patients with mantle cell lymphoma (MCL) who had previously failed on ibrutinib, according to findings from a retrospective, observational study.
Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues enrolled 58 MCL patients across 11 study sites. The patients had a median age of 71 years and 88% of patients had received three or more prior therapies. Most had received ibrutinib as monotherapy and used a lenalidomide-containing therapy next.
The overall response rate was 29% (95% confidence interval, 18%-43%). The rate was similar between patients with MCL refractory to ibrutinib and patients who relapsed/progressed on or following ibrutinib use (32% versus 30%, respectively). There was a 14% complete response, though it varied by subgroup with 8% among MCL patients refractory to ibrutinib and 22% among relapsed/progressed patients. There was a 20-week median duration of response, but 82% of responders were censored so the researchers urged caution in interpreting that finding.
Among the 58 patients, more than 80% reported one or more treatment-emergent adverse events during lenalidomide treatment and 20 patients (34%) had serious events. Nine patients (16%) discontinued the drug because of adverse events.
“Lenalidomide addresses an unmet medical need and widens the therapeutic options in a difficult-to-treat patient population,” the researchers wrote.
Read the full study in the Journal of Hematology Oncology (2017 Nov 2;10[1]:171).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Lenalidomide alone and in combination showed “clinically significant activity” and no new safety signals in patients with mantle cell lymphoma (MCL) who had previously failed on ibrutinib, according to findings from a retrospective, observational study.
Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues enrolled 58 MCL patients across 11 study sites. The patients had a median age of 71 years and 88% of patients had received three or more prior therapies. Most had received ibrutinib as monotherapy and used a lenalidomide-containing therapy next.
The overall response rate was 29% (95% confidence interval, 18%-43%). The rate was similar between patients with MCL refractory to ibrutinib and patients who relapsed/progressed on or following ibrutinib use (32% versus 30%, respectively). There was a 14% complete response, though it varied by subgroup with 8% among MCL patients refractory to ibrutinib and 22% among relapsed/progressed patients. There was a 20-week median duration of response, but 82% of responders were censored so the researchers urged caution in interpreting that finding.
Among the 58 patients, more than 80% reported one or more treatment-emergent adverse events during lenalidomide treatment and 20 patients (34%) had serious events. Nine patients (16%) discontinued the drug because of adverse events.
“Lenalidomide addresses an unmet medical need and widens the therapeutic options in a difficult-to-treat patient population,” the researchers wrote.
Read the full study in the Journal of Hematology Oncology (2017 Nov 2;10[1]:171).
mschneider@frontlinemedcom.com
On Twitter @maryellenny
FROM THE JOURNAL OF HEMATOLOGY & ONCOLOGY
FDA approves brentuximab vedotin for pcALCL, MF

The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of brentuximab vedotin (BV, ADCETRIS).
BV is now approved for adults with primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy.
This is the fourth FDA-approved indication for BV. The drug has regular approval for 2 indications in classical Hodgkin lymphoma and accelerated approval for the treatment of systemic ALCL.
In November 2016, the FDA granted BV breakthrough therapy designation for the treatment of patients with pcALCL and CD30-expressing MF who require systemic therapy and have received one prior systemic therapy. The agency also granted the supplemental biologics license application priority review.
The approval for BV in pcALCL and CD30-expressing MF is based on data from the phase 3 ALCANZA trial and a pair of phase 2 investigator-sponsored trials.
Phase 3 trial
Results from ALCANZA were presented at the 9th Annual T-cell Lymphoma Forum in January and published in The Lancet in June.
There were 128 patients in the intent-to-treat and safety populations. Sixty-four patients (48 with MF and 16 with pcALCL) were randomized to receive BV at 1.8 mg/kg every 3 weeks for up to 48 weeks.
The other 64 patients (49 with MF and 15 with pcALCL) were randomized to receive standard of care (SOC)—methotrexate at 5 mg to 50 mg weekly or bexarotene at a target dose of 300 mg/m² daily for up to 48 weeks.
The study’s primary endpoint was the rate of objective response lasting at least 4 months (ORR4). The ORR4 rate was significantly higher with BV than with SOC—56.3% and 12.5%, respectively (P<0.0001).
For patients with MF, the ORR4 was 50% with BV and 10% with SOC. For patients with pcALCL, the ORR4 was 75% with BV and 20% with SOC.
Overall, the complete response (CR) rates were 15.6% in the BV arm and 1.6% in the SOC arm (P=0.0046).
For patients with MF, the CR rate was 10% with BV and 0% with SOC. For patients with pcALCL, the CR rate was 31% with BV and 7% with SOC.
Progression-free survival (PFS) was significantly longer in the BV arm than the SOC arm. The median PFS was 16.7 months and 3.5 months, respectively. The hazard ratio was 0.270 (P<0.0001).
For patients with MF, the median PFS was 15.9 months with BV and 3.5 months with SOC. For patients with pcALCL, the median PFS was 27.5 months with BV and 5.3 months with SOC.
The most common adverse events (AEs) of any grade (occurring in 15% or more of patients in the BV and SOC arms, respectively) were peripheral neuropathy (67% and 6%), nausea (36% and 13%), diarrhea (29% and 6%), fatigue (29% and 27%), vomiting (17% and 5%), alopecia (15% and 3%), pruritus (17% and 13%), pyrexia (17% and 18%), decreased appetite (15% and 5%), and hypertriglyceridemia (2% and 18%).
Phase 2 trials
Data from the investigator-sponsored trials were published in the Journal of Clinical Oncology in 2015.
The first study was published in July of that year. The trial enrolled 32 patients with MF or Sézary syndrome. Thirty patients were evaluable for efficacy, and more than half had received 3 or more prior systemic therapies.
Patients received BV (1.8 mg/kg) every 3 weeks for a maximum of 16 doses. The primary endpoint was objective clinical response rate.
Seventy percent of patients (21/30) achieved an objective response across all stages of disease. One patient had a CR, 20 had a partial response, 4 had stable disease, 5 had progressive disease, and 2 were not evaluable for response.
The most common related AEs of any grade were peripheral neuropathy (66%), fatigue (47%), nausea (28%), hair loss (22%), and neutropenia (19%). Grade 3/4 related AEs included neutropenia (n=4), rash (n=3), and peripheral neuropathy (n=1).
The second phase 2 trial was published in August 2015. This trial enrolled CD30-positive patients with lymphomatoid papulosis (LyP), pcALCL, and MF.
Fifty-four patients were enrolled, and 48 were evaluable at the time of analysis. Patients had received an infusion of BV (1.8 mg/kg) every 21 days.
Seventy-three percent of patients (35/48) achieved an objective response, including 100% (20/20) with LyP and/or pcALCL and 54% (15/28) with MF. The CR rate was 35% (n=17).
The most common AEs were peripheral neuropathy (67%), fatigue (35%), skin rash (24%), diarrhea (15%), muscle pain (17%), localized skin infection (15%), neutropenia (15%), and hair loss (11%).
Grade 3/4 AEs included neutropenia (n=3), nausea (n=2), unstable angina or myocardial infarction (n=2), infection (n=2), joint pain (n=2), fatigue (n=1), deep vein thrombosis (n=1), pulmonary embolism (n=1), aminotransferase elevation (n=1), and dehydration (n=1). ![]()
FDA approves IV formulation of aprepitant for CINV

The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
The US Food and Drug Administration (FDA) has approved use of an intravenous (IV) formulation of aprepitant (CINVANTI™) to prevent chemotherapy-induced nausea and vomiting (CINV).
CINVANTI is intended to be used in combination with other antiemetic agents to prevent acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic chemotherapy (HEC) and moderately emetogenic chemotherapy (MEC).
CINVANTI is to be used in combination with a 5-HT3 receptor antagonist and dexamethasone.
The full prescribing information is available at www.cinvanti.com.
The US commercial launch of CINVANTI is planned for January 2018.
CINVANTI is the first IV formulation to directly deliver aprepitant, a substance P/neurokinin-1 (NK1) receptor antagonist.
Aprepitant is also the active ingredient in EMEND® capsules, which were approved by the FDA in 2003. EMEND IV®, which was approved in 2008, contains aprepitant’s prodrug, fosaprepitant.
Heron Therapeutics, Inc., developed CINVANTI in an attempt to provide an IV formulation of aprepitant that has the same efficacy as IV fosaprepitant but does not pose the risk of adverse events (AEs) related to polysorbate 80.
“Aprepitant has long been the standard in the NK1 class, and it remains the only single-agent NK1 with proven efficacy in preventing CINV in both the acute and delayed phases in HEC and MEC,” said Rudolph M. Navari, MD, PhD, of the University of Alabama Birmingham School of Medicine.
“Because CINVANTI is a novel, polysorbate 80-free, IV formulation of aprepitant, it enables physicians to provide patients with standard-of-care efficacy without the potential risk of polysorbate 80-related adverse events, such as infusion-site reactions.”
The FDA approved CINVANTI based on data demonstrating the bioequivalence of CINVANTI to EMEND IV.
A phase 1, randomized, 2-way cross-over study comparing the drugs enrolled 100 healthy subjects. The subjects received CINVANTI at 130 mg or EMEND IV at 150 mg, given over 30 minutes on day 1 of periods 1 and 2.
The researchers said 90% confidence intervals for CINVANTI AUC0-t (area under the time-concentration curve from time 0 to the last measurable concentration), AUC0-inf (area under the time-concentration curve from time 0 extrapolated to infinity), and C12h (plasma concentration at 12 hours) “were well within bioequivalence bounds,” which was 80% to 125%.
The team also found the incidence of treatment-emergent AEs was lower with CINVANTI than EMEND IV—21% and 28%, respectively. The same was true for related treatment-emergent AEs—15% and 28%, respectively.
These data were presented at the Hematology/Oncology Pharmacy Association Annual Conference in March/April and the Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) Annual Meeting in June. ![]()
FDA approves brentuximab vedotin for primary cutaneous anaplastic large cell lymphoma
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Approval was based on a 56% objective response rate for brentuximab vedotin versus 12% for physician’s choice in a phase 3 trial (ALCANZA) of 131 patients with mycosis fungoides or pcALCL. All patients had received one prior systemic therapy and were randomized (1:1) to receive either brentuximab vedotin or the physician’s choice of methotrexate or bexarotene, the Food and Drug Administration said in a press statement.
The most common adverse reactions for patients in the brentuximab vedotin arm were anemia, peripheral sensory neuropathy, nausea, diarrhea, fatigue, and neutropenia. The most common adverse event leading to discontinuation of brentuximab vedotin was peripheral neuropathy.
The recommended dose of brentuximab vedotin is 1.8 mg/kg up to a maximum of 180 mg/kg as an IV infusion over 30 minutes every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity, the FDA wrote.
Brentuximab vedotin is marketed as Adcetris by Seattle Genetics.
ALCANZA results were presented at ASH 2016 and published in the Lancet in Aug. 5, 2017.
Ibrutinib sustains efficacy in CLL at 4-year follow-up

NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
NEW YORK, NY—The 4-year follow-up of the RESONATE trial suggests ibrutinib may provide long-term efficacy in previously treated patients with chronic lymphocytic leukemia (CLL).
The median progression-free survival (PFS) has not yet been reached in this trial, regardless of high-risk cytogenetics, according to Jennifer Brown, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
She presented the update at Lymphoma & Myeloma 2017. The follow-up study was awarded the best clinical CLL abstract of the meeting.
In the phase 3 RESONATE study, investigators compared ibrutinib—the first-in-class, once-daily, oral inhibitor of Bruton tyrosine kinase—to ofatumumab in previously treated CLL/small lymphocytic lymphoma (SLL).
The primary analysis showed ibrutinib significantly improved survival, with a 78% reduction in the risk of progression and a 57% reduction in the risk of death.
The phase 3 trial randomized 195 CLL/SLL patients to oral ibrutinib at 420 mg once daily and 196 patients to intravenous ofatumumab at an initial dose of 300 mg followed by 2000 mg for 11 doses over 24 weeks.
One hundred thirty-three patients progressed on ofatumumab and crossed over to receive once-daily ibrutinib.
Patient characteristics
In each arm, the median patient age was 67, more than half of patients had an ECOG status of 1, and more than half had advanced-stage disease.
High-risk genetic abnormalities were common, Dr Brown said, with deletion 11q in a third of patients in the ibrutinib arm and 31% in the ofatumumab arm. Another third in each arm had deletion 17p, while 51% in the ibrutinib arm and 46% in the ofatumumab arm had TP53 mutation.
About a quarter of the patients in each arm had complex karyotype, and 73% and 63% in the ibrutinib and ofatumumab arms, respectively, were IGHV-unmutated.
Survival
Ibrutinib significantly extended PFS compared with ofatumumab. At a median follow-up for ibrutinib of 44 months (range, 0.33 – 53), ibrutinib led to an 87% reduction in the risk of progression or death. The 3-year PFS rate was 59% with ibrutinib and 3% with ofatumumab.
Ibrutinib conferred a benefit in PFS across all baseline patient characteristics.
Among ibrutinib-treated patients, the 3-year PFS was 53% for patients with deletion 17p, 66% for those with deletion 11q but not deletion 17p, and 58% for those with neither abnormality.
Dr Brown noted how closely complex karyotype associates with high-risk cytogenetics. Forty-two percent of patients with 17p deletion had a complex karyotype, as did 23% of patients with 11q deletion and 15% of patients with neither 17p nor 11q deletion.
For IGHV-mutation status, Dr Brown said there is no difference in PFS with this degree of follow-up.
In terms of TP53 mutation status, Dr Brown pointed out a trend toward a worse PFS in those patients with the mutation.
“We actually looked by individual p53 mutation versus 17p deletion, versus both, versus neither, in the 2-year follow-up paper and found that p53 with 17p, both abnormalities, did have worse PFS than neither,” she said.
“This may require further follow-up because we do know that most 17p patients also have a p53 mutation, particularly in the relapsed setting.”
As expected, Dr Brown said, those patients with more than 2 prior therapies had a worse PFS compared to patients with 2 or fewer prior therapies.
Multivariate analysis demonstrated that more than 2 prior lines of therapy or an elevated ß2 microglobulin were associated with decreased PFS with ibrutinib.
When the investigators adjusted the overall survival data for cross-over, ibrutinib was projected to continue the overall survival benefit compared with ofatumumab, with a hazard ratio of 0.37.
Response rates
Dr Brown noted that, early on, there’s quite a significant rate of partial response with lymphocytosis observed in patients on ibrutinib.
This “diminishes dramatically,” she said, but about 5% of patients at 3 and 4 years still have ongoing lymphocytosis.
“Similarly, initially, there’s a very low rate of complete remission, which has risen steadily to 9% at this follow-up,” she said.
And the overall response rate is 91%.
Treatment exposure and toxicity
The median duration of ibrutinib treatment is 41 months, and 46% of patients continue on treatment. Twenty-seven percent of patients discontinued due to progression, and 12% because of adverse events (AEs).
Of the 53 patients who discontinued therapy, 14 had transformation as their primary reason, 9 with diffuse large B-cell lymphoma, 3 with Hodgkin disease, and 2 with prolymphocytic lymphoma.
The most frequent AEs leading to discontinuation included pneumonia (n=3), anemia (n=2), thrombocytopenia (n=2), diarrhea (n=2), and anal incontinence (n=2).
AEs leading to discontinuation decreased over time—6% in year 0 to 1 and 4% in years 2 to 3.
“The most frequent cumulative AEs are similar to what we’ve seen in most prior studies,” Dr Brown said, including diarrhea, fatigue, and cough.
In terms of grade 3 or higher AEs, about a quarter of patients had neutropenia, 17% had pneumonia, and 8% had hypertension.
Six percent of patients had major hemorrhage, and all-grade atrial fibrillation occurred in 11% of patients.
“Now, many of the grade 3 and higher AEs did decline over time during the study,” Dr Brown noted. “You can see this is quite evident for neutropenia as well as pneumonia, and all infections declined from year 1 to subsequent years.”
Hypertension, in contrast, has been fairly steady over the later years, she said, and atrial fibrillation is highest in the first 6 months but then continues at a low rate thereafter.
The investigators believe these long-term results demonstrate that ibrutinib is tolerable and continues to show sustained efficacy in previously treated and high-genomic-risk patients with CLL. In addition, no long-term safety signals have emerged.
This study was sponsored by Pharmacyclics, LLC, an AbbVie company. ![]()
Intervention improves well-being in AYAs with cancer

SAN DIEGO—New research suggests an intervention can improve psychosocial health in adolescents and young adults (AYAs) living with cancer.
The intervention, Promoting Resilience in Stress Management (PRISM), is designed to help patients manage stress, set goals, and change their perspective.
Overall, PRISM improved resilience, enhanced quality of life, increased hope, and lowered distress and depression in the patients studied.
Abby R. Rosenberg, MD, of Seattle Children’s Research Institute in Seattle, Washington, presented these results at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 176*).
“The experience of cancer is stressful in all realms, but we tend to focus more on physical symptoms than the equally important social and emotional challenges,” Dr Rosenberg said.
“This is particularly true for adolescents and young adults who already struggle with normal developmental changes. When you throw cancer into the mix, it can become much harder.”
With this in mind, Dr Rosenberg and her colleagues tested PRISM in AYAs with cancer. The trial included 99 English-speaking patients, ages 12 to 25, who were diagnosed with new or newly recurrent cancer.
The patients were randomized to receive PRISM (n=49) plus standard psychosocial supportive care or standard care alone (n=50). Standard care at Seattle Children’s Research Institute includes a dedicated social worker and access to psychologists, child-life specialists, and other experts in AYA oncology care, as needed.
PRISM targets 4 topics:
- Managing stress with skills based on mindfulness and relaxation
- Setting goals that are specific and realistic, as well as planning for roadblocks
- Positive reframing, or recognizing and replacing negative self-talk
- Making meaning, or identifying benefits, gratitude, purpose, and legacy.
Each of the 4 topics were discussed with patients in separate, one-on-one sessions with a trained research associate. The sessions lasted 30 minutes to an hour. Patients also received boosters and worksheets for practicing the skills discussed in the meetings.
After all 4 sessions had been completed, patients could participate in an optional family meeting. During this meeting, patients could discuss with their family members which aspects of PRISM worked.
Results
Patients completed surveys at study enrollment, 2 months, 4 months, and 6 months. There were 74 participants who were still alive and well enough to complete the 6-month survey—36 in the PRISM group and 38 in the control group.
At the 6-month mark, PRISM was associated with (sometimes significant) improvements in resilience (P=0.02), generic quality of life (P=0.08), cancer-specific quality of life (P=0.01), hope (P=0.34), and distress (P=0.03). (P values are for absolute difference from baseline to 6 months.)
In addition, the incidence of depression at 6 months was lower in the PRISM group than the control group—6% and 21%, respectively (odds ratio=0.09, 95% CI 0.01, 1.09).
All but 4 of the PRISM recipients chose to participate in the family meeting following their one-on-one sessions.
“We included the family meeting because teens told us they wanted to share with their parents, and parents told us they wanted to know what their children had learned,” Dr Rosenberg said. “While the specific impact of this meeting is yet to be determined, we hope it will guide families so that there is continued support of teen or young adult patients.”
Now, Dr Rosenberg and her colleagues would like to test PRISM in other patient populations.
“We need to include a much larger cultural demographic in future studies,” Dr Rosenberg noted. “Beyond that, we also need to determine if this type of intervention could translate to other centers where usual care may not be as comprehensive as what we have here.” ![]()
*Some data in the abstract differ from the presentation.
SAN DIEGO—New research suggests an intervention can improve psychosocial health in adolescents and young adults (AYAs) living with cancer.
The intervention, Promoting Resilience in Stress Management (PRISM), is designed to help patients manage stress, set goals, and change their perspective.
Overall, PRISM improved resilience, enhanced quality of life, increased hope, and lowered distress and depression in the patients studied.
Abby R. Rosenberg, MD, of Seattle Children’s Research Institute in Seattle, Washington, presented these results at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 176*).
“The experience of cancer is stressful in all realms, but we tend to focus more on physical symptoms than the equally important social and emotional challenges,” Dr Rosenberg said.
“This is particularly true for adolescents and young adults who already struggle with normal developmental changes. When you throw cancer into the mix, it can become much harder.”
With this in mind, Dr Rosenberg and her colleagues tested PRISM in AYAs with cancer. The trial included 99 English-speaking patients, ages 12 to 25, who were diagnosed with new or newly recurrent cancer.
The patients were randomized to receive PRISM (n=49) plus standard psychosocial supportive care or standard care alone (n=50). Standard care at Seattle Children’s Research Institute includes a dedicated social worker and access to psychologists, child-life specialists, and other experts in AYA oncology care, as needed.
PRISM targets 4 topics:
- Managing stress with skills based on mindfulness and relaxation
- Setting goals that are specific and realistic, as well as planning for roadblocks
- Positive reframing, or recognizing and replacing negative self-talk
- Making meaning, or identifying benefits, gratitude, purpose, and legacy.
Each of the 4 topics were discussed with patients in separate, one-on-one sessions with a trained research associate. The sessions lasted 30 minutes to an hour. Patients also received boosters and worksheets for practicing the skills discussed in the meetings.
After all 4 sessions had been completed, patients could participate in an optional family meeting. During this meeting, patients could discuss with their family members which aspects of PRISM worked.
Results
Patients completed surveys at study enrollment, 2 months, 4 months, and 6 months. There were 74 participants who were still alive and well enough to complete the 6-month survey—36 in the PRISM group and 38 in the control group.
At the 6-month mark, PRISM was associated with (sometimes significant) improvements in resilience (P=0.02), generic quality of life (P=0.08), cancer-specific quality of life (P=0.01), hope (P=0.34), and distress (P=0.03). (P values are for absolute difference from baseline to 6 months.)
In addition, the incidence of depression at 6 months was lower in the PRISM group than the control group—6% and 21%, respectively (odds ratio=0.09, 95% CI 0.01, 1.09).
All but 4 of the PRISM recipients chose to participate in the family meeting following their one-on-one sessions.
“We included the family meeting because teens told us they wanted to share with their parents, and parents told us they wanted to know what their children had learned,” Dr Rosenberg said. “While the specific impact of this meeting is yet to be determined, we hope it will guide families so that there is continued support of teen or young adult patients.”
Now, Dr Rosenberg and her colleagues would like to test PRISM in other patient populations.
“We need to include a much larger cultural demographic in future studies,” Dr Rosenberg noted. “Beyond that, we also need to determine if this type of intervention could translate to other centers where usual care may not be as comprehensive as what we have here.” ![]()
*Some data in the abstract differ from the presentation.
SAN DIEGO—New research suggests an intervention can improve psychosocial health in adolescents and young adults (AYAs) living with cancer.
The intervention, Promoting Resilience in Stress Management (PRISM), is designed to help patients manage stress, set goals, and change their perspective.
Overall, PRISM improved resilience, enhanced quality of life, increased hope, and lowered distress and depression in the patients studied.
Abby R. Rosenberg, MD, of Seattle Children’s Research Institute in Seattle, Washington, presented these results at the 2017 Palliative and Supportive Care in Oncology Symposium (abstract 176*).
“The experience of cancer is stressful in all realms, but we tend to focus more on physical symptoms than the equally important social and emotional challenges,” Dr Rosenberg said.
“This is particularly true for adolescents and young adults who already struggle with normal developmental changes. When you throw cancer into the mix, it can become much harder.”
With this in mind, Dr Rosenberg and her colleagues tested PRISM in AYAs with cancer. The trial included 99 English-speaking patients, ages 12 to 25, who were diagnosed with new or newly recurrent cancer.
The patients were randomized to receive PRISM (n=49) plus standard psychosocial supportive care or standard care alone (n=50). Standard care at Seattle Children’s Research Institute includes a dedicated social worker and access to psychologists, child-life specialists, and other experts in AYA oncology care, as needed.
PRISM targets 4 topics:
- Managing stress with skills based on mindfulness and relaxation
- Setting goals that are specific and realistic, as well as planning for roadblocks
- Positive reframing, or recognizing and replacing negative self-talk
- Making meaning, or identifying benefits, gratitude, purpose, and legacy.
Each of the 4 topics were discussed with patients in separate, one-on-one sessions with a trained research associate. The sessions lasted 30 minutes to an hour. Patients also received boosters and worksheets for practicing the skills discussed in the meetings.
After all 4 sessions had been completed, patients could participate in an optional family meeting. During this meeting, patients could discuss with their family members which aspects of PRISM worked.
Results
Patients completed surveys at study enrollment, 2 months, 4 months, and 6 months. There were 74 participants who were still alive and well enough to complete the 6-month survey—36 in the PRISM group and 38 in the control group.
At the 6-month mark, PRISM was associated with (sometimes significant) improvements in resilience (P=0.02), generic quality of life (P=0.08), cancer-specific quality of life (P=0.01), hope (P=0.34), and distress (P=0.03). (P values are for absolute difference from baseline to 6 months.)
In addition, the incidence of depression at 6 months was lower in the PRISM group than the control group—6% and 21%, respectively (odds ratio=0.09, 95% CI 0.01, 1.09).
All but 4 of the PRISM recipients chose to participate in the family meeting following their one-on-one sessions.
“We included the family meeting because teens told us they wanted to share with their parents, and parents told us they wanted to know what their children had learned,” Dr Rosenberg said. “While the specific impact of this meeting is yet to be determined, we hope it will guide families so that there is continued support of teen or young adult patients.”
Now, Dr Rosenberg and her colleagues would like to test PRISM in other patient populations.
“We need to include a much larger cultural demographic in future studies,” Dr Rosenberg noted. “Beyond that, we also need to determine if this type of intervention could translate to other centers where usual care may not be as comprehensive as what we have here.” ![]()
*Some data in the abstract differ from the presentation.