User login
Novel targeted cancer drugs cause fewer arrhythmias
ORLANDO – Not all oncology drugs are equal when it comes to their risk of treatment-induced cardiac arrhythmias.
Indeed, compared with anthracycline-based regimens, long the workhorse in treating many forms of cancer, the novel targeted agents – tyrosine kinase inhibitors, immune checkpoint inhibitors, and monoclonal antibodies – were 40% less likely to result in a new arrhythmia diagnosis within 6 months of treatment initiation, in a large, single-center retrospective study reported by Andrew Nickel at the annual meeting of the American College of Cardiology.

Overall, 14% of cancer patients developed a first-ever cardiac arrhythmia within the first 6 months after treatment began. In a Cox multivariate analysis, treatment with a targeted cancer agent was independently associated with a 40% lower risk of arrhythmia, compared with anthracycline-containing therapy. Of note, the incidence of new-onset atrial fibrillation was closely similar in the two groups.
Several patient factors emerged as independent predictors of increased risk of cancer treatment–induced arrhythmia in the multivariate analysis: male sex, with a 1.2-fold increased risk; baseline heart failure, with a 2.2-fold risk; and hypertension, which conferred a 1.6-fold increased risk. These are patient groups in which the novel targeted cancer treatments are a particularly attractive option from the standpoint of mitigating arrhythmia risk, provided their use would be appropriate, he observed.
Mr. Nickel reported having no financial conflicts regarding his study, which was conducted free of commercial support.
SOURCE: Nickel A et al. ACC 18. Abstract 900-06.
ORLANDO – Not all oncology drugs are equal when it comes to their risk of treatment-induced cardiac arrhythmias.
Indeed, compared with anthracycline-based regimens, long the workhorse in treating many forms of cancer, the novel targeted agents – tyrosine kinase inhibitors, immune checkpoint inhibitors, and monoclonal antibodies – were 40% less likely to result in a new arrhythmia diagnosis within 6 months of treatment initiation, in a large, single-center retrospective study reported by Andrew Nickel at the annual meeting of the American College of Cardiology.
Overall, 14% of cancer patients developed a first-ever cardiac arrhythmia within the first 6 months after treatment began. In a Cox multivariate analysis, treatment with a targeted cancer agent was independently associated with a 40% lower risk of arrhythmia, compared with anthracycline-containing therapy. Of note, the incidence of new-onset atrial fibrillation was closely similar in the two groups.
Several patient factors emerged as independent predictors of increased risk of cancer treatment–induced arrhythmia in the multivariate analysis: male sex, with a 1.2-fold increased risk; baseline heart failure, with a 2.2-fold risk; and hypertension, which conferred a 1.6-fold increased risk. These are patient groups in which the novel targeted cancer treatments are a particularly attractive option from the standpoint of mitigating arrhythmia risk, provided their use would be appropriate, he observed.
Mr. Nickel reported having no financial conflicts regarding his study, which was conducted free of commercial support.
SOURCE: Nickel A et al. ACC 18. Abstract 900-06.
ORLANDO – Not all oncology drugs are equal when it comes to their risk of treatment-induced cardiac arrhythmias.
Indeed, compared with anthracycline-based regimens, long the workhorse in treating many forms of cancer, the novel targeted agents – tyrosine kinase inhibitors, immune checkpoint inhibitors, and monoclonal antibodies – were 40% less likely to result in a new arrhythmia diagnosis within 6 months of treatment initiation, in a large, single-center retrospective study reported by Andrew Nickel at the annual meeting of the American College of Cardiology.
Overall, 14% of cancer patients developed a first-ever cardiac arrhythmia within the first 6 months after treatment began. In a Cox multivariate analysis, treatment with a targeted cancer agent was independently associated with a 40% lower risk of arrhythmia, compared with anthracycline-containing therapy. Of note, the incidence of new-onset atrial fibrillation was closely similar in the two groups.
Several patient factors emerged as independent predictors of increased risk of cancer treatment–induced arrhythmia in the multivariate analysis: male sex, with a 1.2-fold increased risk; baseline heart failure, with a 2.2-fold risk; and hypertension, which conferred a 1.6-fold increased risk. These are patient groups in which the novel targeted cancer treatments are a particularly attractive option from the standpoint of mitigating arrhythmia risk, provided their use would be appropriate, he observed.
Mr. Nickel reported having no financial conflicts regarding his study, which was conducted free of commercial support.
SOURCE: Nickel A et al. ACC 18. Abstract 900-06.
REPORTING FROM ACC 2018
Key clinical point: The novel targeted cancer therapies cause markedly fewer cardiac arrhythmias.
Major finding: Cancer patients treated with a tyrosine kinase inhibitor, immune checkpoint inhibitor, or another of the novel targeted therapies were 40% less likely than were those on anthracycline-based therapy to develop a treatment-induced cardiac arrhythmia up to 6 months after treatment initiation.
Study details: This was a retrospective single-center study including more than 5,000 cancer patients.
Disclosures: The presenter reported having no financial conflicts regarding his study, which was conducted free of commercial support.
Source: Nickel A et al. ACC 18, Abstract #900-06.
FDA approves anti-CD38 with VMP in myeloma
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
The who are ineligible for autologous stem cell transplant (ASCT).
The drug is approved in combination with a standard VMP regimen – bortezomib (Velcade), melphalan, and prednisone. The FDA had granted priority review to the drug application in January 2018 based on the results of the phase 3 ALCYONE study (NCT02195479).
Daratumumab, an anti-CD38 monoclonal antibody, reduced the risk of disease progression or death by 50%, compared with VMP alone in the ALCYONE study. The median progression-free survival had not yet been reached in the daratumumab arm; the median progression-free survival was 18.1 months in the VMP-only arm (N Engl J Med. 2018;378:518-28).
Daratumumab is marketed by Janssen Biotech as Darzalex.
Drug granted fast track designations for FL, DLBCL

The US Food and Drug Administration (FDA) has granted 2 fast track designations to 5F9, an anti-CD47 antibody.
The designations are for 5F9 as a treatment for relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).
Data supporting the fast track designations were derived from a phase 1b/2 trial of 5F9 in combination with rituximab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma, including DLBCL and FL.
Forty Seven, Inc., the company developing 5F9, expects to announce initial safety and efficacy data from the phase 1b portion of the trial in the second quarter of 2018.
About fast track designation
The FDA’s fast track drug development program is designed to expedite clinical development and submission of applications for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the drug’s development plan and written communications about issues such as trial design and use of biomarkers.
Drugs that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met.
Fast track drugs may also be eligible for rolling review, which allows a developer to submit individual sections of a drug’s application for review as they are ready, rather than waiting until all sections are complete.
The US Food and Drug Administration (FDA) has granted 2 fast track designations to 5F9, an anti-CD47 antibody.
The designations are for 5F9 as a treatment for relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).
Data supporting the fast track designations were derived from a phase 1b/2 trial of 5F9 in combination with rituximab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma, including DLBCL and FL.
Forty Seven, Inc., the company developing 5F9, expects to announce initial safety and efficacy data from the phase 1b portion of the trial in the second quarter of 2018.
About fast track designation
The FDA’s fast track drug development program is designed to expedite clinical development and submission of applications for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the drug’s development plan and written communications about issues such as trial design and use of biomarkers.
Drugs that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met.
Fast track drugs may also be eligible for rolling review, which allows a developer to submit individual sections of a drug’s application for review as they are ready, rather than waiting until all sections are complete.
The US Food and Drug Administration (FDA) has granted 2 fast track designations to 5F9, an anti-CD47 antibody.
The designations are for 5F9 as a treatment for relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).
Data supporting the fast track designations were derived from a phase 1b/2 trial of 5F9 in combination with rituximab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma, including DLBCL and FL.
Forty Seven, Inc., the company developing 5F9, expects to announce initial safety and efficacy data from the phase 1b portion of the trial in the second quarter of 2018.
About fast track designation
The FDA’s fast track drug development program is designed to expedite clinical development and submission of applications for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates frequent interactions with the FDA review team, including meetings to discuss the drug’s development plan and written communications about issues such as trial design and use of biomarkers.
Drugs that receive fast track designation may be eligible for accelerated approval and priority review if relevant criteria are met.
Fast track drugs may also be eligible for rolling review, which allows a developer to submit individual sections of a drug’s application for review as they are ready, rather than waiting until all sections are complete.
Early results favor combo IL-15/anti-CD20 in indolent NHL
CHICAGO – A combination of an immunostimulatory IL-15-based agent, ALT-803, with a therapeutic monoclonal antibody (mAb) against CD20, was well tolerated and had clinical activity in patients with indolent non-Hodgkin lymphoma (iNHL), according to preliminary findings from a phase 1 study.
“The cancer immunotherapy breakthrough that happened several years ago continues year after year, with a plethora of different modalities of immunotherapy at our disposal,” Todd A. Fehniger, MD, PhD, said at the annual meeting of the American Association for Cancer Research.
Immunotherapy with anti-CD20 mAbs, alone or in combination with chemotherapy, is a standard therapy for iNHL patients. Since iNHL cells express CD20, targeting it with mAbs triggers antitumor responses via cell surface receptors resulting in a potent antibody-dependent cellular toxicity. However, response in patients is highly heterogeneous, with relapse within a few months in a subset of patients. In addition, chemotherapeutic combinations can be toxic and result in serious and long-term complications.
“Relapsed or refractory iNHL is not curable and treatment strategies without long-term complications are needed,” said Dr. Fehniger, associate professor of medicine at Washington University, St. Louis.
In an attempt to address this, Dr. Fehniger and his colleagues combined rituximab, an anti-CD20 antibody, with a relatively new IL-15 agonist immunostimulatory agent called ALT-803.
In the phase 1 trial, the researchers enrolled patients with indolent non-Hodgkin lymphoma who had relapsed after at least 1 prior to CD20 antibody containing therapy. The study was a standard 3+3 dose escalation design with rituximab administered by intravenous infusion, 375 mg/m2 in four weekly doses, followed by a rest and four consolidation doses every 8 weeks for four cycles.
ALT-803 was administered concurrently at dose levels of 1 mcg/kg, 3 mcg/kg, and 6 mcg/kg IV followed by 6 mcg/kg, 10 mcg/kg, 15 mcg/kg, and 20 mcg/kg subcutaneously.
In total, 21 patients were treated: 16 patients had follicular lymphoma, four patients had marginal zone lymphoma, and one patient had small lymphocytic lymphoma. The median prior therapies received was two (range: 1-18) and five patients were treated who were refractory to prior anti-CD20 MAb therapy.
ALT-803 was well tolerated with no dose limiting toxicities or grade 4 or 5 adverse events. No patients discontinued ALT-803 and the recommended phase 2 dose was 20 mcg/kg subcutaneously. Grade 3 adverse events, regardless of attribution to ALT-803, included transient hypertension (14%), anemia (5%), nausea (5%), chills (5%), fever (5%), neutropenia (5%), and hyperglycemia (5%).
“Patients who received [subcutaneous] ALT-803 developed a unique injection site rash reaction that peaked 7-10 days later but resolved typically within 14 days. It was self-limited and resolved on its own,” Dr. Fehniger said.
At the time of the presentation, the best overall response rate was achieved in 11 of 21 patients (52%), with 9 complete responders (43%), and 2 partial responders (10%).
Of the 12 patients treated with ALT-803 subcutaneously, 11 patients had either stable disease, or partial or complete responses. All 11 patients remained on study and were in consolidation or follow-up and have not relapsed, Dr. Fehniger reported.
Among the five rituximab-refractory patients, the researchers observed one complete response, two patients with stable disease (45% and 36% tumor volume decrease), and two patients with partial disease. The durability of the responses can only be understood with longer follow-up, Dr. Fehniger said.
The peripheral blood of the patients was analyzed via flow cytometry and mass cytometry. Over the duration of four weekly doses, there was an increase in percentage (sixfold, P less than .001) and absolute number (10-fold, P less than .001) of natural killer cells at the 15-mcg/kg and 20-mcg/kg subcutaneous dose levels of ALT-803.
These results suggest that further studies of ALT-803 with other therapeutic targeting mAbs, or other immunotherapy modalities, are warranted, the researchers concluded.
Dr. Fehniger reported research funding from Altor BioScience.
SOURCE: Fehniger TA et al. AACR Annual Meeting, Abstract CT146.
CHICAGO – A combination of an immunostimulatory IL-15-based agent, ALT-803, with a therapeutic monoclonal antibody (mAb) against CD20, was well tolerated and had clinical activity in patients with indolent non-Hodgkin lymphoma (iNHL), according to preliminary findings from a phase 1 study.
“The cancer immunotherapy breakthrough that happened several years ago continues year after year, with a plethora of different modalities of immunotherapy at our disposal,” Todd A. Fehniger, MD, PhD, said at the annual meeting of the American Association for Cancer Research.
Immunotherapy with anti-CD20 mAbs, alone or in combination with chemotherapy, is a standard therapy for iNHL patients. Since iNHL cells express CD20, targeting it with mAbs triggers antitumor responses via cell surface receptors resulting in a potent antibody-dependent cellular toxicity. However, response in patients is highly heterogeneous, with relapse within a few months in a subset of patients. In addition, chemotherapeutic combinations can be toxic and result in serious and long-term complications.
“Relapsed or refractory iNHL is not curable and treatment strategies without long-term complications are needed,” said Dr. Fehniger, associate professor of medicine at Washington University, St. Louis.
In an attempt to address this, Dr. Fehniger and his colleagues combined rituximab, an anti-CD20 antibody, with a relatively new IL-15 agonist immunostimulatory agent called ALT-803.
In the phase 1 trial, the researchers enrolled patients with indolent non-Hodgkin lymphoma who had relapsed after at least 1 prior to CD20 antibody containing therapy. The study was a standard 3+3 dose escalation design with rituximab administered by intravenous infusion, 375 mg/m2 in four weekly doses, followed by a rest and four consolidation doses every 8 weeks for four cycles.
ALT-803 was administered concurrently at dose levels of 1 mcg/kg, 3 mcg/kg, and 6 mcg/kg IV followed by 6 mcg/kg, 10 mcg/kg, 15 mcg/kg, and 20 mcg/kg subcutaneously.
In total, 21 patients were treated: 16 patients had follicular lymphoma, four patients had marginal zone lymphoma, and one patient had small lymphocytic lymphoma. The median prior therapies received was two (range: 1-18) and five patients were treated who were refractory to prior anti-CD20 MAb therapy.
ALT-803 was well tolerated with no dose limiting toxicities or grade 4 or 5 adverse events. No patients discontinued ALT-803 and the recommended phase 2 dose was 20 mcg/kg subcutaneously. Grade 3 adverse events, regardless of attribution to ALT-803, included transient hypertension (14%), anemia (5%), nausea (5%), chills (5%), fever (5%), neutropenia (5%), and hyperglycemia (5%).
“Patients who received [subcutaneous] ALT-803 developed a unique injection site rash reaction that peaked 7-10 days later but resolved typically within 14 days. It was self-limited and resolved on its own,” Dr. Fehniger said.
At the time of the presentation, the best overall response rate was achieved in 11 of 21 patients (52%), with 9 complete responders (43%), and 2 partial responders (10%).
Of the 12 patients treated with ALT-803 subcutaneously, 11 patients had either stable disease, or partial or complete responses. All 11 patients remained on study and were in consolidation or follow-up and have not relapsed, Dr. Fehniger reported.
Among the five rituximab-refractory patients, the researchers observed one complete response, two patients with stable disease (45% and 36% tumor volume decrease), and two patients with partial disease. The durability of the responses can only be understood with longer follow-up, Dr. Fehniger said.
The peripheral blood of the patients was analyzed via flow cytometry and mass cytometry. Over the duration of four weekly doses, there was an increase in percentage (sixfold, P less than .001) and absolute number (10-fold, P less than .001) of natural killer cells at the 15-mcg/kg and 20-mcg/kg subcutaneous dose levels of ALT-803.
These results suggest that further studies of ALT-803 with other therapeutic targeting mAbs, or other immunotherapy modalities, are warranted, the researchers concluded.
Dr. Fehniger reported research funding from Altor BioScience.
SOURCE: Fehniger TA et al. AACR Annual Meeting, Abstract CT146.
CHICAGO – A combination of an immunostimulatory IL-15-based agent, ALT-803, with a therapeutic monoclonal antibody (mAb) against CD20, was well tolerated and had clinical activity in patients with indolent non-Hodgkin lymphoma (iNHL), according to preliminary findings from a phase 1 study.
“The cancer immunotherapy breakthrough that happened several years ago continues year after year, with a plethora of different modalities of immunotherapy at our disposal,” Todd A. Fehniger, MD, PhD, said at the annual meeting of the American Association for Cancer Research.
Immunotherapy with anti-CD20 mAbs, alone or in combination with chemotherapy, is a standard therapy for iNHL patients. Since iNHL cells express CD20, targeting it with mAbs triggers antitumor responses via cell surface receptors resulting in a potent antibody-dependent cellular toxicity. However, response in patients is highly heterogeneous, with relapse within a few months in a subset of patients. In addition, chemotherapeutic combinations can be toxic and result in serious and long-term complications.
“Relapsed or refractory iNHL is not curable and treatment strategies without long-term complications are needed,” said Dr. Fehniger, associate professor of medicine at Washington University, St. Louis.
In an attempt to address this, Dr. Fehniger and his colleagues combined rituximab, an anti-CD20 antibody, with a relatively new IL-15 agonist immunostimulatory agent called ALT-803.
In the phase 1 trial, the researchers enrolled patients with indolent non-Hodgkin lymphoma who had relapsed after at least 1 prior to CD20 antibody containing therapy. The study was a standard 3+3 dose escalation design with rituximab administered by intravenous infusion, 375 mg/m2 in four weekly doses, followed by a rest and four consolidation doses every 8 weeks for four cycles.
ALT-803 was administered concurrently at dose levels of 1 mcg/kg, 3 mcg/kg, and 6 mcg/kg IV followed by 6 mcg/kg, 10 mcg/kg, 15 mcg/kg, and 20 mcg/kg subcutaneously.
In total, 21 patients were treated: 16 patients had follicular lymphoma, four patients had marginal zone lymphoma, and one patient had small lymphocytic lymphoma. The median prior therapies received was two (range: 1-18) and five patients were treated who were refractory to prior anti-CD20 MAb therapy.
ALT-803 was well tolerated with no dose limiting toxicities or grade 4 or 5 adverse events. No patients discontinued ALT-803 and the recommended phase 2 dose was 20 mcg/kg subcutaneously. Grade 3 adverse events, regardless of attribution to ALT-803, included transient hypertension (14%), anemia (5%), nausea (5%), chills (5%), fever (5%), neutropenia (5%), and hyperglycemia (5%).
“Patients who received [subcutaneous] ALT-803 developed a unique injection site rash reaction that peaked 7-10 days later but resolved typically within 14 days. It was self-limited and resolved on its own,” Dr. Fehniger said.
At the time of the presentation, the best overall response rate was achieved in 11 of 21 patients (52%), with 9 complete responders (43%), and 2 partial responders (10%).
Of the 12 patients treated with ALT-803 subcutaneously, 11 patients had either stable disease, or partial or complete responses. All 11 patients remained on study and were in consolidation or follow-up and have not relapsed, Dr. Fehniger reported.
Among the five rituximab-refractory patients, the researchers observed one complete response, two patients with stable disease (45% and 36% tumor volume decrease), and two patients with partial disease. The durability of the responses can only be understood with longer follow-up, Dr. Fehniger said.
The peripheral blood of the patients was analyzed via flow cytometry and mass cytometry. Over the duration of four weekly doses, there was an increase in percentage (sixfold, P less than .001) and absolute number (10-fold, P less than .001) of natural killer cells at the 15-mcg/kg and 20-mcg/kg subcutaneous dose levels of ALT-803.
These results suggest that further studies of ALT-803 with other therapeutic targeting mAbs, or other immunotherapy modalities, are warranted, the researchers concluded.
Dr. Fehniger reported research funding from Altor BioScience.
SOURCE: Fehniger TA et al. AACR Annual Meeting, Abstract CT146.
REPORTING FROM THE AACR ANNUAL MEETING
Key clinical point:
Major finding: The ALT-803 plus rituximab combination achieved an overall response rate in 52% of patients, a complete response in 43%, and partial response in 10%.
Study details: A phase 1 study of 21 patients with indolent non-Hodgkin lymphoma.
Disclosures: Dr. Fehniger reported research funding from Altor BioScience LLC.
Source: Fehniger TA et al. AACR Annual Meeting, Abstract CT146.
Novartis CAR T-cell therapy adds a lymphoma indication
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
FDA issues CRL for proposed biosimilar rituximab

The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) saying the agency cannot approve Sandoz’s proposed biosimilar rituximab.
Sandoz submitted the biologics licensing application for the product, known as GP2013, in September 2017.
The company was seeking approval for GP2013 to treat follicular lymphoma (FL), diffuse large B-cell lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis, and microscopic polyangiitis.
The drug already has approval for these indications in Europe. The European Commission approved GP2013 (Rixathon) in June 2017.
As for US approval, Sandoz said it is evaluating the content of the FDA’s CRL and “remains committed to further discussions with FDA in order to bring this important medicine to US patients as soon as possible.”
The company said it “stands behind the robust body of evidence included in the regulatory submission” for GP2013.
Part of this evidence is the ASSIST-FL trial, in which researchers compared GP2013 to the reference product, Roche’s MabThera. Results from this trial were published in The Lancet Haematology and presented at ESMO 2017 Congress.
This phase 3 trial included adults with previously untreated, advanced stage FL. Patients received 8 cycles of cyclophosphamide, vincristine, and prednisone with either GP2013 or reference rituximab. Responders then received GP2013 or rituximab monotherapy as maintenance for up to 2 years.
At a median follow-up of 11.6 months, the overall response rate was 87% (271/311) in the GP2013 arm and 88% in the rituximab arm (274/313). Complete response rates were 15% (n=46) and 13% (n=42), respectively.
Rates of adverse events (AEs) were 93% in the GP2013 arm and 91% in the rituximab arm. Rates of serious AEs were 23% and 20%, respectively. The rate of discontinuation due to AEs was 7% in both arms.
The most common AE was neutropenia, which occurred in 26% of patients in the GP2013 arm and 30% of those in the rituximab arm in the combination phase. Rates of neutropenia in the maintenance phase were 10% and 6%, respectively.
Five-year survival for non-Hodgkin lymphoma tops 71%
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.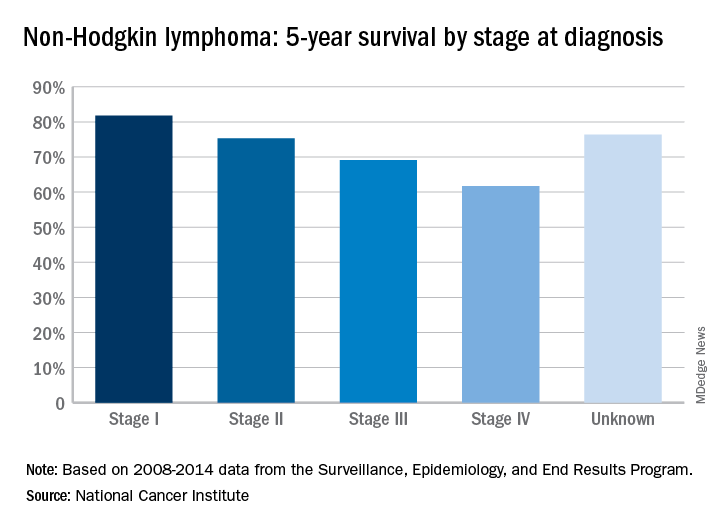
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.
The overall 5-year survival rate for non-Hodgkin lymphoma (NHL) is 71.4%, according to the National Cancer Institute.
That number falls neatly into the middle of the range for survival by stage at diagnosis, with stage I (81.8%) and stage II (75.3%) disease on the high side and stage III (69.1%) and stage IV (61.7%) on the low side, the most recent data from the Surveillance, Epidemiology, and End Results (SEER) Program show. Five-year survival for NHL of unknown stage at diagnosis is 76.4%.
FDA approves CAR T-cell therapy for lymphoma

The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
The US Food and Drug Administration (FDA) has approved tisagenlecleucel (Kymriah®) for its second indication.
The chimeric antigen receptor (CAR) T-cell therapy is now approved to treat adults with relapsed or refractory large B-cell lymphoma after 2 or more lines of systemic therapy.
This includes patients with diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.
The application for tisagenlecleucel in B-cell lymphoma was granted priority review. The FDA aims to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
Tisagenlecleucel is also FDA-approved to treat patients age 25 and younger who have B-cell precursor acute lymphoblastic leukemia that is refractory or in second or later relapse.
Access to tisagenlecleucel
The prescribing information for tisagenlecleucel includes a boxed warning detailing the risk of cytokine release syndrome (CRS) and neurological toxicities for patients receiving tisagenlecleucel.
Because of these risks, tisagenlecleucel is only available through a Risk Evaluation and Mitigation Strategy (REMS) program. The REMS program serves to inform and educate healthcare professionals about the risks associated with tisagenlecleucel treatment.
Novartis, the company marketing tisagenlecleucel, has established a network of certified treatment centers throughout the US. Staff at these centers are trained on the use of tisagenlecleucel and appropriate patient care.
Tisagenlecleucel is manufactured at a Novartis facility in Morris Plains, New Jersey. In the US, the target turnaround time for manufacturing tisagenlecleucel is 22 days.
Tisagenlecleucel costs $475,000 for a single course of treatment. However, Novartis said it is collaborating with the US Centers for Medicare and Medicaid Services on the creation of an appropriate value-based pricing approach.
The company also has a program called KYMRIAH CARES™, which offers financial assistance to eligible patients to help them gain access to tisagenlecleucel.
Phase 2 trial
The FDA approval of tisagenlecleucel for adults with relapsed/refractory B-cell lymphoma is based on results of the phase 2 JULIET trial.
The prescribing information for tisagenlecleucel includes data on 106 patients treated on this trial.
Only 68 of these patients were evaluable for efficacy. They had a median age of 56 (range, 22 to 74), and 71% were male.
Seventy-eight percent of patients had primary DLBCL not otherwise specified, and 22% had DLBCL following transformation from follicular lymphoma. Seventeen percent had high grade DLBCL.
Fifty-six percent of patients had refractory disease, and 44% had relapsed after their last therapy. The median number of prior therapies was 3 (range, 1 to 6), and 44% of patients had undergone autologous transplant.
Ninety percent of patients received lymphodepleting chemotherapy (66% fludarabine and 24% bendamustine) prior to tisagenlecleucel, and 10% did not. The median dose of tisagenlecleucel was 3.5 × 108 CAR+ T cells (range, 1.0 to 5.2 × 108).
The overall response rate was 50%, with 32% of patients achieving a complete response and 18% achieving a partial response. The median duration of response was not reached with a median follow-up of 9.4 months.
In all 106 patients infused with tisagenlecleucel, the most common grade 3/4 adverse events were infections (25%), CRS (23%), neurologic events (18%), febrile neutropenia (17%), encephalopathy (11%), lymphopenia (94%), neutropenia (81%), leukopenia (77%), anemia (58%), thrombocytopenia (54%), hypophosphatemia (24%), hypokalemia (12%), and hyponatremia (11%).
Three patients died within 30 days of tisagenlecleucel infusion. All of them had CRS and either stable or progressive disease. One of these patients developed bowel necrosis.
One patient died of infection. There were no deaths attributed to neurological events, and no fatal cases of cerebral edema.
Predicting response to CAR T-cell therapy in CLL

Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Researchers may have discovered why some patients with advanced chronic lymphocytic leukemia (CLL) don’t respond to chimeric antigen receptor (CAR) T-cell therapy.
The team found that CLL patients with elevated levels of “early memory” T cells prior to receiving CAR T-cell therapy had a partial or complete response to treatment, while patients with lower levels of these T cells did not respond.
The early memory T cells were marked by the expression of CD8 and CD27, as well as the absence of CD45RO.
The researchers validated the association between the early memory T cells and response in a small group of patients, predicting with 100% accuracy which patients would achieve a complete response.
Joseph A. Fraietta, PhD, of the University of Pennsylvania in Philadelphia, and his colleagues reported these findings in Nature Medicine. This research was supported, in part, by Novartis.
For this study, the researchers retrospectively analyzed 41 patients with advanced, heavily pretreated, high-risk CLL who received at least 1 dose of CD19-directed CAR T cells.
Consistent with the team’s previously reported findings, they were not able to identify patient or disease-specific factors that predict who responds best to the therapy.
Therefore, the researchers compared the gene expression profiles and phenotypes of T cells in patients who had a complete response, partial response, or no response to therapy.
The CAR T cells that persisted and expanded in complete responders were enriched in genes that regulate early memory and effector T cells and possess the IL-6/STAT3 signature.
Non-responders, on the other hand, expressed genes involved in late T-cell differentiation, glycolysis, exhaustion, and apoptosis. These characteristics make for a weaker set of T cells to persist, expand, and fight the CLL.
“Pre-existing T-cell qualities have previously been associated with poor clinical response to cancer therapy, as well differentiation in the T cells,” Dr Fraietta said. “What is special about what we have done here is finding that critical cell subset and signature.”
Elevated levels of the IL-6/STAT3 signaling pathway in these early T cells correlated with clinical responses to CAR T-cell therapy.
To validate these findings, the researchers screened for the early memory T cells in a group of 8 CLL patients, before and after CAR T-cell therapy. The team identified the complete responders with 100% specificity and sensitivity.
“With a very robust biomarker like this, we can take a blood sample, measure the frequency of this T-cell population, and decide with a degree of confidence whether we can apply this therapy and know the patient would have a response,” Dr Fraietta said.
“The ability to select patients most likely to respond would have tremendous clinical impact, as this therapy would be applied only to patients most likely to benefit, allowing patients unlikely to respond to pursue other options.”
These findings also suggest the possibility of improving CAR T-cell therapy by selecting for cell manufacturing the subpopulation of T cells responsible for driving responses. However, this approach would come with challenges.
“What we’ve seen in these non-responders is that the frequency of these T cells is low, so it would be very hard to infuse them as starting populations,” said study author J. Joseph Melenhorst, PhD, also of the University of Pennsylvania.
“But one way to potentially boost their efficacy is by adding checkpoint inhibitors with the therapy to block the negative regulation prior to CAR T-cell therapy, which a past, separate study has shown can help elicit responses in these patients.”
The researchers also noted that it’s unclear why some patients’ T cells are suboptimal prior to treatment. However, the team believes this could have to do with prior therapies.
Future studies with a larger group of CLL patients should be conducted to help answer these questions and validate the findings from this study, the researchers said.
Team identifies 5 subtypes of DLBCL

New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.
New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.
New research has revealed 5 genetic subtypes of diffuse large B-cell lymphoma (DLBCL).
Researchers identified a group of low-risk activated B-cell (ABC) DLBCLs, 2 subsets of germinal center B-cell (GCB) DLBCLs, a group of ABC/GCB-independent DLBCLs, and a group of ABC DLBCLs with genetic characteristics found in primary central nervous system lymphoma and testicular lymphoma.
The researchers believe these findings may have revealed new therapeutic targets for DLBCL, some of which could be inhibited by drugs that are already approved or under investigation in clinical trials.
Margaret Shipp, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, and her colleagues conducted this research and reported the results in Nature Medicine.
The team performed genetic analyses on samples from 304 DLBCL patients and observed great genetic diversity. The median number of genetic driver alterations in individual tumors was 17.
The researchers integrated data on 3 types of genetic alterations—recurrent mutations, somatic copy number alterations, and structural variants—to define previously unappreciated DLBCL subtypes.
“Specific genes that were perturbed by mutations could also be altered by changes in gene copy numbers or by chromosomal rearrangements, underscoring the importance of evaluating all 3 types of genetic alterations,” Dr Shipp noted.
“Most importantly, we saw that there were 5 discrete types of DLBCL that were distinguished one from another on the basis of the specific types of genetic alterations that occurred in combination.”
The researchers classified these subtypes as clusters (C) 1 to 5.
C1 consisted of largely ABC-DLBCLs with genetic features of an extra-follicular, possibly marginal zone origin.
C2 included both ABC and GCB DLBCLs with biallelic inactivation of TP53, 9p21.3/CDKN2A, and associated genomic instability.
Most DLBCLs in C3 were of the GCB subtype and were characterized by BCL2 structural variants and alterations of PTEN and epigenetic enzymes.
C4 consisted largely of GCB DLBCLs with alterations in BCR/PI3K, JAK/STAT, and BRAF pathway components and multiple histones.
Most C5 DLBCLs were of the ABC subtype, and the researchers said the major components of the C5 signature—BCL2 gain, concordant MYD88L265P/CD79B mutations, and mutations of ETV6, PIM1, GRHPR, TBL1XR1, and BTG1—were similar to those observed in primary central nervous system and testicular lymphoma.
Dr Shipp and her colleagues also identified a sixth cluster of DLBCLs (dubbed C0) that “lacked defining genetic drivers.”
Finally, the team found that patients with C0, C1, and C4 DLBCLs had more favorable outcomes, while patients with C2, C3, and C5 DLBCLs had less favorable outcomes.
“We feel this research opens the door to a whole series of additional investigations to understand how the combinations of these genetic alterations work together, and then to use that information to benefit patients with targeted therapies,” Dr Shipp said.
She and her colleagues are now working on creating a clinical tool to identify these genetic signatures in patients. The team is also developing clinical trials that will match patients with given genetic signatures to targeted treatments.
Another group of researchers recently identified 4 genetic subtypes of DLBCL.