User login
Group learns how protein promotes AML

A few years ago, researchers discovered that inhibiting the protein BRD4 can treat acute myeloid leukemia (AML). However, the mechanism that explains how the protein works has remained a mystery.
Now, investigators have discovered the larger signaling pathway to which BRD4 belongs. The team said their discovery points to additional therapeutic targets for AML and other malignancies.
The group described this work in Molecular Cell.
BRD4: A retrospective
In 2011, Christopher Vakoc, MD, PhD, of Cold Spring Harbor Laboratory in Cold Spring Harbor, New York, and his colleagues identified potential drug targets for AML using RNA interference. Out of that screen came BRD4 and the realization that leukemia cells were “extremely sensitive” to inhibition of this protein.
In a bit of serendipity, drugs to inhibit BRD4 had just been developed for other purposes. Dr Vakoc and his colleagues tested these drugs and found that one in particular, JQ1, worked well against a mouse model of aggressive AML.
In the past few years, several groups have reported similar therapeutic results in mice using JQ1 and closely related drugs.
“It’s been very satisfying to see that other groups have independently validated our findings,” Dr Vakoc said.
Due to the evidence of their effectiveness in mice, inhibitors of BRD4 moved into clinical trials starting in 2013. Currently, there are 12 active clinical trials targeting BRD4 in leukemia and other cancers, including one sponsored by a company to which Dr Vakoc has licensed JQ1.
“Once we published the first paper in 2011, the main objective in our lab has been to understand why these drugs work,” Dr Vakoc said. “Knowing the mechanism of action of a drug is essential to making the drug better because there will likely be many generations of BRD4 inhibitors.”
JQ1 and BRD4: How they work
In the current study, Dr Vakoc and his colleagues discovered that BRD4 works very closely with transcription factors that bind to DNA and selectively control the activity of certain genes. The transcription factors control blood formation and essentially give blood cells their “identity.”
The researchers found that JQ1 can make leukemia cells shed their leukemia characteristics and differentiate into normal white blood cells. The team also identified an intermediary molecule called p300 between BRD4 and the leukemia-associated transcription factors.
Active areas of research in Dr Vakoc’s lab include exploring other players in the pathway, particularly the molecules that BRD4 controls, and learning more about the transcription factors.
“This new work is leading us to realize that transcription factors are the masters of the biological universe,” Dr Vakoc said. “Clearly, they are driving the cancer phenotype.” ![]()
A few years ago, researchers discovered that inhibiting the protein BRD4 can treat acute myeloid leukemia (AML). However, the mechanism that explains how the protein works has remained a mystery.
Now, investigators have discovered the larger signaling pathway to which BRD4 belongs. The team said their discovery points to additional therapeutic targets for AML and other malignancies.
The group described this work in Molecular Cell.
BRD4: A retrospective
In 2011, Christopher Vakoc, MD, PhD, of Cold Spring Harbor Laboratory in Cold Spring Harbor, New York, and his colleagues identified potential drug targets for AML using RNA interference. Out of that screen came BRD4 and the realization that leukemia cells were “extremely sensitive” to inhibition of this protein.
In a bit of serendipity, drugs to inhibit BRD4 had just been developed for other purposes. Dr Vakoc and his colleagues tested these drugs and found that one in particular, JQ1, worked well against a mouse model of aggressive AML.
In the past few years, several groups have reported similar therapeutic results in mice using JQ1 and closely related drugs.
“It’s been very satisfying to see that other groups have independently validated our findings,” Dr Vakoc said.
Due to the evidence of their effectiveness in mice, inhibitors of BRD4 moved into clinical trials starting in 2013. Currently, there are 12 active clinical trials targeting BRD4 in leukemia and other cancers, including one sponsored by a company to which Dr Vakoc has licensed JQ1.
“Once we published the first paper in 2011, the main objective in our lab has been to understand why these drugs work,” Dr Vakoc said. “Knowing the mechanism of action of a drug is essential to making the drug better because there will likely be many generations of BRD4 inhibitors.”
JQ1 and BRD4: How they work
In the current study, Dr Vakoc and his colleagues discovered that BRD4 works very closely with transcription factors that bind to DNA and selectively control the activity of certain genes. The transcription factors control blood formation and essentially give blood cells their “identity.”
The researchers found that JQ1 can make leukemia cells shed their leukemia characteristics and differentiate into normal white blood cells. The team also identified an intermediary molecule called p300 between BRD4 and the leukemia-associated transcription factors.
Active areas of research in Dr Vakoc’s lab include exploring other players in the pathway, particularly the molecules that BRD4 controls, and learning more about the transcription factors.
“This new work is leading us to realize that transcription factors are the masters of the biological universe,” Dr Vakoc said. “Clearly, they are driving the cancer phenotype.” ![]()
A few years ago, researchers discovered that inhibiting the protein BRD4 can treat acute myeloid leukemia (AML). However, the mechanism that explains how the protein works has remained a mystery.
Now, investigators have discovered the larger signaling pathway to which BRD4 belongs. The team said their discovery points to additional therapeutic targets for AML and other malignancies.
The group described this work in Molecular Cell.
BRD4: A retrospective
In 2011, Christopher Vakoc, MD, PhD, of Cold Spring Harbor Laboratory in Cold Spring Harbor, New York, and his colleagues identified potential drug targets for AML using RNA interference. Out of that screen came BRD4 and the realization that leukemia cells were “extremely sensitive” to inhibition of this protein.
In a bit of serendipity, drugs to inhibit BRD4 had just been developed for other purposes. Dr Vakoc and his colleagues tested these drugs and found that one in particular, JQ1, worked well against a mouse model of aggressive AML.
In the past few years, several groups have reported similar therapeutic results in mice using JQ1 and closely related drugs.
“It’s been very satisfying to see that other groups have independently validated our findings,” Dr Vakoc said.
Due to the evidence of their effectiveness in mice, inhibitors of BRD4 moved into clinical trials starting in 2013. Currently, there are 12 active clinical trials targeting BRD4 in leukemia and other cancers, including one sponsored by a company to which Dr Vakoc has licensed JQ1.
“Once we published the first paper in 2011, the main objective in our lab has been to understand why these drugs work,” Dr Vakoc said. “Knowing the mechanism of action of a drug is essential to making the drug better because there will likely be many generations of BRD4 inhibitors.”
JQ1 and BRD4: How they work
In the current study, Dr Vakoc and his colleagues discovered that BRD4 works very closely with transcription factors that bind to DNA and selectively control the activity of certain genes. The transcription factors control blood formation and essentially give blood cells their “identity.”
The researchers found that JQ1 can make leukemia cells shed their leukemia characteristics and differentiate into normal white blood cells. The team also identified an intermediary molecule called p300 between BRD4 and the leukemia-associated transcription factors.
Active areas of research in Dr Vakoc’s lab include exploring other players in the pathway, particularly the molecules that BRD4 controls, and learning more about the transcription factors.
“This new work is leading us to realize that transcription factors are the masters of the biological universe,” Dr Vakoc said. “Clearly, they are driving the cancer phenotype.” ![]()
FDA grants drug orphan designation for AML

Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for GMI-1271 to treat acute myeloid leukemia (AML).
GMI-1271, an E-selectin antagonist, has shown promise in preclinical research and proven safe in a phase 1 trial of healthy volunteers, according to GlycoMimetics, Inc., the company developing the drug.
Now, the company is recruiting adults with AML in a phase 1/2 study to test GMI-1271 in combination with chemotherapy.
“Having the FDA designate GMI-1271 as an orphan drug for the treatment of AML is an important accomplishment for GlycoMimetics,” said Helen Thackray, MD, vice president of clinical development and chief medical officer at GlycoMimetics. “This is a significant regulatory milestone for our program.”
The FDA’s orphan drug designation program is designed to promote the development of drugs intended to treat diseases affecting fewer than 200,000 people in the US.
Orphan designation provides a drug’s developer with benefits such as a 7-year period of marketing exclusivity if the drug is approved, protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and regulatory fee waivers.
Research with GMI-1271
Preclinical research presented at ASH 2013 suggested that GMI-1271 was able to overcome chemotherapy resistance in AML cells in vitro. The drug also reduced the leukemic burden in mouse models of AML when given in combination with daunorubicin and cytarabine.
Murine research presented at ASH 2014 suggested that, by inhibiting E-selectin, GMI-1271 may increase leukemic stem cells’ sensitivity to chemotherapeutic drugs.
At the same meeting, researchers presented preclinical data on GMI-1271 as a treatment for chronic myeloid leukemia, multiple myeloma, and venous thromboembolism.
In November 2014, GlycoMimetics announced results of phase 1 trial of GMI-1271 in healthy volunteers. Twenty-eight adults were enrolled in cohorts to receive the drug at 3 dose levels.
The company said subjects tolerated GMI-1271 well, and pharmacokinetics were as predicted based on preclinical research.
A multicenter, phase 1/2 trial of GMI-1271 in combination with chemotherapy is now recruiting adult patients with AML. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for GMI-1271 to treat acute myeloid leukemia (AML).
GMI-1271, an E-selectin antagonist, has shown promise in preclinical research and proven safe in a phase 1 trial of healthy volunteers, according to GlycoMimetics, Inc., the company developing the drug.
Now, the company is recruiting adults with AML in a phase 1/2 study to test GMI-1271 in combination with chemotherapy.
“Having the FDA designate GMI-1271 as an orphan drug for the treatment of AML is an important accomplishment for GlycoMimetics,” said Helen Thackray, MD, vice president of clinical development and chief medical officer at GlycoMimetics. “This is a significant regulatory milestone for our program.”
The FDA’s orphan drug designation program is designed to promote the development of drugs intended to treat diseases affecting fewer than 200,000 people in the US.
Orphan designation provides a drug’s developer with benefits such as a 7-year period of marketing exclusivity if the drug is approved, protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and regulatory fee waivers.
Research with GMI-1271
Preclinical research presented at ASH 2013 suggested that GMI-1271 was able to overcome chemotherapy resistance in AML cells in vitro. The drug also reduced the leukemic burden in mouse models of AML when given in combination with daunorubicin and cytarabine.
Murine research presented at ASH 2014 suggested that, by inhibiting E-selectin, GMI-1271 may increase leukemic stem cells’ sensitivity to chemotherapeutic drugs.
At the same meeting, researchers presented preclinical data on GMI-1271 as a treatment for chronic myeloid leukemia, multiple myeloma, and venous thromboembolism.
In November 2014, GlycoMimetics announced results of phase 1 trial of GMI-1271 in healthy volunteers. Twenty-eight adults were enrolled in cohorts to receive the drug at 3 dose levels.
The company said subjects tolerated GMI-1271 well, and pharmacokinetics were as predicted based on preclinical research.
A multicenter, phase 1/2 trial of GMI-1271 in combination with chemotherapy is now recruiting adult patients with AML. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for GMI-1271 to treat acute myeloid leukemia (AML).
GMI-1271, an E-selectin antagonist, has shown promise in preclinical research and proven safe in a phase 1 trial of healthy volunteers, according to GlycoMimetics, Inc., the company developing the drug.
Now, the company is recruiting adults with AML in a phase 1/2 study to test GMI-1271 in combination with chemotherapy.
“Having the FDA designate GMI-1271 as an orphan drug for the treatment of AML is an important accomplishment for GlycoMimetics,” said Helen Thackray, MD, vice president of clinical development and chief medical officer at GlycoMimetics. “This is a significant regulatory milestone for our program.”
The FDA’s orphan drug designation program is designed to promote the development of drugs intended to treat diseases affecting fewer than 200,000 people in the US.
Orphan designation provides a drug’s developer with benefits such as a 7-year period of marketing exclusivity if the drug is approved, protocol assistance, the ability to apply for research funding, tax credits for certain research expenses, and regulatory fee waivers.
Research with GMI-1271
Preclinical research presented at ASH 2013 suggested that GMI-1271 was able to overcome chemotherapy resistance in AML cells in vitro. The drug also reduced the leukemic burden in mouse models of AML when given in combination with daunorubicin and cytarabine.
Murine research presented at ASH 2014 suggested that, by inhibiting E-selectin, GMI-1271 may increase leukemic stem cells’ sensitivity to chemotherapeutic drugs.
At the same meeting, researchers presented preclinical data on GMI-1271 as a treatment for chronic myeloid leukemia, multiple myeloma, and venous thromboembolism.
In November 2014, GlycoMimetics announced results of phase 1 trial of GMI-1271 in healthy volunteers. Twenty-eight adults were enrolled in cohorts to receive the drug at 3 dose levels.
The company said subjects tolerated GMI-1271 well, and pharmacokinetics were as predicted based on preclinical research.
A multicenter, phase 1/2 trial of GMI-1271 in combination with chemotherapy is now recruiting adult patients with AML. ![]()
Modified T cells may treat GVHD
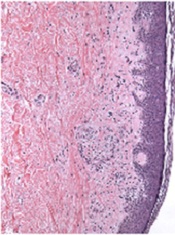
Image from PLOS ONE
A T-cell therapy designed to mitigate graft-vs-host disease (GVHD) is both feasible and safe, according to results of a pilot study published in Molecular Therapy.
To create this therapy, researchers transduced donor T cells with γ-retroviruses carrying a CD34-TK75 fusion gene.
The team said this allows them to track the cells via PET/CT and induce apoptosis by administering the antiviral drug ganciclovir if patients begin showing signs of GVHD.
“If donor T cells expand and cause severe graft-vs-host disease, we can give the patient ganciclovir, and it should kill the T cells and stop the process,” said study author John F. DiPersio, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
Dr DiPersio and his colleagues tested the CD34-TK75-enriched T cells in 8 patients—4 with acute myeloid leukemia and 4 with myelodysplastic syndromes—who relapsed after allogeneic transplant. Six patients underwent [18F]FHBG PET/CT to track the T cells at several time points after infusion.
Patients received T-cell infusions ranging from 0.1 × 106 cells/kg to 1.3 × 106 cells/kg. Seven of the 8 patients received chemotherapy before T-cell infusion.
Four patients achieved a complete remission, 1 had progressive disease, and 3 patients died before the researchers could evaluate them for response.
Two patients developed GVHD—1 before T-cell infusion and 1 after. The patient who developed GVHD before infusion had grade 2 liver GVHD that resolved after an increased dose of steroids. The patient ultimately died of relapsed leukemia.
The patient who developed GVHD after T-cell infusion did not respond to chemotherapy or T-cell infusion. At 64 days after infusion, he exhibited symptoms consistent with grade 4 liver GVHD. He was treated with high-dose steroids and ganciclovir but did not respond. His primary cause of death was relapsed/progressive disease.
The mean overall survival after T-cell infusion was 165 days, 4 patients were still alive at 6 months, and 1 patient lived 408 days. All of the patients ultimately died.
The researchers said they did not detect any replication competent retrovirus or any antibodies against CD34-TK in any of the patients. The team also said there were no toxicities related to the CD34-TK75-enriched T cells.
Among patients who underwent imaging, there was no clear distinction between the [18F]FHBG biodistribution at baseline and later time points. And there was no difference between images in the patient who developed GVHD after infusion and patients who did not.
Past work from Dr DiPersio’s lab showed that leukemic mice that receive donor T cells and go on to develop GVHD show a characteristic migration pattern of the T cells through the body. When the cells gather early in the thymus, the mice develop GVHD. When T cells don’t migrate to the thymus, GVHD doesn’t occur.
In the current study, the patients received a relatively small number of CD34-TK75-enriched T cells, so the researchers were not able to show whether the migration patterns in mice were similar in humans.
However, based on the results of this study, Dr DiPersio and his colleagues are participating in a larger trial in partnership with a company based in Italy. The researchers are planning to include patients from multiple medical centers, and each participant should receive about 50 times more of the CD34-TK75-enriched T cells than were administered in this trial.
Washington University is the only center in the new trial that will be able to perform the imaging studies examining migration patterns of the T cells. ![]()
Image from PLOS ONE
A T-cell therapy designed to mitigate graft-vs-host disease (GVHD) is both feasible and safe, according to results of a pilot study published in Molecular Therapy.
To create this therapy, researchers transduced donor T cells with γ-retroviruses carrying a CD34-TK75 fusion gene.
The team said this allows them to track the cells via PET/CT and induce apoptosis by administering the antiviral drug ganciclovir if patients begin showing signs of GVHD.
“If donor T cells expand and cause severe graft-vs-host disease, we can give the patient ganciclovir, and it should kill the T cells and stop the process,” said study author John F. DiPersio, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
Dr DiPersio and his colleagues tested the CD34-TK75-enriched T cells in 8 patients—4 with acute myeloid leukemia and 4 with myelodysplastic syndromes—who relapsed after allogeneic transplant. Six patients underwent [18F]FHBG PET/CT to track the T cells at several time points after infusion.
Patients received T-cell infusions ranging from 0.1 × 106 cells/kg to 1.3 × 106 cells/kg. Seven of the 8 patients received chemotherapy before T-cell infusion.
Four patients achieved a complete remission, 1 had progressive disease, and 3 patients died before the researchers could evaluate them for response.
Two patients developed GVHD—1 before T-cell infusion and 1 after. The patient who developed GVHD before infusion had grade 2 liver GVHD that resolved after an increased dose of steroids. The patient ultimately died of relapsed leukemia.
The patient who developed GVHD after T-cell infusion did not respond to chemotherapy or T-cell infusion. At 64 days after infusion, he exhibited symptoms consistent with grade 4 liver GVHD. He was treated with high-dose steroids and ganciclovir but did not respond. His primary cause of death was relapsed/progressive disease.
The mean overall survival after T-cell infusion was 165 days, 4 patients were still alive at 6 months, and 1 patient lived 408 days. All of the patients ultimately died.
The researchers said they did not detect any replication competent retrovirus or any antibodies against CD34-TK in any of the patients. The team also said there were no toxicities related to the CD34-TK75-enriched T cells.
Among patients who underwent imaging, there was no clear distinction between the [18F]FHBG biodistribution at baseline and later time points. And there was no difference between images in the patient who developed GVHD after infusion and patients who did not.
Past work from Dr DiPersio’s lab showed that leukemic mice that receive donor T cells and go on to develop GVHD show a characteristic migration pattern of the T cells through the body. When the cells gather early in the thymus, the mice develop GVHD. When T cells don’t migrate to the thymus, GVHD doesn’t occur.
In the current study, the patients received a relatively small number of CD34-TK75-enriched T cells, so the researchers were not able to show whether the migration patterns in mice were similar in humans.
However, based on the results of this study, Dr DiPersio and his colleagues are participating in a larger trial in partnership with a company based in Italy. The researchers are planning to include patients from multiple medical centers, and each participant should receive about 50 times more of the CD34-TK75-enriched T cells than were administered in this trial.
Washington University is the only center in the new trial that will be able to perform the imaging studies examining migration patterns of the T cells. ![]()
Image from PLOS ONE
A T-cell therapy designed to mitigate graft-vs-host disease (GVHD) is both feasible and safe, according to results of a pilot study published in Molecular Therapy.
To create this therapy, researchers transduced donor T cells with γ-retroviruses carrying a CD34-TK75 fusion gene.
The team said this allows them to track the cells via PET/CT and induce apoptosis by administering the antiviral drug ganciclovir if patients begin showing signs of GVHD.
“If donor T cells expand and cause severe graft-vs-host disease, we can give the patient ganciclovir, and it should kill the T cells and stop the process,” said study author John F. DiPersio, MD, PhD, of the Washington University School of Medicine in St Louis, Missouri.
Dr DiPersio and his colleagues tested the CD34-TK75-enriched T cells in 8 patients—4 with acute myeloid leukemia and 4 with myelodysplastic syndromes—who relapsed after allogeneic transplant. Six patients underwent [18F]FHBG PET/CT to track the T cells at several time points after infusion.
Patients received T-cell infusions ranging from 0.1 × 106 cells/kg to 1.3 × 106 cells/kg. Seven of the 8 patients received chemotherapy before T-cell infusion.
Four patients achieved a complete remission, 1 had progressive disease, and 3 patients died before the researchers could evaluate them for response.
Two patients developed GVHD—1 before T-cell infusion and 1 after. The patient who developed GVHD before infusion had grade 2 liver GVHD that resolved after an increased dose of steroids. The patient ultimately died of relapsed leukemia.
The patient who developed GVHD after T-cell infusion did not respond to chemotherapy or T-cell infusion. At 64 days after infusion, he exhibited symptoms consistent with grade 4 liver GVHD. He was treated with high-dose steroids and ganciclovir but did not respond. His primary cause of death was relapsed/progressive disease.
The mean overall survival after T-cell infusion was 165 days, 4 patients were still alive at 6 months, and 1 patient lived 408 days. All of the patients ultimately died.
The researchers said they did not detect any replication competent retrovirus or any antibodies against CD34-TK in any of the patients. The team also said there were no toxicities related to the CD34-TK75-enriched T cells.
Among patients who underwent imaging, there was no clear distinction between the [18F]FHBG biodistribution at baseline and later time points. And there was no difference between images in the patient who developed GVHD after infusion and patients who did not.
Past work from Dr DiPersio’s lab showed that leukemic mice that receive donor T cells and go on to develop GVHD show a characteristic migration pattern of the T cells through the body. When the cells gather early in the thymus, the mice develop GVHD. When T cells don’t migrate to the thymus, GVHD doesn’t occur.
In the current study, the patients received a relatively small number of CD34-TK75-enriched T cells, so the researchers were not able to show whether the migration patterns in mice were similar in humans.
However, based on the results of this study, Dr DiPersio and his colleagues are participating in a larger trial in partnership with a company based in Italy. The researchers are planning to include patients from multiple medical centers, and each participant should receive about 50 times more of the CD34-TK75-enriched T cells than were administered in this trial.
Washington University is the only center in the new trial that will be able to perform the imaging studies examining migration patterns of the T cells. ![]()
Blinatumomab for hard-to-treat form of acute lymphoblastic leukemia
The US Food and Drug Administration (FDA) has granted accelerated approval to blinatumomab for the treatment of adult patients with relapsed/ refractory Philadelphia chromosome-negative precursor B-cell acute lymphoblastic leukemia (BCP-ALL).1 Blinatumomab is the first of a novel class of antibodies to receive regulatory approval; a bispecific antibody targeting both CD19, expressed on the surface of B cells, and CD3, on cytotoxic T cells. The approval was based on the findings of a single-arm, multicenter, open-label study in patients at high-risk of poor outcome, which showed a significant improvement of blinatumomab over other available therapies in this setting.2
Click on the PDF icon at the top of this introduction to read the full article.
The US Food and Drug Administration (FDA) has granted accelerated approval to blinatumomab for the treatment of adult patients with relapsed/ refractory Philadelphia chromosome-negative precursor B-cell acute lymphoblastic leukemia (BCP-ALL).1 Blinatumomab is the first of a novel class of antibodies to receive regulatory approval; a bispecific antibody targeting both CD19, expressed on the surface of B cells, and CD3, on cytotoxic T cells. The approval was based on the findings of a single-arm, multicenter, open-label study in patients at high-risk of poor outcome, which showed a significant improvement of blinatumomab over other available therapies in this setting.2
Click on the PDF icon at the top of this introduction to read the full article.
The US Food and Drug Administration (FDA) has granted accelerated approval to blinatumomab for the treatment of adult patients with relapsed/ refractory Philadelphia chromosome-negative precursor B-cell acute lymphoblastic leukemia (BCP-ALL).1 Blinatumomab is the first of a novel class of antibodies to receive regulatory approval; a bispecific antibody targeting both CD19, expressed on the surface of B cells, and CD3, on cytotoxic T cells. The approval was based on the findings of a single-arm, multicenter, open-label study in patients at high-risk of poor outcome, which showed a significant improvement of blinatumomab over other available therapies in this setting.2
Click on the PDF icon at the top of this introduction to read the full article.
The generalist’s dilemma: training and staying current in the face of increasing specialization
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Elotuzumab plus len-dex extends remissions of multiple myeloma
Adding the investigational monoclonal antibody elotuzumab to a standard regimen for early relapsed/refractory multiple myeloma extended the duration of remissions by a mean of 5 months.
The combination of elotuzumab (elo), lenalidomide (len), and dexamethasone (dex) was associated with a 30% reduction in the risk of disease progression or death compared with len/dex alone, and the benefit of the drug was also seen, although to a lesser degree, in patients with high-risk disease mutations, reported Dr. Sagar Lonial of the Winship Cancer Institute of Emory University, Atlanta.
“We were certainly excited about the fact that there is such a big difference in progression-free survival between the two arms,” he said at a briefing on studies to be presented at the annual meeting of the American Society of Clinical Oncology.
“Patients who received elotuzumab had a longer duration of remission and had a higher overall response rate, and this improvement in clinical parameters occurred without a significant increase in adverse events or toxicity,” he added.
Dr. Julie M. Vose, ASCO president-elect, who comoderated the briefing, said that “although we have had monoclonal antibodies in a number of other malignancies for years, this truly is the first one for myeloma.”
Dr. Lonial reported interim results from ELOQUENT-2, a phase III study comparing the efficacy and safety of the elo/len/dex combination vs. len/dex alone in 646 patients with relapsed/refractory multiple myeloma who had received one, two, or three prior lines of therapy and whose disease was not resistant to len/dex.
The patients were randomized to receive either oral len/dex on a standard schedule, or oral lenalidomide plus oral dexamethasone on weeks when there was no elotuzumab infusion, with both oral and intravenous dexamethasone delivered on weeks when the antibody was delivered by solutional infusion. The regimens were delivered in 28-day cycles until disease progression or unacceptable toxicities occurred.
At 24 months median follow-up, the mean progression-free survival (PFS) for patients on elo/len/dex was 19.4 months, compared with 14.9 months for len/dex alone (P = .0004). One-year PFS rates were 68% for elo/len/dex, compared with 57% for len/dex. Two-year PFS rates were 41% vs. 27%, respectively.
The overall response rate with the antibody-containing combination was 79% compared with 66% for len/dex alone (P = .0002). Dr. Lonial noted that the antibody-containing combination was effective both in patients with average risk, and in those with the high risk cytogenetic abnormalities, including the 17p deletion and the t(4;14) chromosomal translocation, although the response was not as robust in these patients.
A total of 210 patients died during follow-up, 94 who had been treated with elo/len/dex, and 116 who had been treated with len/dex.
Grade 3 or 4 adverse events occurring in 15% or more of patients included neutropenia in 25% of patients on elo/len/dex, and 33% of patients on len/dex, and anemia, which occurred in 15% and 16% of patients, respectively. In all, 10% of patients in the elotuzumab arm had mild infusion reactions after the first few doses.
Based on data from earlier trials, the Food and Drug Administration has granted elotuzumab breakthrough designation for treatment of multiple myeloma that has relapsed or is refractory to at least one prior line of therapy. Breakthrough status entitles the manufacturer to an expedited review of the drug.
Adding the investigational monoclonal antibody elotuzumab to a standard regimen for early relapsed/refractory multiple myeloma extended the duration of remissions by a mean of 5 months.
The combination of elotuzumab (elo), lenalidomide (len), and dexamethasone (dex) was associated with a 30% reduction in the risk of disease progression or death compared with len/dex alone, and the benefit of the drug was also seen, although to a lesser degree, in patients with high-risk disease mutations, reported Dr. Sagar Lonial of the Winship Cancer Institute of Emory University, Atlanta.
“We were certainly excited about the fact that there is such a big difference in progression-free survival between the two arms,” he said at a briefing on studies to be presented at the annual meeting of the American Society of Clinical Oncology.
“Patients who received elotuzumab had a longer duration of remission and had a higher overall response rate, and this improvement in clinical parameters occurred without a significant increase in adverse events or toxicity,” he added.
Dr. Julie M. Vose, ASCO president-elect, who comoderated the briefing, said that “although we have had monoclonal antibodies in a number of other malignancies for years, this truly is the first one for myeloma.”
Dr. Lonial reported interim results from ELOQUENT-2, a phase III study comparing the efficacy and safety of the elo/len/dex combination vs. len/dex alone in 646 patients with relapsed/refractory multiple myeloma who had received one, two, or three prior lines of therapy and whose disease was not resistant to len/dex.
The patients were randomized to receive either oral len/dex on a standard schedule, or oral lenalidomide plus oral dexamethasone on weeks when there was no elotuzumab infusion, with both oral and intravenous dexamethasone delivered on weeks when the antibody was delivered by solutional infusion. The regimens were delivered in 28-day cycles until disease progression or unacceptable toxicities occurred.
At 24 months median follow-up, the mean progression-free survival (PFS) for patients on elo/len/dex was 19.4 months, compared with 14.9 months for len/dex alone (P = .0004). One-year PFS rates were 68% for elo/len/dex, compared with 57% for len/dex. Two-year PFS rates were 41% vs. 27%, respectively.
The overall response rate with the antibody-containing combination was 79% compared with 66% for len/dex alone (P = .0002). Dr. Lonial noted that the antibody-containing combination was effective both in patients with average risk, and in those with the high risk cytogenetic abnormalities, including the 17p deletion and the t(4;14) chromosomal translocation, although the response was not as robust in these patients.
A total of 210 patients died during follow-up, 94 who had been treated with elo/len/dex, and 116 who had been treated with len/dex.
Grade 3 or 4 adverse events occurring in 15% or more of patients included neutropenia in 25% of patients on elo/len/dex, and 33% of patients on len/dex, and anemia, which occurred in 15% and 16% of patients, respectively. In all, 10% of patients in the elotuzumab arm had mild infusion reactions after the first few doses.
Based on data from earlier trials, the Food and Drug Administration has granted elotuzumab breakthrough designation for treatment of multiple myeloma that has relapsed or is refractory to at least one prior line of therapy. Breakthrough status entitles the manufacturer to an expedited review of the drug.
Adding the investigational monoclonal antibody elotuzumab to a standard regimen for early relapsed/refractory multiple myeloma extended the duration of remissions by a mean of 5 months.
The combination of elotuzumab (elo), lenalidomide (len), and dexamethasone (dex) was associated with a 30% reduction in the risk of disease progression or death compared with len/dex alone, and the benefit of the drug was also seen, although to a lesser degree, in patients with high-risk disease mutations, reported Dr. Sagar Lonial of the Winship Cancer Institute of Emory University, Atlanta.
“We were certainly excited about the fact that there is such a big difference in progression-free survival between the two arms,” he said at a briefing on studies to be presented at the annual meeting of the American Society of Clinical Oncology.
“Patients who received elotuzumab had a longer duration of remission and had a higher overall response rate, and this improvement in clinical parameters occurred without a significant increase in adverse events or toxicity,” he added.
Dr. Julie M. Vose, ASCO president-elect, who comoderated the briefing, said that “although we have had monoclonal antibodies in a number of other malignancies for years, this truly is the first one for myeloma.”
Dr. Lonial reported interim results from ELOQUENT-2, a phase III study comparing the efficacy and safety of the elo/len/dex combination vs. len/dex alone in 646 patients with relapsed/refractory multiple myeloma who had received one, two, or three prior lines of therapy and whose disease was not resistant to len/dex.
The patients were randomized to receive either oral len/dex on a standard schedule, or oral lenalidomide plus oral dexamethasone on weeks when there was no elotuzumab infusion, with both oral and intravenous dexamethasone delivered on weeks when the antibody was delivered by solutional infusion. The regimens were delivered in 28-day cycles until disease progression or unacceptable toxicities occurred.
At 24 months median follow-up, the mean progression-free survival (PFS) for patients on elo/len/dex was 19.4 months, compared with 14.9 months for len/dex alone (P = .0004). One-year PFS rates were 68% for elo/len/dex, compared with 57% for len/dex. Two-year PFS rates were 41% vs. 27%, respectively.
The overall response rate with the antibody-containing combination was 79% compared with 66% for len/dex alone (P = .0002). Dr. Lonial noted that the antibody-containing combination was effective both in patients with average risk, and in those with the high risk cytogenetic abnormalities, including the 17p deletion and the t(4;14) chromosomal translocation, although the response was not as robust in these patients.
A total of 210 patients died during follow-up, 94 who had been treated with elo/len/dex, and 116 who had been treated with len/dex.
Grade 3 or 4 adverse events occurring in 15% or more of patients included neutropenia in 25% of patients on elo/len/dex, and 33% of patients on len/dex, and anemia, which occurred in 15% and 16% of patients, respectively. In all, 10% of patients in the elotuzumab arm had mild infusion reactions after the first few doses.
Based on data from earlier trials, the Food and Drug Administration has granted elotuzumab breakthrough designation for treatment of multiple myeloma that has relapsed or is refractory to at least one prior line of therapy. Breakthrough status entitles the manufacturer to an expedited review of the drug.
Key clinical point: The novel antibody elotuzumab extended multiple myeloma remissions when added to standard therapy.
Major finding: Mean progression-free survival at 24-months median follow-up was 19.4 months for elotuzumab plus lenalidomide/dexamethasone, vs. 14.9 months for lenalidomide/dexamethasone alone.
Data source: Interim analysis of a phase III randomized trial of 646 patients with relapsed/refractory multiple myeloma.
Disclosures: Bristol-Myers Squibb and AbbVie sponsored the trial. Dr. Lonial disclosed consulting/advisory roles with BMS and other companies. His coauthors disclosed multiple industry relationships.
Familial factors linked to child’s risk of blood cancer

A new study has linked a father’s age at his child’s birth to the risk that the child will develop a hematologic malignancy as an adult, but this risk only proved significant among children without siblings.
Only-children whose fathers were 35 or older at the child’s birth were significantly more likely to develop hematologic malignancies than only-children with fathers who were younger than 25 at the child’s birth.
There was no association between these cancers and a mother’s age, either among only-children or those with siblings.
A previous study of more than 100,000 women also showed an association between paternal—but not maternal—age at a child’s birth and the risk of hematologic malignancy.
To further investigate the association, Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues analyzed data from women and men enrolled in the American Cancer Society Cancer Prevention Study-II Nutrition Cohort.
The team reported their findings in the American Journal of Epidemiology.
Among the 138,003 participants, there were 2532 cases of hematologic malignancies diagnosed between 1992 and 2009.
Subjects’ mothers tended to be younger at their birth than fathers, with median ages of 27 and 31, respectively. Almost a third of the fathers were 35 or older when a subject was born, compared with 17% of the mothers.
In the categorical analysis, the researchers found a positive association between older paternal age at a subject’s birth and the risk of hematologic malignancies in male, but not female, subjects. The hazard ratio (HR) was 1.35 for male subjects with fathers aged 35 and older compared to those whose fathers were younger than 25.
On the other hand, when paternal age was modeled as a continuous variable, there was no association with the risk of hematologic malignancy for males or females. Likewise, there was no association between maternal age at a subject’s birth and the risk of hematologic malignancy in male or female subjects.
However, among subjects without siblings, there was a significant, positive association with paternal age and the risk of hematologic malignancy (P=0.002).
When the researchers separated only-children by sex, they found a suggestive positive association between paternal age and hematologic malignancy for females (HR=1.40) and a significant association for males (HR=1.84). However, the linear spline was significant for males (P=0.01) and females (P=0.04).
There was no association between paternal age at a subject’s birth and the risk of hematologic malignancy among subjects with at least 1 sibling (HR=1.06).
The researchers said the fact that the association between paternal age and malignancy was significant in subjects with no siblings suggests it may be related to the “hygiene hypothesis”—the idea that exposure to mild infections in childhood, which might be more numerous with more siblings, are important to immune system development and may reduce the risk of immune-related diseases.
It is possible that the combination of having an older father and no siblings may promote cell proliferation in those individuals with an underdeveloped immune system and, as such, favors the development of cancers related to the immune system, the team said.
They added that this study suggests a need for further research to better understand the association between paternal age at a child’s birth and hematologic malignancies.
“The lifetime risk of these cancers is fairly low—about 1 in 20 men and women will be diagnosed with lymphoma, leukemia, or myeloma at some point during their lifetime—so people born to older fathers should not be alarmed,” Dr Teras said.
“Still, the study does highlight the need for more research to confirm these findings and to clarify the biologic underpinning for this association, given the growing number of children born to older fathers in the United States and worldwide.” ![]()
A new study has linked a father’s age at his child’s birth to the risk that the child will develop a hematologic malignancy as an adult, but this risk only proved significant among children without siblings.
Only-children whose fathers were 35 or older at the child’s birth were significantly more likely to develop hematologic malignancies than only-children with fathers who were younger than 25 at the child’s birth.
There was no association between these cancers and a mother’s age, either among only-children or those with siblings.
A previous study of more than 100,000 women also showed an association between paternal—but not maternal—age at a child’s birth and the risk of hematologic malignancy.
To further investigate the association, Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues analyzed data from women and men enrolled in the American Cancer Society Cancer Prevention Study-II Nutrition Cohort.
The team reported their findings in the American Journal of Epidemiology.
Among the 138,003 participants, there were 2532 cases of hematologic malignancies diagnosed between 1992 and 2009.
Subjects’ mothers tended to be younger at their birth than fathers, with median ages of 27 and 31, respectively. Almost a third of the fathers were 35 or older when a subject was born, compared with 17% of the mothers.
In the categorical analysis, the researchers found a positive association between older paternal age at a subject’s birth and the risk of hematologic malignancies in male, but not female, subjects. The hazard ratio (HR) was 1.35 for male subjects with fathers aged 35 and older compared to those whose fathers were younger than 25.
On the other hand, when paternal age was modeled as a continuous variable, there was no association with the risk of hematologic malignancy for males or females. Likewise, there was no association between maternal age at a subject’s birth and the risk of hematologic malignancy in male or female subjects.
However, among subjects without siblings, there was a significant, positive association with paternal age and the risk of hematologic malignancy (P=0.002).
When the researchers separated only-children by sex, they found a suggestive positive association between paternal age and hematologic malignancy for females (HR=1.40) and a significant association for males (HR=1.84). However, the linear spline was significant for males (P=0.01) and females (P=0.04).
There was no association between paternal age at a subject’s birth and the risk of hematologic malignancy among subjects with at least 1 sibling (HR=1.06).
The researchers said the fact that the association between paternal age and malignancy was significant in subjects with no siblings suggests it may be related to the “hygiene hypothesis”—the idea that exposure to mild infections in childhood, which might be more numerous with more siblings, are important to immune system development and may reduce the risk of immune-related diseases.
It is possible that the combination of having an older father and no siblings may promote cell proliferation in those individuals with an underdeveloped immune system and, as such, favors the development of cancers related to the immune system, the team said.
They added that this study suggests a need for further research to better understand the association between paternal age at a child’s birth and hematologic malignancies.
“The lifetime risk of these cancers is fairly low—about 1 in 20 men and women will be diagnosed with lymphoma, leukemia, or myeloma at some point during their lifetime—so people born to older fathers should not be alarmed,” Dr Teras said.
“Still, the study does highlight the need for more research to confirm these findings and to clarify the biologic underpinning for this association, given the growing number of children born to older fathers in the United States and worldwide.” ![]()
A new study has linked a father’s age at his child’s birth to the risk that the child will develop a hematologic malignancy as an adult, but this risk only proved significant among children without siblings.
Only-children whose fathers were 35 or older at the child’s birth were significantly more likely to develop hematologic malignancies than only-children with fathers who were younger than 25 at the child’s birth.
There was no association between these cancers and a mother’s age, either among only-children or those with siblings.
A previous study of more than 100,000 women also showed an association between paternal—but not maternal—age at a child’s birth and the risk of hematologic malignancy.
To further investigate the association, Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues analyzed data from women and men enrolled in the American Cancer Society Cancer Prevention Study-II Nutrition Cohort.
The team reported their findings in the American Journal of Epidemiology.
Among the 138,003 participants, there were 2532 cases of hematologic malignancies diagnosed between 1992 and 2009.
Subjects’ mothers tended to be younger at their birth than fathers, with median ages of 27 and 31, respectively. Almost a third of the fathers were 35 or older when a subject was born, compared with 17% of the mothers.
In the categorical analysis, the researchers found a positive association between older paternal age at a subject’s birth and the risk of hematologic malignancies in male, but not female, subjects. The hazard ratio (HR) was 1.35 for male subjects with fathers aged 35 and older compared to those whose fathers were younger than 25.
On the other hand, when paternal age was modeled as a continuous variable, there was no association with the risk of hematologic malignancy for males or females. Likewise, there was no association between maternal age at a subject’s birth and the risk of hematologic malignancy in male or female subjects.
However, among subjects without siblings, there was a significant, positive association with paternal age and the risk of hematologic malignancy (P=0.002).
When the researchers separated only-children by sex, they found a suggestive positive association between paternal age and hematologic malignancy for females (HR=1.40) and a significant association for males (HR=1.84). However, the linear spline was significant for males (P=0.01) and females (P=0.04).
There was no association between paternal age at a subject’s birth and the risk of hematologic malignancy among subjects with at least 1 sibling (HR=1.06).
The researchers said the fact that the association between paternal age and malignancy was significant in subjects with no siblings suggests it may be related to the “hygiene hypothesis”—the idea that exposure to mild infections in childhood, which might be more numerous with more siblings, are important to immune system development and may reduce the risk of immune-related diseases.
It is possible that the combination of having an older father and no siblings may promote cell proliferation in those individuals with an underdeveloped immune system and, as such, favors the development of cancers related to the immune system, the team said.
They added that this study suggests a need for further research to better understand the association between paternal age at a child’s birth and hematologic malignancies.
“The lifetime risk of these cancers is fairly low—about 1 in 20 men and women will be diagnosed with lymphoma, leukemia, or myeloma at some point during their lifetime—so people born to older fathers should not be alarmed,” Dr Teras said.
“Still, the study does highlight the need for more research to confirm these findings and to clarify the biologic underpinning for this association, given the growing number of children born to older fathers in the United States and worldwide.” ![]()
Team links telomere degeneration and MDS
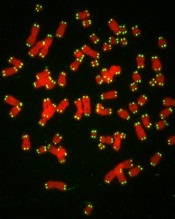
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
Explaining obesity in cancer survivors

Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Researchers have identified several factors that may influence the risk of obesity in childhood cancer survivors.
Previous research showed that obesity rates are elevated in childhood cancer survivors who were exposed to cranial radiation.
But the new study has shown that other types of treatment, a patient’s age, and certain genetic variants are associated with obesity in this population.
Carmen Wilson, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues reported these findings in Cancer.
The researchers evaluated 1,996 childhood cancer survivors treated at St. Jude. The patients’ median age at diagnosis was 7.2 years (range, 0.1-24.8), and their median age at follow-up was 32.4 years (range, 18.9-63.8).
At the time of evaluation, 645 patients (32.3%) were of normal weight, 71 (3.6%) were underweight, 556 (27.9%) were overweight, and 723 (36.2%) were obese.
The prevalence of obesity was highest in male leukemia survivors (42.5%) and females who survived neuroblastoma (43.6%), followed closely by those who survived leukemia (43.1%).
Multivariable analyses showed that 3 factors were independently associated with an increased risk of obesity: older age at the time of evaluation (≥30 years vs <30 years; P<0.001), undergoing cranial radiation (P<0.001), and receiving glucocorticoids (P=0.004).
On the other hand, receiving chest, abdominal, or pelvic radiation was associated with a decreased risk of obesity (P<0.001).
The researchers also identified 166 single nucleotide polymorphisms that were associated with obesity among cancer survivors who had received cranial radiation. The strongest association was in variants of genes involved in neuron growth, repair, and connectivity.
Among survivors who did not receive cranial radiation, only 1 single nucleotide polymorphism—rs12073359, located on chromosome 1—was associated with an increased risk of obesity.
Dr Wilson said these findings might help us identify the childhood cancer survivors who are most likely to become obese. The results may also provide a foundation for future research efforts aimed at characterizing molecular pathways involved in the link between childhood cancer treatment and obesity. ![]()
Inhibitor may benefit certain ALL patients
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents.
*Information in the abstract differs from that presented at the meeting.