User login
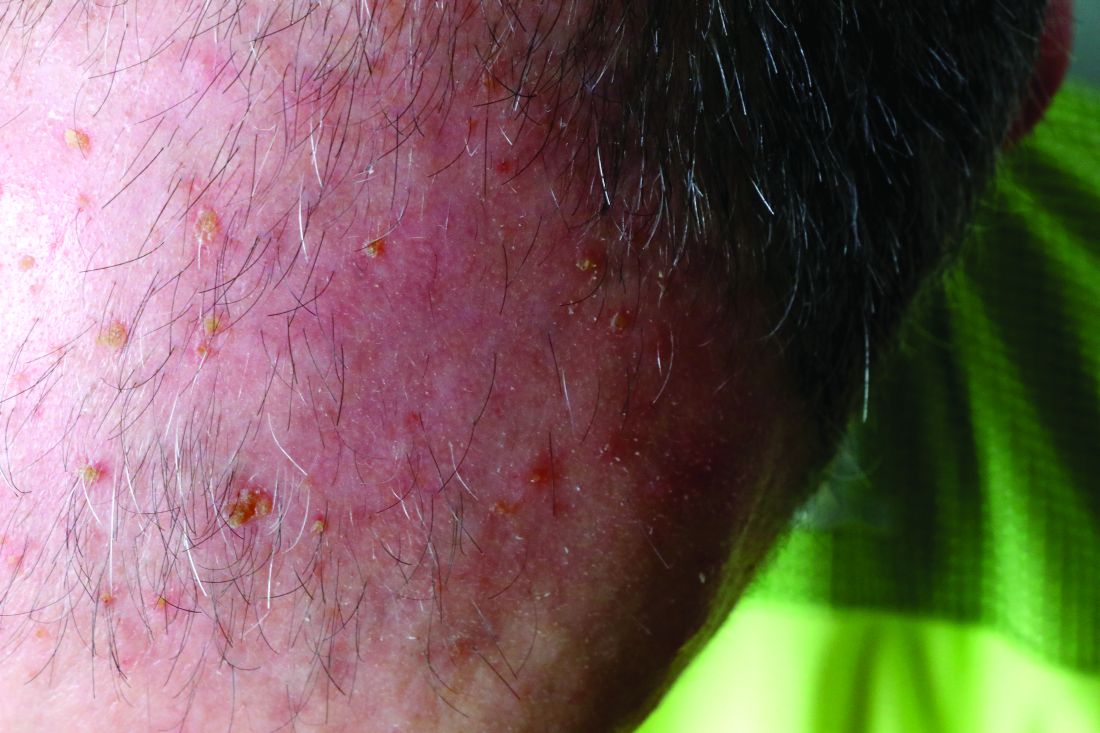
Pigmentation on foot

The FP suspected that this was an acral lentiginous melanoma (ALM).
The FP considered the various ways to biopsy this lesion. Doing a biopsy of the whole lesion was neither practical, nor desirable, given that the lesion was >2 cm and located on the bottom of the patient’s foot. And while incomplete sampling could result in a false negative result, this lesion was so suspicious for melanoma that any well-performed biopsy would likely produce a diagnosis of melanoma. (Of course if melanoma was not diagnosable with partial sampling, a full excisional biopsy would be needed.)
The FP also debated doing a 6-mm punch biopsy vs a shave biopsy; his intent was to get below some of the deeper pigment. Incisional biopsy done as a small ellipse with suturing was also an option. It would also be the most time consuming.
The FP presented the biopsy options to the patient and explained that the shave biopsy would require a dressing and the punch biopsy would require sutures, which would need to be removed at the next visit. Both biopsy methods would likely cause the foot to be uncomfortable to walk on for several days.
The FP and patient decided to do a deep shave biopsy (saucerization). After administering local anesthesia with lidocaine and epinephrine, the FP performed a deep shave biopsy that included about 1 cm of the darkest and thickest portion of the suspected melanoma.
The biopsy results indicated that the patient had an acral lentiginous melanoma, which measured 0.7 mm at its deepest point in the biopsied portion. The patient was referred to Orthopedics for a wide excision and repair. A sentinel node biopsy was not needed because the lesion was less than 1 mm in depth and there was no ulceration. The surgery and healing time were challenging for the patient, but she did not lose her foot and has remained cancer free.
Photo courtesy of Jonathan Karnes, MD and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Squamous cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1103-1111.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com
The FP suspected that this was an acral lentiginous melanoma (ALM).
The FP considered the various ways to biopsy this lesion. Doing a biopsy of the whole lesion was neither practical, nor desirable, given that the lesion was >2 cm and located on the bottom of the patient’s foot. And while incomplete sampling could result in a false negative result, this lesion was so suspicious for melanoma that any well-performed biopsy would likely produce a diagnosis of melanoma. (Of course if melanoma was not diagnosable with partial sampling, a full excisional biopsy would be needed.)
The FP also debated doing a 6-mm punch biopsy vs a shave biopsy; his intent was to get below some of the deeper pigment. Incisional biopsy done as a small ellipse with suturing was also an option. It would also be the most time consuming.
The FP presented the biopsy options to the patient and explained that the shave biopsy would require a dressing and the punch biopsy would require sutures, which would need to be removed at the next visit. Both biopsy methods would likely cause the foot to be uncomfortable to walk on for several days.
The FP and patient decided to do a deep shave biopsy (saucerization). After administering local anesthesia with lidocaine and epinephrine, the FP performed a deep shave biopsy that included about 1 cm of the darkest and thickest portion of the suspected melanoma.
The biopsy results indicated that the patient had an acral lentiginous melanoma, which measured 0.7 mm at its deepest point in the biopsied portion. The patient was referred to Orthopedics for a wide excision and repair. A sentinel node biopsy was not needed because the lesion was less than 1 mm in depth and there was no ulceration. The surgery and healing time were challenging for the patient, but she did not lose her foot and has remained cancer free.
Photo courtesy of Jonathan Karnes, MD and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Squamous cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1103-1111.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com
The FP suspected that this was an acral lentiginous melanoma (ALM).
The FP considered the various ways to biopsy this lesion. Doing a biopsy of the whole lesion was neither practical, nor desirable, given that the lesion was >2 cm and located on the bottom of the patient’s foot. And while incomplete sampling could result in a false negative result, this lesion was so suspicious for melanoma that any well-performed biopsy would likely produce a diagnosis of melanoma. (Of course if melanoma was not diagnosable with partial sampling, a full excisional biopsy would be needed.)
The FP also debated doing a 6-mm punch biopsy vs a shave biopsy; his intent was to get below some of the deeper pigment. Incisional biopsy done as a small ellipse with suturing was also an option. It would also be the most time consuming.
The FP presented the biopsy options to the patient and explained that the shave biopsy would require a dressing and the punch biopsy would require sutures, which would need to be removed at the next visit. Both biopsy methods would likely cause the foot to be uncomfortable to walk on for several days.
The FP and patient decided to do a deep shave biopsy (saucerization). After administering local anesthesia with lidocaine and epinephrine, the FP performed a deep shave biopsy that included about 1 cm of the darkest and thickest portion of the suspected melanoma.
The biopsy results indicated that the patient had an acral lentiginous melanoma, which measured 0.7 mm at its deepest point in the biopsied portion. The patient was referred to Orthopedics for a wide excision and repair. A sentinel node biopsy was not needed because the lesion was less than 1 mm in depth and there was no ulceration. The surgery and healing time were challenging for the patient, but she did not lose her foot and has remained cancer free.
Photo courtesy of Jonathan Karnes, MD and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Karnes J, Usatine R. Squamous cell carcinoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1103-1111.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com

Consider individualized testosterone protocol for transgender acne
but patient compatibility will depend upon several factors, advised Jason A. Park and his associates in a research letter to the editor of the Journal of the American Academy of Dermatology.
In a multivariate logistic regression analysis, Mr. Park and his colleagues at Boston University sought to determine the timing of onset of acne in female-to-male transgender patients.
A total of 55 patients undergoing hormone therapy at the Center for Transgender Medicine and Surgery at Boston Medical Center between January 1, 2010, and December 31, 2017 were selected following a systematic chart review. Patients were excluded who were under the age of 18 years, who had been receiving testosterone therapy for less than 2 years, who presented with acne before start of treatment, or whose medical records were incomplete.
Given evidence in prior studies reporting on an association between elevated androgen levels and increased incidence of acne in this patient group, a median serum testosterone level of 630 ng/dL “was used to differentiate between higher and lower levels.”
Acne was found to develop in 9% of transgender men after 3 months and in 18% after 6 months; 38% of the subjects were found to have developed acne at some point during the study after 24 months of treatment. The authors found that acne was “significantly associated with serum testosterone levels higher than 630 ng/dL.” Increased body mass index (BMI), especially in those with positive smoking status, also was associated with an increased incidence of acne, the authors said.
According to several existing studies, transgender men undergoing testosterone therapy tend to develop increased sebum production and acne. Because the systemic and dermatologic virilization effects of testosterone are unpredictable once treatment has started, and because individual goals also are varied (from maximum virilization to only suppressing feminine secondary sex characteristics), Mr. Park and his colleagues suggested that customization may be ideal, provided they do not clash with individual patient transition goals, priorities, risk factors, and other comorbidities that may be present.
The study was funded by the Medical Student Summer Research Program at Boston University. The authors had no conflicts of interest to report.
SOURCE: Park JA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.12.040.
but patient compatibility will depend upon several factors, advised Jason A. Park and his associates in a research letter to the editor of the Journal of the American Academy of Dermatology.
In a multivariate logistic regression analysis, Mr. Park and his colleagues at Boston University sought to determine the timing of onset of acne in female-to-male transgender patients.
A total of 55 patients undergoing hormone therapy at the Center for Transgender Medicine and Surgery at Boston Medical Center between January 1, 2010, and December 31, 2017 were selected following a systematic chart review. Patients were excluded who were under the age of 18 years, who had been receiving testosterone therapy for less than 2 years, who presented with acne before start of treatment, or whose medical records were incomplete.
Given evidence in prior studies reporting on an association between elevated androgen levels and increased incidence of acne in this patient group, a median serum testosterone level of 630 ng/dL “was used to differentiate between higher and lower levels.”
Acne was found to develop in 9% of transgender men after 3 months and in 18% after 6 months; 38% of the subjects were found to have developed acne at some point during the study after 24 months of treatment. The authors found that acne was “significantly associated with serum testosterone levels higher than 630 ng/dL.” Increased body mass index (BMI), especially in those with positive smoking status, also was associated with an increased incidence of acne, the authors said.
According to several existing studies, transgender men undergoing testosterone therapy tend to develop increased sebum production and acne. Because the systemic and dermatologic virilization effects of testosterone are unpredictable once treatment has started, and because individual goals also are varied (from maximum virilization to only suppressing feminine secondary sex characteristics), Mr. Park and his colleagues suggested that customization may be ideal, provided they do not clash with individual patient transition goals, priorities, risk factors, and other comorbidities that may be present.
The study was funded by the Medical Student Summer Research Program at Boston University. The authors had no conflicts of interest to report.
SOURCE: Park JA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.12.040.
but patient compatibility will depend upon several factors, advised Jason A. Park and his associates in a research letter to the editor of the Journal of the American Academy of Dermatology.
In a multivariate logistic regression analysis, Mr. Park and his colleagues at Boston University sought to determine the timing of onset of acne in female-to-male transgender patients.
A total of 55 patients undergoing hormone therapy at the Center for Transgender Medicine and Surgery at Boston Medical Center between January 1, 2010, and December 31, 2017 were selected following a systematic chart review. Patients were excluded who were under the age of 18 years, who had been receiving testosterone therapy for less than 2 years, who presented with acne before start of treatment, or whose medical records were incomplete.
Given evidence in prior studies reporting on an association between elevated androgen levels and increased incidence of acne in this patient group, a median serum testosterone level of 630 ng/dL “was used to differentiate between higher and lower levels.”
Acne was found to develop in 9% of transgender men after 3 months and in 18% after 6 months; 38% of the subjects were found to have developed acne at some point during the study after 24 months of treatment. The authors found that acne was “significantly associated with serum testosterone levels higher than 630 ng/dL.” Increased body mass index (BMI), especially in those with positive smoking status, also was associated with an increased incidence of acne, the authors said.
According to several existing studies, transgender men undergoing testosterone therapy tend to develop increased sebum production and acne. Because the systemic and dermatologic virilization effects of testosterone are unpredictable once treatment has started, and because individual goals also are varied (from maximum virilization to only suppressing feminine secondary sex characteristics), Mr. Park and his colleagues suggested that customization may be ideal, provided they do not clash with individual patient transition goals, priorities, risk factors, and other comorbidities that may be present.
The study was funded by the Medical Student Summer Research Program at Boston University. The authors had no conflicts of interest to report.
SOURCE: Park JA et al. J Am Acad Dermatol. 2019. doi: 10.1016/j.jaad.2018.12.040.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Destress dermatologic procedures with honesty, distraction, relaxation
according to a report published in Pediatric Dermatology.

For many children, the anticipation of pain and the anxiety about a procedure results in a more painful experience, wrote Andrew M. Armenta of the University of Texas, Galveston, and his colleagues. Preparing children in advance and using cognitive behavioral therapy (CBT) strategies in the moment can help reduce their anxiety.
“CBT is a skill‐based approach that focuses on the present and aims to teach efficient ways of identifying distorted thinking, modifying beliefs, and changing behaviors for a more favorable outcome of real‐life situations,” they wrote.
First, Dr. Armenta and his associates advised, be honest with children about what to expect from a procedure. Evidence does not support phrases such as, “It won’t hurt,” or “It will be over soon,” to reduce anxiety.
Timing the disclosure of a procedure and creating the appropriate setting also can help reduce anxiety. For very young children, short notice of a procedure is often best, with the promise of a small reward or outing afterward. Older children may want some advance notice so they can feel prepared, but their specific concerns should be addressed.
CBT-based techniques include deep breathing and positive coping statements such as “I can do this” for older children, or encouraging them to talk about a family pet or listen to music. Younger children may be distracted with pinwheels, rattles, or songs. “Additionally, in recent years, virtual reality headsets have even proved to be effective distractors, resulting in an overall reduction in both pain and fear,” Dr. Armenta and his associates noted.
Other useful strategies include allowing children to choose their position and location for an injection or procedure when possible. Small children may be able to sit on the lap of an adult, and older children may prefer sitting up to lying down. Avoid physical restraint unless it is absolutely necessary for safety, the researchers emphasized.
Incorporating CBT-based strategies of breathing and distraction with honesty and respectful disclosure of what is being done and why “not only makes practicing pediatric dermatology easier, but also can improve patient adherence to painful procedures,” they said.
No disclosure information was given.
SOURCE: Armenta AM et al. Pediatr Dermatol. 2019. doi: 10.1111/pde.13739.
according to a report published in Pediatric Dermatology.
For many children, the anticipation of pain and the anxiety about a procedure results in a more painful experience, wrote Andrew M. Armenta of the University of Texas, Galveston, and his colleagues. Preparing children in advance and using cognitive behavioral therapy (CBT) strategies in the moment can help reduce their anxiety.
“CBT is a skill‐based approach that focuses on the present and aims to teach efficient ways of identifying distorted thinking, modifying beliefs, and changing behaviors for a more favorable outcome of real‐life situations,” they wrote.
First, Dr. Armenta and his associates advised, be honest with children about what to expect from a procedure. Evidence does not support phrases such as, “It won’t hurt,” or “It will be over soon,” to reduce anxiety.
Timing the disclosure of a procedure and creating the appropriate setting also can help reduce anxiety. For very young children, short notice of a procedure is often best, with the promise of a small reward or outing afterward. Older children may want some advance notice so they can feel prepared, but their specific concerns should be addressed.
CBT-based techniques include deep breathing and positive coping statements such as “I can do this” for older children, or encouraging them to talk about a family pet or listen to music. Younger children may be distracted with pinwheels, rattles, or songs. “Additionally, in recent years, virtual reality headsets have even proved to be effective distractors, resulting in an overall reduction in both pain and fear,” Dr. Armenta and his associates noted.
Other useful strategies include allowing children to choose their position and location for an injection or procedure when possible. Small children may be able to sit on the lap of an adult, and older children may prefer sitting up to lying down. Avoid physical restraint unless it is absolutely necessary for safety, the researchers emphasized.
Incorporating CBT-based strategies of breathing and distraction with honesty and respectful disclosure of what is being done and why “not only makes practicing pediatric dermatology easier, but also can improve patient adherence to painful procedures,” they said.
No disclosure information was given.
SOURCE: Armenta AM et al. Pediatr Dermatol. 2019. doi: 10.1111/pde.13739.
according to a report published in Pediatric Dermatology.
For many children, the anticipation of pain and the anxiety about a procedure results in a more painful experience, wrote Andrew M. Armenta of the University of Texas, Galveston, and his colleagues. Preparing children in advance and using cognitive behavioral therapy (CBT) strategies in the moment can help reduce their anxiety.
“CBT is a skill‐based approach that focuses on the present and aims to teach efficient ways of identifying distorted thinking, modifying beliefs, and changing behaviors for a more favorable outcome of real‐life situations,” they wrote.
First, Dr. Armenta and his associates advised, be honest with children about what to expect from a procedure. Evidence does not support phrases such as, “It won’t hurt,” or “It will be over soon,” to reduce anxiety.
Timing the disclosure of a procedure and creating the appropriate setting also can help reduce anxiety. For very young children, short notice of a procedure is often best, with the promise of a small reward or outing afterward. Older children may want some advance notice so they can feel prepared, but their specific concerns should be addressed.
CBT-based techniques include deep breathing and positive coping statements such as “I can do this” for older children, or encouraging them to talk about a family pet or listen to music. Younger children may be distracted with pinwheels, rattles, or songs. “Additionally, in recent years, virtual reality headsets have even proved to be effective distractors, resulting in an overall reduction in both pain and fear,” Dr. Armenta and his associates noted.
Other useful strategies include allowing children to choose their position and location for an injection or procedure when possible. Small children may be able to sit on the lap of an adult, and older children may prefer sitting up to lying down. Avoid physical restraint unless it is absolutely necessary for safety, the researchers emphasized.
Incorporating CBT-based strategies of breathing and distraction with honesty and respectful disclosure of what is being done and why “not only makes practicing pediatric dermatology easier, but also can improve patient adherence to painful procedures,” they said.
No disclosure information was given.
SOURCE: Armenta AM et al. Pediatr Dermatol. 2019. doi: 10.1111/pde.13739.
FROM PEDIATRIC DERMATOLOGY

Nodules, tumors, and hyperpigmented patches
A 36-year-old African-American man presented to our clinic for evaluation of a chronic “rash” on his trunk, arms, and legs that had been present for 3 years. Empiric therapy for tinea corporis, nummular eczema, and confluent and reticulated papillomatosis had failed to improve his lesions.
Physical examination revealed a widespread skin eruption consisting of well-demarcated, hyperpigmented, scaly patches and plaques distributed throughout the patient’s trunk and extremities. Initial biopsies of the skin lesions (performed at an outside institution prior to current presentation) were nondiagnostic.
Over the next several months, the patient developed numerous cutaneous nodules and tumors within the background of his persistent patches and plaques (FIGURE). These nodules and tumors intermittently disappeared and recurred, seemingly independent of therapies including psoralen and ultraviolet A phototherapy (PUVA), interferon, and methotrexate.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: CD30+ transformed mycosis fungoides
Repeat biopsies ultimately confirmed a diagnosis of mycosis fungoides (MF), a primary cutaneous non-Hodgkin lymphoma of T-cell origin.
Lymphomas occurring in the skin without evidence of disease in other areas of the body at the time of diagnosis are referred to as primary cutaneous lymphomas. They are broadly divided by their cell of origin into cutaneous T-cell and cutaneous B-cell lymphomas. Differentiating primary cutaneous lymphoma from systemic lymphomas with secondary skin involvement is an important distinction, as primary cutaneous lymphomas have different clinical courses and prognoses and require different treatments.1
MF is the most common type of cutaneous T-cell lymphoma (CTCL), yet it remains an uncommon diagnosis as cutaneous lymphomas comprise only 3.9% of all non-Hodgkin lymphomas.2 The CD30+ transformed variant identified in this patient is a rare subtype of MF that is associated with increased morbidity and mortality.3 Other types of CTCL include primary cutaneous anaplastic large cell lymphoma, lymphomatoid papulosis, and adult T-cell leukemia/lymphoma, all of which can resemble MF clinically and are diagnosed by histopathology.
A nonspecific presentation
Patients with MF typically present with a complaint of chronic, slowly progressive skin lesions of various sizes and morphologies including scaly patches, plaques, and tumors, often in skin distributions that are not directly exposed to the sun (often referred to as a “bathing suit” distribution).4 Pruritus typically accompanies these skin lesions, and alopecia and poikiloderma are common findings. Patients with more advanced cases may present with generalized erythroderma. This finding should raise concern for progression to Sézary syndrome, a more aggressive type of CTCL that manifests with leukemic involvement of malignant T-cells matching the T-cell clones found in the skin.2
Alopecia is a common manifestation of MF that can be clinically indistinguishable from alopecia areata.5 While isolated alopecia is unlikely to be a consequence of MF, alopecia in the setting of widespread patches and plaques should raise suspicion for a diagnosis of MF.
Continue to: Diagnostic dilemma
Diagnostic dilemma: Condition mimics benign disorders
This case illustrates the diagnostic dilemma posed by MF due to its propensity to mimic more benign skin disorders, both clinically and histologically.2 One literature review of MF cases in which the diagnosis was not suspected clinically but was eventually confirmed histopathologically found that 25 unique dermatoses can be mimicked clinically by MF.6 The more common conditions that MF can be mistaken for are discussed below. In light of the diagnostic challenge posed by this multitude of morphologic presentations, referral to Dermatology for histopathologic evaluation is a reasonable next step when empiric treatment fails to improve symptoms of these common skin disorders.
Chronic atopic dermatitis (eczema) is characterized by dry skin and severe pruritis, usually associated with lichenified plaques and a history of atopic disease. The pruritic, scaly plaques typical of MF may be misdiagnosed as eczema; however, a distribution involving the “bathing suit” area is more suggestive of MF. Patients with atopic dermatitis are more likely to display a flexural distribution.2
Tinea corporis typically presents as annular, erythematous, pruritic, scaly patches or plaques that are similar to the scaling lesions of MF. Tinea corporis can be distinguished from MF via potassium hydroxide preparation of skin scrapings from an active lesion, which should demonstrate the segmented hyphae characteristic of this infection. If lesions of this type fail to respond to topical antifungal therapy, a skin biopsy may be warranted for further evaluation.
Nummular eczema, like tinea corporis, presents with round, coin-shaped patches that are highly pruritic and can be difficult to distinguish from the pruritic patches of MF. Unlike MF, however, nummular eczema typically is observed in patients with diffusely dry skin7; the lack of this finding should prompt consideration of alternative diagnoses, including MF.
Psoriasis classically presents with symmetrically distributed, well-demarcated plaques with prominent scaling that are usually asymptomatic, but may be associated with pruritis. Psoriatic plaques may be difficult to distinguish clinically from the plaques of MF, but their distribution may offer clues; psoriasis typically follows an extensor surface distribution, whereas MF is more commonly identified in a “bathing suit” or generalized distribution.4
Continue to: Tx based on disease extent, impact on quality of life
Tx based on disease extent, impact on quality of life
Staging of MF is the primary determinant of treatment and involves evaluation of skin, lymph node, viscera, and blood involvement. Early-stage MF has a favorable prognosis and can be effectively managed with skin-directed treatments. These include topical corticosteroids, topical nitrogen mustard agents, topical retinoids, and phototherapy (PUVA).8 Total skin electron beam therapy has been proven effective in the treatment of refractory and extensive MF, and local radiation therapy also is an effective treatment for isolated cutaneous tumors. Prolonged complete remissions of early-stage MF have been achieved using skin-directed treatments.8
In contrast, advanced MF often is treatment refractory and carries a poor prognosis. Systemic treatments such as interferon therapy, oral retinoids, extracorporeal photopheresis, and chemotherapy are added in patients with advanced disease after skin-directed therapy fails to adequately control tumor burden.
Our patient initially was treated with PUVA as well as 6 million U of interferon alfa-2B 3 times weekly, which he eventually elected to discontinue after 8 months because he wanted to father a child. His disease became more widespread after discontinuing these treatments despite interim therapy with clobetasol ointment .05%. His larger nodules and tumors were intermittently treated with local radiation with good response. Methotrexate 15 mg weekly was initiated following his decline after discontinuing PUVA and interferon treatment. Treatment with methotrexate resulted in an overall decrease in his tumor burden and several months of clinical stability.
Approximately 6 months after initiating methotrexate, his condition notably worsened. He developed generalized erythroderma, systemic symptoms including fever, chills, and unintentional 9.07-kg weight loss, in addition to new findings of palmoplantar keratoderma and nail dystrophy. In light of this systemic progression and concern for developing Sézary syndrome, extracorporeal photopheresis, bexarotene (an oral retinoid) 75 mg twice daily, and interferon alfa-2B 5 million U 3 times weekly were initiated. His condition continued to decline despite these therapies, culminating in a hospital admission for sepsis.
The patient recovered, and his regimen was changed to PUVA 3 times weekly in addition to treatment with intravenous brentuximab 1.8 mg/kg, a monoclonal antibody that targets CD30 receptors. Unfortunately, his condition continued to decline on this treatment regimen, which was subsequently abandoned in favor of chemotherapy with gemcitabine 1980 mg/250 mL normal saline. The patient was improving clinically with each cycle of gemcitabine and the plan was to transition him to extracorporeal photopheresis and bexarotene for maintenance therapy; however, the patient succumbed to his disease and passed away at the age of 37.
CORRESPONDENCE
Jonathan Banta, MD, PSC 103, Box 3613, APO, AE 09603 jonathan.c.banta.mil@mail.mil
1. Whittaker S, Knobler R, Ortiz P, et al. Major achievements of the EORTC Cutaneous Lymphoma Task Force (CLTF). EJC Suppl. 2012;10:46-50.
2. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16.
3. Kadin ME, Hughey LC, Wood GS. Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol. 2014;70:374-376.
4. Pimpinelli N, Olsen EA, Santucci M, et al; International Society for Cutaneous Lymphoma. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
5. Bi MY, Curry JL, Christiano AM, et al. The spectrum of hair loss in patients with mycosis fungoides and Sézary syndrome. J Am Acad Dermatol. 2011;64:53-63.
6. Zackheim HS, McCalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
7. Jiamton S, Tangjaturonrusamee C, Kulthanan K. Clinical features and aggravating factors in nummular eczema in Thais. Asian Pac J Allergy Immunol. 2013;31:36-42.
8. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-223.e17.
A 36-year-old African-American man presented to our clinic for evaluation of a chronic “rash” on his trunk, arms, and legs that had been present for 3 years. Empiric therapy for tinea corporis, nummular eczema, and confluent and reticulated papillomatosis had failed to improve his lesions.
Physical examination revealed a widespread skin eruption consisting of well-demarcated, hyperpigmented, scaly patches and plaques distributed throughout the patient’s trunk and extremities. Initial biopsies of the skin lesions (performed at an outside institution prior to current presentation) were nondiagnostic.
Over the next several months, the patient developed numerous cutaneous nodules and tumors within the background of his persistent patches and plaques (FIGURE). These nodules and tumors intermittently disappeared and recurred, seemingly independent of therapies including psoralen and ultraviolet A phototherapy (PUVA), interferon, and methotrexate.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: CD30+ transformed mycosis fungoides
Repeat biopsies ultimately confirmed a diagnosis of mycosis fungoides (MF), a primary cutaneous non-Hodgkin lymphoma of T-cell origin.
Lymphomas occurring in the skin without evidence of disease in other areas of the body at the time of diagnosis are referred to as primary cutaneous lymphomas. They are broadly divided by their cell of origin into cutaneous T-cell and cutaneous B-cell lymphomas. Differentiating primary cutaneous lymphoma from systemic lymphomas with secondary skin involvement is an important distinction, as primary cutaneous lymphomas have different clinical courses and prognoses and require different treatments.1
MF is the most common type of cutaneous T-cell lymphoma (CTCL), yet it remains an uncommon diagnosis as cutaneous lymphomas comprise only 3.9% of all non-Hodgkin lymphomas.2 The CD30+ transformed variant identified in this patient is a rare subtype of MF that is associated with increased morbidity and mortality.3 Other types of CTCL include primary cutaneous anaplastic large cell lymphoma, lymphomatoid papulosis, and adult T-cell leukemia/lymphoma, all of which can resemble MF clinically and are diagnosed by histopathology.
A nonspecific presentation
Patients with MF typically present with a complaint of chronic, slowly progressive skin lesions of various sizes and morphologies including scaly patches, plaques, and tumors, often in skin distributions that are not directly exposed to the sun (often referred to as a “bathing suit” distribution).4 Pruritus typically accompanies these skin lesions, and alopecia and poikiloderma are common findings. Patients with more advanced cases may present with generalized erythroderma. This finding should raise concern for progression to Sézary syndrome, a more aggressive type of CTCL that manifests with leukemic involvement of malignant T-cells matching the T-cell clones found in the skin.2
Alopecia is a common manifestation of MF that can be clinically indistinguishable from alopecia areata.5 While isolated alopecia is unlikely to be a consequence of MF, alopecia in the setting of widespread patches and plaques should raise suspicion for a diagnosis of MF.
Continue to: Diagnostic dilemma
Diagnostic dilemma: Condition mimics benign disorders
This case illustrates the diagnostic dilemma posed by MF due to its propensity to mimic more benign skin disorders, both clinically and histologically.2 One literature review of MF cases in which the diagnosis was not suspected clinically but was eventually confirmed histopathologically found that 25 unique dermatoses can be mimicked clinically by MF.6 The more common conditions that MF can be mistaken for are discussed below. In light of the diagnostic challenge posed by this multitude of morphologic presentations, referral to Dermatology for histopathologic evaluation is a reasonable next step when empiric treatment fails to improve symptoms of these common skin disorders.
Chronic atopic dermatitis (eczema) is characterized by dry skin and severe pruritis, usually associated with lichenified plaques and a history of atopic disease. The pruritic, scaly plaques typical of MF may be misdiagnosed as eczema; however, a distribution involving the “bathing suit” area is more suggestive of MF. Patients with atopic dermatitis are more likely to display a flexural distribution.2
Tinea corporis typically presents as annular, erythematous, pruritic, scaly patches or plaques that are similar to the scaling lesions of MF. Tinea corporis can be distinguished from MF via potassium hydroxide preparation of skin scrapings from an active lesion, which should demonstrate the segmented hyphae characteristic of this infection. If lesions of this type fail to respond to topical antifungal therapy, a skin biopsy may be warranted for further evaluation.
Nummular eczema, like tinea corporis, presents with round, coin-shaped patches that are highly pruritic and can be difficult to distinguish from the pruritic patches of MF. Unlike MF, however, nummular eczema typically is observed in patients with diffusely dry skin7; the lack of this finding should prompt consideration of alternative diagnoses, including MF.
Psoriasis classically presents with symmetrically distributed, well-demarcated plaques with prominent scaling that are usually asymptomatic, but may be associated with pruritis. Psoriatic plaques may be difficult to distinguish clinically from the plaques of MF, but their distribution may offer clues; psoriasis typically follows an extensor surface distribution, whereas MF is more commonly identified in a “bathing suit” or generalized distribution.4
Continue to: Tx based on disease extent, impact on quality of life
Tx based on disease extent, impact on quality of life
Staging of MF is the primary determinant of treatment and involves evaluation of skin, lymph node, viscera, and blood involvement. Early-stage MF has a favorable prognosis and can be effectively managed with skin-directed treatments. These include topical corticosteroids, topical nitrogen mustard agents, topical retinoids, and phototherapy (PUVA).8 Total skin electron beam therapy has been proven effective in the treatment of refractory and extensive MF, and local radiation therapy also is an effective treatment for isolated cutaneous tumors. Prolonged complete remissions of early-stage MF have been achieved using skin-directed treatments.8
In contrast, advanced MF often is treatment refractory and carries a poor prognosis. Systemic treatments such as interferon therapy, oral retinoids, extracorporeal photopheresis, and chemotherapy are added in patients with advanced disease after skin-directed therapy fails to adequately control tumor burden.
Our patient initially was treated with PUVA as well as 6 million U of interferon alfa-2B 3 times weekly, which he eventually elected to discontinue after 8 months because he wanted to father a child. His disease became more widespread after discontinuing these treatments despite interim therapy with clobetasol ointment .05%. His larger nodules and tumors were intermittently treated with local radiation with good response. Methotrexate 15 mg weekly was initiated following his decline after discontinuing PUVA and interferon treatment. Treatment with methotrexate resulted in an overall decrease in his tumor burden and several months of clinical stability.
Approximately 6 months after initiating methotrexate, his condition notably worsened. He developed generalized erythroderma, systemic symptoms including fever, chills, and unintentional 9.07-kg weight loss, in addition to new findings of palmoplantar keratoderma and nail dystrophy. In light of this systemic progression and concern for developing Sézary syndrome, extracorporeal photopheresis, bexarotene (an oral retinoid) 75 mg twice daily, and interferon alfa-2B 5 million U 3 times weekly were initiated. His condition continued to decline despite these therapies, culminating in a hospital admission for sepsis.
The patient recovered, and his regimen was changed to PUVA 3 times weekly in addition to treatment with intravenous brentuximab 1.8 mg/kg, a monoclonal antibody that targets CD30 receptors. Unfortunately, his condition continued to decline on this treatment regimen, which was subsequently abandoned in favor of chemotherapy with gemcitabine 1980 mg/250 mL normal saline. The patient was improving clinically with each cycle of gemcitabine and the plan was to transition him to extracorporeal photopheresis and bexarotene for maintenance therapy; however, the patient succumbed to his disease and passed away at the age of 37.
CORRESPONDENCE
Jonathan Banta, MD, PSC 103, Box 3613, APO, AE 09603 jonathan.c.banta.mil@mail.mil
A 36-year-old African-American man presented to our clinic for evaluation of a chronic “rash” on his trunk, arms, and legs that had been present for 3 years. Empiric therapy for tinea corporis, nummular eczema, and confluent and reticulated papillomatosis had failed to improve his lesions.
Physical examination revealed a widespread skin eruption consisting of well-demarcated, hyperpigmented, scaly patches and plaques distributed throughout the patient’s trunk and extremities. Initial biopsies of the skin lesions (performed at an outside institution prior to current presentation) were nondiagnostic.
Over the next several months, the patient developed numerous cutaneous nodules and tumors within the background of his persistent patches and plaques (FIGURE). These nodules and tumors intermittently disappeared and recurred, seemingly independent of therapies including psoralen and ultraviolet A phototherapy (PUVA), interferon, and methotrexate.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: CD30+ transformed mycosis fungoides
Repeat biopsies ultimately confirmed a diagnosis of mycosis fungoides (MF), a primary cutaneous non-Hodgkin lymphoma of T-cell origin.
Lymphomas occurring in the skin without evidence of disease in other areas of the body at the time of diagnosis are referred to as primary cutaneous lymphomas. They are broadly divided by their cell of origin into cutaneous T-cell and cutaneous B-cell lymphomas. Differentiating primary cutaneous lymphoma from systemic lymphomas with secondary skin involvement is an important distinction, as primary cutaneous lymphomas have different clinical courses and prognoses and require different treatments.1
MF is the most common type of cutaneous T-cell lymphoma (CTCL), yet it remains an uncommon diagnosis as cutaneous lymphomas comprise only 3.9% of all non-Hodgkin lymphomas.2 The CD30+ transformed variant identified in this patient is a rare subtype of MF that is associated with increased morbidity and mortality.3 Other types of CTCL include primary cutaneous anaplastic large cell lymphoma, lymphomatoid papulosis, and adult T-cell leukemia/lymphoma, all of which can resemble MF clinically and are diagnosed by histopathology.
A nonspecific presentation
Patients with MF typically present with a complaint of chronic, slowly progressive skin lesions of various sizes and morphologies including scaly patches, plaques, and tumors, often in skin distributions that are not directly exposed to the sun (often referred to as a “bathing suit” distribution).4 Pruritus typically accompanies these skin lesions, and alopecia and poikiloderma are common findings. Patients with more advanced cases may present with generalized erythroderma. This finding should raise concern for progression to Sézary syndrome, a more aggressive type of CTCL that manifests with leukemic involvement of malignant T-cells matching the T-cell clones found in the skin.2
Alopecia is a common manifestation of MF that can be clinically indistinguishable from alopecia areata.5 While isolated alopecia is unlikely to be a consequence of MF, alopecia in the setting of widespread patches and plaques should raise suspicion for a diagnosis of MF.
Continue to: Diagnostic dilemma
Diagnostic dilemma: Condition mimics benign disorders
This case illustrates the diagnostic dilemma posed by MF due to its propensity to mimic more benign skin disorders, both clinically and histologically.2 One literature review of MF cases in which the diagnosis was not suspected clinically but was eventually confirmed histopathologically found that 25 unique dermatoses can be mimicked clinically by MF.6 The more common conditions that MF can be mistaken for are discussed below. In light of the diagnostic challenge posed by this multitude of morphologic presentations, referral to Dermatology for histopathologic evaluation is a reasonable next step when empiric treatment fails to improve symptoms of these common skin disorders.
Chronic atopic dermatitis (eczema) is characterized by dry skin and severe pruritis, usually associated with lichenified plaques and a history of atopic disease. The pruritic, scaly plaques typical of MF may be misdiagnosed as eczema; however, a distribution involving the “bathing suit” area is more suggestive of MF. Patients with atopic dermatitis are more likely to display a flexural distribution.2
Tinea corporis typically presents as annular, erythematous, pruritic, scaly patches or plaques that are similar to the scaling lesions of MF. Tinea corporis can be distinguished from MF via potassium hydroxide preparation of skin scrapings from an active lesion, which should demonstrate the segmented hyphae characteristic of this infection. If lesions of this type fail to respond to topical antifungal therapy, a skin biopsy may be warranted for further evaluation.
Nummular eczema, like tinea corporis, presents with round, coin-shaped patches that are highly pruritic and can be difficult to distinguish from the pruritic patches of MF. Unlike MF, however, nummular eczema typically is observed in patients with diffusely dry skin7; the lack of this finding should prompt consideration of alternative diagnoses, including MF.
Psoriasis classically presents with symmetrically distributed, well-demarcated plaques with prominent scaling that are usually asymptomatic, but may be associated with pruritis. Psoriatic plaques may be difficult to distinguish clinically from the plaques of MF, but their distribution may offer clues; psoriasis typically follows an extensor surface distribution, whereas MF is more commonly identified in a “bathing suit” or generalized distribution.4
Continue to: Tx based on disease extent, impact on quality of life
Tx based on disease extent, impact on quality of life
Staging of MF is the primary determinant of treatment and involves evaluation of skin, lymph node, viscera, and blood involvement. Early-stage MF has a favorable prognosis and can be effectively managed with skin-directed treatments. These include topical corticosteroids, topical nitrogen mustard agents, topical retinoids, and phototherapy (PUVA).8 Total skin electron beam therapy has been proven effective in the treatment of refractory and extensive MF, and local radiation therapy also is an effective treatment for isolated cutaneous tumors. Prolonged complete remissions of early-stage MF have been achieved using skin-directed treatments.8
In contrast, advanced MF often is treatment refractory and carries a poor prognosis. Systemic treatments such as interferon therapy, oral retinoids, extracorporeal photopheresis, and chemotherapy are added in patients with advanced disease after skin-directed therapy fails to adequately control tumor burden.
Our patient initially was treated with PUVA as well as 6 million U of interferon alfa-2B 3 times weekly, which he eventually elected to discontinue after 8 months because he wanted to father a child. His disease became more widespread after discontinuing these treatments despite interim therapy with clobetasol ointment .05%. His larger nodules and tumors were intermittently treated with local radiation with good response. Methotrexate 15 mg weekly was initiated following his decline after discontinuing PUVA and interferon treatment. Treatment with methotrexate resulted in an overall decrease in his tumor burden and several months of clinical stability.
Approximately 6 months after initiating methotrexate, his condition notably worsened. He developed generalized erythroderma, systemic symptoms including fever, chills, and unintentional 9.07-kg weight loss, in addition to new findings of palmoplantar keratoderma and nail dystrophy. In light of this systemic progression and concern for developing Sézary syndrome, extracorporeal photopheresis, bexarotene (an oral retinoid) 75 mg twice daily, and interferon alfa-2B 5 million U 3 times weekly were initiated. His condition continued to decline despite these therapies, culminating in a hospital admission for sepsis.
The patient recovered, and his regimen was changed to PUVA 3 times weekly in addition to treatment with intravenous brentuximab 1.8 mg/kg, a monoclonal antibody that targets CD30 receptors. Unfortunately, his condition continued to decline on this treatment regimen, which was subsequently abandoned in favor of chemotherapy with gemcitabine 1980 mg/250 mL normal saline. The patient was improving clinically with each cycle of gemcitabine and the plan was to transition him to extracorporeal photopheresis and bexarotene for maintenance therapy; however, the patient succumbed to his disease and passed away at the age of 37.
CORRESPONDENCE
Jonathan Banta, MD, PSC 103, Box 3613, APO, AE 09603 jonathan.c.banta.mil@mail.mil
1. Whittaker S, Knobler R, Ortiz P, et al. Major achievements of the EORTC Cutaneous Lymphoma Task Force (CLTF). EJC Suppl. 2012;10:46-50.
2. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16.
3. Kadin ME, Hughey LC, Wood GS. Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol. 2014;70:374-376.
4. Pimpinelli N, Olsen EA, Santucci M, et al; International Society for Cutaneous Lymphoma. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
5. Bi MY, Curry JL, Christiano AM, et al. The spectrum of hair loss in patients with mycosis fungoides and Sézary syndrome. J Am Acad Dermatol. 2011;64:53-63.
6. Zackheim HS, McCalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
7. Jiamton S, Tangjaturonrusamee C, Kulthanan K. Clinical features and aggravating factors in nummular eczema in Thais. Asian Pac J Allergy Immunol. 2013;31:36-42.
8. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-223.e17.
1. Whittaker S, Knobler R, Ortiz P, et al. Major achievements of the EORTC Cutaneous Lymphoma Task Force (CLTF). EJC Suppl. 2012;10:46-50.
2. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part I. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16.
3. Kadin ME, Hughey LC, Wood GS. Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol. 2014;70:374-376.
4. Pimpinelli N, Olsen EA, Santucci M, et al; International Society for Cutaneous Lymphoma. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
5. Bi MY, Curry JL, Christiano AM, et al. The spectrum of hair loss in patients with mycosis fungoides and Sézary syndrome. J Am Acad Dermatol. 2011;64:53-63.
6. Zackheim HS, McCalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
7. Jiamton S, Tangjaturonrusamee C, Kulthanan K. Clinical features and aggravating factors in nummular eczema in Thais. Asian Pac J Allergy Immunol. 2013;31:36-42.
8. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e1-223.e17.

Pruritic rash on chest and back
A 26-year-old woman presented to our clinic with pruritic, hyperpigmented, symmetric edematous plaques on her upper flank, chest, and lower back (FIGURE) 3 weeks after starting a strict ketogenic (high fat/low carbohydrate) diet for postpartum weight loss. The patient was an otherwise healthy stay-at-home mother with an unremarkable medical history.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Prurigo pigmentosa
We recognized that this was a case of prurigo pigmentosa based on the characteristic pruritic rash that had developed after the patient started a strict ketogenic diet.
Prurigo pigmentosa is a benign, pruritic rash that most commonly presents with erythematous or hyperpigmented, symmetrically distributed urticarial papules and plaques on the chest and back. Females represent approximately 70% of cases with a predominant age range of 11 to 30.1
While the pathophysiology remains unknown, the rash most often is reported in association with ketogenic diets.1 Despite occurring in only a fraction of patients on the ketogenic diet, the characteristic presentation has led to the alternative name of the “keto rash” in online nutritional forums and blogs.
Although prurigo pigmentosa is relatively uncommon (with an unknown incidence), primary care physicians may begin to encounter the characteristic rash more frequently, given the number of articles over the past 5 years in the primary care and nutritional literature highlighting the diet’s health benefits.2 The ketogenic diet is a high-fat, low-carbohydrate diet with preliminary evidence of improved weight loss, cardiovascular health, and glycemic control suggested by a meta-analysis of 13 randomized controlled trials.3 Additionally, the popular press and general public’s rising interest are likely to increase the number of patients on this diet.
A clinical diagnosis
The diagnosis is made clinically, so the appearance of a symmetric pruritic, hyperpigmented rash on the chest and back should prompt the physician to ask about any recent changes in diet. Laboratory analysis is unnecessary, as a complete blood count, basic metabolic panel, and liver function panel are almost always normal. Urinary ketones are present in only 30% to 50% of patients; there is, however, an absence of blood ketones.
Continue to: Other conditions can mimic prurigo pigmentosa
Other conditions can mimic prurigo pigmentosa
Urticaria presents as individual lesions that often have a pale center. The lesions may occur anywhere on the body and generally last less than 24 hours. History may reveal a trigger including drugs, infection, food, or emotional stress in up to 50% of cases.
Irritant contact dermatitis often is associated with a stinging or burning sensation. Irritant and allergic contact dermatitis may have a geometric, or “outside job,” distribution suggestive of external contact, potentially with plants, alkalis, acids, or solvents.5
Confluent and reticulated papillomatosis is a rare asymptomatic dermatosis of unknown etiology that presents as hyperpigmented papules on the upper trunk, neck, and axillae. Most patients lack associated pruritis which is in contrast to prurigo pigmentosa.6
Pityriasis rosea is a viral exanthem that may be associated with constitutional symptoms and often presents initially with a herald patch progressing to a classic “Christmas tree” distribution with a fine collarette of scale. It often is asymptomatic, although some cases may be pruritic.5
Treatment focuses on dietary modification
Primary treatment includes resumption of a normal diet. This often leads to rapid resolution of pruritis. Residual hyperpigmentation may take months to fade.
Continue to: Pharmaceutical intervention may be necessary
Pharmaceutical intervention may be necessary
If additional treatment is required, minocycline 100 to 200 mg/d has been reported most effective, likely due to its anti-inflammatory properties.1,4 Topical corticosteroids and oral antihistamines provide symptomatic relief in some patients.1,4
Our patient had resolution of the pruritis and urticarial lesions within 2 days of resuming a normal diet; however, residual asymptomatic hyperpigmentation persisted. A retrial of the ketogenic diet initiated a flare of the rash in the same distribution. It rapidly resolved with carbohydrate intake.
CORRESPONDENCE
Daniel Croom, MD, 34520 Bob Wilson Dr, Naval Medical Center San Diego, San Diego, CA 92134; daniel.l.croom.mil@mail.mil
1. Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
2. Abbasi J. Interest in the ketogenic diet grows for weight loss and type 2 diabetes. JAMA. 2018;319:215-217.
3. Bueno NB, de Melo IS, de Oliveira SL, et al. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: a meta-analysis of randomised controlled trials. Br J Nutr. 2013;110:1178-1187.
4. Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2012;26:1149-1153.
5. James WD, Berger T, Elston D. Diseases of the skin appendages. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Saunders Elsevier; 2015:747-788.
6. Shevchenko A, Valdes-Rodriguez R, Hsu S, et al. Prurigo pigmentosa: case series and differentiation from confluent and reticulated papillomatosis. JAAD Case Rep. 2018;4:77-80.
A 26-year-old woman presented to our clinic with pruritic, hyperpigmented, symmetric edematous plaques on her upper flank, chest, and lower back (FIGURE) 3 weeks after starting a strict ketogenic (high fat/low carbohydrate) diet for postpartum weight loss. The patient was an otherwise healthy stay-at-home mother with an unremarkable medical history.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Prurigo pigmentosa
We recognized that this was a case of prurigo pigmentosa based on the characteristic pruritic rash that had developed after the patient started a strict ketogenic diet.
Prurigo pigmentosa is a benign, pruritic rash that most commonly presents with erythematous or hyperpigmented, symmetrically distributed urticarial papules and plaques on the chest and back. Females represent approximately 70% of cases with a predominant age range of 11 to 30.1
While the pathophysiology remains unknown, the rash most often is reported in association with ketogenic diets.1 Despite occurring in only a fraction of patients on the ketogenic diet, the characteristic presentation has led to the alternative name of the “keto rash” in online nutritional forums and blogs.
Although prurigo pigmentosa is relatively uncommon (with an unknown incidence), primary care physicians may begin to encounter the characteristic rash more frequently, given the number of articles over the past 5 years in the primary care and nutritional literature highlighting the diet’s health benefits.2 The ketogenic diet is a high-fat, low-carbohydrate diet with preliminary evidence of improved weight loss, cardiovascular health, and glycemic control suggested by a meta-analysis of 13 randomized controlled trials.3 Additionally, the popular press and general public’s rising interest are likely to increase the number of patients on this diet.
A clinical diagnosis
The diagnosis is made clinically, so the appearance of a symmetric pruritic, hyperpigmented rash on the chest and back should prompt the physician to ask about any recent changes in diet. Laboratory analysis is unnecessary, as a complete blood count, basic metabolic panel, and liver function panel are almost always normal. Urinary ketones are present in only 30% to 50% of patients; there is, however, an absence of blood ketones.
Continue to: Other conditions can mimic prurigo pigmentosa
Other conditions can mimic prurigo pigmentosa
Urticaria presents as individual lesions that often have a pale center. The lesions may occur anywhere on the body and generally last less than 24 hours. History may reveal a trigger including drugs, infection, food, or emotional stress in up to 50% of cases.
Irritant contact dermatitis often is associated with a stinging or burning sensation. Irritant and allergic contact dermatitis may have a geometric, or “outside job,” distribution suggestive of external contact, potentially with plants, alkalis, acids, or solvents.5
Confluent and reticulated papillomatosis is a rare asymptomatic dermatosis of unknown etiology that presents as hyperpigmented papules on the upper trunk, neck, and axillae. Most patients lack associated pruritis which is in contrast to prurigo pigmentosa.6
Pityriasis rosea is a viral exanthem that may be associated with constitutional symptoms and often presents initially with a herald patch progressing to a classic “Christmas tree” distribution with a fine collarette of scale. It often is asymptomatic, although some cases may be pruritic.5
Treatment focuses on dietary modification
Primary treatment includes resumption of a normal diet. This often leads to rapid resolution of pruritis. Residual hyperpigmentation may take months to fade.
Continue to: Pharmaceutical intervention may be necessary
Pharmaceutical intervention may be necessary
If additional treatment is required, minocycline 100 to 200 mg/d has been reported most effective, likely due to its anti-inflammatory properties.1,4 Topical corticosteroids and oral antihistamines provide symptomatic relief in some patients.1,4
Our patient had resolution of the pruritis and urticarial lesions within 2 days of resuming a normal diet; however, residual asymptomatic hyperpigmentation persisted. A retrial of the ketogenic diet initiated a flare of the rash in the same distribution. It rapidly resolved with carbohydrate intake.
CORRESPONDENCE
Daniel Croom, MD, 34520 Bob Wilson Dr, Naval Medical Center San Diego, San Diego, CA 92134; daniel.l.croom.mil@mail.mil
A 26-year-old woman presented to our clinic with pruritic, hyperpigmented, symmetric edematous plaques on her upper flank, chest, and lower back (FIGURE) 3 weeks after starting a strict ketogenic (high fat/low carbohydrate) diet for postpartum weight loss. The patient was an otherwise healthy stay-at-home mother with an unremarkable medical history.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Prurigo pigmentosa
We recognized that this was a case of prurigo pigmentosa based on the characteristic pruritic rash that had developed after the patient started a strict ketogenic diet.
Prurigo pigmentosa is a benign, pruritic rash that most commonly presents with erythematous or hyperpigmented, symmetrically distributed urticarial papules and plaques on the chest and back. Females represent approximately 70% of cases with a predominant age range of 11 to 30.1
While the pathophysiology remains unknown, the rash most often is reported in association with ketogenic diets.1 Despite occurring in only a fraction of patients on the ketogenic diet, the characteristic presentation has led to the alternative name of the “keto rash” in online nutritional forums and blogs.
Although prurigo pigmentosa is relatively uncommon (with an unknown incidence), primary care physicians may begin to encounter the characteristic rash more frequently, given the number of articles over the past 5 years in the primary care and nutritional literature highlighting the diet’s health benefits.2 The ketogenic diet is a high-fat, low-carbohydrate diet with preliminary evidence of improved weight loss, cardiovascular health, and glycemic control suggested by a meta-analysis of 13 randomized controlled trials.3 Additionally, the popular press and general public’s rising interest are likely to increase the number of patients on this diet.
A clinical diagnosis
The diagnosis is made clinically, so the appearance of a symmetric pruritic, hyperpigmented rash on the chest and back should prompt the physician to ask about any recent changes in diet. Laboratory analysis is unnecessary, as a complete blood count, basic metabolic panel, and liver function panel are almost always normal. Urinary ketones are present in only 30% to 50% of patients; there is, however, an absence of blood ketones.
Continue to: Other conditions can mimic prurigo pigmentosa
Other conditions can mimic prurigo pigmentosa
Urticaria presents as individual lesions that often have a pale center. The lesions may occur anywhere on the body and generally last less than 24 hours. History may reveal a trigger including drugs, infection, food, or emotional stress in up to 50% of cases.
Irritant contact dermatitis often is associated with a stinging or burning sensation. Irritant and allergic contact dermatitis may have a geometric, or “outside job,” distribution suggestive of external contact, potentially with plants, alkalis, acids, or solvents.5
Confluent and reticulated papillomatosis is a rare asymptomatic dermatosis of unknown etiology that presents as hyperpigmented papules on the upper trunk, neck, and axillae. Most patients lack associated pruritis which is in contrast to prurigo pigmentosa.6
Pityriasis rosea is a viral exanthem that may be associated with constitutional symptoms and often presents initially with a herald patch progressing to a classic “Christmas tree” distribution with a fine collarette of scale. It often is asymptomatic, although some cases may be pruritic.5
Treatment focuses on dietary modification
Primary treatment includes resumption of a normal diet. This often leads to rapid resolution of pruritis. Residual hyperpigmentation may take months to fade.
Continue to: Pharmaceutical intervention may be necessary
Pharmaceutical intervention may be necessary
If additional treatment is required, minocycline 100 to 200 mg/d has been reported most effective, likely due to its anti-inflammatory properties.1,4 Topical corticosteroids and oral antihistamines provide symptomatic relief in some patients.1,4
Our patient had resolution of the pruritis and urticarial lesions within 2 days of resuming a normal diet; however, residual asymptomatic hyperpigmentation persisted. A retrial of the ketogenic diet initiated a flare of the rash in the same distribution. It rapidly resolved with carbohydrate intake.
CORRESPONDENCE
Daniel Croom, MD, 34520 Bob Wilson Dr, Naval Medical Center San Diego, San Diego, CA 92134; daniel.l.croom.mil@mail.mil
1. Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
2. Abbasi J. Interest in the ketogenic diet grows for weight loss and type 2 diabetes. JAMA. 2018;319:215-217.
3. Bueno NB, de Melo IS, de Oliveira SL, et al. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: a meta-analysis of randomised controlled trials. Br J Nutr. 2013;110:1178-1187.
4. Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2012;26:1149-1153.
5. James WD, Berger T, Elston D. Diseases of the skin appendages. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Saunders Elsevier; 2015:747-788.
6. Shevchenko A, Valdes-Rodriguez R, Hsu S, et al. Prurigo pigmentosa: case series and differentiation from confluent and reticulated papillomatosis. JAAD Case Rep. 2018;4:77-80.
1. Kim JK, Chung WK, Chang SE, et al. Prurigo pigmentosa: clinicopathological study and analysis of 50 cases in Korea. J Dermatol. 2012;39:891-897.
2. Abbasi J. Interest in the ketogenic diet grows for weight loss and type 2 diabetes. JAMA. 2018;319:215-217.
3. Bueno NB, de Melo IS, de Oliveira SL, et al. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: a meta-analysis of randomised controlled trials. Br J Nutr. 2013;110:1178-1187.
4. Oh YJ, Lee MH. Prurigo pigmentosa: a clinicopathologic study of 16 cases. J Eur Acad Dermatol Venereol. 2012;26:1149-1153.
5. James WD, Berger T, Elston D. Diseases of the skin appendages. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Saunders Elsevier; 2015:747-788.
6. Shevchenko A, Valdes-Rodriguez R, Hsu S, et al. Prurigo pigmentosa: case series and differentiation from confluent and reticulated papillomatosis. JAAD Case Rep. 2018;4:77-80.

FDA extends Dupixent indication for 12- to 17-year-olds
The Food and Drug Administration has approved dupilumab for adolescents with moderate to severe atopic dermatitis (AD) that has been inadequately controlled with topical prescription treatments “or when those therapies are not advisable.”

Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, was initially approved in March 2017, for the same indication, becoming the first targeted biologic treatment for AD. The adolescent approval was announced by the manufacturer.
While there are several systemic medications used as second-line therapy for treatment of pediatric AD, dupilumab is the first FDA-approved biologic for treatment of the disease in adolescents aged 12-17 years, Dawn Marie R. Davis, MD, a pediatric dermatologist at the Mayo Clinic, Rochester (MN), and current president of the Society for Pediatric Dermatology, said in an interview.
FDA approval should decrease insurance barriers and the need for prior authorization, thus increasing access to the drug, she noted, adding, “I hope it will offer a successful alternative to other advanced therapies, as the medicine works through a different mechanism of action, compared to the current systemic medications available.”
With the expanded indication to include adolescents, “patients with more moderate to severe disease who aren’t well controlled with a topical therapy are going to get treatment that will change their lives for many years to come,” dupilumab investigator Eric L. Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland, said in an interview. “On the whole, patients are likely being undertreated and suffering from the disease more than they need to be,” said Dr. Simpson, “With the advent of this new therapy and the new data, it’s going to change the risk benefit calculation for providers and for patients.”
Results from a phase 3 clinical trial of dupilumab in adolescents with moderate to severe AD were presented last fall at the European Academy of Dermatology and Venereology Congress in Paris. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo. Dr. Simpson, the first author of this study, presented the results at that meeting.
Dr. Simpson said that he hopes dupilumab approval for adolescents and the clinical trial results will help providers recognize when patients are not in good control of their AD, and which patients qualify for a step-up in therapy when treatments such as topical therapy or prednisone are not effective. “There are so many patients out there who qualify for a step-up in therapy,” he commented. “I hope that provides comfort to both patients and providers, that it’s OK to take the next step, because the results show us that, not only it can improve your skin rash, but it can have dramatic effects on all the downstream effects of the condition.”
These downstream effects include not only quality of life and comorbidities of mental health but also the patient’s emotional state. Hopefully, dupilumab can reduce stigmatization of AD and feelings of embarrassment for adolescents at a time in life when “socialization, education, and activity is so important in creating your kind of identity in yourself and your sense of self-worth,” Dr. Simpson said.
“It is important to remember atopic dermatitis is a disease that impacts not only the skin, but the patient as a whole,” said Dr. Davis. “It is an exciting time to be caring for atopic dermatitis patients with the various new medications coming to market.”
The FDA had granted a priority review for the adolescent indication; previously the FDA had granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications
The dosing for adolescents is weight based; two doses are available, 200 mg and 300 mg, administered subcutaneously, every other week after a loading dose. The updated prescribing information is available at https://www.regeneron.com/sites/default/files/Dupixent_FPI.pdf.Dr. Simpson reports relationships with Sanofi and Regeneron Pharmaceuticals. Dr. Davis reports no relevant financial disclosures.
The Food and Drug Administration has approved dupilumab for adolescents with moderate to severe atopic dermatitis (AD) that has been inadequately controlled with topical prescription treatments “or when those therapies are not advisable.”
Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, was initially approved in March 2017, for the same indication, becoming the first targeted biologic treatment for AD. The adolescent approval was announced by the manufacturer.
While there are several systemic medications used as second-line therapy for treatment of pediatric AD, dupilumab is the first FDA-approved biologic for treatment of the disease in adolescents aged 12-17 years, Dawn Marie R. Davis, MD, a pediatric dermatologist at the Mayo Clinic, Rochester (MN), and current president of the Society for Pediatric Dermatology, said in an interview.
FDA approval should decrease insurance barriers and the need for prior authorization, thus increasing access to the drug, she noted, adding, “I hope it will offer a successful alternative to other advanced therapies, as the medicine works through a different mechanism of action, compared to the current systemic medications available.”
With the expanded indication to include adolescents, “patients with more moderate to severe disease who aren’t well controlled with a topical therapy are going to get treatment that will change their lives for many years to come,” dupilumab investigator Eric L. Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland, said in an interview. “On the whole, patients are likely being undertreated and suffering from the disease more than they need to be,” said Dr. Simpson, “With the advent of this new therapy and the new data, it’s going to change the risk benefit calculation for providers and for patients.”
Results from a phase 3 clinical trial of dupilumab in adolescents with moderate to severe AD were presented last fall at the European Academy of Dermatology and Venereology Congress in Paris. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo. Dr. Simpson, the first author of this study, presented the results at that meeting.
Dr. Simpson said that he hopes dupilumab approval for adolescents and the clinical trial results will help providers recognize when patients are not in good control of their AD, and which patients qualify for a step-up in therapy when treatments such as topical therapy or prednisone are not effective. “There are so many patients out there who qualify for a step-up in therapy,” he commented. “I hope that provides comfort to both patients and providers, that it’s OK to take the next step, because the results show us that, not only it can improve your skin rash, but it can have dramatic effects on all the downstream effects of the condition.”
These downstream effects include not only quality of life and comorbidities of mental health but also the patient’s emotional state. Hopefully, dupilumab can reduce stigmatization of AD and feelings of embarrassment for adolescents at a time in life when “socialization, education, and activity is so important in creating your kind of identity in yourself and your sense of self-worth,” Dr. Simpson said.
“It is important to remember atopic dermatitis is a disease that impacts not only the skin, but the patient as a whole,” said Dr. Davis. “It is an exciting time to be caring for atopic dermatitis patients with the various new medications coming to market.”
The FDA had granted a priority review for the adolescent indication; previously the FDA had granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications
The dosing for adolescents is weight based; two doses are available, 200 mg and 300 mg, administered subcutaneously, every other week after a loading dose. The updated prescribing information is available at https://www.regeneron.com/sites/default/files/Dupixent_FPI.pdf.Dr. Simpson reports relationships with Sanofi and Regeneron Pharmaceuticals. Dr. Davis reports no relevant financial disclosures.
The Food and Drug Administration has approved dupilumab for adolescents with moderate to severe atopic dermatitis (AD) that has been inadequately controlled with topical prescription treatments “or when those therapies are not advisable.”
Dupilumab (Dupixent), which inhibits interleukin-4 and interleukin-13 signaling, was initially approved in March 2017, for the same indication, becoming the first targeted biologic treatment for AD. The adolescent approval was announced by the manufacturer.
While there are several systemic medications used as second-line therapy for treatment of pediatric AD, dupilumab is the first FDA-approved biologic for treatment of the disease in adolescents aged 12-17 years, Dawn Marie R. Davis, MD, a pediatric dermatologist at the Mayo Clinic, Rochester (MN), and current president of the Society for Pediatric Dermatology, said in an interview.
FDA approval should decrease insurance barriers and the need for prior authorization, thus increasing access to the drug, she noted, adding, “I hope it will offer a successful alternative to other advanced therapies, as the medicine works through a different mechanism of action, compared to the current systemic medications available.”
With the expanded indication to include adolescents, “patients with more moderate to severe disease who aren’t well controlled with a topical therapy are going to get treatment that will change their lives for many years to come,” dupilumab investigator Eric L. Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland, said in an interview. “On the whole, patients are likely being undertreated and suffering from the disease more than they need to be,” said Dr. Simpson, “With the advent of this new therapy and the new data, it’s going to change the risk benefit calculation for providers and for patients.”
Results from a phase 3 clinical trial of dupilumab in adolescents with moderate to severe AD were presented last fall at the European Academy of Dermatology and Venereology Congress in Paris. In that study, the proportion of patients who achieved a 75% or greater improvement in the Eczema Area and Severity Index at 16 weeks was 38.1% with monthly dupilumab, 41.5% with dupilumab every 2 weeks, and 8.2% with placebo. Dr. Simpson, the first author of this study, presented the results at that meeting.
Dr. Simpson said that he hopes dupilumab approval for adolescents and the clinical trial results will help providers recognize when patients are not in good control of their AD, and which patients qualify for a step-up in therapy when treatments such as topical therapy or prednisone are not effective. “There are so many patients out there who qualify for a step-up in therapy,” he commented. “I hope that provides comfort to both patients and providers, that it’s OK to take the next step, because the results show us that, not only it can improve your skin rash, but it can have dramatic effects on all the downstream effects of the condition.”
These downstream effects include not only quality of life and comorbidities of mental health but also the patient’s emotional state. Hopefully, dupilumab can reduce stigmatization of AD and feelings of embarrassment for adolescents at a time in life when “socialization, education, and activity is so important in creating your kind of identity in yourself and your sense of self-worth,” Dr. Simpson said.
“It is important to remember atopic dermatitis is a disease that impacts not only the skin, but the patient as a whole,” said Dr. Davis. “It is an exciting time to be caring for atopic dermatitis patients with the various new medications coming to market.”
The FDA had granted a priority review for the adolescent indication; previously the FDA had granted Breakthrough Therapy designation for dupilumab in 2016 for the treatment of moderate to severe AD in adolescents and severe AD in children aged 6 months to 11 years who are insufficiently controlled with topical medications
The dosing for adolescents is weight based; two doses are available, 200 mg and 300 mg, administered subcutaneously, every other week after a loading dose. The updated prescribing information is available at https://www.regeneron.com/sites/default/files/Dupixent_FPI.pdf.Dr. Simpson reports relationships with Sanofi and Regeneron Pharmaceuticals. Dr. Davis reports no relevant financial disclosures.
Abdominal Wall Schwannoma
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess. 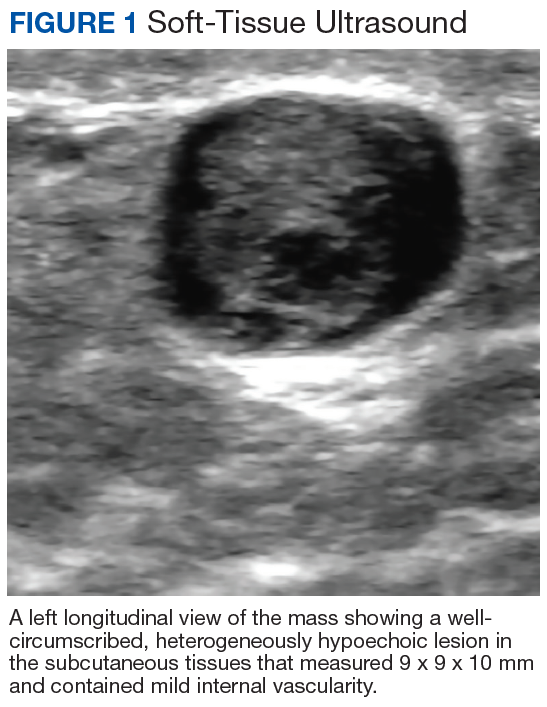
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1). 
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve. 
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12 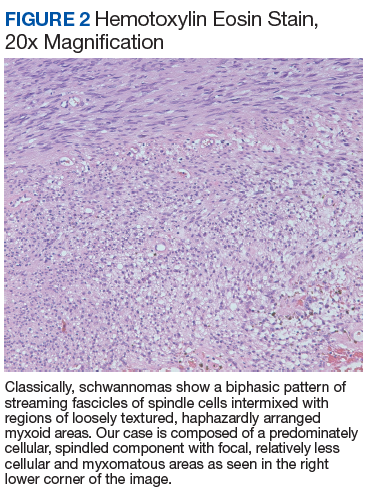
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess.
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1).
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve.
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
Schwannomas are benign tumors exclusively composed of Schwann cells that arise from the peripheral nerve sheath; these tumors theoretically can present anywhere in the body where nerves reside. They tend to occur in the head and neck region (classically an acoustic neuroma) but also occur in other locations, including the retroperitoneal space and the extremities, particularly flexural surfaces. Patients with cutaneous schwannomas are most likely to present to their primary care provider’s office reporting skin findings or localized pain, and providers should be aware of schwannomas on the differential for painful nodular growths.
Case Presentation
A 70-year-old man with type 2 diabetes mellitus presented to the primary care clinic for intermittent, sharp, localized left lower quadrant abdominal wall pain that was gradually progressive over the previous few months. The patient noticed the development of a small nodule 7 to 8 months prior to the visit, at which time the pain was less frequent and less severe. He reported no postprandial association of the pain, nausea, vomiting, diarrhea, constipation, or other gastrointestinal symptoms.
Ten months prior to the presentation, he was involved in a low-impact motor vehicle collision as a pedestrian in which he fell face-first onto the hood of an oncoming car. At that time, he did not note any abdominal trauma or pain. Evaluation at a local emergency department did not reveal any major injuries. In the interim, he had self-administered insulin in his abdominal region, as he had without incident for the previous 2 years. He reported that he was not injecting near the site of the nodule since it had formed. He could not recall whether the location was a previous insulin administration site.
On examination, the patient’s vital signs were normal as were the cardiac and respiratory examinations. An abdominal exam revealed normal bowel sounds and no overlying skin changes or discoloration. Palpation revealed a 1.5 x 1 cm rubbery-to-firm, well-circumscribed subcutaneous nodule along his mid-left abdomen, about 7 cm lateral to the umbilicus. The nodule was sensitive to both light touch and deep pressure. It was firmer than expected for an abdominal wall lipoma. There was no central puncta or pore to suggest an epidermal inclusion cyst. There was no surrounding erythema or induration to suggest an abscess.
The patient was referred for surgery and underwent excisional biopsy of the mass. Pathology revealed a well-circumscribed vascular/spindle-cell lesion consistent with a schwannoma. His postoperative course was uncomplicated. At 4-week follow-up the incision had healed completely and the patient was pain free.
Discussion
Soft-tissue nodules are common—about two-thirds of soft-tissue tumors are classified into 7 diagnostic categories: lipoma and lipoma variants (16%), fibrous histiocytoma (13%), nodular fasciitis (11%), hemangioma (8%), fibromatosis (7%), neurofibroma (5%), and schwannoma (5%).1 Peripheral nerve tumors (schwannomas, neurofibromas) can be associated with pain or paresthesias, and less commonly, neurologic deficits, such as motor weakness. Peripheral nerve tumors have several classifications, such as nonneoplastic vs neoplastic, benign vs malignant, and sheath vs nonsheath origins. Schwannomas are considered part of the neoplastic subset due to their growth; otherwise, they are benign with a sheath origin. In contrast to neurofibromas, benign schwannomas have a slower rate of progression, lower association with pain, and fewer neurologic symptoms.2
The neural sheath is made up of 3 types of cells: the fibroblast, the Schwann cell, and the perineural cell, which lacks a basement membrane. It is the Schwann cell that can give rise to the 3 main types of cutaneous nerve tumors: neuromas, neurofibromas, and schwannomas.3 A nerve that is both entering and exiting a mass is a classic presentation for a peripheral nerve sheath tumor. If the nerve is eccentric to the lesion, then it is consistent with a schwannoma (not a neurofibroma).4 Schwannomas are made exclusively of Schwann cells that arise from the nerve sheath, whereas neurofibromas are made up of all the different cell types that constitute a nerve. Bilateral vestibular schwannomas (acoustic neuromas) are virtually pathognomonic of neurofibromatosis 2 (NF-2), which can manifest as hearing loss, tinnitus, and equilibrium problems. In contrast, neurofibromatosis 1 (NF-1) is more common, characterized by multiple café au lait spots, freckling in the axillary and groin regions, increased risk of cancers overall, and development of pedunculated skin growths, brain, or organ-based neurofibromas.
Diagnosis
A workup generally includes a thorough history and examination as well as imaging. In cases of superficial subcutaneous lesions, an ultrasound is often the imaging modality of choice. However, magnetic resonance imaging (MRI) and computed tomography (CT) scans are frequently used for more deep-seated lesions. There can be significant differences between malignant and benign neural lesions on MRI and CT in terms of contrast-uptake and heterogeneity of tissue, but the visual features are not consistent. Best estimates for MRI suggest 61% sensitivity and 90% specificity for the diagnosis of high-grade malignant peripheral nerve sheath tumors based on imaging alone.5
Definitive diagnosis requires surgical excision. Fine-needle aspiration can be used to diagnose subcutaneous nodules, but there is a possibility that degenerative changes and nuclear atypia seen on a smaller sample may be confused with a more aggressive sarcoma. For example, long-standing schwannomas are often called ancient, meaning that they break down over time, and the atypia they display is a regressive phenomenon.6 Therefore, a small or limited tissue sampling may not be representative of the entire lesion.7 As such, patients will likely need referral for surgical removal to determine the exact nature of the growth.
Although schwannomas are uncommon overall, the highest incidence is in the fourth decade of life with a slight predominance in females. They are often incidentally found as a palpable mass but can be symptomatic with paresthesias, pain, or neurologic changes—particularly when identified in the retroperitoneum or along joints. Schwannomas are most commonly found in the retroperitoneum (32%), mediastinum (23%), head and neck (18%), and extremities (16%).8 The majority of cases (about 90%) are sporadic; whereas 2% are related to NF-2.9 The abdominal wall schwannoma is rare. Our review of English-language literature in PubMed and EMBASE found only 5 other case reports (Table 1).
On physical examination, superficial lesions are freely movable except for a single point of attachment, which is generally along the long axis of the nerve.
Pathology
On gross pathology examination, schwannomas have a well-circumscribed smooth external surface. On microscopy, schwannomas are truly encapsulated, uninodular, spindle-cell proliferations arranged in a streaming pattern within a background of thick, hyalinized blood vessels. Classic schwannomas typically exhibit a biphasic pattern of alternating areas of high and low cellularity and are named for Swedish neurologist Nils Antoni. The more cellular regions are referred to as Antoni A areas and consist of streaming fascicles of compact spindle cells that often palisade around acellular eosinophilic areas of fibrillary processes known as Verocay bodies.
In contrast, the lower cellularity regions (Antoni B areas) consist of multipolar, loosely textured cells with abundant cytoplasm, haphazardly arranged processes, and an overall myxoid appearance.11 Schwannomas are known to have widely variable proportions of Antoni A and Antoni B areas; in this case, the excised specimen was noted to have predominately Antoni A areas without well-defined Verocay bodies and only scattered foci showing some suggestion of the hypocellular Antoni B architecture (Figure 2).9,12
Immunohistochemical stains for S100 and SOX10 (used to identify cells derived from a neural crest lineage) were strongly positive, which is characteristic of schwannomas.13 Although there have only been rare reports of extracranial schwannomas undergoing malignant transformation, it is critical to rule out the possibility of a de novo malignant peripheral nerve sheath tumor (MPNST).13 In general, MPNSTs tend to be more cellular, have brisk mitotic activity, areas of necrosis, hyperchromatic nuclei, and conspicuous pleomorphism. Mitotic figures, which can be concerning for malignant potential if present in high number, were noted occasionally in our patient; however, occasional mitosis may be seen in classic schwannomas. Clinically, MPNSTs have a poor prognosis. Based on case reports, disease-specific survival at 10 years is 31.6% for localized disease and only 7.5% for metastatic disease.14 In this case, there was no evidence of any of the high-grade features of a malignant peripheral nerve sheath tumor, thus supporting the diagnosis of schwannoma (neurilemmoma).
Treatment
Schwannomas are exclusively treated by excision. Prognosis is good with low recurrence rates. It is unknown what the recurrence rates are for completely resected abdominal wall schwannomas since there are so few reports in the literature. For other well-known entities, such as vestibular schwannoma (acoustic neuromas), the recurrence rates are generally 2% to 3%.15 Transformation of schwannomas into MPNSTs are so unusual that they are only described in single case reports.
Conclusion
Soft-tissue masses are a common complaint. Most are benign and do not require excision unless it interferes with the quality of life of the patient or if the diagnosis is uncertain. It is important to be aware of schwannomas in the differential diagnosis of soft-tissue masses. Diagnosis may be achieved through the combination of imaging and biopsy, but the definitive diagnosis is made on complete excision of the mass.
Acknowledgments
Contributors: Michael Lewis, MD, Department of Pathology, VA Greater Los Angeles Healthcare System. Written permission also was obtained from the patient.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
1. Kransdorf MJ. Benign soft-tissue tumors in a large referral population: distribution of specific diagnoses by age, sex, and location. AJR Am J Roentgenol. 1995;164(2):395-402.
2. Valeyrie-Allanore L, Ismaili N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumors: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153(1):79-82.
3. Patterson JW. Neural and neuroendocrine tumors. In: Weedon’s Skin Pathology. 4th ed. Elsevier; 2016:1042-1049.
4. Balzarotti R, Rondelli F, Barizzi J, Cartolari R. Symptomatic schwannoma of the abdominal wall: a case report and review of the literature. Oncol Lett. 2015;9(3):1095-1098.
5. Wasa J, Nishida Y, Tsukushi S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568-1574.
6. Dodd LG, Marom EM, Dash RC, Matthews MR, McLendon RE. Fine-needle aspiration cytology of “ancient” schwannoma. Diagn Cytopathol. 1999;20(5):307-311.
7. Powers CN, Berardo MD, Frable WJ. Fine-needle aspiration biopsy: pitfalls in the diagnosis of spindle-cell lesions. Diagn Cytopathol. 1994;10(3):232-240; discussion 241.
8. White W, Shiu MH, Rosenblum MK, Erlandson RA, Woodruff JM. Cellular schwannoma: a clinicopathologic study of 57 patients and 58 tumors. Cancer. 1990;66(6):1266-1275.
9. Goldblum JR, Weiss SW, Folpe AL. Benign tumors of peripheral nerves. In: Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Elsevier; 2014:813-828.
10. Naversen DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Deramatol. 1993;28(2, pt 2):298-300.
11. Burger PC, Scheithauer BW. Diagnostic Pathology: Neuropathology. 1st ed. Salt Lake City, UT: Amirsys; 2012.
12. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, eds. World Health Organization Histological Classification of Tumours of the Central Nervous System. Vol. 1. Paris, France: International Agency for Research on Cancer; 2016.
13. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994;18(9)82-895.
14. Zou C, Smith KD, Liu J, et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014-1022.
15. Ahmad RA, Sivalingam S, Topsakal V, Russo A, Taibah A, Sanna M. Rate of recurrent vestibular schwannoma after total removal via different surgical approaches. Ann Otol Rhinol Laryngol. 2012;121(3):156-161.
16. Bhatia RK, Banerjea A, Ram M, Lovett BE. Benign ancient schwannoma of the abdominal wall: an unwanted birthday present. BMC Surg. 2010;10:1-5.
17. Mishra A, Hamadto M, Azzabi M, Elfagieh M. Abdominal wall schwannoma: case report and review of the literature. Case Rep Radiol. 2013;2013:456863.
18. Liu Y, Chen X, Wang T, Wang Z. Imaging observations of a schwannoma of low malignant potential in the anterior abdominal wall: a case report. Oncol Lett. 2014;8(3):1159-1162.
19. Ginesu GC, Puledda M, Feo CF et al. Abdominal wall schwannoma. J Gastrointest Surg. 2016;20(10):1781-1783.
Sudden-onset rash on the trunk and limbs • morbid obesity • family history of diabetes mellitus • Dx?
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.

THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION

Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; cks048@ha.org.hk
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.
THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION
Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; cks048@ha.org.hk
THE CASE
A 37-year-old man presented with a sudden-onset, nonpruritic, nonpainful, papular rash of 1 month’s duration on his trunk and both arms and legs. Two weeks prior to the current presentation, he consulted a general practitioner, who treated the rash with a course of unknown oral antibiotics; the patient showed no improvement. He recalled that on a few occasions, he used his fingers to express a creamy discharge from some of the lesions. This temporarily reduced the size of those papules.
His medical history was unremarkable except for morbid obesity. He did not drink alcohol regularly and was not taking any medications prior to the onset of the rash. He had no family history of hyperlipidemia, but his mother had a history of diabetes mellitus.
Physical examination showed numerous discrete erythematous papules with a creamy center on his trunk and his arms and legs. The lesions were more numerous on the extensor surfaces of the arms and legs. Some of the papules coalesced to form small plaques (FIGURE). There was no scaling, and the lesions were firm in texture. The patient’s face was spared, and there was no mucosal involvement. The patient was otherwise systemically well.
THE DIAGNOSIS
Based on the morphology, distribution, and abrupt onset of the diffuse nonpruritic papules in this morbidly obese (but otherwise systemically well) middle-aged man, a clinical diagnosis of eruptive xanthoma was suspected. Subsequent blood testing revealed an elevated serum triglyceride level of 47.8 mmol/L (reference range, <1.7 mmol/L), elevated serum total cholesterol of 7.1 mmol/L (reference range, <6.2 mmol/L), and low serum high-density lipoprotein cholesterol of 0.7 mmol/L (reference range, >1 mmol/L in men). He also had an elevated fasting serum glucose level of 12.9 mmol/L (reference range, 3.9–5.6 mmol/L) and an elevated hemoglobin A1c (glycated hemoglobin) level of 10.9%.
Subsequent thyroid, liver, and renal function tests were normal, but the patient had heavy proteinuria, with an elevated urine albumin-to-creatinine ratio of 355.6 mg/mmol (reference range, ≤2.5 mg/mmol). The patient was referred to a dermatologist, who confirmed the clinical diagnosis without the need for a skin biopsy.
DISCUSSION
Eruptive xanthoma is characterized by an abrupt onset of crops of multiple yellowish to brownish papules that can coalesce into small plaques. The lesions can be generalized, but tend to be more densely distributed on the extensor surfaces of the arms and legs, buttocks, and thighs.5 Eruptive xanthoma often is associated with hypertriglyceridemia, which can be primary—as a result of a genetic defect caused by familial hypertriglyceridemia—or secondary, associated with poorly controlled diabetes mellitus, morbid obesity, excessive alcohol consumption, nephrotic syndrome, hypothyroidism, primary biliary cholangitis, and drugs like estrogen replacement therapies, corticosteroids, and isotretinoin.6 Pruritus and tenderness may or may not be present, and the Köbner phenomenon may occur.7
Continue to: The differential diagnosis
The differential diagnosis for eruptive xanthoma includes xanthoma disseminatum, non–Langerhans cell histiocytoses (eg, generalized eruptive histiocytosis), and cutaneous mastocytosis.1
Xanthoma disseminatum is an extremely rare, but benign, disorder of non–Langerhans cell origin. The average age of onset is older than 40 years. The rash consists of multiple red-yellow papules and nodules that most commonly present in flexural areas. Forty percent to 60% of patients have mucosal involvement, and rarely the central nervous system is involved.8
Generalized eruptive histiocytosis is another rare non–Langerhans cell histiocytosis that occurs mainly in adults and is characterized by widespread, symmetric, red-brown papules on the trunk, arms, and legs, and rarely the mucous membranes.9
Cutaneous mastocytosis, especially xanthelasmoid mastocytosis, consists of multiple pruritic, yellowish, papular or nodular lesions that may mimic eruptive xanthoma. It occurs mainly in children and rarely in adults.10
Confirming the diagnosis, initiating treatment
The diagnosis of eruptive xanthoma can be confirmed by skin biopsy if other differential diagnoses cannot be ruled out or the lesions do not resolve with treatment. Skin biopsy will reveal lipid-laden macrophages (known as foam cells) deposited in the dermis.7
Continue to: Treatment of eruptive xanthoma
Treatment of eruptive xanthoma involves management of the underlying causes of the condition. In most cases, dietary control, intensive triglyceride-lowering therapies, and treatment of other secondary causes of hypertriglyceridemia result in complete resolution of the lesions within several weeks.5
Our patient’s outcome
Our patient’s sudden-onset rash alerted us to the presence of type 2 diabetes mellitus, hypertriglyceridemia, and heavy proteinuria, which he was not aware of previously. We counselled him about stringent low-sugar, low-lipid diet control and exercise, and we started him on metformin and gemfibrozil. He was referred to an internal medicine specialist for further assessment and management of his severe hypertriglyceridemia and heavy proteinuria.
The rash started to wane 1 month after the patient started the metformin and gemfibrozil, and his drug regimen was changed to combination therapy with metformin/glimepiride and fenofibrate/simvastatin 6 weeks later when he was seen in the medical specialty clinic. Fundus photography performed 1 month after starting oral antidiabetic therapy showed no diabetic retinopathy or lipemia retinalis.
After 3 months of treatment, his serum triglycerides and hemoglobin A1c levels dropped to 3.8 mmol/L and 8.7%, respectively. The rash also resolved considerably, with only residual papules on the abdomen. This rapid clinical response to treatment of the underlying hypertriglyceridemia and diabetes further supported the clinical diagnosis of eruptive xanthoma.
THE TAKEAWAY
Eruptive xanthoma is relatively rare, but it is important for family physicians to recognize this clinical presentation as a potential indicator of severe hypertriglyceridemia. Recognizing hypertriglyceridemia early is important, as it can be associated with an increased risk for acute pancreatitis. Moreover, eruptive xanthoma might be the sole presenting symptom of underlying diabetes mellitus or familial hyperlipidemia, both of which can lead to a significant increase in cardiovascular risk if uncontrolled.
CORRESPONDENCE
Chan Kam Sum, MBChB, FRACGP, Tseung Kwan O Jockey Club General Out-patient Clinic, 99 Po Lam Road North, G/F, Tseung Kwan O, Kowloon, Hong Kong; cks048@ha.org.hk
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.
1. Tang WK. Eruptive xanthoma. [case reports]. Hong Kong Dermatol Venereol Bull. 2001;9:172-175.
2. Frew J, Murrell D, Haber R. Fifty shades of yellow: a review of the xanthodermatoses. Int J Dermatol. 2015;54:1109-1123.
3. Zak A, Zeman M, Slaby A, et al. Xanthomas: clinical and pathophysiological relations. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014;158:181-188.
4. Sandhu S, Al-Sarraf A, Taraboanta C, et al. Incidence of pancreatitis, secondary causes, and treatment of patients referred to specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis. 2011;10:157.
5. Holsinger JM, Campbell SM, Witman P. Multiple erythematous-yellow, dome-shaped papules. Am Fam Physician. 2010;82:517.
6. Loeckermann S, Braun-Falco M. Eruptive xanthomas in association with metabolic syndrome. Clin Exp Dermatol. 2010;35:565-566.
7. Merola JF, Mengden SJ, Soldano A, et al. Eruptive xanthomas. Dermatol Online J. 2008;14:10.
8. Park M, Boone B, Devas S. Xanthoma disseminatum: case report and mini-review of the literature. Acta Dermatovenerol Croat. 2014;22:150-154.
9. Attia A, Seleit I, El Badawy N, et al. Photoletter to the editor: generalized eruptive histiocytoma. J Dermatol Case Rep. 2011;5:53-55.
10. Nabavi NS, Nejad MH, Feli S, et al. Adult onset of xanthelasmoid mastocytosis: report of a rare entity. Indian J Dermatol. 2016;61:468.

BTK inhibitor calms pemphigus vulgaris with low-dose steroids
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
WASHINGTON – An investigational molecule that blocks the downstream proinflammatory effects of B cells controlled disease activity and induced clinical remission in patients with pemphigus by 12 weeks.
At the end of a 24-week, open-label trial, a key driver of the sometimes-fatal blistering disease, Deedee Murrell, MD, said at the annual meeting of the American Academy of Dermatology.
The clinical efficacy plus a favorable safety profile supports the further development of the molecule, designed and manufactured by Principia Biopharma in San Francisco. The company is currently recruiting for a pivotal phase 3 trial of PRN1008 in 120 patients with moderate to severe pemphigus vulgaris.
Despite the recent approval of rituximab (Rituxan) for moderate to severe pemphigus, there remains an unmet need for a quick-acting, steroid-sparing, anti-inflammatory treatment, said Dr. Murrell, professor and head of the department of dermatology at the University of New South Wales, Sydney.
“We need something to use instead of high-dose steroids while we are waiting for rituximab to kick in, which can take 3 months,” and rituximab, which depletes B cells, puts patients at risk for infection, she said. “We need something that has rapid onset, is steroid sparing, safe for chronic administration, avoids B-cell depletion, and is convenient.”
Blocking the BTK receptor on B cells puts the brakes on the B-cell mediated inflammatory pathway, preventing activation of monocytes, macrophages, mast cells, basophils, and neutrophils. At the same time, however, it does not deplete the B-cell population, said Dr. Murrell, the lead investigator.
The BELIEVE study comprised 27 patients with mild to severe pemphigus of an average 6 years’ duration. Most (18) had relapsing disease; the remainder had newly diagnosed pemphigus. A majority (16) had severe disease, as measured by a score of 15 or more on the Pemphigus Disease Activity Index (PDAI). Almost all (23) were positive for antidesmoglein antibodies. Only one patient was negative for antibodies.
The mean corticosteroid dose at baseline was 14 mg/day, although that ranged from no steroids to 30 mg/day.
The study consisted of a 12-week treatment phase and a 12-week follow-up phase. During treatment, patients could take no more than 0.5 mg/kg of prednisone daily, although with 400 mg PRN1008 twice a day. They were allowed to undertake rescue immunosuppression if they experienced a disease flare.
The primary endpoint was disease control by day 29 as evidenced by no new lesions. Secondary endpoints were complete remission, minimization of prednisone, quality of life, antibody levels, and clinician measures including the PDAI and the Autoimmune Bullous Skin Disorder Intensity Score.
By the end of week 4, 54% of patients had achieved the primary endpoint. The benefit continued to expand, with 73% reaching that response by the end of week 12. During this period, the mean prednisone dose was 12 mg/day.
Among the 24 patients who completed the study, complete remission occurred in 17% by week 12. However, patients continued to respond through the follow-up period, even after the study medication was stopped. By week 24, 25% of these patients experienced a complete remission. At the point of remission, the mean steroid dose was 8 mg/day. The median duration of remission was 2 months after stopping PRN1008.
The PDAI fell by a median of 70% by week 12 and was maintained at that level by the end of week 24. The median level of antidesmoglein autoantibodies fell by up to 65%. Again, the improvement continued throughout the off-drug follow-up period. In subgroup analyses, PRN1008 was more effective in patients with moderate to severe disease than those with mild disease (80% response vs. 64%). It was equally effective in those with newly diagnosed disease (75% vs. 72%) and regardless of antibody level at baseline.
The adverse event profile was relatively benign. Most side effects were mild and transient, and included upper abdominal pain, headache, and nausea. There were two mild infections and one serious infection, which presented in a patient with a long-standing localized cellulitis that activated and was associated a high fever. It was culture negative and PRN1008 was restarted without issue.
There was also one serious adverse event and one death, both unrelated to the study drug. One patient developed a pancreatic cyst that was discovered on day 29. The patient dropped out of the study to have elective surgery. The death occurred in a patient who developed acute respiratory failure on day 8 of treatment, caused by an undiagnosed congenital pulmonary sequestration. The patient died of a brain embolism shortly after lung surgery.
Dr. Murrell designed the study and was an investigator. She reported a financial relationship with Principia, as well as with numerous other pharmaceutical companies.
SOURCE: Murrell D et al. AAD 2019, Session S034.
REPORTING FROM AAD 2019
Fluorouracil beats other actinic keratosis treatments in head-to-head trial
A head-to-head comparison of four commonly used field-directed treatments for actinic keratosis (AK) found that 5% fluorouracil cream was the most effective at reducing the size of lesions.

In a study published in the March 7 issue of the New England Journal of Medicine, researchers reported the outcomes of a multicenter, single-blind trial in 602 patients with five or more AK lesions in one continuous area on the head measuring 25-100 cm2. Patients were randomized to treatment with either 5% fluorouracil cream, 5% imiquimod cream, methyl aminolevulinate photodynamic therapy (MAL-PDT), or 0.015% ingenol mebutate gel.
Overall, 74.7% of patients who received fluorouracil cream achieved treatment success – defined as at least a 75% reduction in lesion size at 12 months after the end of treatment – compared with 53.9% of patients treated with imiquimod, 37.7% of those treated with MAL-PDT, and 28.9% of those treated with ingenol mebutate. The differences between fluorouracil and the other treatments was significant.
Maud H.E. Jansen, MD, and Janneke P.H.M. Kessels, MD, of the department of dermatology at Maastricht (the Netherlands) University Medical Center and their coauthors pointed out that, while there was plenty of literature about different AK treatments, there were few head-to-head comparisons and many studies were underpowered or had different outcome measures.
Even when the analysis was restricted to patients with grade I or II lesions, fluorouracil remained the most effective treatment, with 75.3% of patients achieving treatment success, compared with 52.6% with imiquimod, 38.7% with MAL-PDT, and 30.2% with ingenol mebutate.
There were not enough patients with more severe grade III lesions to enable a separate analysis of their outcomes; 49 (7.9%) of patients in the study had at least one grade III lesion.
The authors noted that many previous studies had excluded patients with grade III lesions. “Exclusion of patients with grade III lesions was associated with slightly higher rates of success in the fluorouracil, MAL-PDT, and ingenol mebutate groups than the rates in the unrestricted analysis,” they wrote. The inclusion of patients with grade III AK lesions in this trial made it “more representative of patients seen in daily practice,” they added.
Treatment failure – less than 75% clearance of actinic keratosis at 3 months after the final treatment – was seen after one treatment cycle in 14.8% of patients treated with fluorouracil, 37.2% of patients on imiquimod, 34.6% of patients given photodynamic therapy, and 47.8% of patients on ingenol mebutate therapy.
All these patients were offered a second treatment cycle, but those treated with imiquimod, PDT, and ingenol mebutate were less likely to undergo a second treatment.
The authors suggested that the higher proportion of patients in the fluorouracil group who were willing to undergo a second round of therapy suggests they may have experienced less discomfort and inconvenience with the therapy to begin with, compared with those treated with the other regimens.
Full adherence to treatment was more common in the ingenol mebutate (98.7% of patients) and MAL-PDT (96.8%) groups, compared with the fluorouracil (88.7%) and imiquimod (88.2%) groups. However, patients in the fluorouracil group reported greater levels of patient satisfaction and improvements in health-related quality of life than did patients in the other treatment arms of the study.
No serious adverse events were reported with any of the treatments, and no patients stopped treatment because of adverse events. However, reports of moderate or severe crusts were highest among patients treated with imiquimod, and moderate to severe vesicles or bullae were highest among those treated with ingenol mebutate. Severe pain and severe burning sensation were significantly more common among those treated with MAL-PDT.
While the study had some limitations, the results “could affect treatment choices in both dermatology and primary care,” the authors wrote, pointing out how common AKs are in practice, accounting for 5 million dermatology visits in the United States every year. When considering treatment costs, “fluorouracil is also the most attractive option,” they added. “It is expected that a substantial cost reduction could be achieved with more uniformity in care and the choice for effective therapy.”
The study was supported by the Netherlands Organization for Health Research and Development. Five of the 11 authors declared conference costs, advisory board fees, or trial supplies from private industry, including from manufacturers of some of the products in the study. The remaining authors had no disclosures.
SOURCE: Jansen M et al. N Engl J Med. 2019;380:935-46.
A head-to-head comparison of four commonly used field-directed treatments for actinic keratosis (AK) found that 5% fluorouracil cream was the most effective at reducing the size of lesions.
In a study published in the March 7 issue of the New England Journal of Medicine, researchers reported the outcomes of a multicenter, single-blind trial in 602 patients with five or more AK lesions in one continuous area on the head measuring 25-100 cm2. Patients were randomized to treatment with either 5% fluorouracil cream, 5% imiquimod cream, methyl aminolevulinate photodynamic therapy (MAL-PDT), or 0.015% ingenol mebutate gel.
Overall, 74.7% of patients who received fluorouracil cream achieved treatment success – defined as at least a 75% reduction in lesion size at 12 months after the end of treatment – compared with 53.9% of patients treated with imiquimod, 37.7% of those treated with MAL-PDT, and 28.9% of those treated with ingenol mebutate. The differences between fluorouracil and the other treatments was significant.
Maud H.E. Jansen, MD, and Janneke P.H.M. Kessels, MD, of the department of dermatology at Maastricht (the Netherlands) University Medical Center and their coauthors pointed out that, while there was plenty of literature about different AK treatments, there were few head-to-head comparisons and many studies were underpowered or had different outcome measures.
Even when the analysis was restricted to patients with grade I or II lesions, fluorouracil remained the most effective treatment, with 75.3% of patients achieving treatment success, compared with 52.6% with imiquimod, 38.7% with MAL-PDT, and 30.2% with ingenol mebutate.
There were not enough patients with more severe grade III lesions to enable a separate analysis of their outcomes; 49 (7.9%) of patients in the study had at least one grade III lesion.
The authors noted that many previous studies had excluded patients with grade III lesions. “Exclusion of patients with grade III lesions was associated with slightly higher rates of success in the fluorouracil, MAL-PDT, and ingenol mebutate groups than the rates in the unrestricted analysis,” they wrote. The inclusion of patients with grade III AK lesions in this trial made it “more representative of patients seen in daily practice,” they added.
Treatment failure – less than 75% clearance of actinic keratosis at 3 months after the final treatment – was seen after one treatment cycle in 14.8% of patients treated with fluorouracil, 37.2% of patients on imiquimod, 34.6% of patients given photodynamic therapy, and 47.8% of patients on ingenol mebutate therapy.
All these patients were offered a second treatment cycle, but those treated with imiquimod, PDT, and ingenol mebutate were less likely to undergo a second treatment.
The authors suggested that the higher proportion of patients in the fluorouracil group who were willing to undergo a second round of therapy suggests they may have experienced less discomfort and inconvenience with the therapy to begin with, compared with those treated with the other regimens.
Full adherence to treatment was more common in the ingenol mebutate (98.7% of patients) and MAL-PDT (96.8%) groups, compared with the fluorouracil (88.7%) and imiquimod (88.2%) groups. However, patients in the fluorouracil group reported greater levels of patient satisfaction and improvements in health-related quality of life than did patients in the other treatment arms of the study.
No serious adverse events were reported with any of the treatments, and no patients stopped treatment because of adverse events. However, reports of moderate or severe crusts were highest among patients treated with imiquimod, and moderate to severe vesicles or bullae were highest among those treated with ingenol mebutate. Severe pain and severe burning sensation were significantly more common among those treated with MAL-PDT.
While the study had some limitations, the results “could affect treatment choices in both dermatology and primary care,” the authors wrote, pointing out how common AKs are in practice, accounting for 5 million dermatology visits in the United States every year. When considering treatment costs, “fluorouracil is also the most attractive option,” they added. “It is expected that a substantial cost reduction could be achieved with more uniformity in care and the choice for effective therapy.”
The study was supported by the Netherlands Organization for Health Research and Development. Five of the 11 authors declared conference costs, advisory board fees, or trial supplies from private industry, including from manufacturers of some of the products in the study. The remaining authors had no disclosures.
SOURCE: Jansen M et al. N Engl J Med. 2019;380:935-46.
A head-to-head comparison of four commonly used field-directed treatments for actinic keratosis (AK) found that 5% fluorouracil cream was the most effective at reducing the size of lesions.
In a study published in the March 7 issue of the New England Journal of Medicine, researchers reported the outcomes of a multicenter, single-blind trial in 602 patients with five or more AK lesions in one continuous area on the head measuring 25-100 cm2. Patients were randomized to treatment with either 5% fluorouracil cream, 5% imiquimod cream, methyl aminolevulinate photodynamic therapy (MAL-PDT), or 0.015% ingenol mebutate gel.
Overall, 74.7% of patients who received fluorouracil cream achieved treatment success – defined as at least a 75% reduction in lesion size at 12 months after the end of treatment – compared with 53.9% of patients treated with imiquimod, 37.7% of those treated with MAL-PDT, and 28.9% of those treated with ingenol mebutate. The differences between fluorouracil and the other treatments was significant.
Maud H.E. Jansen, MD, and Janneke P.H.M. Kessels, MD, of the department of dermatology at Maastricht (the Netherlands) University Medical Center and their coauthors pointed out that, while there was plenty of literature about different AK treatments, there were few head-to-head comparisons and many studies were underpowered or had different outcome measures.
Even when the analysis was restricted to patients with grade I or II lesions, fluorouracil remained the most effective treatment, with 75.3% of patients achieving treatment success, compared with 52.6% with imiquimod, 38.7% with MAL-PDT, and 30.2% with ingenol mebutate.
There were not enough patients with more severe grade III lesions to enable a separate analysis of their outcomes; 49 (7.9%) of patients in the study had at least one grade III lesion.
The authors noted that many previous studies had excluded patients with grade III lesions. “Exclusion of patients with grade III lesions was associated with slightly higher rates of success in the fluorouracil, MAL-PDT, and ingenol mebutate groups than the rates in the unrestricted analysis,” they wrote. The inclusion of patients with grade III AK lesions in this trial made it “more representative of patients seen in daily practice,” they added.
Treatment failure – less than 75% clearance of actinic keratosis at 3 months after the final treatment – was seen after one treatment cycle in 14.8% of patients treated with fluorouracil, 37.2% of patients on imiquimod, 34.6% of patients given photodynamic therapy, and 47.8% of patients on ingenol mebutate therapy.
All these patients were offered a second treatment cycle, but those treated with imiquimod, PDT, and ingenol mebutate were less likely to undergo a second treatment.
The authors suggested that the higher proportion of patients in the fluorouracil group who were willing to undergo a second round of therapy suggests they may have experienced less discomfort and inconvenience with the therapy to begin with, compared with those treated with the other regimens.
Full adherence to treatment was more common in the ingenol mebutate (98.7% of patients) and MAL-PDT (96.8%) groups, compared with the fluorouracil (88.7%) and imiquimod (88.2%) groups. However, patients in the fluorouracil group reported greater levels of patient satisfaction and improvements in health-related quality of life than did patients in the other treatment arms of the study.
No serious adverse events were reported with any of the treatments, and no patients stopped treatment because of adverse events. However, reports of moderate or severe crusts were highest among patients treated with imiquimod, and moderate to severe vesicles or bullae were highest among those treated with ingenol mebutate. Severe pain and severe burning sensation were significantly more common among those treated with MAL-PDT.
While the study had some limitations, the results “could affect treatment choices in both dermatology and primary care,” the authors wrote, pointing out how common AKs are in practice, accounting for 5 million dermatology visits in the United States every year. When considering treatment costs, “fluorouracil is also the most attractive option,” they added. “It is expected that a substantial cost reduction could be achieved with more uniformity in care and the choice for effective therapy.”
The study was supported by the Netherlands Organization for Health Research and Development. Five of the 11 authors declared conference costs, advisory board fees, or trial supplies from private industry, including from manufacturers of some of the products in the study. The remaining authors had no disclosures.
SOURCE: Jansen M et al. N Engl J Med. 2019;380:935-46.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE